Introduction
Pancreatic cancer is one of the most difficult types
of cancer and is a major cause of cancer-related deaths worldwide,
because only 20% of patients have resectable-stage cancer at the
time of diagnosis. In addition, pancreatic cancer is largely
resistant to systemic chemotherapy (1,2). In
patients with metastatic pancreatic cancer, the 5-year survival
rate is only 2%. Recently, novel molecular agents targeting
specific biologic pathways that are activated and involved in the
pathogenesis of cancer have been developed to treat various types
of cancer. Since the epidermal growth factor receptor (EGFR) is
overexpressed in many pancreatic cancers, EGFR-targeting therapy
appears promising (3). In a recent
randomized phase III clinical trial, erlotinib, an EGFR tyrosine
kinase inhibitor (EGFR-TKI) in combination with gemcitabine
demonstrated improvement in survival compared to gemcitabine
monotherapy (median overall survival, 6.24 vs. 5.91 months;
P=0.038) (4). Therefore, erlotinib
in combination with gemcitabine has been approved as a major
chemotherapy drug for advanced pancreatic cancer, based on
high-level evidence (5,6). However, the median overall survival
rate for these patients indicates extremely poor prognosis compared
to that of other types of cancers. Thus, more effective therapeutic
strategies are required to improve therapeutic outcome.
Macroautophagy (hereafter, autophagy) is
evolutionally well-conserved in eukaryotes as the major degradation
mechanism whereby cellular proteins and organelle are sequestered
into a double-membrane vesicle (autophagosome) and are delivered to
lysosomes. They are then membrane-fused (autolysosome) and cargos
are degraded for metabolic recycling by lysosomal hydrolytic
enzymes, including protease (7,8). It
has become apparent that many aggressive cancer cells have
upregulated autophagy and are dependent on autophagy for survival
(9). EGFR tyrosine kinase
(EGFR-TK) is overexpressed or somatic mutationally activated in a
broad range of human cancers, including NSCLC and pancreatic
cancers (10). Activated EGFR
suppresses autophagy via activation of mammalian targets of
rapamycin (mTORs), as well as tyrosine phosphorylation of Beclin-1
(11–13). In response to ligand binding, EGFR
becomes a dimer and is activated via tyrosine phosphorylation.
Phosphorylated EGFR subsequently activates intracellular
pro-survival and anti-apoptotic signals. One of the downstream
activated EGFR signals involves the PI3K/AKT/mTOR pathway, made up
of well-established negative regulators for autophagy (14). In response to nutrient and growth
factor availability, mTOR promotes cell growth while suppressing
autophagy. In addition, the activated EGFR binds the autophagy
initiating protein Beclin-1, leading to multisite tyrosine
phosphorylation and enhanced binding to autophagy inhibitors,
including Rubicon (15). Thus,
exposure to EGFR-TKIs such as gefitinib (GEF) and erlotinib (ERL)
induces autophagy via cancellation of the PI3K/AKT/mTOR pathway, as
well as blocking phosphorylation of Beclin-1, which subsequently
leads to dissociation of Rubicon from Beclin-1 to initiating
autophagy (13,15). A recent report confirmed that
inactivated endosomal, but not cell surface EGFR interacts with
LAPTM4B, resulting in a complex formation for recruiting Rubicon
from Beclin-1 (16). As described
above, dissociation of Rubicon from Beclin-1 is essential for
autophagy initiation. Cancer cells with higher EGFR expression
could thus be supported and survive by efficient autophagy
induction under various metabolic stresses, such as low nutrient
and hypoxic conditions due to insufficient vascularization
(15,16). Therefore, if autophagy induction in
response to EGFR-TKI promotes tumor cell survival, combined
treatment with an autophagy inhibitor may promote tumor cell
death.
We and others have reported that macrolide
antibiotics such as clarithromycin (CAM) and azithromycin (AZM)
inhibit autophagy flux (17–19).
We also reported that combined treatment with the proteasome
inhibitor bortezomib and CAM for simultaneous blocking of the
ubiquitin-proteasome system and the autophagy-lysosome system
resulted in pronounced apoptosis induction of myeloma cells along
with accumulation of intracellular ubiquitinated proteins and
aggresome, which results in pronounced endoplasmic reticulum (ER)
stress loading (20). Under
accumulation of unfolded proteins inside the ER lumen, a series of
adaptive responses occur: i) attenuation of translation for
repression of unfolded protein accumulation; ii) induction of
chaperon proteins for proper refolding; and iii) exporting of
unfolded proteins to outside ER, following their polyubiquitination
and degradation by proteasome [ER-associated degradation (ERAD)].
However, when ER stress loading exceeds these adaptive capacities,
apoptosis is induced via transcriptional activation of a
proapoptotic transcription factor such as C/EBP homologous protein
(CHOP)/GADD153 and the other mechanisms (21,22).
Many lines of evidence now confirm that apoptosis induction via ER
stress loading in cancer cells is a novel potential therapeutic
strategy (23).
We recently reported that combined treatment using
GEF and CAM resulted in pronounced cytotoxicity as well as ER
stress loading in NSCLC (24). We
also reported that macrolide antibiotics, including CAM and AZM,
enhanced bortezomib-induced cytotoxicity in multiple myeloma cells
(19). As EGFR-TKI induced
cytoprotective autophagy in NSCLC, we hypothesized that the
efficiency of macrolides in blocking autophagy might directly
affect the enhanced cytotoxicity in refractory pancreatic cancer
cells. In the present study, we used a novel 12-membered EM900,
which shows anti-inflammatory and/or immunomodulatory effects,
without possessing antibacterial activity (25), as well as CAM and AZM to compare
their effects of: i) enhancing GEF-induced cytotoxicity; ii)
blocking autophagy flux; and iii) ER stress loading in pancreatic
cancer cell lines. Our data clearly exhibited a positive
correlation between blocking efficiency in autophagy inhibition and
pronounced EGFR-TKI-induced cytotoxicity. This result suggests the
possibility of using a macrolide in combination with EGFR-TKI
therapy for pancreatic cancer.
Materials and methods
Reagents
GEF and erlotinib hydrochloride (ERL) were purchased
from Biomolecules (Hamburg, Germany), and gemcitabine hydrochloride
was purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo,
Japan). They were dissolved in dimethyl sulfoxide (DMSO) at a
concentration of 10 mM as a stock solution. CAM and AZM were
purchased from Tokyo Chemical Industry. Non-antibiotic macrolide
EM900 [(8R,9S)-8,9- dihydro-6,9- epoxy-8,9- anhydropseudo-
erythromycin A] was synthesized as previously reported (25). CAM, AZM and EM900 were dissolved in
ethanol to prepare stock solutions of 5 mM. E-64d and pepstatin A,
which are inhibitors of lysosomal proteases, and pan-caspase
inhibitor Z-VAD-FMK were all purchased from Peptide Institute
(Osaka, Japan). Necrostatin-1, a specific inhibitor of RIPK1, was
purchased from Enzo Life Sciences (Farmingdale, NY, USA).
Cell lines and culture conditions
For the present study, leukemia cell line K562 and
pancreatic cancer cell lines BxPC-3, Capan-1 and PANC-1 were
obtained from the American Type Culture Collection (ATCC; Manassas,
VA, USA). BxPC-3 cells and K562 cells were maintained in RPMI-1640
medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10%
fetal bovine serum (FBS; Biowest, Kiltimagh, Ireland), 2 mM
L-glutamine, penicillin (100 U/ml), and streptomycin (100 μg/ml)
(Wako Pure Chemicals Industries, Osaka, Japan). PANC-1 cells were
maintained in Dulbecco's modified Eagle's medium (Sigma)
supplemented with 10% FBS, penicillin (100 U/ml) and streptomycin
(100 μg/ml). Capan-1 cells were maintained in Iscove's modified
Dulbecco's medium (Life Technologies, Carlsbad, CA, USA)
supplemented with 20% FBS, penicillin (100 U/ml) and streptomycin
(100 μg/ml). All cell lines were cultured in a humidified incubator
containing 5% CO2 and 95% air at 37°C.
Assessment of the viable number of
cells
The number of viable cells was assessed using
CellTiter Blue, a cell viability assay kit (Promega, Madison, WI,
USA), with fluorescence measurements at 570 nm for excitation and
590 nm for fluorescence emission.
Immunoblotting
Immunoblotting was performed as previously described
(19). Cells were lysed with RIPA
lysis buffer (Nacalai Tesque, Kyoto, Japan) supplemented with a
protease and phosphatase inhibitor cocktail (Nacalai Tesque).
Cellular proteins were quantified using a DC protein assay kit of
Bio-Rad Laboratories (Richmond, CA, USA). Equal amounts of proteins
were loaded onto the gels, separated by SDS-PAGE, and transferred
onto Immobilon-P membranes (Millipore Corp., Bedford, MA, USA).
These membranes were probed with first antibodies (Abs) such as
anti-LC3B antibody (Ab) (Novus Biologicals LLC, Littleton, CO,
USA), anti-p62 (D-3) monoclonal (m) Ab (sequestsome-1), anti-β
actin (C4) mAb, anti-GAPDH (6C5) mAb (Santa Cruz Biotechnology,
Santa Cruz, CA, USA), anticleaved-caspase-3 Ab, and anti-PARP Ab
(Cell Signaling Technology, Danvers, MA, USA). Immunoreactive
proteins were detected using horseradish peroxidase-conjugated
second Abs and an enhanced chemiluminescence reagent (ECL)
(Millipore). Densitometry was performed using a Molecular Imager
ChemiDoc XRS System (Bio-Rad Laboratories).
Gene expression analysis
Real-time polymerase chain reaction (PCR) for gene
expression analysis was performed as previously described in detail
(19).
Electron microscopy
Cells were fixed with 2.5% glutaralde-hyde in 0.1 M
phosphate buffer (pH 7.3) for 1 h. The samples were further fixed
in 1% osmium tetroxide for 1 h, dehydrated in graded ethanol
(30–100%), and embedded in Quetol 812 epoxy resin (Nisshin EM Co.,
Ltd., Tokyo, Japan). Ultrathin sections were cut using an Ultracut
J microtome (Reichert Jung, Vienna, Austria). The sections were
stained with lead nitrate and uranium acetate, and subjected to
electron microscopic analysis using a scanning electron microscope
JEM-1200EX II (JEOL, Tokyo, Japan).
Stable transfection of GFP-LC3 plasmid in
K562 cells
For the present study, a pEGFP-LC3 plasmid vector
(no. 2490) was purchased from Addgene (Cambridge, MA, USA). K562
cells were transfected with plasmid DNA using Lipofectamine 2000
(Life Technologies) according to the manufacturer's instructions.
Briefly, 4 μg of pEGFP-LC3 plasmids solution and 10 μl of
Lipofectamine 2000 were incubated in 500 μl of serum-free Opti-MEM
(Life Technologies) for 20 min and mixed into 2×106
cells cultured in 1.5 ml of antibiotic-free RPMI-1640 with 10% FBS
in a 60-mm dish. Forty-eight hours after transfection, the cells
were seeded into a 96-well plate and selected for limited dilution
cloning in the presence of 1 μg/ml of G418 for 1 month.
K562/GFP-LC3 clone #2 was used for the following experiments.
After treatment with macrolide, K562/GFP-LC3 cells
were spread and fixed on slide glasses using a Cytospin 4
Centrifuge (Thermo Fisher Scientific, Inc., Rockford, IL, USA) to
make slide glass preparations. Analysis by confocal microscopy for
the assessment of autophagosomes was performed using a confocal
laser scanning fluorescence microscope LSM 700 (Carl Zeiss, Jena,
Germany).
Statistical analysis
The data are expressed as mean ± SD. Statistical
analysis was performed using the Mann-Whitney U test
(two-tailed).
Results
Cell growth inhibition in pancreatic
cancer cell lines after treatment with EGFR-TKI
Pancreatic cancer cell lines PANC-1 and Capan-1,
both of which have activating EGFR mutations, and BxPC-3 expressing
wild-type EGFR, were cultured in the presence of the EGFR-TKIs GEF
and erlotinib hydrochloride (ERL) at various concentrations
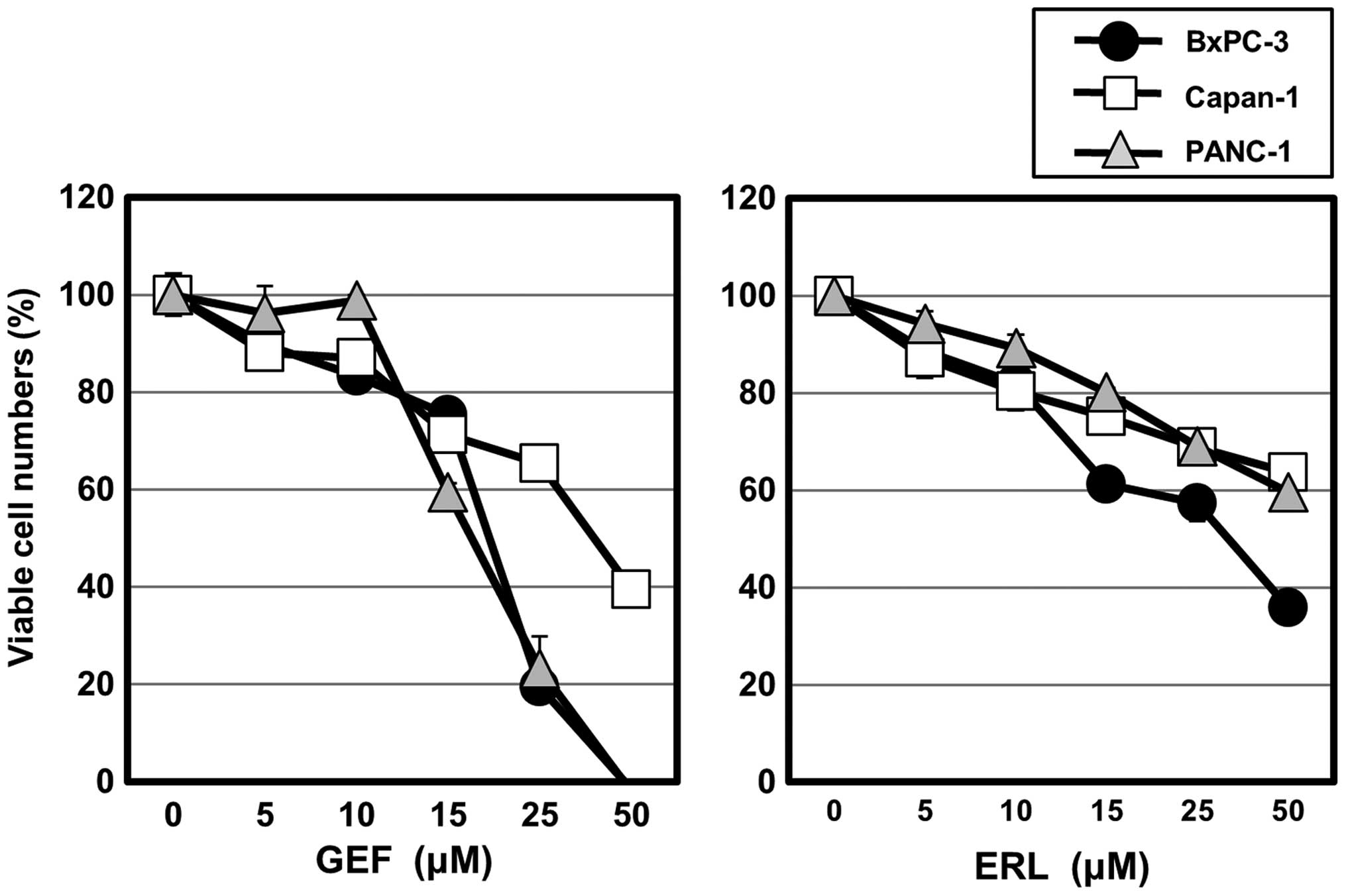
(Fig. 1). Although both GEF and
ERL inhibited cell growth in these cell lines in a dose-dependent
manner, GEF was more effective than erlotinib in the cytotoxic
effect in our culture system. Therefore, the following in
vitro experiments were performed using mainly GEF. A
concentration of 50% cell growth inhibition (IC50) after
48-h treatment with GEF was 20.4 μM for PANC-1, 39.5 μM for Capan-1
and 19.5 μM for BxPC-3.
Autophagy induction in response to GEF in
pancreatic cancer cell lines
Conversion from the soluble cytosolic LC3B-I to the
membrane-bound LC3B-II via conjugation of phosphatidyl ethanolamine
represents the formation of autophagosomes. Therefore, increased
expression of LC3B-II is a good marker for autophagosome evaluation
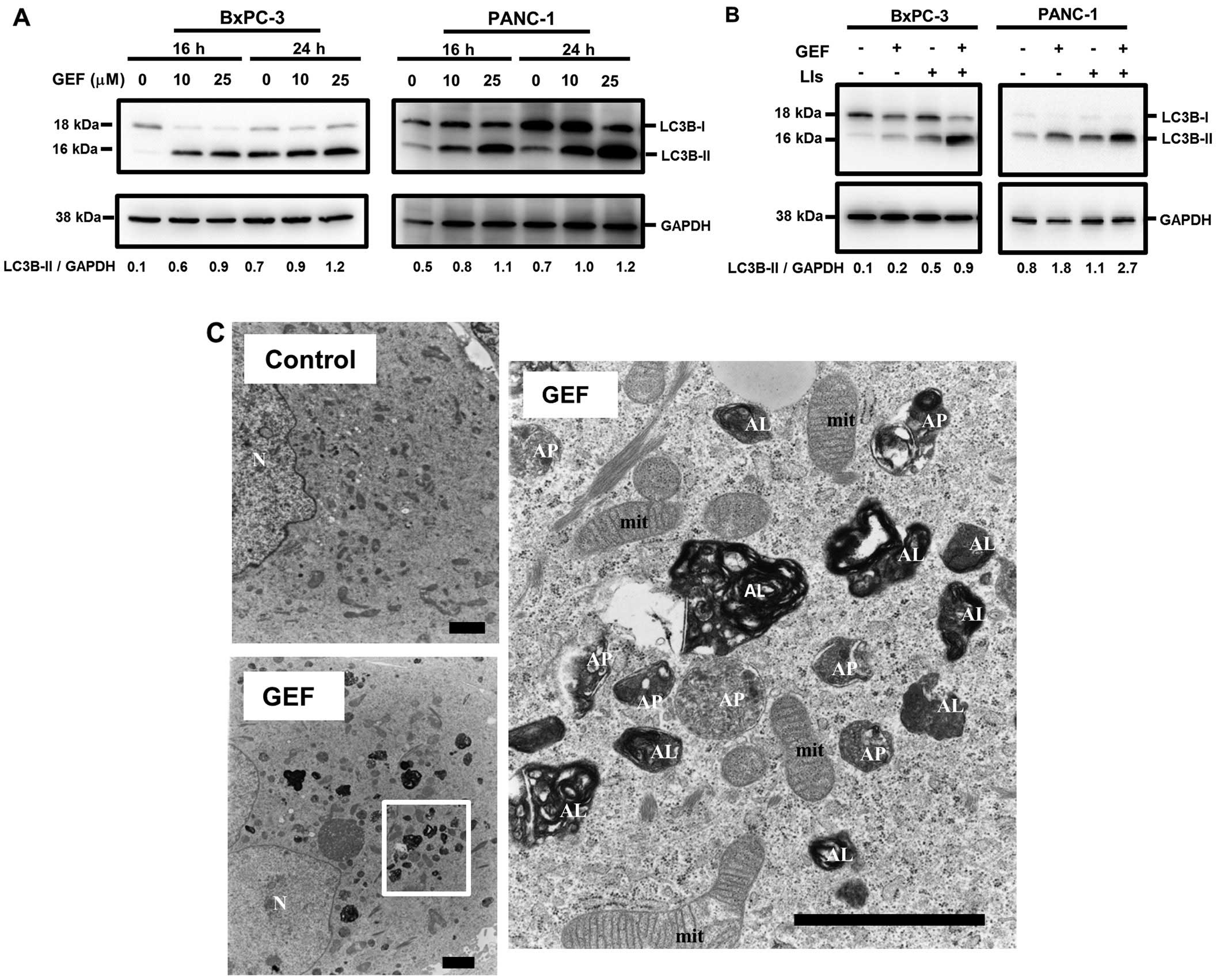
(27). Treatment with GEF induced
the increased expression of LC3B-II in a dose- and a time-dependent
manner in PANC-1 cells and BxPC-3 cells (Fig. 2A). Not shown is that ERL-treatment
also increased the expression of LC3B-II as well as GEF. In
addition, combined treatment with GEF and lysosomal inhibitors such
as E-64d and pepstatin A, which are used for blocking autophagy
flux, further enhanced LC3B-II expression compared to treatment
with GEF alone or with only lysosomal inhibitors (Fig. 2B). Furthermore, electron microscopy
indicated a marked increase of autophagosomes and autolysosomes in
PANC-1 cells after treatment with GEF (Fig. 2C). These data indicate that GEF
induces autophagy in pancreatic cell lines as well as other cancer
cell lines, including NSCLC cells previously reported by us and
others (25,26).
Enhanced cytotoxicity of EGFR-TKI by
combined treatment with macrolides in pancreatic cancer cell
lines
Using NSCLC cell lines, we have reported that
GEF-induced autophagy functions as cytoprotective, and simultaneous
treatment with GEF and CAM results in enhanced cytotoxicity
(25). In the present study, we
selected three macrolides (CAM, AZM and EM900) and examined their
efficacy for GEF-induced cytotoxicity as well as blocking autophagy
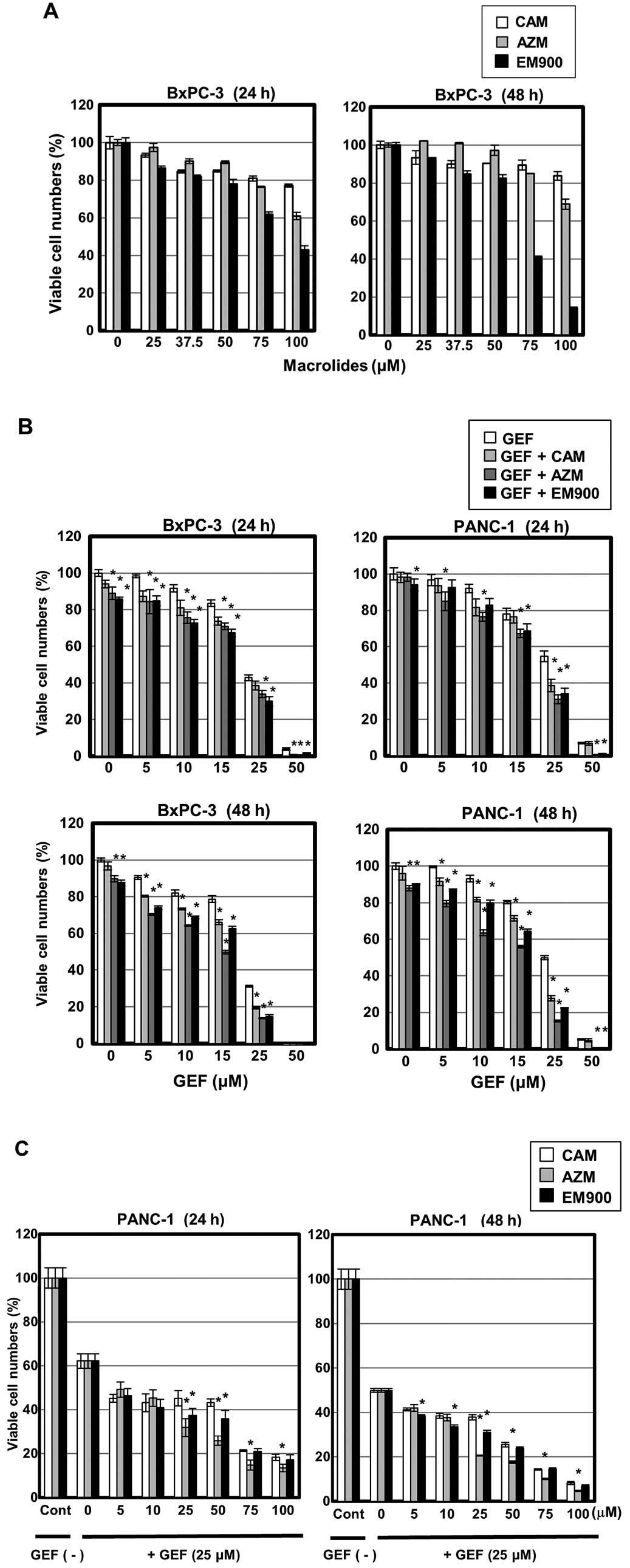
flux in pancreatic cancer cell lines. First, treatment with up to
50 μM CAM, AZM or EM900 resulted in little cytotoxicity in BxPC-3
cells. More than 75 μM EM900 exhibited some cytotoxic effect
(Fig. 3A). Next, BxPC-3 and PANC-1
cells were treated with GEF at various concentrations in the
presence of 50 μM CAM, AZM or EM900. Significant enhancement of
GEF-induced cytotoxicity was observed (Fig. 3B). During 48-h exposure, AZM was
more potent than CAM or EM900 for enhancing GEF-induced
cytotoxicity. At 25 and 50 μM GEF, EM900 was superior to CAM. The
concentration of GEF was therefore fixed at 25 μM, and PANC-1 cells
were treated with macrolides at various concentrations. It was
noteworthy that all three macrolides at >5 μM resulted in
enhanced reduction of the viable cell number as comparing with
treating the cells with GEF alone (Fig. 3C). Additionally, AZM was superior
to CAM and EM900 in enhancing the killing effect of 25 μM GEF.
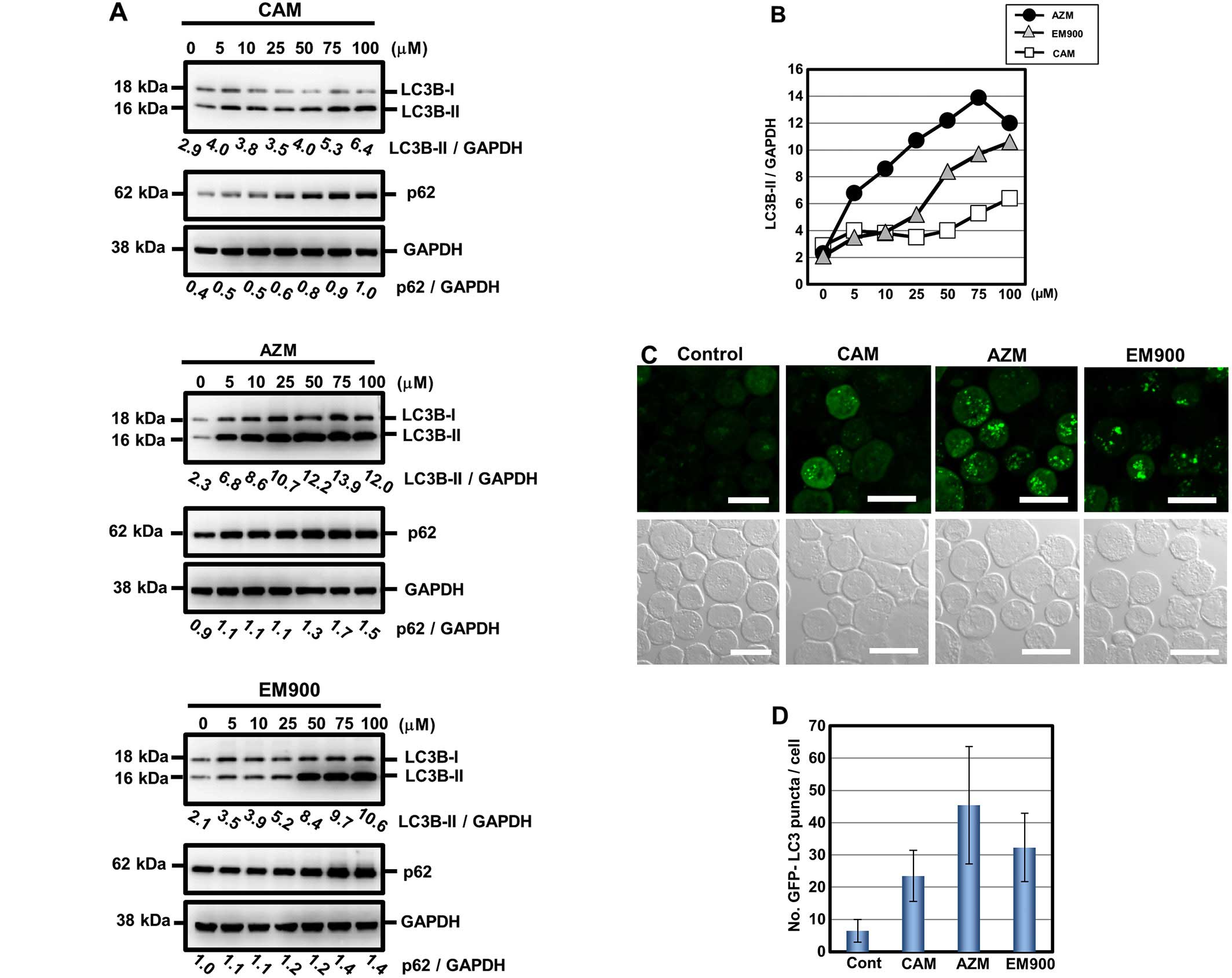
Comparing the blocking effect of
autophagy flux among CAM, AZM and EM900 in PANC-1 cells
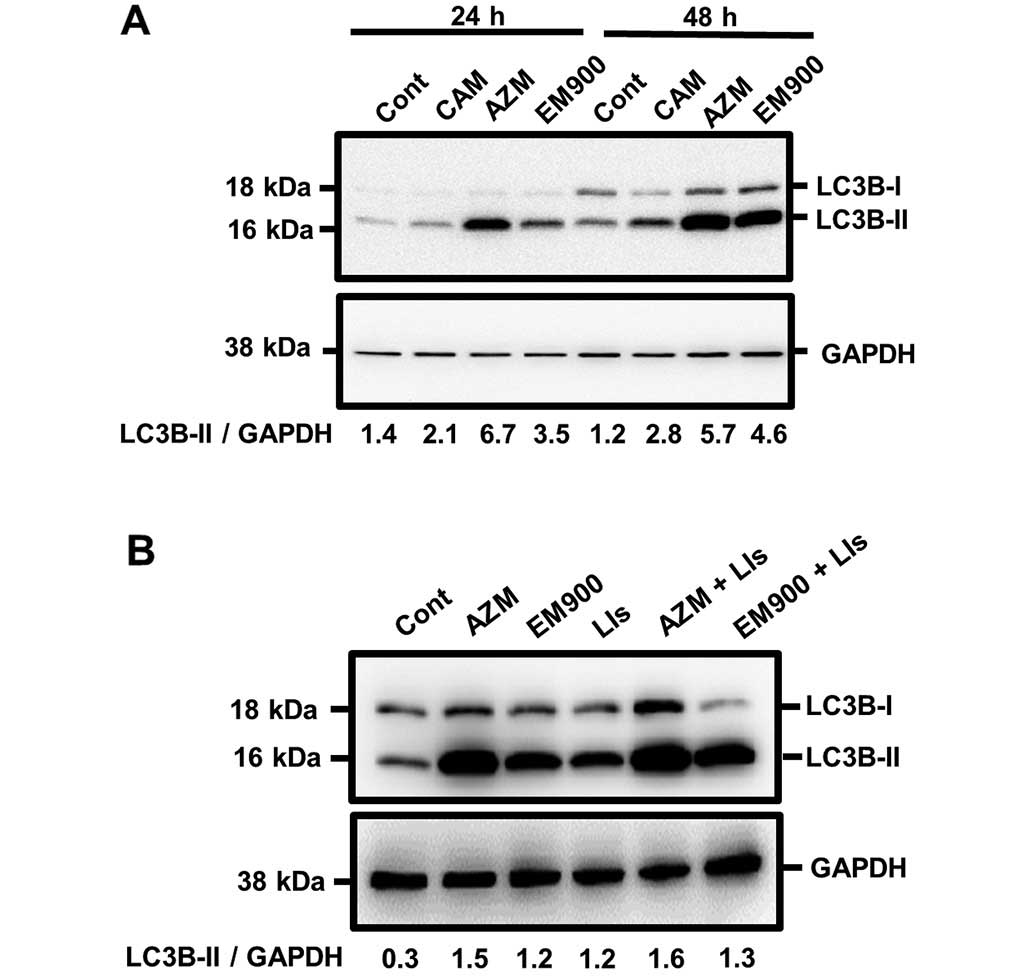
Based on the above data, we further assessed the
blocking effect of macrolides on autophagy flux in pancreatic
cancer cell lines. After treatment with 50 μM macrolides for 24 and
48 h, PANC-1 cells exhibited increased expression of LC3B-II
(Fig. 4A). However, unlike GEF
treatment (Fig. 2B), combined
treatment with lysosomal inhibitors and a macrolide did not
increase LC3B-II expression further, compared with treatment with
either a macrolide alone or only lysosome inhibitors. These results
indicate that all tested macrolides effectively block autophagy
flux (Fig. 4B). For comparison of
efficiency on blocking autophagy, PANC-1 cells were treated with a
macrolide at various concentrations for 24 h. Immunoblotting with
anti-LC3B Ab clearly indicated accumulation of autophagosome in a
dose-dependent manner for all three macrolides (Fig. 5A). P62, a substrate of autophagy
(28), also accumulated after
macrolide treatment in a dose-dependent manner. To standardize for
comparison among the three macrolides, the expression ratios of
LC3B-II to GAPDH were divided by that of untreated controls and
plotted (Fig. 5B). AZM is the most
potent in blocking efficiency in autophagy; EM900 is the second
most potent and superior to CAM. It was noteworthy that the
inhibitory efficacy of autophagy in macrolides correlated well with
their enhancing effect for GEF-induced cytotoxicity (Fig. 3B and C). To confirm their blocking
efficiency, K562 cells with a stably transfected GFP-LC3 gene
(K562/GFP-LC3) were cultured in the presence of these macrolides at
50 μM for 24 h. As indicated in Fig.
5C and D, the number of puncta of GFP-LC3 pre-cell, which
indicates autophagosome accumulation in response to the macrolide,
was consistent with the result presented in Fig. 5A and B. Increase in the number of
GFP-LC3 puncta was in the order of AZM, EM900 and CAM. These data
together strongly suggested that enhanced GEF-induced cytotoxicity
by combining macrolides is due to their effect in blocking
autophagy flux.
Non-apoptotic cell death induction after
treatment with GEF with/without macrolides
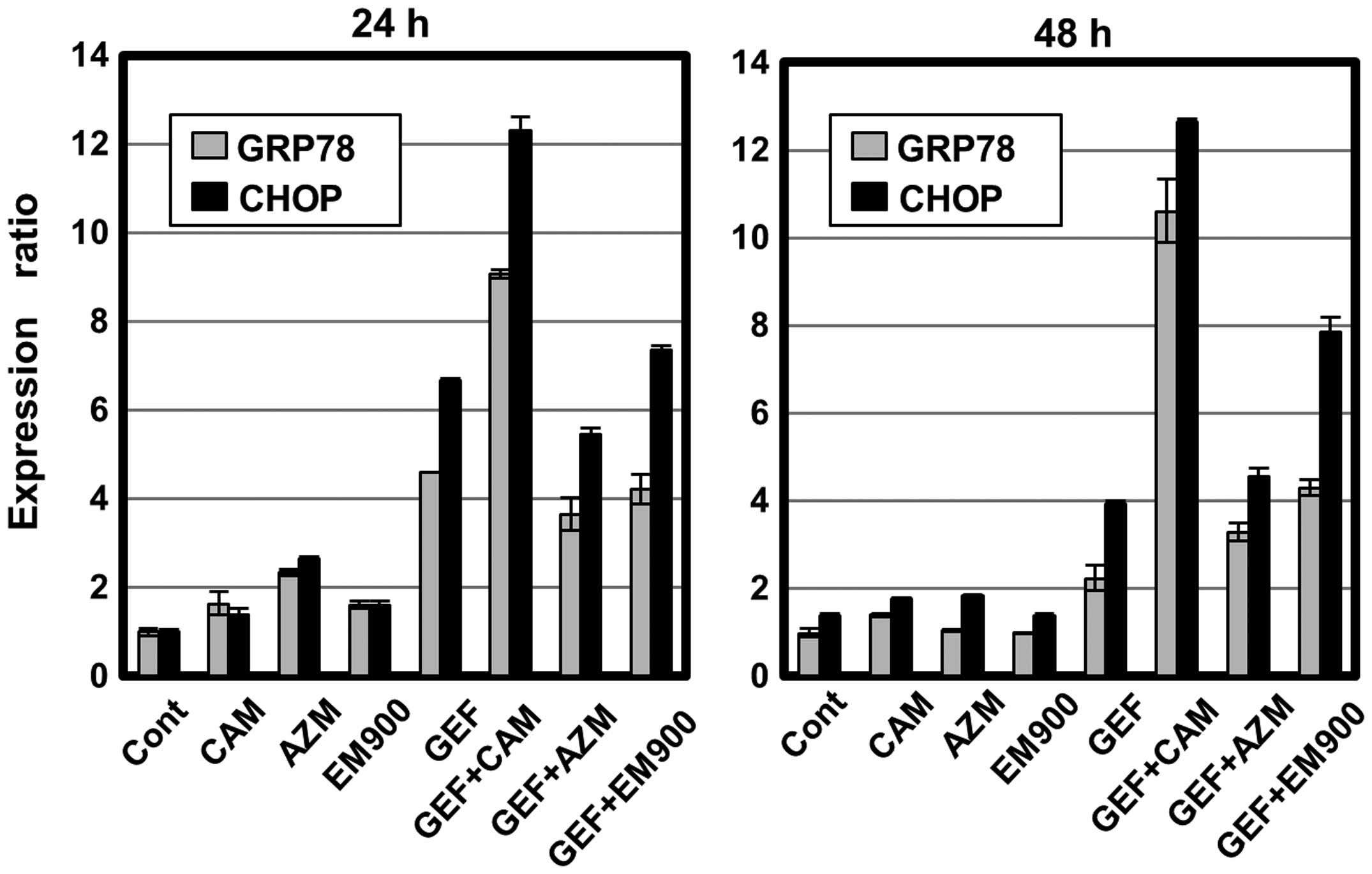
We next assessed ER stress loading after treatment
with GEF and macrolides, because apoptosis is induced when ER
stress is overloaded beyond cellular adaptive capacity (21,22).
Real-time PCR indicated that GEF treatment upregulated ER
stress-related genes such as GRP78 and CHOP, whereas, treatment
with macrolides had little effect (Fig. 6). Combined treatment with GEF plus
CAM resulted in pronounced expressions of GRP78 and CHOP, which is
a pro-apoptotic transcription factor that upregulates various
pro-apoptotic genes such as Bax and Bim (21). Unexpectedly, concomitant AZM or
EM900 plus GEF did not significantly enhance ER stress loading
compared with treatment of the cells with GEF alone. A discrepancy
between the efficiency of autophagy inhibition and ER stress
loading led us to examine whether the enhanced cytotoxicity is
completely mediated though apoptosis.
As indicated in Fig.
7A, treating PANC-1 cells with GEF in the presence or absence
of EM900 produced no apoptotic features but did produce necrosis.
However, cells treated with gemcitabine (gem), which is the most
widely used medicine for treating patients with metastatic
pancreatic cancer, produced nuclear chromatin condensation in some
cells (arrows). Electron microscopy also indicated that, although
gem-treated PANC-1 cells exhibited a sharp nuclear notch as well as
nuclear heterochromatin formation GEF treatment with/without AZM
did not produce prominent nuclear change, but did produce
accumulation of a large number of autophagosomes and autolysosomes,
and increased swollen mitochondria with destruction of cristae
structure inside them (Fig. 7B).
Furthermore, immunoblotting with anti-cleaved capase-3 Ab and
anti-PARP Ab indicated no cleavage of caspase-3 and PARP after
treatment with GEF plus AZM or EM900 (Fig. 7C). All these data suggest that
enhanced cytotoxicity by combining GEF plus macrolide as well as
GEF-induced cytotoxicity itself does not involve apoptosis in
pancreatic cancer cell lines. This result was also supported by the
data presented in Fig. 7D; in the
presence of necrostatin-1, a potent and selective inhibitor of
necroptosis via blocking RIP-1 kinase (29), GEF- and GEF plus EM900-induced cell
death was significantly attenuated, while Z-VAD-FMK, a pan-caspase
inhibitor for apoptosis inhibition, had little effect. In contrast,
gemcitabine-induced cytotoxicity was significantly suppressed in
the presence of Z-VAD-FMK, but not in the presence of
necrostatin-1. Thus, a combination of GEF plus macrolide induces
mainly non-apoptosis cell death such as necroptosis, whereas GEM
induces apoptosis, in PANC-1 cells.
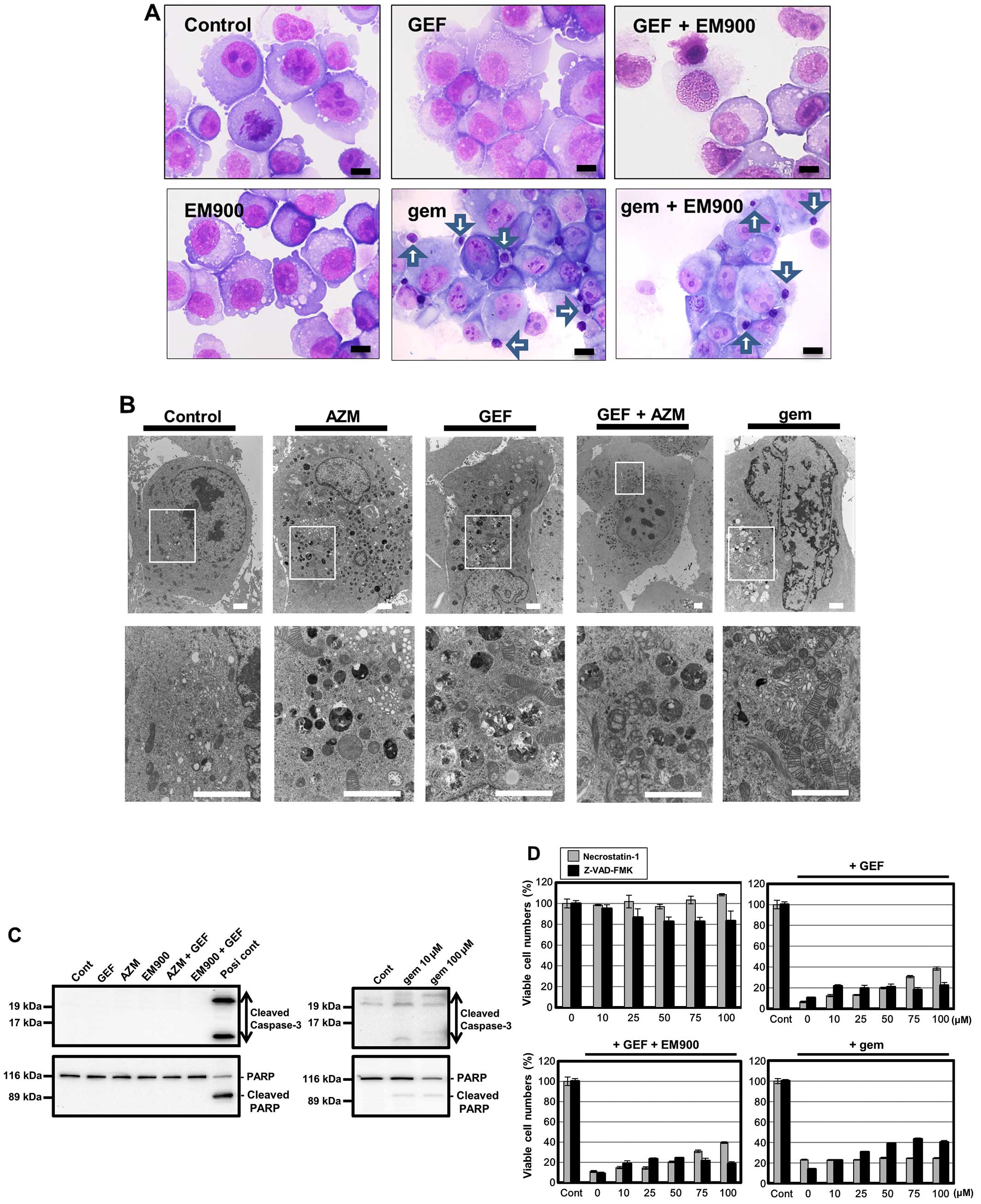 | Figure 7Morphological changes in PANC-1 cells
after treatment with GEF and macrolides. (A) May-Giemsa staining:
May-Giemsa staining was performed after treatment with either GEF
(25 μM) or gemcitabine (100 μM) in the presence or absence of EM900
(50 μM) for 48 h. Scale bars, 10 μm. Arrows indicate nuclear
chromatin condensations. (B) Electron microscopy: PANC-1 cells were
processed for electron microscopy after treatment with GEF (25 μM),
AZM (50 μM), GEF+AZM, and gemcitabine (100 μM) for 48 h. Lower
panels are at higher magnifications of the squared areas in each
upper panel. Scale bars, 2 μm. (C) PANC-1 cells were treated with
GEF (25 μM), AZM (50 μM), EM900 (50 μM), GEF+AZM, and gemcitabine
(10 and 100 μM) for 48 h. Cellular proteins were separated by 15%
SDS-PAGE for caspase-3 and 7.5% for PARP and immunoblotted with
either anti-cleaved caspase-3 Ab or anti-PARP Ab. Cell lysate
derived from HL-60 cells treated with vitamin K2 was used as a
positive control for apoptosis induction (42). (D) PANC-1 cells
were treated with various concentrations of either necrostatin-1 or
Z-VAD-FMK in the presence or absence of GEF (50 μM), GEF (50 μM)
plus EM900 (50 μM), or gem (300 μM) for 48 h. The number of viable
cells was assessed using CellTiter Blue. |
Discussion
In the present study, we demonstrated that
macrolides such as AZM, CAM and EM900 enhance GEF-induced
cytotoxicity in pancreatic cancer cell lines. In addition, the
efficiency of macrolides in blocking autophagy flux correlated well
with their enhancement of GEF-induced cytotoxicity (Figs. 3 and 4). Among the three macrolides, AZM was
the most potent for enhancing the GEF cytotoxicity as well as for
inhibiting autophagy. Many previous reports suggest that autophagy
induction by GEF is a metabolic stress response and functions as
cytoprotective, which might be crucial in resistance to EGFR-TKIs
(11–13). Therefore, it is reasonable that
blocking efficiency directly influences the cytotoxic effect of
GEF. However, the precise mechanism for blocking autophagy by
macrolides must still be clarified.
Based on the chemical structure of macrolides, a
common target appears to be involved in the process of autophagy.
Autophagy inhibition by AZM was originally reported by Renna et
al in 2011 (18). Long-term
oral administration of AZM against chronic sinopulmonary infection
in adult patients with cystic fibrosis is associated with the
development of infection with non-tuberculous mycobacteria. They
found that, in primary human macrophages, AZM concentrations
achieved during therapeutic dosing blocked autophagosome clearance
by preventing lysosomal acidification, thereby impairing autophagic
and phagosomal degradation. As a consequence, AZM treatment
inhibited intracellular killing of mycobacteria within macrophages,
which resulted in chronic non-tuberculous mycobacteria infection
(18). Electron microscopy
indicated accumulation of a large number of autophagosomes and
autolysosomes in PANC-1 cells after treatment with AZM (Fig. 7B). This result indicated that the
membrane fusion between autophagosome and lysosome (autolysosome
formation) is not impaired. Lysosomes generate and maintain their
pH gradients by vacuolar (V)-type ATPase activity for pumping
protons into the lysosome lumen (31). Therefore, preventing lysosomal
acidification by AZM as indicated by a previous report suggests
that V-type ATPase is a candidate target molecule for AZM. Indeed,
bafilomycin A1 and concanamycin A are macrolide
compounds that have a selective inhibitory effect on V-type ATPase,
and they are frequently used in experiments in vitro for
blocking autophagy (32). However,
both macrolides have reportedly prevented maturation of autophagic
vacuoles by inhibiting fusion between autophagosomes and lysosomes
(32). Valosin-containing protein
(VCP)/p97 appears to be another potential target (33). A recent report demonstrated that
AZM and CAM interact with VCP, based on AZM- and CAM-immobilized
affinity chromatography (33).
Pathophysiologically, germline mutations in VCP genes
account for a spectrum of pathological phenotypes that include
inclusion body myopathy with Paget's disease of the bone and
frontotemporal dementia, hereditary spastic paraplegia (IBMPFD),
and 1–2% of familial amyotrophic lateral sclerosis (34,35).
In addition, VCP/p97 plays a critical role in a broad range of
cellular activities, including ERAD, the ubiquitin-proteasome
system, prevention of polyglutamine aggregation and autophagosome
maturation in autophagy (35,36).
VCP mutation (p.Glu185Lys) segregating in the autosomal
dominant Charcot-Marie-Tooth disease type 2 family was also
identified (37). Notably,
functional studies confirmed that the Glu185Lys variant impaired
autophagic function, leading to accumulation of immature
autophagosomes (37).
Identification of the target molecule(s) of macrolide for autophagy
inhibition as well as assessment of the affinity to the macrolides
used in this study helps to clarify the phenomena analyzed in this
study.
Despite the positive relationship between blocking
efficiency and enhancement of the cytotoxicity of GEF,
discrepancies in ER stress loading were observed (Fig. 5 vs. 6). CAM and GEF was the most potent
combination for upregulation of pro-apoptotic transcription factor
CHOP, whereas concomitant AZM or EM900 plus GEF did not
significantly enhance ER stress loading, compared with GEF alone
(Fig. 6). Other than ER
stress-mediated apoptosis, alternative pathway(s) such as
non-apoptotic cell death might contribute to the pronounced
cytotoxic effect. We previously reported that a murine embryonic
fibroblast (MEF) cell line derived from CHOP knockout mice still
exhibited enhanced cytotoxicity with combined treatment of GEF and
CAM, although they were much less sensitive to GEF plus CAM than
the CHOP+/+ MEF cell line (24). The same phenomenon was observed in
PC-9 cell knocked down CHOP by siRNA versus control PC-9 cells
(24). Although cytotoxicity was
enhanced, a series of experiments performed for assessing apoptosis
did not yield any apparent features of apoptosis (Fig. 7A–C). However, in the presence of
necrostatin-1, a specific inhibitor for necroptosis via blocking
RIP1 kinase (29), GEF- and GEF
plus macrolide-induced cell death was partially but significantly
suppressed (Fig. 7D). This result
suggests that necroptosis might contribute to enhanced
cytotoxicity. Unlike NSCLC cell lines, PANC-1 and BxPC-3 cells did
not exhibit prominent apoptotic features even after treatment with
GEF alone (24). In addition,
treatment with gemcitabine resulted in only slight apoptotic
change, such as weak cleavages of caspase-3 and PARP, as well as
partial chromatin condensation without nuclear fragmentation
(Fig. 7A–C). Also, their features
were much weaker than those in other cell lines (e.g., NSCLC,
breast cancer and leukemia cell lines) (data not shown). Therefore,
pancreatic cancer cell lines used in the present study may be
rather resistant to apoptosis induction. This result might
represent the difficult clinical features of pancreatic cancer
patients with chemoresistance.
It is necessary to determine the most effective way
to interconnect autophagy inhibition and non-apoptotic cell death.
Currently, no study has demonstrated necroptosis induction by
EGFR-TKIs. However, it has been confirmed that non-apoptotic cell
death, including necroptosis, alternatively becomes prominent when
cells are resistant to apoptosis induction (38–40).
In PANC-1 cells, a higher concentration of gemcitabine (100 μM) was
required for apoptosis induction, indicating difficulty in inducing
apoptosis in them, unlike other cancer cell lines (Fig. 7D). It was reported that autophagy
suppresses RIP kinase-dependent necrosis (necroptosis) in RCC4
cells, a human renal cell carcinoma cell line (41). Inhibition of mTOR with the specific
mTOR inhibitor CCL-779 stimulated autophagy, leading to elimination
of RIP kinases via autophagy-mediated degradation. However, with
the simultaneous inhibition of autophagy using chloroquine and mTOR
with CCL-779, necroptosis was induced (41). This result is somewhat similar to
our observation, since EGFR-TKI inhibits the axis of the
PI3K/AKT/mTOR pathway. In their system, autophagy of mitochondria
(mitophagy) is required for cell survival, since mTOR inhibition
turns off the Nrf2 antioxidant defense (41). Thus, simultaneous mTOR and
autophagy inhibition leads to an imbalance between ROS production
by mitochondria and the defense by Nrf2, which causes necroptosis.
Pancreatic cell lines appear to have higher basal autophagy with
increased mitochondria than other cancer cell lines (Figs. 2A and 7B). For clinical use of a macrolide as a
potent ‘sensitizer’ in EGFR-TKI therapy, it will also be important
to establish the underlying molecular mechanism of enhanced
non-apoptotic cell death, as well as to identify the target(s) of
macrolides.
Acknowledgements
The present study was supported by funds provided
through an MEXT-supported program of the Strategic Research
Foundation at Private Universities (S1411011, 2014–2018) from the
Ministry of Education, Culture, Sports, Science and Technology of
Japan to M.H. and K.M., Grants-in-Aid for Scientific Research (C)
from The Ministry of Education, Culture, Sports, Science and
Technology to T.I. (no. 24591018) and to K.M. (no. 26460478), and a
Grant-in-Aid from Tokyo Medical University Cancer Research to
K.M.
References
|
1
|
Gresham GK, Wells GA, Gill S, Cameron C
and Jonker DJ: Chemotherapy regimens for advanced pancreatic
cancer: A systematic review and network meta-analysis. BMC Cancer.
14:4712014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Von Hoff DD, Ervin T, Arena FP, Chiorean
EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et
al: Increased survival in pancreatic cancer with nab-paclitaxel
plus gemcitabine. N Engl J Med. 369:1691–1703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ueda S, Ogata S, Tsuda H, Kawarabayashi N,
Kimura M, Sugiura Y, Tamai S, Matsubara O, Hatsuse K and Mochizuki
H: The correlation between cytoplasmic overexpression of epidermal
growth factor receptor and tumor aggressiveness: Poor prognosis in
patients with pancreatic ductal adenocarcinoma. Pancreas. 29:e1–e8.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moore MJ, Goldstein D, Hamm J, Figer A,
Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, et al;
National Cancer Institute of Canada Clinical Trials Group.
Erlotinib plus gemcitabine compared with gemcitabine alone in
patients with advanced pancreatic cancer: A phase III trial of the
National Cancer Institute of Canada Clinical Trials Group. J Clin
Oncol. 25:1960–1966. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang ZY, Yuan JQ, Di MY, Zheng DY, Chen
JZ, Ding H, Wu XY, Huang YF, Mao C and Tang JL: Gemcitabine plus
erlotinib for advanced pancreatic cancer: A systematic review with
meta-analysis. PLoS One. 8:e575282013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Diaz Beveridge R, Alcolea V, Aparicio J,
Segura Á, García J, Corbellas M, Fonfría M, Giménez A and Montalar
J: Management of advanced pancreatic cancer with gemcitabine plus
erlotinib: Efficacy and safety results in clinical practice. JOP.
15:19–24. 2014.PubMed/NCBI
|
|
7
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Galluzzi L, Pietrocola F, Levine B and
Kroemer G: Metabolic control of autophagy. Cell. 159:1263–1276.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
White E: Deconvoluting the
context-dependent role for autophagy in cancer. Nat Rev Cancer.
12:401–410. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ciardiello F and Tortora G: EGFR
antagonists in cancer treatment. N Engl J Med. 358:1160–1174. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Han W, Pan H, Chen Y, Sun J, Wang Y, Li J,
Ge W, Feng L, Lin X, Wang X, et al: EGFR tyrosine kinase inhibitors
activate autophagy as a cytoprotective response in human lung
cancer cells. PLoS One. 6:e186912011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jutten B and Rouschop KM: EGFR signaling
and autophagy dependence for growth, survival, and therapy
resistance. Cell Cycle. 13:42–51. 2014. View Article : Google Scholar :
|
|
13
|
Fung C, Chen X, Grandis JR and Duvvuri U:
EGFR tyrosine kinase inhibition induces autophagy in cancer cells.
Cancer Biol Ther. 13:1417–1424. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim YC and Guan KL: mTOR: A pharmacologic
target for autophagy regulation. J Clin Invest. 125:25–32. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wei Y, Zou Z, Becker N, Anderson M,
Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, et
al: EGFR-mediated Beclin 1 phosphorylation in autophagy
suppression, tumor progression, and tumor chemoresistance. Cell.
154:1269–1284. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tan X, Thapa N, Sun Y and Anderson RA: A
kinase-independent role for EGF receptor in autophagy initiation.
Cell. 160:145–160. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nakamura M, Kikukawa Y, Takeya M, Mitsuya
H and Hata H: Clarithromycin attenuates autophagy in myeloma cells.
Int J Oncol. 37:815–820. 2010.PubMed/NCBI
|
|
18
|
Renna M, Schaffner C, Brown K, Shang S,
Tamayo MH, Hegyi K, Grimsey NJ, Cusens D, Coulter S, Cooper J, et
al: Azithromycin blocks autophagy and may predispose cystic
fibrosis patients to mycobacterial infection. J Clin Invest.
121:3554–3563. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Moriya S, Che XF, Komatsu S, Abe A,
Kawaguchi T, Gotoh A, Inazu M, Tomoda A and Miyazawa K: Macrolide
antibiotics block autophagy flux and sensitize to bortezomib via
endoplasmic reticulum stress-mediated CHOP induction in myeloma
cells. Int J Oncol. 42:1541–1550. 2013.PubMed/NCBI
|
|
20
|
Moriya S, Komatsu S, Yamasaki K, Kawai Y,
Kokuba H, Hirota A, Che XF, Inazu M, Gotoh A, Hiramoto M, et al:
Targeting the integrated networks of aggresome formation,
proteasome, and autophagy potentiates ER stress-mediated cell death
in multiple myeloma cells. Int J Oncol. 46:474–486. 2015.
|
|
21
|
Verfaillie T, Salazar M, Velasco G and
Agostinis P: Linking ER stress to autophagy: Potential implications
for cancer therapy. Int J Cell Biol. 2010:9305092010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang M and Kaufman RJ: The impact of the
endoplasmic reticulum protein-folding environment on cancer
development. Nat Rev Cancer. 14:581–597. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tabas I and Ron D: Integrating the
mechanisms of apoptosis induced by endoplasmic reticulum stress.
Nat Cell Biol. 13:184–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sugita S, Ito K, Yamashiro Y, Moriya S,
Che XF, Yokoyama T, Hiramoto M and Miyazawa K: EGFR-independent
autophagy induction with gefitinib and enhancement of its cytotoxic
effect by targeting autophagy with clarithromycin in non-small cell
lung cancer cells. Biochem Biophys Res Commun. 461:28–34. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sugawara A, Sueki A, Hirose T, Nagai K,
Gouda H, Hirono S, Shima H, Akagawa KS, Omura S and Sunazuka T:
Novel 12-membered non-antibiotic macrolides from erythromycin A;
EM900 series as novel leads for anti-inflammatory and/or
immunomodulatory agents. Bioorg Med Chem Lett. 21:3373–3376. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dragowska WH, Weppler SA, Wang JC, Wong
LY, Kapanen AI, Rawji JS, Warburton C, Qadir MA, Donohue E, Roberge
M, et al: Induction of autophagy is an early response to gefitinib
and a potential therapeutic target in breast cancer. PLoS One.
8:e765032013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jiang P and Mizushima N: LC3- and
p62-based biochemical methods for the analysis of autophagy
progression in mammalian cells. Methods. 75:13–18. 2015. View Article : Google Scholar
|
|
29
|
Degterev A, Hitomi J, Germscheid M, Ch'en
IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al:
Identification of RIP1 kinase as a specific cellular target of
necrostatins. Nat Chem Biol. 4:313–321. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sui X, Kong N, Zhu M, Wang X, Lou F, Han W
and Pan H: Cotargeting EGFR and autophagy signaling: A novel
therapeutic strategy for non-small-cell lung cancer. Mol Clin
Oncol. 2:8–12. 2014.PubMed/NCBI
|
|
31
|
Mindell JA: Lysosomal acidification
mechanisms. Annu Rev Physiol. 74:69–86. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Harada M, Sakisaka S, Yoshitake M, Kin M,
Ohishi M, Shakado S, Mimura Y, Noguchi K, Sata M and Tanikawa K:
Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPases,
inhibits the receptor-mediated endocytosis of asialoglycoproteins
in isolated rat hepatocytes. J Hepatol. 24:594–603. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nujić K, Smith M, Lee M, Belamarić D,
Tomašković L, Alihodžić S, Malnar I, Polančec D, Schneider K and
Eraković Haber V: Valosin containing protein (VCP) interacts with
macrolide antibiotics without mediating their anti-inflammatory
activities. Eur J Pharmacol. 677:163–172. 2012. View Article : Google Scholar
|
|
34
|
Watts GD, Wymer J, Kovach MJ, Mehta SG,
Mumm S, Darvish D, Pestronk A, Whyte MP and Kimonis VE: Inclusion
body myopathy associated with Paget disease of bone and
frontotemporal dementia is caused by mutant valosin-containing
protein. Nat Genet. 36:377–381. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Meyer H and Weihl CC: The VCP/p97 system
at a glance: Connecting cellular function to disease pathogenesis.
J Cell Sci. 127:3877–3883. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Meyer H, Bug M and Bremer S: Emerging
functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat
Cell Biol. 14:117–123. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gonzalez MA, Feely SM, Speziani F,
Strickland AV, Danzi M, Bacon C, Lee Y, Chou TF, Blanton SH, Weihl
CC, et al: A novel mutation in VCP causes Charcot-Marie-Tooth Type
2 disease. Brain. 137:2897–2902. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nikoletopoulou V, Markaki M, Palikaras K
and Tavernarakis N: Crosstalk between apoptosis, necrosis and
autophagy. Biochim Biophys Acta. 1833.3448–3459. 2013.
|
|
39
|
Vanden Berghe T, Linkermann A,
Jouan-Lanhouet S, Walczak H and Vandenabeele P: Regulated necrosis:
The expanding network of non-apoptotic cell death pathways. Nat Rev
Mol Cell Biol. 15:135–147. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Feoktistova M and Leverkus M: Programmed
necrosis and necroptosis signalling. FEBS J. 282:19–31. 2015.
View Article : Google Scholar
|
|
41
|
Bray K, Mathew R, Lau A, Kamphorst JJ, Fan
J, Chen J, Chen HY, Ghavami A, Stein M, DiPaola RS, et al:
Autophagy suppresses RIP kinase-dependent necrosis enabling
survival to mTOR inhibition. PLoS One. 7:e418312012. View Article : Google Scholar : PubMed/NCBI
|