Introduction
Hepatocellular carcinoma (HCC) is the most common
cancer and the third leading cause of cancer-related deaths
worldwide (1). Yet, the
pathogenesis mechanisms of HCC remain unclear. One of the potential
molecular mechanisms involves the long non-coding RNAs (lncRNAs)
(2,3).
LncRNAs play an important role in tumorigenesis and
investigation of their biological functions and molecular
mechanisms is important for understanding the development and
progression of cancer (4). The
lncRNA HOTAIR is overexpressed in HCC tissues compared to
corresponding non-cancerous tissues and high HOTAIR expression
tightly correlated with the presence of liver metastasis (5). lncRNA maternally-expressed gene 3
(MEG3) is an imprinted gene, located on human chromosome 14q32.3
(6). The downregulation of MEG3
has been observed in various human cancers (7–9).
MEG3 might function as a tumor suppressor (10,11).
However, the reasons for its downregulation and its function in HCC
are not well understood.
Adenosine and their analogues can interfere with the
synthesis of nucleic acids and exert genotoxic activity by
incorporating with and altering the DNA or RNA macromolecules,
which makes them exciting candidates for anticancer therapy
(12–14). Although MEG3 plays a role in
inhibiting tumor growth, no data are available on its function
under genotoxic stress-induced apoptosis. In our previous study, we
demonstrated that adenosine induces hepatoma cell apoptosis and the
mechanism is involved in DNA demethylation and genotoxic effects
(15,16). In this study, we treated HepG2
cells with adenosine or 5-Aza-2′-deoxy-cytidine (5-Aza-CdR), a
selective DNA methyltransferase inhibitor, and observed the
methylation status of MEG3 promoter and MEG3 mRNA expression. In
addition, an improved MS2-BioTRAP (MS2 biotin tagged RNA affinity
purification) technique was applied to screen MEG3-binding protein
in HEK293 cell line. The antitumor effect of adenosine and MEG3 in
HCC was observed and the possible molecular mechanisms were
investigated.
Materials and methods
Cell culture and experimental groups
HepG2, Huh-7 and HEK293 cells, obtained from the
American Type Culture Collection, were cultured in Dulbecco's
modified Eagle's medium supplemented with 10% (v/v) fetal bovine
serum, penicillin (final concentration, 100 U/ml), and streptomycin
(final concentration, 100 mg/ml) under a humidified atmosphere of
5% CO2 and 95% air at 37°C. Cells were passaged at
70–80% confluence. HCC cells were divided into two groups for the
experiment of overexpression MEG3: pcDNA3.1 (control group) and
pcDNA3.1-MEG3 vector (experimental group).
Treatment of HepG2 cells with adenosine
or 5-Aza-CdR
A total of 1.0×105 HepG2 cells were
inoculated into each well of a 6-well plate. After the cells
attached, the original medium was replaced with fresh medium
containing 4.0 mmol/l adenosine for 48 h or 20 μmol/l 5-Aza-cdR for
72 h. The medium with the same concentration of 5-Aza-CdR was
replaced daily. Cells were collected and prepared for detection of
MEG3 expression.
Detection of DNA methylation by
methylation-specific PCR (MSP)
A total of 1.0×105 HCC cells were
inoculated into each well of a 6-well plate and treated as
described above (16). DNA from
the cells was extracted using an EZNA Tissue DNA kit according to
the manufacturer's instructions. The methylation status of the MEG3
gene was determined by MSP assay. For the MSP, the CpGenome Turbo
Bisulfite Modification kit was used in accordance with the
manufacturer's instructions. The sequences of the
methylation-specific primers of MEG3 were as follows: the
methylated pair (M), 5′-GTTAGTAATCGGG TTTGTCGGC (forward) and
5′-AATCATAACTCCGAACAC CCGCG(reverse);the unmethylated
pair(U),5′-GAGGATGGT TAGTTATTGGGGT (forward) and 5′-CCACCATAACCA
ACACCCTATAATCACA (reverse). MEG3 DNA was amplified in a DNA Engine
Peltier thermal cycler (Bio-Rad) by 1 cycle of 95°C for 15 min
followed by 5 cycles of 94°C for 30 sec, 70°C for 30 sec and 72°C
for 30 sec; 5 cycles of 94°C for 30 sec, 65°C for 30 sec and 72°C
for 30 sec; 30 cycles of 94°C for 30 sec, 60°C for 30 sec and 72°C
for 30 sec; and 1 cycle of 72°C for 7 min. The PCR products were
identified by electrophoresis through a 2.0% agarose gel and
ethidium bromide staining.
Plasmid identification and cell
transfection and bacterial transformation
PcDNA3.1-MEG3 plasmid was constructed as previously
described (16). PcDNA3.1-MEG3
plasmids were extracted from E. coli according to the
manufacturer's specifications. Both restriction enzymes,
XhoI and BamHI, were used to cut the pcDNA3.1-MEG3
plasmid DNA for 1 h at 37°C. Subsequently, DNA fragments after
digestion were identified by the agarose gel electrophoresis and
quantified by capturing the ethidium bromide absorbance with a
fluorescence microscope (Olympus BX51, Tokyo, Japan).
For ectopic MEG3 expression, the full-length MEG3
cDNA was subcloned into the pcDNA3.1 plasmid vector and transfected
into HepG2 or HuH7 cells as previously described (16). PcDNA3.1 was transfected to cells in
parallel as a control. The cells were then subjected to RNA/protein
extraction and further functional assays.
RNA extraction and qRT-PCR analysis
HepG2 and Huh7 cells were transfected with
pcDNA3.1-MEG3 vector or pcDNA3.1 for 48 h. The total RNA was
isolated with TRIzol reagent, according to the manufacturer's
protocol. For reverse transcriptase analysis, 1 μg of total RNA was
reverse-transcribed using random primers under standard conditions
using an EasyScript First-Strand cDNA Synthesis Super Mix kit.
Real-time PCR amplification with one microliter of the reverse
transcriptase reaction mixture was performed on the ABI 7500
real-time PCR system (Applied Biosystems, Foster City, CA, USA)
using SYBR Premix Ex Taq following the manufacturer's instructions.
PCR conditions were 95°C for 30 sec, followed by 40 cycles of 95°C
for 3 sec and 60°C for 30 sec. Gene-specific primers are listed in
Table I. 18s rRNA was used as a
housekeeper gene for the qRT-PCR reactions. Fold change of MEG3,
p53, MDM2, caspase-3 and cyclin D1 was calculated by the
2−ΔΔCt method.
 | Table IPrimer sequences for quantitative
real-time PCR. |
Table I
Primer sequences for quantitative
real-time PCR.
| Gene | Primer
sequences |
|---|
| MEG3 | F:
5′-CTCAGGCAGGATCTGGCATA-3prime;
R: 5prime;-CCTGGAGTGCTGTTGGAGAA-3prime; |
| p53 | F:
5prime;-TGAAGCTCCCAGAATGCCAG-3prime;
R: 5prime;-GGGAGTACGTGCAAGTCACA-3prime; |
| MDM-2 | F:
5prime;-ATCAGGCAGGGGAGAGTGAT-3prime;
R: 5prime;-CAATTCTCACGAAGGGCCCA-3prime; |
| Caspase-3 | F:
5prime;-AGAGCTGGACTGCGGTATTGGAG-3prime;
R: 5prime;-GAACCATGACCCGTCCCTTG-3prime; |
| Cyclin D1 | F:
5prime;-GTGCATCTACACCGACAACTCCA-3prime;
R: 5prime;-GTGCATCTACACCGACAACTCCA-3prime; |
| 18S | F:
5prime;-AAACGGCTACCACATCCAAG-3prime;
R: 5prime;-CAATTACAGGGCCTCGAAAG-3prime; |
Protein extraction and western blot
analysis
Cells were washed with ice-cold PBS, harvested and
lysed in RIPA buffer supplemented with protease inhibitor cocktail.
Cell lysates were centrifuged at 12,000 rpm at 4°C for 10 min. The
supernatant was boiled for 5 min and subjected to 12% SDS-PAGE and
transferred to a nitrocellulose membrane. The membranes were
blocked with 5% fat-free milk for 1 h at room temperature, followed
by incubation with primary antibodies against β-actin (1:3,000),
Mdm2 (1:500), cyclin D1 (1:500), total caspase-3 (1:800), cleaved
caspase-3 (1:800) and ILF3 (1:1,000) at 4°C overnight. The next
day, the membranes were incubated with biotin-conjugated secondary
antibody for 1 h at room temperature, followed by incubation with
an HRP complex at room temperature for 30 min. Bands were
visualized with an ECL detection system (Thermo Scientific,
Waltham, MA, USA). Protein expression was analyzed by the Quantity
One software (Bio-Rad, Hercules, CA, USA) and normalized to that of
β-actin.
Identification of proteins associated
with lncRNA MEG3
An improved MS2-BioTRAP (MS2 biotin tagged RNA
affinity purification) technique was used which relies on the
interaction of the bacteriophage MS2 coat-protein with a specific
viral RNA stem-loop structure that was inserted into or grafted
onto the 3′ end of the lncRNA MEG3 (17). Twelve copies of the MS2
coat-protein binding site were introduced into the region encoding
the MEG3 to generate a MEG3-MS2bs expression plasmid. This
expression plasmid and a second plasmid encoding a Flag-tagged MS2
coat-protein were transiently introduced into cultured HEK293
cells. In brief, the pcDNA3.1-Flag-MS2 vector, pcDNA3.1-MS2bs
vector and pcDNA3.1-MEG3-MS2bs vector were constructed as
previously described by us (16).
HEK293 cells are cultured and harvest at 80% confluence, followed
by co-transfection with pcDNA3.1-Flag-MS2 vector and
pcDNA3.1-MEG3-MS2bs using Lipofectamine 2000. Cells co-transfected
with pcDNA3.1 and pcDNA3.1-MEG3-MS2bs vector were used as a
control. Cells were further incubated for 48 h, then collected for
RIP experiments. The protein components were determined by mass
spectrometry, and the candidate proteins were confirmed by western
blot analysis.
RNA binding protein
immunoprecipitation
HEK293 cells were harvested and lysed in RIP lysis
buffer with protein inhibitor, RNA inhibitor and DNase I on ice.
Cell lysates were preincubated with the antibody. RNAs were
immunoprecipitated with the antibody against RBP and protein A/G
magnetic beads. The magnetic bead bound complexes were immobilized
with a magnet and unbound material was washed off. Then, RNAs were
extracted, precipitated and turned into a cDNA library by ImProm-II
reverse Transcription system (Promega, USA). The remaining beads
were boiled and separated by SDS-PAGE gel for western blot
analysis.
Statistical analysis
All the experiments described were performed at
least in triplicate. The data are expressed as mean ± SD.
Statistical significance was calculated using either the Student's
t-test, or one-way analysis of variance (ANOVA) with a post-hoc
test of multiple comparisons. A p<0.05 was considered
statistically significant.
Results
Downregulation of MEG3 is due to
hypermethylation in the MEG3 promoter region
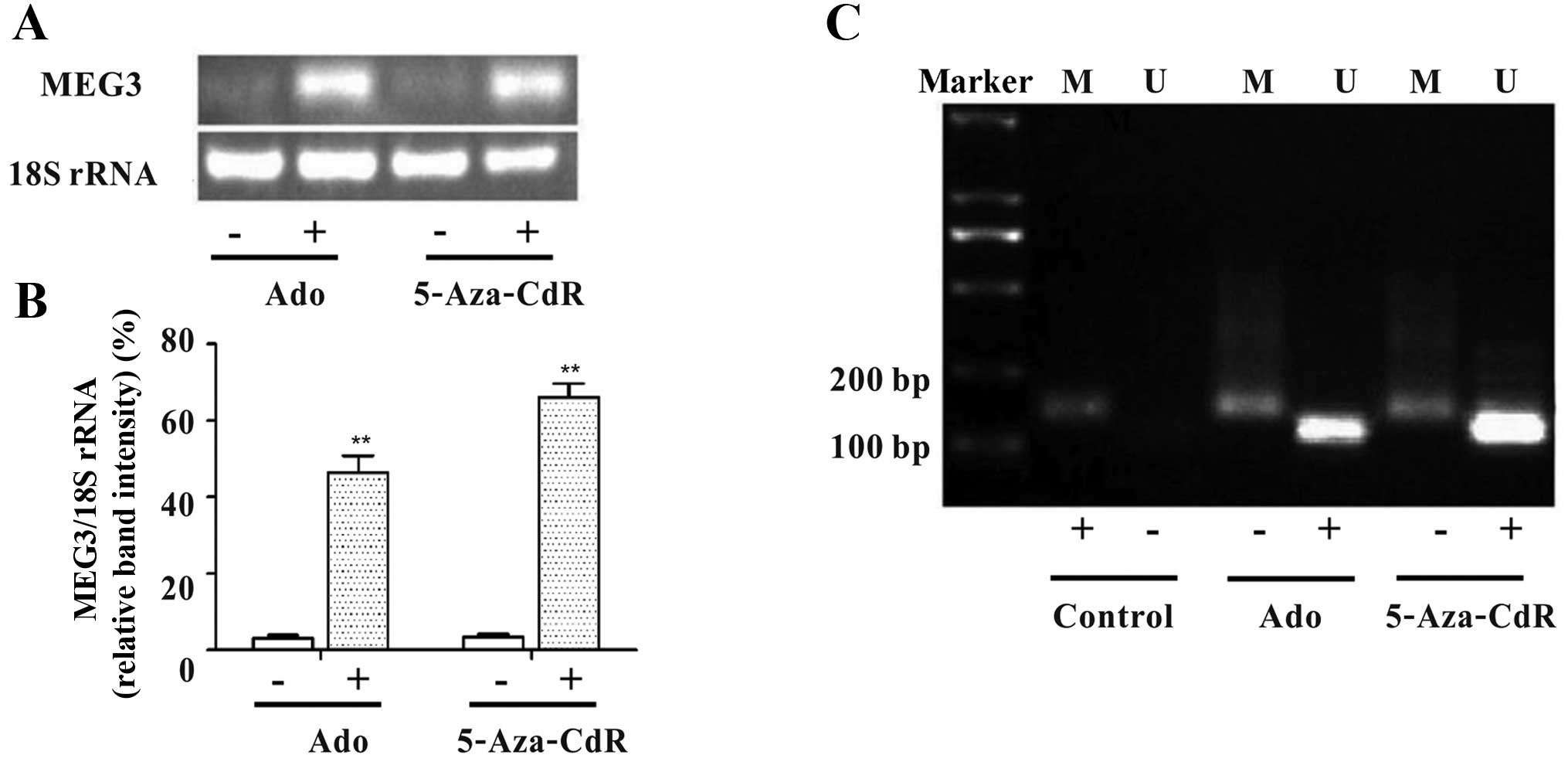
As shown in Fig. 1A and
B, the mRNA relative expression level of MEG3 significantly
increased after treatment with adenosine or 5-Aza-CdR (Fig. 1A and B). The unmethylated pattern
(U) and a methylated pattern (M) were observed in the cells treated
with adenosine or 5-Aza-CdR by MSP. The two DNA methyltransferase
inhibitors, which reverse the abnormal methylation status of the
MEG3 promoter, increased the expression of unmethylated product
(120 bp, Fig. 1C). The results
indicate that the methylation status of MEG3 promoter may lead to
downregulation of MEG3 expression in HepG2 cells.
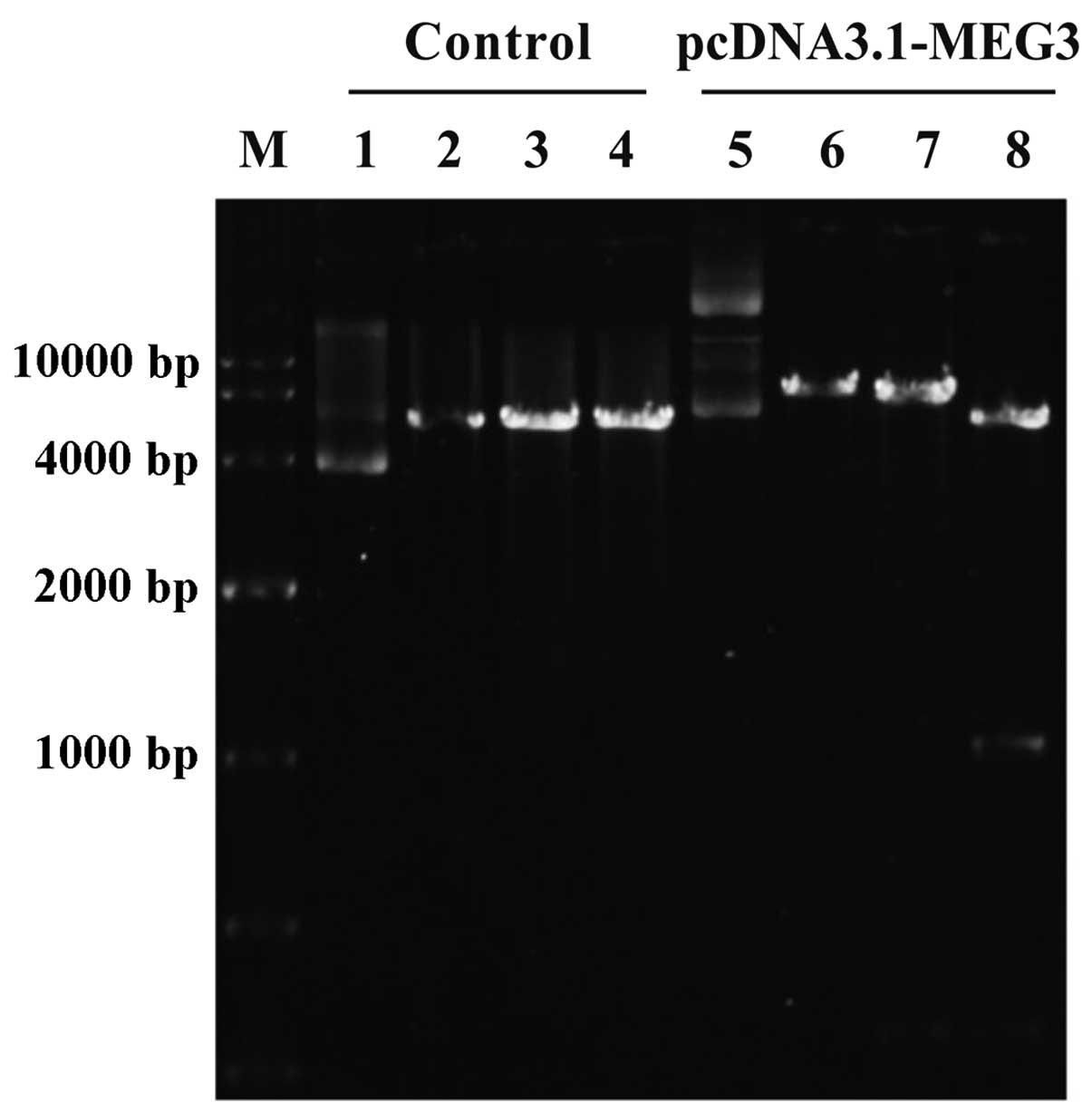
PcDNA3.1-MEG3 plasmid is constructed and
identified
PcDNA3.1-MEG3 plasmid that expressed lncRNA MEG3 was
constructed. LncRNA MEG3 gene was inserted into the pcDNA3.1
vector, which was confirmed by XhoI and BamHI
restriction enzyme digestion. The particular sequence of MEG3
insertion into vectors was the same as designed and the fragment
was not present in the control vector, showing pcDNA 3.1-MEG3
plasmid was successfully constructed (Fig. 2).
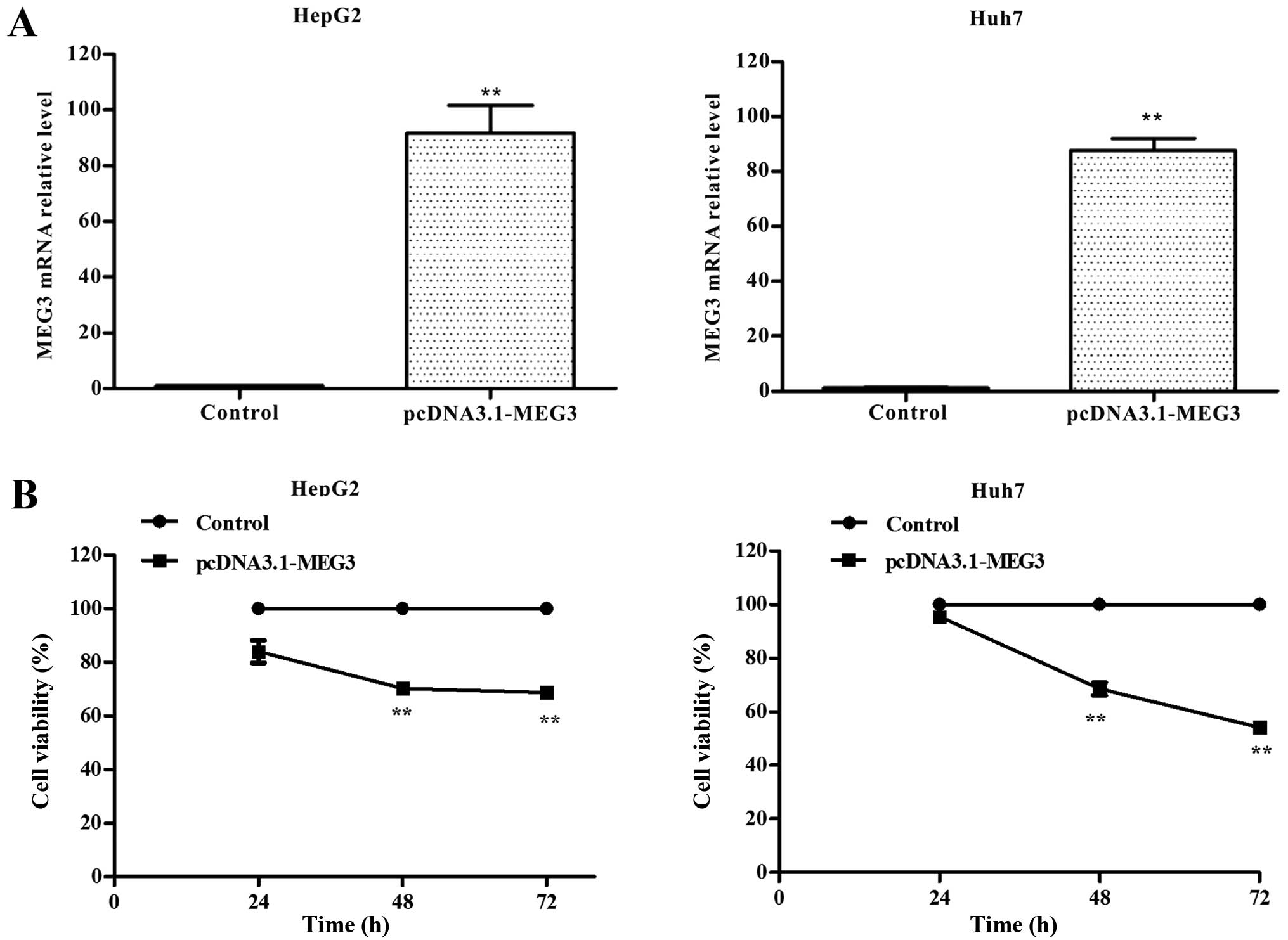
Ectopic expression of MEG3 and its
effects on cell growth inhibition
HepG2 and Huh7 cells were transiently transfected
with either pcDNA3.1 (control) or pcDNA3.1-MEG3, and MEG3 mRNA
expression was measured by qRT-PCR. Compared to the control group,
MEG3 mRNA expression levels increased by >90-fold or 80-fold in
MEG3-transfected HepG2 or Huh7 cells (Fig. 3A), showing the high level mRNA
expression of MEG3 was obtained in cells.
The effect of MEG3 on the proliferation of hepatoma
cells was measured using the MTT assay. Ectopic expression of MEG3
significantly decreased cell vitality rate by 29.6 and 31.2% after
48 and 72 h in HepG2 cells, respectively, compared with the control
group (p<0.01). Ectopic expression of MEG3 significantly
decreased cell vitality rate by 31.5 and 45.9% after 48 and 72 h in
Huh7 cells, respectively, compared with the control group (Fig. 3B, p<0.01). These results
suggested that ectopic expression of MEG3 can inhibit tumor cell
proliferation.
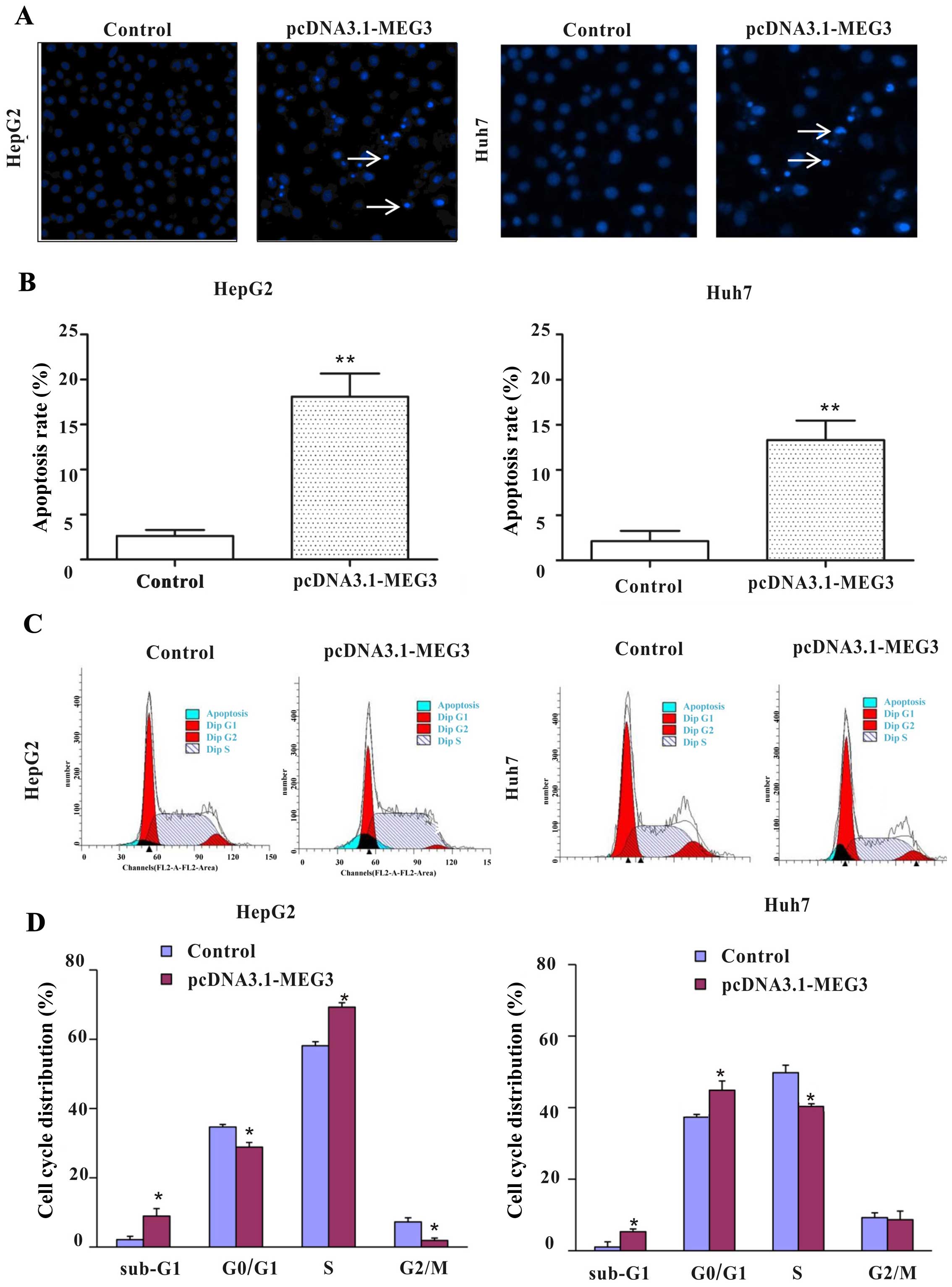
MEG3 induces cell apoptosis and cell
cycle arrest in vitro
To detect whether apoptosis was a contributing
factor to cell growth inhibition, hepatoma cells were stained with
Hoechst 33258 after transfection with plasmids. The nuclei of the
control group were uniform and without condensation or
fragmentation. Compared with the control group (pcDNA3.1), nuclei
in cells with ectopic expression of MEG3 became condensed and
shrunken, and typical apoptotic bodies appeared. The percentages of
apoptosis cells after MEG3 plasmid transfection are 18.1 and 13.3%
in HepG2 and Huh7 cells, respectively, which is significantly
higher than that of the control group (Fig. 4C, p<0.01).
In order to further investigate whether MEG3-induced
apoptosis is related to the cell cycle, the cell cycle distribution
was detected by flow cytometry. Ectopic expression of MEG3
significantly decreased the cells of G0/G1 and G2/M phase, but
increased the percentage of S phase from 58.08 to 69.27% in HepG2
cells. Ectopic expression of MEG3 significantly increased the
percentage of G0/G1 phase from 38.50 to 48.89% and decreased the
percentage of G2/S phase in Huh7 cells (Fig. 4C and D, p<0.05). Statistical
results showed that ectopic expression of MEG3 increased the
percentage of apoptotic cells (sub-G1 phase) in both cell types
(p<0.05, Fig. 4D), which is
consistent with the result of fluorescence microscopy observations
(Fig. 4B). Taken together, these
results confirm that cell growth inhibition and cell cycle arrest
caused by MEG3 is facilitated by the induction of apoptosis.
MEG3-triggered apoptosis is mediated by
p53 pathway and cell cycle arrest
To probe the mechanism by which MEG3 initiated the
cell cycle arrest, the investigation was focused on the cell cycle
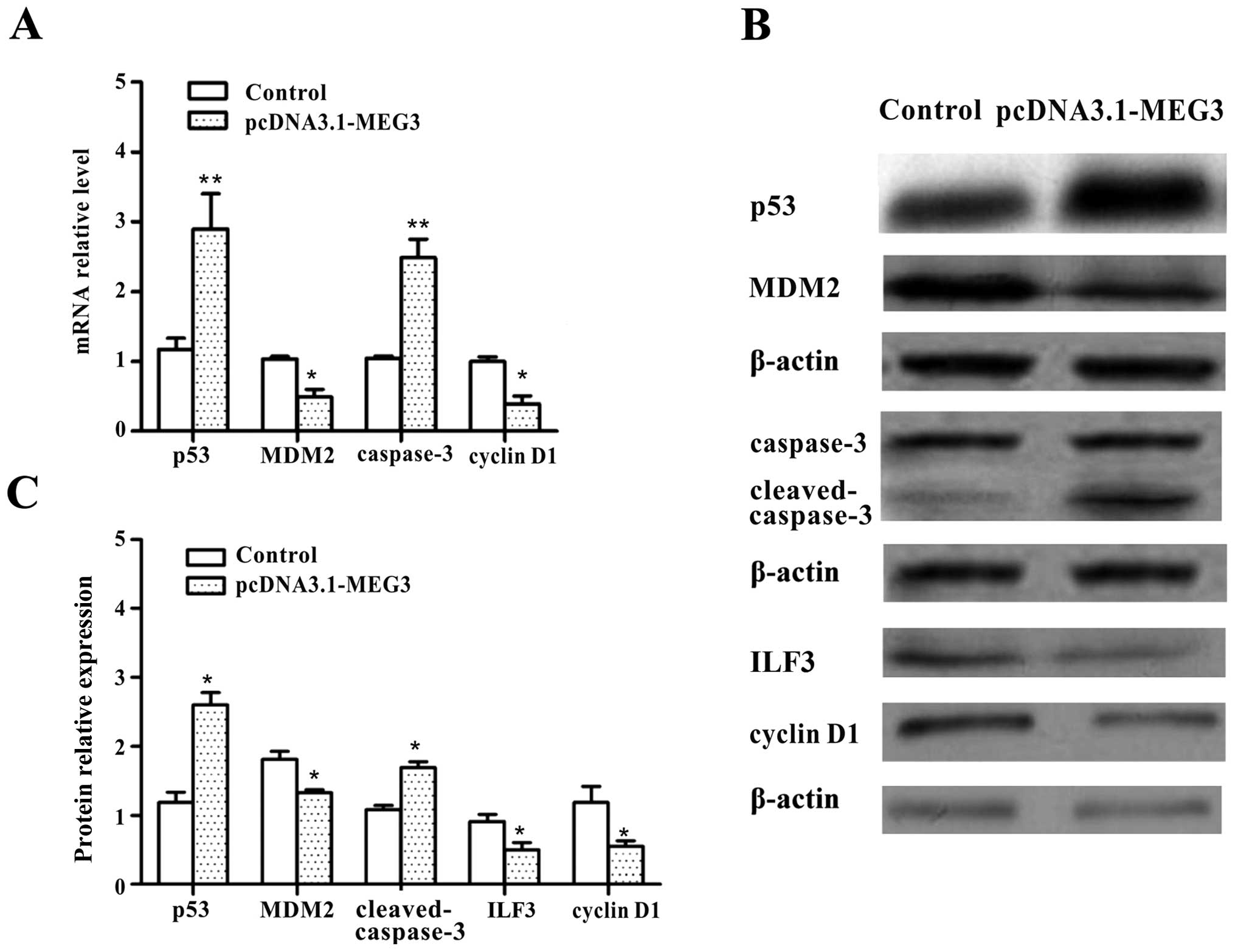
checkpoint, the cyclins (cyclin D1), p53 and other relative
factors. Ectopic expression of MEG3 resulted in marked reductions
in the mRNA and protein expression of cyclin D1 and MDM2. By
contrast, the expression of p53 and caspase-3 mRNA and protein
levels significantly increased in HepG2 cells (Fig. 5A–C, p<0.05). These data confirm
that MEG3 functions as a tumor suppressor gene by activating the
p53-mediated apoptosis pathway and causes cell cycle arrest in
HepG2 cells.
ILF3 was also investigated by western blot analysis.
The result showed that the expression level of ILF3 decreased after
transfection with pcDNA3.1-MEG3, compared with the control group
(Fig. 5B and C, p<0.05). The
data indicated that ILF3 protein expression is negatively regulated
by MEG3 in HepG2 cells.
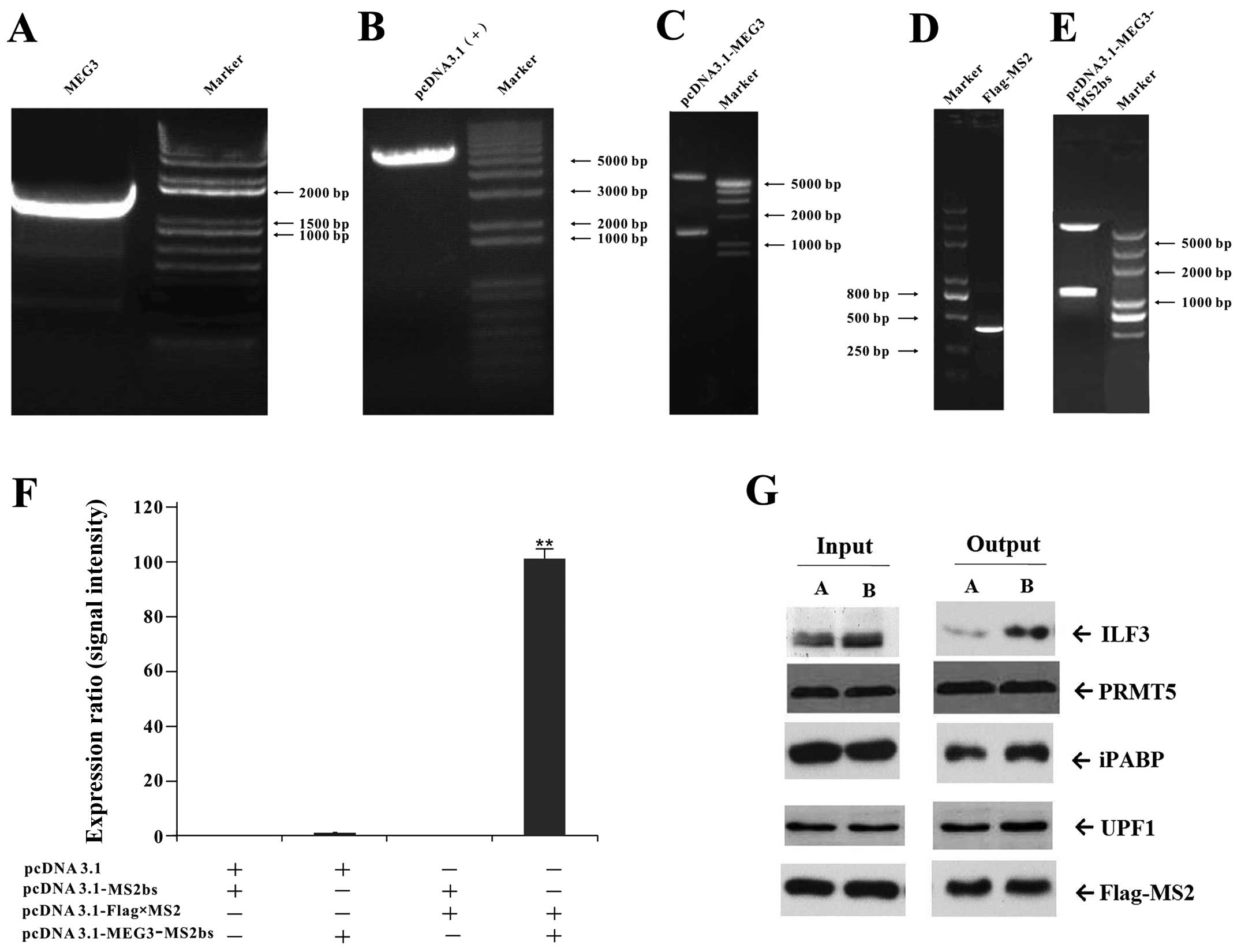
Screening of MEG3-binding protein
MS2-BioTRAP (MS2 biotin tagged RNA affinity
purification) experiment was carried out (Fig. 6). Sequence analysis and restriction
endonuclease analysis of target genes demonstrated that the
position and size of target gene cDNA insertion were consistent
with the design (Fig. 6A–E). The
pcDNA3.1-Flag-MS2 and pcDNA3.1-MEG3-MS2bs vectors were
co-transfected into HEK293 cells for the RIP experiment. The
expression intensity of MEG3 mRNA in co-transfected group with
pcDNA3.1-Flag-MS2 and pcDNA3.1-MEG3-MS2bs is ~98-fold higher than
that of the control group, showing it is an effective method to
study RNA-protein interaction (Fig.
6F). The protein components were determined by mass
spectrometry, and the candidate proteins were confirmed by RNA
pulldown and western blot analysis. Of the four candidate proteins
selected in the co-expression network by GO analysis, ILF3 protein
expression obviously increased, showing ILF3 is a MEG3-binding
protein (Fig. 6G).
Discussion
DNA methylation is a major regulatory mechanism of
tumor epigenetic control and it has an important role in gene
transcription and splicing. It has been demonstrated that DNA
promoter hypermethylation is associated with inactivation of
several tumor suppressor genes, and these aberrant DNA methylations
are always accompanied by a progressive dysregulation of DNMT
expression (18). It is reported
that the promoter region of lncRNA MEG3 is rich in CpG
dinucleotides. The methylation pattern of the 2.1 kb 5-flanking
region of MEG3 contains functionally important sequences for gene
expression (19). In general
conditions, MEG3 is expressed in normal tissues, with high
expression in the brain and pituitary gland (10). Many tumor tissues only express low
MEG3, or even no MEG3 (9,20). In pituitary tumors and leukemia,
MEG3 promoter hypermethylation is also associated with the loss of
MEG3 expression (21,22). In our previous study, adenosine
that has been confirmed to have cytotoxic effects on tumor cells
also showed a demethylation role in HepG2 cells (15). In order to investigate the
relationship between methylation status of MEG3 promoter and the
expression of MEG3 gene, we treated hepatoma cells with adenosine
or another selective DNMT inhibitor the 5-Aza-CdR. Our results
showed that both adenosine and 5-Aza-CdR increased the expression
of the unmethylated product and MEG3 mRNA expression (Fig. 1), thus revealing that MEG3 is
correlated with a typical hypermethylation status in the promoter
region in HepG2 cells, and that we are the first to demonstrate
that adenosine can trigger MEG3 activation.
In order to further identify that MEG3 has an
antitumor role, pcDNA3.1-MEG3 plasmid vectors were constructed and
transfected into hepatoma cells. The results showed that ectopic
expression of MEG3 inhibited cell growth in a time-dependant manner
(Fig. 3B) and increased cell
apoptosis in HepG2 and Huh7 cell lines (Fig. 4A and C).
The cell cycle is divided into four distinct phases,
G1, S, G2 and M, and is tightly controlled by a series of key
components including cyclins, the cyclin-dependent kinases. In this
study, MEG3 induced cell cycle arrest significantly at the S phases
and increased cell cycle arrest at the G0/G1 phases in HepG2 cells,
accompanying by the decreased cyclin D1 expression, showing that
cyclin D1 is essential for cell cycle transition of G1 to S phase
and the decrease of cyclin D1 may lead to hampering cells from
G0/G1 phase (23). However, we
also observed that MEG3 caused different cell cycle arrest in two
hepatoma cells (Fig. 4) and we
considered the difference might be related to different p53 gene
types, for HepG2 is a wild-type p53 and Huh7 is mutant p53 cell
line.
Among all apoptosis-related genes, p53 is of
particular importance (24). Tight
regulation of the anti-proliferative activities of p53 is mostly
achieved by the MDM2 ubiquitin ligase, which targets p53 for
degradation (25). On the one
hand, MDM2/p53 forms an autoregulatory feedback loop that may be
needed to maintain a critical MDM2/p53 ratio within cells (26). On the other hand, an excess of MDM2
protein can abrogate the transcriptional activation of p53
(25). Factors that differentially
regulate ubiquitination of MDM2 and p53 may affect cell fate
profoundly as they can change the MDM2/p53 ratio (27). Because wild-type p53 is a tumor
suppressive factor while mutant p53 does not have this function, we
chose wild-type p53 HepG2 cells for our research. Fig. 5 shows that ectopic expression of
MEG3 decreased MDM2, and increased p53, and cleaved-caspase-3
expression in HepG2 cells, suggesting that downregulation of MDM2
contributes to p53 activation (28,29).
In addition, MEG3 might induce apoptosis through p53-independent
pathways since exogenous MEG3 also inhibits Huh7 cell
proliferation.
ILF3 is known to be a transcription factor and has
two main isoforms (NF90 and NF110) (30,31).
They participate in many aspects of RNA metabolism, including
transcription, degradation and translation. In this study, an
improved MS2-BioTRAP technique was used to screen the MEG3-binding
proteins. Of the four candidate proteins selected in the
co-expression network, ILF3 was identified as the binding protein
by RIP, RNA pulldown and western blot analysis in HEK293 cells
(Fig. 6). Zhu et al
demonstrated that MEG3 can interact with DNA domain of p53 protein
and regulate p53 target genes (32). Shamanna et al reported that
knockdown of ILF3 by RNA interference led to elevated levels of p53
proteins in HPV-derived HeLa and SiHa cells (33). We speculated that MEG3 and ILF3
might form a complex which participates in the regulation of target
genes (such as p53) and induce cell apoptosis. In this study, we
observed that the expression level of ILF3 decreased after
pcDNA3.1-MEG3 transfection in HepG2 cells. The mechanisms of ILF3
downregulation are due to increased ubiquitination and degradation,
or other reasons that need to be further investigated.
In conclusion, this study demonstrates that the
antitumor effect of adenosine is involved in MEG3 activation and
the hypermethylation of MEG3 promoter region may contribute to its
low expression. MEG3 negatively regulates hepatoma cell growth by
affecting cell cycle progression and inducing cell apoptosis.
Moreover, the present study identified that ILF3 is a MEG3 binding
protein and might participate in the anticancer regulation of
MEG3.
Acknowledgements
The authors are grateful for funding support from
the National Nature Science Foundation of China (no. 30972925) and
the Guangdong Natural Science Foundation in China (no.
2014A030313470). The authors would like to thank Professor En-min
Li; and Dong-yang Huang and Yu-cai Fu for excellent technical
support.
References
|
1
|
Bosetti C, Turati F and La Vecchia C:
Hepatocellular carcinoma epidemiology. Best Pract Res Clin
Gastroenterol. 28:753–770. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ponting CP, Oliver PL and Reik W:
Evolution and functions of long noncoding RNAs. Cell. 136:629–641.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mitra SA, Mitra AP and Triche TJ: A
central role for long non-coding RNA in cancer. Front Genet.
3:172012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Qin R, Chen Z, Ding Y, Hao J, Hu J and Guo
F: Long non-coding RNA MEG3 inhibits the proliferation of cervical
carcinoma cells through the induction of cell cycle arrest and
apoptosis. Neoplasma. 60:486–492. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang Z, Zhou L, Wu LM, Lai MC, Xie HY,
Zhang F and Zheng SS: Overexpression of long non-coding RNA HOTAIR
predicts tumor recurrence in hepatocellular carcinoma patients
following liver transplantation. Ann Surg Oncol. 18:1243–1250.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Miyoshi N, Wagatsuma H, Wakana S,
Shiroishi T, Nomura M, Aisaka K, Kohda T, Surani MA, Kaneko-Ishino
T and Ishino F: Identification of an imprinted gene, Meg3/Gtl2 and
its human homologue MEG3, first mapped on mouse distal chromosome
12 and human chromosome 14q. Genes Cells. 5:211–220. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou Y, Zhong Y, Wang Y, Zhang X, Batista
DL, Gejman R, Ansell PJ, Zhao J, Weng C and Klibanski A: Activation
of p53 by MEG3 non-coding RNA. J Biol Chem. 282:24731–24742. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou S, Wang J and Zhang Z: An emerging
understanding of long noncoding RNAs in kidney cancer. J Cancer Res
Clin Oncol. 140:1989–1995. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhou Y, Zhang X and Klibanski A: MEG3
noncoding RNA: A tumor suppressor. J Mol Endocrinol. 48:R45–R53.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang X, Zhou Y, Mehta KR, Danila DC,
Scolavino S, Johnson SR and Klibanski A: A pituitary-derived MEG3
isoform functions as a growth suppressor in tumor cells. J Clin
Endocrinol Metab. 88:5119–5126. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jia LF, Wei SB, Gan YH, Guo Y, Gong K,
Mitchelson K, Cheng J and Yu GY: Expression, regulation and roles
of miR-26a and MEG3 in tongue squamous cell carcinoma. Int J
Cancer. 135:2282–2293. 2014. View Article : Google Scholar
|
|
12
|
Kang W, Seol HJ, Seong DH, Kim J, Kim Y,
Kim SU, Nam DH and Joo KM: Adenosine potentiates the therapeutic
effects of neural stem cells expressing cytosine deaminase against
meta-static brain tumors. Oncol Rep. 30:1101–1106. 2013.PubMed/NCBI
|
|
13
|
Wu LF, Wei BL, Guo YT, Ye YQ, Li GP, Pu ZJ
and Feng JL: Apoptosis induced by adenosine involves endoplasmic
reticulum stress in EC109 cells. Int J Mol Med. 30:797–804.
2012.PubMed/NCBI
|
|
14
|
Wu LF, Ye YQ, Huang GY, Li HB, Li GP, Pu
ZJ, Wei BL and Feng JL: Involvement of endoplasmic reticulum stress
in adenosine-induced human hepatoma HepG2 cell apoptosis. Oncol
Rep. 26:73–79. 2011.PubMed/NCBI
|
|
15
|
Xiang MQ, Liu LX, De W, Ye YQ, Pu ZJ and
Wu LF: The mechanism of demethylation on adenosine and
homocysteine-induced apoptosis in HepG2 cells. Chin Pharmacol Bull.
30:973–979. 2014.
|
|
16
|
Wu LF, Guo YT, Zhang QH, Xiang MQ, Deng W,
Ye YQ, Pu ZJ, Feng JL and Huang GY: Enhanced antitumor effects of
adenoviral-mediated siRNA against GRP78 gene on adenosine-induced
apoptosis in human hepatoma HepG2 cells. Int J Mol Sci. 15:525–544.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gong C, Popp MW and Maquat LE: Biochemical
analysis of long non-coding RNA-containing ribonucleoprotein
complexes. Methods. 58:88–93. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lo PH, Tanikawa C, Katagiri T, Nakamura Y
and Matsuda K: Identification of novel epigenetically inactivated
gene PAMR1 in breast carcinoma. Oncol Rep. 33:267–273. 2015.
|
|
19
|
Zhao J, Dahle D, Zhou Y, Zhang X and
Klibanski A: Hyper-methylation of the promoter region is associated
with the loss of MEG3 gene expression in human pituitary tumors. J
Clin Endocrinol Metab. 90:2179–2186. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang X, Gejman R, Mahta A, Zhong Y, Rice
KA, Zhou Y, Cheunsuchon P, Louis DN and Klibanski A: Maternally
expressed gene 3, an imprinted noncoding RNA gene, is associated
with meningioma pathogenesis and progression. Cancer Res.
70:2350–2358. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gejman R, Batista DL, Zhong Y, Zhou Y,
Zhang X, Swearingen B, Stratakis CA, Hedley-Whyte ET and Klibanski
A: Selective loss of MEG3 expression and intergenic differentially
methylated region hypermethylation in the MEG3/DLK1 locus in human
clinically nonfunctioning pituitary adenomas. J Clin Endocrinol
Metab. 93:4119–4125. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Benetatos L, Hatzimichael E, Dasoula A,
Dranitsaris G, Tsiara S, Syrrou M, Georgiou I and Bourantas KL: CpG
methylation analysis of the MEG3 and SNRPN imprinted genes in acute
myeloid leukemia and myelodysplastic syndromes. Leuk Res.
34:148–153. 2010. View Article : Google Scholar
|
|
23
|
Shirali S, Aghaei M, Shabani M, Fathi M,
Sohrabi M and Moeinifard M: Adenosine induces cell cycle arrest and
apoptosis via cyclin D1/Cdk4 and Bcl-2/Bax pathways in human
ovarian cancer cell line OVCAR-3. Tumour Biol. 34:1085–1095. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shan X, Fu YS, Aziz F, Wang XQ, Yan Q and
Liu JW: Ginsenoside Rg3 inhibits melanoma cell proliferation
through down-regulation of histone deacetylase 3 (HDAC3) and
increase of p53 acetylation. PLoS One. 9:e1154012014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Haupt Y, Maya R, Kazaz A and Oren M: Mdm2
promotes the rapid degradation of p53. Nature. 387:296–299. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu X, Bayle JH, Olson D and Levine AJ: The
p53-mdm-2 auto-regulatory feedback loop. Genes Dev. 7A:1126–1132.
1993. View Article : Google Scholar
|
|
27
|
Michael D and Oren M: The p53-Mdm2 module
and the ubiquitin system. Semin Cancer Biol. 13:49–58. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Clegg HV, Itahana Y, Itahana K, Ramalingam
S and Zhang Y: Mdm2 RING mutation enhances p53 transcriptional
activity and p53-p300 interaction. PLoS One. 7:e382122012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lu KH, Li W, Liu XH, Sun M, Zhang ML, Wu
WQ, Xie WP and Hou YY: Long non-coding RNA MEG3 inhibits NSCLC
cells proliferation and induces apoptosis by affecting p53
expression. BMC Cancer. 13:4612013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Monsalve M, Wu Z, Adelmant G, Puigserver
P, Fan M and Spiegelman BM: Direct coupling of transcription and
mRNA processing through the thermogenic coactivator PGC-1. Mol
Cell. 6:307–316. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ohno M, Kunimoto M, Nishizuka M, Osada S
and Imagawa M: Ku proteins function as corepressors to regulate
farnesoid X receptor-mediated gene expression. Biochem Biophys Res
Commun. 390:738–742. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhu J, Liu S, Ye F, Shen Y, Tie Y, Zhu J,
Wei L, Jin Y, Fu H, Wu Y, et al: Long noncoding RNA MEG3 interacts
with p53 protein and regulates partial p53 target genes in hepatoma
cells. PLoS One. 10:e01397902015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shamanna RA, Hoque M, Pe'ery T and Mathews
MB: Induction of p53, p21 and apoptosis by silencing the NF90/NF45
complex in human papilloma virus-transformed cervical carcinoma
cells. Oncogene. 32:5176–5185. 2013. View Article : Google Scholar
|