Introduction
Despite the advances in our understanding of cancer
biology, diagnosis and therapy in the past decades, pancreatic
cancer is still one of the deadliest types of human cancer, with a
median survival rate of less than 6 months (1,2).
Worldwide, there were an estimated 338,000 new cases of pancreatic
cancer and 330,000 pancreatic cancer death in 2012 (3). It is the only type of cancer with an
annual mortality rate which is so close to its annual incidence
rate. This mainly stems from its late diagnosis, and its resistance
to the current forms of therapy (4). Since its introduction in 1997,
gemcitabine is the gold standard for the treatment of locally
advanced and metastatic pancreatic cancer (5,6). In
addition, of the numerous agents investigated, only the addition of
erlotinib (OSI-744), an epidermal growth factor receptor (EGFR)
tyrosine kinase inhibitor, to gemcitabine therapy led to a modest,
nonetheless significant prolonged median overall survival in
pancreatic cancer patients (7–9). The
limited clinical benefit of erlotinib stems from the fact that the
majority of pancreatic cancer patients simply do not respond to
this treatment or acquire drug resistance following a short course
of therapy (10). While the
addition of two more effective chemotherapy combinations in the
last decade, such as FOLFIRINOX (fluorouracil, irinotecan and
oxaliplatin) and the combination of gemcitabine with Nab
(nanoparticle albumin-bound)-paclitaxel improved the median
survival rates for patients with metastatic disease, such
combinational approaches are only suitable for patients with good
performance status, with gemcitabine monotherapy remaining the only
option for patients with poor performance status (6,11).
The extremely limited progress in improving survival
outcomes in pancreatic cancer during the last decades, underlines
the need not only for the development of more effective inhibitors
for existing targets such as EGFR, but also it is imperative to
develop new targeted agents and combination therapies for
overcoming drug resistance.
We have shown previously that afatinib, an
irreversible ErbB family blocker, is superior at inhibiting the
growth of a panel of human pancreatic cancer cell lines compared to
first generation reversible EGFR inhibitors such as erlotinib
(OSI-774) or gefitinib (12,13).
As drug-resistance is a major cause of treatment failure,
development of drug-resistant pancreatic cancer models could help
in unravelling the molecular mechanisms of acquired resistance and
facilitate the discovery of novel and more effective approaches for
the treatment of pancreatic cancer patients. We have shown
previously that of seven pancreatic cancer cell lines investigated,
BxPc3 cells exhibited the highest sensitivity to targeted agents
afatinib and erlotinib (12). In
this study, we developed three variants of the human pancreatic
cancer cell line BxPc3, resistant to afatinib, erlotinib and
gemcitabine and investigated the possible molecular alterations
accompanying the acquisition of a drug-resistant phenotype. In
particular, we determined the expression levels of EGFR ligands,
putative cancer stem cells (CSCs) and epithelial mesenchymal
transition (EMT) markers, as well as the expression levels and
activation status of several receptor tyrosine kinases (RTKs)
including the HER family of receptors and several downstream cell
signalling molecules in the parental pancreatic cancer and its
drug-resistant variants. We also investigated the therapeutic
potential of several agents in the treatment of such drug-resistant
variants.
Materials and methods
Cell culture and tumour cell lines
BxPc3 pancreatic cancer cell line was purchased from
the American Type Culture Collection (ATCC). BxPc3 cells were
cultured routinely at 37°C in a humidified atmosphere (5%
CO2) in RPMI-1640 medium (Sigma-Aldrich, Dorset, UK)
supplemented with 10% foetal bovine serum (PAA, UK), antibiotics
penicillin (50 U/ml), streptomycin (0.05 mg/ml) and neomycin (0.1
mg/ml) and glutamine at a final concentration of 2 mM
(Sigma-Aldrich), as previously described (14).
Antibodies and other reagents
Primary mouse antibodies HM50.67A and HM43.16B, were
raised against the external domain of the HER-2 and EGFR,
respectively (15). Mouse MAB3481
(anti-HER-3), MAB11311 (anti-HER-4), anti-insulin like growth
factor receptor I (IGF-IR) mAbs and anti-E-cadherin were purchased
from R&D Systems (Abingdon, UK). Secondary FITC-conjugated
rabbit anti-mouse mAb STAR9B was obtained from AbD Serotec (UK).
Gemcitabine was acquired from Healthcare at Home (UK) while The
irreversible pan-HER family blocker afatinib was developed by
Boehringer Ingelheim (Austria) as previously described (16,17).
OSI-774 was kindly provided by OSI-Phamarceutical (USA).
Doxycycline, 5-fluorouracil (5-FU), oxaliplatin, and mouse
anti-EGFR antibody were purchased from Sigma-Aldrich. Mouse
antibodies against β-actin and vimentin as well as rabbit
antibodies against AKT, Mitogen-activated protein kinase (MAPK),
phospho-MAPK (Thr202/Tyr204), phospho AKT (S473), Signal transducer
and activator of transcription 3 (STAT3), p-STAT3 (Y705), Src,
p-Src (Y416), c-MET (mouse), p-MET (Y1234/1235), p-EGFR (Y1086,
1068, 1143, 1173, 1045), p-HER3 (Y1289), HER3, HER2 and p-HER2
(Y1221/1222) were purchased from Cell Signalling, UK. The mouse
anti-p-IGF-IR (Y1161) antibody and STAT3 inhibitor Stattic were
purchased from Santa Cruz Biotechnology Inc. (Insight
Biotechnology, UK).
Establishment of drug-resistant cell
lines
We showed previously that of all pancreatic cancer
cell lines investigated, BxPc3 cells exhibited the highest overall
sensitivity to treatment with either HER-targeted or
chemotherapeutic agents (12).
Drug-resistant pancreatic cancer variants were developed by the
treatment of BxPc3 cells with escalating doses of afatinib,
erlotinib or gemcitabine. Cells were cultured routinely in small
cell culture flasks (25 cm2) in growth medium/10% FBS in
the presence of increasing doses of an inhibitor for a period of
over 6 months. Once tumour cells were able to maintain exponential
growth at the presence of at least 3× the IC50
concentration of the drug, they were passed 15 times in drug-free
medium and drug sensitivity was determined again to ensure that
drug resistance acquisition was permanent.
Migration studies
For migration studies, 200 μl of cell suspension at
a density of 2×105 cells/ml were mixed with 50 μl of
serum free medium alone or with the inhibitors and then seeded into
Transwell inserts (pore size 8 μm) of 24-well plates (Becton
Dickinson Ltd., UK). The lower chamber was filled with 750 ml of
growth medium supplemented with 10% FBS (as chemoattractant) and
cells were incubated at 37°C for 6 h. Following incubation,
non-migrated cells were removed from the Transwell insert (upper
surface of the membrane) using a cotton swab, and cells were fixed
with ice-cold methanol for 10 min at room temperature. Cells were
stained with haematoxylin and were then washed. The number of cells
that had migrated through the membrane was counted under a
microscope at ×100 magnification. Five fields were counted in total
for each sample. Results are expressed as the average number of
migrated cells.
Flow cytometry
The cell surface expression of putative pancreatic
CSCs (CD44, CD24 and CD133), HER family members (EGFR, HER-2, HER-3
and HER-4), IGF-IR and c-MET was assessed by flow cytometry as
previously described (12). A
minimum of 10,000 events were recorded following excitation with an
argon laser at 488 nm using the FL-1 detector (525 nm) of a BD
FACsCalibur flow cytometer (Becton Dickinson Ltd.). Mean
fluorescence intensity values were calculated using the CellQuest
Pro software (Becton Dickinson Ltd.) and compared with those of
negative controls (no primary antibody).
Growth inhibition studies
The effect of the various agents, on the growth of
human cancer cell lines was investigated using the sulforhodamine B
(SRB; Sigma-Aldrich) colorimetric assay as previously described
(12). Interactions between the
different agents when used in combination were assessed, using the
combination index (CI) as described by Chou and Talalay (18). For each combination the two drugs
were mixed at their 8X IC50 followed by 8 doubling
dilutions. Interpretation of the results was based on the proposed
descriptions for presenting the degrees of antagonism or synergism
by Calcusyn software. In general, CI<0.9 indicates a synergistic
effect while CI between 0.90–1.10 denotes an additive effect.
CI>1.1 indicates antagonistic effects. Data analysis was
performed using the Calcusyn software (Biosoft, Cambridge, UK).
Determination of autocrine ligand
production by tumour cells
The level of autocrine EGFR ligands [EGF, TGF-α,
beta-cellulin, amphiregulin and heparin-binding EGF (HB-EGF)]
secreted by the tumour cells into the culture supernatant was
determined using the R&D Duoset ELISA kit following the
manufacturer's instructions (R&D Systems). Briefly, tumour
cells were grown in wells in a 6-well plate with 5 ml of growth
medium supplemented with 10% FBS until almost confluent. Growth
medium was replaced with fresh serum-free medium and incubated
overnight at 37°C. Supernatants were collected from each well and
then the number of cells in each well was determined for all
samples.
A standard curve was created for each ligand
investigated, using a four parameter logistic (4-PL) curve-fit.
Concentration of ligands in cell supernatants was determined from
each standard curve using GraphPad prism 6 software (GraphPad
Software, Inc., La Jolla, CA, USA).
Analysis of receptor tyrosine kinase
(RTK) phosphorylation status by the Proteome Profiler 96 Human
Phospho-RTK Array 1
The basal phosphorylation status of 16 RTKs (EGFR,
HER-2, HER-3, HER-4, HGF-R (c-MET), IGF-IR, INS-R, M-CSF R, MSP-R,
PDGFRa, PDGFRb, SCF R, Tie-2, VEGFR1, VEGFR2, VEGFR3) was
investigated in BxPc3 parental cells and its drug-resistant
variants using the Proteome Profiler 96 Human Phospho-RTK Array 1
(Catalog # ARZ001) following the manufacturer's instructions
(R&D Systems). The plate was imaged using a G-box imaging
system (Invitrogen, UK) and data analysis was performed using
Q-view software (Quansys Biosciences, Logan, UT, USA).
Western blotting
Parental cancer cells and drug-resistant variants
were grown to near confluency in 6-well culture plates containing 5
ml of 10% FBS RPMI growth medium. Cells were washed once with 5 ml
of RPMI/0.5% FBS, lysed using 400 μl of lithium dodecyl sulfate
(LDS) lysis buffer (Invitrogen) containing protease inhibitor
cocktail (Sigma-Aldrich). Cell lysates were heated at 90°C for 5
min, protein samples (25 μg) were separated on 4% to 12% Bis-Tris
gels (Invitrogen) and transferred to polyvinylidene difluoride
(PVDF) membranes (Invitrogen). The PVDF membranes were probed with
antibodies against several cell signalling molecules at optimal
concentrations according to the manufacturer's instructions. The
specific signals were detected using the WesternBreeze
chemiluminescence kit (Alkaline phosphatase conjugated secondary
antibody) (Invitrogen). Results were visualized using the G-box
imaging system (Syngene, Cambridge, UK).
Mutational analysis by next generation
sequencing
Characterization of somatic mutations in the
parental BxPc3 cell line and its drug-resistant clones was
performed by using the Ion AmpliSeq™ Cancer Hotspot Panel v2
(CHPv2) (Life Technologies, Paisley, UK) following the
manufacturer's instructions. Ion AmpliSeq CHPv2 is a next
generation sequencing assay which can identify numerous somatic
mutations [2855 hot spots/catalogue of somatic mutations in cancer
(COSMIC) mutations] from 50 genes including EGFR, HER2, KRAS, p53,
PIK3CA, PTEN and c-MET among others. For a detailed list of all
genes and hotspots included in the assay please see Table I). Genomic DNA was extracted from
each cancer cell line using the AllPrep DNA/RNA/Protein Mini kit
(Qiagen, UK) according to the manufacturer's instructions. Analysis
of sequencing data was performed using the Ion Reporter software
(Life Technologies) and confirmed by using NextGENe®
software (Softgenetics, UK).
 | Table IChanges in the sensitivity of drug
resistant variants to various inhibitors. |
Table I
Changes in the sensitivity of drug
resistant variants to various inhibitors.
|
IC50 |
|---|
|
|
|---|
| Cell line | Gemcitabine | 5-FU | Doxycycline | Oxaliplatin | Afatinib | Erlotinib | Gefitinib | NVP-AEW541 | Crizotinib |
|---|
| BxPc3 parental | 7.5 nM | 655 nM | 11 μM | 3.8 μM | 17 nM | 1.45 μM | 2.3 μM | 1.25 μM | 1.71 μM |
| BxPc3AFR | 386 nM | 555 nM | 10.68 μM | 3.2 μM | 1.32 μM | 4.3 μM | 8.3 μM | 1.63 μM | 1.46 μM |
| Fold change |
51.46a | 0.84 | 0.97 |
0.84a |
77.64a |
2.96a |
3.55a | 1.30 | 0.85 |
| BxPc3GEMR | 663 nM | 613.5 nM | 7.2 μM | 1.34 μM | 1.2 μM | 6.1 μM | 5.545 μM | 1.34 μM | 1.24 μM |
| Fold change |
88.4a | 0.93 | 0.75 |
0.35a |
70.88a |
4.20a |
2.37a | 1.07 | 0.72 |
| BxPc3OSIR | 507.5 nM | 1.2 μM | 7.3 μM | 11.25 μM | 3.1 μM | 5.25 μM | 6.4 μM | 3.25 μM | 1.61 μM |
| Fold change |
67.66a |
1.83a | 0.66 |
2.96a |
182.3a |
3.62a |
2.74a |
2.58a | 0.93 |
Statistical analysis
The unpaired two-tailed Student's t-test was used
for comparing mean values between two groups. Data are presented as
mean ± SD. p<0.05 was considered statistically significant.
Results
Establishment of drug-resistant
pancreatic cancer cell lines
In this study, we established three variant forms of
BxPc3 cells; with acquired resistance to gemcitabine (BxPc3GEMR),
afatinib (BxPc3AFR) and erlotinib (BxPc3OSIR). IC50
values for each drug in the parental cells and their drug-resistant
variants, following at least 15 passages in drug-free medium, are
presented in Table I. The
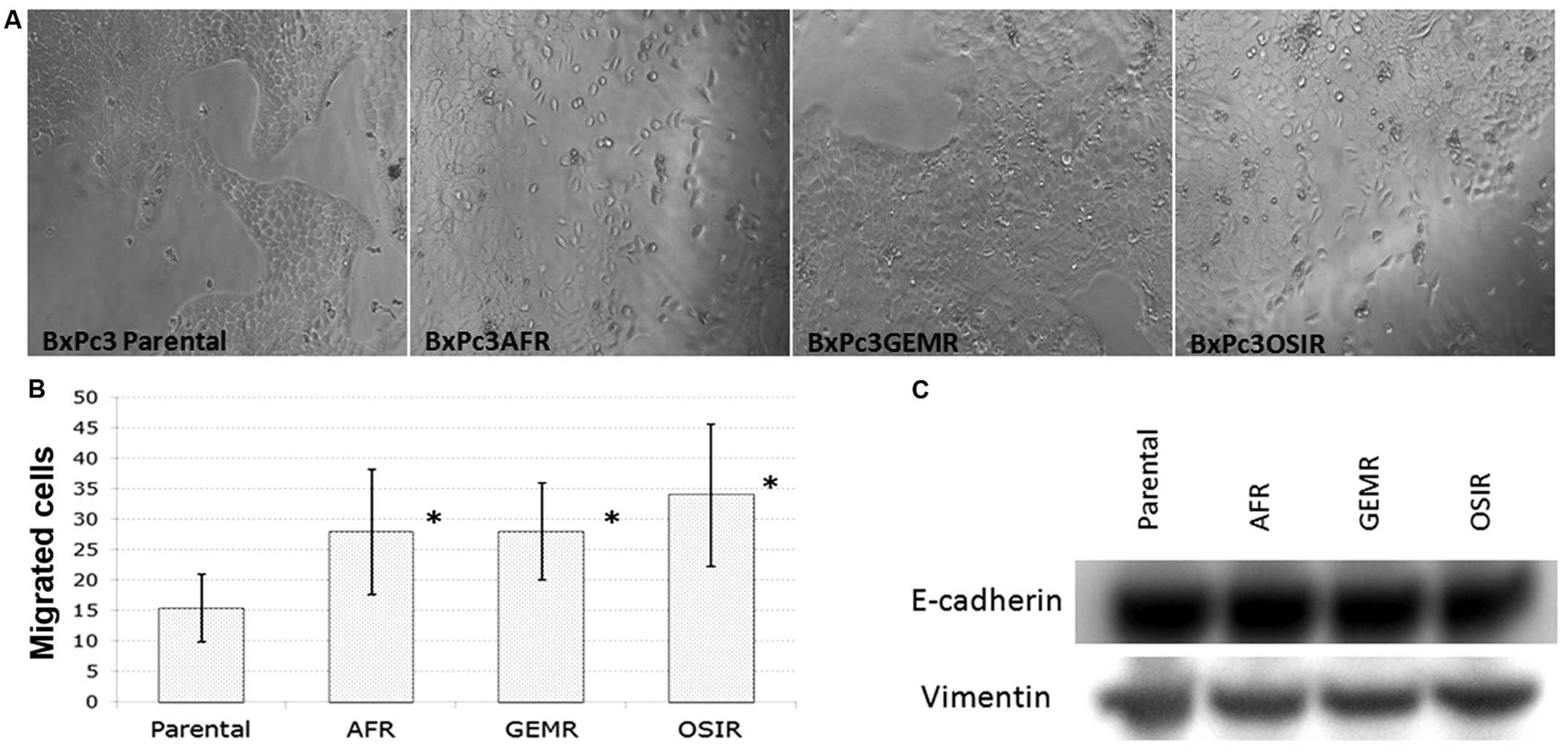
morphology of the drug-resistant variants is presented in Fig. 1A. All BxPc3 derived drug-resistant
cell lines, gained increased migration ability compared to the
parental cell line indicating a higher metastatic potential
(Fig. 1B). However, there were no
changes in the expression of EMT markers (vimentin and E-cadherin)
in the drug-resistant variants compared to the parental cell line
(Fig. 1C).
Growth response of parental and
drug-resistant pancreatic cancer cell variants to treatment with
various agents following acquisition of drug resistance
All three drug-resistant variants exhibited
significant changes in their sensitivity to treatment with several
agents compared to their parental counterpart (Table I). For example, BxPc3AFR variant
with acquired resistance to afatinib (77.64-fold change,
p<0.05), also developed resistance to erlotinib (2.96-fold
change, p<0.05), gefitinib (3.55-fold change, p<0.05) and
gemcitabine (51.46-fold change, p<0.05) (Table I). Similarly, BxPc3GEMR and
BxPc3OSIR variants demonstrated a reduced sensitivity to treatment
with gemcitabine, afatinib, erlotinib and gefitinib. For example,
BxPc3OSIR variant in addition to erlotinib, became highly resistant
to treatment with afatinib with 182-fold increase in its
IC50 value (p<0.05) (Table I). However, the changes in
sensitivity to other agents differed considerably in each of these
cell lines. For example, in comparison to the parental BxPc3 cells,
both BxPc3AFR and BxPc3GEMR cell lines became more sensitive to
treatment with oxaliplatin, while BxPc3OSIR cells became less
sensitive (Table I).
Acquisition of resistance to treatment
with afatinib and gemcitabine is accompanied by upregulation of
EGFR ligand amphiregulin (AR) in pancreatic cancer variants
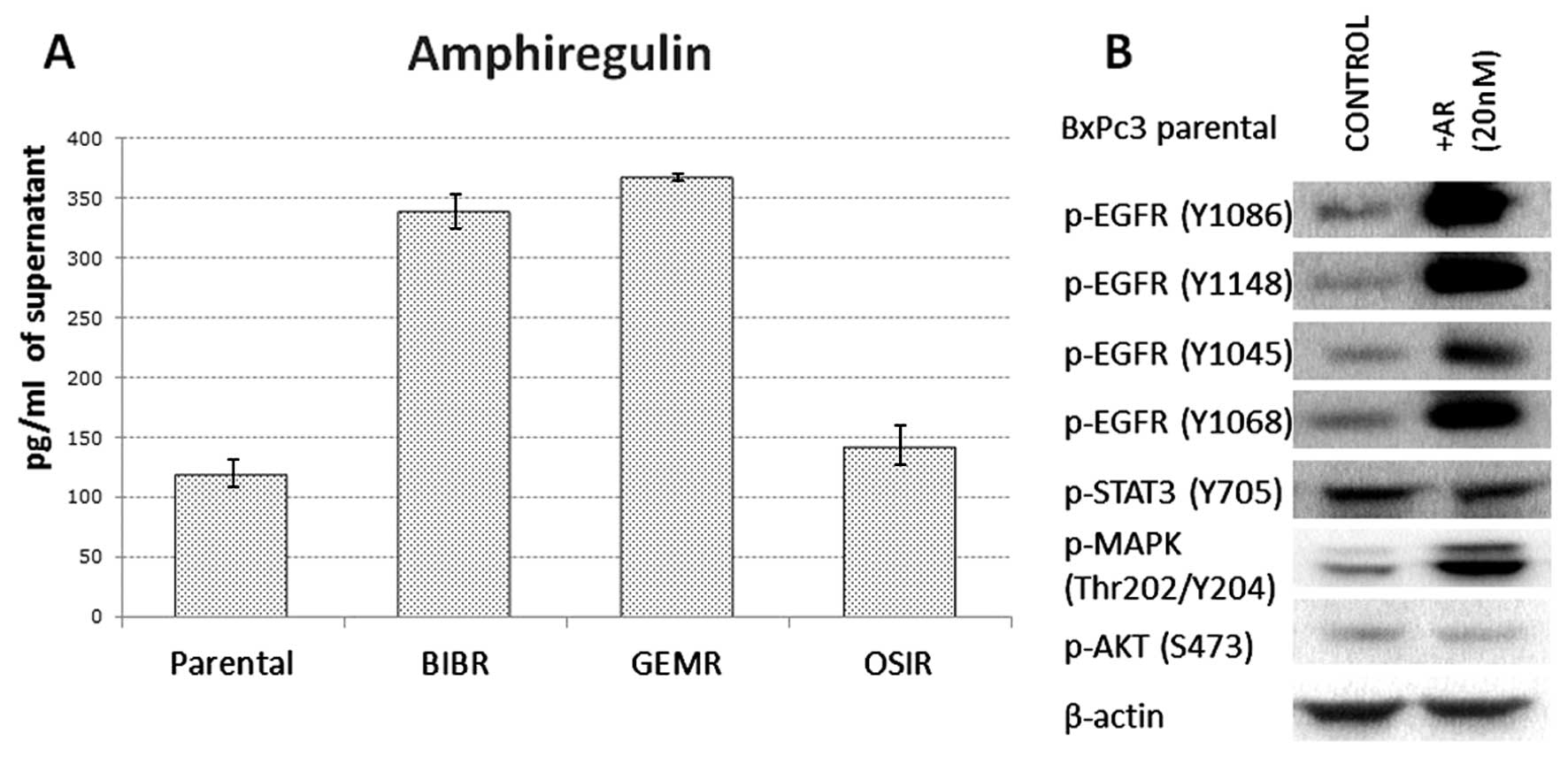
Of all growth factors investigated, BxPc3 parental
cell line was found to secrete only AR. Of note, acquired
resistance to afatinib and gemcitabine in pancreatic cancer cells
was accompanied by a 3-fold increase in secretion of AR. The
concentration of AR secreted by BxPc3, BxPc3AFR, BxPc3GEMR and
BxPc3OSIR was 120, 338.5, 367.8 and 142.9 pg/ml, respectively
(Fig. 2A). Treatment of parental
BxPc3 cells with 20 nM of AR for 15 min was accompanied by
increased phosphorylation of all EGFR tyrosine residues and MAPK
but it had no effect on the phosphorylation status of STAT3 and AKT
(Fig. 2B).
Expression levels of putative pancreatic
CSC markers, HER family members, IGF-IR and c-MET in drug
resistance variants
The cell surface expression of putative pancreatic
cancer CSC markers CD44, CD24 and CD133, as well as all members of
the HER family, IGF-IR and c-MET was analysed in the parental BxPc3
cell line and its drug-resistant variants by flow cytometry. Both
BxPc3AFR and BxPc3OSIR variants exhibited a statistically
significant increase in CD44 expression with a 3.14- and 2.23-fold
change (p<0.05), respectively, compared to the parental BxPc3
cells. No differences were observed in the expression of CD24 while
all cell lines were found to be negative for CD133 (Table II). There was a small decrease in
the expression of several markers in BxPc3GEMR, however, none of
these was found to be statistically significant (Table II).
| Table IIChanges in the expression of CSC
markers (CD44, CD24 and CD133), HER family members, IGF-IR and
c-MET in drug resistant established cell lines relative to their
parental counterparts. |
Table II
Changes in the expression of CSC
markers (CD44, CD24 and CD133), HER family members, IGF-IR and
c-MET in drug resistant established cell lines relative to their
parental counterparts.
| Cell line | EGFR | HER-2 | HER-3 | HER-4 | IGF-IR | c-MET | CD44 | CD24 | CD133 |
|---|
| BxPc3AFR | 0.97 | 0.59 | 1.07 | N/A | 1.04 | 0.96 |
3.14a | 0.69 | N/A |
| BxPc3GEMR | 0.69 |
0.66a | 0.70 | N/A | 0.76 | 0.84 | 0.86 | 0.60 | N/A |
| BxPc3OSIR | 1.17 | 0.78 | 1.34 | N/A | 1.07 | 0.80 |
2.23a | 0.83 | N/A |
Upregulation of phosphorylated c-MET and
STAT3 in drug-resistant variants
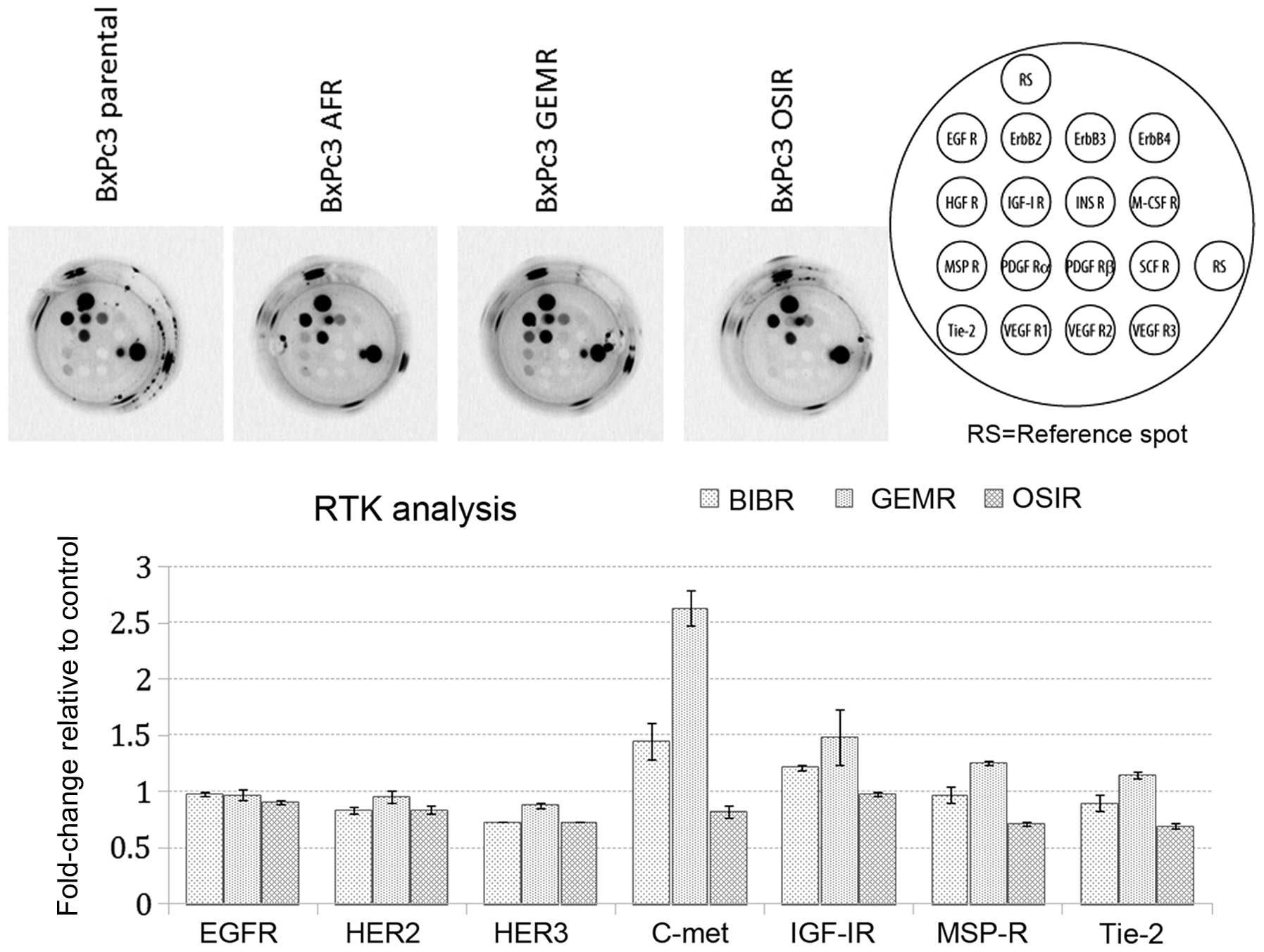
Next, we investigated the basal phosphorylation
status of 16 RTKs in BxPc3 parental cells and its drug-resistant
variants. Our aim was to determine whether acquisition of
resistance was accompanied by any changes in the phosphorylation
status of major RTKs. Noteworthy, detectable levels of
phosphorylated c-MET receptor were observed only in BxPc3AFR and
BxPc3GEMR variants (Fig. 3). The
upregulation of p-c-MET in the BxPc3AFR and BxPc3GEMR-resistant
variants was confirmed by subsequent western blot analysis, which
also showed upregulation of p-c-MET in the BxPc3OSIR variant which
was not detectable by the RTK assay (Fig. 4).
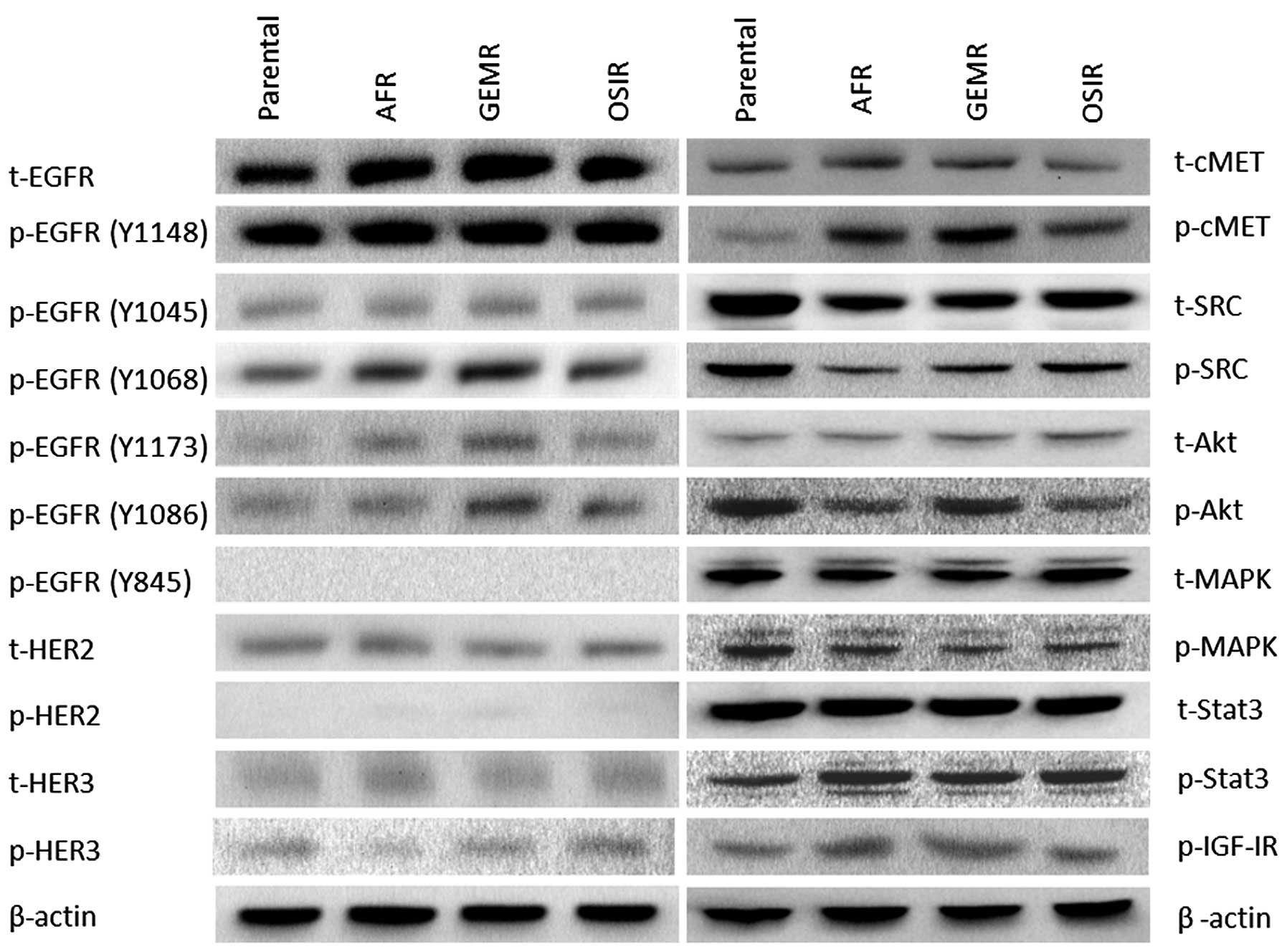
A detailed analysis of the phosphorylation status of
several tyrosine residues of the EGFR, revealed several differences
between the drug-resistant variants and the parental cell line
(Fig. 4). While there were no
major differences in the phosphorylation status of EGFR tyrosine
residues Y1148 and Y1045 between the parental cell line and the
drug-resistant variants, a significant increase in phosphorylation
levels of Y1068 in all three BxPC3 variants. In addition, the
phosphorylation of EGFR residues Y1173 and Y1086 was increased in
some drug-resistant variants (Fig.
4). Next, we examined the activation status of several
downstream signalling molecules including SRC, MAPK, AKT and STAT3
in the parental cell line and its drug-resistant variants showing
downregulation of pAKT in BxPc3AFR and BxPc3OSIR but not BxPc3GEMR
cells. In addition, there was an upregulation of pSTAT3 in all
drug-resistant variants compared to the parental cell line.
Interestingly, total and phosphorylated SRC was downregulated in
all resistant clones (Fig. 4).
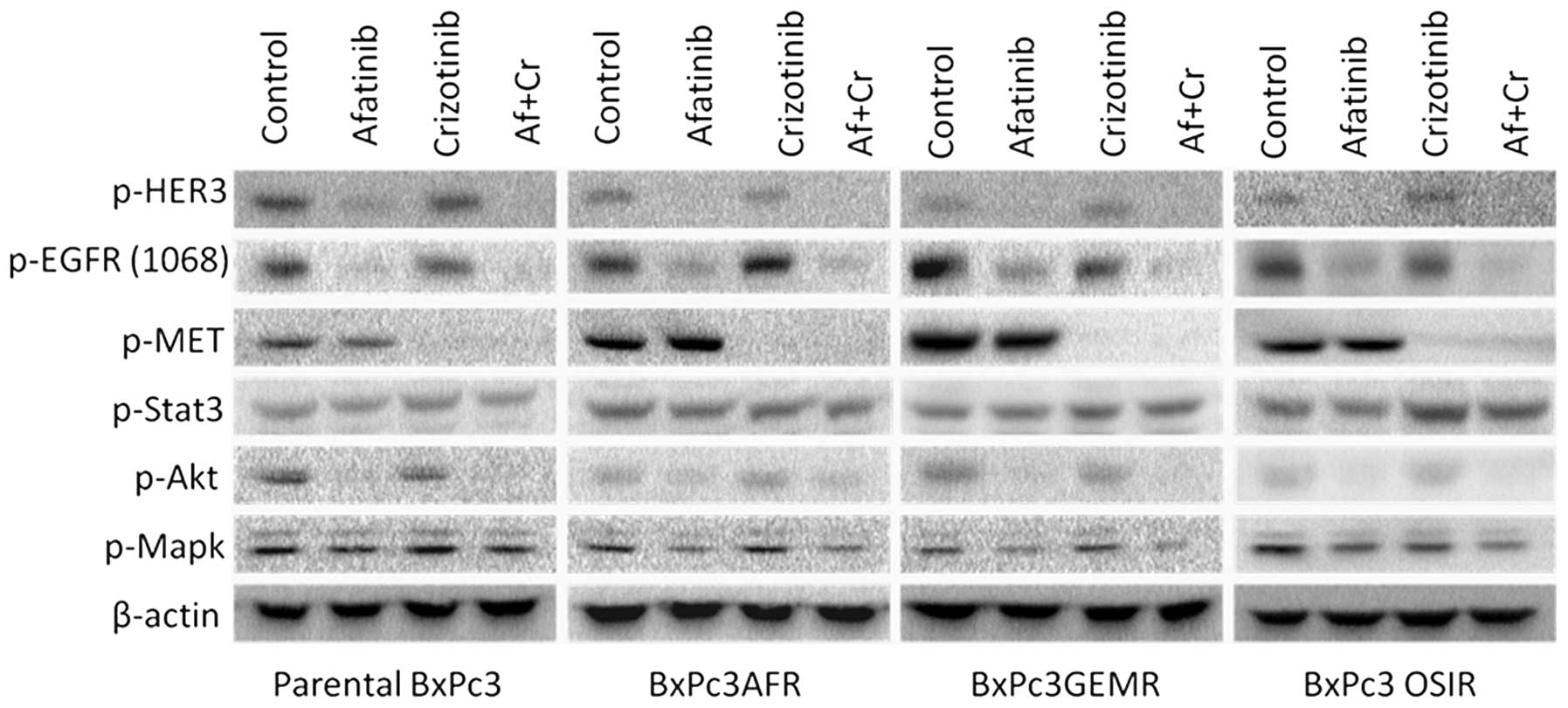
Effect of afatinib and crizotinib
treatment on the phosphorylation status of EGFR, HER3, p-c-MET and
downstream molecules STAT3, AKT and MAPK
Since upregulation of phorphorylated c-MET and STAT3
was found in all drug-resistant variants, we examined the effect of
crizotinib, a c-MET inhibitor, when used alone or in combination
with afatinib on the activation of downstream molecules STAT3, AKT
and MAPK. At 400 nM, crizotinib blocked completely the activation
of c-MET but had no effect on the activation status of either STAT3
or HER family members, EGFR (Y1068) and HER3 (Fig. 5). Similarly, treatment with
afatinib had no effect on the phosphorylation levels of STAT3, or
c-MET, however, it was more effective at blocking the
phosphorylation of EGFR at tyrosine 1068 in the parental cell line
compared to the drug-resistant variants suggesting that STAT3
activation was a result of a c-MET and HER family
members-independent mechanism (Fig.
5).
Next generation sequencing using the
CHPv2 revealed no differences between the parental cell line and
drug-resistant clones
Next generation sequencing data analysis revealed 13
SNVs in the parental and the drug-resistant variants, 3 of which
were found to be intronic. Of the 10 exonic SNVs only 3 led to
amino acid substitutions, 2 of which in the TP53 gene and one in
the KDR gene (Table III).
| Table IIIMutations of BxPc3 cell line as
determined using next generation sequencing (cancer hotspot v2
amplicons). |
Table III
Mutations of BxPc3 cell line as
determined using next generation sequencing (cancer hotspot v2
amplicons).
| Locus | Genotype | Control
genotype | Type | Gene | Location | AA change |
|---|
| chr4:1807894 | A/A | G | SNV | FGFR3 | Exonic | Synonymous |
| chr4:55141050 |
AGCCCGGATGGACATG/AGCCCGGATGGACATG |
AGCCCAGATGGACATG | SNV | PDGFRA | Exonic | Synonymous |
| chr4:55152040 | C/T | C | SNV | PDGFRA | Exonic | Synonymous |
| chr4:55593481 | A/G | A | SNV | KIT | Exonic | Synonymous |
| chr4:55972974 | A/A | T | SNV | KDR | Exonic | p.Gln472His |
| chr4:55980239 | T/T | C | SNV | KDR | Intronic | N/A |
| chr5:112175769 | CAG/CAG | CGG | SNV | APC | exonic | Synonymous |
| chr7:55249063 | G/A | G | SNV | EGFR | Exonic | Synonymous |
| chr11:534242 | A/G | A | SNV | HRAS | Exonic | Synonymous |
| chr13:28610183 | G/G | A | SNV | FLT3 | Intronic | N/A |
| chr17:7578190 | C/C | T | SNV | TP53 | Exonic | p.Tyr181Cys |
| chr17:7579470 | CGC/CGC | CGG | SNV | TP53 | Exonic | p.Pro33Arg |
| chr22:24176287 | A/A | G | SNV | SMARCB1 | Intronic | N/A |
Drug-resistant variants exhibited
increased sensitivity to treatment with Stat-3 inhibitor
Stattic
As there were higher levels of phosphorylation of
STAT3 in all drug-resistant variants, we sought to investigate the
inhibitory effect of STAT3 inhibitor Stattic, when used alone or in
combination with afatinib or gemcitabine. All drug-resistant
variants exhibited higher sensitivity to STAT3 inhibition with
IC50 values of 750 nM (p=0.012), 760 nM (p=0.012) and
1.26 μM (not significant, p=0.22) in BxPc3AFR, BxPc3GEMR and
BxPc3OSIR, respectively, compared to the IC50 value of
1.51 μM in the parental cell line. Of note, treatment of the
parental BxPc3 cell line with a combination of afatinib with STAT3
led to synergism with a CI value range of 0.5–0.6 while the same
combination had an additive effect in the drug-resistant variants
(Table IV). Similarly, the
combination of Crizotinib with afatinib led to a strong synergistic
effect in the parental cell line, it had an additive or slight
synergistic effect in all drug-resistant variants (Table IV).
| Table IVIC50 values for STAT3
inhibitor stattic and combination index (CI) values of afatinib
plus stattic or afatinib plus crizotinib in the parental BxPc3 cell
line and its drug-resistant variants (three independent
experiments). |
Table IV
IC50 values for STAT3
inhibitor stattic and combination index (CI) values of afatinib
plus stattic or afatinib plus crizotinib in the parental BxPc3 cell
line and its drug-resistant variants (three independent
experiments).
| Cell line | Stattic
IC50 range | Afatinib + Stattic
CI range (effect) | Afatinib +
Crizotinib CI range (effect) |
|---|
| BxPc3 | 1.36–1.9 μM | 0.503–0.603
(Synergism) | 0.15–0.22 (Strong
synergism) |
| BxPc3AFR | 703–774
nMa | 0.86–0.98
(Additive) | 0.73–0.87 (Slight
synergism) |
| BxPc3GEMR | 691–775
nMa | 1.05–1.08
(Additive) | 0.96–1.07
(Additive) |
| BxPc3OSIR | 1.22–1.27 μM | 0.72–0.85
(Additive/Slight synergism) | 0.7–0.88
(Additive/Slight synergism) |
Discussion
Drug resistance is one of the greatest challenges in
clinical oncology (6). Molecular
changes that lead to a decreased influx of the drug [low expression
of the human equilibrative nucleoside transporter-1(hENT1)] or a
low rate conversion of gemcitabine to its active metabolites,
caused by a low efficacy of deoxycytidine kinase (dCK) are some of
the mechanisms which have been identified so far for acquired
resistance to gemcitabine (19,20).
Similarly, a number of mechanisms have been identified for acquired
resistance to HER inhibitors, including the HER family member
modification (e.g. EGFR T790M mutation), the activation of
alternative signalling pathways (e.g. c-MET, IGF-IR), production of
autocrine ligands or mutations in downstream signalling molecules
such as K-RAS (10,21,22).
In our previous study, we investigated and reported
the growth response of a panel of seven human pancreatic tumour
cells lines (BxPC-3, AsPC-1, FA6, PANC-1, CAPAN-1, MiaPaca2, PT-45)
to the treatment with a wide range of agents including afatinib,
and erlotinib. We have shown that of these agents, afatinib was
more effective than erlotinib in inhibiting the growth of these
cancer cell lines in vitro. More importantly, of all the
cell lines examined in that study, BxPC3 cells were the most
sensitive to treatment with both afatinib and erlotinib, with
IC50 values of 11 and 1,200 nM, respectively (12). Since KRAS mutations have
already been established as a mechanism of resistance to EGFR
inhibitors, and in BxPC-3 cells it is the only one with a wild-type
KRAS gene and consequently most sensitive to treatment with
both afatinib and erlotinib, we developed variants of BxPC-3 cells
with acquired resistance to these drugs.
In this study, we sought to investigate molecular
changes accompanying the acquisition of drug resistance to
HER-targeted therapy or gemcitabine in pancreatic cancer, and to
determine therapeutic interventions that could overcome this
phenomenon. We found that acquired resistance to one agent such as
gemcitabine was accompanied by reduced sensitivity to afatinib and
erlotinib and vice versa, indicating the acquisition of a drug
cross-resistance phenotype (Table
II). However, the changes in sensitivity to other
chemotherapeutic agents did not follow the same pattern in the cell
lines. For example, while BxPc3GEMR and BxPc3AFR cells showed an
increase in sensitivity to oxaliplatin treatment, the
IC50 value in BxPc3OSIR for oxaliplatin was increased by
almost 3-fold (p<0.05). Similarly, while there was no
significant change in the sensitivity of BxPc3AFR cells to
treatment with doxycycline, both BxPc3GEMR and BxPc3OSIR cells were
found to have a significantly lower IC50 for doxycycline
compared to the parental cell line indicating that different
mechanisms could be contributing to the acquisition of drug
resistance in these cell lines (Table III).
Numerous studies have identified cells with stem
cell characteristics, that represent a small subpopulation within
haematological or solid tumours known as cancer stem cells (CSCs)
which have the capacity of self-renewal, differentiation, and high
tumourigenicity (23). According
to the CSC model, current therapeutic strategies can eliminate the
majority of tumour cells. However, due to their high intrinsic drug
resistance, CSCs can escape conventional treatments and lead to
tumour recurrence. The innate resistance of CSCs to treatment with
conventional therapies stems from specific traits which confer high
resistance to therapeutic agents, such as high detoxification
capacity, increased DNA repair capability, increased drug efflux
due to high expression of ABC transporters and infrequent
replication (24,25). One of the most well established
mechanisms involved in acquisition of multi-drug resistance (MDR)
is the over-expression of drug efflux proteins, mainly the
ATP-binding cassette (ABC) transporters. The ABC superfamily
consists of 48 members which can use energy to facilitate the
transport of various agents and therefore, can confer a multidrug
phenotype (26,27).
Therefore, we started to examine the expression
levels of several CSC markers including CD133, CD24 and CD44 as
well as some of the basic members of ABC transporters such as
P-glycoprotein (P-gp) in the developed drug-resistant variants
(28–30). Noteworthy, of all markers
investigated, CD44 expression was found to be increased in BxPc3AFR
and BxPc3OSIR drug-resistant variants (Table IV). However, the percentage of the
population of CD44 positive cells in these drug-resistant variants
was above 99%, indicating that the upregulation of CD44 was not
restricted to a small subpopulation of these cells. Since CD44 can
associate directly with HER family members and enhance their
activation and subsequent mitogenic signals, we hypothesized that
CD44 overexpression could be involved in the acquisition of
resistance in these cell lines (31–33).
However, addition of a blocking anti-CD44 antibody to treatment of
these cell lines with HER inhibitors erlotinib and afatinib, failed
to re-sensitize them to the latter (data not shown). In addition,
with the exception of a small increase (not significant) in ABCG2
expression in BxPc3AFR cells, there were no major changes in the
expression of ABC transporters investigated (P-gp, MRP-2 and ABCG2)
in all drug-resistant variants (data not shown).
The phosphorylation status of various tyrosine
residues of EGFR exhibited several differences between the parental
cell line and the drug-resistant variants. For example, even though
phosphorylation levels of Y1148 and Y1045 were similar between the
parental cell line and its drug-resistant variants, an increase of
phosphorylation of Y1068 and Y1173 was observed in the BxPc3AFR and
BxPc3GEMR clones (Fig. 4).
Furthermore, afatinib at 400 nM, led to an almost complete
inhibition of EGFR phosphorylation at tyrosine 1068 only in the
parental BxPc3 cells (Fig. 5). In
addition, we found a 3-fold increase in the autocrine production of
amphiregulin in the BxPc3AFR and BxPc3GMER variants but not
BxPc3OSIR compared to the parental cell line, indicating that the
differences in the phosphorylation of EGFR tyrosine residues or the
low efficacy of afatinib could result from the presence of an
amphiregulin autocrine loop. However, addition of an anti-EGFR
antibody (ICR62 at 200 nM) to afatinib treatment failed to
re-sensitize them to the latter, indicating that AR overexpression
alone could not explain the acquisition of resistance to anti-HER
treatment in these cell lines (data not shown).
Next generation sequencing using the CHPv2 pool of
primers, revealed no differences between the parental cell line and
its drug-resistant variants in 2855 hot spots/COSMIC mutations from
50 genes including EGFR, HER2, KRAS, p53, PIK3CA, PTEN and c-MET
among others. However, the possibility of mutations in these genes
leading to drug resistance cannot be excluded and could be
addressed only by full gene-sequencing for these biomarkers
(Table III).
Several studies have shown that c-MET signalling can
be involved in the acquisition of drug resistance to either HER
inhibitors or gemcitabine through overexpression of the receptor or
hyperactivation of the c-MET/HGF signalling axis (34–38).
Of note, despite increased phosphorylation of c-MET in all
drug-resistant variants, growth inhibition analysis showed no
significant change in the sensitivity to the c-MET inhibitor
crizotinib. In addition, while treatment with crizotinib (400 nM)
blocked the phosphorylation of c-MET completely, it had no effect
on the phosphorylation status of STAT3, MAPK or AKT (Table I and Fig. 5).
Activation of STAT3 has also been implicated in
resistance to HER inhibitors or cytotoxics in several malignancies
such as lung cancer, non-Hodgkin's lymphoma and multiple myeloma
(39–42). In non-small cell lung cancer cells,
Kim et al found that resistance to afatinib is mediated by
the activation of STAT3 via the IL-6R/JAK1 signalling axis
(43). More recently, STAT3 has
been found to have a critical role in conferring resistance to
anoikis and promote metastasis in pancreatic cancer cells (44). STAT3 has been shown to be activated
in an EGFR-dependent mechanism through association of STAT3 with
the EGFR tyrosine residues 1068 and 1086 which act as docking
sites, as well as EGFR-independent mechanisms, including the IL-6
receptor, SRC family kinases and JAK (45). In our study, while short term
treatment with AR led to a significant increase of phosphorylation
in all EGFR tyrosine residues tested in the parental cell line, it
had no effect on STAT3 phosphorylation (Fig. 2B). In addition, as mentioned above,
even though afatinib inhibited the phosphorylation of EGFR at Y1068
in all drug-resistant variants, it had no effect on the activation
status of STAT3, indicating that upregulation of p-STAT3 was a
result of an EGFR independent mechanism and warrants further
investigation (Fig. 5).
Of note, we found that treatment with the STAT3
inhibitor Stattic, had a higher inhibitory effect on the
drug-resistant variants compared with the parental cell line
(Table IV). Other studies have
also indicated that inhibition of STAT3 in some cancer types can
re-sensitize cells that have acquired resistance to EGFR inhibitors
or chemotherapeutics (40,46). Currently, the therapeutic potential
of STAT3 inhibition is being pursued in two separate lung cancer
studies: i) A phase I/II trial of Ruxolitinib (a JAK1/2 inhibitor)
in combination with erlotinib in patients with lung adenocarcinoma
(EGFR-mutant), with acquired resistance to erlotinib
(ClinicalTrials.gov identifier: NCT02155465) and ii) a phase I
trial of afatinib plus ruxolitinib in non-small lung cancer (NSCLC)
patients (ClinicalTrials.gov identifier: NCT02145637) will provide
more information on this new therapeutic concept. In our
preclinical study, when Stattic was used in combination with
afatinib, it led to an additive effect in all drug-resistant
pancreatic cancer variants (Table
IV). In contrast, the combination of Stattic with gemcitabine
led to an antagonistic effect (data not shown). In addition, while
the sensitivity of the drug-resistant variants to treatment with
the c-MET inhibitor crizotinib remained unchanged, a combination of
afatinib with the latter produced an additive or synergistic effect
indicating the therapeutic potential of this combination in
overcoming resistance of pancreatic cancer cells to HER inhibitors
and warrants further investigation.
In conclusion, our results indicate that the
activation of STAT3 may play an important role in the acquisition
of resistance to gemcitabine and HER inhibitors in pancreatic
cancer and warrant further investigations on the therapeutic
potential of STAT3 inhibitors in such a setting.
Acknowledgements
This study was supported by Kingston University. We
are grateful to OSI-Pharmaceutical (USA) for kindly providing
OSI-774 for use in our study. F. Scola is employee of Boehringer
Ingelheim, where afatinib was developed and produced. All other
authors declare they have no conflict of interest.
Abbreviations:
|
EGFR
|
epidermal growth factor receptor
|
|
FOLFIRINOX
|
fluorouracil, irinotecan and
oxaliplatin
|
|
CSC
|
cancer stem cells
|
|
EMT
|
epithelial mesenchymal transition
|
|
RTK
|
receptor tyrosine kinases
|
|
ATCC
|
American Type Culture Collection
|
|
MAPK
|
mitogen-activated protein kinase
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
IGF-IR
|
insulin like growth factor receptor
I
|
|
5-FU
|
5-fluorouracil
|
|
HB-EGF
|
heparin-binding EGF
|
|
PVDF
|
polyvinylidene difluoride
|
|
dCK
|
deoxycytidine kinase
|
|
MDR
|
multi-drug resistance
|
|
ABC transporters
|
ATP-binding cassette transporters
|
|
P-gp
|
P-glycoprotein
|
|
NSCLC
|
non-small lung cancer
|
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ioannou N, Seddon AM, Dalgleish A,
Mackintosh D and Modjtahedi H: Expression pattern and targeting of
HER family members and IGF-IR in pancreatic cancer. Front Biosci
(Landmark Ed). 17:2698–2724. 2012. View
Article : Google Scholar
|
|
3
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
4
|
Alberts SR, Gores GJ, Kim GP, Roberts LR,
Kendrick ML, Rosen CB, Chari ST and Martenson JA: Treatment options
for hepatobiliary and pancreatic cancer. Mayo Clin Proc.
82:628–637. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Burris HA III, Moore MJ, Andersen J, Green
MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo
AM, Tarassoff P, et al: Improvements in survival and clinical
benefit with gemcitabine as first-line therapy for patients with
advanced pancreas cancer: A randomized trial. J Clin Oncol.
15:2403–2413. 1997.PubMed/NCBI
|
|
6
|
Thota R, Pauff JM and Berlin JD: Treatment
of metastatic pancreatic adenocarcinoma: A review. Oncology
(Williston Park). 28:70–74. 2014.
|
|
7
|
Moore MJ, Goldstein D, Hamm J, Figer A,
Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, et al;
National Cancer Institute of Canada Clinical Trials Group.
Erlotinib plus gemcitabine compared with gemcitabine alone in
patients with advanced pancreatic cancer: A phase III trial of the
National Cancer Institute of Canada Clinical Trials Group. J Clin
Oncol. 25:1960–1966. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kelley RK and Ko AH: Erlotinib in the
treatment of advanced pancreatic cancer. Biologics. 2:83–95.
2008.PubMed/NCBI
|
|
9
|
Modjtahedi H and Dean C: The receptor for
EGF and its ligands - expression, prognostic value and target for
therapy in cancer (Review). Int J Oncol. 4:277–296. 1994.PubMed/NCBI
|
|
10
|
Chong CR and Jänne PA: The quest to
overcome resistance to EGFR-targeted therapies in cancer. Nat Med.
19:1389–1400. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Garrido-Laguna I and Hidalgo M: Pancreatic
cancer: From state-of-the-art treatments to promising novel
therapies. Nat Rev Clin Oncol. 12:319–334. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ioannou N, Dalgleish AG, Seddon AM,
Mackintosh D, Guertler U, Solca F and Modjtahedi H: Anti-tumour
activity of afatinib, an irreversible ErbB family blocker, in human
pancreatic tumour cells. Br J Cancer. 105:1554–1562. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Modjtahedi H, Cho BC, Michel MC and Solca
F: A comprehensive review of the preclinical efficacy profile of
the ErbB family blocker afatinib in cancer. Naunyn Schmiedebergs
Arch Pharmacol. 387:505–521. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ioannou N, Seddon AM, Dalgleish A,
Mackintosh D and Modjtahedi H: Treatment with a combination of the
ErbB (HER) family blocker afatinib and the IGF-IR inhibitor,
NVP-AEW541 induces synergistic growth inhibition of human
pancreatic cancer cells. BMC Cancer. 13:412013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cunningham MP, Thomas H, Fan Z and
Modjtahedi H: Responses of human colorectal tumor cells to
treatment with the anti-epidermal growth factor receptor monoclonal
antibody ICR62 used alone and in combination with the EGFR tyrosine
kinase inhibitor gefitinib. Cancer Res. 66:7708–7715. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Solca F: Pharmacology and molecular
mechanisms of BIBW2992 a potent irreversible dual EGFR/HER-2 kinase
inhibitor of cancer therapy. Target Oncol. 2:s152007.
|
|
17
|
García-Echeverría C, Pearson MA, Marti A,
Meyer T, Mestan J, Zimmermann J, Gao J, Brueggen J, Capraro HG,
Cozens R, et al: In vivo antitumor activity of NVP-AEW541-A novel,
potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell.
5:231–239. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim MP and Gallick GE: Gemcitabine
resistance in pancreatic cancer: Picking the key players. Clin
Cancer Res. 14:1284–1285. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nakano Y, Tanno S, Koizumi K, Nishikawa T,
Nakamura K, Minoguchi M, Izawa T, Mizukami Y, Okumura T and Kohgo
Y: Gemcitabine chemoresistance and molecular markers associated
with gemcitabine transport and metabolism in human pancreatic
cancer cells. Br J Cancer. 96:457–463. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Morgillo F, Cantile F, Fasano M, Troiani
T, Martinelli E and Ciardiello F: Resistance mechanisms of tumour
cells to EGFR inhibitors. Clin Transl Oncol. 11:270–275. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lin L and Bivona TG: Mechanisms of
resistance to epidermal growth factor receptor inhibitors and novel
therapeutic strategies to overcome resistance in NSCLC patients.
Chemother Res Pract. 2012:8172972012.PubMed/NCBI
|
|
23
|
Yu Z, Pestell TG, Lisanti MP and Pestell
RG: Cancer stem cells. Int J Biochem Cell Biol. 44:2144–2151. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gottesman MM, Fojo T and Bates SE:
Multidrug resistance in cancer: Role of ATP-dependent transporters.
Nat Rev Cancer. 2:48–58. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dean M, Fojo T and Bates S: Tumour stem
cells and drug resistance. Nat Rev Cancer. 5:275–284. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lage H: An overview of cancer multidrug
resistance: A still unsolved problem. Cell Mol Life Sci.
65:3145–3167. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dean M: ABC transporters, drug resistance,
and cancer stem cells. J Mammary Gland Biol Neoplasia. 14:3–9.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li C, Heidt DG, Dalerba P, Burant CF,
Zhang L, Adsay V, Wicha M, Clarke MF and Simeone DM: Identification
of pancreatic cancer stem cells. Cancer Res. 67:1030–1037. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lonardo E, Hermann PC and Heeschen C:
Pancreatic cancer stem cells - update and future perspectives. Mol
Oncol. 4:431–442. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Simeone DM: Pancreatic cancer stem cells:
Implications for the treatment of pancreatic cancer. Clin Cancer
Res. 14:5646–5648. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Marhaba R and Zöller M: CD44 in cancer
progression: Adhesion, migration and growth regulation. J Mol
Histol. 35:211–231. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sherman LS, Rizvi TA, Karyala S and Ratner
N: CD44 enhances neuregulin signaling by Schwann cells. J Cell
Biol. 150:1071–1084. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bourguignon LYW, Zhu H, Zhou B, Diedrich
F, Singleton PA and Hung MC: Hyaluronan promotes CD44v3-Vav2
interaction with Grb2-p185(HER2) and induces Rac1 and Ras signaling
during ovarian tumor cell migration and growth. J Biol Chem.
276:48679–48692. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ozasa H, Oguri T, Maeno K, Takakuwa O,
Kunii E, Yagi Y, Uemura T, Kasai D, Miyazaki M and Niimi A:
Significance of c-MET overexpression in cytotoxic anticancer
drug-resistant small-cell lung cancer cells. Cancer Sci.
105:1032–1039. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hage C, Rausch V, Giese N, Giese T,
Schönsiegel F, Labsch S, Nwaeburu C, Mattern J, Gladkich J and Herr
I: The novel c-Met inhibitor cabozantinib overcomes gemcitabine
resistance and stem cell signaling in pancreatic cancer. Cell Death
Dis. 4:e6272013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen Y and Fu L: Mechanisms of acquired
resistance to tyrosine kinase inhibitors. Acta Pharm Sin B.
1:197–207. 2011. View Article : Google Scholar
|
|
37
|
Moschetta M, Basile A, Ferrucci A,
Frassanito MA, Rao L, Ria R, Solimando AG, Giuliani N, Boccarelli
A, Fumarola F, et al: Novel targeting of phospho-cMET overcomes
drug resistance and induces antitumor activity in multiple myeloma.
Clin Cancer Res. 19:4371–4382. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bardelli A, Corso S, Bertotti A, Hobor S,
Valtorta E, Siravegna G, Sartore-Bianchi A, Scala E, Cassingena A,
Zecchin D, et al: Amplification of the MET receptor drives
resistance to anti-EGFR therapies in colorectal cancer. Cancer
Discov. 3:658–673. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ and
Settleman J: Drug resistance via feedback activation of Stat3 in
oncogene-addicted cancer cells. Cancer Cell. 26:207–221. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Alas S and Bonavida B: Inhibition of
constitutive STAT3 activity sensitizes resistant non-Hodgkin's
lymphoma and multiple myeloma to chemotherapeutic drug-mediated
apoptosis. Clin Cancer Res. 9:316–326. 2003.PubMed/NCBI
|
|
41
|
Wu K, Chang Q, Lu Y, Qiu P, Chen B, Thakur
C, Sun J, Li L, Kowluru A and Chen F: Gefitinib resistance resulted
from STAT3-mediated Akt activation in lung cancer cells.
Oncotarget. 4:2430–2438. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tang J, Guo F, Du Y, Liu X, Qin Q, Liu X,
Yin T, Jiang L and Wang Y: Continuous exposure of non-small cell
lung cancer cells with wild-type EGFR to an inhibitor of EGFR
tyrosine kinase induces chemoresistance by activating STAT3. Int J
Oncol. 46:2083–2095. 2015.PubMed/NCBI
|
|
43
|
Kim SM, Kwon OJ, Hong YK, Kim JH, Solca F,
Ha SJ, Soo RA, Christensen JG, Lee JH and Cho BC: Activation of
IL-6R/JAK1/STAT3 signaling induces de novo resistance to
irreversible EGFR inhibitors in non-small cell lung cancer with
T790M resistance mutation. Mol Cancer Ther. 11:2254–2264. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fofaria NM and Srivastava SK: STAT3
induces anoikis resistance, promotes cell invasion and metastatic
potential in pancreatic cancer cells. Carcinogenesis. 36:142–150.
2015. View Article : Google Scholar :
|
|
45
|
Yu H, Lee H, Herrmann A, Buettner R and
Jove R: Revisiting STAT3 signalling in cancer: New and unexpected
biological functions. Nat Rev Cancer. 14:736–746. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sen M, Joyce S, Panahandeh M, Li C, Thomas
SM, Maxwell J, Wang L, Gooding WE, Johnson DE and Grandis JR:
Targeting Stat3 abrogates EGFR inhibitor resistance in cancer. Clin
Cancer Res. 18:4986–4996. 2012. View Article : Google Scholar : PubMed/NCBI
|