Introduction
Breast cancer is the most common cancer and the
leading cause of cancer-related death in women worldwide (1). It is not considered to be only one
disease but a heterogeneous group of different diseases of distinct
molecular subtypes. Breast cancer can be categorized into four
subtypes, luminal A, luminal B, basal-like and HER2-positive
(2,3). Three biomarkers, estrogen receptor
(ER), progesterone receptor (PR), and human epidermal growth factor
receptor 2 (HER2) are often used for subtyping of breast cancer
pathologically (2). Although the
overall mortality rate for breast cancer patients has declined in
developed countries, the patients diagnosed of basal-like subtype
have a poorer short-term prognosis than those diagnosed with other
breast cancer subtypes. The majority of basal-like breast cancer
cases are also referred to as ‘triple-negative’ because they are
characterized as the lack of the expression of these three
biomarkers. Currently, no targeted therapies are available for
these tumors. Thus, there is great unmet medical need for the
development of novel molecular targeted therapeutic strategies.
Soy products have long been suggested to be useful
in the prevention of cancer development (4,5).
Epidemiological studies have shown the chemopreventive effect of
soy intake for breast cancer (4,6).
Especially, studies have shown that Asian women, who consume a diet
high in soy products, have a lesser incidence of breast cancer and
risk of breast cancer recurrence than the women in western counties
(6).
Genistein, a phytoestrogen, is the major
isoflavonoid contained in soybeans and is considered the active
micronutrient responsible for the chemopreventive effect of soy. It
has a broad spectrum of anticancer properties in triple negative
breast cancer (TNBC) cells. Genistein can inhibit cell growth
(7), induce apoptosis (8) and G2/M phase arrest (9), and decrease cell invasiveness
(10) in TNBC cells. Although a
number of studies have been performed to dissect the molecular
mechanisms of the effects of genistein on TNBC cells (8–13),
they cannot comprehend its pleiotropic effects because of the
limited throughput of traditional biochemical methods.
Multiplexed quantitation via isobaric labeling
reagents [e.g., tandem mass tags (TMT) (14) and isobaric tags for relative and
absolute quantitation (iTRAQ) (15)] combined with phosphopeptide
enrichment has been employed to dissect complex signaling pathways
using mass spectrometry. Nirujogi et al combined an 8-plex
TMT labeling strategy with titanium dioxide-based phosphopeptide
enrichment to examine the changes of the phosphoproteome in the
brains of rats after the exposure to VX, a nerve agent (16). Roitinger and colleagues combined a
4-plex iTRAQ labeling strategy with a phosphopeptide enrichment
pipeline (immobilized metal affinity chromatography coupled with
metal oxide affinity chromatography) to characterize the DNA damage
response signaling pathway in Arabidopsis thaliana (17). Herein, we employed a TMT-based
quantitative phosphoproteomic approach to identify
genistein-regulated changes of phosphorylation in TNBC cell line,
MDA-MB-231 after the short-term treatment. All together, we
identified 5,445 phosphorylation sites on 2,008 phosphoproteins out
of 3,452 proteins identified. The TMT-based quantitation revealed
332 genistein-regulated phosphorylation events. Bioinformatics
analysis revealed that genistein can modulate phosphorylation on
proteins involved in regulation of the cell cycle and DNA damage
response. They include critical components of DNA replication fork,
cohesin complex, kinetochores, and the BRCA1 complex. Manual
literature curation provides evidence that genistein-induced
changes on these proteins could contribute to its anticancer
effects. Overall, our data set provides a valuable resource for
further investigation on the anticancer molecular mechanism of
genistein in TNBC cells.
Materials and methods
Cell line and reagents
The breast cancer cell line MDA-MB-231 was
maintained in DMEM supplemented with FBS, L-glutamine, penicillin,
streptomycin at 37°C in 5% CO2. Genistein was purchased
from Sigma-Aldrich (St. Louis, MO, USA). Titanspheres
(TiO2, 5 μm beads) were from GL Sciences Inc. (Torrance,
CA, USA). L-1-Tosylamide-2-phenylethyl chloromethyl ketone (TPCK)
treated trypsin was from Worthington Biochemical Corp. (Lakewood,
NJ, USA). All other reagents used in this study were from Fisher
Scientific (Pittsburgh, PA, USA).
Cell lysis, protein digestion and TMT
labeling
MDA-MB-231 cells were plated at 3×106
cells per 150-mm plate overnight. Three populations of cells were
subjected to different treatments with 40 μM genistein for 0, 3 or
24 h, respectively. The treatments were carried out in biological
duplicates. After treatment, cells were washed with PBS, collected
and lysed in lysis buffer (4% SDS, 50 mM triethylammonium
bicarbonate (TEABC), 10 mM sodium fluoride, 1 mM sodium
orthovanadate and 1 mM β-glycerophosphate, 2.5 mM sodium
pyrophosphate) by sonication. After centrifugation at 16,000 × g at
15°C for 20 min, the supernatant was collected and the protein
concentration was determined using bicinchoninic acid (BCA) assay
(Pierce, Waltham, MA, USA). An equal amount of protein (400 μg)
from each condition was reduced by DTT at a final concentration of
5 mM at 60°C for 20 min and alkylated using 10 mM iodoacetamide for
10 min at room temperature (RT) in the dark.
The samples were then subjected to the
filter-assisted sample preparation protocol (18) with minor modifications. Briefly,
after reduction and alkylation, the protein was mixed with UA
solution (8 M urea in 50 mM TEABC, pH 8.0) to dilute the
concentration of SDS to 0.1% and transferred to an Amicon Ultra
centrifugal filter unit (30 kDa cut-off, Millipore). The unit was
centrifuged at 14,000 × g at 20°C for 15 min. The concentrate was
diluted with 400 μl of UA solution and then subjected to
centrifugation. This step was repeated once. The resulting
concentrate was then diluted with 400 μl of 50 mM TEABC, pH 8.0
followed by centrifugation. This step was repeated once. The
concentrate was transferred to a fresh tube and subjected to
tryptic digestion overnight. The resulting peptides were dried
completely in a vacuum concentrator and kept at −80°C.
Tandem mass tag (TMT) labeling was carried out
according to the manufacturer instructions. Briefly, tryptic
peptides from each sample was reconstituted in 100 μl of 50 mM
TEABC buffer and mixed with the TMT reagent reconstituted in 41 μl
of anhydrous acetonitrile (ACN), incubated at RT for 1 h. All the
labeled peptides from each sample were equally mixed, dried
completely in a vacuum concentrator and kept at −80°C.
Fractionation of peptides by basic
reversed-phase liquid chromatography (bRPLC)
TMT-labeled peptide mixture were resuspended in 1 ml
of 10 mM TEABC, pH 8.0 and loaded on a XBridge BEH C18 column,
130Å, 5 μm, 4.6 mm × 250 mm (Waters, Milford, MA, USA), and
fractionated on an Agilent 1100 Series HPLC system by basic
reversed-phase chromatography at a flow rate of 400 μl/min. Mobile
phase consisted of 10 mM TEABC, pH 8.0 (buffer A) and 10 mM TEABC,
90% acetonitrile, pH 8.0 (buffer B). After loading 1 ml of sample
(2.4 mg of protein) onto the column, the peptides were separated
using the following gradient: 2 min isocratic hold at 0% B, 0 to
15% solvent B in 8 min; 15 to 28.5% solvent B in 33 min; 28.5 to
34% solvent B in 5.5 min; 34 to 60% solvent B in 13 min, for a
total gradient time of 64.5 min. Using 96X 1 ml well plates
(Fisher, #7701-5200), fractions were collected every 0.6 min for a
total of 96 fractions through the elution profile of the
separation. Collection (5%) from each well were merged into 6
fractions and dried by vacuum centrifugation for the LC-MS/MS
analysis of the proteomic changes in cells. The rest collected from
each well was merged into 12 fractions and dried by vacuum
centrifugation for TiO2-based phosphopeptide
enrichment.
Phosphopeptide enrichment strategy
The 12 fractions of TMT-labeled peptides were
subjected to TiO2-based phosphopeptide enrichment as
described by Larsen et al (19) with minor modification. Briefly,
TiO2 beads were pretreated by incubation with
2,5-dihydroxybenzoic acid (DHB) solution (80% v/v ACN, 3% v/v TFA,
5% w/v DHB) for 20 min at room temperature. Each fraction was
resuspended in DHB solution and incubated with pretreated
TiO2 beads (Peptides: TiO2=1:1).
Phosphopeptide-bound TiO2 beads were washed twice with
400 μl of washing solution (80% v/v ACN, 3% v/v TFA). Peptides were
eluted three times with 20 μl of 4% v/v ammonia into 20 μl of 20%
v/v TFA and dried completely by vacuum centrifugation. The dried
peptides were resuspended in 50 μl 0.15% TFA, and desalted using
C18 Stage Tips (20).
Liquid chromatography tandem mass
spectrometry
LC-MS/MS analysis of peptides and phosphopeptides
was carried out using a reversed phase liquid chromatography system
interfaced with an LTQ-Orbitrap Velos mass spectrometer. The mass
spectrometer was operated in the ‘high-high’ mode, where mass
spectra of both precursor and product ions were acquired in the
high resolution Orbitrap analyzer (Thermo Scientific). The peptides
were loaded onto an analytical column (10 cm × 75 μm, Magic C18 AQ
5 μm, 120 Å) by 0.1% v/v formic acid and eluted using an ACN
gradient (0–60% v/v) containing 0.1% v/v formic acid. The settings
were: i) Precursor scans (FTMS) from 350–1,800 m/z at 30,000
resolution; and ii) MS2 scan (FTMS) of HCD fragmentation of the 10
most intense ions (isolation width: 1.20 m/z; normalized collision
energy: 40.0; activation time=0.1 msec; FT first mass value: 110.00
(fixed)) at 15,000 resolution.
Mass spectrometry data analysis
The tandem mass spectra were searched using
Andromeda algorithm (21) against
a human UniProt database (released in February, 2014) through the
MaxQuant platform (version 1.4.1.2) (22,23).
The search parameters included: 6plex TMT; a maximum of two missed
cleavages; fixed modification: carbamidomethylation of cysteine;
variable modification: protein N-term acetylation, oxidation of
methionine, and phosphorylation of serine, threonine and tyrosine.
The first search and main search for peptide tolerance were set to
20 ppm and 4.5 ppm, respectively. The FTMS MS/MS tolerance was set
to 50 ppm. The maximum number of modifications per peptide was set
to 5 and the maximum charge was set at 7. The reward type of the
target-decoy analysis was chosen. The peptide-spectrum match (PSM)
false discovery rate (FDR), protein FDR and the site decoy fraction
were set to 0.01. The minimum peptide length was set to 7. The
minimal scores for unmodified and modified peptides were 0 and 40,
respectively. The minimal delta score for unmodified and modified
peptides were 0 and 17, respectively. The minimum of unique and
razor peptides for identification was set to 1. The reporter ion
intensities for each PSM with PIF (precursor ion fraction) >0.75
were calculated using MaxQuant. The quantitation of identified
proteins was determined by normalized reporter ion intensities
using at least 1 razor/unique non-phosphopeptide. The quantitation
of each identified phosphosites were determined by normalized
reporter ion intensities using the least modified peptide and
normalized. The probability of phosphorylation for each Ser/Thr/Tyr
site on each peptide was calculated using Andromeda (MaxQuant). The
mass spectrometry proteomics data and the supplementary data have
been deposited to the ProteomeXchange Consortium (24) via the PRIDE partner repository with
the dataset identifier PXD002735 (http://www.ebi.ac.uk/pride/archive/projects/PXD002735/files).
Bioinformatics analysis
Molecular function and cellular localization of
phosphoproteins were obtained from the PANTHER Classification
System (25,26). Enrichment analysis of biological
process GO term of phosphoproteins was performed using the DAVID
functional annotation tool (27,28).
Results
Mass spectrometric analysis of
genistein-treated TNBC cells
Previously, several studies have shown that
genistein inhibits cell growth and induces apoptosis in a
dose-dependent manner (8,11,12,29)
during the treatment time ranging from 24 h to 6 days. However, the
short-term effects of genistein on the signaling network in TNBC
MDA-MB-231 cells during the first 24-h treatment, especially at 3-h
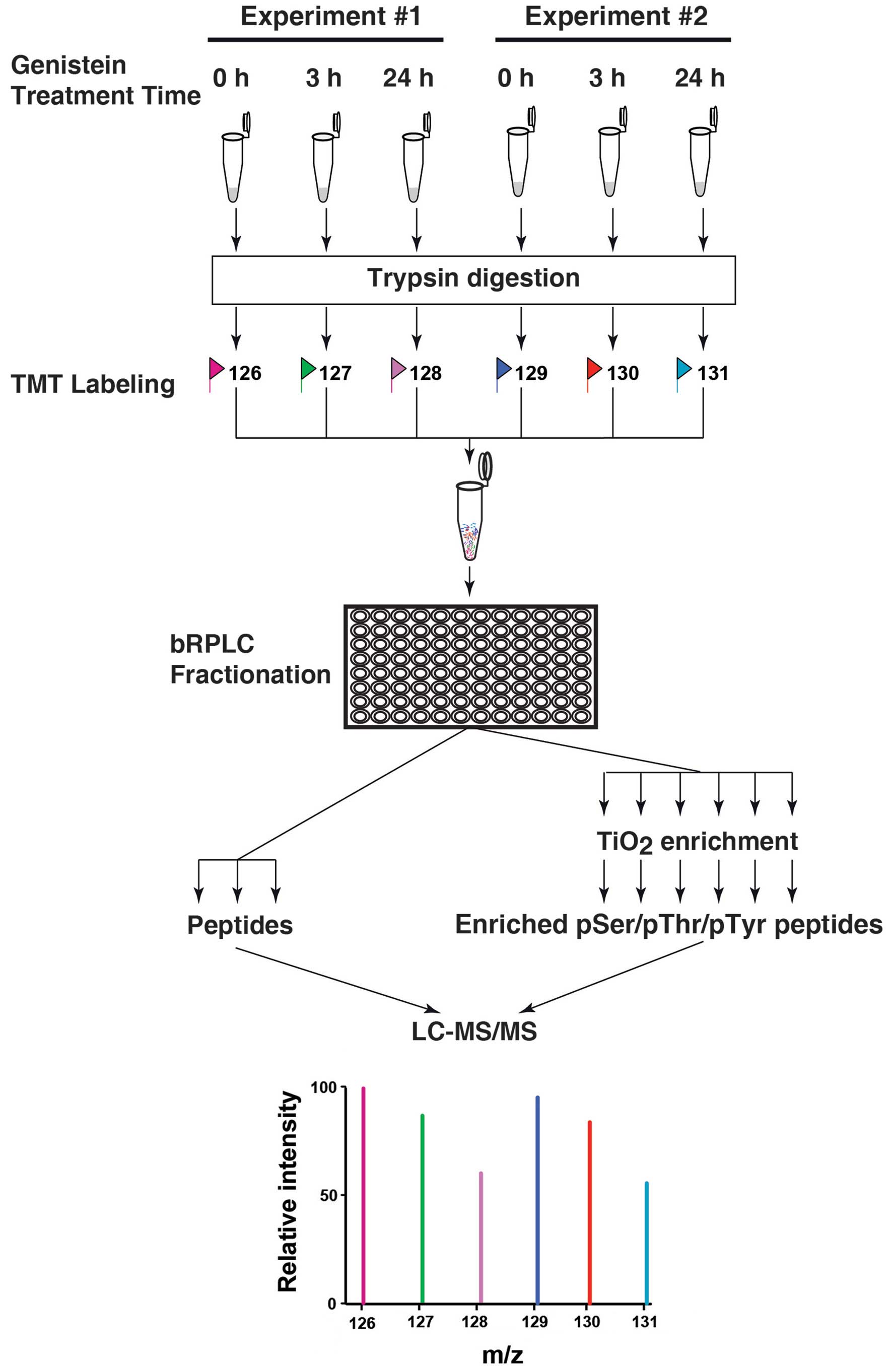
time point, have not been investigated. By combining a 6-plex TMT
labeling strategy and a TiO2-based phosphopeptide
enrichment strategy coupling with high resolution Fourier transform
mass spectrometry, we systematically measure the changes of the
cellular phosphoproteome in biological duplicate experiments at two
different time points, 3-h and 24-h treatments, as shown in
Fig. 1.
Eighteen LC-MS/MS runs generated a total of 247,126
mass spectra. The mass spectra were searched against a Uniprot
database (released in February, 2014) using Andromeda (21) through MaxQuant (Version 1.4.1.2)
(23). Using a FDR cut-off of 1%
at both peptide and protein levels, a target-decoy analysis
generated 27,666 peptide-spectrum-matches, of which 14,462 were
phosphopeptide-spectrum-matches. After excluding reverse and
contaminating matches, we identified a total of 3,452 proteins and
12,813 peptides. Among these identified peptides, 4,562 were
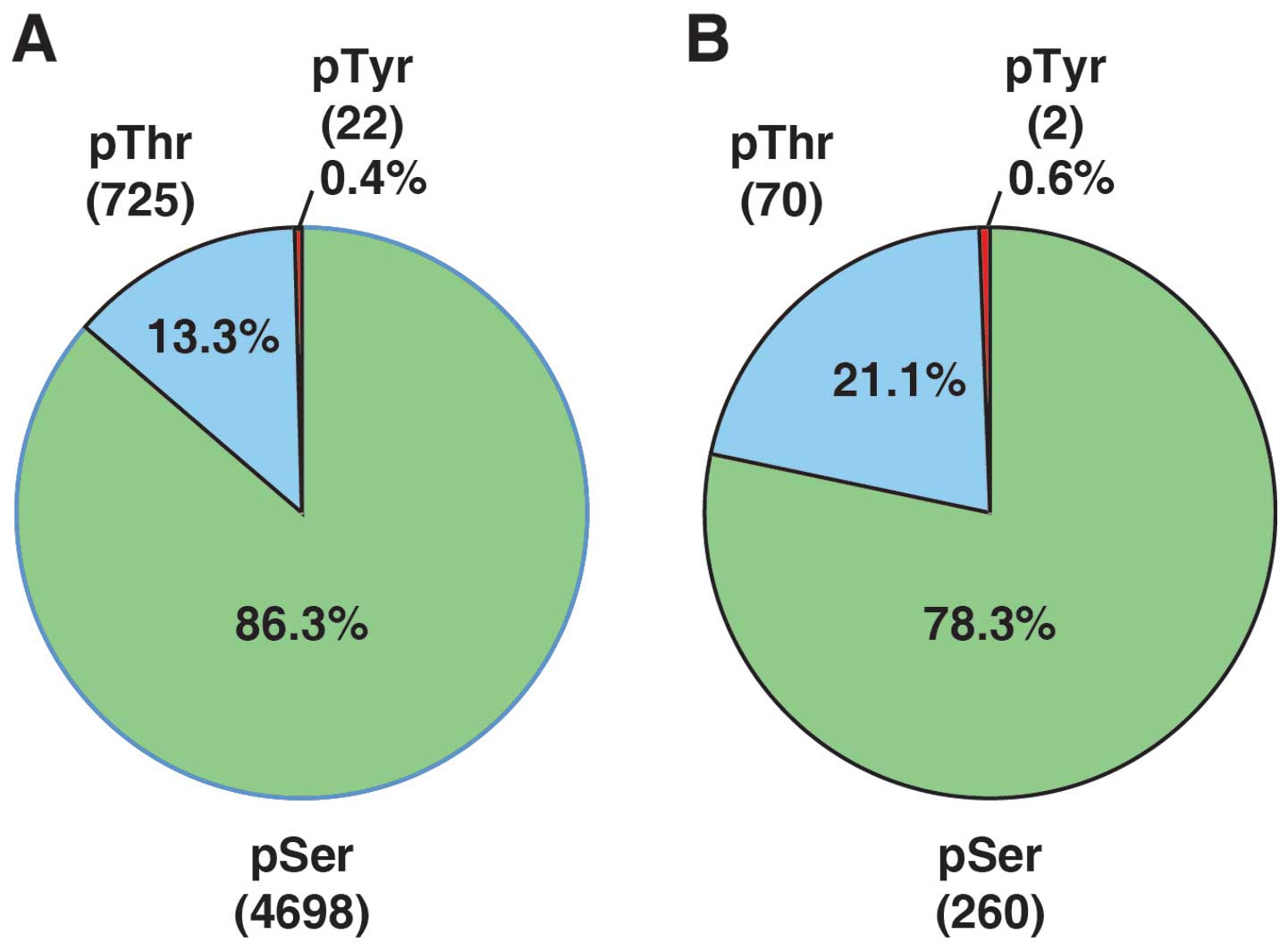
phosphopeptides that contained 5,445 phosphorylation sites (4,698
phosphoserine, 725 phosphothreonine, 22 phosphotyrosine sites).
These phosphopeptides were mapped to 2,008 phosphoproteins.
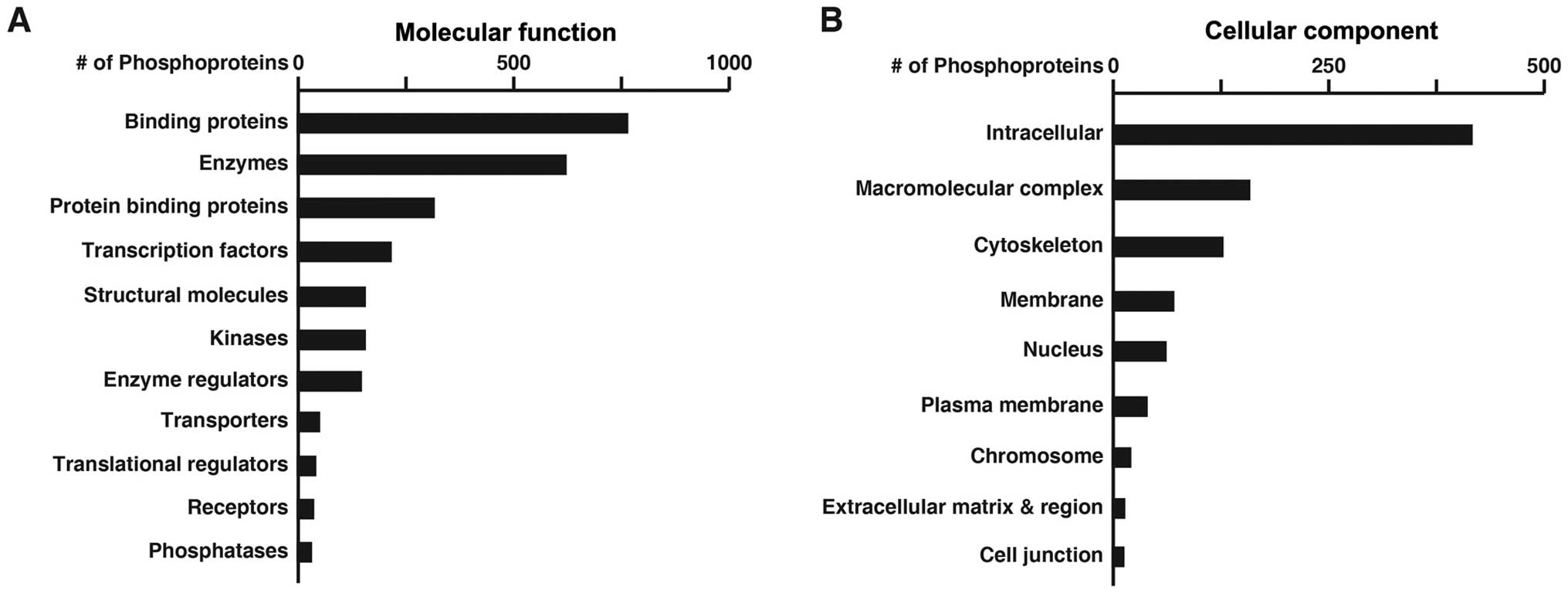
Fig. 2 shows the numbers of these
phosphoproteins categorized by molecular function (A) or cellular
component (B) based on the annotation by the PANTHER classification
system.
The quantitation of proteins and phosphosites was
determined by the reporter ion intensities using the MaxQuant
platform. The reporter ion intensities derived from mass spectra
with precursor ion fraction (PIF) of >0.75 were considered for
the quantitation. The changes of identified proteins were carried
out using the quantitation of non-phosphopeptides, while changes in
phosphosites were calculated using the least-modified
phosphopeptides and then normalized to the changes of the
corresponding protein, if possible. We were able to quantitate
2,087 proteins and 4,750 phosphosites (Fig. 3 and data not shown). Table I summarizes the statistics of the
quantitative data on the fold-changes of proteins and
phosphorylation in genistein-treated MDA-MB-231 cells. The narrow
range of 95% confidence interval suggests that the majority of
quantitated proteins remained unchanged during the course of
genistein treatment in cells. Previously, TMT-based quantitative
proteomics studies have chosen 1.5-fold change as a cut-off for
significant changes (16,30). Thus, we also chose 1.5-fold cut-off
for increased protein/phosphorylation level and a 0.67-fold cut-off
for decreased protein/phosphorylation level. Only the changes of
phosphorylation that showed consistency in both biological
replicate experiments were considered for further analysis. By
using this cut-off, 2,078 out of 2,087 proteins remained unchanged
during the 24 h treatment of TNBC cells with genistein, while the
expression level of only 8 proteins were modulated by genistein
(Table II). We also identified
332 genistein-regulated phosphorylation sites in our analysis
(Table III and data not shown).
Fig. 4 shows the distribution of
phosphor-serine, -threonine and -tyrosine sites among identified
phosphosites (A) and genistein-regulated phosphosites (B).
 | Table IStatistical analysis of quantitative
proteomics and phosphoproteomics data. |
Table I
Statistical analysis of quantitative
proteomics and phosphoproteomics data.
| A, Fold change of
the protein level of in MDA-MB-231 cells after treatment with
genistein |
|---|
|
|---|
| Experiment | Time point (h) | Median | Mean (log10
transformed) | Standard deviation
(log10 transformed) | 95% Confidence
interval |
|---|
| #1 | 3 | 1.03 | 0.00889 | 0.0752 | 0.858–1.213 |
| 24 | 1.01 | 0.00504 | 0.0604 | 0.880–1.162 |
| #2 | 3 | 1.02 | 0.00182 | 0.0951 | 0.807–1.250 |
| 24 | 1.02 | 0.00475 | 0.0732 | 0.854–1.196 |
|
| B, Fold change of
the phosphorylation level in MDA-MB-231 cells after treatment with
genistein |
|
| Experiment | Time point (h) | Median | Mean (log10
transformed) | Standard deviation
(log10 transformed) | 95% Confidence
interval |
|
| #1 | 3 | 0.99 | 0.00011 | 0.1301 | 0.741–1.350 |
| 24 | 0.98 | −0.00636 | 0.0941 | 0.793–1.224 |
| #2 | 3 | 0.99 | −0.00048 | 0.1270 | 0.746–1.338 |
| 24 | 0.98 | −0.00332 | 0.0889 | 0.809–1.218 |
| Table IIThe genistein-regulated proteins in
MDA-MB-231 cells. |
Table II
The genistein-regulated proteins in
MDA-MB-231 cells.
| | | Fold change |
|---|
| | |
|
|---|
| Gene symbol | Protein | Accession no. | 3 h | 24 h |
|---|
| CCNB1 | G2/mitotic-specific
cyclin-B1 | Q9NRN7 | 2.2 | 1.1 |
| CSTB | Cystatin-B | P04080 | 2.6 | 2.0 |
| EEF1B2 | Elongation factor
1-β | P24534 | 1.7 | 1.1 |
| PRC1 | Protein regulator
of cytokinesis 1 | O43663 | 1.6 | 1.0 |
| TK1 | Thymidine kinase,
cytosolic | P04183 | 1.7 | 1.1 |
| TPX2 | Targeting protein
for Xklp2 | Q9ULW0 | 1.6 | 1.1 |
|
HIST1H1C | Histone H1.2 | P16403 | 0.57 | 0.87 |
|
TNKS1BP1 | 182 kDa
tankyrase-1-binding protein | Q9C0C2 | 0.57 | 0.63 |
| Table IIIA partial list of genistein-regulated
phosphoproteins involved in regulation of the cell cycle in
MDA-MB-231 cells. |
Table III
A partial list of genistein-regulated
phosphoproteins involved in regulation of the cell cycle in
MDA-MB-231 cells.
| | | | | Fold change |
|---|
| | | | |
|
|---|
| Gene symbol | Protein | Uniprot ID | Phosphosite | Identified
peptides | 3 h | 24 h |
|---|
| AKAP12 | A-kinase anchor
protein 12 | Q02952 | S612 | EGVTPWApSFK | 0.61 | 0.91 |
| AKAP9 | A-kinase anchor
protein 9 | Q99996 | S45 | AQSDGQSPpSKK | 1.70 | 0.98 |
| ANLN | Actin-binding
protein anillin | Q9NQW6 | S72 |
RCpSDNTEVEVSNLENK | 0.56 | 0.91 |
|
ARHGAP19 | Rho
GTPase-activating protein 19 | Q14CB8 | S422 | pSFSGLIK | 1.72 | 1.05 |
| BRCA1 | Breast cancer type
1 susceptibility protein | P38398 | S753 | DLMLpSGER | 1.99 | 1.24 |
| S1524 | NYPpSQEELIK | 2.16 | 1.48 |
|
C6orf106 | Uncharacterized
protein C6orf106 | Q9H6K1 | S215 |
KVEGNFNPFApSPQK | 1.86 | 1.09 |
| CCNY; | Cyclin-Y-like
protein 1; | Q8ND76; | S73; S95 |
ASTIFLSKpSQTDVR | 1.56 | 1.20 |
| CCNYL1 | Cyclin-Y | Q8N7R7 | | | | |
| CDC20 | Cell division cycle
protein 20 homolog | Q12834 | T106 | ENQPENSQpTPTKK | 2.12 | 1.14 |
| T70 | VQTpTPSKPGGDR | 2.27 | 1.19 |
| CDC6 | Cell division
control protein 6 homolog | Q99741 | S54 | ALPLpSPR | 1.72 | 1.19 |
| CDCA3 | Cell division
cycle-associated protein 3 | Q99618 | S94 | QLpSEVFETEDSK | 1.88 | 0.86 |
| CDK1; | Cyclin-dependent
kinase 1; | P06493; | T14; T14; T14 | IGEGpTpYGVVYK | 2.83 | 1.21 |
| CDK2; | Cyclin-dependent
kinase 2; | P24941; | Y15; Y15; Y15 | IGEGpTpYGVVYK | 2.29 | 1.12 |
| CDK3 | Cyclin-dependent
kinase 3 | Q00526 | Y19; Y19; Y19 | IGEGpTYGVVpYK | 2.44 | 1.16 |
| CDK12 | Cyclin-dependent
kinase 12 | Q9NYV4 | T893 | LYNSEESRPYpTNK | 1.59 | 1.93 |
| CDK16 | Cyclin-dependent
kinase 16 | Q00536 | S153 | RVpSLSEIGFGK | 1.48 | 1.57 |
| CENPF | Centromere protein
F | P49454 | S1747 | LQLQGLDLpSSR | 0.54 | 1.06 |
| CENPV | Centromere protein
V | Q7Z7K6 | S47 | SApSQAGSK | 1.93 | 1.87 |
| S45 | pSASQAGSK | 0.24 | 0.81 |
| CLASP1 | CLIP-associating
protein 1 isoform 5 | Q7Z460-4 | S687 | VVSQpSQPGpSR | 1.79 | 1.64 |
| CLASP2 | CLIP-associating
protein 2 | O75122 | S455 | MVSQpSQPGpSR | 2.13 | 1.85 |
| CSNK1D | Casein kinase I
isoform δ | P48730 | S331 | GLPSTApSGR | 0.71 | 0.57 |
| FAM83D | Protein FAM83D | Q9H4H8 | S462 | GTQpSTEGpSPVSK | 1.78 | 0.99 |
| FANCD2 | Fanconi anemia
group D2 protein | Q9BXW9 | S717 | DGGPVTpSQESGQK | 2.20 | 1.25 |
| GINS2 | DNA replication
complex GINS protein PSF2 | Q9Y248 | S182 |
TNLQPLESTQpSQDF | 2.42 | 2.07 |
| GNL3L | Guanine
nucleotide-binding protein-like 3-like protein | Q9NVN8 | S465 | LLHpSPMTK | 1.70 | 1.20 |
| GSK3B | Glycogen synthase
kinase-3β | P49841 | S9 |
TTpSFAESCKPVQQPSAFGSMK | 1.86 | 1.66 |
| H2AFY | Core histone
macro-H2A.1; Histone H2A | O75367 | T129 | GKLEAIIpTPPPAK | 0.66 | 0.94 |
| HAUS6 | HAUS augmin-like
complex subunit 6 | Q7Z4H7 | S552 | AVLSDpSPQLSEGK | 0.63 | 0.84 |
| HJURP | Holliday junction
recognition protein | Q8NCD3 | S412 | WLIpSPVK | 1.60 | 0.88 |
| HP1BP3 | Heterochromatin
protein 1-binding protein 3 isoform 3 | Q5SSJ5-3 | S3 | MApSpSPRPK | 0.47 | 0.89 |
| HP1BP3 | Heterochromatin
protein 1-binding protein 3 isoform 3 | Q5SSJ5-3 | S4 | MApSpSPRPK | 0.47 | 0.89 |
| ID4 | DNA-binding protein
inhibitor ID-4 | P47928 | S5 | AVpSPVRPSGR | 1.82 | 1.17 |
| INCENP | Inner centromere
protein | Q9NQS7 | T219 |
TLSPTPASATAPTSQGIPpTpSDEESTPKK | 1.69 | 1.32 |
| INCENP | Inner centromere
protein | Q9NQS7 | T292 |
VLAPILPDNFSpTPTGSR | 0.54 | 0.88 |
| ING5 | Inhibitor of growth
protein 5 | Q8WYH8 | T147 | RpTSEEDTPK | 0.60 | 0.97 |
| KIF11 | Kinesin-like
protein KIF11 | P52732 | T926 | LDIPTGTpTPQR | 1.68 | 0.98 |
| KIF1A | Kinesin-like
protein KIF1A | Q12756 | S1094 | DVLpSPLRPSR | 1.98 | 0.97 |
| KIF20A | Kinesin-like
protein KIF20A | O95235 | S825 | LQGQVpSAK | 0.64 | 0.98 |
| KIF20B | Kinesin-like
protein KIF20B | Q96Q89 | S1740 |
FGDFLQHpSPSILQSK | 1.64 | 0.99 |
| T1644 | HPGCTpTPVTVK | 1.56 | 0.99 |
| LMNB1 | Lamin-B1 | P20700 | S23 | AGGPTpTPLpSPTR | 0.46 | 0.82 |
| T20 | AGGPTpTPLpSPTR | 0.27 | 0.74 |
| LMNB2 | Lamin-B2 | Q03252 | S17 | AGGPApTPLpSPTR | 0.51 | 0.77 |
| S404 |
ATSSSSGpSLSATGR | 0.63 | 0.88 |
| T14 | AGGPApTPLpSPTR | 0.51 | 0.77 |
| MAP4 |
Microtubule-associated protein 4 | P27816 | S636 |
KCpSLPAEEDSVLEK | 0.47 | 1.02 |
| MARCKS | Myristoylated
alanine-rich C-kinase substrate | P29966 | S145 |
AEDGApTPpSPSNETPK | 0.64 | 0.88 |
| S147 |
AEDGATPSPpSNETPK | 0.52 | 0.79 |
| T143 |
AEDGApTPpSPSNETPK | 0.55 | 0.93 |
| MCM3 | DNA replication
licensing factor MCM3 | P25205 | S728 | TADpSQETK | 5.6 | 2.04 |
| MCM6 | DNA replication
licensing factor MCM6 | Q14566 | S762 |
EIESEIDpSEEELINK | 9.62 | 2.19 |
| MDC1 | Mediator of DNA
damage checkpoint protein 1 | Q14676 | S1086 |
QDGpSQEAPEAPLSSELEPFHPKPK | 2.84 | 2.17 |
| S955 | GEPEGGpSQDQK | 4.36 | 3.35 |
| MELK | Maternal embryonic
leucine zipper kinase | Q14680 | S356 |
SNNWpSLEDVTASDK | 1.67 | 1.09 |
| S529 | VFGpSLER | 1.72 | 1.03 |
|
MIS18BP1 | Mis18-binding
protein 1 | C9J2Q8 | S110 | ANYEpSPGK | 1.52 | 1.01 |
| NASP | Nuclear
autoantigenic sperm protein | P49321 | S421 | LVPpSQEETK | 3.08 | 0.49 |
| NPM1 | Nucleophosmin | P06748 | S218 | DSKPSpSpTPR | 0.53 | 0.87 |
| S254 | MQApSIEK | 0.40 | 0.81 |
| T219 | DSKPSpSpTPR | 0.54 | 0.88 |
| T199 | SIRDpTPAK | 0.63 | 0.81 |
| NUP214 | Nuclear pore
complex protein Nup214 | P35658 | S678 |
ITPPAAKPGpSPQAK | 1.52 | 1.08 |
| ORC2 | Origin recognition
complex subunit 2 | Q13416 | T226 |
VVSAPVGKEpTPSKR | 1.67 | 1.04 |
| POLA2 | DNA polymerase α
subunit B | Q14181 | S152 |
pSPHQLLSPSSFpSPSATPSQK | 1.78 | 1.07 |
| T127 | AISpTPETPLTK | 1.78 | 1.02 |
| PTTG1;
PTTG2 | Securin;
Securin-2 | O95997; Q9NZH5 | S165; S165 | LFQLGPPpSPVK | 2.06 | 1.02 |
| RANBP2 | E3 SUMO-protein
ligase RanBP2 | P49792 | S955 | FESPATGILpSPR | 1.95 | 1.24 |
| RB1 |
Retinoblastoma-associated protein | P06400 | S807 |
IPGGNIYIpSPLKpSPYK | 1.66 | 1.08 |
| S811 |
IPGGNIYIpSPLKpSPYK | 1.66 | 1.08 |
| T821 |
ISEGLPpTPTKMpTPR | 1.76 | 1.09 |
| T823 |
ISEGLPTPpTKMpTPR | 1.80 | 1.08 |
| T826 |
ISEGLPpTPTKMpTPR | 1.77 | 1.09 |
| RBM10 | RNA-binding protein
10 | P98175 | S50 | EYGpSQEGK | 2.55 | 1.52 |
| RECQL5 | ATP-dependent DNA
helicase Q5 | O94762 | S815 |
YTGEEDGAGGHpSPAPPQTEECLR | 2.11 | 1.50 |
| REXO4 | RNA exonuclease
4 | Q9GZR2 | S111 | KETpSPQVK | 0.61 | 0.91 |
| RFC1 | Replication factor
C subunit 1 | P35251 | S29 | TKpSDEETLK | 9.99 | 11.91 |
| SMC1A | Structural
maintenance of chromosomes protein 1A | Q14683 | S957 |
GTMDDISQEEGSpSQGEDSVSGSQR | 2.34 | 1.38 |
| S358 | MEEEpSQpSQGR | 1.81 | 1.16 |
| S360 | MEEEpSQpSQGR | 3.13 | 1.78 |
| SMC3 | Structural
maintenance of chromosomes protein 3 | Q9UQE7 | S1065 | KGDVEGpSQpSQDEG
EGSGESER | 5.91 | 3.47 |
| S1067 |
KGDVEGpSQpSQDEGEGSGESER | 2.14 | 1.63 |
| S1083 |
GSGpSQSSVPSVDQFTGVGIR | 3.22 | 1.69 |
| TOP1 | DNA topoisomerase
1 | P11387 | S97 | VRApSGDAK | 0.58 | 0.81 |
| TOP2A | DNA topoisomerase
2-α | P11388 | S1377 | SVVpSDLEADDVK | 2.26 | 1.17 |
| S1106 |
VPDEEENEEpSDNEKETEK | 1.62 | 1.13 |
| TOPBP1 | DNA topoisomerase
2-binding protein 1 | Q92547 | S888 | NAVALSApSPQLK | 2.72 | 1.44 |
| TP53BP1 | Tumor suppressor
p53-binding protein 1 | Q12888 | S1068 |
GNLLHFPSpSQGEEEKEK | 1.97 | 1.63 |
| TP53BP1 | Tumor suppressor
p53-binding protein 1 | Q12888 | S398 |
QDKPMDTSVLpSEEGGEPFQK | 2.06 | 1.35 |
| UIMC1 | BRCA1-A complex
subunit RAP80 | Q96RL1 | S597 | ADQGDGPEGpSGR | 2.63 | 1.32 |
| S101 |
EVNpSQEEEEEELLR | 1.86 | 1.33 |
Genistein regulates a number of
biological processes during the cell cycle in TNBC cells
To identify the biological processes regulated by
genistein in cells, genistein-regulated phosphoproteins identified
in our study were analyzed in the DAVID bioinformatics resources
(27,28) using the biological process ontology
terms. The analysis revealed that proteins involved in the cell
cycle, mitosis, DNA replication and cell division were
significantly enriched in genistein-regulated phosphoproteins
(Table III and data not shown).
Although it has been shown that genistein can induce G2/M cell
cycle arrest and decrease the expression of CDK1 in MDA-MB-231
cells after 48 h of treatment (11), the results here indicated that
genistein regulates various phases during the cell cycle. To
understand the effects of genistein on regulation of the cell
cycle, manual literature curation was performed to find the roles
of the phosphorylation of these proteins during the progress of the
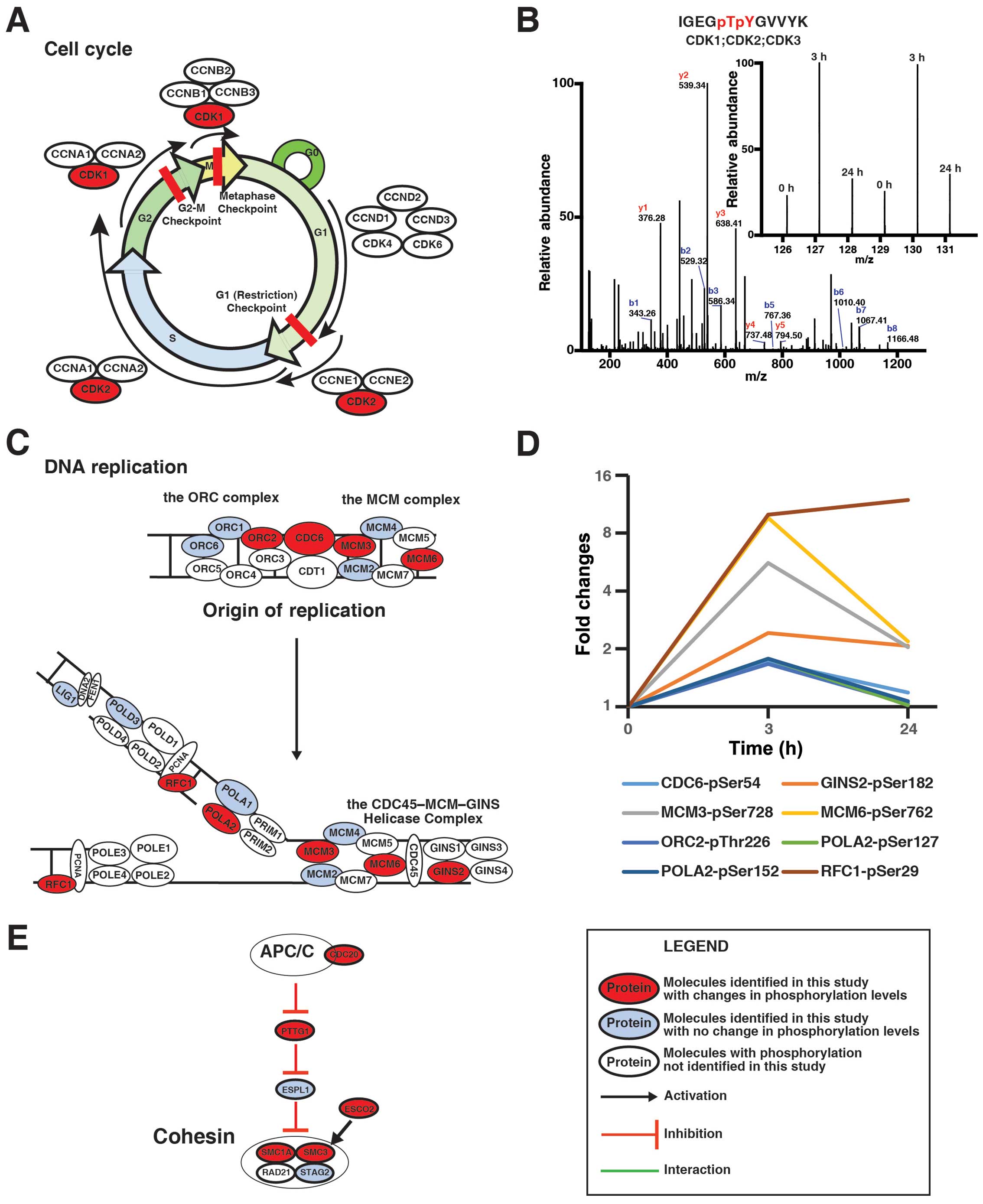
cell cycle. Fig. 5A shows that
three critical CDKs during the progress of the cell cycle were
regulated by genistein, while Fig.
5B demonstrates that 3 h after the treatment, genistein
increased the amount of the phosphopeptide ‘IGEGpTpYGVVYK’, which
is shared among CDK1, CDK2, and CDK3. Fig. 5C illustrates that 8 protein
critical for DNA replication were regulated by genistein in
MDA-MB-231 cells. The quantitative analysis revealed that the
phosphorylation levels of these proteins were increased after 3-h
treatment with genistein and that the phosphorylation on MCM3,
MCM5, and GINS2 even remains higher after 24-h treatment with
genistein (Fig. 5D). Fig. 5E shows that genistein decreased
phosphorylation level of ESCO2 and increased phosphorylation level
of CDC20, PTTG1, SMC1A and SMC3 in cells, suggesting that genistein
can inhibit cleavage of the cohesin complex during mitosis.
Table III summarized the changes
of phosphorylation of the proteins involved in the cell cycle
progress. Although the effects of genistein-induced changes of
phosphorylation of these proteins cannot be detailed here, for the
first time, our data revealed that genistein regulates the cell
cycle progression in a more comprehensive manner than previously
reported (11).
Genistein activates the DNA damage
response pathway in TNBC cells
The enrichment analysis by DAVID revealed that 23
genistein-regulated phosphoproteins are involved in response to DNA
damage (data not shown). To understand phosphorylation-based
regulation of DNA damage response pathway by genistein, we
constructed a DNA damage response pathway by manual literature
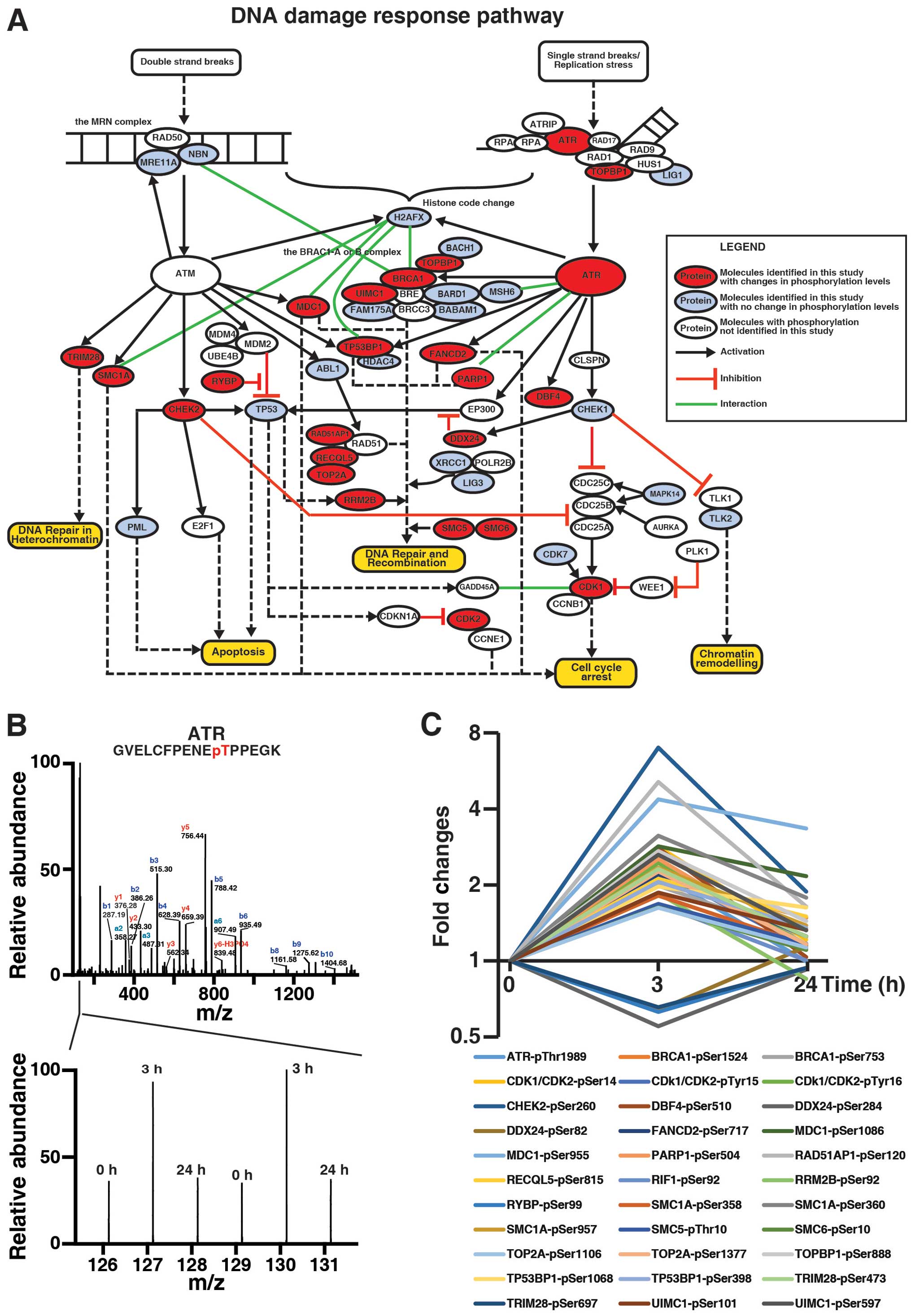
curation, as shown in Fig. 6A.
Here, for the first time, we were able to demonstrate that ATR was
hyperphosphorylated at Thr1989, an autophosphorylation site crucial
for ATR activation (31), by
genistein at 3 h but not 24 h (Fig. 6B
and C), consistent with previous report that genistein
metabolite activates ATR in cells (32). In addition, our data also showed
that TOPBP1, which activates the ATR-ATRIP complex during stressed
DNA replication (33,34), was hyperphosphorylated at Ser888 by
genistein at 3 h but not 24 h (Fig.
6C). Thus, our data suggest that genistein may induce
activation of ATR signaling in MDA-MB-231 cells in an acute manner.
Noteworthy, in our analysis, BRCA1 and RAP80 (UIMC1) were
hyperphosphorylated by genistein at 3 h, suggesting that genistein
may activate the BRCA1-A and -B complexes via the ATR signaling
pathway. Fig. 6C illustrates
genistein-induced changes of 23 signaling molecules, which were
quantitated by TMT-based quantitative phosphoproteomics, in
ATM/ATR-mediated DNA damage response. Taken together, our analysis
revealed the complexity of genistein-induced DNA damage response,
which is more than reported previously by Vauzour and colleagues,
especially during the acute treatment phase (32).
Discussion
Triple-negative breast cancer accounts for
approximately 12–17% of breast cancer cases in women. Patients with
triple-negative tumors have relatively poor clinical outlook,
evidenced by lower 5-year survival rate and higher distant
recurrence rate than other breast cancer patients. These patients
cannot be treated with endocrine therapy or therapy targeting
over-expressed receptors, due to no or less expression of
ER/HER2/PR compared with other subtypes of breast cancer (35,36).
There is an urgent need to develop a novel therapeutic strategy for
these patients. Genistein, one of the major isoflavones in soy
products, has been reported to inhibit proliferation of TNBC cells
(8,11,12,29,37,38).
A number of epidemiological studies reported a significant inverse
association between soy intake and risk of breast cancer incidence
and recurrence (4,5). Thus, genistein can serve as a
potential attractive therapeutic agent for TNBC.
Previously, a number of groups have attempted to
understand the anti-proliferative effects of genistein in TNBC
cells, especially MDA-MB-231 cells (8,10–13,29).
Genistein may inhibit cell growth and induce apoptosis via multiple
signaling pathways. Genistein can upregulate the expression of Bax
and p21WAF1 and downregulate the expression of Bcl-2 and p53
(8). It can inhibit NF-κB activity
via the Akt signaling pathway (13) or the Notch-1 pathway (29) or the MEK5/ERK5 pathway (12). Genistein also induces
phosphorylation of ERK1/2 (11).
However, we did not detect the reported changes of signaling
molecules involved in these signaling pathways at a protein level.
Especially, our analysis did not detect significant changes in the
proteome induced by genistein in MDA-MB-231 cells, out of 2087
proteins, only the expression levels of 8 proteins were shown to be
changed by genistein (Table II).
In contrast, for the first time, our data focusing on the acute
genistein treatment phase showed that phosphorylation of a number
of signaling molecules involved in the regulation of the cell cycle
were regulated by genistein in TNBC cells.
In eukaryotes, the cell cycle can be divided into
the five different phases: G0, G1, S, G2, and M phases (Fig. 5A). Two classes of proteins, cyclins
and cyclin-dependent kinases (CDKs) determine the progress of the
cell cycle. Among them, CDK1 and CDK2 are two critical kinases
controlling the correct transition of the different phases in the
cell cycle (Fig. 5A).
Phosphorylation on the Thr14 and Tyr15 sites inhibits CDK1 kinase
activity, preventing the G2/M transition (39). Genistein can induce G2/M phase
arrest in MDA-MB-231 cells (9).
Here, for the first time, we were able to detect the increased
phosphorylation at the Thr14 and Tyr15 sites on CDK1 in MDA-MB-231
cells after 3 h exposure to genistein, although we cannot exclude
the possibility that genistein could increase the conserved Thr14
and Tyr15 sites on CDK2 and CDK3 (Fig.
5B). The data suggest that genistein executes its effects on
regulation of the cell cycle in a more acute and direct manner than
what previously described (11).
Mitosis and cytokinesis occur during the M phase of
the cell cycle. Our data demonstrated that genistein regulates
phosphorylation of a number of proteins involved in mitosis and
cytokinesis (Table III). The
cohesin complex, consisting of SMC1A, SMC3, RAD21, and STAG2, can
form a large protein ring to hold sister chromatids together before
anaphase during mitosis (40).
When anaphase starts, the anaphase promoting complex (APC) marks an
inhibitory protein securin (PTTG1) with ubiquitination for
degradation. The destruction of securin allows the activation of a
protease called separase (ESPL1), which cleave the cohesin complex
to allow the separation of sister chromatids. Phosphorylation of
CDC20 by Bub1 mediates inhibition of APC by spindle checkpoint
(41). ESCO2 is a cohesin
acetyltransferase and required for the establishment of sister
chromatid cohesin (42,43). As shown in Fig. 5E, our data indicated that genistein
decreased phosphorylation level of ESCO2 and increased
phosphorylation level of CDC20, PTTG1, SMC1A and SMC3 in cells,
suggesting that genistein can inhibit cleavage of the cohesin
complex during mitosis.
The kinetochore is the protein complex assembled at
each centromere on sister chromatids that serves as the attachment
site for spindle microtubules to pull sister chromatids apart
during mitosis. CLASP1 and CLASP2 belong to a family of
microtubule-associated proteins and are required at kinetochore for
attached microtubules (44,45).
Our data showed that phosphorylation at the Ser687 site on CLASP1
and at the Ser455 site on CLASP2 were elevated by genistein at 3-h
and 24-h time point. CENPF is assembled onto kinetochores at late
G2 phase and is involved in chromosome segregation during mitosis
(46). Phosphorylation at the
Ser1747 site on CENPF was found to be decreased by genistein at 3-h
time point and to return to the normal at 24-h point. CENPV was
identified from mitotic chromosome scaffolds by mass spectrometry
and has been shown to be required for centromere organization,
chromosome alignment and cytokinesis (47). Of note, genistein was found to
increase phosphorylation at the Ser47 site on CENPV at 3-h and 24-h
time points and to decrease phosphorylation at the Ser45 site on
CENPV at 3-h, but not 24-h time point. The human Augmin complex
(HAUS) plays a critical role in microtubule attachment to the
kinetochore and central spindle formation (48). HAUS6, a subunit of the HAUS
complex, was found to be hypophosphorylated by genistein at Ser552
at 3-h, but not 24-h time point. MIS18BP1, also called KNL-2, is
required for CENP-A incorporation into chromatin. MIS18BP1 and
CENP-A localize to centromeres throughout the cell cycle. The
depletion of MIS18BP1 in cells causes defect of the kinetochore
assembly and chromosome segregation (49). At 3-h time point, phosphorylation
of MIS18BP1 at the Ser110 was elevated by genistein, but then
decreased to the normal at 24-h. HJURP, a CENP-A chaperon, is also
required for CENP-A localization to centromeres and incorporation
of newly synthesized CENP-A into centromeres (50). It is sufficient to form a
functional de novo kinetochore (51). Phosphorylation of HJURP was
observed to be elevated by genistein at 3-h time point. Spindly
(SPDL1), a mitotic checkpoint protein, is required for localization
of dynein and dynactin to the mitotic kinetochore (52). Farnesylation plays a role in
targeting Spindly to kinetochores. Here we reported the elevated
phosphorylation of Spindly at Ser555 by genistein at 3-h time
point. Our quantitative data on the phosphorylation of these
proteins suggest that genistein may regulate the formation of the
kinetochore by phosphorylation during mitosis, which has not been
reported previously. Taken together, our data suggest that
genistein may have a more direct effect on regulation of the cell
cycle in a complex manner.
ATM (ataxia-telangiectasia mutated) and ATR (ataxia
telangiectasia and Rad3-related protein) are two critical protein
kinases to sense DNA lesions induced by DNA damage or DNA
replication stress and then to elicit the cellular responses that
include cell cycle arrest, DNA repair and apoptosis (53). Whereas ATM mostly responds to
double-strand DNA breaks, ATR is activated by a broad spectrum of
DNA damage, including persistent single-stranded DNA during
stressed replication fork. Genistein can induce the phosphorylation
of ATM/Chk2 in irradiated MDA-MB-231 cells (54). Our data showed that at 3 h,
genistein can activate ATR by hyperphosphorylation at Thr1989, an
autophosphorylation site crucial for ATR activation (31). TOPBP1, an interacting protein of
DNA topoisomerase II β, binds to single-stranded DNA and activates
the ATR-ATRIP complex during stressed DNA replication (33,34).
Noteworthy, our data also showed that at 3 h, genistein can induce
hyperphosphorylation of TOPBP1 at Ser888. Thus, our data suggest
acute effects of genistein on ATR signaling in MDA-MB-231
cells.
BRCA1 is a human tumor suppressor gene and
its mutations lead to an increased risk for breast cancer. The
BRCA1 protein product plays critical roles in DNA repair,
cell cycle checkpoint control, and maintenance of genomic stability
(55,56). BRCA1 executes its functions by
forming at least three different protein complexes (57). The BRCA1-A complex, which contains
the BRCA1-BARD1 heterodimer, UIMC1, BRCC3, BRE, FAM175A and BABAM1,
recognizes and binds K63-linked polyubiquitin chains present on
histone H2A and H2AX at DNA damage sites to promote DNA repair
(58). The BRCA1-B complex
consisting of the BRCA1-BARD1 heterodimer, BACH1 and TOPBP1, binds
DNA lesions during S phase at DNA damage sites and is required for
progression through S phase (59).
The BRCA1-C complex, which also contains BRCA1-BARD1 heterodimer,
RBBP8, and the MRN complex (MRE11A/RAD50/NBS1), binds to the
double-stranded DNA breaks during S and G2 phase of the cell cycle
and is required for DNA damage-induced Chk1 phosphorylation and the
G2/M transition checkpoint (59,60).
In our analysis, BRCA1 were hyperphosphorylated at Ser753 and
Ser1524 by genistein at 3-h. While phosphorylation at Ser1524 is
required for ATM-mediated S phase checkpoint and response to
double-stranded DNA breaks (61,62),
the role of phosphorylation at Ser753 remains unknown. Moreover,
BRCA1-A complex subunit RAP80 (UIMC1), and TOPBP1, a component of
the BRCA1 B complex, were also hyperphosphorylated by genistein at
3-h. Phosphorylation at Ser101 of RAP80 (UIMC1) is critical to
target the BRCA1-A complex to specific ubiquitin structure at DNA
damage sites (63,64). Thus, our data suggest that
genistein may activate the BRCA1-A and -B complexes via the ATR
signaling pathway. Taken together, we concluded that genistein may
affect more acutely the active DNA damage response pathway in a
complex manner.
The difference of our data from previous reports may
be due to the difference of the time points chosen in each of the
studies. We aimed to identify the early signaling events (i.e. 3-h)
in cells after the treatment with genistein, while previous reports
were focused on the long-term effects of genistein on signaling
pathways (>24 h). Our approach allows us to demonstrate that
genistein may induce G2/M cell cycle arrest and apoptosis by
regulating the cell cycle and DNA damage response more acutely and
directly than indicated by previous reports. Notably, our data also
revealed that genistein can induce hyperphosphorylation of
components of the MCM complex, which could lead to inhibition of
DNA replication (65). Thus,
genistein may induce G2/M cell cycle arrest via stressed DNA
replication.
While we describe the phosphoprotein changes in the
core components in the cell cycle and the DNA damage response
signaling pathways, it is important to keep in mind that these
protein modifications are not independent of the other components
of these pathways. In fact, these core components interact with
many other signaling molecules to mediate the biological effects of
genistein in cells. Overall, our studies revealed the complexity of
the genistein effects on regulation of the cell cycle and opened
promising avenues for future investigation into question regarding
early effects in TNBC cells following treatment with genistein.
Acknowledgements
This study was supported by the Fundamental Research
Funds for the Cancer Hospital, Chinese Academy of Medical Sciences
(Y.F., no. JK2014B10), National Basic Research Program of China
(973 Program, Y.J., no. 2015CB553902), the Fundamental Research
Funds for the Central Universities (Z.H., no. 33320140103), the
National Natural Science Foundation of China (J.W., General
Program: 81372829), and Beijing Municipal Natural Science
Foundation (J.W., no. 7142140).
Abbreviations:
|
TMT
|
tandem mass tag
|
|
TNBC
|
triple-negative breast cancer
|
|
ATR
|
ataxia telangiectasia and Rad3-related
protein
|
|
ER
|
estrogen receptor
|
|
PR
|
progesterone receptor
|
|
HER2
|
human epidermal growth factor receptor
2
|
|
ORC
|
origin recognition complex
|
|
MCM
|
mini chromosome maintenance
|
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Reis-Filho JS and Pusztai L: Gene
expression profiling in breast cancer: Classification,
prognostication, and prediction. Lancet. 378:1812–1823. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Perou CM and Børresen-Dale AL: Systems
biology and genomics of breast cancer. Cold Spring Harb Perspect
Biol. 3:a0032932011. View Article : Google Scholar
|
|
4
|
Adlercreutz CH, Goldin BR, Gorbach SL,
Höckerstedt KA, Watanabe S, Hämäläinen EK, Markkanen MH, Mäkelä TH,
Wähälä KT and Adlercreutz T: Soybean phytoestrogen intake and
cancer risk. J Nutr. 125(Suppl): S757–S770. 1995.
|
|
5
|
Messina MJ, Persky V, Setchell KD and
Barnes S: Soy intake and cancer risk: A review of the in vitro and
in vivo data. Nutr Cancer. 21:113–131. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nechuta SJ, Caan BJ, Chen WY, Lu W, Chen
Z, Kwan ML, Flatt SW, Zheng Y, Zheng W, Pierce JP, et al: Soy food
intake after diagnosis of breast cancer and survival: An in-depth
analysis of combined evidence from cohort studies of US and Chinese
women. Am J Clin Nutr. 96:123–132. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fioravanti L, Cappelletti V, Miodini P,
Ronchi E, Brivio M and Di Fronzo G: Genistein in the control of
breast cancer cell growth: Insights into the mechanism of action in
vitro. Cancer Lett. 130:143–152. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li Y, Upadhyay S, Bhuiyan M and Sarkar FH:
Induction of apoptosis in breast cancer cells MDA-MB-231 by
genistein. Oncogene. 18:3166–3172. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cappelletti V, Fioravanti L, Miodini P and
Di Fronzo G: Genistein blocks breast cancer cells in the G(2)M
phase of the cell cycle. J Cell Biochem. 79:594–600. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Horia E and Watkins BA: Complementary
actions of docosahexaenoic acid and genistein on COX-2, PGE2 and
invasiveness in MDA-MB-231 breast cancer cells. Carcinogenesis.
28:809–815. 2007. View Article : Google Scholar
|
|
11
|
Li Z, Li J, Mo B, Hu C, Liu H, Qi H, Wang
X and Xu J: Genistein induces G2/M cell cycle arrest via stable
activation of ERK1/2 pathway in MDA-MB-231 breast cancer cells.
Cell Biol Toxicol. 24:401–409. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li Z, Li J, Mo B, Hu C, Liu H, Qi H, Wang
X and Xu J: Genistein induces cell apoptosis in MDA-MB-231 breast
cancer cells via the mitogen-activated protein kinase pathway.
Toxicol In Vitro. 22:1749–1753. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gong L, Li Y, Nedeljkovic-Kurepa A and
Sarkar FH: Inactivation of NF-kappaB by genistein is mediated via
Akt signaling pathway in breast cancer cells. Oncogene.
22:4702–4709. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thompson A, Schäfer J, Kuhn K, Kienle S,
Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK and Hamon
C: Tandem mass tags: A novel quantification strategy for
comparative analysis of complex protein mixtures by MS/MS. Anal
Chem. 75:1895–1904. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ross PL, Huang YN, Marchese JN, Williamson
B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, et
al: Multiplexed protein quantitation in Saccharomyces cerevisiae
using amine-reactive isobaric tagging reagents. Mol Cell
Proteomics. 3:1154–1169. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nirujogi RS, Wright JD Jr, Manda SS, Zhong
J, Na CH, Meyerhoff J, Benton B, Jabbour R, Willis K, Kim MS, et
al: Phosphoproteomic analysis reveals compensatory effects in the
piriform cortex of VX nerve agent exposed rats. Proteomics.
15:487–499. 2015. View Article : Google Scholar
|
|
17
|
Roitinger E, Hofer M, Köcher T, Pichler P,
Novatchkova M, Yang J, Schlögelhofer P and Mechtler K: Quantitative
phosphoproteomics of the ataxia telangiectasia-mutated (ATM) and
ataxia telangiectasia-mutated and rad3-related (ATR) dependent DNA
damage response in Arabidopsis thaliana. Mol Cell Proteomics.
14:556–571. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wiśniewski JR, Zougman A, Nagaraj N and
Mann M: Universal sample preparation method for proteome analysis.
Nat Methods. 6:359–362. 2009. View Article : Google Scholar
|
|
19
|
Larsen MR, Thingholm TE, Jensen ON,
Roepstorff P and Jørgensen TJ: Highly selective enrichment of
phosphorylated peptides from peptide mixtures using titanium
dioxide micro-columns. Mol Cell Proteomics. 4:873–886. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rappsilber J, Ishihama Y and Mann M: Stop
and go extraction tips for matrix-assisted laser
desorption/ionization, nanoelectrospray, and LC/MS sample
pretreatment in proteomics. Anal Chem. 75:663–670. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cox J, Neuhauser N, Michalski A, Scheltema
RA, Olsen JV and Mann M: Andromeda: A peptide search engine
integrated into the MaxQuant environment. J Proteome Res.
10:1794–1805. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cox J, Matic I, Hilger M, Nagaraj N,
Selbach M, Olsen JV and Mann M: A practical guide to the MaxQuant
computational platform for SILAC-based quantitative proteomics. Nat
Protoc. 4:698–705. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cox J and Mann M: MaxQuant enables high
peptide identification rates, individualized p.p.b.-range mass
accuracies and proteome-wide protein quantification. Nat
Biotechnol. 26:1367–1372. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vizcaíno JA, Deutsch EW, Wang R, Csordas
A, Reisinger F, Ríos D, Dianes JA, Sun Z, Farrah T, Bandeira N, et
al: ProteomeXchange provides globally coordinated proteomics data
submission and dissemination. Nat Biotechnol. 32:223–226. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mi H, Muruganujan A and Thomas PD: PANTHER
in 2013: Modeling the evolution of gene function, and other gene
attributes, in the context of phylogenetic trees. Nucleic Acids
Res. 41:D377–D386. 2013. View Article : Google Scholar :
|
|
26
|
Mi H, Muruganujan A, Casagrande JT and
Thomas PD: Large-scale gene function analysis with the PANTHER
classification system. Nat Protoc. 8:1551–1566. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang W, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar :
|
|
28
|
Huang W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar
|
|
29
|
Pan H, Zhou W, He W, Liu X, Ding Q, Ling
L, Zha X and Wang S: Genistein inhibits MDA-MB-231 triple-negative
breast cancer cell growth by inhibiting NF-κB activity via the
Notch-1 pathway. Int J Mol Med. 30:337–343. 2012.PubMed/NCBI
|
|
30
|
Wang Z, Liang S, Lian X, Liu L, Zhao S,
Xuan Q, Guo L, Liu H, Yang Y, Dong T, et al: Identification of
proteins responsible for adriamycin resistance in breast cancer
cells using proteomics analysis. Sci Rep. 5:93012015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu S, Shiotani B, Lahiri M, Maréchal A,
Tse A, Leung CC, Glover JN, Yang XH and Zou L: ATR
autophosphorylation as a molecular switch for checkpoint
activation. Mol Cell. 43:192–202. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vauzour D, Vafeiadou K, Rice-Evans C,
Cadenas E and Spencer JP: Inhibition of cellular proliferation by
the genistein metabolite 5,7,3′,4′-tetrahydroxyisoflavone is
mediated by DNA damage and activation of the ATR signalling
pathway. Arch Biochem Biophys. 468:159–166. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tuul M, Kitao H, Iimori M, Matsuoka K,
Kiyonari S, Saeki H, Oki E, Morita M and Maehara Y: Rad9, Rad17,
TopBP1 and claspin play essential roles in heat-induced activation
of ATR kinase and heat tolerance. PLoS One. 8:e553612013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kumagai A, Lee J, Yoo HY and Dunphy WG:
TopBP1 activates the ATR-ATRIP complex. Cell. 124:943–955. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Foulkes WD, Smith IE and Reis-Filho JS:
Triple-negative breast cancer. N Engl J Med. 363:1938–1948. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tichy JR, Deal AM, Anders CK, Reeder-Hayes
K and Carey LA: Race, response to chemotherapy, and outcome within
clinical breast cancer subtypes. Breast Cancer Res Treat.
150:667–674. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rajah TT, Peine KJ, Du N, Serret CA and
Drews NR: Physiological concentrations of genistein and
17β-estradiol inhibit MDA-MB-231 breast cancer cell growth by
increasing BAX/BCL-2 and reducing pERK1/2. Anticancer Res.
32:1181–1191. 2012.PubMed/NCBI
|
|
38
|
Santell RC, Kieu N and Helferich WG:
Genistein inhibits growth of estrogen-independent human breast
cancer cells in culture but not in athymic mice. J Nutr.
130:1665–1669. 2000.PubMed/NCBI
|
|
39
|
Krek W and Nigg EA: Differential
phosphorylation of vertebrate p34cdc2 kinase at the G1/S and G2/M
transitions of the cell cycle: Identification of major
phosphorylation sites. EMBO J. 10:305–316. 1991.PubMed/NCBI
|
|
40
|
Hagstrom KA and Meyer BJ: Condensin and
cohesin: More than chromosome compactor and glue. Nat Rev Genet.
4:520–534. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tang Z, Shu H, Oncel D, Chen S and Yu H:
Phosphorylation of Cdc20 by Bub1 provides a catalytic mechanism for
APC/C inhibition by the spindle checkpoint. Mol Cell. 16:387–397.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Whelan G, Kreidl E, Wutz G, Egner A,
Peters JM and Eichele G: Cohesin acetyltransferase Esco2 is a cell
viability factor and is required for cohesion in pericentric
heterochromatin. EMBO J. 31:71–82. 2012. View Article : Google Scholar :
|
|
43
|
Vega H, Waisfisz Q, Gordillo M, Sakai N,
Yanagihara I, Yamada M, van Gosliga D, Kayserili H, Xu C, Ozono K,
et al: Roberts syndrome is caused by mutations in ESCO2, a human
homolog of yeast ECO1 that is essential for the establishment of
sister chromatid cohesion. Nat Genet. 37:468–470. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Maiato H, Fairley EA, Rieder CL, Swedlow
JR, Sunkel CE and Earnshaw WC: Human CLASP1 is an outer kinetochore
component that regulates spindle microtubule dynamics. Cell.
113:891–904. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Maia AR, Garcia Z, Kabeche L, Barisic M,
Maffini S, Macedo-Ribeiro S, Cheeseman IM, Compton DA, Kaverina I
and Maiato H: Cdk1 and Plk1 mediate a CLASP2 phospho-switch that
stabilizes kinetochore-microtubule attachments. J Cell Biol.
199:285–301. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liao H, Winkfein RJ, Mack G, Rattner JB
and Yen TJ: CENP-F is a protein of the nuclear matrix that
assembles onto kinetochores at late G2 and is rapidly degraded
after mitosis. J Cell Biol. 130:507–518. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tadeu AM, Ribeiro S, Johnston J, Goldberg
I, Gerloff D and Earnshaw WC: CENP-V is required for centromere
organization, chromosome alignment and cytokinesis. EMBO J.
27:2510–2522. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lawo S, Bashkurov M, Mullin M, Ferreria
MG, Kittler R, Habermann B, Tagliaferro A, Poser I, Hutchins JR,
Hegemann B, et al: HAUS, the 8-subunit human Augmin complex,
regulates centrosome and spindle integrity. Curr Biol. 19:816–826.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Maddox PS, Hyndman F, Monen J, Oegema K
and Desai A: Functional genomics identifies a Myb domain-containing
protein family required for assembly of CENP-A chromatin. J Cell
Biol. 176:757–763. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dunleavy EM, Roche D, Tagami H, Lacoste N,
Ray-Gallet D, Nakamura Y, Daigo Y, Nakatani Y and
Almouzni-Pettinotti G: HJURP is a cell-cycle-dependent maintenance
and deposition factor of CENP-A at centromeres. Cell. 137:485–497.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Barnhart MC, Kuich PH, Stellfox ME, Ward
JA, Bassett EA, Black BE and Foltz DR: HJURP is a CENP-A chromatin
assembly factor sufficient to form a functional de novo
kinetochore. J Cell Biol. 194:229–243. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Barisic M, Sohm B, Mikolcevic P, Wandke C,
Rauch V, Ringer T, Hess M, Bonn G and Geley S: Spindly/CCDC99 is
required for efficient chromosome congression and mitotic
checkpoint regulation. Mol Biol Cell. 21:1968–1981. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Maréchal A and Zou L: DNA damage sensing
by the ATM and ATR kinases. Cold Spring Harb Perspect Biol.
5:52013. View Article : Google Scholar
|
|
54
|
Liu X, Sun C, Jin X, Li P, Ye F, Zhao T,
Gong L and Li Q: Genistein enhances the radiosensitivity of breast
cancer cells via G(2)/M cell cycle arrest and apoptosis. Molecules.
18:13200–13217. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Narod SA and Foulkes WD: BRCA1 and BRCA2:
1994 and beyond. Nat Rev Cancer. 4:665–676. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Venkitaraman AR: Cancer susceptibility and
the functions of BRCA1 and BRCA2. Cell. 108:171–182. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang B: BRCA1 tumor suppressor network:
Focusing on its tail. Cell Biosci. 2:62012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wang B, Hurov K, Hofmann K and Elledge SJ:
NBA1, a new player in the Brca1 A complex, is required for DNA
damage resistance and checkpoint control. Genes Dev. 23:729–739.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Greenberg RA, Sobhian B, Pathania S,
Cantor SB, Nakatani Y and Livingston DM: Multifactorial
contributions to an acute DNA damage response by
BRCA1/BARD1-containing complexes. Genes Dev. 20:34–46. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yu X and Chen J: DNA damage-induced cell
cycle checkpoint control requires CtIP, a phosphorylation-dependent
binding partner of BRCA1 C-terminal domains. Mol Cell Biol.
24:9478–9486. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Xu B, O'Donnell AH, Kim ST and Kastan MB:
Phosphorylation of serine 1387 in Brca1 is specifically required
for the Atm-mediated S-phase checkpoint after ionizing irradiation.
Cancer Res. 62:4588–4591. 2002.PubMed/NCBI
|
|
62
|
Cortez D, Wang Y, Qin J and Elledge SJ:
Requirement of ATM-dependent phosphorylation of brca1 in the DNA
damage response to double-strand breaks. Science. 286:1162–1166.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kim H, Chen J and Yu X: Ubiquitin-binding
protein RAP80 mediates BRCA1-dependent DNA damage response.
Science. 316:1202–1205. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sobhian B, Shao G, Lilli DR, Culhane AC,
Moreau LA, Xia B, Livingston DM and Greenberg RA: RAP80 targets
BRCA1 to specific ubiquitin structures at DNA damage sites.
Science. 316:1198–1202. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ilves I, Tamberg N and Botchan MR:
Checkpoint kinase 2 (Chk2) inhibits the activity of the
Cdc45/MCM2-7/GINS (CMG) replicative helicase complex. Proc Natl
Acad Sci USA. 109:13163–13170. 2012. View Article : Google Scholar : PubMed/NCBI
|