Introduction
The environment surrounding cells or tissues is
considered to be an important factor affecting their biological
progress, including cell growth, proliferation, tumorigenesis, and
cell death. During tumorigenesis, cancer cells are under extremely
stressful conditions (e.g., nutrient starvation, acidic, or hypoxic
conditions) because of their uncontrolled growth and proliferation
(1–3). Hypoxia, a condition of a poor oxygen
supply, plays a vital role in tumorigenesis, angiogenesis,
metabolism, proliferation, metastasis, and cell death (4–9).
When cells are initially exposed to hypoxia, they stimulate
particular adaptive responses such as autophagy, which regulate a
series of physiological and cellular mechanisms necessary for
survival (10–15). Further severe hypoxia eventually
results in apoptotic cell death (14,16–18).
Although cancer cells are usually much more sensitive than normal
cells to such stress, they seem to have developed adaptive
mechanisms at different molecular levels to withstand this extreme
environmental condition and to delay or suppress cell death, thus
aggravating tumorigenesis (19–21).
Therefore, the precise understanding of the molecular mechanism of
hypoxia-induced cancer cell growth and death appears to be very
important in cancer research and treatment.
In an effort to identify the proteins responsible
for the resistance of cancer cells to hypoxia-induced cell death,
we initially focused on TRIP-Br1, the transcriptional regulator
interacting with PHD-bromodomain 1 (also known as SERTAD1, p34SEI-1
or SEI-1), a member of the TRIP-Br family. TRIP-Br1 is known to be
involved in various important biological functions, such as
transcription, cell cycle progression, metabolism, metastasis,
tumorigenesis, and cell death (22–31).
However, little is known about its cellular function under hypoxic
condition. It is now widely accepted that many oncoproteins
suppress autophagy and apoptosis, whereas tumor suppressors mostly
induce them (32,33). In our previous study, we showed
that the expression of TRIP-Br1 oncoprotein significantly increased
in response to nutrient starvation, rending cancer cells resistant
to cell death by suppressing three types of representative
programed cell deaths (type I apoptosis, type II autophagy-induced
cell death, and type III/IV necrosis/necroptosis) (34). Therefore, it was hypothesize that
TRIP-Br1 might inhibit cell death under hypoxic condition.
In this study, we attempted to clarify how TRIP-Br1
contributes to the survival of cancer cells under hypoxic condition
during tumor growth.
Materials and methods
Cell lines, cell cultures, and
materials
Six breast cancer cell lines (MCF7, MDA-MB-231,
T47D, Hs578D, BT549, and MDA-MB-435) and two fibroblast normal cell
lines (MEF and NIH3T3) were cultured in Dulbecco's modified Eagle's
medium (DMEM; Welgene Inc., Daegu, Korea) supplemented with 10%
fetal bovine serum (FBS; Gibco BRL, Carlsbad, CA, USA) and 1%
antibiotic-antimycotic (Gibco BRL). MCF10A normal breast epithelial
cells were grown in DMEM/F12 medium (Invitrogen, Carlsbad, CA, USA,
cat. 11330-032) supplemented with 20 ng/ml of epithelial growth
factor (EGF; Sigma-Aldrich, cat. E9644), 100 ng/ml of cholera toxin
(Sigma-Aldrich, cat. C-8052), 10 μg/ml of insulin (Sigma-Aldrich,
cat. I-9278), 0.5 mg/ml of hydrocortisone (Sigma-Aldrich, cat.
H-0888), 5% horse serum (Invitrogen, cat. 16050-122), and 1%
antibiotic-antimycotic. All cells were cultured at 37°C in a
humidified atmosphere composed of 5% CO2. Cell lines
were purchased from the American Type Culture Collection (ATCC).
Cobalt chloride (CoCl2) and LY294004 were purchased from
Sigma-Aldrich (cat. STBC9672V) and Calbiochem, San Diego, CA, USA
(cat. 440202), respectively.
Western blot analysis
Immunoblotting analysis was performed as previously
described (35). Antibodies used
in this study were TRIP-Br1 (Enzo Life Sciences, cat. ALX-804-645),
TRIP-Br3 (Abcam, cat. ab107944), HIF-1α (Cell Signaling Technology,
cat. 3716S), PARP (Cell Signaling Technology, cat. 9542), Bax
(Santa Cruz Biotechnology, cat. sc-20067), XIAP (Cell Signaling
Technology, cat. 2042), SQSTM1/p62 (Cell Signaling Technology, cat.
5114S), LC3 (Enzo Life Sciences, cat. ALX-803-082), and γ-tubulin
(Santa Cruz Biotechnology, cat. sc-7396).
Reverse transcription polymerase chain
reaction (RT-PCR) analysis
Total RNA was extracted from MCF7, MDA-MB-231, and
MCF10A cells by using an RNeasy mini kit (Qiagen, Hilden, Germany).
For reverse transcription, 1 μg of RNA from each sample was
subjected to cDNA synthesis using the TOPscript™
RT2XPreMIXkit(Enzynomics, cat.RT203-50-F, Korea) according to the
manufacturer's instructions. Each gene product was amplified using
10 ng of cDNA, the corresponding pair of primers, and an AccuPower
PCR PreMix system (Bioneer, cat. K-2016, Korea), in which the
β-actin gene product was used as an internal control. The
oligonucleotide sequences for RT-PCR analysis were pRT-HIF-1α-F/R:
5′-CATGGAAGGTATTGCACTGC-3′/5′-TGGCAAGCATCCTGTACTGT-3′;
pRT-TRIP-Br1-F/R:
5′-AGGACCTCAGCCACATTGAG-3′/5′-GGTGCCCAAAGTTCATTGTC-3′;
pRT-TRIP-Br3-F/R:
5′-CTGGTGAAGTTGCAGCTTTG-3′/5′-GGCAAAGGTCAGAAACTGGA-3′;
pRT-β-actin-F/R:
5′-AGGTCGGAGTCAACGGATTTG-3′/5′-GTGATGGCATGGACTGTGGT3′.
Suppression of TRIP-Br1 gene and
overexpression of TRIP-Br1 and HIF-1α genes
To repress TRIP-Br1 expression, cells were
transfected with scrambled small interfering RNA (scRNA) or
TRIP-Br1 silencing siRNA (siTRIP-Br1) (Santa Cruz Biotechnology,
cat. sc-62988). These cells were then incubated in Opti-MEM
(Invitrogen, cat. 31985) at 37°C for 6 h and the transfection
medium was replaced with fresh growth medium. For ectopic
overexpression of TRIP-Br1 and HIF-1α, MCF7 and MDA-MB-231 cells
were transfected with 8 μg of TRIP-Br1 (pTRIP-Br1) or HIF-1α
(pHIF-1α) overexpressing plasmids and their corresponding control
vectors (pEGFP or pCMV-tag2B) by using Lipofectamine 2000
(Invitrogen, cat. 52887, Korea) for 48 h. The pTRIP-Br1 and pHIF-1α
plasmids were kindly provided by Dr Rikiro Fukunaga (Osaka
University, Japan) and Dr Young Yang (Sookmyung Women's University,
Republic of Korea), respectively.
Analysis of apoptosis and autophagy
Apoptosis and autophagy were mainly analyzed by
employing western blotting with corresponding markers or regulatory
proteins: PARP, Bax, and XIAP for apoptosis; SQSTM1/p62 and LC3 for
autophagy. Cell viability was evaluated by means of the MTT assay
following our previous method (36).
Results
Upregulated TRIP-Br1 expression in
overcrowded and hypoxic conditions
During tumorigenesis, the uncontrolled growth of
cancer cells gives rise to oxygen deficiency and nutrient
starvation and eventually cancer cells are under much more
stressful condition compared to normal cells. To elucidate how
cancer cells overcome these stressful environments and continue
growing, we cultured six breast cancer cell lines (MCF7,
MDA-MB-231, T47D, Hs578D, BT549, and MDA-MB-435) and normal cell
lines (MCF10A, MEF, and NIH3T3) in complete media until cell
densities reached ~80% confluence to serve as the normal control
(NC) or until the cells reached a very high cell density and became
overcrowded (OC). Our previous data revealed that overcrowded
condition increased TRIP-Br1, but decreased TRIP-Br3 at the protein
level (34,37). We proposed that a balance between
TRIP-Br1 and TRIP-Br3 levels seems to be crucial to cell survival
and death (34,37).
TRIP-Br3 (also known as SEI-3/CDCA4/Hepp) is a
putative tumor suppressor whereas TRIP-Br1 is an oncoprotein even
though both of them belong to the same TRIP-Br family (35,38–40).
Therefore, the levels of TRIP-Br1 and TRIP-Br3 expression were
determined under condition of overcrowding. Noteworthy, TRIP-Br1
expression increased significantly in all six cancer cell lines but
only slightly in the three normal cell lines, which were not
overcrowded, probably because their growth and proliferation were
controlled (i.e., in a monolayer with ~90% confluence) (Fig. 1A). In contrast, TRIP-Br3 expression
decreased in both the cancer cells and the normal cells, in which
TRIP-Br3 expression decreased to a greater extent in the normal
cells than the cancer cells (Fig.
1A). In an effort to find what kind of stressful condition(s)
is responsible for the TRIP-Br1 upregulation and TRIP-Br3
downregulation under the overcrowded condition, we previously
showed that decreased nutrients (such as serum, glucose, and amino
acids) affect TRIP-Br1 and TRIP-Br3 expression (34,37).
An overcrowded environment leads to another type of extreme stress
than that caused by hypoxia, as well as nutrient starvation. Our
data showed that HIF-1α expression, a marker of hypoxia, increased
as a result of overcrowding, suggesting the presence of hypoxia in
an overcrowded environment (Fig.
1A).
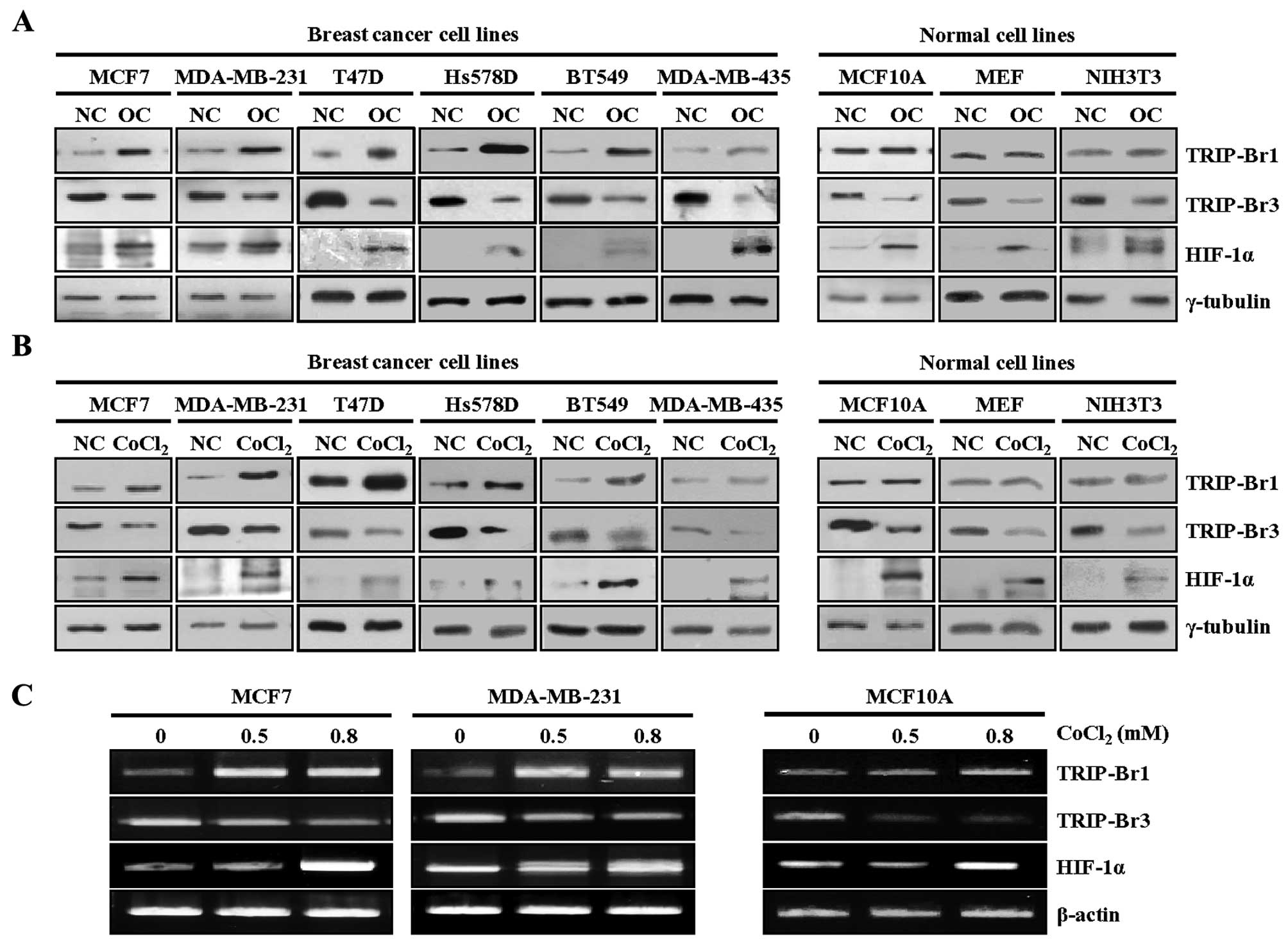 | Figure 1Upregulated TRIP-Br1 expression in
overcrowded and hypoxic conditions. (A) Six breast cancer cell
lines (MCF7, MDA-MB-231, T47D, Hs578D, BT549, and MDA-MB-435) and
three normal cell lines (MCF10A, MEF, and NIH3T3) were cultured in
complete media until the cells either reached ~80% confluence
(indicated as ‘NC’) or were overcrowded at high levels of cell
confluence by being cultured in a complete medium for long time
(indicated as ‘OC’). TRIP-Br1 and TRIP-Br3 expression levels were
checked by means of western blot analysis, in which HIF-1α was used
as a marker for hypoxic condition. (B) The six breast cancer cell
lines and three normal cell lines were incubated for 24 h in growth
media under either normoxic or hypoxic (0.8 mM of CoCl2)
conditions and levels of TRIP-Br1 and TRIP-Br3 expression were
measured at the protein level. (C) MCF7 and MDA-MB-231 breast
cancer cells and MCF10A normal cells were cultured in media with
the concentration of CoCl2 as indicated for 24 h, after
which TRIP-Br1 and TRIP-Br3 expression levels were measured at the
transcriptional level by using RT-PCR, with β-actin used as the
internal control. |
Assuming that TRIP-Br1 and TRIP-Br3 expression might
be affected at least in part by overcrowding-induced hypoxia, we
examined their expression levels under cobalt chloride
(CoCl2)-mediated hypoxic condition. The same cell lines
were treated with 0.8 mM of CoCl2, a well-established
chemical inducer of hypoxia-like responses (4). TRIP-Br1 expression was significantly
increased in all six cancer cell lines, but no significant change
was detected in the three normal cell lines when they were exposed
to CoCl2-generated hypoxia (Fig. 1B). In contrast, TRIP-Br3 expression
decreased in both cancer cells and normal cells (Fig. 1B). These results suggest that the
hypoxic condition is responsible for TRIP-Br1 upregulation and
TRIP-Br3 downregulation. In a further study, the expression levels
of TRIP-Br1 and TRIP-Br3 in response to hypoxia were also tested at
the transcriptional level and similar result was obtained (Fig. 1C).
Taken together, our data strongly suggest that
hypoxic condition induces TRIP-Br1 upregulation and TRIP-Br3
downregulation at both the transcriptional and protein levels.
Inhibitory role of TRIP-Br1 in
hypoxia-induced cell death
TRIP-Br1 expression significantly increased in the
cancer cell lines but not in the normal cell lines under
CoCl2-generated hypoxic condition. We then sought to
determine the cellular function of TRIP-Br1 upregulation.
Previously, we showed that TRIP-Br1 functions as an oncoprotein by
inhibiting cell death in response to anticancer drug treatment and
nutrient/serum starvation (25,34).
Considering these results, it was presumed that TRIP-Br1 might
function in the same way under hypoxic condition. This hypothesis
was tested by determining the effect of TRIP-Br1 on cell death.
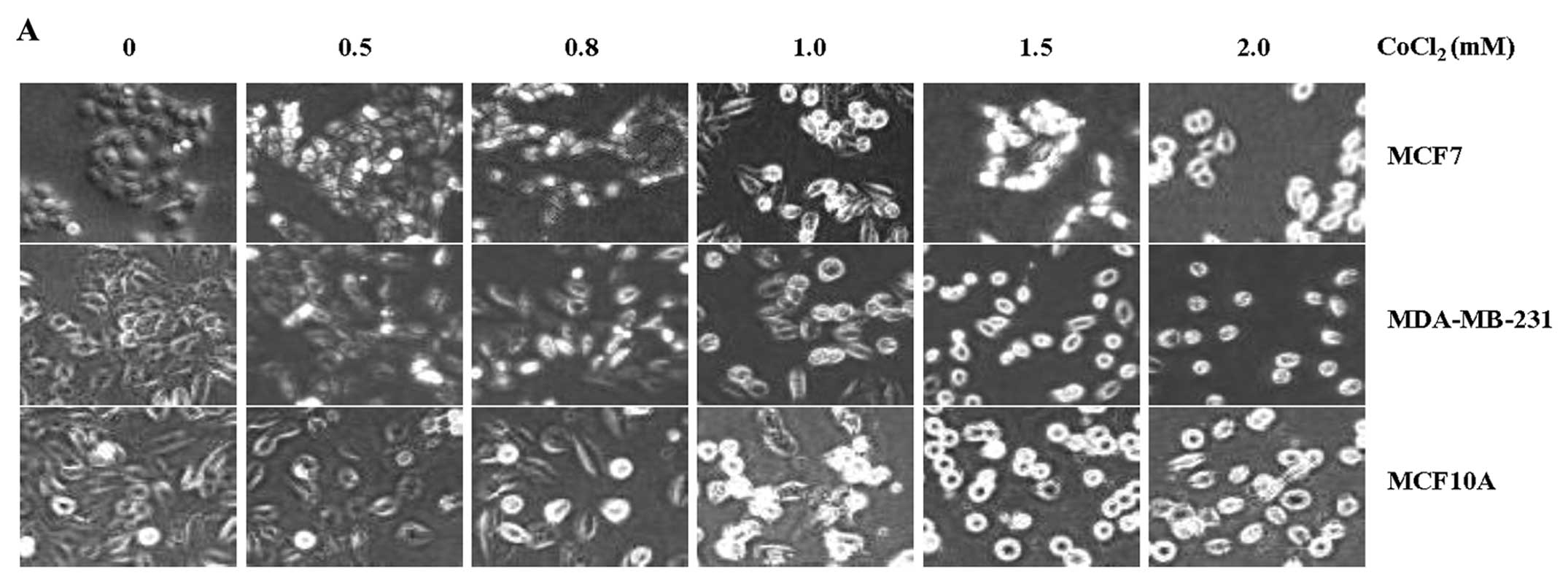
Among the above cell lines, we chose three major
representative cell lines, MCF7, MDA-MB-231, and normal MCF10A for
further studies. Since all of the cell lines we used showed very
similar results in the expression and functions of TRIP-Br1 upon
treatment of various stresses including anticancer drug, nutrient
depletion, and hypoxia (31,34),
we chose three representative cell lines for further studies. These
were incubated in growth media with different concentrations of
CoCl2 for 24 h. The phenotypes of cell death in response
to hypoxia were photographed under a microscope and the survival
rates were measured (Fig. 2A and
B). Our data showed that the rate of cell death was increased
in response to CoCl2-induced hypoxia in a dose-dependent
manner (Fig. 2B). Of note, the
MCF7 and MDA-MB-231 cancer cells with high levels of TRIP-Br1
protein survived for relatively longer periods of time in response
to CoCl2 treatment, as compared with MCF10A normal cells
(Fig. 2A and B).
In addition, our data also showed that hypoxia
induced apoptosis and autophagy. These results were tested by means
of western blot analysis with apoptosis-related regulatory proteins
in the CoCl2-mediated oxygen-insufficient condition. In
Fig. 2C, cleaved PARP and Bax
expression levels were increased in the MCF7, MDA-MB-231, and
MCF10A cells in proportion to the CoCl2 concentration.
In addition, hypoxia-stimulated autophagy was also examined because
this phenomenon is important for achieving cellular homeostasis in
response to a variety of stimuli including hypoxia (12,15,41,42).
The level of SQSTM1/p62 expression was decreased in response to
CoCl2-induced hypoxia in a dose-dependent manner
(Fig. 2C). The conversion ratio
from LC3-I to LC3-II was enhanced in response to
CoCl2-mediated hypoxia (Fig. 2C). These data obviously suggest
that hypoxia can stimulate apoptosis and autophagy, in which
TRIP-Br1 expression increased, implying an inhibitory role of
TRIP-Br1 in apoptosis and autophagy.
Despite hypoxia being more toxic to cancer cells
than to normal cells, many cancer cells are still able to surmount
this stressful condition by modulating a cascade of regulatory
systems or proteins. We showed that TRIP-Br1 gene expression was
significantly increased only in the cancer cells but not in the
normal cells under CoCl2-evoked hypoxic condition. Our
previous report also showed that TRIP-Br1 provides an
anti-apoptotic function to cancer cells in response to anticancer
drugs and nutrient starvation (25,34).
Considering these results, it was hypothesized that TRIP-Br1
upregulation might contribute to the enhanced survival of cancer
cells under hypoxic condition. This hypothesis is supported by the
finding that TRIP-Br1 knock-down in MCF7 and MDA-MB-231 cancer
cells accelerated cell death after they were exposed to
CoCl2 (Fig. 2D). This
possibility was also examined using western blot analysis, in which
the levels of cleaved PARP and Bax expression increased after
TRIP-Br1 silencing in CoCl2-containing media and even in
complete media (Fig. 2E). In our
previous study, we proposed that TRIP-Br1 could inhibit apoptosis
by stabilizing the well-known inhibitor of apoptosis, X-linked
inhibitor of apoptosis protein (XIAP) (25). We also showed that TRIP-Br1 can
suppress three types of programed cell deaths (apoptosis,
autophagy, and necrosis/necroptosis) via XIAP stabilization
(34). Therefore, XIAP expression
levels were measured and found to be greatly decreased after
TRIP-Br1 silencing in CoCl2-containing media, indicating
that TRIP-Br1 can inhibit apoptosis by stabilizing XIAP (Fig. 2E). TRIP-Br1 knock-down also
decreased SQSTM1/p62 protein levels and increased the LC3
conversion rate from LC3-I to LC3-II, indicating that TRIP-Br1
represses autophagy as well apoptosis under hypoxic condition
(Fig. 2E).
Taken together, our data suggest that TRIP-Br1 may
function as an oncogene by rendering cancer cells resistant to
apoptosis through the stabilization of XIAP under stressful hypoxic
conditions.
Effect of PI3K/AKT signaling pathway on
TRIP-Br1 expression under CoCl2-induced hypoxic
condition
We have shown that hypoxia stimulated much higher
levels of TRIP-Br1 expression in cancer cells as compared with
normal cells, conferring on cancer cells the ability to develop an
enhanced adaptive mechanism for resisting cell death. Our next
question was what kind of mechanism is in charge of TRIP-Br1
upregulation in response to hypoxia. It has been widely accepted
that oncogenic protein usually stimulates cancer cell proliferation
by triggering the activation of a string of signaling pathways,
such as the phosphoinositol-3-kinase (PI3K)/AKT signaling pathway.
We previously proposed that inhibition of the PI3K/AKT signaling
pathway increased TRIP-Br1 but decreased TRIP-Br3 expression under
serum starvation condition (34,37).
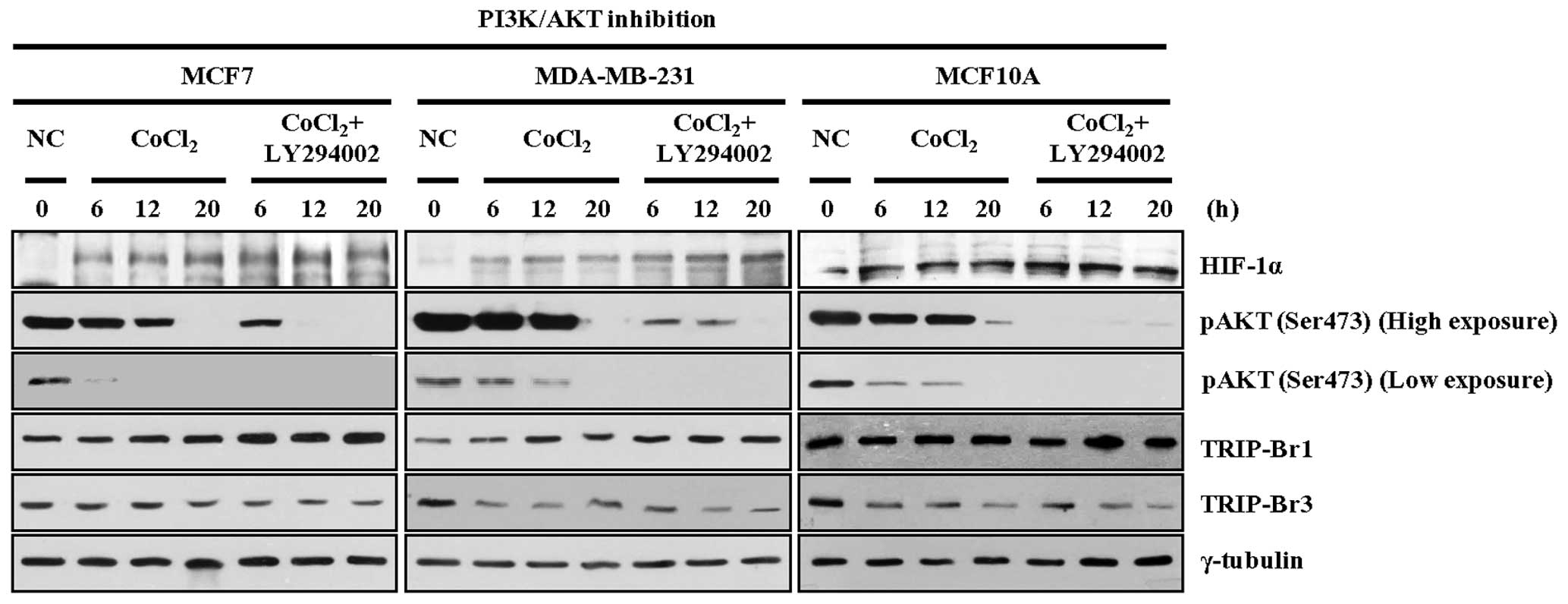
Therefore, we examined the effect of this pathway on TRIP-Br1 and
TRIP-Br3 expression levels by treating MCF7, MDA-MB-231, and MCF10A
cells with LY294002, a PI3K/AKT inhibitor, in the media along with
CoCl2. It was found that CoCl2-mediated
hypoxia inhibited AKT phosphorylation on the 473 serine residue. Of
note, inhibition of the PI3K/AKT pathway enhanced TRIP-Br1
upregulation or TRIP-Br3 down-regulation under hypoxic condition
(Fig. 3).
These results suggest that hypoxia-induced blockage
of the PI3K/AKT signaling pathway is at least partly responsible
for the changes in TRIP-Br1 and TRIP-Br3 expression levels in an
oxygen-deprived environment.
HIF-1α-mediated TRIP-Br1 upregulation
under hypoxic condition
It is well known that HIF-1α is a central regulator
of the expression of a broad range of genes, including those
involved in tumorigenesis (7,8,43,44).
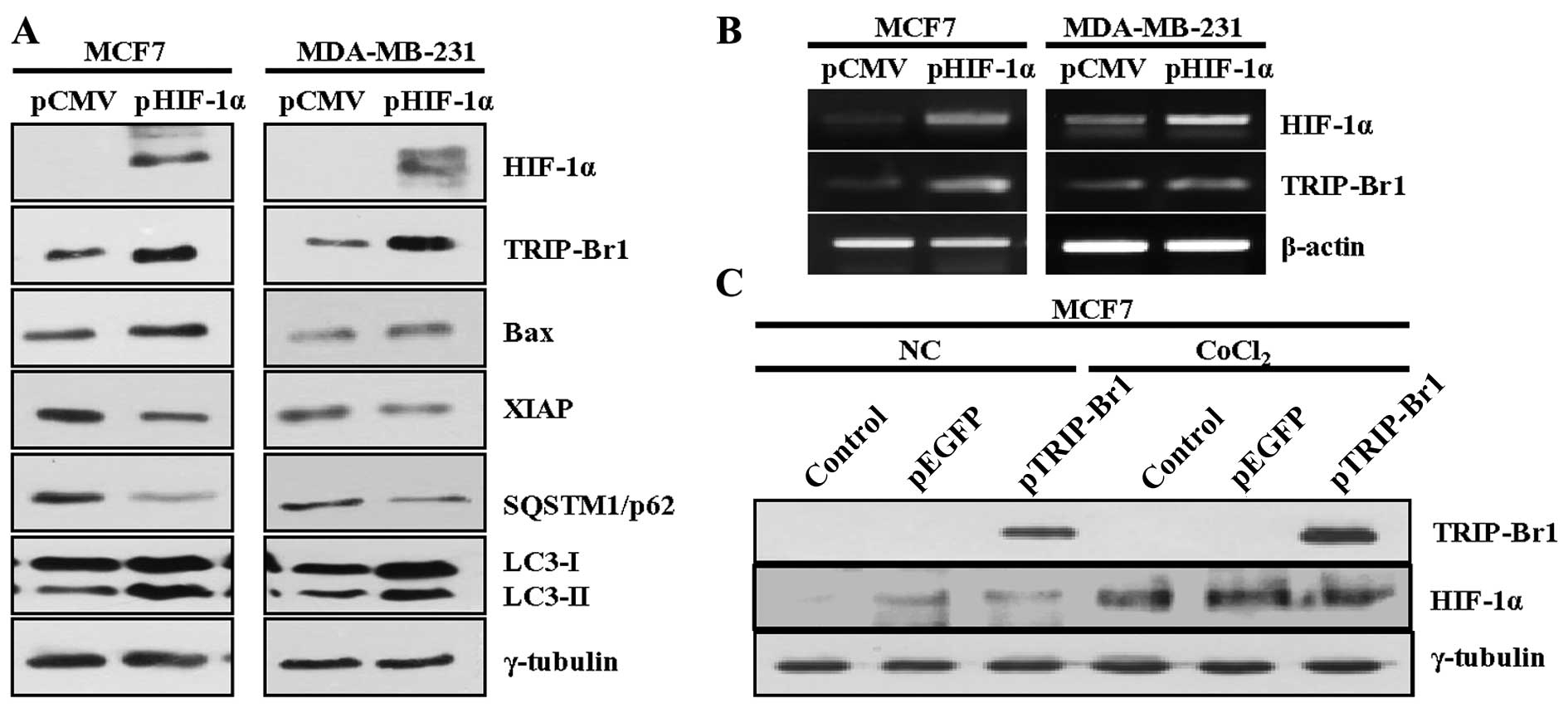
Thus, when we investigated whether or not HIF-1α could affect
oncogenic TRIP-Br1 expression, we found that the transfection of
HIF-1α-overexpressing plasmid into the MCF7 and MDA-MB-231 cells
induced TRIP-Br1 upregulation, as was the case under hypoxic
condition at both the protein and the transcriptional levels
(Fig. 4A and B). Our data also
revealed that HIF-1α overexpression induced apoptosis and autophagy
in the MCF7 and MDA-MB-231 cancer cells (Fig. 4A). In contrast, ectopic
overexpression of the TRIP-Br1 gene had no effect on HIF-1α
expression (Fig. 4C).
These data suggest that HIF-1α is at least partly
responsible for the TRIP-Br1 upregulation under hypoxic
condition.
Discussion
Hypoxia has been a major topic in the area of cancer
research for many decades. It is commonly found in many cases of
solid tumors, in which the tumor cells struggle to adapt to this
stressful environment by developing a broad range of cellular
mechanisms (7,8,12,15,18,44).
In order to identify the mechanism to explain how cancer cells can
survive and continue to grow despite being deprived of oxygen, we
initially focused on the role of TRIP-Br1 oncoprotein in
hypoxia.
Under oxygen-poor conditions, TRIP-Br1 was found to
be significantly upregulated in the breast cancer cell lines
selected for testing, but was only slightly upregulated in the
normal cell lines. This finding underlies the fact that breast
cancer cells and normal cells seem to react differently to hypoxia
through the regulation of TRIP-Br1 expression. In our study, oxygen
deprivation caused injury to cancer and normal cells, inducing
apoptosis. However, TRIP-Br1 enables cancer cells to adapt to the
stress of hypoxia, thus rending them resistant to cell death.
Interestingly, even though TRIP-Br1 and TRIP-Br3 belong to the same
TRIP-Br family, their levels of expression changed in opposite
change in expression level under hypoxic conditions. These results
imply the similar but different cellular functions of TRIP-Br1 and
TRIP-Br3. TRIP-Br3 is known to function as a putative tumor
suppressor, exerting cellular effects in tumorigenesis that differ
from the effects of TRIP-Br1 (37–39).
However, our previous study showed that both TRIP-Br1 and TRIP-Br3
can inhibit apoptosis in condition of serum starvation (34,37).
As we have now seen in the case of hypoxia, TRIP-Br1 expression
levels increased while TRIP-Br3 expression decreased (37). In normal cells, TRIP-Br3 proteins
are rapidly and greatly degraded. Rapid decrease in TRIP-Br3
expression triggers the ubiquitination and degradation of XIAP
protein, eventually leading to cell death (37). In cancer cells, however, TRIP-Br3
expression is slightly downregulated (37). Therefore, we proposed that TRIP-Br3
and TRIP-Br1 may coordinately regulate apoptosis in normal and
cancer cells by competitively interacting with XIAP. Considering
our data as a whole, we speculate that TRIP-Br1 and TRIP-Br3 may be
under a similar regulatory control system in hypoxic stressful
environments. We also suspect that they may act as adopter
proteins, functioning differently by changing their binding
partners, although this possibility needs to be studied
further.
Attention has also been focused on the effect of
HIF-1α on TRIP-Br1 expression, because the expression of many genes
is known to be regulated by HIF-1α, a key regulator of a wide range
of cellular responses to hypoxia in mammalian cells (5,7,11,43).
Our data showed that TRIP-Br1 expression was stimulated by HIF-1α
induction in both CoCl2-caused hypoxia and exogenous
insertion at the transcriptional and translational levels. This
finding provides a clue to the notion that HIF-1α may contribute to
the TRIP-Br1-mediated resistance of tumor cells to apoptosis caused
by hypoxia. Nevertheless, it remains to be determined how HIF-1α
promotes TRIP-Br1 upregulation, directly or indirectly, under
conditions of oxygen insufficiency.
In summary, our results demonstrate that TRIP-Br1
confers resistance to hypoxia-induced cell death in cancer cells.
Thus, targeting TRIP-Br1-mediated cell death under hypoxic
condition may provide vital information to those working in cancer
research and in the development of effective anticancer drugs.
Acknowledgements
This work was supported by the grant from Sookmyung
Women's University (2013).
Abbreviations:
|
TRIP-Br1
|
transcriptional regulator interacting
with the PHD-bromodomain 1
|
|
TRIP-Br3
|
transcriptional regulator interacting
with the PHD-bromodomain 3
|
|
XIAP
|
X-linked inhibitor of apoptosis
protein
|
|
PI3K
|
phosphoinositol-3-kinase
|
|
HIF-1α
|
hypoxia-inducible factor 1α
|
References
|
1
|
Leontieva OV and Blagosklonny MV:
Yeast-like chronological senescence in mammalian cells: Phenomenon,
mechanism and pharmacological suppression. Aging (Albany, NY).
3:1078–1091. 2011. View Article : Google Scholar
|
|
2
|
Rubin H: Multistage carcinogenesis in cell
culture. Dev Biol (Basel). 106:61–66; discussion 67, 143–160.
2001.
|
|
3
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Piret JP, Mottet D, Raes M and Michiels C:
CoCl2, a chemical inducer of hypoxia-inducible factor-1,
and hypoxia reduce apoptotic cell death in hepatoma cell line
HepG2. Ann NY Acad Sci. 973:443–447. 2002. View Article : Google Scholar
|
|
5
|
Semenza GL: HIF-1: mediator of
physiological and patho-physiological responses to hypoxia. J Appl
Physiol (1985). 88:1474–1480. 2000.
|
|
6
|
Lee KA, Roth RA and LaPres JJ: Hypoxia,
drug therapy and toxicity. Pharmacol Ther. 113:229–246. 2007.
View Article : Google Scholar
|
|
7
|
Carmeliet P, Dor Y, Herbert JM, Fukumura
D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R,
Maxwell P, et al: Role of HIF-1alpha in hypoxia-mediated apoptosis,
cell proliferation and tumour angiogenesis. Nature. 394:485–490.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rankin EB and Giaccia AJ: The role of
hypoxia-inducible factors in tumorigenesis. Cell Death Differ.
15:678–685. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rofstad EK and Danielsen T:
Hypoxia-induced metastasis of human melanoma cells: Involvement of
vascular endothelial growth factor-mediated angiogenesis. Br J
Cancer. 80:1697–1707. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kothari S, Cizeau J, McMillan-Ward E,
Israels SJ, Bailes M, Ens K, Kirshenbaum LA and Gibson SB: BNIP3
plays a role in hypoxic cell death in human epithelial cells that
is inhibited by growth factors EGF and IGF. Oncogene. 22:4734–4744.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Suzuki H, Tomida A and Tsuruo T:
Dephosphorylated hypoxia-inducible factor 1alpha as a mediator of
p53-dependent apoptosis during hypoxia. Oncogene. 20:5779–5788.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tafani M, Schito L, Anwar T, Indelicato M,
Sale P, Di Vito M, Morgante E, Beraldi R, Makovec F, Letari O, et
al: Induction of autophagic cell death by a novel molecule is
increased by hypoxia. Autophagy. 4:1042–1053. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Scarlatti F, Granata R, Meijer AJ and
Codogno P: Does autophagy have a license to kill mammalian cells?
Cell Death Differ. 16:12–20. 2009. View Article : Google Scholar
|
|
14
|
Bursch W, Karwan A, Mayer M, Dornetshuber
J, Fröhwein U, Schulte-Hermann R, Fazi B, Di Sano F, Piredda L,
Piacentini M, et al: Cell death and autophagy: Cytokines, drugs,
and nutritional factors. Toxicology. 254:147–157. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang H, Bosch-Marce M, Shimoda LA, Tan
YS, Baek JH, Wesley JB, Gonzalez FJ and Semenza GL: Mitochondrial
autophagy is an HIF-1-dependent adaptive metabolic response to
hypoxia. J Biol Chem. 283:10892–10903. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ou XM, Chen K and Shih JC: Monoamine
oxidase A and repressor R1 are involved in apoptotic signaling
pathway. Proc Natl Acad Sci USA. 103:10923–10928. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Izuishi K, Kato K, Ogura T, Kinoshita T
and Esumi H: Remarkable tolerance of tumor cells to nutrient
deprivation: Possible new biochemical target for cancer therapy.
Cancer Res. 60:6201–6207. 2000.PubMed/NCBI
|
|
18
|
Piret JP, Lecocq C, Toffoli S, Ninane N,
Raes M and Michiels C: Hypoxia and CoCl2 protect HepG2
cells against serum deprivation- and t-BHP-induced apoptosis: A
possible anti-apoptotic role for HIF-1. Exp Cell Res. 295:340–349.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Heyman SN, Leibowitz D, Mor-Yosef Levi I,
Liberman A, Eisenkraft A, Elcalai R, Khamaisi M and Rosenberger C:
Adaptive response to hypoxia and remote ischemia preconditioning: A
new HIF era in clinical medicine. Acta Physiol (Oxf). Oct
9–2015.(Epub ahead of print).
|
|
20
|
Hirst DG and Wood PJ: The adaptive
response of mouse tumours to anaemia and retransfusion. Int J
Radiat Biol Relat Stud Phys Chem Med. 51:597–609. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Acker T and Plate KH: A role for hypoxia
and hypoxia-inducible transcription factors in tumor physiology. J
Mol Med Berl. 80:562–575. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hsu SI, Yang CM, Sim KG, Hentschel DM,
O'Leary E and Bonventre JV: TRIP-Br: A novel family of PHD zinc
finger- and bromodomain-interacting proteins that regulate the
transcriptional activity of E2F-1/DP-1. EMBO J. 20:2273–2285. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sim KG, Zang Z, Yang CM, Bonventre JV and
Hsu SI: TRIP-Br links E2F to novel functions in the regulation of
cyclin E expression during cell cycle progression and in the
maintenance of genomic stability. Cell Cycle. 3:1296–1304. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sim KG, Cheong JK and Hsu SI: The TRIP-Br
family of transcriptional regulators is essential for the execution
of cyclin E-mediated cell cycle progression. Cell Cycle.
5:1111–1115. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hong SW, Kim CJ, Park WS, Shin JS, Lee SD,
Ko SG, Jung SI, Park IC, An SK, Lee WK, et al: p34SEI-1 inhibits
apoptosis through the stabilization of the X-linked inhibitor of
apoptosis protein: p34SEI-1 as a novel target for anti-breast
cancer strategies. Cancer Res. 69:741–746. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li J, Muscarella P, Joo SH, Knobloch TJ,
Melvin WS, Weghorst CM and Tsai MD: Dissection of CDK4-binding and
transactivation activities of p34(SEI-1) and comparison between
functions of p34(SEI-1) and p16(INK4A). Biochemistry.
44:13246–13256. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sugimoto M, Nakamura T, Ohtani N, Hampson
L, Hampson IN, Shimamoto A, Furuichi Y, Okumura K, Niwa S, Taya Y,
et al: Regulation of CDK4 activity by a novel CDK4-binding protein,
p34(SEI-1). Genes Dev. 13:3027–3033. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liew CW, Boucher J, Cheong JK, Vernochet
C, Koh HJ, Mallol C, Townsend K, Langin D, Kawamori D, Hu J, et al:
Ablation of TRIP-Br2, a regulator of fat lipolysis, thermogenesis
and oxidative metabolism, prevents diet-induced obesity and insulin
resistance. Nat Med. 19:217–226. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fernandez-Marcos PJ, Pantoja C,
Gonzalez-Rodriguez A, Martin N, Flores JM, Valverde AM, Hara E and
Serrano M: Normal proliferation and tumorigenesis but impaired
pancreatic function in mice lacking the cell cycle regulator sei1.
PLoS One. 5:e87442010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jung S, Ohk J, Jeong D, Li C, Lee S, Duan
J, Kim C, Lim JS, Yang Y, Kim KI, et al: Distinct regulatory effect
of the p34SEI-1 oncoprotein on cancer metastasis in
HER2/neu-positive and -negative cells. Int J Oncol. 45:189–196.
2014.PubMed/NCBI
|
|
31
|
Hong SW, Shin JS, Lee YM, Kim DG, Lee SY,
Yoon DH, Jung SY, Hwang JJ, Lee SJ, Cho DH, et al: p34 (SEI-1)
inhibits ROS-induced cell death through suppression of ASK1. Cancer
Biol Ther. 12:421–426. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gozuacik D and Kimchi A: Autophagy as a
cell death and tumor suppressor mechanism. Oncogene. 23:2891–2906.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen S, Rehman SK, Zhang W, Wen A, Yao L
and Zhang J: Autophagy is a therapeutic target in anticancer drug
resistance. Biochim Biophys Acta. 1806:220–229. 2010.PubMed/NCBI
|
|
34
|
Jung S, Li C, Duan J, Lee S, Kim K, Park
Y, Yang Y, Kim KI, Lim JS, Cheon CI, et al: TRIP-Br1 oncoprotein
inhibits autophagy, apoptosis, and necroptosis under
nutrient/serum-deprived condition. Oncotarget. 6:29060–29075.
2015.PubMed/NCBI
|
|
35
|
Jung S, Li C, Jeong D, Lee S, Ohk J, Park
M, Han S, Duan J, Kim C, Yang Y, et al: Oncogenic function of
p34SEI-1 via NEDD4-1-mediated PTEN ubiquitination/degradation and
activation of the PI3K/AKT pathway. Int J Oncol. 43:1587–1595.
2013.PubMed/NCBI
|
|
36
|
Choi EJ, Kim T and Lee MS: Pro-apoptotic
effect and cytotoxicity of genistein and genistin in human ovarian
cancer SK-OV-3 cells. Life Sci. 80:1403–1408. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li C, Jung S, Lee S, Jeong D, Yang Y, Kim
KI, Lim JS, Cheon CI, Kim C, Kang YS, et al: Nutrient/serum
starvation derived TRIP-Br3 down-regulation accelerates apoptosis
by destabilizing XIAP. Oncotarget. 6:7522–7535. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hayashi R, Goto Y, Ikeda R, Yokoyama KK
and Yoshida K: CDCA4 is an E2F transcription factor family-induced
nuclear factor that regulates E2F-dependent transcriptional
activation and cell proliferation. J Biol Chem. 281:35633–35648.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tategu M, Nakagawa H, Hayashi R and
Yoshida K: Transcriptional co-factor CDCA4 participates in the
regulation of JUN oncogene expression. Biochimie. 90:1515–1522.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bennetts JS, Fowles LF, Berkman JL, van
Bueren KL, Richman JM, Simpson F and Wicking C: Evolutionary
conservation and murine embryonic expression of the gene encoding
the SERTA domain-containing protein CDCA4 (HEPP). Gene.
374:153–165. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bohensky J, Shapiro IM, Leshinsky S,
Terkhorn SP, Adams CS and Srinivas V: HIF-1 regulation of
chondrocyte apoptosis: Induction of the autophagic pathway.
Autophagy. 3:207–214. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pursiheimo JP, Rantanen K, Heikkinen PT,
Johansen T and Jaakkola PM: Hypoxia-activated autophagy accelerates
degradation of SQSTM1/p62. Oncogene. 28:334–344. 2009. View Article : Google Scholar
|
|
43
|
Weidemann A and Johnson RS: Biology of
HIF-1alpha. Cell Death Differ. 15:621–627. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Greijer AE and van der Wall E: The role of
hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J
Clin Pathol. 57:1009–1014. 2004. View Article : Google Scholar : PubMed/NCBI
|