Multiple myeloma (MM) is the second most prevalent
hematological malignancy worldwide, with a median onset of 60 years
of age (1–6). MM is currently incurable, albeit
clinically manageable and typically manifests with an accumulation
of terminally differentiated monoclonal plasma cells (PCs) in the
bone marrow (3). It is
distinguished from solitary plasmacytoma by the presence of
aberrant PCs at numerous skeletal sites (7,8).
MM can be ‘secretory’ or ‘non-secretory’ depending
on the serum/urine levels of secreted monoclonal immunoglobulin.
‘Secretory MM’ is characterized by the presence of abnormal levels
of monoclonal proteins (M-protein) or paraproteins in circulation
and urine. ‘Non-secretory’ MM accounts for 1% of all MM cases and
lacks the hallmark of increased serum or urine M-protein or
paraprotein. Consequently, the diagnosis of non-secretory MM
depends rather on an increase in tumor burden and evidence of end
organ damage (9,10). The complex spectrum of
physiological impairment attributed to MM include lytic bone
lesions, osteoporosis, compression fractures, bone pain and
ultimately patient immobility. The abundance of malignant
monoclonal PCs also severely compromises patient immunity and
hematopoiesis (11).
The inclusion of immunomodulatory drugs (IMiDs) as
part of high dose chemotherapy together with systemic and
cytogenetic prognostic markers have improved patient survival in
MM. Thalidomide, and its derivatives are currently approved for use
across all phases of MM therapy. These drugs possess
immunomodulatory, anti-angiogenic, anti-inflammatory and
anti-proliferative capacity (12).
Over the past few decades, a 30–40% complete response rate and an
increase in median survival of 4–5 years have been achieved with
these drugs in combination with auto-transplants in younger de
novo patients (13).
Most MM patients respond successfully to initial
induction therapy, however, all the patients eventually relapse,
forcing a review of the treatment regimen (14). A significant contributor to
treatment failure leading to clinical relapse is the emergence of
multi-drug resistance (MDR) (15).
MDR is the phenomenon whereby the cancer cells become resistant to
a wide variety of structurally and functionally unrelated drugs
following exposure to a single chemotherapeutic agent (16–18).
Existing measures for assessing the clinical state of MM patients
include serum markers [immunoglobulins, β2-microglobulin
(B2M), free light chain assays, creatinine, C-reactive
protein (CRP) and thymidine kinase] followed by confirmation with
invasive bone marrow biopsy. However, these do not offer a direct
measurement of the presence or the evolution of proteins
responsible for drug resistance on malignant PCs. MM is
characterized by the presence of multiple clones with differing
degrees of drug sensitivity at the time of diagnosis. Consequently,
despite complex chemotherapeutic regimes (19), therapeutic response is
unpredictable and extremely variable with MM patients. Furthermore,
bone marrow biopsy cannot assess the patchy tumor infiltrates in
multiple sites associated with MM and provides an indirect measure
of tumor burden distributed throughout the skeletal system. This
impacts the quality of life for the patient and translates to
highly heterogeneous patient survival rates ranging from a few
weeks to more than 10 years (20).
Aside from the significant physical and emotional
costs associated with the emergence of MDR and subsequent relapse,
there are also significant financial costs incurred with the
management of MDR. The drugs used at relapse are typically novel,
costly and with associated side effects. The estimated cost of an
effective melphalan, prednisone and velcade regimen approximates
$119,102 (US), while a novel superior regimen utilizing melphalan
and prednisone combined with lenalidomide maintenance can reach as
high as $248,358 (US) (21).
Consequently, MM remains one of the most costly cancers to treat
when total treatment costs are considered (21–24).
Here, we review the factors limiting the successful
treatment outcome in the complex multiple myeloma clinical setting.
We focus on the persistent issue of drug resistant clones in MM and
the major role played by ATP-binding cassette (ABC) transporters
along with other resistance mechanisms in relapse in the era of
novel therapeutics.
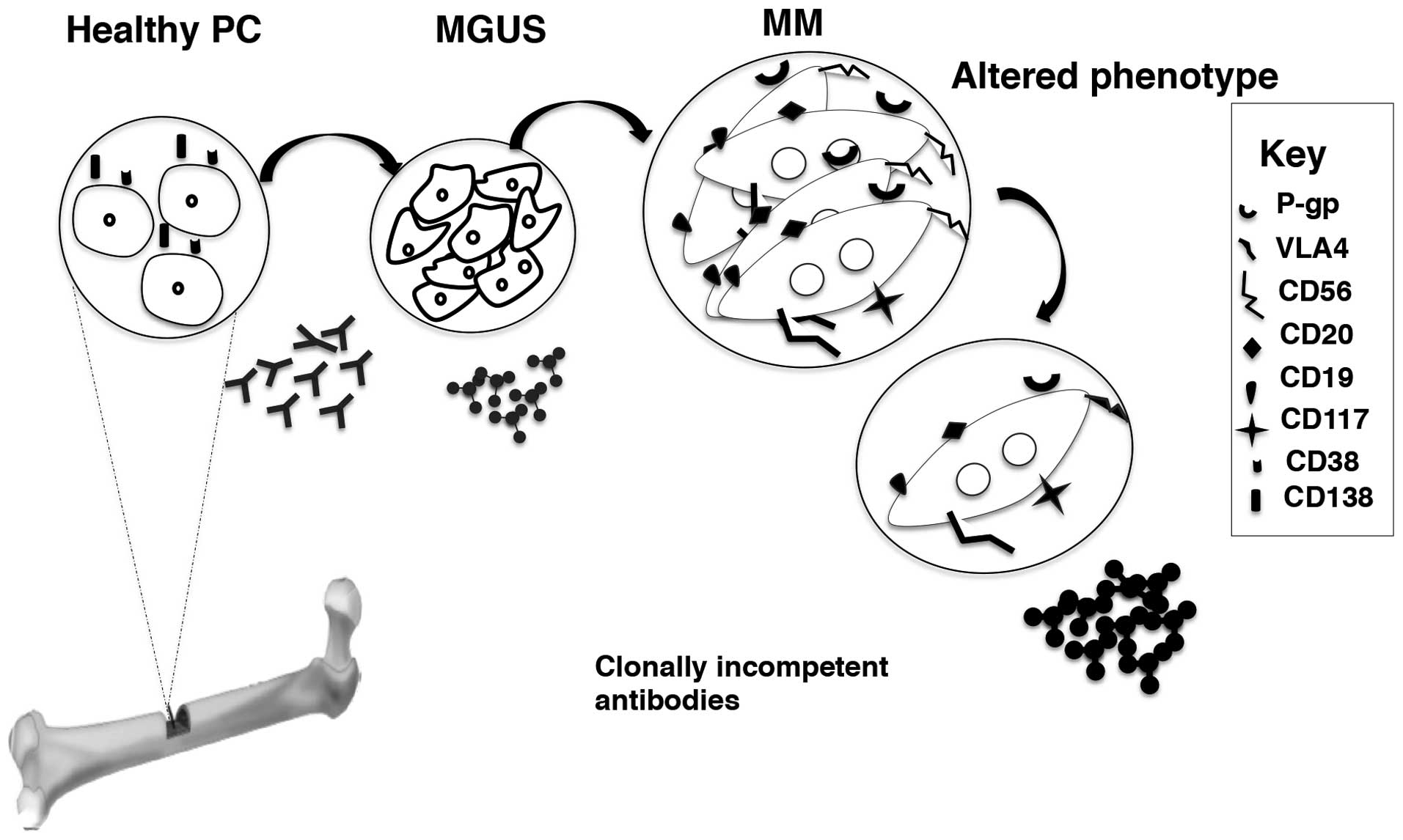
MM is a hematological malignancy characterized by
the accumulation of aberrant PCs in the bone marrow (25). PCs are terminally differentiated
activated B cells retained in the G1 phase of the cell cycle
(26). PCs express surface markers
that are reflective of their elaborate maturation and
differentiation process. PCs typically can be distinguished from
naïve B cells by the lack of CD10, CD19 and CD20 expression on
their surface (27). Two specific
surface antigens on PCs are CD38 and CD138 (28–31).
CD38 is an ectoenzyme important in signal transduction, cell
adhesion and calcium signaling, and is expressed across all PC
developmental stages (28). CD138
is a trans membrane proteoglycan that facilitates cell
binding, cell signaling, cell-cell and cell-extracellular matrix
interactions (32). Amongst the
typical markers expressed on PCs, CD45 is considered an early PC
marker (plasma blasts) (27).
According to the maturation stages, PCs are grouped into plasma
blasts (CD138− CD45++), early PC's
(CD138+ CD45+) and mature PC's
(CD138++ CD45− or weak CD45 expression) based
on the antigen expression on their surface (5).
Monoclonal Gammopathy of Undetermined Significance
(MGUS) is a benign condition that can precede malignant
transformation to MM (36).
Clinically, MGUS is characterized by excessive PC growth whilst
retaining a stable M-protein profile (37). Serum M-protein levels of <3g/dl,
small amounts of monoclonal light chains in urine, the absence of
end organ damage, absence of lytic bone lesions, anemia and
hypocalcaemia define the pre-malignant condition MGUS (38). The rate of transition from MGUS to
MM is ~1%/year (36,38).
The exact cause of malignant transformation of PCs
remains unknown. However, ras mutations are absent in pre-malignant
MGUS and are observed in MM (39).
It has been suggested that the myeloma clone arises from a
pre-switched B cell (40),
preconditioned as a result of prior exposure to certain triggers
(i.e. viruses, chemicals and radiation). Other reasons proposed are
an incompetent immune system, age and a family history of
lymphato-hematopoietic cancer (36).
In malignant cells, the genotype is aberrant with
frequent chromosomal deletions or hyperdiploidy (chromosomes
3,5,7,9,11,15,19 and 21) that results in abnormal functions of cell
cycle regulatory genes (cyclin D1, D2 and D3)
(41). Malignant PCs also present
with aberrant phenotypes at diagnosis. Surface markers such as
CD56, CD117 and CD20 are found in decreasing order of expression on
aberrant PCs. Isolated strong CD56 expression is common in MM and
can be used to distinguish MM from MGUS, while CD56−
phenotype is said to be associated with a high risk subtype with
chromosomal abnormality [t(11;14)] in terms of survival (42–44).
Malignant PCs also display an increased expression of various
adhesion molecules compared to non-malignant PCs. Fibronectin
receptor, very late antigen 4 (VLA-4), the lymphocyte homing
receptor CD44 and neural cell adhesion molecule (N-CAM and CD56)
are abundantly expressed on malignant PCs (45). In contrast, VLA-5, the laminin
receptor VLA-6, and the vitronectin receptor CD51 are weakly
expressed (45). In advanced MM,
mature PCs escape the bone marrow niche and are found in
circulation (46). Interestingly,
only 50% of the circulating population expresses CD138, which is
widely considered to be an exclusive mature PC marker of
hematopoietic origin (27,47). Consequently, circulating PCs are
also classified according to the presence or absence of CD138 apart
from the maturation based classification of normal PCs mentioned
above (27). The circulating
CD138− PCs are thought to be plasma blasts as they
express CD45, CD20, CD19 and human leukocyte antigen (HLA)-class II
and more actively proliferating (5,47,48)
(Fig. 1).
Other clinical manifestations alone or in
combination are also considered at diagnosis. These include
elevated calcium levels or hypercalcemia >11.5 mg/dl/>2.65
mmol/l indicating defective bone physiology, renal insufficiency
signified by creatinine >2 mg/dl/177 μmol/l or more, anemic
hemoglobin levels of <10 g/dl or 2 g/dl < normal levels or
hemoglobin <12.5 mmol/l or 1.25 mmol/l < normal levels, and
bone lesions or pain (9).
International uniform response criteria by International Myeloma
Working Group recommends that amyloidosis and/or systemic light
chain deposition disease (LCDD) should be correspondingly
categorized as ‘myeloma with documented amyloidosis’ or ‘myeloma
with documented LCDD’, requiring confirmation through bone marrow
biopsy to ascertain the existence of ≥30% PCs and/or
myeloma-related bone disease. Following diagnosis, MM patients are
usually placed on induction therapy with conventional or novel
agents followed by autologous stem cell transplant depending on
eligibility of each patient (52).
Response to treatment is subsequently evaluated through regular
monitoring of serum and urine M-protein levels by immune fixation
and confirmed by periodic bone marrow aspiration (53).
MM is a highly heterogeneous disease with respect to
survival and clinical manifestations (54), hence it is difficult to accommodate
every criterion in one staging system (55). In 2005, the International Myeloma
Working Group established the International Staging System (ISS)
for MM (Table I) (54). Until 2005, MM staging predominantly
relied on the Durie-Salmon Staging (DSS) system, which was
established in 1975 (56). DSS
correlates various biochemical factors with tumor burden for
staging of malignancy. This makes it difficult to achieve consensus
across various laboratories (57).
The advantage of ISS is that it is a statistical model, which
emphasizes the duration of survival based on the measure of two
parameters, B2M and serum albumin (55). ISS uses B2M as a measure
of the rate of myeloma growth with serum albumin indicative of
tumor burden (54,55,58).
Since its launch, ISS has been validated, is statistically easier
to assess and is more robust compared to the DSS system (55,59).
Myeloma, unlike other hematological malignancies, is
uniquely characterized by intricate cytogenetic and molecular
genetic abnormalities resonant of epithelial tumors (60). A de novo patient usually
presents hyperdiploid with multiple trisomies or hypodiploid with
one of several types of immunoglobulin heavy chain (IgH)
translocations (61). The
importance of cytogenetic markers and gene profiling on therapeutic
decision making is becoming increasingly evident in MM (62).
Chromosomal abnormalities associated with
immunoglobulin heavy chain translocations result in abnormal gene
regulation in MM (63). Cell cycle
regulatory genes are impaired in MM and the dysregulation of
cyclin D1, D2 or D3 is considered to be an
initial oncogenic pathway in MM and MGUS (64). 25% of IgH translocations in MM
directly affect cyclin D1 (11q13), cyclin D2 t(4;14),
cyclin D3 (6p21) or musculoaponeurotic fibro-sarcoma
(MAF) oncogene (c-MAF, 16q23 or MAF oncogene
homolog B (MAFB), 20q11 (41,64).
The recurrent translocations associated with MM are
t(4;14)(p16;q32), and t(14;16) (q32;q23) which are correlated with
a negative prognosis (61).
Myeloma patients frequently present with chromosomal deletions of
13q14 and 17p13 (63). Several
other genetic components such as tumor suppressor genes (p53,
phosphatase and tension homolog-PTEN), retinoblastoma protein-Rb
protein) and transcription factor, myelocytomatosis viral oncogene
homolog (c-myc) also show abnormalities in MM, however, the
exact origin of these genetic and epigenetic changes in the course
of MM pathogenesis is not known yet (39).
Monoclonal protein (M-protein or paraprotein)
production, is a salient feature of secretory MM (70). Based on immunoglobulin heavy chain
structure, MM is classified into IgG, IgD and IgE subtypes of which
IgG MM is most common (11).
Paraproteinemia and an associated hyperviscosity
syndrome, arising from elevated systemic M-protein levels are
typically associated with MM (11,71).
Approximately 25% of MM patients present with paraproteinuria
resulting in renal insufficiency, while ~50% have renal failure
(11,37,50,72)
resulting from direct damage and blockage to the kidney (73). Other MM associated renal
complications include, myeloma cast nephropathy, amyloidosis,
fibrillary glomerulonephritis, immunotactoid glomerular nephritis
and light chain deposition disease (72).
MM patients are immunocompromized due to the
defective hematopoiesis and the aberrant PCs producing clonally
incompetent M-proteins. This is in addition to the gradual
reduction in immune competence coinciding with late middle age
(40). Yaccoby et al
proposed limited mobility in the aged population resulting in
reduced exposure to antigens as the potential reason for the
reduced differentiation rate of the memory B-lymphocytes to PCs
(74,75).
The manipulative tumor cells strategically elude the
immune watch and facilitate tumor survival. One such mechanism is
the phenomenon of ‘trogocytosis’ in which the surface antigen
exchange occurs in lymphocytes creating unique cell phenotypes with
specific function (76). The
immune synapse facilitates unique cell types to maintain
intracellular signaling in T cell subsets and aid in tumor-induced
immune suppression (77). The
phenomenon of trogocytosis is more common in MM compared to other
mature B cell malignancies and T cells are more proficient in
acquiring antigens from malignant PCs (78). Impaired immune system in MM
patients also leads to recurrent infections with a life-changing
impact on patients and care givers (79).
One of the characteristic features of MM is the
tendency of aberrant PCs to be confined to the bone marrow.
Malignant PCs favor a microenvironment analogous to normal
long-lived PCs (3,74,75)
and tend to migrate to peripheral blood only in the terminal stage
of the disease (3,45,74,75).
These malignant PCs evolve ‘autocrine growth supporting loops’ at
this terminal stage which facilitate microenvironment independent
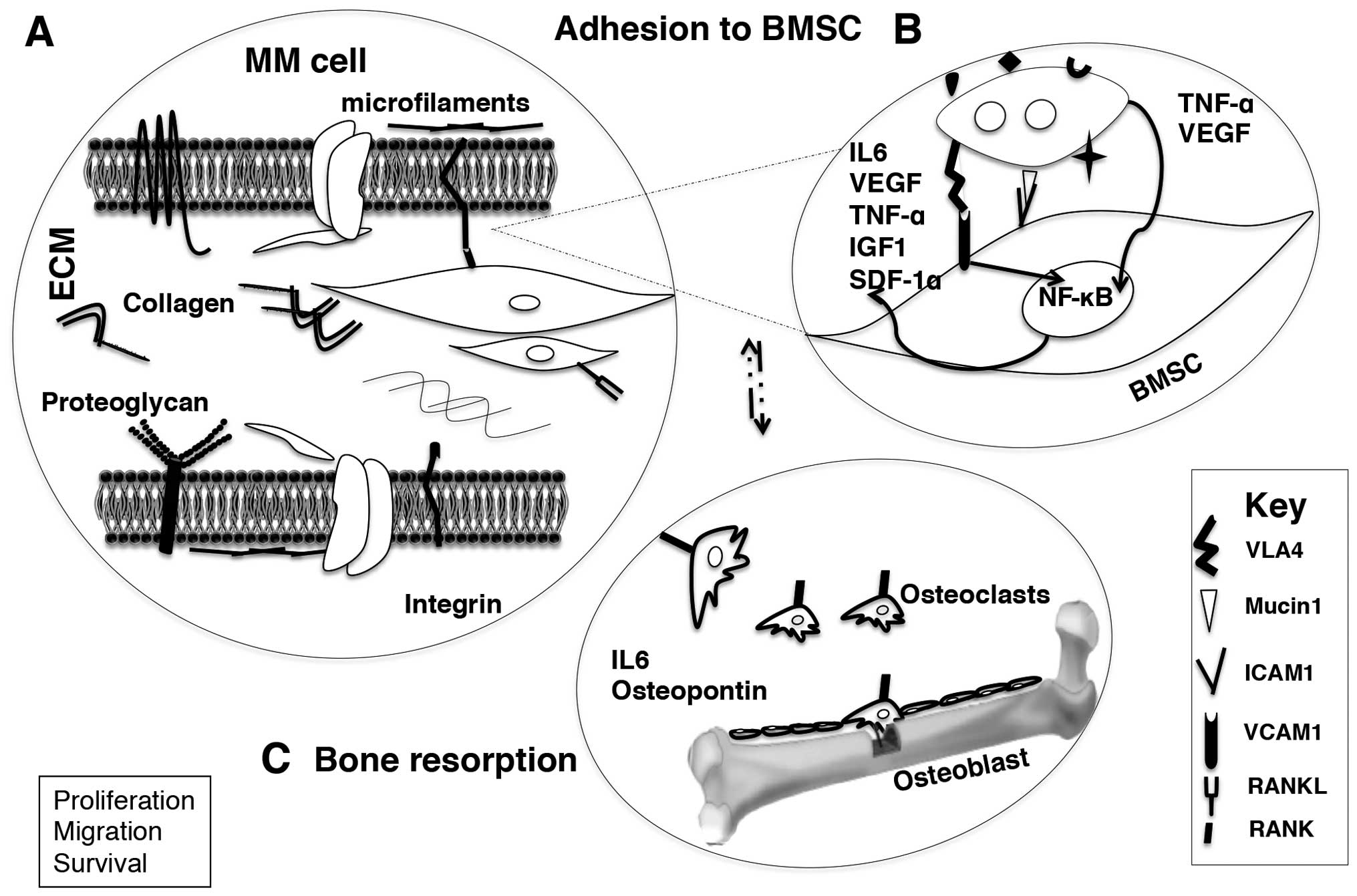
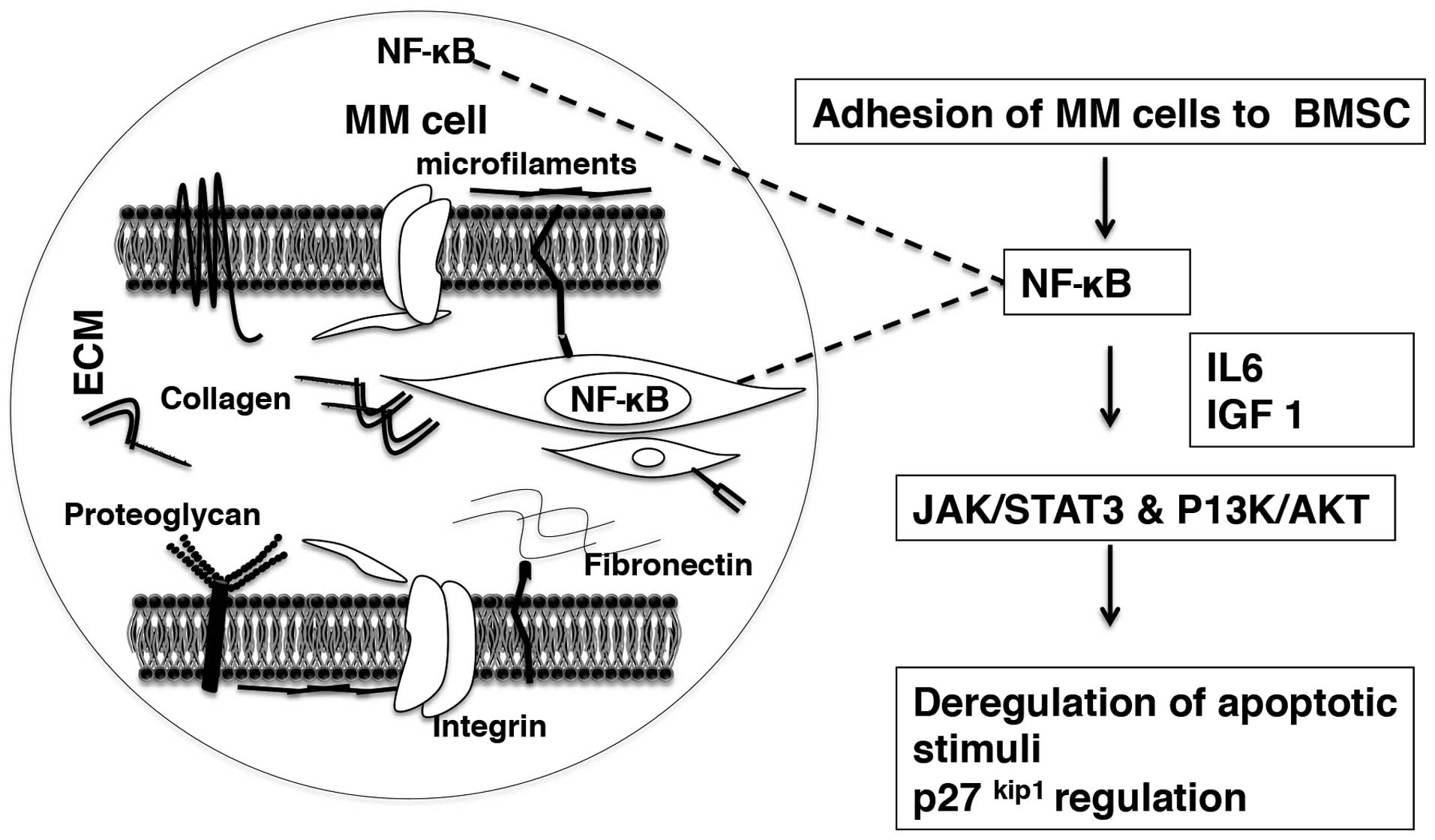
survival (35). The adhesion of MM
cell with bone marrow stromal cell orchestrates homing via adhesion
to the endothelium, invasion through the sub-endothelial membrane,
and chemotactic migration within the bone marrow stroma (35,45)
(Fig. 2).
Aberrant PC interaction with bone marrow stromal
cells (BMSCs) and extra cellular matrix (ECM), subsequently alter
the normal microenvironment to tumor advantage (80,81).
Cytokines such as interleukin 6 (IL6), vascular endothelial growth
factor (VEGF), tumor necrosis factor-α (TNF-α), insulin-like growth
factor 1 (IGF1) support the growth of MM cells (82,83).
Along with IL6 and IGF1, IL21 promote the tumor survival while VEGF
plays a role in MM cell migration with stromal cell derived
factor-1α (SDF-1α) (84–87). The initial binding between MM cells
and bone marrow stromal cells is mediated via adhesion receptor
integrins (integrin α4β1, VLA4), through their ligands [vascular
cell adhesion molecule 1 (VCAM1)] (88,89).
The binding, further, upregulates cytokine and/or chemokine release
from stromal cells to the microenvironment (Fig. 2A). In addition, the transcription
factor nuclear factor-κB (NF-κB) plays a significant role in the
initiation of various cell-signaling pathways in MM cell and BMSC
following the adhesion (84,90).
The adhesion of MM cell to stroma triggers NF-κB and
mitogen-activated-protein kinase (MAPK) signaling cascade in BMSC,
which in turn results in a change in phenotype of MM, and BMSC with
co-expression of adhesion molecules. Subsequently, cytokines
secreted from MM cells trigger inflammatory cytokine production and
NF-κB activation in BMSC (IL6, TNF-α and VEGF). The inflammatory
cytokines from BMSCs trigger signaling pathways in MM cells (MAPK,
phosphatidyl inositol 3 kinase/protein kinase B (P13/AKT), Janus
kinase/signal transducer and activation of transcription 3
(JAK/STAT3) pathways which enhance proliferation, cell cycle
modulation and tumor survival via activation of antiapoptotic
signals (91–93) (Fig.
2B).
Osteolytic lesions, compromise mobility, can result
in spinal cord compression and moderate to severe nerve damage in
MM. In fact, morbidity and mortality in MM is mostly associated
with osteolytic lesions (80,81).
Abe et al (81)
demonstrated that peripheral blood mononuclear cell-derived
osteoclasts enhance MM cell survival and growth in primary MM, as
well as MM cell lines than stromal cells (75,80,81).
Receptor activator of nuclear factor κB (RANK) on the surface of
osteoclasts and the ligand (RANKL) expressed on the BMSC activate
the osteoclasts while osteoprotegerin on BMSCs a decoy ligand of
RANK prevents RANK-RANKL communication (89). Manipulative MM cells stimulate
RANKL expression on BMSCs simultaneously reduce osteoprotegerin
expression which accordingly promotes osteoclastogenesis.
Consequent adhesion of MM cells to osteoclasts enhances the
production of osteopontin and IL6, which augments MM cell growth
and survival (88,89) (Fig.
2C).
Treatment of MM typically involves combination
chemotherapy including cyclophosphamide or melphalan, a steroid
(dexamethasone or prednisolone), a novel agent [e.g. proteasome
inhibitor, immunomodulatory drug (IMiDs)] and may be followed by
autologous stem cell transplant depending on the age at diagnosis
(2). Treatment of progressive MM
consists of induction, maintenance and supportive regimens
(50). In patients below 65 years
of age, autologous stem cell transplant (ASCT) is considered
(13). In many cases a single
autologous stem cell transplant can result in progression-free
survival in comparison with chemotherapy alone (94).
The IMiDs and the proteasome inhibitors (e.g.
bortezomib and carfilzomib) have provided significant improvements
in survival and quality of life in MM (95). IMiDs are structural and functional
analogs of thalidomide that have potent immunomodulatory
properties, anti-myeloma activity and better tolerability profiles
(96). Thalidomide was the first
immunomodulatory agent approved for use in MM. It is highly active
against MM, however, is limited by considerable toxicity,
particularly in older patients (97). Lenalidomide, an analog of
thalidomide, possesses more potent activity with less toxicity and
consequently is preferred for use across phases of MM treatment
(98).
Thalidomide monotherapy when used for induction
therapy produces a low response rate of ~35% (99,100). In the context of relapsed
disease, thalidomide monotherapy results in a median event-free
survival of 6–12 months and median overall survival of 14 months
(101). Thalidomide's combination
with dexamethasone improves the rate to 60–75% and is associated
with a high incidence of grade 3–4 toxicity (102–104). For relapsed MM, the addition of
an alkylating agent (cyclophosphamide or melphalan) further
increases the response rate to 75–80% (105,106). In comparison with the response
rates achieved using novel agents such as bortezomib or
lenalidomide, thalidomide monotherapy is not superlative. In
addition, combination of thalidomide with cytotoxic agents such as
doxorubicin or cyclophosphamide, improves the response rate and
quality of response further. Consequently, a three-combination
regimen is more commonly used when thalidomide induction is
considered (104). However, for
consolidation/maintenance therapy, the impact of thalidomide on
therapeutic outcome remains unclear. Results obtained from the
British Myeloma Research Council Myeloma IX study demonstrates that
thalidomide is associated with shorter post-relapse survival
suggesting that thalidomide maintenance may induce drug resistance
compromising duration of response and survival especially in
patients with high risk genotype [t(4;14), t(14,16),
t(14,20), 1q21amp, del(17p)] (107,108).
Other novel agents like thalidomide derivatives
(lenalidomide) and proteasome inhibitor (bortezomib) combination
chemotherapy increases the overall response rate to 90% or above
(109–112).
A complete remission or complete response (CR) in MM
is clinically defined as negative serum and urine immunofixation,
no plasmacytoma and ≤5% PCs in bone marrow for at least 2 months
(113), whereas partial response
is stated by >50% reduction of serum M-protein and >90% of
Bence Jones protein (113,114).
The malignant PCs enter a static phase with
typically lower levels of proliferative markers such as thymidine
kinase, high sensitive CRP marking the remission status of MM
patient after successful induction therapy (115–117). However, MM cells eventually
overcome this passive phase and become aggressive within a short
space of time (118). This
complex process is said to include loss of immune regulation,
clonal evolution, cytokine deviance, oncogene stimulation and/or
tumor suppressor gene anomaly (118). The mechanisms underlying
initiation, a prolonged asymptomatic stage, progression and
aggressive transformation of PCs are not yet clear (118). The failure of the current
chemotherapeutic regimen to eliminate the malignant clone in MM is
considered to be one of the major causes of consecutive relapse
(118). Relapse from a complete
response is clinically defined by the reappearance of the serum or
urine M-protein (paraproteinemia), ≥5% bone marrow PCs, new lytic
bone lesions and/or soft tissue plasmacytomas, an increase in the
size of residual bone lesions and/or the development of
hypercalcaemia (corrected serum calcium >11.5 mg/dl) not
attributed to another cause (114,119).
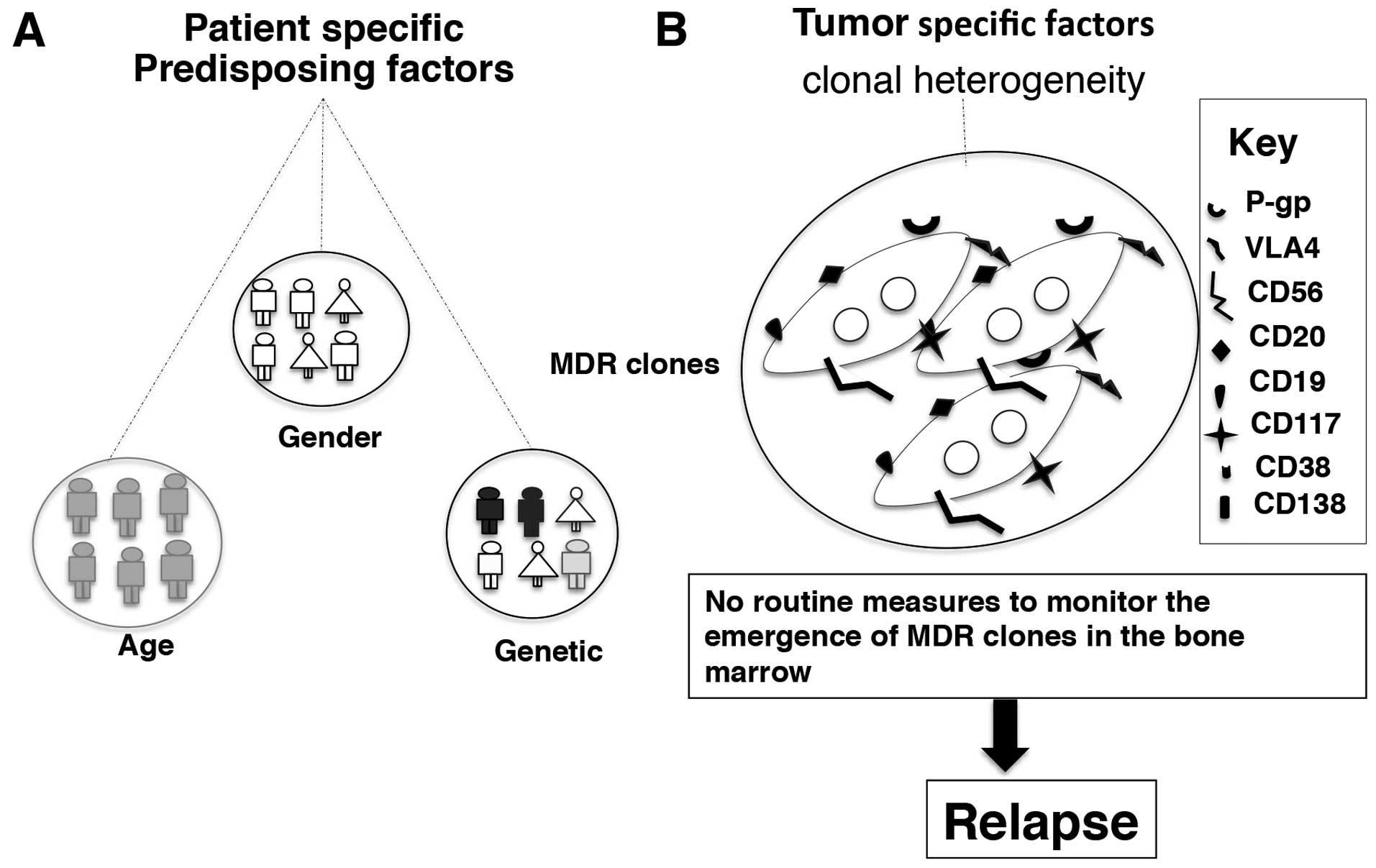
The incidence and risk of developing MM increases
with age, with predominantly 80% of affected patients being above
the age of 60 (1,5,120).
The classic disease manifestations in MM such as anemia, bone pain
and associated fracture and renal involvement imitate the
complications associated with ageing process (36). Consequently, patients discount the
warning signals, which results in delayed diagnosis, which severely
compromises the accessible therapeutic decisions for the elderly
patients. Myeloma is more common in men than women for reasons yet
unknown (5) (Fig. 3A).
The incidence MM is lowest among those of Asian
descent, is intermediate in Caucasians and is highest in African
Americans (25,121,122). Various independent studies have
suggested that there may be a greater genetic predisposition to
MGUS in Africans and African Americans than in Caucasians (123). Although the reason for this
genetic pre-disposition is not known, a small number of studies
have revealed that the variation in the prevalence of
immunoglobulin subtypes and the overexpression of either κ or λ
free light chain ratios in different races may contribute to the
differential cytogenetic susceptibility between races (123). The presence of a rare deletion of
193 bp in the long arm of the pseudogene [poly(ADP-ribose)
polymerase-allele B] of chromosome 13 (negative prognosis in MM) is
more frequent in African Americans than Caucasians (69). Although the etiology of MM remains
unknown, a family history of hematological disorders, either alone
or combined with exposure to certain viruses, radiation and
chemicals, is a proposed risk factor (36) (Fig.
3A).
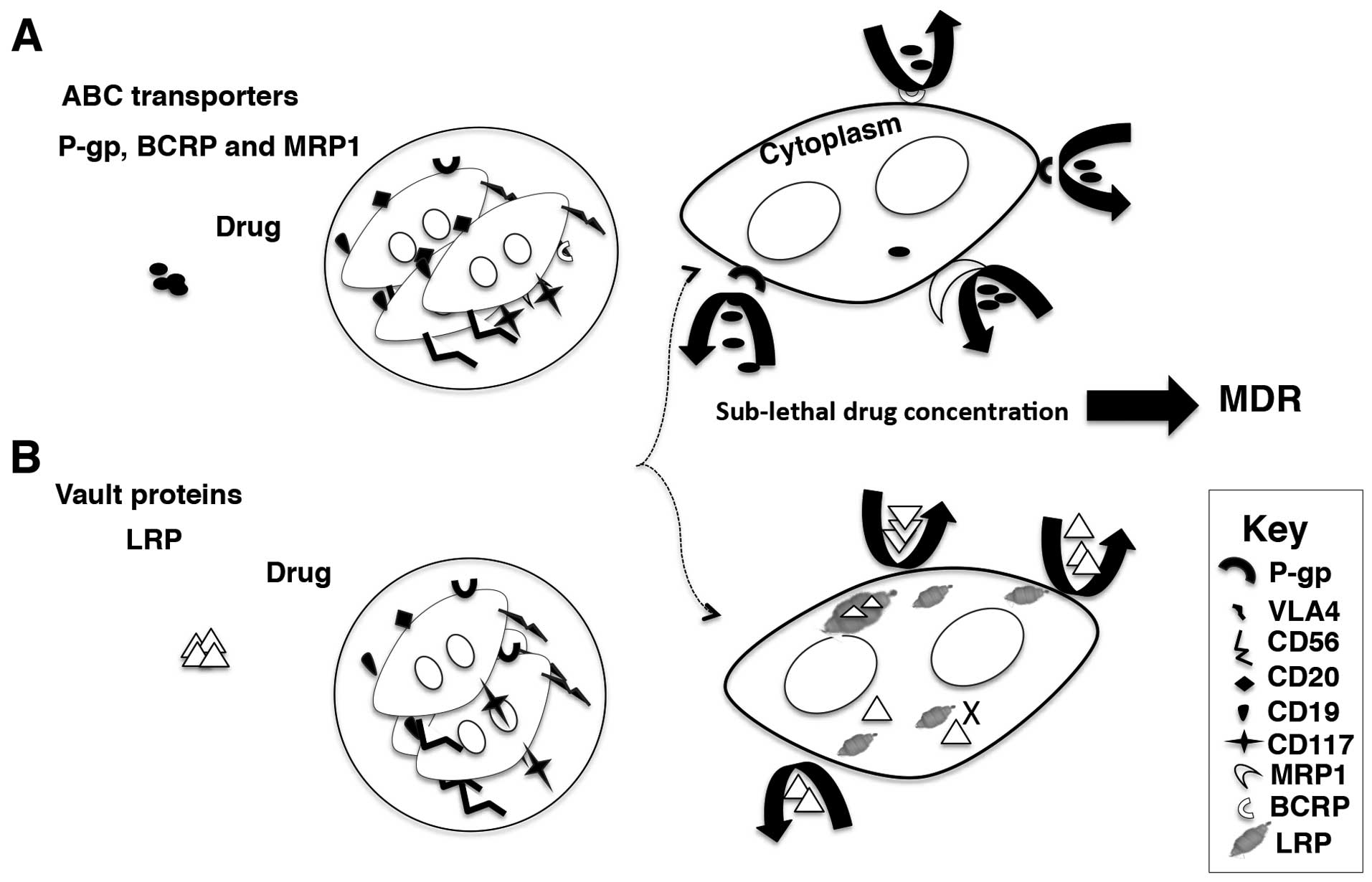
Numerous studies have confirmed the presence of
tumor-initiating cells (stem cells) in the bone marrow and their
role in disease relapse (48,124). The primary bone marrow contain a
small population of clonotypic B cells with an immature phenotype
(CD138−) known as ‘side population’ or MM initiating
cells with stem-cell characteristics besides the malignant ‘main
population’ (48). These cells
contain more quiescent cells than ‘main population’ cells in cell
cycle analysis. The MM stem cells or ‘side population’ (SP) cells
are enriched source of cancer stem cells and characteristically
show low staining of Hoechst 33342 dye, have high clonogenic
potential and possess self renewal capacity (48,125). The SP cells contain hypermutated
Ig genes, overexpress members of the ABC transporter family such as
permeability-glycoprotein (P-gp), multi drug resistance-related
protein 1 (MRP1) and breast cancer related protein (BCRP) much like
the stem cells (126). The
self-renewal capacity of the clonotypic MM cells is mainly
attributed to the abnormal signaling pathways found in MM such as
Hedgehog, Notch and Wnt signaling pathways (126).
The overexpression of drug efflux pumps is known to
compromise the treatment outcome in MM (124). As mentioned, the side population
has high expression of MDR proteins. The inability of
chemotherapeutics to eradicate MM clones is a major limitation in
MM management and a major cause of relapse (127). The detrimental MM clone is
persistent during the remission phase and possess high
proliferating potential once activated (118). The presence of drug efflux pumps
further adds to the deleterious potential of the aforementioned MM
clone and cause inevitable relapse (124) (Fig.
3B).
Primary or acquired drug resistance is a major
obstacle in MM therapy. In the past, conventional chemotherapeutic
treatment of MM, was primarily focused on alkylator and
corticosteroid based regimens (VAD regimen-vincristine, adriamycin
or doxorubicin, dexamethasone) (128). The current therapeutic regimen
includes IMiDs, proteasome inhibitors to improve outcome in MM
patients. However, overexpression of MDR genes, topoisomerases and
glutathione transferases mediate drug resistance in MM and many
cancers (129). Cell adhesion
mediated drug resistance (CAM-DR) and overexpression of
anti-apoptotic proteins are typical resistance mechanisms also
contributing to relapse in MM (130,131).
Topoisomerase II (topo II) is a 170–173 kDa
homodimeric protein involved in DNA replication, recombination and
gene transcription (132,133). Topo II is an ideal drug target
and anthracyclins (doxorubicin), anthracenedions (mitoxantrone) and
intercalating agents (acridines) are the main topoisomerase
inhibitors used in MM therapy. These drugs interact with topo II to
form a temporary complex, which prevents chromosome segregation and
DNA synthesis (129). Point
mutations in essential domains of the malignant PCs modify the drug
target topo II by epigenetic changes such as hypermethylation at
the CpG. Island of promoter region affecting the gene expression
(129). Structural changes to
topo II (α to β) also contribute to drug resistance to topo II
inhibitors used in MM therapy (129). The sub-cellular localization of
topo II is also crucial in determining the drug effectiveness and
is governed by the adhesion molecule- mediated resistance mechanism
in MM (134). Turner et al
demonstrated that tumor density plays a role in topo II resistance
in such a way that in high density MM tumors, majority of the topo
II is transported away from the DNA to the cytoplasm and the drugs
fail to form cleavable complexes resulting in poor therapeutic
outcome (135).
Glutathione (γ-glutamylcysteinyl-glycine) is a
tripeptide thiol present throughout the mammalian organ system
specifically in the liver and kidney. Physiologically, glutathione
plays a critical role in clearance of xenobiotics, harmful
radiations and free radicals (129,136). Glutathione transferases (GST) are
a family of detoxification enzymes catalyzing the non-covalent or
covalent conjugation of glutathione with the diverse detrimental
electrophilic compounds. GSTs also sequester toxic compounds and
protect the cells from the oxidative stress through inherent
organic peroxidase activity. The cytosolic and microsomal GST forms
in humans are differentiated as GST-π, -α and -μ of which GST-π
form is the most common enzyme. The conjugation with glutathione
makes the toxic compounds water soluble facilitating an easy
expulsion from the cells. In the malignant status, this effective
detoxification mechanism also becomes unfavorable. Active GSTs are
either increased in the cell or the expression levels of the
isozymes are altered to protect the tumor by catalyzing the toxic
chemotherapeutics (136,137). Alkylating agents, melphalan and
cyclophosphamide used in myeloma therapy are inactivated by GST
catalysis resulting in poor therapeutic outcome (129). In addition, high percentage of
co-expression of GST-π (82%) with P-gp which is another class of
MDR protein (72%) in MM relapse is reported by Petrini et al
(138). This implicates
co-operation of two distinct MDR pathways in coordinating poor
therapeutic response.
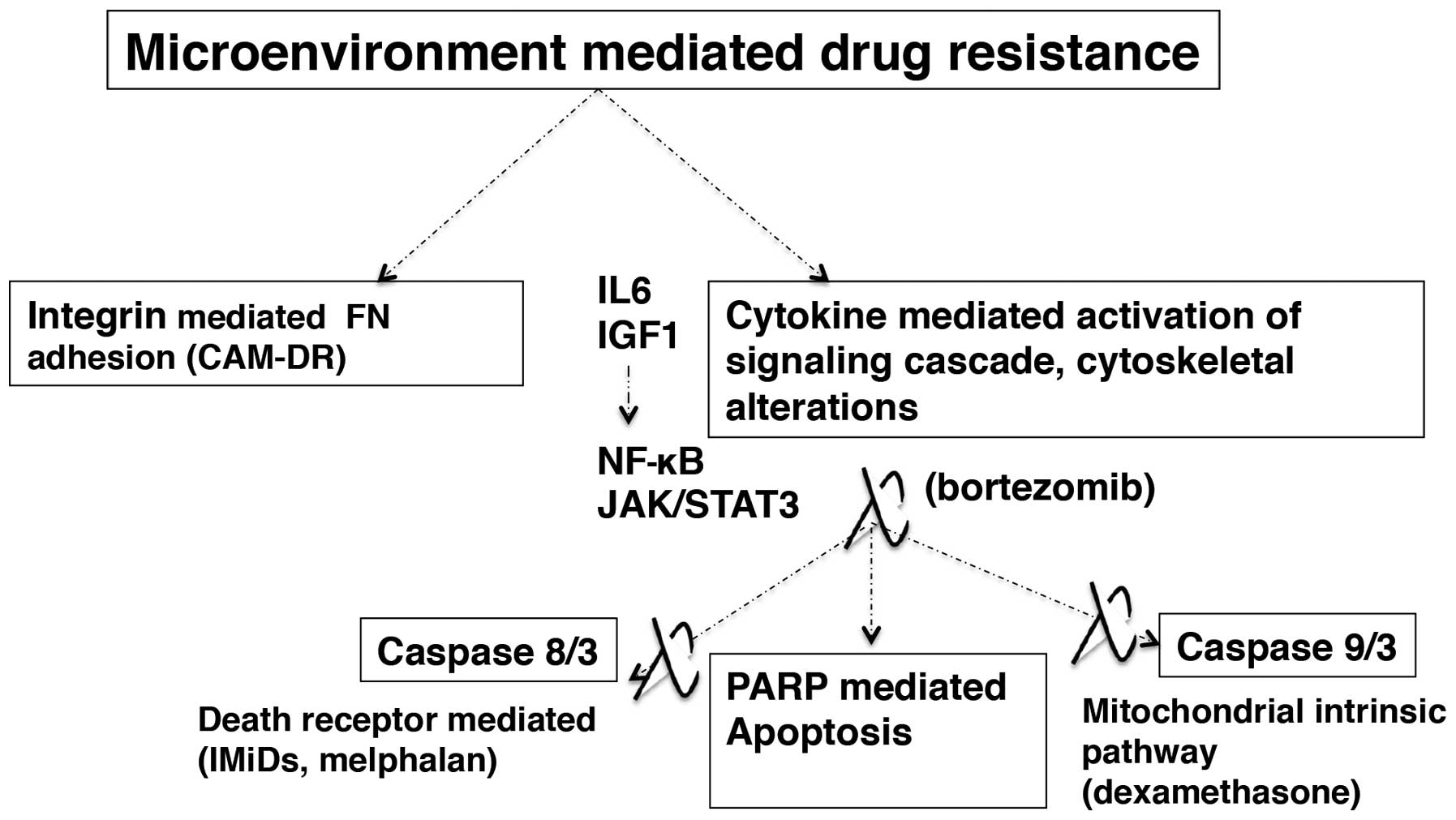
Apart from microenvironment-mediated pathogenesis
mentioned above, components of the bone marrow microenvironment
contribute to treatment unresponsiveness in MM (139,140). The microenvironment related
resistance mechanisms could be classified as integrin mediated
adhesion to ECM (fibronectin) disrupting the apoptotic stimuli
through cytokine-mediated upregulation of cell signaling and
caspase mediated apoptotic cascade in MM cell.
Microenvironment-dependent drug resistance in MM is considered as a
bonus mechanism in MM cells by which the drug resistant cells are
selected early on during initial therapy and they later acquire
more explicit drug resistance during the course of chemotherapy
(141).
The MM cell-BM microenvironment cytokines regulate
apoptosis and MM cell survival through their participation in
P13K/AKT and JAK/STAT3 signaling pathways (84). Novel and conventional
chemotherapeutics in MM target the caspase-mediated apoptosis
pathways. Caspase-8/3 mediated death receptor pathway (IMiDs,
melphalan) and caspase-9/3 mediated mitochondrial intrinsic pathway
(dexamethasone) follow subsequent poly-(ADP-ribose) polymerase
(PARP) cleavage resulting in apoptotic death of MM cells (92,143–145). The proteasome inhibitor class
(bortezomib) targets both caspase-8/3 and caspase-9/3 pathways
(146). The IL6 mediated
activation of JAK/STAT3 signaling cascade results in upregulation
of myeloid cell leukemia sequence 1 (MCL1) and B cell
lymphoma/leukemia family (Bcl-XL) leading to dexamethasone
resistance (147). P13K/AKT
signaling and NF-κB activation in MM cells are coordinated by IL6
and IGF1 by inducing inhibitors of drug-induced apoptosis resulting
in treatment unresponsiveness and eventual survival of the tumor
(148,149) (Fig.
5).
Cancer cells often develop cross-resistance (to a
large variety of chemically and pharmacologically unrelated drugs
leading to the phenomenon of multiple (or multi-drug) resistance
(MDR) (17). MDR is mainly
attributed to the over-expression of ATP-dependent efflux
transporters belonging to the ABC superfamily (17) (Fig.
6). The overexpression of the ATP binding cassette (ABC)
transporters on the plasma membranes of malignant PCs contribute to
MDR in MM. P-gp, MRP1 lung resistance protein (LRP) and BCRP are
all members of the ABC superfamily of membrane transporters and
mediate MDR in MM through their drug efflux capacity (130).
In MM therapy, maximal response rates and improved
survival is achieved through combination thalidomide therapy.
Combination chemotherapy is however compromised by the
overexpression of the multidrug transporters (P-gp, MRP1, BCRP and
LRP) on malignant cells, which maintain intracellular drug
accumulation deficits in resistant cells. Although there is no
current evidence to suggest that thalidomide itself is a substrate
of the these drug efflux pumps, the drugs used in combination as
part of the recommended regimens are themselves substrates of one
or more of these efflux transporters (Table II) (151–166). This contributes to compromised
therapeutic effects, reduced rates of response and overall survival
of the tumor.
The main components of conventional induction
regimen in MM, the vinca-alkaloid (melphalan), anthracyclines
(doxorubicin, daunarubicin) are common substrates of ABC
transporters such as P-gp or MRP1 (Table II). Chemotherapeutic resistance in
MM patients is frequently associated with the overexpression of
P-gp (167). At least 5% of cases
of untreated MM presents with P-gp which can compromise the
induction therapy outcome significantly (168). In addition, the circulating B
cells or the ‘side population’ in MM express P-gp comprising the
resident MDR clone, which leads to MM relapse (169). Nuessler et al also
reported that 33% of patients at relapse or progressive MM are
positive for functional P-gp (170).
Breast cancer resistance protein is another ABC
transporter family member with a molecular weight of 72 kDa
typically expressed at pharmacological barriers (181,182). Structurally, BCRP encoded by the
ABCG2 gene, consists of a single nucleotide binding domain
(NBD) and one trans membrane domain (TMD). Consequently,
BCRP requires at least two NBDs to function as a drug efflux pump
and usually exists as an oligomer (183). BCRP was initially described in
MCF-7/AdVrp human MDR breast cancer cell line that did not express
P-gp or MRP1 (184,185). In MM, BCRP shows impaired
function and is not associated with drug resistance in de
novo patients (184).
However, BCRP is closely associated with the compounding problem of
clonogenic potential of MM cells leading to relapse. The ‘MM stem
cells’ or ‘side population’ (Hoechst 33342 low staining) have
higher BCRP mRNA levels and functional activity compared to the
rest of the MM cells (main population) (186). Functional BCRP expression in MM
is inversely proportional to promoter methylation in ABCG2
gene in such a way that unmethylated promoter site results in
moderate or high BCRP (ABCG2) expression (187). Numerous polymorphisms for BCRP
have been reported in literature (V12M, Q141K, F208S, S248P, F431L,
S441N and F489L) however they have not been linked to MM yet
(188).
LRP is a 110-kDa protein expressed in the kidneys,
adrenal glands, heart, lungs, muscles, thyroid, prostate, bone
marrow and testis. Most vaults are complex ribonucleoprotein
particles comprising two large molecular weight proteins and a
small RNA in addition to the 110 kDa LRP. They are mostly present
in cytoplasm, with a small fraction present in the nuclear membrane
and nuclear pore complex (189).
They are assumed to translocate substances across the nucleus and
cytoplasm and are said to be involved in MDR (130,190). Raaijmakers et al reported
the prevalence of LRP in untreated MM patients (153). This study established the
relevance of LRP as an independent predictor in comparison with
current markers (PC labeling index, serum B2M or lactate
dehydrogenase level) for therapeutic response and survival in MM
patients treated with melphalan (melphalan and prednisone)
(153). Thus, screening for LRP
prior to treatment to identify the positive population is
recommended in therapeutic design in de novo MM to
circumvent LRP mediated drug resistance (153). There are currently more than 100
polymorphisms identified for LRP (191). LRP expression rather than
polymorphic state have been correlated with therapeutic response
(192–195).
In the past few decades, substantial research has
focused on the development and trial of agents, which can reverse
the drug efflux capacity of ABC transporters, in particular P-gp in
cancer (15,130,196,197). Indeed, the pharmacological
inhibition of P-gp activity has been a major focus in many MM
clinical studies (198). In an
attempt to circumvent acquired MDR, several inhibitors have been
used to improve treatment outcome of patients with MM (16,199–201). The cyclosporin A reversal effect
has been evident in phase II studies with MM and acute myeloid
leukemia, although phase III clinical trials failed to give the
expected response in progression-free survival and overall survival
(197). Since the initial
successful clinical trials, verapamil and cyclosporin were combined
with vincristine, adriamycin and dexamethasone (VAD) in MM,
however, these have had disappointing results mainly due to lack of
improved efficacy or dose related toxicity (15,196,197).
In conclusion, management of MM relies on
combination therapy and different drug resistance mechanisms, topo
IIα and GST-π-dependent resistance, specifically the drug efflux
pathways pose a significant challenge in MM clinical setting.
Conservative regime in MM, are mostly substrates of
ABC-transporters, topo IIα and GST-π-dependent resistance
mechanisms (170). Recent studies
have reported that the novel agents are also substrates of ABC
transporters specifically P-gp (154,157).
Herein, we explored the relevant innate and
acquired challenges associated with the therapeutic management of
MM including the role of MDR in therapeutic failure. Many cases of
MM with late middle age onset fail to be accurately diagnosed early
as recurrent infections, tiredness and bone/joint pain is often
associated with normal ageing-related complications.
MM is currently an incurable and chronic disease,
with ‘non-secretory myeloma’ exclusively dependent on frequent bone
marrow aspiration for the assessment of molecular, cytogenetic
markers including aberrant PC population, and categorizing complete
response. Secretory myeloma is partially dependent on bone marrow
aspiration for the confirmation of the clinical status (9). This is largely because the malignancy
is restricted to the bone marrow and is rarely seen in peripheral
blood (202). Current risk
stratification in MM is also primarily dependent on cytogenetic
markers and is assessed using invasive bone marrow biopsy.
Nevertheless, the BM biopsy does not provide a sensitive assessment
of genetic abnormalities in multiple tumor sites throughout the
skeletal system of MM patients. Therefore, even invasive biopsy is
not comprehensive in risk profiling patients with MM.
The current ISS, although, presents with distinct
advantages over its predecessors, the precise indication of the
higher ISS stage (stage III) is inconclusive in terms of whether it
suggests tumor burden/aggressiveness or the level of end-organ
damage or both (55). There are
several reliable systemic markers present for prognosis, like
B2M, M-protein, however these markers are insufficient
in gauging the transition from the indolent phase of MM to an
aggressive disease state (118).
In the case of ‘non-secretory myeloma’, diagnosis and prognosis are
further limited as it lacks the typical hallmark of the
disease.
T cells proficiently acquire antigens from MM cells
over any other cell type and create novel cell types through
trogocytosis (76,77). It is not understood clearly if the
novel T regulatory cell types provide new ligands for receptors and
regulate signaling pathways, however, this mechanism enables the
malignant PCs to effectively evade the immune system recognition
and thereby stimulate tumor growth (203).
We have very little understanding regarding the
intricate cycle of dormant and malignant phase of PCs in MM or in
other words how PCs escape the plateau phase in the remission
status and become aggressive again in relapse. This phenomenon
underlines the fact that even aggressive therapy is not successful
in eliminating the neoplastic origin of MM (118). As discussed, the MM stem cell
population (SP cells) and circulating CD138− PCs are
said to have an aggressive proliferative and dissemination capacity
(5,27). In addition, they characteristically
have self-renewal potential and overexpress the ABC transporters on
their surface (124). The
persevering MM clone with MDR phenotype potentially lead to
treatment failure and currently this aspect is not routinely
monitored in the clinical setting. Another complicating aspect of
MM is the high heterogeneity in survival amongst patients. MDR
phenotype, genetics of MM, including specific IgH translocations
and individual immune profiles are potential players with a role in
the disparity in survival amongst MM patients. Present systemic
markers do not assist greatly in risk stratification, thus, it is
more reliant on the cytogenetic markers in this aspect. Therefore,
inclusion of more systemic markers, alone or in combination that
would aid in early detection, tailor an individualized approach to
optimize a prognostic surveillance at diagnosis and after primary
surgery is highly recommended in MM (204–207).
The derivatives of thalidomide (IMiDs) have
improved overall survival and have increased the cost of treatment
significantly. However, in a phase 1 clinical trial conducted in
2011, involving 21 patients with refractive myeloma who were
treated with lenalidomide and temsirolimus (mTOR pathway
inhibitor-CCI-779), a high concentration of the drug was detected
in the blood causing toxicity. The patients experienced unusual
side effects such as electrolyte imbalance, rashes, fatigue, and
neutropenia. Further investigation of the pharmacokinetic profiles
of CCI-779 and lenalidomide suggested a drug-drug interaction,
hinting that the disposition of CCI-779 is arguably mediated by
CYP3A4/5 and P-gp (208–210). The clinical trial assessed
toxicity or adverse effects and response to treatment by serum and
urine M-protein quantification every four weeks. There was only
limited documented clinical evidence suggesting lenalidomide and
P-gp interaction and this possibility was investigated through
in vitro studies to determine whether lenalidomide can be
transported by P-gp. The in vitro studies proved that
lenalidomide is actively transported by P-gp and this effect was
reversed by CCI-779 and verapamil. In addition, ABCB1
silencing RNA or short interfering RNA (siRNA) knockdown studies
in vitro also showed more lenalidomide uptake, supporting
lenalidomide and P-gp drug-drug interaction (154).
In light of emerging studies that these novel drugs
that have been incorporated to MM therapeutic management are
substrates of ABC transporters, the situation warrants a
re-evaluation of the manipulative power of MM cells (154). It is evident that opting for more
aggressive chemotherapy has brought some promise of prolonging
remission and survival in MM. However, this recent study serves as
a reminder of aggressive chemotherapy pitfalls of side effects,
toxicity and eventual development of MDR phenotype in patients
(211). More importantly out of
the innate MM complications contributing to treatment failure, MDR
is an element that can be modulated and targeted, which, therefore
invites specific attention.
The single-nucleotide polymorphism in MRP1
(rs4148356, R723Q) has also been shown to impact on the clinical
outcomes of MM patients (178).
The MRP1 mutation Arg723Gln has an effect on the protein expression
and trafficking, significantly reducing MRP1-mediated resistance to
a wide spectrum of drugs. The presence of R723Q results in extended
time to progression, progression-free survival and overall survival
in MM patients. This has been ascribed to the differential ability
of the isoform in trafficking glutathione and/or regulating its
expression (178). It is
currently unknown whether polymorphisms of BCRP play a role in MM
treatment outcome (216).
The integrin mediated (CAM-DR) drug resistance
mechanism is considered to enable MM cells to survive the initial
drug toxicity, which in the course of therapy aids in selective
expression of classical drug resistance pathways such as
ABC-transporter overexpression in MM cells (65,68,142).
Current measures of therapeutic response rely on
invasive bone marrow biopsy, immunofixation, serum protein
electrophoresis, quantitation, measurement of free light chain and
CT/MRI scans (217). A full blood
count, biochemistry screen, B2M and light chain assays
are other prominent systemic markers along with radiology used for
staging, diagnosis and monitoring in MM (1,58).
None of the above markers, however, provide a direct assessment of
the emergence of MDR or detect the expression and evolution of
resistance markers, polymorphic variants of resistance markers or
nucleic acid signatures, which may contribute to disease
progression and individual therapeutic responsiveness.
Cancer biology in general is an intricate process,
especially in MM, where individual immunological and tumor profiles
change dynamically during the course of treatment. Despite our
knowledge of the MM landscape, the intrinsic challenge of
heterogeneity provides a significant complication in the management
of MM, necessitating individualized analysis of MM pathogenesis and
routine monitoring of evolution of drug resistance.
|
1
|
Malpas JS: Management of multiple myeloma.
BMJ. 2:163–165. 1969. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kyle RA and Rajkumar SV: Treatment of
multiple myeloma: A comprehensive review. Clin Lymphoma Myeloma.
9:278–288. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Katz BZ: Adhesion molecules - The
lifelines of multiple myeloma cells. Semin Cancer Biol. 20:186–195.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Barlogie B, Alexanian R and Jagannath S:
Plasma cell dyscrasias. JAMA. 268:2946–2951. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Reid S, Yang S, Brown R, Kabani K, Aklilu
E, Ho PJ, Woodland N and Joshua D: Characterisation and relevance
of CD138-negative plasma cells in plasma cell myeloma. Int J Lab
Hematol. 32:e190–e196. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dimopoulos MA and Terpos E: Multiple
myeloma. Ann Oncol. 21(Suppl 7): vii143–vii150. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kyle RA: Multiple myeloma: How did it
begin? Mayo Clin Proc. 69:680–683. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kyle RA: Multiple myeloma: An odyssey of
discovery. Br J Haematol. 111:1035–1044. 2000. View Article : Google Scholar
|
|
9
|
Durie BG, Harousseau JL, Miguel JS, Bladé
J, Barlogie B, Anderson K, Gertz M, Dimopoulos M, Westin J,
Sonneveld P, et al; International Myeloma Working Group.
International uniform response criteria for multiple myeloma.
Leukemia. 20:1467–1473. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rajkumar SV, Dimopoulos MA, Palumbo A,
Blade J, Merlini G, Mateos MV, Kumar S, Hillengass J, Kastritis E,
Richardson P, et al: International Myeloma Working Group updated
criteria for the diagnosis of multiple myeloma. Lancet Oncol.
15:e538–e548. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Martin NH: The immunoglobulins: A review.
J Clin Pathol. 22:117–131. 1969. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Quach H, Ritchie D, Stewart AK, Neeson P,
Harrison S, Smyth MJ and Prince HM: Mechanism of action of
immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia.
24:22–32. 2010. View Article : Google Scholar
|
|
13
|
Barlogie B, Shaughnessy J, Tricot G,
Jacobson J, Zangari M, Anaissie E, Walker R and Crowley J:
Treatment of multiple myeloma. Blood. 103:20–32. 2004. View Article : Google Scholar
|
|
14
|
Merchionne F, Perosa F and Dammacco F: New
therapies in multiple myeloma. Clin Exp Med. 7:83–97. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sonneveld P, Schoester M and de Leeuw K:
Clinical modulation of multidrug resistance in multiple myeloma:
Effect of cyclosporine on resistant tumor cells. J Clin Oncol.
12:1584–1591. 1994.PubMed/NCBI
|
|
16
|
Sonneveld P, Durie BG, Lokhorst HM, Marie
JP, Solbu G, Suciu S, Zittoun R, Löwenberg B and Nooter K; The
Leukaemia Group of the EORTC and the HOVON. Modulation of
multidrug-resistant multiple myeloma by cyclosporin. Lancet.
340:255–259. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gong J, Jaiswal R, Mathys JM, Combes V,
Grau GE and Bebawy M: Microparticles and their emerging role in
cancer multidrug resistance. Cancer Treat Rev. 38:226–234. 2012.
View Article : Google Scholar
|
|
18
|
Biedler JL and Riehm H: Cellular
resistance to actinomycin D in Chinese hamster cells in vitro:
Cross-resistance, radioautographic, and cytogenetic studies. Cancer
Res. 30:1174–1184. 1970.PubMed/NCBI
|
|
19
|
Turesson I, Velez R, Kristinsson SY and
Landgren O: Patterns of improved survival in patients with multiple
myeloma in the twenty-first century: A population-based study. J
Clin Oncol. 28:830–834. 2010. View Article : Google Scholar :
|
|
20
|
Decaux O, Lodé L, Magrangeas F, Charbonnel
C, Gouraud W, Jézéquel P, Attal M, Harousseau JL, Moreau P,
Bataille R, et al; Intergroupe Francophone du Myélome. Prediction
of survival in multiple myeloma based on gene expression profiles
reveals cell cycle and chromosomal instability signatures in
high-risk patients and hyperdiploid signatures in low-risk
patients: A study of the Intergroupe Francophone du Myélome. J Clin
Oncol. 26:4798–4805. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Garrison LP Jr, Wang ST, Huang H,
Ba-Mancini A, Shi H, Chen K, Korves C, Dhawan R, Cakana A, van de
Velde H, et al: The cost-effectiveness of initial treatment of
multiple myeloma in the U.S. with bortezomib plus melphalan and
prednisone versus thalidomide plus melphalan and prednisone or
lenalidomide plus melphalan and prednisone with continuous
lenalidomide maintenance treatment. Oncologist. 18:27–36. 2013.
View Article : Google Scholar :
|
|
22
|
Gaultney JG, Franken MG, Tan SS, Redekop
WK, Huijgens PC, Sonneveld P and Uyl-de Groot CA: Real-world health
care costs of relapsed/refractory multiple myeloma during the era
of novel cancer agents. J Clin Pharm Ther. 38:41–47. 2013.
View Article : Google Scholar
|
|
23
|
Goodwin JA, Coleman EA, Sullivan E, Easley
R, McNatt PK, Chowdhury N and Stewart CB: Personal Financial
Effects of Multiple Myeloma and Its Treatment. Cancer Nurs.
36:301–308. 2013. View Article : Google Scholar
|
|
24
|
Durie B, Binder G, Pashos C, Khan Z,
Hussein M and Borrello I: Total cost comparison in
relapsed/refractory multiple myeloma. J Med Econ. 16:614–622. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bergsagel D: The incidence and
epidemiology of plasma cell neoplasms. Stem Cells. 13(Suppl 2):
1–9. 1995.PubMed/NCBI
|
|
26
|
Chen-Kiang S: Cell-cycle control of plasma
cell differentiation and tumorigenesis. Immunol Rev. 194:39–47.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Caraux A, Klein B, Paiva B, Bret C,
Schmitz A, Fuhler GM, Bos NA, Johnsen HE, Orfao A and Perez-Andres
M; Myeloma Stem Cell Network. Circulating human B and plasma cells.
Age-associated changes in counts and detailed characterization of
circulating normal CD138− and CD138+ plasma
cells. Haematologica. 95:1016–1020. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Alessio M, Roggero S, Funaro A, De Monte
LB, Peruzzi L, Geuna M and Malavasi F: CD38 molecule: Structural
and biochemical analysis on human T lymphocytes, thymocytes, and
plasma cells. J Immunol. 145:878–884. 1990.PubMed/NCBI
|
|
29
|
Ruiz-Argüelles GJ and San Miguel JF: Cell
surface markers in multiple myeloma. Mayo Clin Proc. 69:684–690.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kara IO, Sahin B, Paydas S and Cetiner S:
Flow cytometric evaluation of bone marrow plasma cells using CD19,
CD45, CD56, CD38, and CD138 and correlation with bone marrow
infiltration ratio in multiple myeloma patients. Saudi Med J.
25:1587–1592. 2004.PubMed/NCBI
|
|
31
|
Rawstron AC: Immunophenotyping of plasma
cells. Curr Protoc Cytom. 2006.Chapter 6:Unit6.23
|
|
32
|
Bayer-Garner IB, Sanderson RD, Dhodapkar
MV, Owens RB and Wilson CS: Syndecan-1 (CD138) immunoreactivity in
bone marrow biopsies of multiple myeloma: Shed syndecan-1
accumulates in fibrotic regions. Mod Pathol. 14:1052–1058. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tokoyoda K, Hauser AE, Nakayama T and
Radbruch A: Organization of immunological memory by bone marrow
stroma. Nat Rev Immunol. 10:193–200. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Moser K, Tokoyoda K, Radbruch A, MacLennan
I and Manz RA: Stromal niches, plasma cell differentiation and
survival. Curr Opin Immunol. 18:265–270. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vande Broek I, Vanderkerken K, Van Camp B
and Van Riet I: Extravasation and homing mechanisms in multiple
myeloma. Clin Exp Metastasis. 25:325–334. 2008. View Article : Google Scholar
|
|
36
|
Alexander DD, Mink PJ, Adami HO, Cole P,
Mandel JS, Oken MM and Trichopoulos D: Multiple myeloma: A review
of the epidemiologic literature. Int J Cancer. 120(Suppl 12):
40–61. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Brigden ML: The search for meaning in
monoclonal protein. Is it multiple myeloma or monoclonal gammopathy
of undetermined significance? Postgrad Med. 106:135–142; quiz 185.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kyle RA, Therneau TM, Rajkumar SV, Offord
JR, Larson DR, Plevak MF and Melton LJ III: A long-term study of
prognosis in monoclonal gammopathy of undetermined significance. N
Engl J Med. 346:564–569. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fonseca R, Barlogie B, Bataille R, Bastard
C, Bergsagel PL, Chesi M, Davies FE, Drach J, Greipp PR, Kirsch IR,
et al: Genetics and cytogenetics of multiple myeloma: A workshop
report. Cancer Res. 64:1546–1558. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pilarski LM and Belch AR: Circulating
monoclonal B cells expressing P glycoprotein may be a reservoir of
multidrug-resistant disease in multiple myeloma. Blood. 83:724–736.
1994.PubMed/NCBI
|
|
41
|
Bergsagel PL, Kuehl WM, Zhan F, Sawyer J,
Barlogie B and Shaughnessy J Jr: Cyclin D dysregulation: An early
and unifying pathogenic event in multiple myeloma. Blood.
106:296–303. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hundemer M, Klein U, Hose D, Raab MS,
Cremer FW, Jauch A, Benner A, Heiss C, Moos M, Ho AD, et al: Lack
of CD56 expression on myeloma cells is not a marker for poor
prognosis in patients treated by high-dose chemotherapy and is
associated with translocation t(11;14). Bone Marrow Transplant.
40:1033–1037. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chang H, Samiee S and Yi QL: Prognostic
relevance of CD56 expression in multiple myeloma: A study including
107 cases treated with high-dose melphalan-based chemotherapy and
autologous stem cell transplant. Leuk Lymphoma. 47:43–47. 2006.
View Article : Google Scholar
|
|
44
|
Van Camp B, Durie BG, Spier C, De Waele M,
Van Riet I, Vela E, Frutiger Y, Richter L and Grogan TM: Plasma
cells in multiple myeloma express a natural killer cell-associated
antigen: CD56 (NKH-1; Leu-19). Blood. 76:377–382. 1990.PubMed/NCBI
|
|
45
|
Van Riet I and Van Camp B: The involvement
of adhesion molecules in the biology of multiple myeloma. Leuk
Lymphoma. 9:441–452. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rawstron AC, Owen RG, Davies FE, Johnson
RJ, Jones RA, Richards SJ, Evans PA, Child JA, Smith GM, Jack AS,
et al: Circulating plasma cells in multiple myeloma:
Characterization and correlation with disease stage. Br J Haematol.
97:46–55. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
O'Connell FP, Pinkus JL and Pinkus GS:
CD138 (syndecan-1), a plasma cell marker immunohistochemical
profile in hematopoietic and nonhematopoietic neoplasms. Am J Clin
Pathol. 121:254–263. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Matsui W, Huff CA, Wang Q, Malehorn MT,
Barber J, Tanhehco Y, Smith BD, Civin CI and Jones RJ:
Characterization of clonogenic multiple myeloma cells. Blood.
103:2332–2336. 2004. View Article : Google Scholar
|
|
49
|
Kyle RA and Rajkumar SV: Criteria for
diagnosis, staging, risk stratification and response assessment of
multiple myeloma. Leukemia. 23:3–9. 2009. View Article : Google Scholar :
|
|
50
|
Palumbo A, Attal M and Roussel M: Shifts
in the therapeutic paradigm for patients newly diagnosed with
multiple myeloma: Maintenance therapy and overall survival. Clin
Cancer Res. 17:1253–1263. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lonial S and Kaufman JL: Non-secretory
myeloma: a clinician's guide. Oncology (Williston Park).
27:924–930. 2013.
|
|
52
|
Cavo M, Rajkumar SV, Palumbo A, Moreau P,
Orlowski R, Bladé J, Sezer O, Ludwig H, Dimopoulos MA, Attal M, et
al; International Myeloma Working Group. International Myeloma
Working Group consensus approach to the treatment of multiple
myeloma patients who are candidates for autologous stem cell
transplantation. Blood. 117:6063–6073. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fernández de Larrea C, Delforge M, Davies
F and Bladé J: Response evaluation and monitoring of multiple
myeloma. Expert Rev Hematol. 7:33–42. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Greipp PR, San Miguel J, Durie BG, Crowley
JJ, Barlogie B, Bladé J, Boccadoro M, Child JA, Avet-Loiseau H,
Kyle RA, et al: International staging system for multiple myeloma.
J Clin Oncol. 23:3412–3420. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hari PN, Zhang MJ, Roy V, Pérez WS, Bashey
A, To LB, Elfenbein G, Freytes CO, Gale RP, Gibson J, et al: Is the
International Staging System superior to the Durie-Salmon staging
system? A comparison in multiple myeloma patients undergoing
autologous transplant. Leukemia. 23:1528–1534. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Durie BG and Salmon SE: A clinical staging
system for multiple myeloma. Correlation of measured myeloma cell
mass with presenting clinical features, response to treatment, and
survival. Cancer. 36:842–854. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Salmon SE and Durie BG: Cellular kinetics
in multiple myeloma. A new approach to staging and treatment. Arch
Intern Med. 135:131–138. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Palumbo A, Bringhen S, Falco P, Cavallo F,
Ambrosini MT, Avonto I, Gay F, Caravita T, Bruno B and Boccadoro M:
Time to first disease progression, but not beta2-microglobulin,
predicts outcome in myeloma patients who receive thalidomide as
salvage therapy. Cancer. 110:824–829. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hungria VT, Maiolino A, Martinez G,
Colleoni GW, Coelho EO, Rocha L, Nunes R, Bittencourt R, Oliveira
LC, Faria RM, et al; International Myeloma Working Group Latin
America. Confirmation of the utility of the International Staging
System and identification of a unique pattern of disease in
Brazilian patients with multiple myeloma. Haematologica.
93:791–792. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kuehl WM and Bergsagel PL: Multiple
myeloma: Evolving genetic events and host interactions. Nat Rev
Cancer. 2:175–187. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sawyer JR: The prognostic significance of
cytogenetics and molecular profiling in multiple myeloma. Cancer
Genet. 204:3–12. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Stewart AK and Fonseca R: Prognostic and
therapeutic significance of myeloma genetics and gene expression
profiling. J Clin Oncol. 23:6339–6344. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Liebisch P and Döhner H: Cytogenetics and
molecular cytogenetics in multiple myeloma. Eur J Cancer.
42:1520–1529. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kuehl WM and Bergsagel PL: Early genetic
events provide the basis for a clinical classification of multiple
myeloma. Hematology Am Soc Hematol Educ Program. 2005:346–352.
2005.
|
|
65
|
Pichiorri F, De Luca L and Aqeilan RI:
MicroRNAs: New players in multiple myeloma. Front Genet. 2:222011.
View Article : Google Scholar
|
|
66
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Petrocca F, Visone R, Onelli MR, Shah MH,
Nicoloso MS, de Martino I, Iliopoulos D, Pilozzi E, Liu CG, Negrini
M, et al: E2F1-regulated microRNAs impair TGFbeta-dependent
cell-cycle arrest and apoptosis in gastric cancer. Cancer Cell.
13:272–286. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Roccaro AM, Sacco A, Thompson B, Leleu X,
Azab AK, Azab F, Runnels J, Jia X, Ngo HT, Melhem MR, et al:
MicroRNAs 15a and 16 regulate tumor proliferation in multiple
myeloma. Blood. 113:6669–6680. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Cao J, Hong CH, Rosen L, Vescio RA,
Smulson M, Lichtenstein AK and Berenson JR: Deletion of genetic
material from a poly(ADP-ribose) polymerase-like gene on chromosome
13 occurs frequently in patients with monoclonal gammopathies.
Cancer Epidemiol Biomarkers Prev. 4:759–763. 1995.PubMed/NCBI
|
|
70
|
Lopes da Silva R, Monteiro A and Veiga J:
Non-secretory multiple myeloma relapsing as extramedullary liver
plasmacytomas. J Gastrointestin Liver Dis. 20:81–83.
2011.PubMed/NCBI
|
|
71
|
Mehta J and Singhal S: Hyperviscosity
syndrome in plasma cell dyscrasias. Semin Thromb Hemost.
29:467–471. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Brown JH and Doherty CC: Renal replacement
therapy in multiple myeloma and systemic amyloidosis. Postgrad Med
J. 69:672–678. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Goldschmidt H, Lannert H, Bommer J and Ho
AD: Multiple myeloma and renal failure. Nephrol Dial Transplant.
15:301–304. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Yaccoby S: The phenotypic plasticity of
myeloma plasma cells as expressed by dedifferentiation into an
immature, resilient, and apoptosis-resistant phenotype. Clin Cancer
Res. 11:7599–7606. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Yaccoby S: Advances in the understanding
of myeloma bone disease and tumour growth. Br J Haematol.
149:311–321. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Brown R, Kabani K, Favaloro J, Yang S, Ho
PJ, Gibson J, Fromm P, Suen H, Woodland N, Nassif N, Hart D and
Joshua D: CD86+ or HLA-G+ myeloma cells are
associated with poor prognosis and once acquired by trogocytosis
create novel Tregacq cells. Blood. 120:2055–2063. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Osborne DG and Wetzel SA: Trogocytosis
results in sustained intracellular signaling in CD4(+) T cells. J
Immunol. 189:4728–4739. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Cook G: Has the T cell bitten off more
than it can chew? Blood. 120:1966–1967. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Nau KC and Lewis WD: Multiple myeloma:
Diagnosis and treatment. Am Fam Physician. 78:853–859.
2008.PubMed/NCBI
|
|
80
|
Tanaka Y1, Abe M, Hiasa M, Oda A, Amou H,
Nakano A, Takeuchi K, Kitazoe K, Kido S, Inoue D, et al: Myeloma
cell-osteoclast interaction enhances angiogenesis together with
bone resorption: a role for vascular endothelial cell growth factor
and osteopontin. Clin Cancer Res. 13:816–823. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Abe M, Hiura K, Wilde J, Shioyasono A,
Moriyama K, Hashimoto T, Kido S, Oshima T, Shibata H, Ozaki S, et
al: Osteoclasts enhance myeloma cell growth and survival via
cell-cell contact: A vicious cycle between bone destruction and
myeloma expansion. Blood. 104:2484–2491. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Hideshima T, Chauhan D, Schlossman R,
Richardson P and Anderson KC: The role of tumor necrosis factor
alpha in the pathophysiology of human multiple myeloma: Therapeutic
applications. Oncogene. 20:4519–4527. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Ge NL and Rudikoff S: Insulin-like growth
factor I is a dual effector of multiple myeloma cell growth. Blood.
96:2856–2861. 2000.PubMed/NCBI
|
|
84
|
Hideshima T and Anderson KC: Molecular
mechanisms of novel therapeutic approaches for multiple myeloma.
Nat Rev Cancer. 2:927–937. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
85
|
Brenne AT, Ro TB, Waage A, Sundan A,
Borset M and Hjorth-Hansen H: Interleukin-21 is a growth and
survival factor for human myeloma cells. Blood. 99:3756–3762. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Podar K, Tai YT, Davies FE, Lentzsch S,
Sattler M, Hideshima T, Lin BK, Gupta D, Shima Y, Chauhan D, et al:
Vascular endothelial growth factor triggers signaling cascades
mediating multiple myeloma cell growth and migration. Blood.
98:428–435. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Hideshima T, Chauhan D, Hayashi T, Podar
K, Akiyama M, Gupta D, Richardson P, Munshi N and Anderson KC: The
biological sequelae of stromal cell-derived factor-1alpha in
multiple myeloma. Mol Cancer Ther. 1:539–544. 2002.PubMed/NCBI
|
|
88
|
Sanz-Rodríguez F and Teixidó J:
VLA-4-dependent myeloma cell adhesion. Leuk Lymphoma. 41:239–245.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Michigami T, Shimizu N, Williams PJ,
Niewolna M, Dallas SL, Mundy GR and Yoneda T: Cell-cell contact
between marrow stromal cells and myeloma cells via VCAM-1 and
alpha(4) beta(1)-integrin enhances production of
osteoclast-stimulating activity. Blood. 96:1953–1960.
2000.PubMed/NCBI
|
|
90
|
Abdi J, Chen G and Chang H: Drug
resistance in multiple myeloma: Latest findings and new concepts on
molecular mechanisms. Oncotarget. 4:2186–2207. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Ogata A, Chauhan D, Teoh G, Treon SP,
Urashima M, Schlossman RL and Anderson KC: IL-6 triggers cell
growth via the Ras-dependent mitogen-activated protein kinase
cascade. J Immunol. 159:2212–2221. 1997.PubMed/NCBI
|
|
92
|
Hideshima T, Nakamura N, Chauhan D and
Anderson KC: Biologic sequelae of interleukin-6 induced PI3-K/Akt
signaling in multiple myeloma. Oncogene. 20:5991–6000. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Burger R, Le Gouill S, Tai YT,
Shringarpure R, Tassone P, Neri P, Podar K, Catley L, Hideshima T,
Chauhan D, et al: Janus kinase inhibitor INCB20 has
antiproliferative and apoptotic effects on human myeloma cells in
vitro and in vivo. Mol Cancer Ther. 8:26–35. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Kumar A, Galeb S and Djulbegovic B:
Treatment of patients with multiple myeloma: An overview of
systematic reviews. Acta Haematol. 125:8–22. 2011. View Article : Google Scholar
|
|
95
|
Andhavarapu S and Roy V: Immunomodulatory
drugs in multiple myeloma. Expert Rev Hematol. 6:69–82. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Knight R: IMiDs: A novel class of
immunomodulators. Semin Oncol. 32(Suppl 5): S24–S30. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Ludwig H, Adam Z, Tóthová E, Hajek R,
Labar B, Egyed M, Spicka I, Gisslinger H, Drach J, Kuhn I, et al:
Thalidomide maintenance treatment increases progression-free but
not overall survival in elderly patients with myeloma.
Haematologica. 95:1548–1554. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Palumbo A, Miguel JS, Sonneveld P, Moreau
P, Drach J, Morgan G and Einsele H: Lenalidomide: A new therapy for
multiple myeloma. Cancer Treat Rev. 34:283–291. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Richardson P and Anderson K: Thalidomide
and dexamethasone: A new standard of care for initial therapy in
multiple myeloma. J Clin Oncol. 24:334–336. 2006. View Article : Google Scholar
|
|
100
|
Rajkumar SV, Dispenzieri A, Fonseca R,
Lacy MQ, Geyer S, Lust JA, Kyle RA, Greipp PR, Gertz MA and Witzig
TE: Thalidomide for previously untreated indolent or smoldering
multiple myeloma. Leukemia. 15:1274–1276. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Prince HM, Schenkel B and Mileshkin L: An
analysis of clinical trials assessing the efficacy and safety of
single-agent thalidomide in patients with relapsed or refractory
multiple myeloma. Leuk Lymphoma. 48:46–55. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Cavo M, Zamagni E, Tosi P, Cellini C,
Cangini D, Tacchetti P, Testoni N, Tonelli M, de Vivo A, Palareti
G, et al: First-line therapy with thalidomide and dexamethasone in
preparation for autologous stem cell transplantation for multiple
myeloma. Haematologica. 89:826–831. 2004.PubMed/NCBI
|
|
103
|
Rajkumar SV, Blood E, Vesole D, Fonseca R
and Greipp PR; Eastern Cooperative Oncology Group. Phase III
clinical trial of thalidomide plus dexamethasone compared with
dexamethasone alone in newly diagnosed multiple myeloma: A clinical
trial coordinated by the Eastern Cooperative Oncology Group. J Clin
Oncol. 24:431–436. 2006. View Article : Google Scholar
|
|
104
|
Wu P, Davies FE, Horton C, Jenner MW,
Krishnan B, Alvares CL, Saso R, McCormack R, Dines S, Treleaven JG,
et al: The combination of cyclophosphomide, thalidomide and
dexamethasone is an effective alternative to cyclophosphamide -
vincristine - doxorubicin - methylprednisolone as induction
chemotherapy prior to autologous transplantation for multiple
myeloma: A case-matched analysis. Leuk Lymphoma. 47:2335–2338.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Dimopoulos MA, Hamilos G, Zomas A, Gika D,
Efstathiou E, Grigoraki V, Poziopoulos C, Xilouri I, Zorzou MP,
Anagnostopoulos N, et al: Pulsed cyclophosphamide, thalidomide and
dexamethasone: An oral regimen for previously treated patients with
multiple myeloma. Hematol J. 5:112–117. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
García-Sanz R, González-Fraile MI, Sierra
M, López C, González M and San Miguel JF: The combination of
thalidomide, cyclophosphamide and dexamethasone (ThaCyDex) is
feasible and can be an option for relapsed/refractory multiple
myeloma. Hematol J. 3:43–48. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Morgan GJ, Jackson GH, Davies FE, Drayson
MT, Owen RG, Gregory WM, Cohen DC, Szubert AJ, Bell SE, Ross F and
Child JA: Maintenance thalidomide may improve progression free but
not overall survival; results from the Myeloma IX Maintenance
Randomisation. Blood (ASH Annual Meeting Abstracts).
112:6562008.
|
|
108
|
Morgan GJ, Gregory WM, Davies FE, Bell SE,
Szubert AJ, Brown JM, Coy NN, Cook G, Russell NH, Rudin C, Roddie
H, Drayson MT, Owen RG, Ross FM, Jackson GH and Child JA; National
Cancer Research Institute Haematological Oncology Clinical Studies
Group. The role of maintenance thalidomide therapy in multiple
myeloma: MRC myeloma IX results and meta-analysis. Blood. 119:7–15.
2012. View Article : Google Scholar
|
|
109
|
Oakervee HE, Popat R, Curry N, Smith P,
Morris C, Drake M, Agrawal S, Stec J, Schenkein D, Esseltine DL, et
al: PAD combination therapy (PS-341/bortezomib, doxorubicin and
dexamethasone) for previously untreated patients with multiple
myeloma. Br J Haematol. 129:755–762. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Popat R, Oakervee HE, Hallam S, Curry N,
Odeh L, Foot N, Esseltine DL, Drake M, Morris C and Cavenagh JD:
Bortezomib, doxorubicin and dexamethasone (PAD) front-line
treatment of multiple myeloma: Updated results after long-term
follow-up. Br J Haematol. 141:512–516. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Mateos MV, Hernández JM, Hernández MT,
Gutiérrez NC, Palomera L, Fuertes M, Díaz-Mediavilla J, Lahuerta
JJ, de la Rubia J, Terol MJ, et al: Bortezomib plus melphalan and
prednisone in elderly untreated patients with multiple myeloma:
Results of a multicenter phase 1/2 study. Blood. 108:2165–2172.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Rajkumar SV, Hayman SR, Lacy MQ,
Dispenzieri A, Geyer SM, Kabat B, Zeldenrust SR, Kumar S, Greipp
PR, Fonseca R, et al: Combination therapy with lenalidomide plus
dexamethasone (Rev/Dex) for newly diagnosed myeloma. Blood.
106:4050–4053. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Bladé J, Samson D, Reece D, Apperley J,
Björkstrand B, Gahrton G, Gertz M, Giralt S, Jagannath S and Vesole
D; Myeloma Subcommittee of the EBMT. European Group for Blood and
Marrow Transplant. Criteria for evaluating disease response and
progression in patients with multiple myeloma treated by high-dose
therapy and haemopoietic stem cell transplantation. Br J Haematol.
102:1115–1123. 1998. View Article : Google Scholar
|
|
114
|
Alexanian R, Delasalle K, Wang M, Thomas S
and Weber D: Curability of multiple myeloma. Bone Marrow Res.
2012:9164792012. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Boccadoro M, Gavarotti P, Fossati G,
Pileri A, Marmont F, Neretto G, Gallamini A, Volta C, Tribalto M,
Testa MG, et al: Low plasma cell 3(H) thymidine incorporation in
monoclonal gammopathy of undetermined significance (MGUS),
smouldering myeloma and remission phase myeloma: A reliable
indicator of patients not requiring therapy. Br J Haematol.
58:689–696. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Brown RD, Joshua DE, Nelson M, Gibson J,
Dunn J and MacLennan IC: Serum thymidine kinase as a prognostic
indicator for patients with multiple myeloma: results from the MRC
(UK) V Trial. Br J Haematol. 84:238–241. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Lust JA, Lacy MQ, Zeldenrust SR,
Dispenzieri A, Gertz MA, Witzig TE, Kumar S, Hayman SR, Russell SJ,
Buadi FK, et al: Induction of a chronic disease state in patients
with smoldering or indolent multiple myeloma by targeting
interleukin 1{beta}-induced interleukin 6 production and the
myeloma proliferative component. Mayo Clin Proc. 84:114–122. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Joshua DE, Gibson J and Brown RD:
Mechanisms of the escape phase of myeloma. Blood Rev. 8:13–20.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Lonial S, Mitsiades CS and Richardson PG:
Treatment options for relapsed and refractory multiple myeloma.
Clin Cancer Res. 17:1264–1277. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Alexanian R, Barlogie B and Dixon D:
High-dose glucocorticoid treatment of resistant myeloma. Ann Intern
Med. 105:8–11. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
McPhedran P, Heath CW Jr and Garcia J:
Multiple myeloma incidence in metropolitan Atlanta, Georgia: Racial
and seasonal variations. Blood. 39:866–873. 1972.PubMed/NCBI
|
|
122
|
Clark DW and MacMahon B: The incidence of
multiple myeloma. J Chronic Dis. 4:508–515. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Greenberg AJ, Vachon CM and Rajkumar SV:
Disparities in the prevalence, pathogenesis and progression of
monoclonal gammopathy of undetermined significance and multiple
myeloma between blacks and whites. Leukemia. 26:609–614. 2012.
View Article : Google Scholar
|
|
124
|
Matsui W, Wang Q, Barber JP, Brennan S,
Smith BD, Borrello I, McNiece I, Lin L, Ambinder RF, Peacock C, et
al: Clonogenic multiple myeloma progenitors, stem cell properties,
and drug resistance. Cancer Res. 68:190–197. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Du J, Liu S, He J, Liu X, Qu Y, Yan W, Fan
J, Li R, Xi H, Fu W, et al: MicroRNA-451 regulates stemness of side
population cells via PI3K/Akt/mTOR signaling pathway in multiple
myeloma. Oncotarget. 6:14993–15007. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Agarwal JR and Matsui W: Multiple myeloma:
A paradigm for translation of the cancer stem cell hypothesis.
Anticancer Agents Med Chem. 10:116–120. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Pilarski LM, Mant MJ and Belch AR: Drug
resistance in multiple myeloma: Novel therapeutic targets within
the malignant clone. Leuk Lymphoma. 32:199–210. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Koskela K, Pelliniemi TT and Remes K: VAD
regimen in the treatment of resistant multiple myeloma: Slow or
fast infusion? Leuk Lymphoma. 10:347–351. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Harris AL and Hochhauser D: Mechanisms of
multidrug resistance in cancer treatment. Acta Oncol. 31:205–213.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Sonneveld P, Lokhorst HM and Vossebeld P:
Drug resistance in multiple myeloma. Semin Hematol. 34(Suppl 5):
34–39. 1997.PubMed/NCBI
|
|
131
|
Tucci M, Quatraro C, Dammacco F and
Silvestris F: Role of active drug transporters in refractory
multiple myeloma. Curr Top Med Chem. 9:218–224. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Wang JC: DNA topoisomerases. Annu Rev
Biochem. 65:635–692. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Liu LF: DNA topoisomerase poisons as
antitumor drugs. Annu Rev Biochem. 58:351–375. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Oloumi A, MacPhail SH, Johnston PJ, Banáth
JP and Olive PL: Changes in subcellular distribution of
topoisomerase IIalpha correlate with etoposide resistance in
multicell spheroids and xenograft tumors. Cancer Res. 60:5747–5753.
2000.PubMed/NCBI
|
|
135
|
Turner JG, Marchion DC, Dawson JL, Emmons
MF, Hazlehurst LA, Washausen P and Sullivan DM: Human multiple
myeloma cells are sensitized to topoisomerase II inhibitors by CRM1
inhibition. Cancer Res. 69:6899–6905. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Campling BG, Baer K, Baker HM, Lam YM and
Cole SP: Do glutathione and related enzymes play a role in drug
resistance in small cell lung cancer cell lines? Br J Cancer.
68:327–335. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Garel MC, Domenget C, Caburi-Martin J,
Prehu C, Galacteros F and Beuzard Y: Covalent binding of
glutathione to hemoglobin. I. Inhibition of hemoglobin S
polymerization. J Biol Chem. 261:14704–14709. 1986.PubMed/NCBI
|
|
138
|
Petrini M, Di Simone D, Favati A, Mattii
L, Valentini P and Grassi B: GST-pi and P-170 co-expression in
multiple myeloma. Br J Haematol. 90:393–397. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Manier S, Sacco A, Leleu X, Ghobrial IM
and Roccaro AM: Bone marrow microenvironment in multiple myeloma
progression. J Biomed Biotechnol. 2012:1574962012. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Meads MB, Gatenby RA and Dalton WS:
Environment-mediated drug resistance: A major contributor to
minimal residual disease. Nat Rev Cancer. 9:665–674. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Shain KH and Dalton WS: Cell adhesion is a
key determinant in de novo multidrug resistance (MDR): New targets
for the prevention of acquired MDR. Mol Cancer Ther. 1:69–78.
2001.
|
|
142
|
Damiano JS, Cress AE, Hazlehurst LA, Shtil
AA and Dalton WS: Cell adhesion mediated drug resistance (CAM-DR):
Role of integrins and resistance to apoptosis in human myeloma cell
lines. Blood. 93:1658–1667. 1999.PubMed/NCBI
|
|
143
|
Mitsiades CS, Treon SP, Mitsiades N, Shima
Y, Richardson P, Schlossman R, Hideshima T and Anderson KC:
TRAIL/Apo2L ligand selectively induces apoptosis and overcomes drug
resistance in multiple myeloma: Therapeutic applications. Blood.
98:795–804. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Mitsiades N, Mitsiades CS, Poulaki V,
Chauhan D, Richardson PG, Hideshima T, Munshi NC, Treon SP and
Anderson KC: Apoptotic signaling induced by immunomodulatory
thalidomide analogs in human multiple myeloma cells: Therapeutic
implications. Blood. 99:4525–4530. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Chauhan D, Pandey P, Hideshima T, Treon S,
Raje N, Davies FE, Shima Y, Tai YT, Rosen S, Avraham S, et al: SHP2
mediates the protective effect of interleukin-6 against
dexamethasone-induced apoptosis in multiple myeloma cells. J Biol
Chem. 275:27845–27850. 2000.PubMed/NCBI
|
|
146
|
Hideshima T, Richardson P, Chauhan D,
Palombella VJ, Elliott PJ, Adams J and Anderson KC: The proteasome
inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes
drug resistance in human multiple myeloma cells. Cancer Res.
61:3071–3076. 2001.PubMed/NCBI
|
|
147
|
Dalton WS and Jove R: Drug resistance in
multiple myeloma: Approaches to circumvention. Semin Oncol.
26(Suppl 13): 23–27. 1999.PubMed/NCBI
|
|
148
|
Mitsiades CS, Mitsiades N, Poulaki V,
Schlossman R, Akiyama M, Chauhan D, Hideshima T, Treon SP, Munshi
NC, Richardson PG, et al: Activation of NF-kappaB and upregulation
of intracellular anti-apoptotic proteins via the IGF-1/Akt
signaling in human multiple myeloma cells: Therapeutic
implications. Oncogene. 21:5673–5683. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Hideshima T, Chauhan D, Richardson P,
Mitsiades C, Mitsiades N, Hayashi T, Munshi N, Dang L, Castro A,
Palombella V, et al: NF-kappa B as a therapeutic target in multiple
myeloma. J Biol Chem. 277:16639–16647. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Nefedova Y, Landowski TH and Dalton WS:
Bone marrow stromal-derived soluble factors and direct cell contact
contribute to de novo drug resistance of myeloma cells by distinct
mechanisms. Leukemia. 17:1175–1182. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Kühne A, Tzvetkov MV, Hagos Y, Lage H,
Burckhardt G and Brockmöller J: Influx and efflux transport as
determinants of melphalan cytotoxicity: Resistance to melphalan in
MDR1 over-expressing tumor cell lines. Biochem Pharmacol. 78:45–53.
2009. View Article : Google Scholar
|
|
152
|
Doyle L and Ross DD: Multidrug resistance
mediated by the breast cancer resistance protein BCRP (ABCG2).
Oncogene. 22:7340–7358. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Raaijmakers HG, Izquierdo MA, Lokhorst HM,
de Leeuw C, Belien JA, Bloem AC, Dekker AW, Scheper RJ and
Sonneveld P: Lung-resistance-related protein expression is a
negative predictive factor for response to conventional low but not
to intensified dose alkylating chemotherapy in multiple myeloma.
Blood. 91:1029–1036. 1998.PubMed/NCBI
|
|
154
|
Hofmeister CC, Yang X, Pichiorri F, Chen
P, Rozewski DM, Johnson AJ, Lee S, Liu Z, Garr CL, Hade EM, et al:
Phase I trial of lenalidomide and CCI-779 in patients with relapsed
multiple myeloma: Evidence for lenalidomide-CCI-779 interaction via
P-glycoprotein. J Clin Oncol. 29:3427–3434. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Tong Z, Yerramilli U, Surapaneni S and
Kumar G: The interactions of lenalidomide with human uptake and
efflux transporters and UDP-glucuronosyltransferase 1A1: Lack of
potential for drug-drug interactions. Cancer Chemother Pharmacol.
73:869–874. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Jiang Y: Pharmacokinetic and
pharmacodynamic studies of lenalidomide and pomalidomide.
Electronic Thesis or Dissertation. Retrieved from https://etd.ohiolink.edu/.
|
|
157
|
O'Connor R, Ooi MG, Meiller J, Jakubikova
J, Klippel S, Delmore J, Richardson P, Anderson K, Clynes M,
Mitsiades CS, et al: The interaction of bortezomib with multidrug
transporters: Implications for therapeutic applications in advanced
multiple myeloma and other neoplasias. Cancer Chemother Pharmacol.
71:1357–1368. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Nakamura T, Tanaka K, Matsunobu T, Okada
T, Nakatani F, Sakimura R, Hanada M and Iwamoto Y: The mechanism of
cross-resistance to proteasome inhibitor bortezomib and overcoming
resistance in Ewing's family tumor cells. Int J Oncol. 31:803–811.
2007.PubMed/NCBI
|
|
159
|
Zimmermann C, Gutmann H and Drewe J:
Thalidomide does not interact with P-glycoprotein. Cancer Chemother
Pharmacol. 57:599–606. 2006. View Article : Google Scholar
|
|
160
|
Dilger K, Alberer M, Busch A, Enninger A,
Behrens R, Koletzko S, Stern M, Beckmann C and Gleiter CH:
Pharmacokinetics and pharmacodynamic action of budesonide in
children with Crohn's disease. Aliment Pharmacol Ther. 23:387–396.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Karssen AM, Meijer OC, van der Sandt IC,
De Boer AG, De Lange EC and De Kloet ER: The role of the efflux
transporter P-glycoprotein in brain penetration of prednisolone. J
Endocrinol. 175:251–260. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Shapiro AB and Ling V: Positively
cooperative sites for drug transport by P-glycoprotein with
distinct drug specificities. Eur J Biochem. 250:130–137. 1997.
View Article : Google Scholar
|
|
163
|
Cole SP and Deeley RG: Transport of
glutathione and glutathione conjugates by MRP1. Trends Pharmacol
Sci. 27:438–446. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Robey RW, Shukla S, Finley EM, Oldham RK,
Barnett D, Ambudkar SV, Fojo T and Bates SE: Inhibition of
P-glycoprotein (ABCB1)- and multidrug resistance-associated protein
1 (ABCC1)-mediated transport by the orally administered inhibitor,
CBT-1((R)). Biochem Pharmacol. 75:1302–1312. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Kitazono M, Sumizawa T, Takebayashi Y,
Chen ZS, Furukawa T, Nagayama S, Tani A, Takao S, Aikou T and
Akiyama S: Multidrug resistance and the lung resistance-related
protein in human colon carcinoma SW-620 cells. J Natl Cancer Inst.
91:1647–1653. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Kang W and Weiss M: Digoxin uptake,
receptor heterogeneity, and inotropic response in the isolated rat
heart: A comprehensive kinetic model. J Pharmacol Exp Ther.
302:577–583. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Abbaszadegan MR, Futscher BW, Klimecki WT,
List A and Dalton WS: Analysis of multidrug resistance-associated
protein (MRP) messenger RNA in normal and malignant hematopoietic
cells. Cancer Res. 54:4676–4679. 1994.PubMed/NCBI
|
|
168
|
Duhem C, Ries F and Dicato M: What does
multidrug resistance (MDR) expression mean in the clinic?
Oncologist. 1:151–158. 1996.PubMed/NCBI
|
|
169
|
Pilarski LM and Belch AR: Intrinsic
expression of the multidrug transporter, P-glycoprotein 170, in
multiple myeloma: Implications for treatment. Leuk Lymphoma.
17:367–374. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Nuessler V, Gieseler F, Gullis E,
Pelka-Fleischer R, Stötzer O, Zwierzina H and Wilmanns W:
Functional P-gp expression in multiple myeloma patients at primary
diagnosis and relapse or progressive disease. Leukemia. 11(Suppl
5): S10–S14. 1997.
|
|
171
|
Drain S, Flannely L, Drake MB, Kettle P,
Orr N, Bjourson AJ, Catherwood MA and Alexander HD: Multidrug
resistance gene expression and ABCB1 SNPs in plasma cell myeloma.
Leuk Res. 35:1457–1463. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Li YH, Wang YH, Li Y and Yang L: MDR1 gene
polymorphisms and clinical relevance. Yi Chuan Xue Bao. 33:93–104.
2006.PubMed/NCBI
|
|
173
|
Drain S, Catherwood MA, Orr N, Galligan L,
Rea IM, Hodkinson C, Drake MB, Kettle PJ, Morris TC and Alexander
HD: ABCB1 (MDR1) rs1045642 is associated with increased overall
survival in plasma cell myeloma. Leuk Lymphoma. 50:566–570. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Buda G, Maggini V, Galimberti S, Martino
A, Giuliani N, Morabito F, Genestreti G, Iacopino P, Rizzoli V,
Barale R, et al: MDR1 polymorphism influences the outcome of
multiple myeloma patients. Br J Haematol. 137:454–456. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Barrand MA, Bagrij T and Neo SY: Multidrug
resistance-associated protein: A protein distinct from
P-glycoprotein involved in cytotoxic drug expulsion. Gen Pharmacol.
28:639–645. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Davey RA, Longhurst TJ, Davey MW, Belov L,
Harvie RM, Hancox D and Wheeler H: Drug resistance mechanisms and
MRP expression in response to epirubicin treatment in a human
leukaemia cell line. Leuk Res. 19:275–282. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Versantvoort CH, Broxterman HJ, Bagrij T,
Scheper RJ and Twentyman PR: Regulation by glutathione of drug
transport in multidrug-resistant human lung tumour cell lines
overexpressing multidrug resistance-associated protein. Br J
Cancer. 72:82–89. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Buda G, Ricci D, Huang CC, Favis R, Cohen
N, Zhuang SH, Harousseau JL, Sonneveld P, Bladé J and Orlowski RZ:
Polymorphisms in the multiple drug resistance protein 1 and in
P-glycoprotein 1 are associated with time to event outcomes in
patients with advanced multiple myeloma treated with bortezomib and
pegylated liposomal doxorubicin. Ann Hematol. 89:1133–1140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Grant CE, Valdimarsson G, Hipfner DR,
Almquist KC, Cole SP and Deeley RG: Overexpression of multidrug
resistance-associated protein (MRP) increases resistance to natural
product drugs. Cancer Res. 54:357–361. 1994.PubMed/NCBI
|
|
180
|
Lehne G: P-glycoprotein as a drug target
in the treatment of multidrug resistant cancer. Curr Drug Targets.
1:85–99. 2000. View Article : Google Scholar
|
|
181
|
Bart J, Hollema H, Groen HJ, de Vries EG,
Hendrikse NH, Sleijfer DT, Wegman TD, Vaalburg W and van der Graaf
WT: The distribution of drug-efflux pumps, P-gp, BCRP, MRP1 and
MRP2, in the normal blood-testis barrier and in primary testicular
tumours. Eur J Cancer. 40:2064–2070. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Cooray HC, Blackmore CG, Maskell L and
Barrand MA: Localisation of breast cancer resistance protein in
microvessel endothelium of human brain. Neuroreport. 13:2059–2063.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Xu J, Liu Y, Yang Y, Bates S and Zhang JT:
Characterization of oligomeric human half-ABC transporter
ATP-binding cassette G2. J Biol Chem. 279:19781–19789. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Raaijmakers MH, de Grouw EP, Heuver LH,
van der Reijden BA, Jansen JH, Scheffer G, Scheper RJ, de Witte TJ
and Raymakers RA: Impaired breast cancer resistance protein
mediated drug transport in plasma cells in multiple myeloma. Leuk
Res. 29:1455–1458. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Doyle LA, Yang W, Abruzzo LV, Krogmann T,
Gao Y, Rishi AK and Ross DD: A multidrug resistance transporter
from human MCF-7 breast cancer cells. Proc Natl Acad Sci USA.
95:15665–15670. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Jakubikova J, Adamia S, Kost-Alimova M,
Klippel S, Cervi D, Daley JF, Cholujova D, Kong SY, Leiba M, Blotta
S, et al: Lenalidomide targets clonogenic side population in
multiple myeloma: Pathophysiologic and clinical implications.
Blood. 117:4409–4419. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Turner JG, Gump JL, Zhang C, Cook JM,
Marchion D, Hazlehurst L, Munster P, Schell MJ, Dalton WS and
Sullivan DM: ABCG2 expression, function, and promoter methylation
in human multiple myeloma. Blood. 108:3881–3889. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Tamura A, Wakabayashi K, Onishi Y, Takeda
M, Ikegami Y, Sawada S, Tsuji M, Matsuda Y and Ishikawa T:
Re-evaluation and functional classification of non-synonymous
single nucleotide polymorphisms of the human ATP-binding cassette
transporter ABCG2. Cancer Sci. 98:231–239. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Izquierdo MA, Scheffer GL, Flens MJ,
Shoemaker RH, Rome LH and Scheper RJ: Relationship of LRP-human
major vault protein to in vitro and clinical resistance to
anticancer drugs. Cytotechnology. 19:191–197. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Feller N, Kuiper CM, Lankelma J, Ruhdal
JK, Scheper RJ, Pinedo HM and Broxterman HJ: Functional detection
of MDR1/P170 and MRP/P190-mediated multidrug resistance in tumour
cells by flow cytometry. Br J Cancer. 72:543–549. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Sherry ST, Ward MH, Kholodov M, Baker J,
Phan L, Smigielski EM and Sirotkin K: dbSNP: The NCBI database of
genetic variation. Nucleic Acids Res. 29:308–311. 2001. View Article : Google Scholar :
|
|
192
|
Henríquez-Hernández LA, Moreno M, Rey A,
Lloret M and Lara PC: MVP expression in the prediction of clinical
outcome of locally advanced oral squamous cell carcinoma patients
treated with radiotherapy. Radiat Oncol. 7:1–6. 2012. View Article : Google Scholar
|
|
193
|
Litviakov NV, Cherdyntseva NV, Tsyganov
MM, Denisov EV, Garbukov EY, Merzliakova MK, Volkomorov VV,
Vtorushin SV, Zavyalova MV, Slonimskaya EM, et al: Changing the
expression vector of multidrug resistance genes is related to
neoadjuvant chemotherapy response. Cancer Chemother Pharmacol.
71:153–163. 2013. View Article : Google Scholar
|
|
194
|
van den Heuvel-Eibrink MM, Sonneveld P and
Pieters R: The prognostic significance of membrane
transport-associated multidrug resistance (MDR) proteins in
leukemia. Int J Clin Pharmacol Ther. 38:94–110. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Herlevsen M, Oxford G, Owens CR, Conaway M
and Theodorescu D: Depletion of major vault protein increases
doxorubicin sensitivity and nuclear accumulation and disrupts its
sequestration in lysosomes. Mol Cancer Ther. 6:1804–1813. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Sikic BI, Fisher GA, Lum BL, Halsey J,
Beketic-Oreskovic L and Chen G: Modulation and prevention of
multidrug resistance by inhibitors of P-glycoprotein. Cancer
Chemother Pharmacol. 40(Suppl 1): S13–S19. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Sikic BI: Pharmacologic approaches to
reversing multidrug resistance. Semin Hematol. 34(Suppl 5): 40–47.
1997.PubMed/NCBI
|
|
198
|
Yang HH, Ma MH, Vescio RA and Berenson JR:
Overcoming drug resistance in multiple myeloma: The emergence of
therapeutic approaches to induce apoptosis. J Clin Oncol.
21:4239–4247. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Fisher GA, Lum BL, Hausdorff J and Sikic
BI: Pharmacological considerations in the modulation of multidrug
resistance. Eur J Cancer. 32A:1082–1088. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Dalton W and Lehnert M: Dexverapamil: A
clinical approach to circumvention of multidrug resistance. J
Cancer Res Clin Oncol. 121:R1. 1995. View Article : Google Scholar
|
|
201
|
Berenson JR, Crowley JJ, Grogan TM,
Zangmeister J, Briggs AD, Mills GM, Barlogie B and Salmon SE:
Maintenance therapy with alternate-day prednisone improves survival
in multiple myeloma patients. Blood. 99:3163–3168. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Urashima M, Chen BP, Chen S, Pinkus GS,
Bronson RT, Dedera DA, Hoshi Y, Teoh G, Ogata A, Treon SP, et al:
The development of a model for the homing of multiple myeloma cells
to human bone marrow. Blood. 90:754–765. 1997.PubMed/NCBI
|
|
203
|
Brown R, Suen H, Favaloro J, Yang S, Ho
PJ, Gibson J and Joshua D: Trogocytosis generates acquired
regulatory T cells adding further complexity to the dysfunctional
immune response in multiple myeloma. OncoImmunology. 1:1658–1660.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Duffy MJ: Serum tumor markers in breast
cancer: Are they of clinical value? Clin Chem. 52:345–351. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Allin KH, Nordestgaard BG, Flyger H and
Bojesen SE: Elevated pre-treatment levels of plasma C-reactive
protein are associated with poor prognosis after breast cancer: A
cohort study. Breast Cancer Res. 13:R552011. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Guo L, Abraham J, Flynn DC, Castranova V,
Shi X and Qian Y: Individualized survival and treatment response
predictions for breast cancers using phospho-EGFR, phospho-ER,
phospho-HER2/neu, phospho-IGF-IR/In, phospho-MAPK, and
phospho-p70S6K proteins. Int J Biol Markers. 22:1–11.
2007.PubMed/NCBI
|
|
207
|
Sargent DJ, Conley BA, Allegra C and
Collette L: Clinical trial designs for predictive marker validation
in cancer treatment trials. J Clin Oncol. 23:2020–2027. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Kumar G, Lau H and Laskin O: Lenalidomide:
In vitro evaluation of the metabolism and assessment of cytochrome
P450 inhibition and induction. Cancer Chemother Pharmacol.
63:1171–1175. 2009. View Article : Google Scholar
|
|
209
|
Lampen A, Zhang Y, Hackbarth I, Benet LZ,
Sewing KF and Christians U: Metabolism and transport of the
macrolide immunosuppressant sirolimus in the small intestine. J
Pharmacol Exp Ther. 285:1104–1112. 1998.PubMed/NCBI
|
|
210
|
Sattler M, Guengerich FP, Yun CH,
Christians U and Sewing KF: Cytochrome P-450 3A enzymes are
responsible for biotransformation of FK506 and rapamycin in man and
rat. Drug Metab Dispos. 20:753–761. 1992.PubMed/NCBI
|
|
211
|
Joshua DE, Brown RD and Gibson J: Multiple
myeloma: Why does the disease escape from plateau phase? Br J
Haematol. 88:667–671. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Fung KL and Gottesman MM: A synonymous
polymorphism in a common MDR1 (ABCB1) haplotype shapes protein
function. Biochim Biophys Acta. 1794:860–871. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Hoffmeyer S, Burk O, von Richter O, Arnold
HP, Brockmöller J, Johne A, Cascorbi I, Gerloff T, Roots I,
Eichelbaum M, et al: Functional polymorphisms of the human
multidrug-resistance gene: Multiple sequence variations and
correlation of one allele with P-glycoprotein expression and
activity in vivo. Proc Natl Acad Sci USA. 97:3473–3478. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Dumontet C, Landi S, Reiman T, Perry T,
Plesa A, Bellini I, Barale R, Pilarski LM, Troncy J, Tavtigian S,
et al: Genetic polymorphisms associated with outcome in multiple
myeloma patients receiving high-dose melphalan. Bone Marrow
Transplant. 45:1316–1324. 2010. View Article : Google Scholar
|
|
215
|
Maggini V, Buda G, Martino A, Presciuttini
S, Galimberti S, Orciuolo E, Barale R, Petrini M and Rossi AM: MDR1
diplotypes as prognostic markers in multiple myeloma. Pharmacogenet
Genomics. 18:383–389. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Natarajan K, Xie Y, Baer MR and Ross DD:
Role of breast cancer resistance protein (BCRP/ABCG2) in cancer
drug resistance. Biochem Pharmacol. 83:1084–1103. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Dispenzieri A, Kyle R, Merlini G, Miguel
JS, Ludwig H, Hajek R, Palumbo A, Jagannath S, Blade J, Lonial S,
et al; International Myeloma Working Group. International Myeloma
Working Group guidelines for serum-free light chain analysis in
multiple myeloma and related disorders. Leukemia. 23:215–224. 2009.
View Article : Google Scholar
|