Introduction
Oral squamous cell carcinoma (OSCC) is a highly
lethal disease, owing to lack of effective diagnostic biomarkers
and therapeutic targets (1,2).
Despite recent advances in diagnosis and treatment, the 5-year
survival rate of patients with OSCC is ≤50% (3,4).
Over the past two decades, numerous prognostic and predictive
markers for the clinical outcomes of OSCC have been proposed
(5), however, few have been
applied in clinical practice owing to the lack of understanding of
the mechanism and the non-reproducibility of the initial findings
(2,6).
RACK1, characterized by highly conserved internal
WD-40 repeats (Trp-Asp) (7) was
originally identified as an anchoring protein for the conventional
protein kinase C (PKC) (8). In
recent years, the understanding of RACK1 function has increased
remarkably. First identified as an anchoring protein for activated
PKC, it is now known to serve as an anchoring or adaptor protein in
various crosstalk signal pathways. RACK1 has been broadly approved
as a multifaceted scaffolding protein involved in multiple
biological events via interaction with different partners (9,10),
including cell migration (11),
and angiogenesis (12). RACK1 also
modulates kinases, phosphodiesterases, and phosphatases by
regulating their activities, subcellular distributions, and
association with substrates, contributing significantly to the
regulation of the signal transduction network (13).
RACK1 exerts dual functions in cell growth. For
example, RACK1 could promote cell growth via PKCβII/eIF4E (14), MKK7/JNK (15), SHH signaling (16), PI3K and GSK3β (17), but inhibits cell growth by
promoting β-catenin (18) and
ΔNp63 (19) degradation and
inhibition of Src activity (20,21).
RACK1 also has opposing roles in apoptosis. It could promote
degradation of pro-apoptotic molecules, such as Fem1b and BimEL
(22–24) or E1A to reduce apoptosis levels
(25). It could increase apoptosis
by dissociation of the Bax/Bcl-XL complex (22), advance Bax oligomerization, and
inactive AKT and enhance the expression of pro-apoptotic Bim
(26). Thus, RACK1 was reported to
be a cancer inhibitor with low expression in gastric cancer
(18), but a cancer promoter with
high expression in multiple kinds of cancer, including
non-small-cell lung cancer (NSCLC) (16), pulmonary adenocarcinoma (27,28),
hepatocellular carcinoma (HCC) (15), glioma, esophageal squamous cell
carcinoma (ESCC) (29), and OSCC
(30).
In our previous studies, RACK1 was found abnormally
overexpressed in OSCC by comparative proteomics and it is an
excellent predictor for poor clinical outcome (30,31).
In addition, knockdown of the RACK1 gene could inhibit the
proliferation, and motility of OSCC cells and induced by decreased
protein levels of pEGFR, HER2, and MMP2/9 (11). However, to our knowledge, there has
been no prior study on the mechanism of RACK1 regulation of cell
growth and the effects of RACK1 on the behavior of OSCC cells in
vivo.
The aim of this study was to elucidate the mechanism
by which RACK1 regulates cell growth in OSCC using in vitro
and in vivo models. We selected OSCC cells stably infected
with lentivirus based RACK1-sh, to investigate the antitumor
efficiency of RACK1 depletion, and employed a xenograft mouse model
and clinical cohort to uncover the potential mechanism. We provide
the basis for the potential use of RACK1 as a novel therapeutic
target for OSCC in the future.
Materials and methods
Clinical tissue samples
The Institutional Review Boards of the West China
Hospital of Stomatology, Sichuan University approved this study.
The study was approved by the ethics committee both of the West
China Hospital of Stomatology and the Guangdong Provincial
Stomatological Hospital and was conducted in agreement with the
Helsinki Declaration.
Four normal tissues from plastic surgery, 8 oral
leukoplakia tissues and 15 excised primary OSCC specimens were
included in this study. The only selection criterion was epithelial
continuity for normal and premalignant tissue (Table I).
 | Table 1Clinicopathological factors in 27
patients with normal oral mucosa, oral leukoplakia, and OSCC. |
Table 1
Clinicopathological factors in 27
patients with normal oral mucosa, oral leukoplakia, and OSCC.
| Characteristic | Normal oral mucosa
(N=4)
No. | Oral leukoplakia
with mild to moderate epithelial hyperplasia (N=4)
No. | Oral leukoplakia
with severe epithelial hyperplasia (N=4)
No. | OSCC
(N=15)
No. |
|---|
| Age |
| <60 years | 2 | 2 | 3 | 6 |
| ≥60 years | 2 | 2 | 1 | 9 |
| Gender |
| Male | 1 | 2 | 4 | 10 |
| Female | 3 | 2 | 0 | 5 |
| Smoking
history |
| Never | 3 | 2 | 1 | 7 |
| Ever | 1 | 2 | 3 | 8 |
| Drinking
history |
| Never | 3 | 2 | 0 | 5 |
| Ever | 1 | 2 | 4 | 10 |
| Cell
differentiation |
| High | NA | NA | NA | 6 |
| Moderate or
low | NA | NA | NA | 9 |
| Primary site |
| Ventral
tongue/floor of mouth | 2 | 1 | 3 | 4 |
| Buccal mucosa | 1 | 3 | 0 | 6 |
| Gingiva | 1 | 0 | 1 | 3 |
| Othersa | 0 | 0 | 0 | 2 |
| Tumor stage |
| T1 or T2 | NA | NA | NA | 7 |
| T3 or T4 | NA | NA | NA | 8 |
| Nodal stage |
| N0 | NA | NA | NA | 11 |
| N1–3 | NA | NA | NA | 8 |
| Clinical TMN
stage |
| I or II | NA | NA | NA | 9 |
| III or IV | NA | NA | NA | 6 |
| Surgical
method |
| Local | 4 | 3 | 3 | 3 |
| Unilateral or
bilateral or other | 0 | 1 | 1 | 12 |
Cell lines
The cell lines 293T and Cal-27 were purchased from
American Type Culture Collection (Manassas, VA, USA). HSC-3 and
HSC-4 cells were purchased from the Cell Bank of Japanese
Collection of Research Bioresource (JCRB, Shinjuku, Japan). UM1 and
UM2 were provided by Dr Xiaofeng Zhou (Center for Molecular Biology
of Oral Diseases, College of Dentistry, University of Illinois at
Chicago). The human immortalized oral keratinocyte cell line
HOK16E6E7 was provided by Dr Xuan Liu (Charles R. Drew University
of Medicine and Science). Five oral squamous cell carcinoma cell
lines (HSC-3, CAL-27, UM1, UM2 and HSC-4) and 293T cells were
maintained in Dulbecco's modified Eagle's medium (Invitrogen)
containing 10% fetal calf serum (Invitrogen), 100 U/l penicillin,
and 10 mg/l streptomycin. HOK16E6E7 cells were cultured in
keratinocyte growth medium containing 0.15 mM calcium and
supplemented with epidermal growth factor (Invitrogen).
shRNA plasmids and transfection
Three lentiviral-based shRNA plasmids were used:
RACK1-specific shRNAs pLV-sh-RACK (RACK1-sh1/2 target the sequence
TCGAGATAAGACCATCATCAT and CAAGCTGAAGACCAACCACAT of RACK1,
respectively), and negative control pLV-sh-NC (NS-sh) were
purchased from Chengdu Bomei Biotechnology Inc.(Chengdu,
China).Transfection regent Lipofectamine 2000 (Invitrogen,
Carlsbad, CA, USA), was used according to the manufacturer's
instructions. Plasmid transfection was performed as previously
described (32).
Stable cell line generation
Lentivirus delivering RACK1-sh or NS-sh was prepared
used 293T cells. HSC-3 cells were infected with either RACK1-sh or
NS-sh. Selective culture medium containing puromycin was used to
select cells with stable expression of sh-RACK1. The expression of
RACK1 was detected by Q-PCR and western blot analysis.
Q-PCR
The nucleotide sequences of the sense and antisense
primers used for RACK1 amplification were
5′-GCTCTGCCATAAACTTCTAGCGTGTGC-3′ and
5′-CTGTGCTTCTGGAGGCAAGGATGGCCA-3′, respectively. The primers for
GAPDH amplification were purchased from Invitrogen. Q-PCR was
performed as previously described (30,31).
Western blot analysis
The protein expression levels of RACK1, AKT, p-AKT,
mTOR, p-mTOR, S6, and p-S6 in the OSCC cells were examined by
western blot analysis. RACK1 antibody was purchased from Santa Cruz
(Santa Cruz, CA, USA) and AKT, p-AKT (Ser-473), mTOR, p-mTOR
(Ser-2448), S6, and p-S6 (Ser235/236) antibodies were purchased
from Cell Signaling (Danvers, MA, USA), western blot analysis was
done as previously described (30,31).
Cell cycle analysis
Cell cycle was assessed as previously described
(33). Briefly, 2×106
HSC-3 cells of each group were harvested, washed twice, and fixed
with cold 70% ethanol at 4°C overnight. The cells were then washed
and digested with RNase, then stained with 800 μl propidium iodide
(50 μg/ml) at room temperature for 30 min. Cell cycle analysis was
done by using FACS Aria flow cytometry system (BD Biosciences).
Data were analyzed using BD FACS Diva software.
In vivo tumor-formation assay
Animal studies were approved by the Animal Care and
Use Committee, State Key Laboratory of Oral Diseases, in compliance
with the Guide for the U.S. Public Health Service's policy on
humane care and use of laboratory animals. The in vivo
tumor-formation was done as previously described (34). Briefly, female BALB/c nude mice 6
weeks of age weighing 20–22 g were used in the study of tumor
formation. The animals were monitored every 3 days for tumor
development. All the mice were sacrificed after the last
measurement. Tumor tissues were cut into 4-mm sections,
deparaffinized, rehydrated, and treated with a peroxidase block,
before processing for H&E and immunohistochemistry. Results of
animal experiments were expressed as mean ± SD of five tumors
analyzed.
Immunohistochemistry assay
Tumor tissues were cut into 4-mm sections,
deparaffinized, rehydrated, and treated with a peroxidase block.
After heat-based antigen retrieval, sections were incubated with 5%
normal goat or horse serum (Zymed, San Francisco, CA, USA) to block
non-specific sites before incubation with anti-RACK1 immunoserum
diluted 1:100 or monoclonal antibody Ki67 diluted 1:200 (from Dako
and Abcam, respectively), or mouse CD34 monoclonal antibody diluted
1:200 (BD Biosciences, San Jose, CA, USA), or anti-VEGF antibody
(R&D Systems, Abingdon, UK), or anti-RhoA antibody (Cell
Signaling Technology, Beverly, MA, USA), anti-mLYVE-1 antibody
(R&D Systems), or anti-E-cadherin antibody (Cell Signaling
Technology) was incubated at 4°C overnight. Sections incubated
without primary antibodies were used as negative controls.
For clinical tumor tissues, the RACK1 and pAKT
staining were determined based on the staining intensity (scale,
1–3) and percentage of tumor staining (scale, 1–3) as previously
described (34). The total
staining was expressed as a product of the two numbers (resulting
in a staining scale of 1–9). The evaluation was performed by two
independent investigators.
Statistical analysis
The values given are mean ± SEM. The significance of
difference between the experimental groups and controls was
assessed by Student's t-test. The difference was considered
significant at the P-value of <0.05.
Results
RACK1 silencing induces G1 and G2 phase
cell cycle arrest in OSCC cells
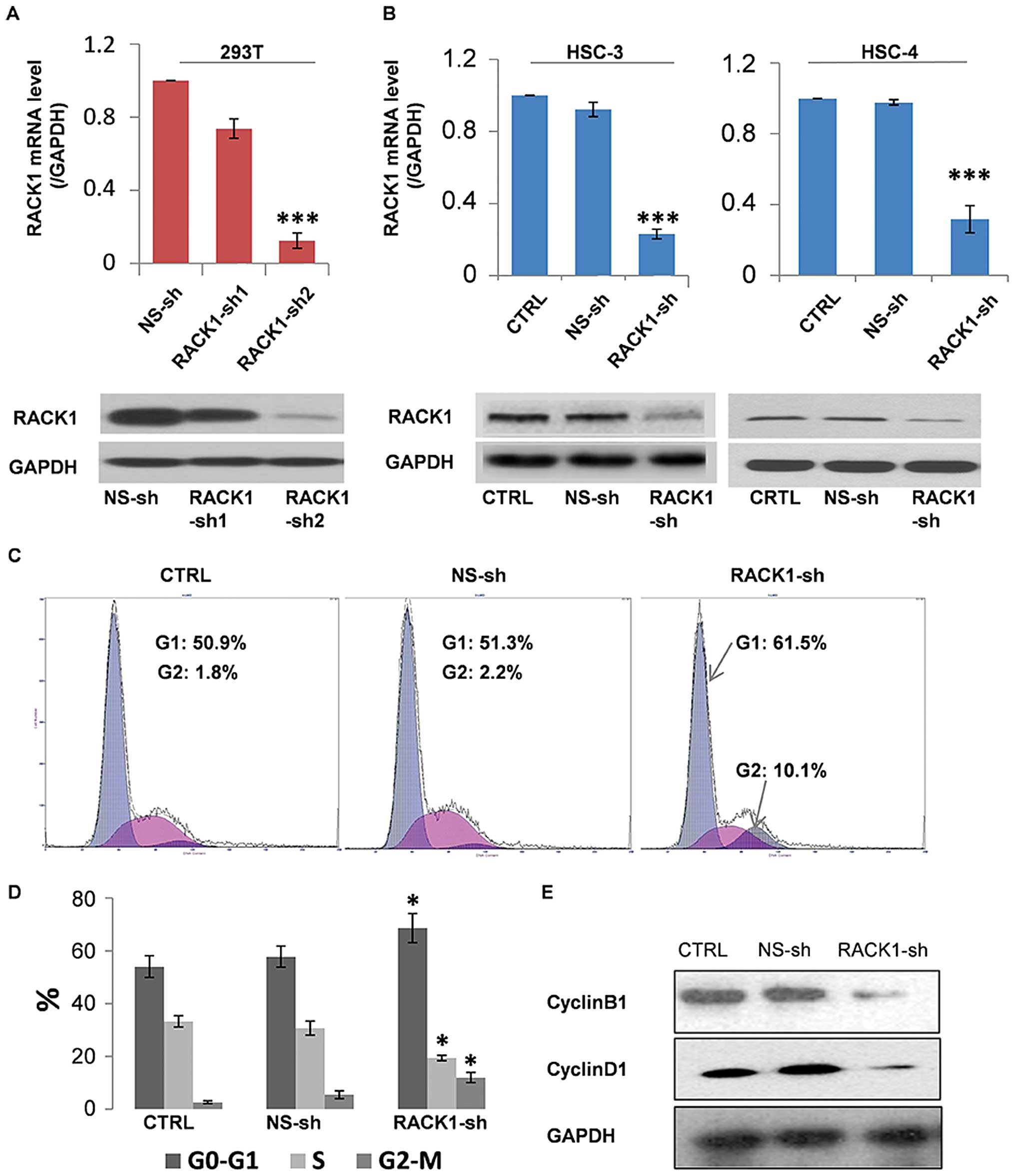
Two shRNAs (RACK1-sh1 and RACK1-sh2) designed
against different regions of RACK1 were transfected into 293T.
RACK1-sh2 had proven effective in the inhibition of RACK1
expression, as confirmed by both RT-PCR and western blotting. Thus,
RACK1-sh2 (RACK1-sh) plasmid and stably transfected RACK1-sh cells
were utilized for the following experiments (Fig. 1A). Then we successfully screened
for stable RACK1 low expression HSC-3 and HSC-4 cell lines with
RACK1-sh (Fig. 1B). RACK1
expression in HSC-4 cells is lower than HSC-3 cells, and HSC-4
cells were hard to conform to a mouse model in our preliminary
experiments (date not shown). Therefore, we selected HSC-3 cells
for the next experiments.
To study the potential mechanisms by which RACK1
silencing inhibits HSC-3 cell growth, the effect of RACK1 shRNA on
cell cycle was evaluated using flow cytometry. As shown in Fig. 1C, the percentages of G0-G1 phase
cells in two control groups were 50–55%, whereas that in the RACK1
shRNA transfected group was 65%. Approximately 20% of the cells in
the RACK1 shRNA-transfected group were in S-phase, compared with
30–35% of cells in the two control groups. Approximately 15% of
cells in the RACK1 shRNA-transfected group were in G2-M phase,
compared with 1–2% of cells in the two control groups (Fig. 1D). Therefore, RACK1 silencing may
arrest the cell cycle at the G1 and G2 phase by inhibiting the G1→S
and G2→M transition in HSC-3 cells. Since cell proliferation is
normally controlled by cell cycle regulatory proteins, we performed
western blotting to investigate the effect of RACK1 silencing on
cell cycle regulators. The expression levels of Cyclin B1 and
Cyclin D1 were downregulated (Fig.
1E). These results indicate that RACK1 silencing might regulate
the cell cycle in HSC-3 cells by modulating the expression of cell
cycle regulators, thus inhibiting the processes of oral
carcinogenesis.
RACK1 contributes to the progression of
OSCC in vivo
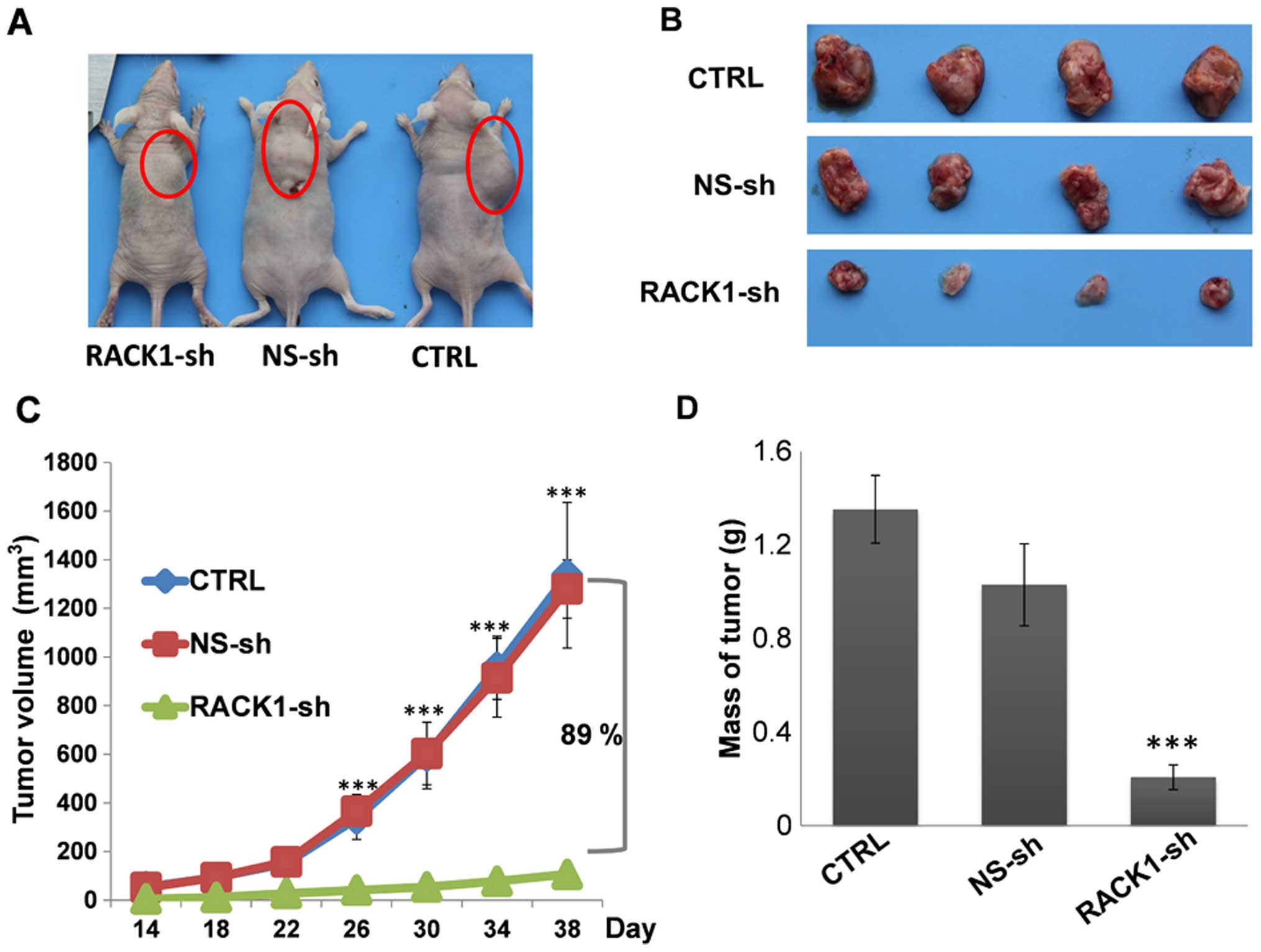
Next, we investigated the effect of RACK1 on the
tumorigenicity of OSCC cells using an in vivo xenograft
model. As shown in Fig. 2, tumors
formed in the mice transplanted with RACK1-sh silenced HSC-3 cells
were smaller in both size and weight than the tumors in mice
transplanted with control cells. The average tumor volume in the
RACK1-sh group was reduced by 89.1%. To investigate the potential
mechanisms underlying the effects of RACK1 silencing in
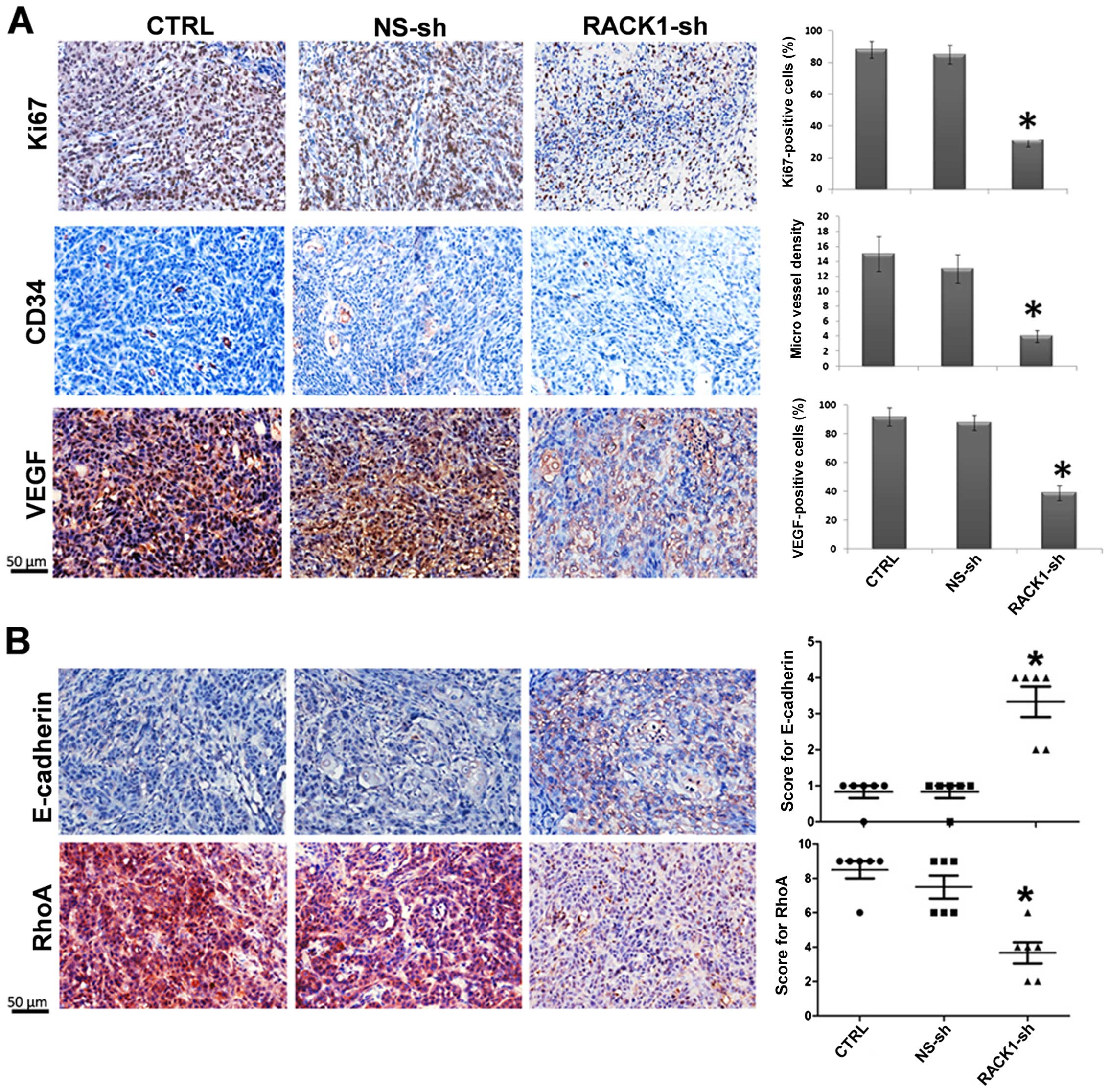
vivo, we examined tumor cell proliferation. Marked reduction in
the proliferation marker Ki-67 expression was found in the RACK1
silenced group (Fig. 3A).
Angiogenesis in tumors was detected by microvessel density (MVD)
CD34 staining, VEGF staining was also used as a marker for
angiogenesis in tumors. As shown in Fig. 3A, these analyses showed significant
decrease in the average number of microvessels per vascular hot
spot and, VEGF expression in the RACK1-sh group compared with those
in the two control groups. Additionally, the expression of RhoA
decreased, and E-cadherin increased in the RACK1 silenced group
(Fig. 3B). Both proteins are
important for tumor metastasis, this is basically consistent with
our above results. Taken together, these results suggest that RACK1
contributes to the progression of OSCC in vivo.
Cell line and clinical relevance of
RACK1-induced AKT activation in human OSCC
Our previous study showed that activated AKT
correlates with poor clinical outcomes for OSCC (35), and single-nucleotide polymorphisms
(SNPs) in AKT1 (rs3803300) are associated with progression-free
survival time of OSCC patients (36). AKT is considered as an important
key protein regulating tumor progression in various types of human
cancer. Peng et al showed knocking down RACK1 in
vitro decreased AKT activity in glioma (37). We further investigated whether
RACK1 expression and AKT activation are relevant in human OSCC cell
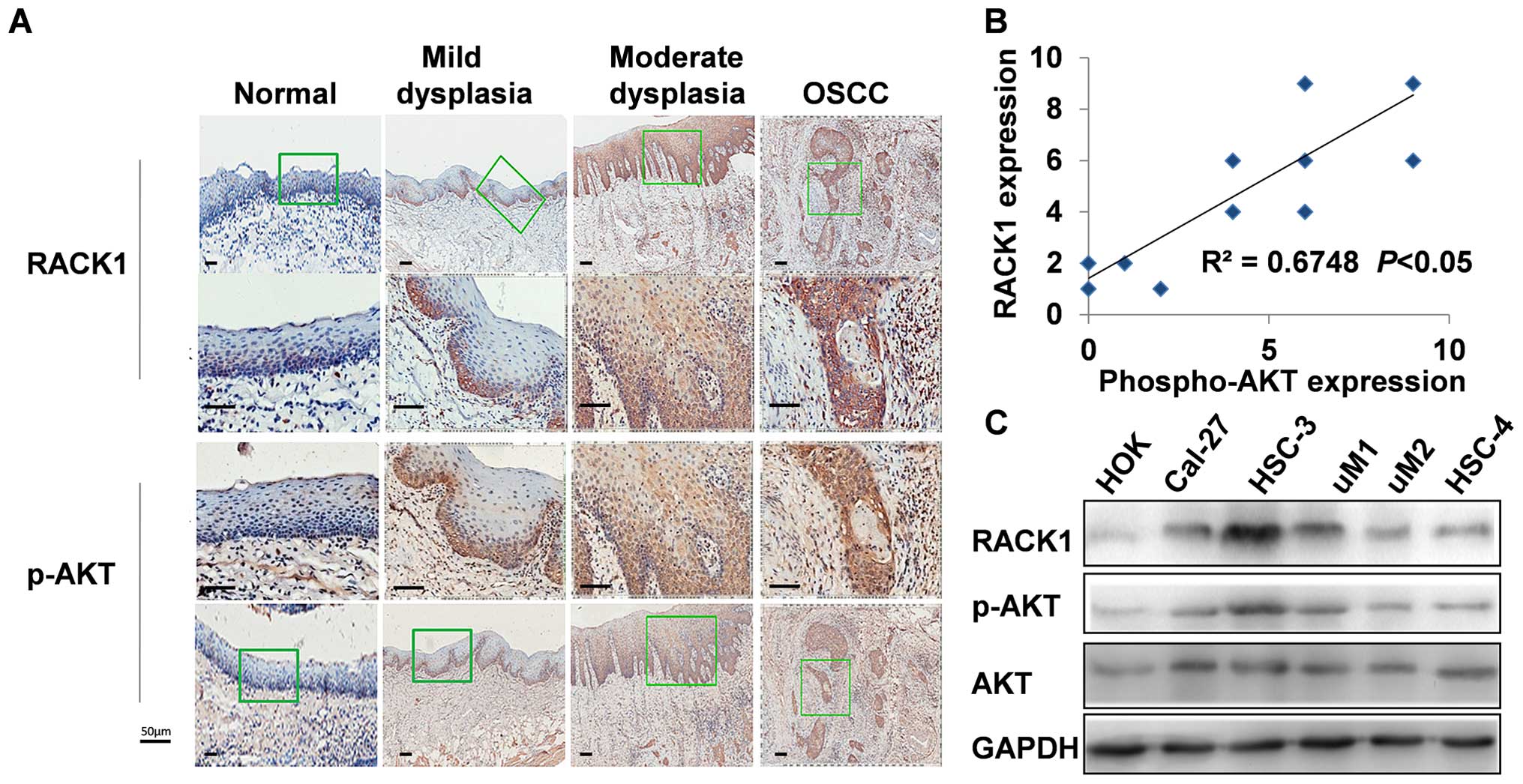
lines and clinical samples. As shown in Fig. 4A, RACK1 and p-AKT showed similar
expression in different stages of oral carcinogenic tissues.
Furthermore, the level of RACK1 expression in 15 collected clinical
OSCC samples correlated positively with p-AKT expression (Fig. 4B, R = 0.6748, P<0.05). To
further confirm the result, OSCC cells were harvested from 6 OSCC
cell lines (HSC-3, HSC-4, SCC-5, CAL-27, SCC-25, and SCC-9),
together with HOK, an immortalized oral keratinocyte cell line, and
then equal amounts of cell lysates were examined using western blot
analysis. Consistently, expression of p-AKT had a positive
correlation with the level of RACK1 in OSCC cell lines (Fig. 4C).
RACK1 contributes to activation of
AKT/mTOR in OSCC
A growing body of evidence demonstrates that the
AKT/mTOR signaling pathway plays a central role in both cell cycle
and angiogenesis in various types of human cancer, including OSCC
(38). According to published
data, the mTOR pathway is aberrantly activated in most OSCC tumors
(69.5%), and in turn phosphorylates and activates proteins causing
aberrant signals, leading to the translation of proteins required
for tumor progression. Therefore, we investigated whether RACK1 is
involved in regulation of the AKT/mTOR signaling pathway in OSCC.
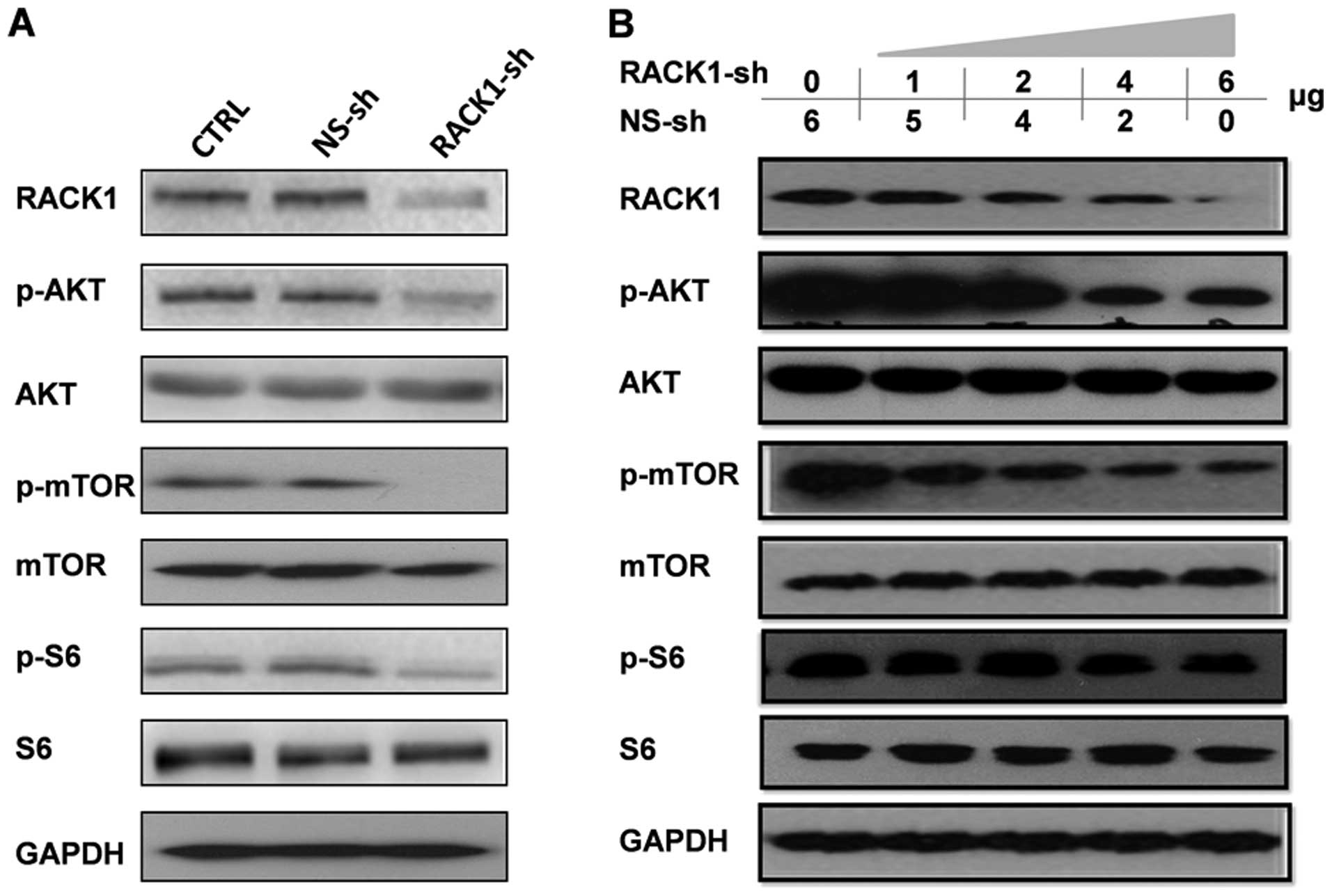
As shown in Fig. 5A, silencing of
RACK1 reduced the phosphorylation of AKT and mTOR, as well as the
well-characterized mTOR downstream protein S6, but did not reduce
Pen AKT, mTOR, and S6 in OSCC cells. Moreover, there was a
dose-dependent effect on the decreased phosphorylation of the
AKT/mTOR pathway in transient RACK1 silencing in 293T cell lines
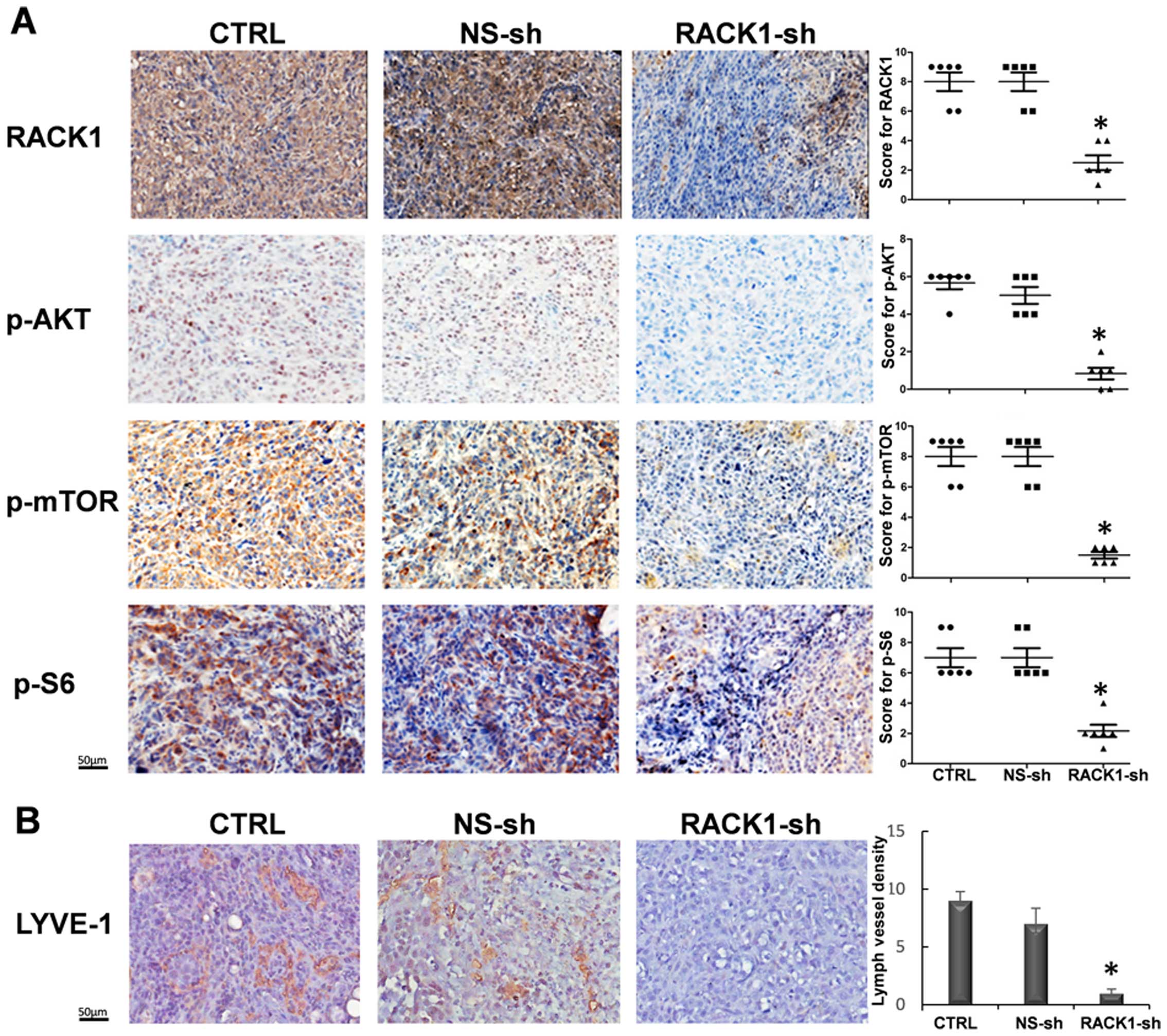
(Fig. 5B). Additionally, depleted
RACK1 could significantly reduce the protein levels of p-AKT,
p-mTOR, and p-S6 in OSCC xenografted tumors in vivo
(Fig. 6A), which was consistent
with our previous findings in vitro. Additionally, the
effect of RACK1 on the lymphangiogenesis in OSCC was also assessed,
as the mTOR function on the lymphangiogenesis was widely described
(39,40). We investigated the expression of
LYVE-1 in the RACK1 silenced group, which was diminished greatly
comparing with the two control groups (Fig. 6B), hinting RACK1 promotes
lymphangiogenesis of OSCC. Collectively, these results indicate
that RACK1 contributes to the activation of AKT/mTOR in OSCC.
Discussion
OSCC is the most common malignancy of HNSCC, and has
a poor survival rate (1,2). Current treatments for OSCC patients
are not satisfactory, and novel therapeutic strategies are urgently
required. Biotherapy was wildly proposed, and it became the fourth
method of treating HNSCC in the last decade (41,42).
Among these biotherapies, gene therapy is the one that has been
most rapidly developed. To date, several gene therapy drugs have
already been used in the clinic to achieve the desired results
(43,44). In this study, we found that
specific targeting of RACK1 could inhibit the cell cycle by
decreasing Cyclin B1 and Cyclin D1. In a mouse xenograft model of
OSCC, an 89% decrease in tumorigenicity was found in the RACK1
silenced group, and intratumoral expression of Ki67, CD34, and VEGF
in RACK1 stably silenced group was significantly decreased when
compared with the two control groups. Moreover, the expression of
RhoA was decrease, whereas E-cadherin was increased in the RACK1
stably silenced group. These proteins are associated with cell
motility, consistent with our previous report that silencing RACK1
by RNA interference could effectively inhibit cell motility ability
in OSCC (11).
AKT, or protein kinase B (PKB), is a
serine-threonine kinase which functions as a downstream target and
effector of phosphatidylinositol 3-kinase (PI3K). Abnormal
activation or expression of AKT can perturb cellular signaling
cascades, resulting in the occurrence of human diseases (45–47).
Recently, AKT has been considered an important oncogene because it
is abnormally activated in several types of human cancers,
especially those that have poor prognosis (48). In our previous OSCC tissue
microarray study, p-AKT was highly expressed in OSCC tissues. The
expression of p-AKT correlated with lymph node metastasis and
recurrence and the 5-year survival rate (35). Both RACK1 and p-AKT were suggested
as predictive markers. We hypothesized that there may be a positive
relationship between RACK1 and p-AKT in OSCC. Further, we
postulated that RACK1 might increase cell growth and angiogenesis
through p-AKT and its downstream pathway. Thus, we analyzed the
expression of RACK1 and p-AKT in a panel of OSCC cell lines and
immortalized oral keratinocyte cells.
To explore the mechanism of RACK1 silencing in OSCC
cell growth inhibition both in vitro and in vivo, we
quantitatively re-assessed expression of RACK1 and p-AKT in a large
precancerous and cancer patient cohort. We detected a similar
tendency between RACK1 and p-AKT in different stages of oral
carcinogenetic tissues. We deduced that the expression of p-AKT is
positively correlated with the level of RACK1 in OSCC cell lines
based on western blot analysis.
Next, we aimed to identify the downstream pathway of
AKT involved in the RACK1 silencing effect. mTOR is one of the
major targets of activated AKT, which in turn regulates a number of
downstream molecules, such as ribosomal protein pS6. AKT is the key
regulator of the AKT/mTOR/S6 pathway, which ultimately controls
fundamental cell processes such as cell cycle, cell proliferation,
and angiogenesis (49,50). The AKT/ mTOR/S6 pathway was
suggested to contribute to the premalignant potential of OSCC
(38,51). Recent findings indicate that
multiple genetic and epigenetic alterations converge on the
persistent activation of AKT/mTOR signaling in most HNSCC lesions.
Therefore, we investigated whether the AKT/mTOR/S6 pathway was
influenced in this case. We found that specific transient knockdown
of RACK1 upregulated the levels of p-AKT, p-mTOR, and p-S6 in a
dose-dependent manner. Moreover, RACK1 knockdown inhibited the
phosphorylation of AKT, mTOR, and S6 in vivo. In addition,
mTOR inhibitors exerted a remarkably increased antitumor activity,
particularly in HNSCC cells and inhibition of mTOR diminished
lymphangiogenesis in the primary tumors (39,40).
Our results also support these previous studies. In other words,
RACK1 might play a significant role in lymphomagenesis with
potential mTOR effect. Thus, we propose that RACK1 may have
potential as a target for therapy in a range of tumor types. Our
results indicate that targeted RACK1 therapy in OSCC cells results
in cell growth inhibition in vitro and OSCC xenografts
suppression in vivo.
In conclusion, this study demonstrates that RACK1
promotes cell growth of OSCC, regulating cell growth and enhancing
the progression of OSCC in vivo, at least in part via
activation of the AKT/mTOR/S6 signaling pathway. This study reveals
a novel mechanism by which RACK1 contributes to the poor prognosis
of OSCC, and suggests a potential novel therapeutic target.
Acknowledgements
This study was supported by grants from National
Natural Science Foundation of China (81321002), Nonprofit Industry
Research Specific Fund of National Health and Family Planning
Commission of China (201502018), National Natural Science
Foundations of China (81302371), ISTCPC (2012DFA31370), Doctoral
Fund of Ministry of Education of China (20130181120084), and
National Natural Science Foundation of China (81472533).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Panzarella V, Pizzo G, Calvino F,
Compilato D, Colella G and Campisi G: Diagnostic delay in oral
squamous cell carcinoma: The role of cognitive and psychological
variables. Int J Oral Sci. 6:39–45. 2014. View Article : Google Scholar :
|
|
3
|
Choi S and Myers JN: Molecular
pathogenesis of oral squamous cell carcinoma: Implications for
therapy. J Dent Res. 87:14–32. 2008. View Article : Google Scholar
|
|
4
|
Ratajczak-Wrona W, Jablonska E, Antonowicz
B, Dziemianczyk D and Grabowska SZ: Levels of biological markers of
nitric oxide in serum of patients with squamous cell carcinoma of
the oral cavity. Int J Oral Sci. 5:141–145. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Principe S, Hui AB, Bruce J, Sinha A, Liu
FF and Kislinger T: Tumor-derived exosomes and microvesicles in
head and neck cancer: Implications for tumor biology and biomarker
discovery. Proteomics. 13:1608–1623. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li X, Amazit L, Long W, Lonard DM, Monaco
JJ and O'Malley BW: Ubiquitin- and ATP-independent proteolytic
turnover of p21 by the REGgamma-proteasome pathway. Mol Cell.
26:831–842. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hu L, Lu F, Wang Y, Liu Y, Liu D, Jiang Z,
Wan C, Zhu B, Gan L, Wang Y, et al: RACK1, a novel
hPER1-interacting protein. J Mol Neurosci. 29:55–63. 2006.
View Article : Google Scholar
|
|
8
|
Besson A, Wilson TL and Yong VW: The
anchoring protein RACK1 links protein kinase Cepsilon to integrin
beta chains. Requirements for adhesion and motility. J Biol Chem.
277:22073–22084. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Daniels CC, Rovnak J and Quackenbush SL:
Walleye dermal sarcoma virus Orf B functions through receptor for
activated C kinase (RACK1) and protein kinase C. Virology.
375:550–560. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wehner P, Shnitsar I, Urlaub H and
Borchers A: RACK1 is a novel interaction partner of PTK7 that is
required for neural tube closure. Development. 138:1321–1327. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li J, Guo Y, Feng X, Wang Z, Wang Y, Deng
P, Zhang D, Wang R, Xie L, Xu X, et al: Receptor for activated C
kinase 1 (RACK1): A regulator for migration and invasion in oral
squamous cell carcinoma cells. J Cancer Res Clin Oncol.
138:563–571. 2012. View Article : Google Scholar
|
|
12
|
Berns H, Humar R, Hengerer B, Kiefer FN
and Battegay EJ: RACK1 is up-regulated in angiogenesis and human
carcinomas. FASEB J. 14:2549–2558. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li JJ and Xie D: RACK1, a versatile hub in
cancer. Oncogene. 34:1890–1898. 2015. View Article : Google Scholar
|
|
14
|
Link AJ, Eng J, Schieltz DM, Carmack E,
Mize GJ, Morris DR, Garvik BM and Yates JR III: Direct analysis of
protein complexes using mass spectrometry. Nat Biotechnol.
17:676–682. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ruan Y, Sun L, Hao Y, Wang L, Xu J, Zhang
W, Xie J, Guo L, Zhou L, Yun X, et al: Ribosomal RACK1 promotes
chemoresistance and growth in human hepatocellular carcinoma. J
Clin Invest. 122:2554–2566. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shi S, Deng YZ, Zhao JS, Ji XD, Shi J,
Feng YX, Li G, Li JJ, Zhu D, Koeffler HP, et al: RACK1 promotes
non-small-cell lung cancer tumorigenicity through activating sonic
hedgehog signaling pathway. J Biol Chem. 287:7845–7858. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu J, Meng J, Du Y, Huang Y, Jin Y, Zhang
J, Wang B, Zhang Y, Sun M and Tang J: RACK1 promotes the
proliferation, migration and invasion capacity of mouse
hepatocellular carcinoma cell line in vitro probably by PI3K/Rac1
signaling pathway. Biomed Pharmacother. 67:313–319. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Deng YZ, Yao F, Li JJ, Mao ZF, Hu PT, Long
LY, Li G, Ji XD, Shi S, Guan DX, et al: RACK1 suppresses gastric
tumorigenesis by stabilizing the β-catenin destruction complex.
Gastroenterology. 142:812–823.e15. 2012. View Article : Google Scholar
|
|
19
|
Li Y, Peart MJ and Prives C: Stxbp4
regulates DeltaNp63 stability by suppression of RACK1-dependent
degradation. Mol Cell Biol. 29:3953–3963. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sutton P, Borgia JA, Bonomi P and Plate
JM: Lyn, a Src family kinase, regulates activation of epidermal
growth factor receptors in lung adenocarcinoma cells. Mol Cancer.
12:762013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang D, Wang Q, Zhu T, Cao J, Zhang X,
Wang J, Wang X, Li Y, Shen B and Zhang J: RACK1 promotes the
proliferation of THP1 acute myeloid leukemia cells. Mol Cell
Biochem. 384:197–202. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Harada H, Quearry B, Ruiz-Vela A and
Korsmeyer SJ: Survival factor-induced extracellular
signal-regulated kinase phosphorylates BIM, inhibiting its
association with BAX and proapoptotic activity. Proc Natl Acad Sci
USA. 101:15313–15317. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang W, Cheng GZ, Gong J, Hermanto U,
Zong CS, Chan J, Cheng JQ and Wang LH: RACK1 and CIS mediate the
degradation of BimEL in cancer cells. J Biol Chem. 283:16416–16426.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Subauste MC, Ventura-Holman T, Du L,
Subauste JS, Chan SL, Yu VC and Maher JF: RACK1 downregulates
levels of the pro-apoptotic protein Fem1b in apoptosis-resistant
colon cancer cells. Cancer Biol Ther. 8:2297–2305. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sang N, Severino A, Russo P, Baldi A,
Giordano A, Mileo AM, Paggi MG and De Luca A: RACK1 interacts with
E1A and rescues E1A-induced yeast growth inhibition and mammalian
cell apoptosis. J Biol Chem. 276:27026–27033. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mamidipudi V and Cartwright CA: A novel
pro-apoptotic function of RACK1: Suppression of Src activity in the
intrinsic and Akt pathways. Oncogene. 28:4421–4433. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nagashio R, Sato Y, Matsumoto T, Kageyama
T, Satoh Y, Shinichiro R, Masuda N, Goshima N, Jiang SX and Okayasu
I: Expression of RACK1 is a novel biomarker in pulmonary
adeno-carcinomas. Lung Cancer. 69:54–59. 2010. View Article : Google Scholar
|
|
28
|
Zhong X, Li M, Nie B, Wu F, Zhang L, Wang
E and Han Y: Overexpressions of RACK1 and CD147 associated with
poor prognosis in stage T1 pulmonary adenocarcinoma. Ann Surg
Oncol. 20:1044–1052. 2013. View Article : Google Scholar
|
|
29
|
Hu F, Tao Z, Wang M, Li G, Zhang Y, Zhong
H, Xiao H, Xie X and Ju M: RACK1 promoted the growth and migration
of the cancer cells in the progression of esophageal squamous cell
carcinoma. Tumour Biol. 34:3893–3899. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang Z, Jiang L, Huang C, Li Z, Chen L,
Gou L, Chen P, Tong A, Tang M, Gao F, et al: Comparative proteomics
approach to screening of potential diagnostic and therapeutic
targets for oral squamous cell carcinoma. Mol Cell Proteomics.
7:1639–1650. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Z, Zhang B, Jiang L, Zeng X, Chen Y,
Feng X, Guo Y and Chen Q: RACK1, an excellent predictor for poor
clinical outcome in oral squamous carcinoma, similar to Ki67. Eur J
Cancer. 45:490–496. 2009. View Article : Google Scholar
|
|
32
|
Zhou Y, Zhu X, Lu R, Dan H, Wang F, Wang
J, Li J, Feng X, Wang H, Ji N, et al: Vesicular stomatitis virus
matrix protein (VSVMP) inhibits the cell growth and tumor
angiogenesis in oral squamous cell carcinoma. Oral Oncol.
48:110–116. 2012. View Article : Google Scholar
|
|
33
|
Jiang L, Zeng X, Wang Z, Ji N, Zhou Y, Liu
X and Chen Q: Oral cancer overexpressed 1 (ORAOV1) regulates cell
cycle and apoptosis in cervical cancer HeLa cells. Mol Cancer.
9:202010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li J, Feng X, Sun C, Zeng X, Xie L, Xu H,
Li T, Wang R, Xu X, Zhou X, et al: Associations between proteasomal
activator PA28γ and outcome of oral squamous cell carcinoma:
Evidence from cohort studies and functional analyses. EBio Med.
2:849–856. 2015.
|
|
35
|
Li Y, Wang J, Wang F, Wang H, Wang J, Zeng
X, Liao G, Dan H and Chen Q: Tissue microarray analysis reveals the
expression and prognostic significance of phosphorylated
AktThr308 in oral squamous cell carcinoma. Oral Surg
Oral Med Oral Pathol Oral Radiol. 116:591–597. 2013. View Article : Google Scholar
|
|
36
|
Wang Y, Lin L, Xu H, Li T, Zhou Y, Dan H,
Jiang L, Liao G, Zhou M, Li L, et al: Genetic variants in AKT1 gene
were associated with risk and survival of OSCC in Chinese Han
population. J Oral Pathol Med. 44:45–50. 2015. View Article : Google Scholar
|
|
37
|
Peng R, Jiang B, Ma J, Ma Z, Wan X, Liu H,
Chen Z, Cheng Q and Chen R: Forced downregulation of RACK1 inhibits
glioma development by suppressing Src/Akt signaling activity. Oncol
Rep. 30:2195–2202. 2013.PubMed/NCBI
|
|
38
|
Kapoor V, Zaharieva MM, Das SN and Berger
MR: Erufosine simultaneously induces apoptosis and autophagy by
modulating the Akt-mTOR signaling pathway in oral squamous cell
carcinoma. Cancer Lett. 319:39–48. 2012. View Article : Google Scholar
|
|
39
|
Patel V, Marsh CA, Dorsam RT, Mikelis CM,
Masedunskas A, Amornphimoltham P, Nathan CA, Singh B, Weigert R,
Molinolo AA, et al: Decreased lymphangiogenesis and lymph node
metastasis by mTOR inhibition in head and neck cancer. Cancer Res.
71:7103–7112. 2011. View Article : Google Scholar
|
|
40
|
Wang Z, Martin D, Molinolo AA, Patel V,
Iglesias-Bartolome R, Degese MS, Vitale-Cross L, Chen Q and Gutkind
JS: mTOR co-targeting in cetuximab resistance in head and neck
cancers harboring PIK3CA and RAS mutations. J Natl Cancer Inst.
106:1062014. View Article : Google Scholar
|
|
41
|
Thomas SM and Grandis JR: The current
state of head and neck cancer gene therapy. Hum Gene Ther.
20:1565–1575. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu S, Xu X, Zeng X, Li L, Chen Q and Li
J: Tumor-targeting bacterial therapy: A potential treatment for
oral cancer (Review). Oncol Lett. 8:2359–2366. 2014.PubMed/NCBI
|
|
43
|
Li Y, Li LJ, Zhang ST, Wang LJ, Zhang Z,
Gao N, Zhang YY and Chen QM: In vitro and clinical studies of gene
therapy with recombinant human adenovirus-p53 injection for oral
leukoplakia. Clin Cancer Res. 15:6724–6731. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang S, Li Y, Li L, Zhang Y, Gao N, Zhang
Z and Zhao H: Phase I study of repeated intraepithelial delivery of
adenoviral p53 in patients with dysplastic oral leukoplakia. J Oral
Maxillofac Surg. 67:1074–1082. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Emamian ES, Hall D, Birnbaum MJ,
Karayiorgou M and Gogos JA: Convergent evidence for impaired
AKT1-GSK3beta signaling in schizophrenia. Nat Genet. 36:131–137.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
46
|
George S, Rochford JJ, Wolfrum C, Gray SL,
Schinner S, Wilson JC, Soos MA, Murgatroyd PR, Williams RM, Acerini
CL, et al: A family with severe insulin resistance and diabetes due
to a mutation in AKT2. Science. 304:1325–1328. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Altomare DA and Testa JR: Perturbations of
the AKT signaling pathway in human cancer. Oncogene. 24:7455–7464.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Franke TF: PI3K/Akt: Getting it right
matters. Oncogene. 27:6473–6488. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Molinolo AA, Amornphimoltham P, Squarize
CH, Castilho RM, Patel V and Gutkind JS: Dysregulated molecular
networks in head and neck carcinogenesis. Oral Oncol. 45:324–334.
2009. View Article : Google Scholar :
|
|
50
|
Khan KH, Yap TA, Yan L and Cunningham D:
Targeting the PI3K-AKT-mTOR signaling network in cancer. Chin J
Cancer. 32:253–265. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Prodromidis G, Nikitakis NG and Sklavounou
A: Immunohistochemical Analysis of the Activation Status of the
Akt/ mTOR/pS6 Signaling Pathway in Oral Lichen Planus. Int J Dent.
2013:7434562013. View Article : Google Scholar
|