Introduction
In a previous study, we performed a genome-wide
linkage analysis of 24 core family members in a rare multi-cancer
family. The family included 103 members spanning six generations,
and 15 members had been diagnosed with various tumor types,
including colorectal cancer (CRC), breast cancer, endometrial
carcinoma and gastric cancer. The disease susceptibility locus in
this family was mapped to chromosome 3q24-26. A novel mutant in the
TSC22D2 gene located in chromosome band 3q24-26 co-segregated with
the cancer phenotype, as demonstrated by exome sequencing (1).
TSC22D2 is a member of the TSC-22 domain family
comprising putative transcription factors that are characterized by
a carboxy-terminal leucine zipper and an adjacent TSC-box. However,
there are few studies describing TSC22D2, and its role in tumor
development remains largely unknown.
Accumulating evidence shows that many TSC22D2 family
proteins interact with other proteins to form macromolecular
complexes and are involved in a broad range of biological
processes. For instance, TSC22D1 (referred as TSC-22) can suppress
tumor growth by binding to p53 (2)
and can enhance TGF-β signaling by associating with Smad4 (3); TSC22D3 (usually called GILZ) plays
multiple biological functions dependent on its interaction with
other proteins, including NF-κB, Ras and p53 (4–6).
Accordingly, the identification of the TSC22D2-associated proteins
or macromolecular complexes is important to precisely understand
the underlying mechanism of TSC22D2 in human carcinogenesis.
In this study, we investigated the role of TSC22D2
in colorectal cancer, the most common malignancy in the
multi-cancer family. We found TSC22D2 was significantly
downregulated in CRC. Further functional study showed that TSC22D2
overexpression can inhibit CRC cell growth. To precisely define the
underlying mechanism by which TSC22D2 influenced CRC cell growth,
we used co-immunoprecipitation (co-IP) and mass spectrometry
approaches to identify the proteins or macromolecular complexes
that interact with TSC22D2, and gained 142 candidate
TSC22D2-binding proteins that were associated with a variety of
cellular processes. Moreover, we determined that TSC22D2 physically
associates with pyruvate kinase isoform M2 (PKM2), a glycolytic
enzyme reported to be associated with the growth and survival of
multiple cancer cell types (7–9), and
demonstrated that TSC22D2 overexpression reduces nuclear PKM2
levels and represses the expression of cyclin D1 (a downstream
target gene of nuclear PKM2).
Materials and methods
CRC samples
Surgical cancer tissue specimens and adjacent normal
mucosa (≥5 cm away from the tumor margins) were obtained from 14
patients with colorectal cancer who had undergone surgery at the
Third Xiangya Hospital of Central South University. Informed
written consent was obtained from each patient, and the research
protocols were approved by the Medical Ethics Committee of Xiangya
Medical College. No patients had received preoperative adjuvant
therapy. After collection, all tissue samples were immediately
frozen in liquid nitrogen until use.
Plasmid construction
The full length TSC22D2 gene was cloned into the
pIRESneo3-Flag vector with NheI and StuI (Takara,
Dalian, China) sites. The TSC22D2 gene was amplified using the
following primers: TSC22D2 sense primer (5′-ACG
TGCTAGCGCCACCATGTCCAAGATGCCGGCCAA-3′), TSC22D2 anti-sense primer
(5′-ACTGAGGCCTTTATGCTG AGGAGACATTCG-3′). Full-length PKM2 was
generously provided by Professor Xianghuo He (Shanghai Medical
College, Fudan University, Shanghai, China) and has been previously
described (10).
Cell lines and cell culture
The SW480 (human colorectal carcinoma) and HEK293
(human embryonic kidney) cell lines were cultured in Dulbecco’s
modified Eagle’s medium (Life Technologies, Grand Island, NY, USA)
with 10% fetal bovine serum (FBS) supplemented with 100 U/ml
penicillin and 100 mg/ml streptomycin. The HeLa human cervical
carcinoma cells were grown in 1640 medium (Life Technologies)
supplemented with 10% FBS, 100 U/ml penicillin and 100 mg/ml
streptomycin.
Cells were transfected using the Lipofectamine 3000
reagent (Life Technologies) according to the manufacturer’s
instructions. HEK293, SW480 and HeLa cells stably expressing
Flag-tagged TSC22D2 (Flag-TSC22D2) or the control vector (Flag-NC)
were obtained by puromycin (600–1,000 ng/ml) (Life Technologies)
selection for one month. All cells were maintained under standard
culture conditions (37°C, 5% CO2).
RNA isolation and quantitative real-time
PCR
Total RNAs were isolated using the TRIzol reagent
(Invitrogen, Carlsbad, CA, USA), and were converted to cDNA using
the GoScript™ Reverse Transcription System (Promega, Madison, WI,
USA) according to the manufacturer’s instructions. All real-time
PCR reactions were performed with SYBR Green (Takara) using the
Bio-Rad CFX Connect Real-Time system (Bio-Rad, Hercules, CA, USA).
The following PCR program was used: denaturation at 95°C for 30
sec, followed by 40 cycles consisting of denaturation at 95°C for 5
sec, annealing at 60°C for 30 sec, and extension at 72°C for 30
sec. A melting curve analysis was applied to assess the specificity
of the amplified PCR products. The amount of each target gene was
quantified by the comparative CT method using GAPDH as
the normalization control. The following primers were synthesized
from Life Technologies and used to amplify TSC22D2, CCND1 and
GAPDH: TSC22D2 forward primer (5′-TGAT GGTGATGAAGACAGTGC-3′),
TSC22D2 reverse primer (5′-GGGTTGGGAGTTGGGATAAT-3′); CCND1 forward
primer (5′-CAACCTCCTCAACGACC-3′), CCND1 reverse primer
(5′-CTTCTGTTCCTCGCAGAC-3′); GAPDH forward primer
(5′-AACGGATTTGGTCGTATTGG-3′), GAPDH reverse primer
(5′-TTGATTTTGGAGGGATCTCG-3′). All experiments were carried out in
triplicate.
Total protein, subcellular fractionation
and western blot analysis
Nuclear and cytosolic extracts were fractionated
using the NE-PER nuclear and cytoplasmic extraction kit (Thermo
Scientific, Bremen, Germany) according to the manufacturer’s
protocol. To isolate total protein, cells were harvested by
scraping and transferred to SDS sample buffer supplemented with a
protease inhibitor cocktail and the PhosSTOP phosphatase inhibitor
(Roche, Pleasanton, CA, USA). Equal levels of protein from the
samples were separated using SDS-PAGE gel electrophoresis and
transferred to a PVDF membrane (Millipore Corp., Billerica, MA,
USA). The membrane was blocked with PBST containing 5% skim milk
for 1 h and then incubated overnight with the indicated primary
antibodies at 4°C. The membrane was washed three times in PBST and
subsequently incubated with an HRP-conjugated secondary antibody
for 2 h at 37°C. The membranes were stripped of the primary
antibodies and re-probed with additional antibodies as necessary.
Bound antibodies were visualized using the enhanced
chemiluminescence kit (Millipore).
The following antibodies were used in this study:
monoclonal mouse anti-Flag (F1804, Sigma, St. Louis, MO, USA),
anti-HA (H3663, Sigma), polyclonal rabbit anti-human TSC22D2
(AV39137, Sigma), polyclonal rabbit anti-human PKM2 (Cell
Signaling, Danvers, MA, USA) and anti-CCND1 (Cell Signaling). An
antibody against GAPDH (Cell Signaling) served as an endogenous
control for equal loading, and anti-H3 (Beyotime, China) served as
a nuclear control.
Cell proliferation assays
Cell proliferation was measured by counting viable
cells using the Z2 Particle Counter and Size Analyzer Beckman
Coulter (Miami, FL, USA). Briefly, SW480 and HeLa cells stably
expressing Flag-tagged TSC22D2 (Flag-TSC22D2) or the control vector
(Flag-NC) were seeded at 1.2×104 cells per well in
24-well plates in quadruplicate, the number of viable cells in each
well was measured at 0, 1, 2, 3, 4 and 5 days.
Colony formation assays
For the colony formation assays, SW480 and HeLa
cells were seeded in a 6-well plate at a density of 500 and 200
cells per well respectively, and incubated at 37°C with 5%
CO2 for 2 weeks. The medium was changed every 3–4 days.
At the end of the incubation, the medium was removed, and the cells
were washed twice with PBS, fixed with 4% paraformaldehyde for 20
min, stained with crystal violet for 30 min at room temperature,
washed and imaged. Colonies of >50 cells were identified using
an inverted microscope (Olympus IX50; Olympus Corp., Tokyo, Japan)
and counted.
Flow cytometry cell cycle assays
SW480 and HeLa stable cells were plated at a density
of 4×105 cells per well in 6-well plates and cultured in
medium without serum (starvation treatment) for 12 h to synchronize
cell cycle progression. Cells were incubated in serum-containing
growth medium for an additional 24–36 h and subsequently
trypsinized, washed, fixed with 70% ice cold ethanol overnight at
4°C. DNA was stained by incubating the cells in PBS containing PI
(50 μg/ml) and RNase A (50 μg/ml). The cell cycle distribution was
determined by flow cytometric analysis using a MoFlo™ XDP
High-Performance Cell Sorter (Beckman Coulter, Brea, CA, USA) and
the data were analyzed using Summit v.5.2 software.
Co-immunoprecipitation (co-IP)
assays
HEK293 Flag-NC, HEK293 Flag-TSC22D2, SW480 Flag-NC,
SW480 Flag-TSC22D2 stably transfected cells and HEK293T cells
transiently cotransfected with Flag-TSC22D2 and HA-PKM2 were seeded
in 100-mm dishes. The cells were lysed in modified lysis buffer (50
mM Tris-HCl pH 7.5, 100 mM NaCI, 50 mM NaF, 1 mM
Na3VO4, 30 mM sodium pyrophosphate, 0.5%
NP-40 and 0.5 mM PMSF (Sigma) supplemented with an EDTA-free
protease inhibitor cocktail (Roche). Lysates were incubated with 5
μg of primary antibody overnight at 4°C. Then, 40 μl of a 1:1
slurry of protein A/G Plus-agarose (Santa Cruz, CA, USA) and a
specific antibody were added to the cells for ≥4 h at 4°C. The
immunoprecipitates were washed four times with lysis buffer and
boiled with sample loading buffer and then analyzed by western
blotting.
Mass spectrometry
Proteins were resolved on a 10% polyacrylamide gel,
stained with Coomassie Brilliant Blue (R250) and subjected to mass
spectrometry. Briefly, the stained SDS-PAGE gels were scanned, and
stained bands were excised, cut into small (<1 mm3)
pieces and washed three times by repetitive dehydration and
hydration with acetonitrile and ammonium bicarbonate, respectively.
Proteins were in-gel reduced in the presence of 25 mM DTT and 25 mM
NH4HCO3 for 1 h at 56°C, immediately
alkylated using 50 mM IAA and 25 mM NH4HCO3
for 30 min at room temperature in the dark and digested overnight
at 37°C with 5 μg of trypsin in 25 mM
NH4HCO3. Digested peptides were recovered,
dried and resuspended in 50% CAN and 0.1% TFA. The peptide mixture
was analyzed by nano-liquid chromatography-tandem mass spectrometry
using an LTQ Velos-Orbitrap MS (Thermo Scientific, Waltham, MA,
USA) coupled to an Ultimate RSLC nano-LC system (Dionex). Briefly,
Raw MS data files were processed using the Proteome Discoverer
v.1.4 (Thermo Scientific). Processed files were searched against
the Swiss-Prot human database using the Sequest HT search engine.
Mass tolerances for precursor and fragment ions were set to 10 ppm
and 0.8 Da, respectively, in the database searches. Peptide
identification with false discovery rates <1% (q-value <0.01)
were discarded.
Immunofluorescence
TSC22D2, HEK293, SW480 and HeLa cells grown on
culture slides were maintained in a 35-mm dish. After cultured for
24 h, the cells were washed with phosphate-buffered saline (PBS),
fixed with 4% paraformaldehyde in PBS for 10 min, permeabilized
with 0.25% Triton X-100 for 30 min and blocked in 5% bovine serum
albumin (BSA) for 1 h at room temperature. Fixed cells were then
incubated with rabbit anti-human TSC22D2 antibody (1:1,000) at 4°C
overnight, washed three times in PBS and stained with Alexa Fluor
488 donkey anti-rabbit IgG (Invitrogen, 1:1,000) for 2 h in the
dark at room temperature. The cells were then incubated with DAPI
to stain the nuclei and visualized using a confocal laser scanning
microscope (Olympus Corp.).
For the colocalization assays, HEK293T transfected
with Flag-TSC22D2 and HA-PKM2 were incubated with the anti-Flag
mouse monoclonal antibody (1:1,000) and the anti-PKM2 rabbit
monoclonal antibody (1:1,000) at 4°C overnight and subsequently
stained with Alexa Fluor 488 donkey anti-mouse IgG (Invitrogen,
1:1,000) and Alexa Fluor 594 donkey anti-rabbit IgG (Invitrogen,
1:1,000) for 2 h at room temperature. After incubation with DAPI to
stain the nuclei, the cells were visualized using a confocal laser
scanning microscope (UltraView Vox; Perkin-Elmer, Waltham, MA,
USA).
Statistical analysis
The experiments were repeated at least three times.
All data are presented as the mean ± standard deviation. Paired
Student’s t-test (two-tailed) and unpaired Student’s t-test
(two-tailed) were performed to compare the means of two samples
unless otherwise indicated. A p-value <0.05 was considered
statistically significant. All statistical analyses were performed
using the GraphPad Prism software (Graphpad Software, Inc.).
Results
Expression of TSC22D2 is decreased in
colorectal cancer samples
We focused on human colorectal cancer in this study
because it was the most frequent malignancy in the multi-cancer
family. We first evaluated the expression of TSC22D2 transcripts in
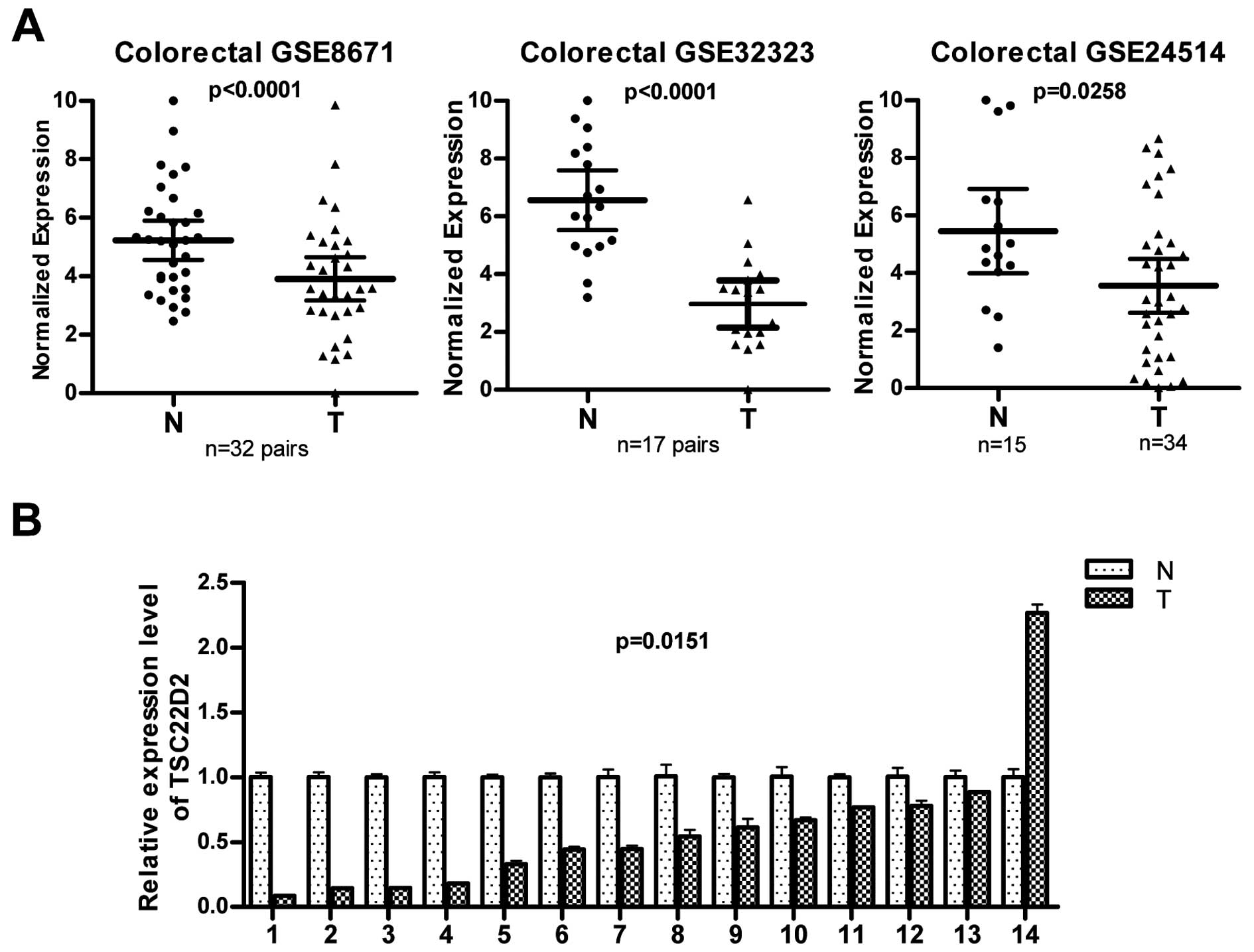
colorectal cancer by interrogating the public gene expression GEO
databases (GSE8671, GSE32323 and GSE24514) and found that the
expression of TSC22D2 was lower in human colorectal cancer samples
than in non-tumor samples (Fig.
1A). To confirm these observations, the expression of TSC22D2
was evaluated in 14 pairs of human CRC samples (primary tumor
tissues and paired adjacent non-tumor tissues). TSC22D2 expression
was reduced in 13 of 14 (92.9%) of colorectal tumors compared with
the paired adjacent non-tumor tissues (Fig. 1B, p=0.015), consistent with the
data provided by the GEO database. These results suggest that
TSC22D2 plays a suppressor role in CRC tumorigenesis.
TSC22D2 suppresses CRC cell growth
To investigate the function of TSC22D2 in colorectal
cancer, the full-length TSC22D2 gene tagged with three Flag tags
was cloned into the pIRESneo3 plasmid and stably transfected into
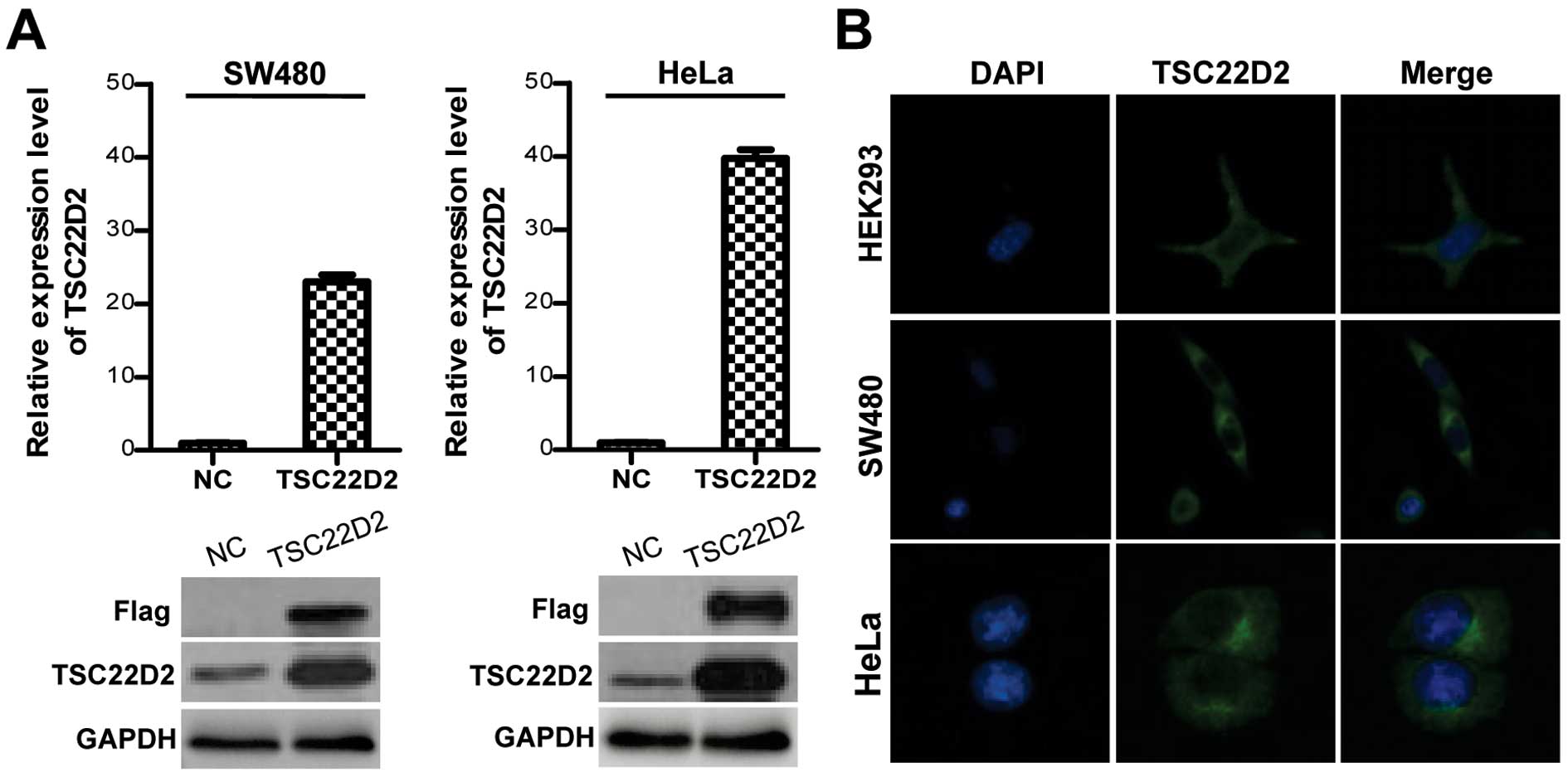
SW480 and HeLa cells. The expression of TSC22D2 was confirmed by
qRT-PCR and western blot analysis (Fig. 2A). Immunofluorescence assays
demonstrated that TSC22D2 predominantly localized to the cytoplasm
under steady state conditions (Fig.
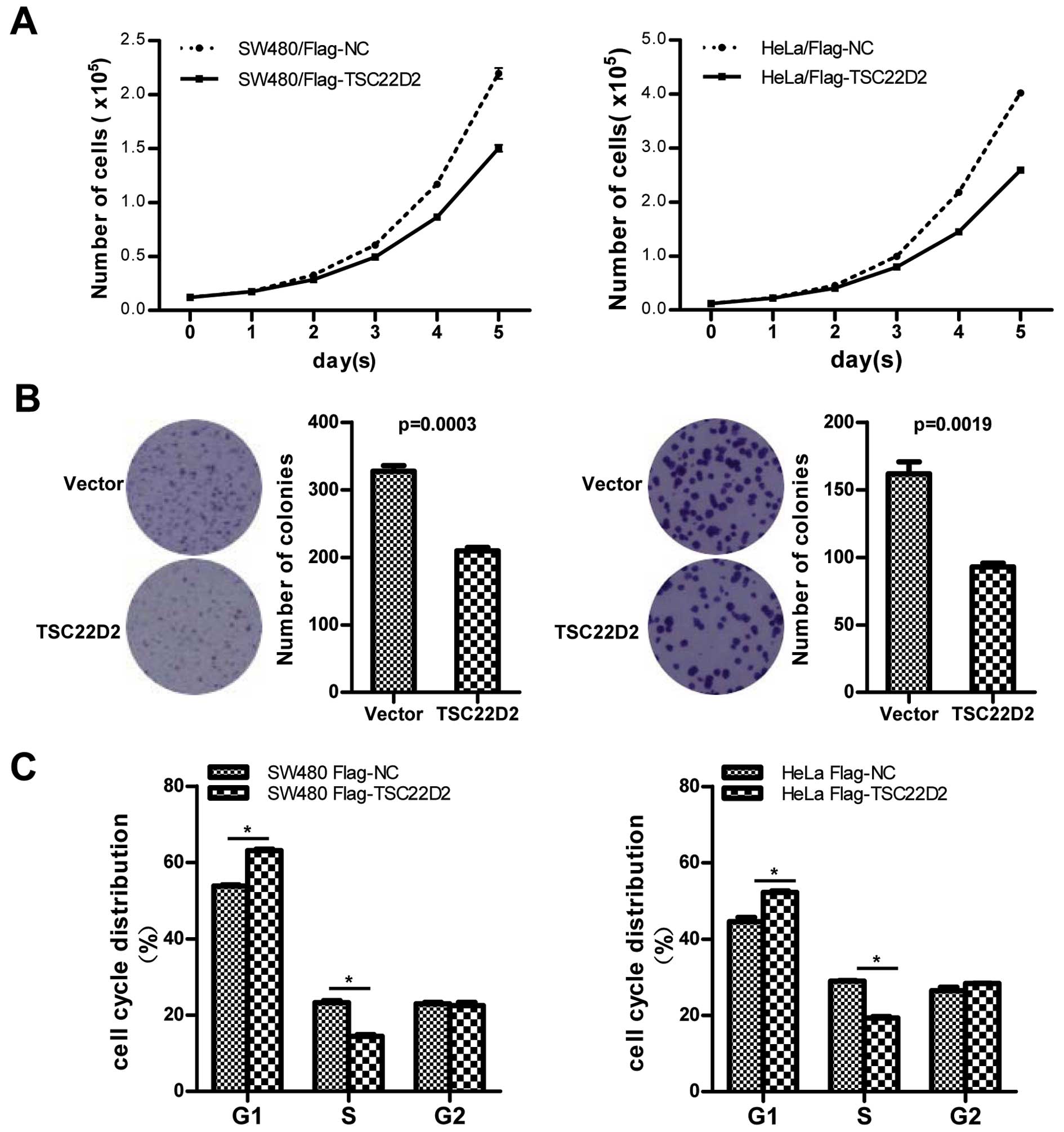
2B). Cell proliferation assays and clone formation assays were
conducted to investigate the effect of TSC22D2 on cell
proliferation. TSC22D2-overexpressing cells exhibited a
significantly slower growth rate (Fig.
3A) and a reduced number of clones (Fig. 3B) compared with control cells,
indicating that TSC22D2 functions as a suppressor in CRC cell
growth. Furthermore, flow cytometry (FCM) revealed that TSC22D2
induced a substantial increase in the proportion of cells at the
G0/G1 phase and a concomitant decrease in the proportion of cells
at the S phase of the cell cycle (Fig.
3C). These results indicate that TSC22D2 induces cell cycle
arrest at the G0/G1 phase.
Identification of TSC22D2 binding
partners
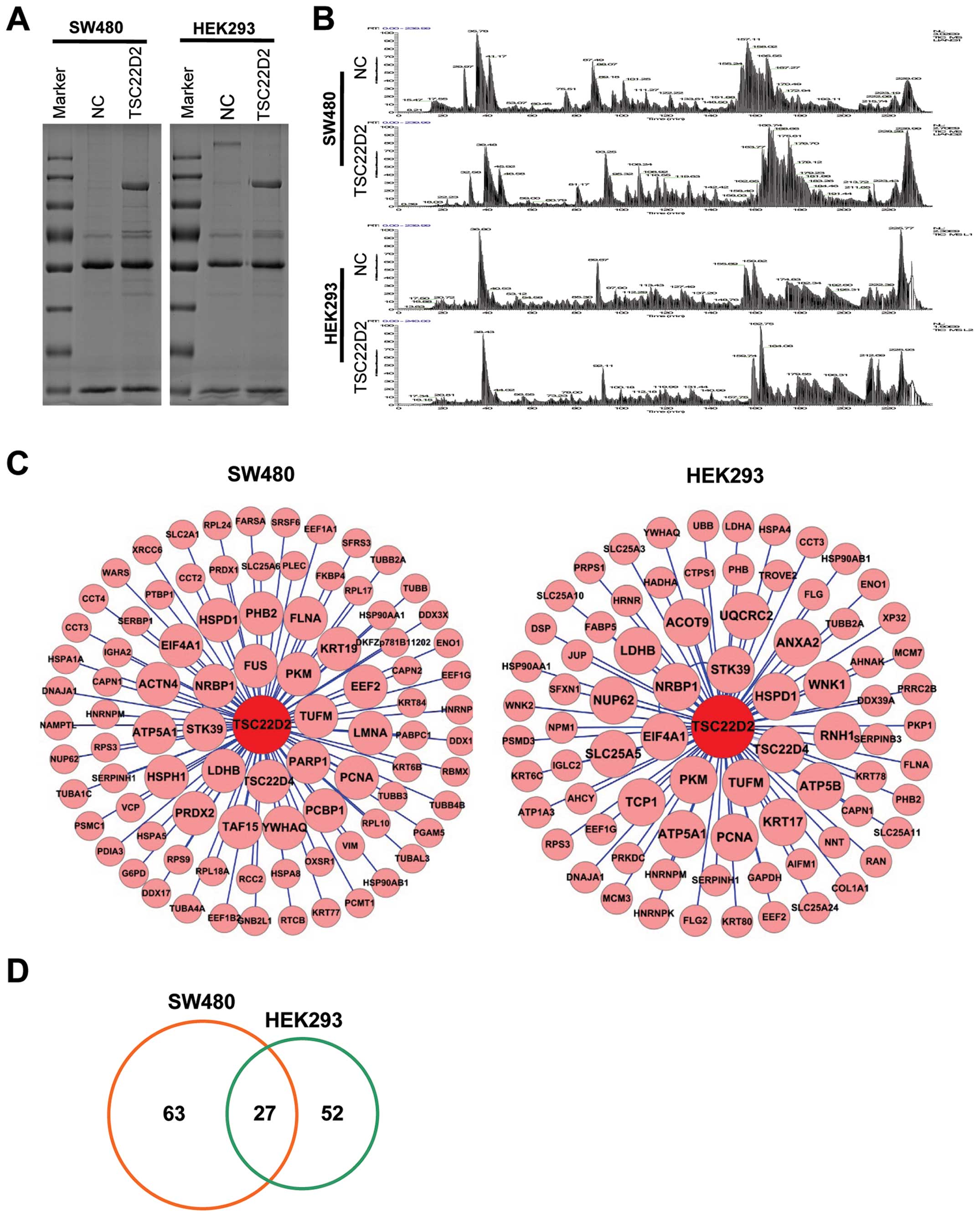
To explore the underlying mechanism by which TSC22D2
exerts its tumor-suppressive function, we performed co-IP assays
(Fig. 4A) and LC-MS/MS analysis
(Fig. 4B) in SW480 and HEK293
cells stably transfected with TSC22D2. We identified 90 and 79
candidate proteins in SW480 and HEK293 samples, respectively
(Fig. 4C). There were 27
overlapping results for proteins associated with metabolism (PKM2,
ATP5A1, LDH and ENO1), stress response and inflammation (STK39,
HSPD1, HSP90AA1 and HSP90AB1), cell proliferation (NRBP1, PKM2,
YWHAQ, LDHB, PCNA and PHB2), apoptosis (HSPD1, CAPN1, DNAJA1,
HSP90AA1 and RPS3) and migration/metastasis (YWHAQ, HSPD1 and PHB2)
between the 2 cell lines (Fig. 4D
and Table I). These findings
suggest that TSC22D2 plays a role in these biological pathways in
cancer cells.
 | Table ITSC22D2-interacting proteins
identified in both SW480 and HEK293 cells. |
Table I
TSC22D2-interacting proteins
identified in both SW480 and HEK293 cells.
| Gene symbol | Description | Confirmed role in
cancer |
|---|
| TSC22D2 | Fesponse to osmotic
stress | |
| NRBP1 | Subcellular
trafficking between the endoplasmic reticulum and Golgi
apparatus | Potentially plays a
suppressive role in tumor progression |
| TSC22D4 | Sequence-specific
DNA binding transcription factor activity | |
| TUFM | Protein translation
in mitochondria and oxidative phosphorylation; regulates type I
interferon and autophagy | |
| PKM2 | Involved in
glycolysis; transcription factor activity | Important for tumor
cell proliferation and survival |
| ATP5A1 | A subunit of
mitochondrial ATP synthase; involved in energy metabolism | |
| STK39 | A serine/threonine
kinase that might function in the cellular stress response
pathway | |
| YWHAQ | Belongs to the
14-3-3 family of proteins, which mediate signal transduction by
binding to phosphoserine-containing proteins | Coordinates the
regulation of proliferation, survival and metastasis |
| HSPD1 | Putative signaling
molecule in the innate immune system; implicated in mitochondrial
protein import and macromolecular assembly | Regulates tumor
cell apoptosis, survival and metastasis |
| LDHB | Catalyzes the
interconversion of pyruvate and lactate and the concomitant
interconversion of NADH and NAD+ in a post-glycolysis
process | A metabolic marker
in cancer and is potentially associated with cell growth |
| PCNA | Cofactor of DNA
polymerase delta; DNA damage response | Important for
cancer cell proliferation |
| PHB2 | Transcriptional
repression; likely to be involved in regulating mitochondrial
respiration activity and aging; associated with the cell cycle | Required for cancer
cell proliferation, and potentially regulates cell migration |
| EIF4A1 | RNA helicase;
translation initiation factor activity; participates in the TGF-β
pathway and p70S6K signaling | |
| ENO1 | Functions as a
glycolytic enzyme and a transcriptional repressor | Promotes cell
proliferation and possesses oncogenic activity |
| FLNA | A
well-characterized actin cross-linking protein; anchors various
transmembrane proteins to the actin cytoskeleton and serves as a
scaffold for a wide range of cytoplasmic signaling proteins | Has a
tumor-promoting effect when localized to the cytoplasm, and
suppresses tumor growth and inhibits metastasis when localized to
the nucleus |
| CAPN1 | Catalyzes limited
proteolysis; involved in ERK signaling and apoptosis | Participates in
apoptosis |
| CCT3 | Molecular
chaperone; assists in the folding of proteins stimulated by ATP
hydrolysis | |
| DNAJA1 | Positive regulation
of viral replication; plays a role in protein transport into
mitochondria via its role as a co-chaperone; protects cells from
apoptosis | Suppresses the
anti-apoptosis state in cancer |
| EEF1G | Translation
elongation factor activity; likely to play a role in anchoring the
translation complex to other cellular components | |
| EEF2 | Promotes the
GTP-dependent translocation of the nascent protein chain from the
A-site to the P-site of the ribosome; an essential factor for
protein synthesis | Functions as an
oncogene in cancer cell growth |
| HNRNPM | Associated with
pre-mRNAs in the nucleus and appears to influence pre-mRNA
processing and other aspects of mRNA metabolism and transport | Regulates cell
cycle progression, promotes cell growth and invasion; promotes
TGFβ-induced EMT and metastasis |
HSP90AA1
HSP90AB1 | Mediates
LPS-induced inflammatory response; molecular chaperone that
promotes the maturation, structural maintenance and proper
regulation of specific target proteins involved in multiple
processes, including cell cycle control and signal
transduction | Is involved in cell
apoptosis and chemosensitivity
Methylated HSP90AB1 accelerates cancer cell proliferation |
| NUP62 | Forms a gateway
that regulates the flow of macromolecules between the nucleus and
the cytoplasm (involved in nuclear-cytoplasmic transport) | Nup62 knockdown
reduces cancer cell growth and viability |
| RPS3 | A component of the
40S small ribosomal subunit; plays a role in the repair of damaged
DNA | Regulates cell
growth, apoptosis and GLI2-mediated migration and invasion |
| SERPINH1 | A collagen-specific
molecular chaperone that plays a role in collagen biosynthesis | Enhances cell
growth, migration and invasion |
| TUBB2A | Key participant in
various processes, including mitosis and intracellular
transport | |
TSC22D2 physically associates with
PKM2
We selected PKM2, a glycolytic enzyme that catalyzes
the conversion of phosphoenopyruvate (PEP) and ADP to pyruvate and
ATP for further analysis due to its relatively high mass
spectrometry score and its previously reported role in cancer cell
growth and survival (7–9).
The interaction between PKM2 and TSC22D2 was further
confirmed by IP analysis of HEK293T cells transfected with
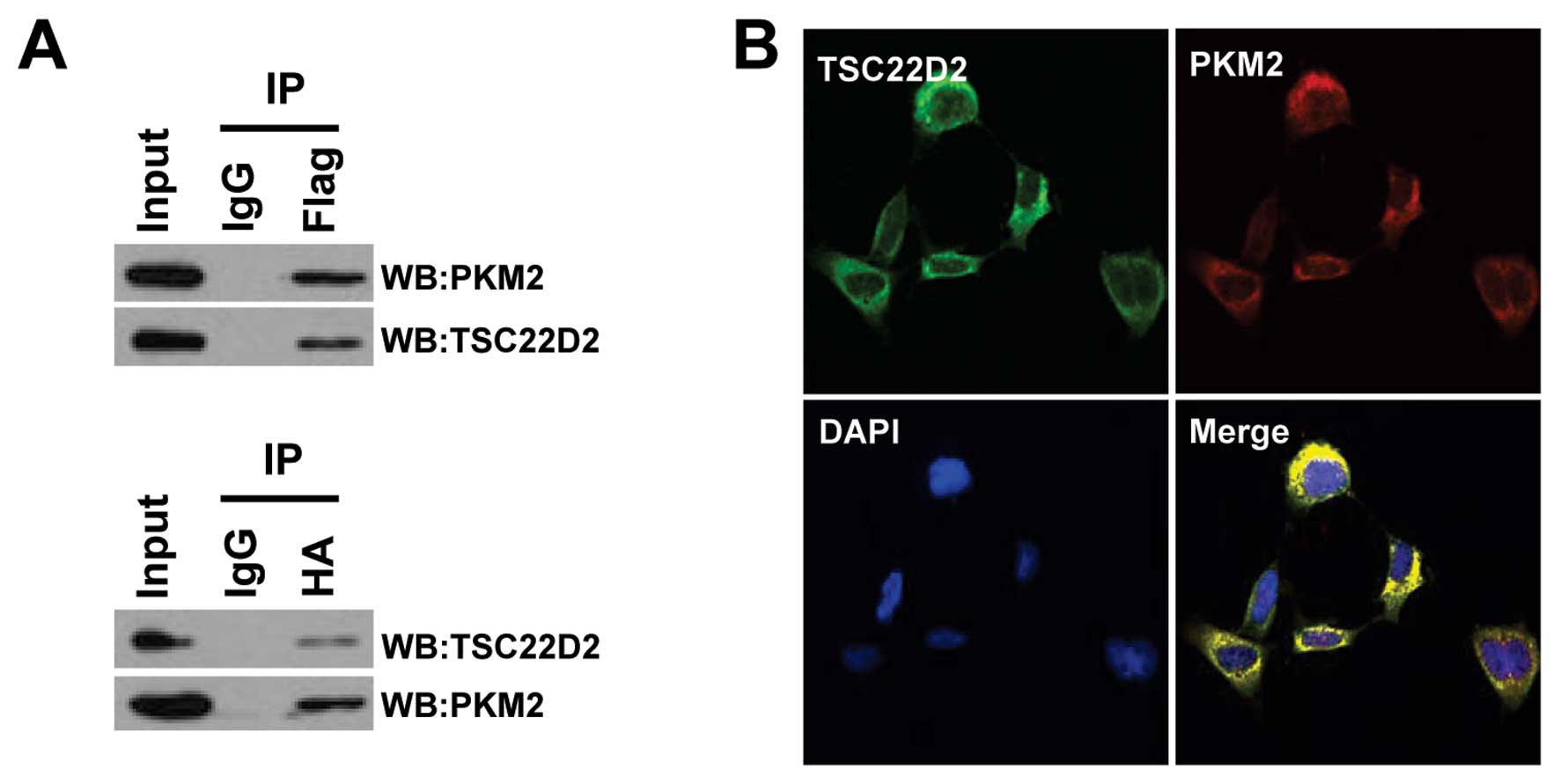
HA-tagged PKM2 and Flag-tagged TSC22D2 (Fig. 5A). To determine whether TSC22D2 and
PKM2 co-localize in cells, we performed immunofluorescence staining
with anti-Flag and anti-PKM2 antibodies in HEK293 cells. The
results revealed that TSC22D2 and PKM2 co-localized primarily in
the cytoplasm (Pearson’s correlation, 0.954) (Fig. 5B). Taken together, these data
suggest that TSC22D2 physically associates with PKM2.
TSC22D2 reduces the level of nuclear PKM2
and suppresses the expression of cyclin D1
Accumulating evidence indicates that PKM2 is crucial
for aerobic glycolysis and that it provides a growth advantage to
tumors (11,12). To determine if the effect of
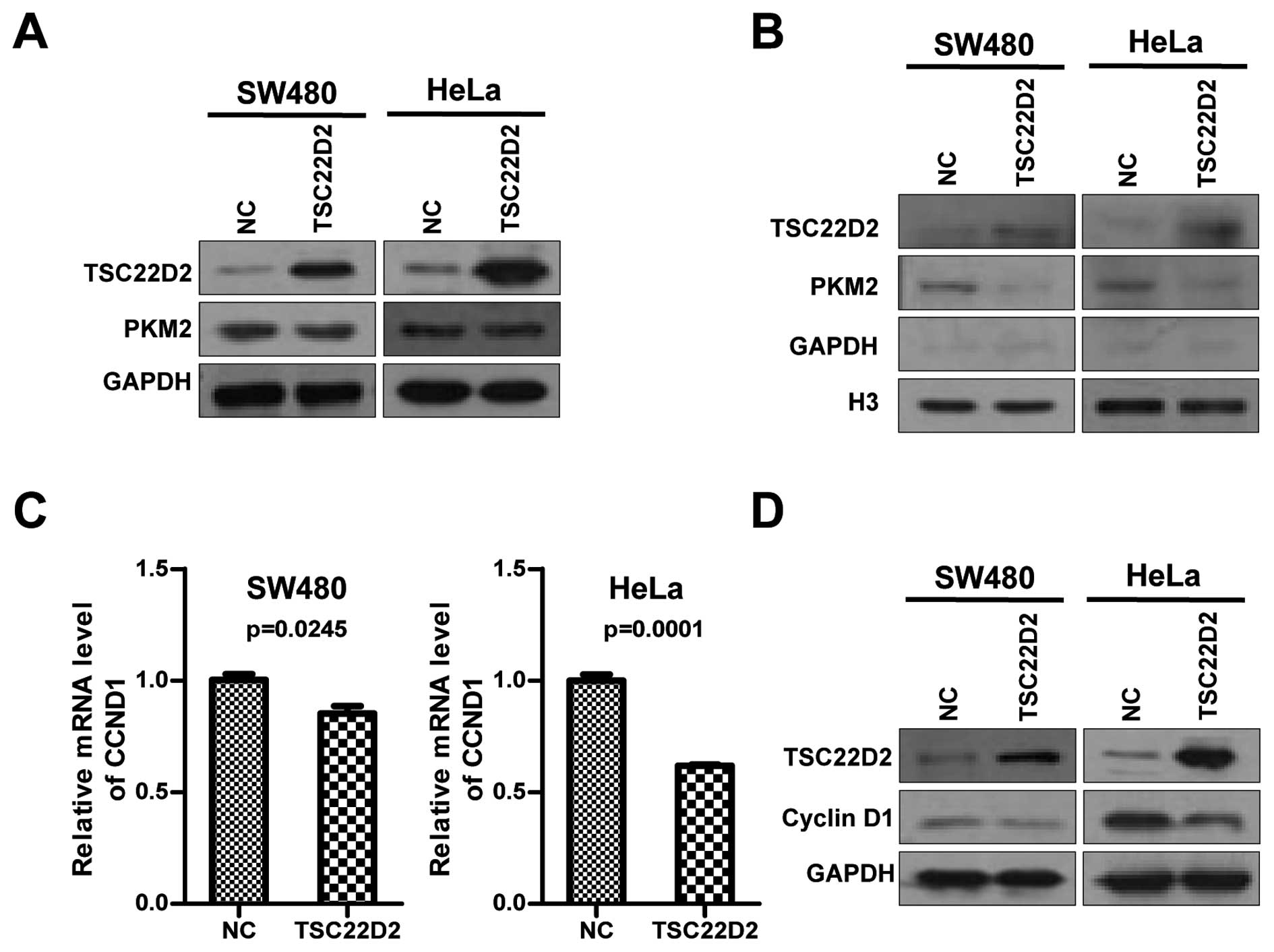
TSC22D2 on cell growth is mediated by PKM2, we evaluated the
expression of PKM2 and observed that there were no significant
changes in PKM2 expression at the mRNA and protein level in cells
overexpressing TSC22D2 (Fig.
6A).
PKM2 predominantly localizes to the cytosol and
plays an important role in metabolic functions. Recently, several
independent studies demonstrated that PKM2 can translocate to the
nucleus and induce the expression of gene products required for
tumorigenesis by activating multiple transcription factors
(13–18). Thus, we asked whether TSC22D2 could
affect the nuclear function of PKM2. A subcellular fractionation
analysis was performed, and a slight but significant decrease in
the nuclear levels of PKM2 was observed in TSC22D2-overexpressing
cells compared with the control cells (Fig. 6B).
Cyclin D1, a key regulator required for the G1/S
cell cycle transition, was reported to be a downstream gene of
nuclear PKM2 (15,16). Therefore, we next investigated the
effect of TSC22D2 on cyclin D1 expression. Intriguingly, TSC22D2
repressed the expression of cyclin D1 at both mRNA and protein
levels (Fig. 6C and D), suggesting
that TSC22D2 might inhibit cell growth by the influence on nuclear
PKM2 and cyclin D1.
Discussion
We previously identified a novel cancer-associated
gene, TSC22D2, by performing genome-wide linkage analysis and exome
sequencing in samples derived from a multi-cancer family. TSC22D2
is a member of the TSC-22 domain family of proteins that are
characterized by a carboxy-terminal leucine zipper and an adjacent
TSC-box. However, its role in tumorigenesis remains largely
unclear. In this study, we demonstrated that TSC22D2 is expressed
at low levels in CRC and that TSC22D2 overexpression significantly
inhibited the growth rate of cancer cells. Taken together, these
results suggest that TSC22D2 might play a suppressive role in
tumorigenesis.
Members of the TSC22 domain family are reported to
interact with other proteins to form macromolecular complexes
associated with a broad range of biological processes. TSC22D1
(also referred as TSC-22) binds to p53 and protects it from
poly-ubiquitination-mediated degradation (2), it can enhance TGF-β signaling by
associating with Smad4 (3).
TSC22D3 (commonly referred to as GILZ) exhibits multiple biological
functions that are dependent on its interaction with other
proteins, including NF-κB, Ras and p53 (4–6).
TSC22D4 is capable of heterodimerizing with apoptosis-inducing
factor (AIF) and might participate in apoptosis (19). However, proteins that interact with
TSC22D2 have not been identified.
In this study, we conducted co-IP assays combined
with LC-MS/MS approaches to identify TSC22D2-interacting proteins.
The results were enriched for proteins associated with the
regulation of gene transcription, suggesting that TSC22D2 plays a
role in transcription. TSC22 domain proteins have a classic leucine
zipper and a TSC-Box structure. The leucine zipper might be a
characteristic of a novel category of DNA binding proteins
(20–22) and the TSC-box is essential for
nuclear localization and transcriptional activity (23). Based on these observations, TSC22
domain proteins are considered to be putative transcription
factors. They primarily localize to the cytoplasm, but in response
to specific stimuli, they can also translocate to the nucleus.
Previous studies have demonstrated that TSC22D1 can translocate to
the nucleus and suppress cell division in response to
anti-proliferative stimuli (24,25).
TSC22D1 and TSC22D3 transcriptional activity has been previously
reported, and the transcriptional activity of TSC22D2 has yet to be
further confirmed.
Our analysis revealed that TSC22D2 interacts with
multiple proteins associated with metabolism (PKM2, ATP5A1, LDHB
and ENO1), the stress response and inflammation (STK39, HSPD1,
HSP90AA1 and HSP90AB1), cell proliferation (NRBP1, PKM2, YWHAQ,
LDHB, PCNA and PHB2), apoptosis (HSPD1, CAPN1, DNAJA1, HSP90AA1 and
RPS3) and migration/metastasis (YWHAQ, HSPD1 and PHB2) (Table I), implying that the function of
TSC22D2 in tumor cells might be mediated by these processes.
Moreover, the interaction between NRBP1 and TSC22D2 was
inadvertently previously reported (26). In addition, TSC22 domain proteins
have been shown to homodimerize and heterodimerize with other
family members (27). In this
study, western blotting and mass spectrometry analyses revealed
that TSC22D2 primarily exists as a dimer, but that it might also
heterodimerize with TSC22D4.
Given that TSC22D2 was capable of inhibiting the
growth of cancer cells, we focused on the PKM2 gene, which was
previously reported to be required for tumor growth (7–9).
Pyruvate kinase M2 (PKM2) is a pyruvate kinase that regulates the
final rate-limiting step of glycolysis by catalyzing the transfer
of a phosphate group from phosphoenolpyruvate (PEP) to ADP to
generate pyruvate and ATP. PKM2 exhibits active pyruvate kinase
activity as well as protein kinase activity that is primarily
associated with transcriptional regulation (reviewed in refs.
11,12). Elevated PKM2 expression is
currently considered to be a common characteristic of cancers.
PKM2 primarily localized to the cytoplasm. In
addition to its well characterized role in glycolysis, PKM2 can
translocate to the nucleus to stimulate the activity of multiple
transcription factors, including HIF-1, STAT3, β-catenin and Oct-4,
thereby increasing the expression of gene products that are
required for tumor growth (16–18,28).
For example, activation of EGFR in human glioblastoma cells induces
the nuclear translocation of PKM2. In addition, the interaction
between PKM2 and β-catenin transactivates factors that induce HDAC3
removal from the CCND1 promoter, histone H3 acetylation and cyclin
D1 expression (16). An increasing
body of evidence indicates that the nuclear translocation of PKM2
promotes the Warburg effect and tumorigenesis (13–18).
In this study, we found that TSC22D2 did not regulate the total
PKM2 protein levels, but that it decreased the level of nuclear
PKM2. Although some studies have suggested that PKM2-interacting
proteins impair PKM2 nuclear translocation by altering the balance
between its monomer, dimer and tetramer forms or by mediating
epigenetic modifications (14,29),
further studies are required to confirm the mechanism by which
TSC22D2 regulates the nuclear PKM2 level.
In conclusion, this is the first study to report the
tumor suppressor role of TSC22D2 in cancer and to identify putative
TSC22D2-interacting proteins using interactome analysis. The
candidate interactions that we identified provide important clues
that will facilitate future studies exploring TSC22D2 functions.
The interaction between TSC22D2 and PKM2 may partially account for
the growth suppressor function of TSC22D2. It is important to note
that further studies of TSC22D2 complexes are required for gaining
additional insight into the functions and precise mechanisms
associated with TSC22D2 in cancer.
Acknowledgements
We are grateful for the gifts of the HA-PKM2
plasmids from Professor Xianghuo He. We also would like to thank
the High Resolution Mass Spectrometry Laboratory of Advanced
Research Center in Central South University for the technological
assistance in proteomic examinations. This study was supported in
part by grants from The National Natural Science Foundation of
China (81272298, 81372907, 81301757, 81472531, 81402009, 81572787
and 81528019), the Natural Science Foundation of Hunan Province
(14JJ1010 and 2015JJ1022) and the project from Central South
University (2013zzts071).
Abbreviations:
|
TSC22D2
|
TSC22 domain family member 2
|
|
PKM2
|
pyruvate kinase isoform M2
|
|
CRC
|
colorectal cancer
|
References
|
1
|
Li Q, Chen P, Zeng Z, Liang F, Song Y,
Xiong F, Li X, Gong Z, Zhou M, Xiang B, et al: Yeast two-hybrid
screening identified WDR77 as a novel interacting partner of
TSC22D2. Tumor Biol. (In press).
|
|
2
|
Yoon CH, Rho SB, Kim ST, Kho S, Park J,
Jang IS, Woo S, Kim SS, Lee JH and Lee SH: Crucial role of TSC-22
in preventing the proteasomal degradation of p53 in cervical
cancer. PLoS One. 7:e420062012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Choi SJ, Moon JH, Ahn YW, Ahn JH, Kim DU
and Han TH: Tsc-22 enhances TGF-beta signaling by associating with
Smad4 and induces erythroid cell differentiation. Mol Cell Biochem.
271:23–28. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ayroldi E, Migliorati G, Bruscoli S,
Marchetti C, Zollo O, Cannarile L, D’Adamio F and Riccardi C:
Modulation of T-cell activation by the glucocorticoid-induced
leucine zipper factor via inhibition of nuclear factor kappaB.
Blood. 98:743–753. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ayroldi E, Petrillo MG, Bastianelli A,
Marchetti MC, Ronchetti S, Nocentini G, Ricciotti L, Cannarile L
and Riccardi C: L-GILZ binds p53 and MDM2 and suppresses tumor
growth through p53 activation in human cancer cells. Cell Death
Differ. 22:118–130. 2015. View Article : Google Scholar
|
|
6
|
Ayroldi E, Zollo O, Bastianelli A,
Marchetti C, Agostini M, Di Virgilio R and Riccardi C: GILZ
mediates the antiproliferative activity of glucocorticoids by
negative regulation of Ras signaling. J Clin Invest. 117:1605–1615.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Christofk HR, Vander Heiden MG, Harris MH,
Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL and
Cantley LC: The M2 splice isoform of pyruvate kinase is important
for cancer metabolism and tumour growth. Nature. 452:230–233. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Anastasiou D, Yu Y, Israelsen WJ, Jiang
JK, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A, et al:
Pyruvate kinase M2 activators promote tetramer formation and
suppress tumorigenesis. Nat Chem Biol. 8:839–847. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hitosugi T, Kang S, Vander Heiden MG,
Chung TW, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ, et
al: Tyrosine phosphorylation inhibits PKM2 to promote the Warburg
effect and tumor growth. Sci Signal. 2:ra732009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen Z, Wang Z, Guo W, Zhang Z, Zhao F,
Zhao Y, Jia D, Ding J, Wang H, Yao M, et al: TRIM35 interacts with
pyruvate kinase isoform M2 to suppress the Warburg effect and
tumorigenicity in hepatocellular carcinoma. Oncogene. 34:3946–3956.
2015. View Article : Google Scholar
|
|
11
|
Chaneton B and Gottlieb E: Rocking cell
metabolism: Revised functions of the key glycolytic regulator PKM2
in cancer. Trends Biochem Sci. 37:309–316. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Luo W and Semenza GL: Emerging roles of
PKM2 in cell metabolism and cancer progression. Trends Endocrinol
Metab. 23:560–566. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lv L, Xu YP, Zhao D, Li FL, Wang W, Sasaki
N, Jiang Y, Zhou X, Li TT, Guan KL, et al: Mitogenic and oncogenic
stimulation of K433 acetylation promotes PKM2 protein kinase
activity and nuclear localization. Mol Cell. 52:340–352. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo
F, Lyssiotis CA, Aldape K, Cantley LC and Lu Z: ERK1/2-dependent
phosphorylation and nuclear translocation of PKM2 promotes the
Warburg effect. Nat Cell Biol. 14:1295–1304. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang W, Xia Y, Hawke D, Li X, Liang J,
Xing D, Aldape K, Hunter T, Alfred Yung WK and Lu Z: PKM2
phosphorylates histone H3 and promotes gene transcription and
tumorigenesis. Cell. 150:685–696. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang W, Xia Y, Ji H, Zheng Y, Liang J,
Huang W, Gao X, Aldape K and Lu Z: Nuclear PKM2 regulates β-catenin
transactivation upon EGFR activation. Nature. 480:118–122. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Luo W, Hu H, Chang R, Zhong J, Knabel M,
O’Meally R, Cole RN, Pandey A and Semenza GL: Pyruvate kinase M2 is
a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell.
145:732–744. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee J, Kim HK, Han YM and Kim J: Pyruvate
kinase isozyme type M2 (PKM2) interacts and cooperates with Oct-4
in regulating transcription. Int J Biochem Cell Biol. 40:1043–1054.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lim J, Hao T, Shaw C, Patel AJ, Szabó G,
Rual JF, Fisk CJ, Li N, Smolyar A, Hill DE, et al: A
protein-protein interaction network for human inherited ataxias and
disorders of Purkinje cell degeneration. Cell. 125:801–814. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Landschulz WH, Johnson PF and McKnight SL:
The leucine zipper: A hypothetical structure common to a new class
of DNA binding proteins. Science. 240:1759–1764. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kouzarides T and Ziff E: Leucine zippers
of fos, jun and GCN4 dictate dimerization specificity and thereby
control DNA binding. Nature. 340:568–571. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Busch SJ and Sassone-Corsi P: Dimers,
leucine zippers and DNA-binding domains. Trends Genet. 6:36–40.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hashiguchi A, Hitachi K, Inui M,
Okabayashi K and Asashima M: TSC-box is essential for the nuclear
localization and antiproliferative effect of XTSC-22. Dev Growth
Differ. 49:197–204. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hino S, Kawamata H, Uchida D, Omotehara F,
Miwa Y, Begum NM, Yoshida H, Fujimori T and Sato M: Nuclear
translocation of TSC-22 (TGF-beta-stimulated clone-22) concomitant
with apoptosis: TSC-22 as a putative transcriptional regulator.
Biochem Biophys Res Commun. 278:659–664. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nakamura M, Kitaura J, Enomoto Y, Lu Y,
Nishimura K, Isobe M, Ozaki K, Komeno Y, Nakahara F, Oki T, et al:
Transforming growth factor-β-stimulated clone-22 is a
negative-feedback regulator of Ras/Raf signaling: Implications for
tumorigenesis. Cancer Sci. 103:26–33. 2012. View Article : Google Scholar
|
|
26
|
Gluderer S, Brunner E, Germann M,
Jovaisaite V, Li C, Rentsch CA, Hafen E and Stocker H: Madm (Mlf1
adapter molecule) cooperates with Bunched A to promote growth in
Drosophila. J Biol. 9:92010. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kester HA, Blanchetot C, den Hertog J, van
der Saag PT and van der Burg B: Transforming growth
factor-beta-stimulated clone-22 is a member of a family of leucine
zipper proteins that can homo- and heterodimerize and has
transcriptional repressor activity. J Biol Chem. 274:27439–27447.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gao X, Wang H, Yang JJ, Liu X and Liu ZR:
Pyruvate kinase M2 regulates gene transcription by acting as a
protein kinase. Mol Cell. 45:598–609. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang HJ, Hsieh YJ, Cheng WC, Lin CP, Lin
YS, Yang SF, Chen CC, Izumiya Y, Yu JS, Kung HJ, et al: JMJD5
regulates PKM2 nuclear translocation and reprograms HIF-1α-mediated
glucose metabolism. Proc Natl Acad Sci USA. 111:279–284. 2014.
View Article : Google Scholar
|