Introduction
Colorectal cancer (CRC) is the third leading cause
of cancer death in Japan (1). The
number of patients with CRC and the number of deaths due to CRC
have increased in Japan because of the popularity of the western
lifestyle (2,3). Some dietary ingredients, such as
fatty acids, are risk factors for CRC (4). Linoleic acid (LA) increases the risk
of colorectal carcinogenesis by generating prostaglandin E2, which
induces chronic persistent inflammation of the mucosa (5). LA also induces dormancy in CRC cells
(6). Trans fatty acids
(TFAs) have been evaluated as a possible CRC risk for decades
(7); however, recent studies have
clearly demonstrated that TFAs are associated with colon
carcinogenesis. Patients who do not use non-steroidal
anti-inflammatory drugs and postmenopausal women showed higher CRC
risks from TFAs (8). Compared with
patients in the lowest quartile of TFA consumption, those in the
highest quartile showed an increased odds ratio of 1.86 for the
prevalence of colorectal adenomas (9). Moreover, elaidic acid (EA), the major
TFA (10), is increased in the
plasma of colon adenoma patients (11). Additionally, EA promotes colon
carcinogenesis in Min mice (12)
and induces the proliferation of Ehrlich ascites sarcoma cells
(13). Thus, TFAs, particularly
EA, possess carcinogenic properties; however, the related
intracellular signals have not been fully determined.
Long-chain fatty acids (LCFAs) bind to specific
membrane-bound receptors, namely G-protein coupled receptor 40
(GPR40) and GPR120 (14). GPR40
and GPR120 bind to various LCFAs. EA is an LCFA containing 18
carbon chain with a trans configuration and is a structural
isomer of oleic acid (OA). OA also binds to GPR40 and 120, which is
coupled with Gq, to stimulate calcium ion influx and inositol
phosphate synthesis in smooth muscle cells (15), inducing smooth muscle contraction.
OA inhibits c-Jun N-terminus kinase and nuclear factor κB signaling
(16), which suppress tumor
necrosis factor α-induced insulin resistance and inflammation.
However, no information is available regarding EA
receptors. EA is a well-known dietary risk factor of cardiovascular
disorders such as atherosclerosis and myocardial infarction
(17,18). OA is present at low concentrations
in CRC tissues (19), whereas EA
is increased in the plasma of colon adenoma patients (11). OA exerts protective effects against
cardiovascular disorders by decreasing low-density lipoprotein
cholesterol levels (20). In
contrast, EA exerts proinflammatory effects (16,21),
suggesting the presence of different intracellular signaling
pathways for EA and OA.
In the present study, we examined the effect of EA
on the metastasis of CRC cells and determined the receptors and
signaling pathways of EA to explain its role in CRC.
Materials and methods
Cell culture and reagents
Mouse colon cancer cell line CT26 was kindly
provided by Dr I. J. Fidler (MD Anderson Cancer Center, Houston,
TX, USA) and human colon cancer cell line HT29 was purchased from
Dainihon Pharmacy, Co., Ltd., (Tokyo, Japan). Cells were routinely
maintained in Dulbecco's modified Eagle's medium (DMEM;
Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal
bovine serum (FBS; Sigma-Aldrich) in 5% CO2 at 37°C.
Cell morphology was evaluated daily by performing microscopic
examination. Each cell line was routinely tested for mycoplasma
contamination by performing genomic PCR. Before manuscript
submission, viability of each cell line was tested by performing
trypan blue exclusion assay.
EA (CAS no. 112-79-8; Wako Pure Chemical Industries
Ltd., Osaka, Japan), OA (CAS no. 112-80-1; Wako Pure Chemical
Industries), methyl-β-cyclodextrin (MbCD; Sigma-Aldrich), human
epidermal growth factor (EGF; Peprotech EC Ltd., London, UK), PP1
(SRC inhibitor; Sigma-Aldrich), Wnt inhibitor (IWP3; Miltenyi
Biotec GmbH, Bergisch Gladbach, Germany) (22), ERK inhibitor
(3-[2-aminoethyl]-5-[{4-ethoxyphenyl}methylene]-2,4-thiazolidinedione;
Santa Cruz Biotechnology, Santa Cruz, CA, USA) and p38 inhibitor
(SB 203580; Abcam, Cambridge, MA, USA) were purchased.
Assessment of cell growth and
apoptosis
The cells (1×104/well) were seeded in a
12-well dish. Cell growth was assessed by performing
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay or by counting the number of cells with autocytometer
(Sysmex, Kobe, Japan) after 48 h, as previously described (23). Apoptosis and necrosis were assessed
using an apoptosis/necrosis detection kit (Enzo Life Sciences,
Plymouth Meeting, PA, USA). Annexin V-EnzoGold and 7-AAD present in
the kit fluorescently label apoptotic and necrotic cells,
respectively. Apoptosis or necrosis was determined by examining
2000 cells under a fluorescent microscope (Leica Microsystems,
Tokyo, Japan). All the experiments were performed in
triplicate.
Sphere assay
The cells (1×107/well) were seeded on
stem cell medium II (Sigma-Aldrich) in a bacteriological 3.5-cm
dish (Becton-Dickinson Labware, Bedford, MA, USA) for 1 week.
Number of spheres formed was counted by performing dark-field
microscopy (Nikon, Tokyo, Japan).
Reverse transcriptase-polymerase chain
reaction
Mouse mRNA expression was assessed by performing
reverse transcriptase-polymerase chain reaction (RT-PCR) with 0.5
µg total RNA extracted using RNeasy kit (Qiagen, Hilden,
Germany). Primer sets used for amplifying mouse CD133 and
nucleostemin (NS; also known as guanine nucleotide-binding
protein-like 3) are as follows: CD133 (NCBI reference
sequence: NM_008935) forward, 5′-GAA AAG TTG CTC TGC GAA CC-3′ and
CD133 reverse 5′-TCT CAA GCT GAA AAG CAG CA-3′; NS
(NCBI reference sequence: NM_153547) forward, 5′-CAG GAT GCT GAC
GAT CAA GA-3′ and NS reverse 5′-TTG ATT GCT CAG GTG ACA GC-3′.
These primer sets were synthesized by Sigma Genosys (Ishikari,
Japan). PCR products obtained were electrophoresed on a 2% agarose
gel and stained with ethidium bromide. β-actin (ACTB) mRNA
(GenBank accession no. NM_001101) was amplified for use as an
internal control.
Immunohistochemical analysis
Cell spheres formed were fixed in 10% buffered
formalin for 12 h at 4°C and were embedded in 1% agar. The agar
specimens were processed into a paraffin-embedded block and
4-µm-thick sections were cut using a microtome. Consecutive
tissue sections were immunohistochemically stained by following the
immunoperoxidase technique, as previously described (24). Immunoblotting analysis was
performed using 0.2 µg/ml anti-MIB1 and anti-NS antibodies
(Dako Corp., Carpinteria, CA, USA) after performing antibody
retrieval, in which the sections were heated three times in
phosphate buffer (pH 6.0) in a microwave for 15 min each.
Immunoreactivity was determined using 0.2 µg/ml secondary
antibodies (Medical and Biological Laboratories, Co., Ltd., Nagoya,
Japan). Color development was performed using diamine benzidine
hydrochloride (Dako) and counterstaining was performed using
Mayer's hematoxylin (Sigma-Aldrich). The number of NS- or
MIB1-positive nuclei were counted and expressed as the mean number
of positive nuclei from 50 high-power fields of tumor
specimens.
Animal models
BALB/c male mice (4-weeks old) were purchased from
Japan SLC (Shizuoka, Japan). The mice were maintained according to
the institutional guidelines approved by the Committee for Animal
Experimentation of Nara Medical University and the current
regulations and standards established by the Ministry of Health,
Labor and Welfare. Each experimental group included five mice.
A subcutaneous tumor model was established by
inoculating CT26 cells (1×107 cells) suspended in Hank's
balanced salt solution (Sigma-Aldrich) into the scapular
subcutaneous tissue of the mice, which were fed with standard diet
(CE-2; CLEA Japan, Tokyo, Japan). The fatty acid composition did
not contain TFAs). For analyzing cancer metastasis to the liver,
lungs and peritoneum, cancer cells (1×106 cells) were
inoculated into the spleen, tail vain and peritoneal cavity,
respectively. EA or OA (10 mg/mouse) was administered by gavage
every 7 days, and metastatic status was assessed after 4 weeks. The
dosage of 10 mg/mouse/week (equivalent to 0.5 g/kg body
weight/week) is approximately equal to 0.53 g/kg body weight/week,
which is according to the Nutrition Facts label of foods 4.6 g per
day (25). Peritoneal metastasis
was determined by measuring the weight of the mesenterium with
tumors, which was separated from the intestinal tube. The liver and
lung metastases were determined by counting the number of tumors on
organ surface.
Western blot analysis
Whole-cell lysates were prepared as previously
described (24). Cell fractions
were extracted by processing the cells with Cell Fractionation kit
(Abcam), according to the manufacturer's instruction.
Proteins (50 µg) present in cell lysates were
separated by performing sodium dodecyl sulfate-polyacrylamide gel
electrophoresis on 12.5% gels and were electrotransferred onto
nitrocellulose membranes for immunoblotting analysis. The membranes
were incubated with primary antibodies, followed by incubation with
peroxidase-conjugated IgG antibodies (MBL). Tubulin antibody
(Proteintech Group, Inc., Rosemont, IL, USA) was used to measure
the amount of protein loaded per lane. Immune complexes were
visualized using CSA system (Dako). Antibodies against GPR40 and
GPR120 (Abnova Corp., Taipei City, Taiwan); EGFR, E-cadherin (ECD)
and β-catenin (Transduction Laboratories, Lexington, KY, USA);
phosphorylated EGFR, NS, phosphorylated ERK1/2
(pThr204), phosphorylated SRC (pTyr530),
Notch1, APC, Wnt5a and CD44 (Santa Cruz Biotechnology);
phosphorylated p38 (pThr180/pTyr182; Cusabio,
College Park, MD, USA); CD133 and Snail1 (pSer246) (Biorbyt,
Cambridge, UK); and phosphorylated β-catenin (pSer33/37;
LifeSpan Biosciences, Inc., Seattle WA, USA) were used as primary
antibodies.
Small interfering RNA
Stealth Select RNAi oligonucleotides (siRNAs)
against human and mouse GPR40 and GPR120 and against mouse EGFR
were purchased from Santa Cruz Biotechnology. AllStars Negative
Control siRNA was used as a control (Qiagen, Valencia, CA, USA).
Cells were transfected with 20 nM siRNA by using Lipofectamine 2000
(Invitrogen Corp., Carlsbad, CA, USA), according to the
manufacturer's recommendations.
Surface expression assay
Surface expression assay was performed to determine
the effect of agonist-mediated GPR internalization. Briefly,
monolayer-cultured CT26 cells in a 96-well dish were preincubated
with peroxidase-labeled antibody against GPR40 or GPR120, followed
by treatment with EA, OA (35 µM) or vehicle. At each
time-point, the cells were counted by performing the MTT assay and
were fixed in 4% paraformaldehyde for 15 min at room temperature.
Surface levels of GPRs were measured by performing
tetramethylbenzene reaction, followed by treatment with
H2SO4 and measurement of absorbance at 450
nm.
Statistical analysis
Statistical analyses of experimental data were
performed by unpaired Student's t-test and one-way ANOVA with post
hoc test. Normal distribution of the data samples was confirmed by
the Kolmogorov-Smirnov normality test. A two-sided P<0.05 was
considered statistically significant.
Results
EA enhances metastasis
As shown in Fig. 1,
CT26 mouse CRC cells were inoculated into the subcutaneous tissue
(Fig. 1A), spleen (liver
metastasis) (Fig. 1B), tail vein
(lung metastasis) (Fig. 1C) and
peritoneum (Fig. 1D) of BALB/c
mice by gavage administration with EA, OA, or control vehicle.
EA-treated mice showed higher growth of subcutaneous tumors and
metastases to the liver, lungs and peritoneum than control mice. In
contrast, OA-treated mice only showed increased peritoneal
metastasis compared with the control mice. Tumors of EA-treated
mice showed increased mRNA expression of CD133 and NS
(Fig. 1E). Moreover, the number of
NS-positive tumor cells increased in the tumors of EA-treated mice
but not in the tumors of control or OA-treated mice (Fig. 1F and G). These results suggest that
EA increases the stemness of CT26 cells to enhance their metastatic
potential.
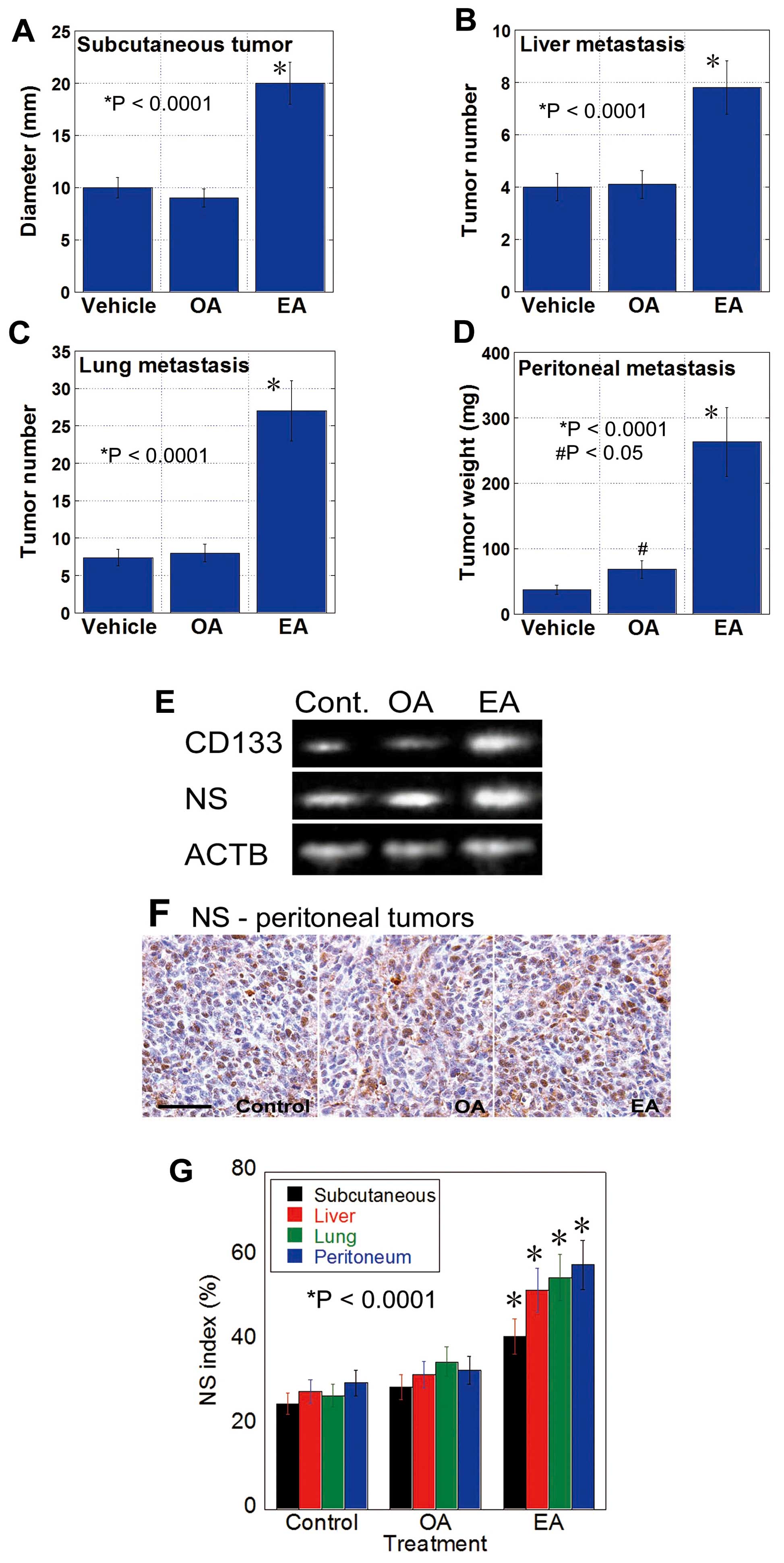 | Figure 1Gavage administration of EA enhances
the metastasis of CT26 cells. (A-D) Metastasis of CT26 mouse CRC
cells in mice treated with EA or OA by gavage (10 mg/mouse in 30%
ethanol) or vehicle (30% ethanol). Tumors were assessed at 4 weeks
after cancer cell inoculation. Tumor diameter (A), tumor number at
the surface of the liver (B) and lung (C), and tumor weight (D) of
the mesenterium were measured for subcutaneous tumor, metastases to
the liver, lung and peritoneum, respectively. Error bar, SD.
*Statistical difference from control was calculated by
the unpaired Student's t-test. (E) Expression of CD133 and NS in
subcutaneous tumors at week 4 was examined by RT-PCR. (F)
Expression of NS was also examined by immunostaining peritoneal
tumors at week 4; scale bar, 100 µm. (G) NS index, i.e., the
percentage of NS-positive nuclei in tumor cells, was calculated by
examining 500 tumor cells by immunostaining. Error bar, SD.
*Statistical difference from control was calculated by
the unpaired Student's t-test. |
Intracellular signaling pathways of
EA
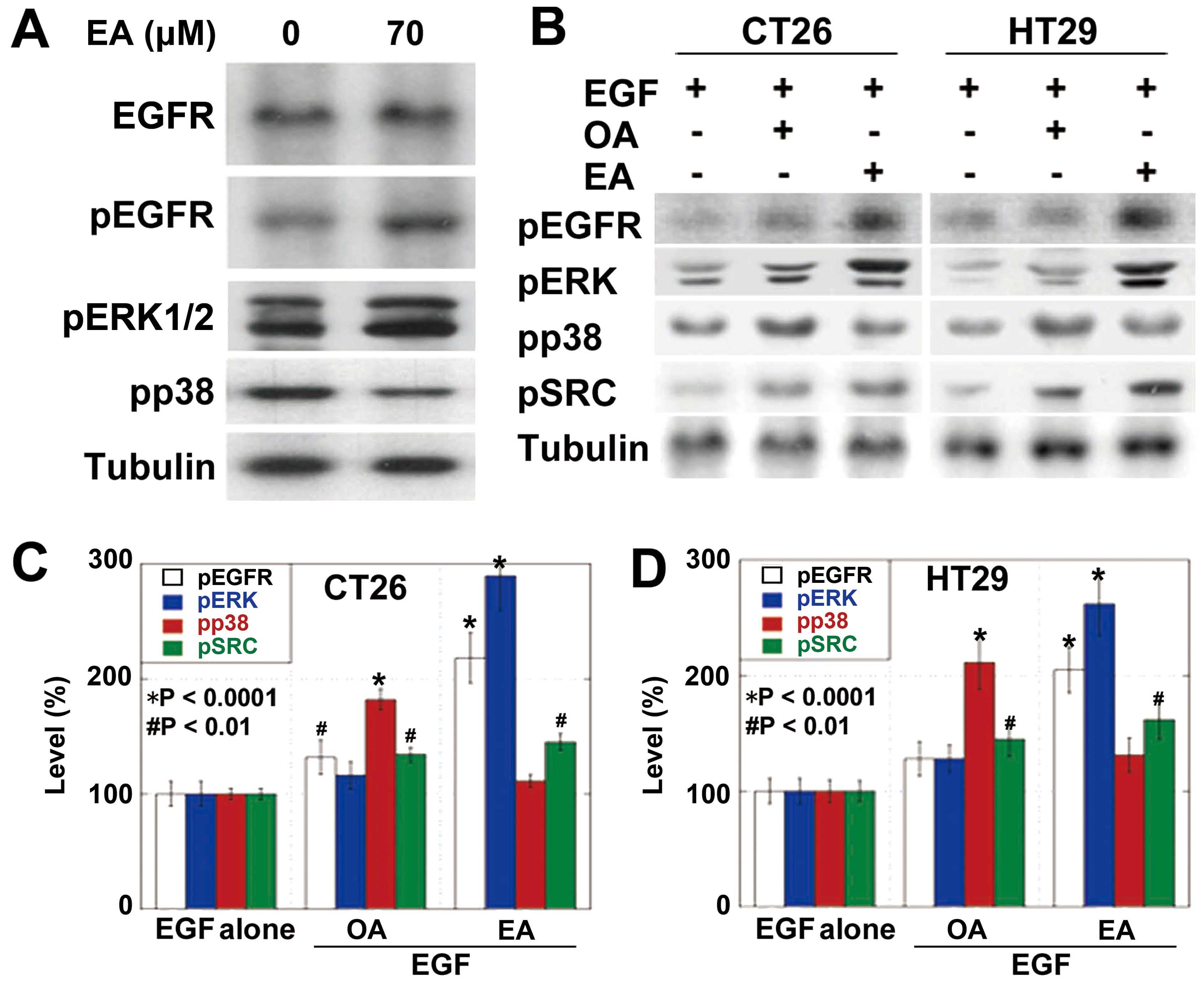
Next, we examined the intracellular signals of EA
(Fig. 2). EA treatment induced the
phosphorylation of EGFR, ERK1/2 but not p38 (Fig. 2A). The effects of EA and OA on EGFR
activation were compared (Fig.
2B–D). Phosphorylation of EGFR and ERK1/2 induced by OA was
less pronounced than that induced by EA in both CRC cells. In
contrast, phosphorylation of p38 induced by OA was more pronounced
than that induced by EA. Importantly, EA treatment induced SRC
phosphorylation at higher levels than those by OA treatment in both
cell types.
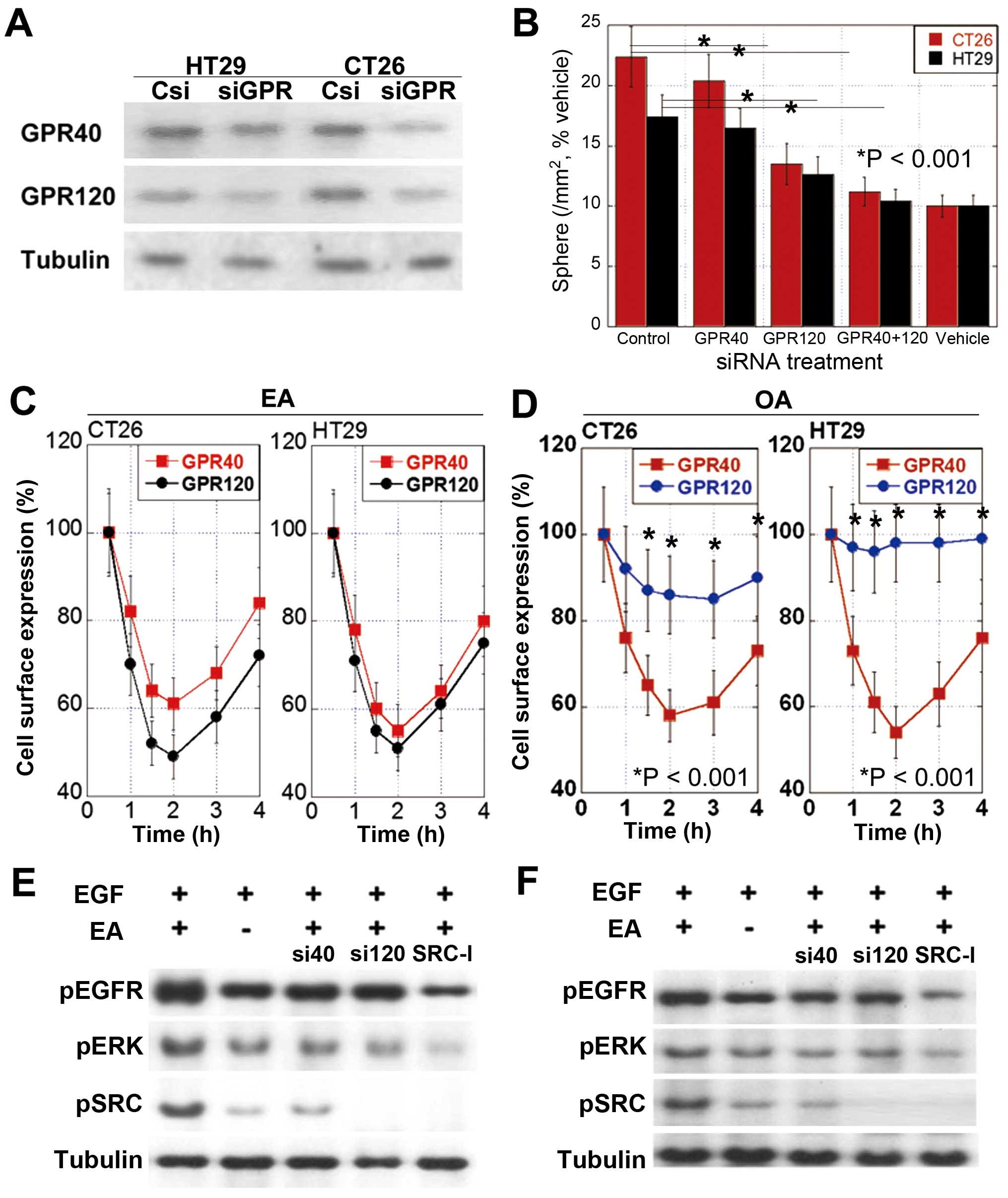
Receptor of EA
GPR40 and GPR120 function as receptors of long chain
fatty acids (26). We observed
that CRC cells expressed different levels of GPR40 and GPR120
(Fig. 3A) and that CT26 cells
showed higher GPR120:GPR40 ratio (5.3) than HT29 cells (1.2).
We knocked down genes encoding GPR40 and/or GPR120
and examined the effects of gene knockdown on the number of
sphere-forming cells (Fig. 3B).
Knockdown of the gene encoding GPR120 decreased the number of
sphere-forming cells more profoundly than the knockdown of the gene
encoding GPR40. Concurrent knockdown of both GPR40- and
GPR120-encoding genes blocked EA-induced increase in the number of
sphere-forming cells.
Binding affinity of EA to GPR40 or GPR120 was
confirmed by examining the cell surface expression of these GPRs.
Cell surface expression of both GPR40 and GPR120 was down-regulated
by internalization for 2 h after EA treatment, after which it
recovered (Fig. 3C). Results of
the surface expression assay indicate that unlike EA, OA may
preferentially bind to GPR40 (Fig.
3D).
EGFR transactivation by EA
Proto-oncogene tyrosine protein kinase (SRC) plays
an important role in OA-induced EGFR transactivation (27). Therefore, we examined the role of
SRC in EA-induced signal transduction. We observed that SRC
inhibition suppressed the phosphorylation of EGFR and ERK1/2 in
CT26 cells after concurrent treatment with EA and EGF (Fig. 3E). Moreover, knockdown of the gene
encoding GPR40 or GPR120 decreased the phosphorylation of EGFR and
ERK1/2 (Fig. 3E). Knockdown of the
gene encoding GPR120 inhibited the phosphorylation of EGFR and
ERK1/2 more profoundly than the knockdown of the gene encoding
GPR40. We also observed these effects in HT29 cells (Fig. 3F). These findings suggest that EA
activates GPR120 and (to a lesser extent) GPR40 in CRC cells for
transactivating EGFR through SRC.
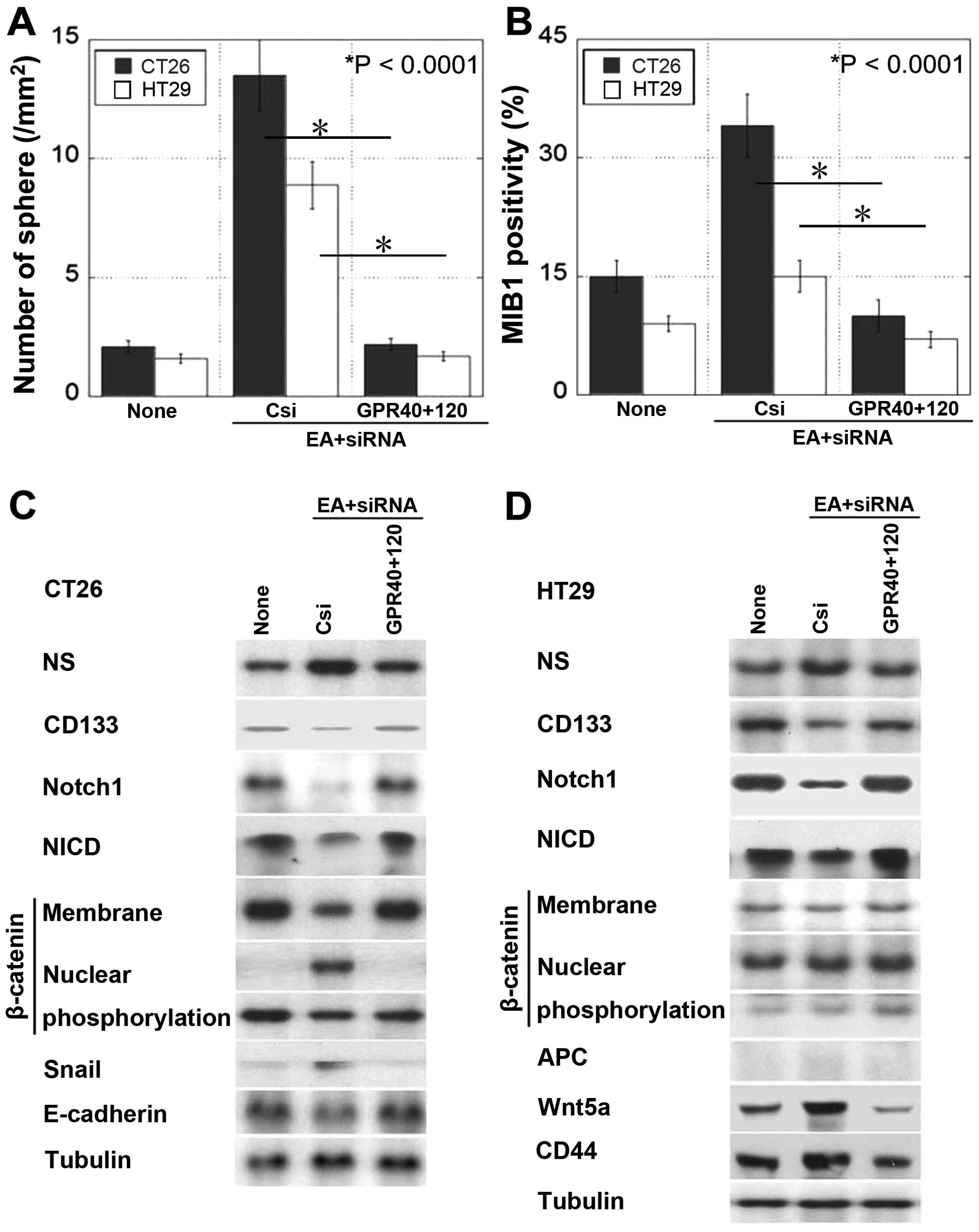
Effect of EA on cancer stem
cell-associated signals
Next, we examined sphere formation as a feature of
cancer stem cells (Fig. 4A and B).
We observed that the number of spheres and MIB1-positive
proliferating cells increased after EA treatment and decreased
after knocking down the genes encoding GPR40 and GPR120. Treatment
of CT26 cells with EA altered stem cell marker expression, i.e., it
increased NS expression and decreased CD133 expression (Fig. 4C). Moreover, EA treatment decreased
Notch and Notch intracellular domain (NICD) expression and induced
β-catenin nuclear translocation associated with EMT, as indicated
by the upregulation of Snail expression and downregulation of
E-cadherin expression. Thus, EA signaling inhibited Notch signaling
and activated canonical Wnt pathway in CT26 cells. In contrast, EA
treatment of HT29 cells harboring truncated APC (28), which showed constitutively
activated canonical Wnt signaling, increased NS expression and
decreased CD133 expression (Fig.
4D). In addition, EA treatment of HT29 cells decreased Notch
and NICD expression and increased Wnt5a and CD44 expression. These
results indicate that EA signaling inhibits Notch signaling and
activates non-canonical Wnt signaling in HT29 cells.
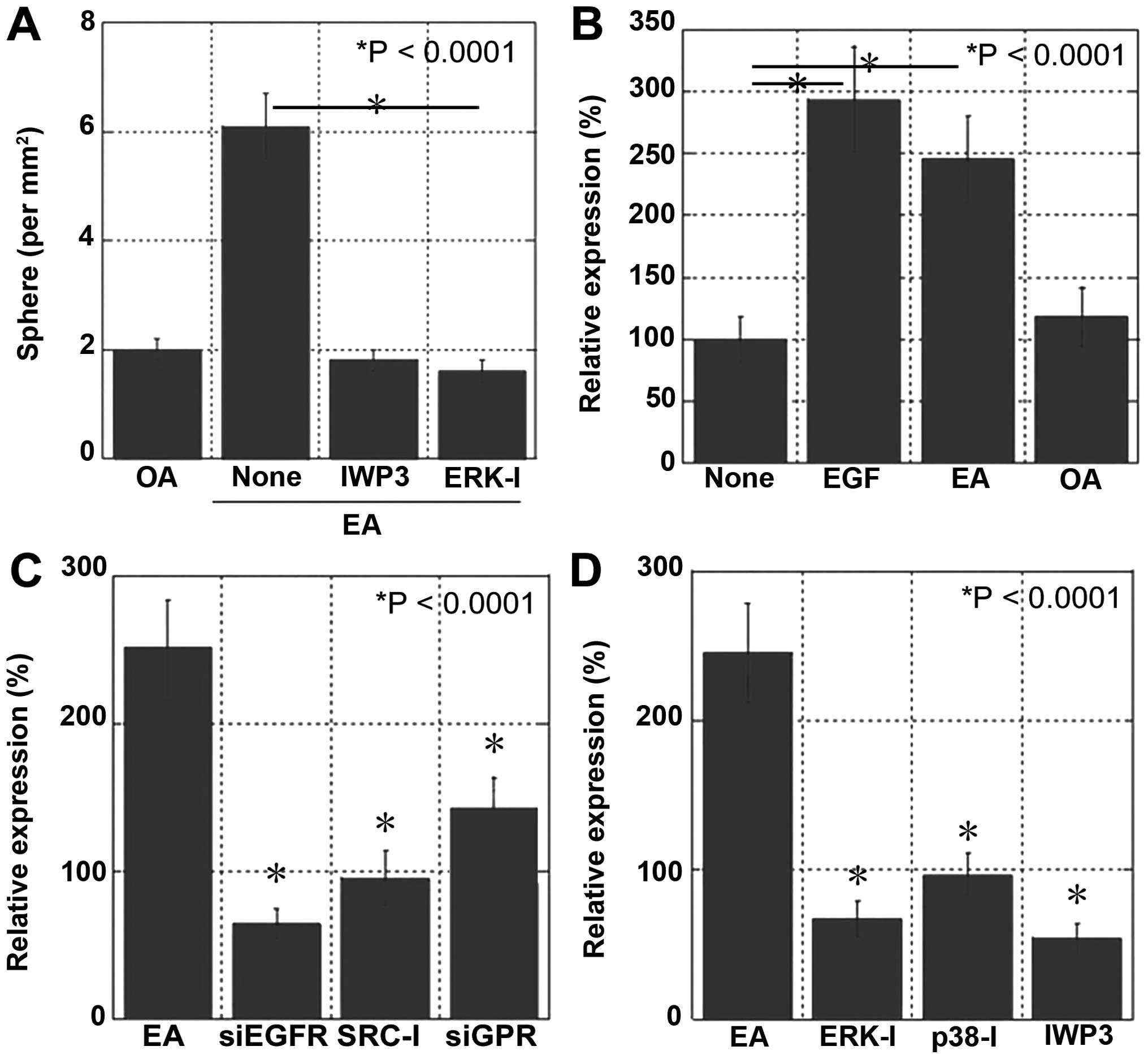
EA signaling and stemness
To confirm that EA induced the stemness and
metastatic ability of CRC cells, we examined the effects of Wnt and
ERK1/2 inhibitors by performing the sphere assay (Fig. 5A). Treatment with Wnt and ERK1/2
inhibitors completely abolished EA-induced increase in sphere
number and liver metastasis.
To confirm EA signaling pathway, NS expression in
CT26 cells was examined at the ligand (Fig. 5B), receptor (Fig. 5C), and post-receptor signaling
levels (Fig. 5D). At the ligand
level, EA treatment increased NS expression to a similar level as
that after EGF treatment, suggesting that EGFR transactivation
induced by EA was as effective as that induced by EGF. At the
receptor level, inhibition of the crosstalk linker SRC and
knockdown of GPR40 and GPR120 suppressed EA-induced NS expression,
which was as effective as that induced by the knockdown of EGFR. At
the post-receptor signaling level, inhibition of ERK1/2, p38 and
Wnt suppressed EA-induced NS expression.
Discussion
The results of the present study indicate that EA
strongly enhances cancer metastasis by increasing the stemness and
the epithelial-mesenchymal transition (EMT) of colon cancer
cells.
GPRs function as receptors of LCFAs. Particularly,
GPR40 and GPR120 are receptors for unsaturated LCFAs such as OA or
LA (26). In the present study,
the binding of EA to GPR40 and GPR120 was confirmed in surface
protein internalization assays. Knockdown of the genes encoding
these two receptors did not completely suppress the EA-induced
increase in the proliferation of cancer spheres. However, further
studies are needed to examine other potential receptors that
effectively prevent EA-induced proliferation of cancer spheres.
GPR40 and GPR120 perform different functions under
different conditions. In islet β cells, GPR40 activation increases
insulin resistance, whereas GPR120 activation increases insulin
sensitivity (29). The motility
and invasion of melanoma, lung and pancreatic cancer cell lines are
enhanced by GPR40, but suppressed by GPR120 (30–32).
The results of the present study indicate that GPR40
and GPR120 are cooperatively involved in EA-induced EGFR
transactivation. In OA-treated MCF7 cells, GPR40 and GPR120 induce
EGFR transactivation (27).
Transactivation may involve the participation of both GPR40 and
GPR120. OA-activated EGFR induces activator protein 1 to enhance
cell proliferation and matrix metalloproteinase expression
(27). We observed that EA
crosstalk from GPRs to EGFR was mediated by SRC. A previous study
reported that the crosstalk between GPRs and EGFR activated
phospholipases C, D and A2, which subsequently activated protein
kinase C, extracellular receptor kinase and Akt (33). Moreover, our data showed that
EA-activated EGFR induced Wnt to enhance stemness and EMT. The
causes of the underlying differences downstream of transactivated
EGFR by EA and OA are unclear. Therefore, further studies are
necessary to explore other receptors or signaling pathways involved
downstream of EGFR.
In wild-type APC-containing CT26 cells, EA signaling
through EGFR induced EMT and increased stemness by activating the
canonical Wnt pathway. In contrast, in APC-null HT29 cells, EA-EGFR
signaling increased stemness by activating the non-canonical Wnt
pathway. This is consistent with our previous findings that EGFR
increased stemness by activating Wnt signaling through the nuclear
translocation of β-catenin (22).
In the present study, we found that EA, GPRs, SRC-EGFR signaling,
and phosphorylation of extracellular receptor kinase 1/2 and p38
were involved in EA-induced Wnt activation. Thus, our results
indicate that EMT and stemness enhance the metastatic potential of
CRC cells. Notably, EA treatment upregulated NS, but not CD133 and
increased MIB1-positive stem cells in the sphere. These findings
suggest that cancer stem cells activated by EA are heterogeneous
and show a preference for proliferative stem cells. Moreover, our
results suggest that inhibition of EA signaling is a relevant
therapeutic strategy for inhibiting EA-induced metastasis of CRC
cells.
EA have been reported as EA incorporated into
mitochondrial membrane reducing early closing and refractoriness of
voltage-dependent anion channels (34). EA affects global DNA methylation by
inducing a pro-inflammatory transcriptional profile different from
that induced by OA in vitro. Moreover, maternal EA
administration induces DNA hypermethylation in mouse progeny
(35). The macrophages from rats
fed a diet containing hydrogenated fat showed upregulation of
cytosolic phospholipase A2, nuclear factor-κB p65 and Toll-like
receptor-2 and -4, and downregulation of peroxisome proliferator
activated receptor-γ and adiponectin receptor-1 and -2, enhancing
pro-inflammatory phenotypes (36).
In human, administration of an EA-rich diet to healthy volunteers
resulted in decreased mitogen-induced CD69 expression on
CD8+ T cells and phagocytic activity on neutrophils
(37). Such epigenetic,
pro-inflammatory and anti-cytotoxic immune properties may affect
carcinogenic and pro-metastatic potential of EA in addition to
enhancing the stemness of cancer cells.
Gavage administration of EA had a marked
pro-metastatic effect in the syngeneic tumor model using CT26 CRC
cells and BALB/c mice. Because the mice were fed with standard diet
containing no TFAs, the enhanced metastatic ability is thought to
be associated with EA administration. EA is a major component of
dietary TFAs (10), and the dosage
used in the experiments was in accordance with the recommendations
by the Nutritional Label in America in 2003 (25). Indeed, our data showed that EA
strongly promotes cancer metastasis. Metastasis is a major problem
in cancer treatment and makes it difficult to cure cancer (38). Prevention of metastasis should be
emphasized in the prevention of carcinogenesis. The ubiquitous
usage of TFAs in western-style diets suggests that TFAs play a
major role in the carcinogenesis and metastasis of CRC. The control
of TFA-associated cancer development and progression is urgently
needed.
Abbreviations:
|
CRC
|
colorectal cancer
|
|
TFA
|
Trans fatty acid
|
|
LCFA
|
long chain fatty acid
|
|
EA
|
elaidic acid
|
|
OA
|
oleic acid
|
|
MbCD
|
methyl-β-cyclodextrin
|
|
EGF
|
epidermal growth factor
|
|
GPR
|
G-protein coupled receptor
|
|
EGFR
|
EGF receptor
|
|
Src
|
proto-oncogene tyrosine-protein kinase
Src
|
|
APC
|
adenomatous polyposis coli
|
|
NS
|
nucleostemine
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
NICD
|
Notch intracellular domain
|
|
FDA
|
Food and Drug Administration
|
|
EMT
|
epithelial-mesenchymal transition
|
Acknowledgments
The authors thank Ms. Tomomi Masutani for her expert
assistance with the preparation of this manuscript. The present
study was supported by the MEXT KAKENHI grant nos. 13200228,
14478268, 13394212, 13209774 and 16675788.
References
|
1
|
Wakao F, Nishimoto H, Kataonoda K, Tsukuma
H and Mikami H: Cancer Statistics in Japan, 2013. National Cancer
Research Institute; Tokyo: 2013
|
|
2
|
Kimura Y, Kono S, Toyomura K, Nagano J,
Mizoue T, Moore MA, Mibu R, Tanaka M, Kakeji Y, Maehara Y, et al:
Meat, fish and fat intake in relation to subsite-specific risk of
colorectal cancer: The Fukuoka Colorectal Cancer Study. Cancer Sci.
98:590–597. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mizoue T, Tanaka K, Tsuji I, Wakai K,
Nagata C, Otani T, Inoue M and Tsugane S; Research Group for the
Development and Evaluation of Cancer Prevention Strategies in
Japan: Alcohol drinking and colorectal cancer risk: An evaluation
based on a systematic review of epidemiologic evidence among the
Japanese population. Jpn J Clin Oncol. 36:582–597. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bultman SJ: Interplay between diet, gut
microbiota, epigenetic events, and colorectal cancer. Mol Nutr Food
Res. May 3–2016.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Moonen HJ, Dommels YE, van Zwam M, van
Herwijnen MH, Kleinjans JC, Alink GM and de Kok TM: Effects of
polyunsaturated fatty acids on prostaglandin synthesis and
cyclooxygenase-mediated DNA adduct formation by heterocyclic
aromatic amines in human adenocarcinoma colon cells. Mol Carcinog.
40:180–188. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ohmori H, Sasahira T, Fujii K, Luo Y,
Shimomoto T and Kuniyasu H: Linoleic acid-induced growth
suppression induces quiescent cancer cell nests in nude mice
Pathobiol. 75:226–232. 2008.
|
|
7
|
Valenzuela A and Morgado N: Trans fatty
acid isomers in human health and in the food industry. Biol Res.
32:273–287. 1999. View Article : Google Scholar
|
|
8
|
Slattery ML, Benson J, Ma KN, Schaffer D
and Potter JD: Trans-fatty acids and colon cancer. Nutr Cancer.
39:170–175. 2001. View Article : Google Scholar
|
|
9
|
Vinikoor LC, Schroeder JC, Millikan RC,
Satia JA, Martin CF, Ibrahim J, Galanko JA and Sandler RS:
Consumption of trans-fatty acid and its association with colorectal
adenomas. Am J Epidemiol. 168:289–297. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Precht D and Molkentin J: Trans fatty
acids: Implications for health, analytical methods, incidence in
edible fats and intake (a review). Nahrung. 39:343–374. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pickens CA, Lane-Elliot A, Comstock SS and
Fenton JI: Altered saturated and monounsaturated plasma
phospholipid fatty acid profiles in adult males with colon
adenomas. Cancer Epidemiol Biomarkers Prev. 25:498–506. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Molin M, Berstad P, Benth JS, Alexander J,
Paulsen JE and Almendingen K: Effect of different degrees of
hydrogenated fish oil on intestinal carcinogenesis in
Min/+ mice. Anticancer Res. 33:477–483. 2013.PubMed/NCBI
|
|
13
|
Awad AB: Trans fatty acids in tumor
development and the host survival. J Natl Cancer Inst. 67:189–192.
1981.PubMed/NCBI
|
|
14
|
Hara T, Kashihara D, Ichimura A, Kimura I,
Tsujimoto G and Hirasawa A: Role of free fatty acid receptors in
the regulation of energy metabolism. Biochim Biophys Acta.
1841:1292–1300. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mizuta K, Zhang Y, Mizuta F, Hoshijima H,
Shiga T, Masaki E and Emala CW Sr: Novel identification of the free
fatty acid receptor FFAR1 that promotes contraction in airway
smooth muscle. Am J Physiol Lung Cell Mol Physiol. 309:L970–L982.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Perdomo L, Beneit N, Otero YF, Escribano
Ó, Díaz-Castroverde S, Gómez-Hernández A and Benito M: Protective
role of oleic acid against cardiovascular insulin resistance and in
the early and late cellular atherosclerotic process. Cardiovasc
Diabetol. 14:752015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Remig V, Franklin B, Margolis S, Kostas G,
Nece T and Street JC: Trans fats in America: A review of their use,
consumption, health implications, and regulation. J Am Diet Assoc.
110:585–592. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mozaffarian D, Aro A and Willett WC:
Health effects of trans-fatty acids: Experimental and observational
evidence. Eur J Clin Nutr. 63(Suppl 2): S5–S21. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang J, Zhang L, Ye X, Chen L, Zhang L,
Gao Y, Kang JX and Cai C: Characteristics of fatty acid
distribution is associated with colorectal cancer prognosis.
Prostaglandins Leukot Essent Fatty Acids. 88:355–360. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huth PJ, Fulgoni VL III and Larson BT: A
systematic review of high-oleic vegetable oil substitutions for
other fats and oils on cardiovascular disease risk factors:
Implications for novel high-oleic soybean oils. Adv Nutr.
6:674–693. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Okada Y, Tsuzuki Y, Sato H, Narimatsu K,
Hokari R, Kurihara C, Watanabe C, Tomita K, Komoto S, Kawaguchi A,
et al: Trans fatty acids exacerbate dextran sodium sulphate-induced
colitis by promoting the up-regulation of macrophage-derived
proinflammatory cytokines involved in T helper 17 cell
polarization. Clin Exp Immunol. 174:459–471. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sasaki T, Kuniyasu H, Luo Y, Kato D,
Shinya S, Fujii K, Ohmori H and Yamashita Y: Significance of
epithelial growth factor in the epithelial-mesenchymal transition
of human gallbladder cancer cells. Cancer Sci. 103:1165–1171. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kuniyasu H, Yano S, Sasaki T, Sasahira T,
Sone S and Ohmori H: Colon cancer cell-derived high mobility group
1/amphoterin induces growth inhibition and apoptosis in
macrophages. Am J Pathol. 166:751–760. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kuniyasu H, Oue N, Wakikawa A, Shigeishi
H, Matsutani N, Kuraoka K, Ito R, Yokozaki H and Yasui W:
Expression of receptors for advanced glycation end-products (RAGE)
is closely associated with the invasive and metastatic activity of
gastric cancer. J Pathol. 196:163–170. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
U.S. Food and Drug Administration: FDA
takes step to further reduce trans fats in processed foods.
2013.
|
|
26
|
Hirasawa A, Hara T, Katsuma S, Adachi T
and Tsujimoto G: Free fatty acid receptors and drug discovery. Biol
Pharm Bull. 31:1847–1851. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Soto-Guzman A, Robledo T, Lopez-Perez M
and Salazar EP: Oleic acid induces ERK1/2 activation and AP-1 DNA
binding activity through a mechanism involving Src kinase and EGFR
transactivation in breast cancer cells. Mol Cell Endocrinol.
294:81–91. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang J, Zhang W, Evans PM, Chen X, He X
and Liu C: Adenomatous polyposis coli (APC) differentially
regulates beta-catenin phosphorylation and ubiquitination in colon
cancer cells. J Biol Chem. 281:17751–17757. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Scheen AJ: Investigational insulin
secretagogues for type 2 diabetes. Expert Opin Investig Drugs.
25:405–422. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fukushima K, Takahashi K, Fukushima N,
Honoki K and Tsujiuchi T: Different effects of GPR120 and GPR40 on
cellular functions stimulated by
12-O-tetradecanoylphorbol-13-acetate in melanoma cells. Biochem
Biophys Res Commun. 475:25–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fukushima K, Yamasaki E, Ishii S,
Tomimatsu A, Takahashi K, Hirane M, Fukushima N, Honoki K and
Tsujiuchi T: Different roles of GPR120 and GPR40 in the acquisition
of malignant properties in pancreatic cancer cells. Biochem Biophys
Res Commun. 465:512–515. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kita T, Kadochi Y, Takahashi K, Fukushima
K, Yamasaki E, Uemoto T, Hirane M, Fukushima N, Honoki K and
Tsujiuchi T: Diverse effects of G-protein-coupled free fatty acid
receptors on the regulation of cellular functions in lung cancer
cells. Exp Cell Res. 342:193–199. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rozengurt E: Mitogenic signaling pathways
induced by G protein-coupled receptors. J Cell Physiol.
213:589–602. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tewari D and Bera AK: Modulation of the
voltage-dependent anion channel of mitochondria by elaidic acid.
Biochem Biophys Res Commun. 477:490–494. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Flores-Sierra J, Arredondo-Guerrero M,
Cervantes-Paz B, Rodríguez-Ríos D, Alvarado-Caudillo Y, Nielsen FC,
Wrobel K, Wrobel K, Zaina S and Lund G: The trans fatty acid
elaidate affects the global DNA methylation profile of cultured
cells and in vivo. Lipids Health Dis. 15:752016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rao YP, Kumar PP and Lokesh BR: Molecular
mechanisms for the modulation of selected inflammatory markers by
dietary rice bran oil in rats fed partially hydrogenated vegetable
fat. Lipids. 51:451–467. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dlouhý P, Kucera P, Kraml P, Pompachová A,
Potocková J, Smejkalová V, Mokrejs P, Jacek M and Andel M:
Short-term dietary intake of C18:1 trans fatty acids decreases the
function of cellular immunity in healthy young men. Ann Nutr Metab.
53:129–136. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fidler IJ: The pathogenesis of cancer
metastasis: The 'seed and soil' hypothesis revisited. Nat Rev
Cancer. 3:453–458. 2003. View Article : Google Scholar : PubMed/NCBI
|