Introduction
Triptolide, one of the members from diterpenoid
family is obtained during the phytochemical investigation of
Tripterygium wilfordii Hook F (1). It has a long history of use in
traditional Chinese medicine for various diseases. Biological
screening of triptolide has revealed its potential as anticancer
agent, an immunosuppressor and as contraceptive (1–3).
Triptolide treatment induces apoptosis in cancer cells through
cytochrome c transfer into the cytosol and inhibition of
expression of anti-apoptotic proteins (4). The apoptotic proteins including
caspases play a vital role in the induction of apoptosis in cancer
cells leading to alteration in cell morphology and biochemical
changes (5–8).
Hepatocellular carcinoma is a commonly detected
cancer and the third leading cause of cancer related deaths
throughout the world (9,10). Studies have revealed that every
year more than 600,000 new cases of primary liver cancer are
detected (9,10). Despite the improvement in the
5-year survival rate of liver cancer patients, the rate of
prognosis of liver cancer patients is very low (11). Liver cancer because of resistance
to currently available chemotherapeutics and radiotherapy easily
shows metastasis in other tissues such as lungs, lymph nodes, bones
and adrenal glands (12).
Therefore, development of novel and effective treatment strategies
for liver cancer is required.
Tumor suppressor gene (p53) controls the function of
several other genes involved in various cellular processes such as
apoptosis induction, arrest of cell cycle and repair of DNA damage
(13–16). Thus, p53 is involved in the
formation and progress of various types of cancer. Activation of
p53 gene takes place during cell stress through phosphorylation.
Among various activation sites available on p53 the most important
site of its activation is the phosphorylation at serine-15
(17,18). Studies have shown that during
stress by oxidants and radiations p53 is phosphorylated which then
induces cell apoptosis (19–21).
On activation p53 induces the expression of various other genes
such as p21 and Bax which are well known for their role in cellular
apoptosis (22–25). Thus, activation of p53 in cancer
cells can play an important role in the arrest of cancer
development and progress. In the present study, effect of
triptolide on the viability and apoptosis of HepG2 and QSG7701
liver cancer cells as well the underlying mechanism was
investigated. The present study demonstrated that triptolide
induced inhibition of liver cancer cell viability involving
activation of the tumor suppressor gene p53.
Materials and methods
Cell culture and reagents
HepG2 and QSG7701 liver cancer cell lines were
obtained from the American Type Culture Collection (ATCC; Manassas,
VA, USA). The cells were maintained in Dulbecco's modified eagle's
medium (DMEM) containing 10% fetal bovine serum (FBS). Cell culture
was carried out in a standard cell culture medium containing
L-glutamine (2 mM), streptomycin (100 µg/ml) and penicillin
(100 U/ml) in an incubator with humidified 5% CO2 and
95% air atmosphere. Triptolide was obtained from Sigma-Aldrich (St.
Louis, MO, USA) and its stock solution in dimethyl sulfoxide (DMSO)
was stored at −10°C. Pifithrin-α was obtained from Calbiochem (La
Jolla, CA, USA).
Cell viability assay
Viability of HepG2 and QSG7701 cells was determined
using a CCK-8 kit (Dojindo Molecular Technologies Inc., Rockville,
MD, USA). HepG2 and QSG7701 cells were seeded at a density of
2×106 cells/well into 96-well plates and treated with
10–100 µM concentrations of triptolide for 48 h. After
incubation, the CCK-8 solution (10 µl) was added to each
well of the plate and the plates were incubated in incubator at
37°C for 3 h. Absorbance was recorded three times independently
using a microplate reader (Bio-Rad Laboratories, Richmond, CA, USA)
at 450 nm.
Apoptosis analysis using fluorescent
microscopy and flow cytometry
HepG2 cells put into the tissue culture slides were
allowed to undergo attachment for 48 h. The cells were then treated
with various concentrations of triptolide for 48 h, washed with
phosphate-buffered saline (PBS) and subsequently put into the 1X
binding buffer (500 µl). A total of 5 µl each of
Annexin V-FITC and propidium iodide were added into the plates and
the cells were incubated under dark conditions for 5 min. The
plates were analyzed using flow cytometry (BD FACSCalibur; BD
Biosciences, San Jose, CA, USA) to determine the apoptotic
cells.
Western blot analysis
HepG2 cells after incubation with various
concentrations of triptolide were harvested and then were washed
with ice-cold PBS. The cells were treated with RIPA lysis buffer
(Roche Diagnostics, Shanghai, China) under ice-cold conditions for
45 min. The lysates were centrifuged at 12,000 rpm for 15 min at
4°C to collect the supernatant. Bradford method was used for the
determination of the protein concentration in lysate. The proteins
were separated by electrophoresis using 8% SDS-polyacrylamide gel
and subsequently were transferred to NC membrane (Millipore,
Billerica, MA, USA). The membrane incubation was performed with 10%
dry milk in TBST to block non-specific binding sites. The membrane
incubation with primary antibodies against Bax, DR5, p53, p53,
histone-H2A.X and p-H2A.X (Cell Signaling Technology, Danvers, MA,
USA) was performed overnight. The membranes were then washed with
PBS and were incubated with secondary antibodies for 1 h.
Expression of the desired proteins was examined using an ECL kit
(Millipore) according to the instructions from the manufacturer.
The bands were visualized using autoradiography with Hyperfilm
(Millipore).
Isolation of nuclei
Into the culture plates (100 mm diameter) HepG2
cells were seeded at a density of 2×106/plate and grown
to confluence. The cells were then treated with triptolide for
different durations. The cells were collected, PBS washed and
subjected to fractionation into separate nuclear and cytosolic
fractions using kit from Qiagen (Valencia, CA, USA). The sample
purity of the nucleus and cytosol was checked using Lamin B and
GAPDH as markers, respectively.
Analysis of DNA binding potential
HepG2 cells seeded into the plates were treated with
triptolide for analysis of DNA binding potential of p53. The
nuclear fractions obtained as mentioned above were examined using
the TransAM p53 kit according to the protocol provided by the
manufacturer (Active Motif, Carlsbad, CA, USA).
RT-PCR assay
Following treatment with triptolide for different
durations HepG2 cells were collected to isolate total cell RNA
using Isogen (Nippon Gene, Co., Ltd., Tokyo, Japan). Incubation of
the RNA with DNase I was carried out to digest DNA that was
contaminated. The RNA samples were purified by extraction using
phenol and precipitation in ethyl alcohol. Ready-To-Go RT-PCR Beads
(Amersham Pharmacia Biotech, Uppsala, Sweden) were used for the
preparation of cDNA. AmpliTaq Gold® DNA Polymerase, LD
(Applied Biosystems, Foster City, CA, USA) was employed for CR of
cDNA using the primers: p21, 5′-TTAGCAGCGGAACAAGGAGT-3′ and
5′-ATTCAGCATTGTGGGAGGAG-3′ upstream and downstream, respectively);
Bax, 5′-ATCCAGGATCGAGCAGGGCG-3′ and 5′-GGTTCTGATCAGTTCCGGCA-3′;
DR5, 5′-AAGACCCTTGTGCTCGTTG-3′ and 5′-TCACCTGAATCACACCTGG-3′;
GAPDH, 5′-TGAAGGTCGGAGTCAACGGATTTGGGT-3′ and
5′-CATGTGGGCCATGAGGTCCACCAC-3′. Proteins were treated at 90°C for
15 min followed by amplification. Amplification of the cDNA was
performed by the sequence of 94°C for 50 sec, 55°C for 50 sec and
72°C for 50 sec. Quenching of the reaction was done using
elongation step at 75°C for 5 min. The 2% agarose gel and ethidium
stain was used for the analysis of PCR products.
ELISA assay for apoptosis analysis
HepG2 cells were seeded at a density of
2×106 into 60-mm cultural dishes (Falcon) and allowed to
attain confluence. The cells were then treated with triptolide for
48 h. Following incubation, the cells were collected, washed with
PBS and lysed. The Cell Death Detection ELISA Plus kit (Roche
Diagnostics, Indianapolis, IN, USA) was used for the measurement of
DNA degradation into the nucleosomal fragments.
p53 silencing using RNA
HepG2 cells seeded at a density of 2×106
into 60-mm cultural dishes were subjected to incubation with RNA
for silencing p53 in transfection reagent (Mirus Bio, Madison, WI,
USA). The plates were incubated for 48 h and then treated with
triptolide for 48 h. After incubation, apoptosis induction in HepG2
cell cultures was analyzed using ELISA assay. Western blot assay
was used to examine the expression of various proteins.
Statistical analysis
The data presented are the means ± standard error or
division of the mean (SEM or SD). Student's t-tests and two-way
ANOVA were used for the analysis of the data using the statistical
program in GraphPad Prism Version 4.0 (GraphPad Software, Inc., San
Diego, CA, USA). All experiments were performed in triplicate. A
P<0.05 was taken to indicate statistically significant
difference.
Results
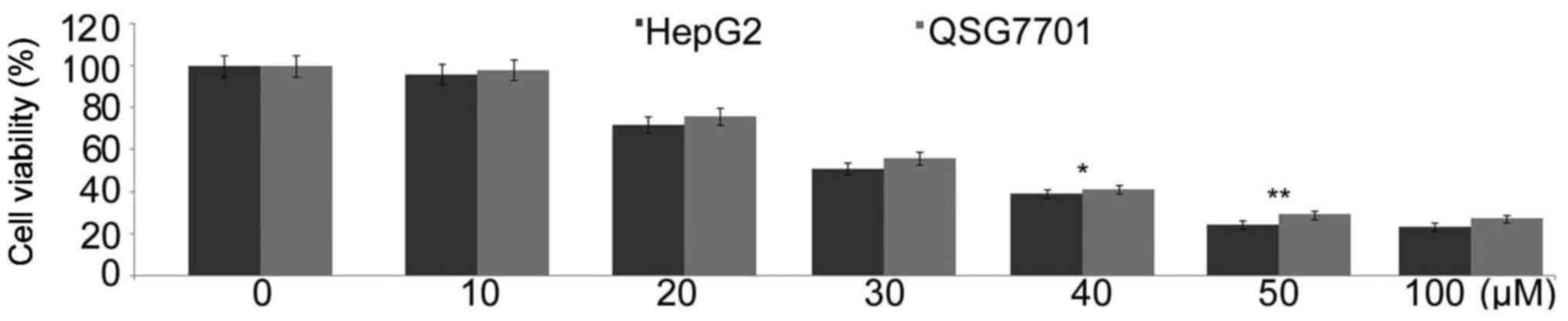
Triptolide treatment inhibits HepG2 and
QSG7701 cell growth and proliferation
In HepG2 and QSG7701 cells treatment with various
doses of triptolide for 48 h caused a concentration-dependent
proliferation reduction. Treatment with range of triptolide
concentrations from 10 to 100 µM led to a significant
viability reduction at 50 µM of 48 h treatment in both HepG2
and QSG7701 cell lines (Fig.
1).
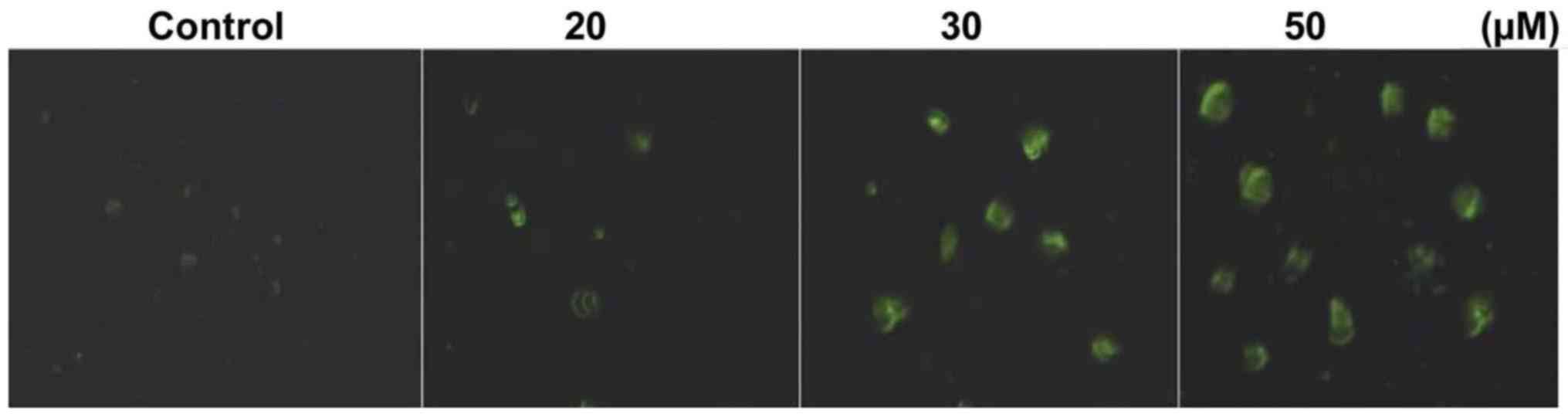
Triptolide treatment of HepG2 cells
induces apoptosis
Annexin V-FITC staining showed that triptolide
treatment of HepG2 cells for 48 h induced apoptosis. HepG2 cells
were treated with 10–50 µM concentrations of triptolide for
48 h and then were analyzed by fluorescent microscopy. Triptolide
treatment of HepG2 cells at 50 µM concentrations induced
apoptosis in 56.45% cells compared to only 2.36% cells in the
control cultures (Fig. 2).
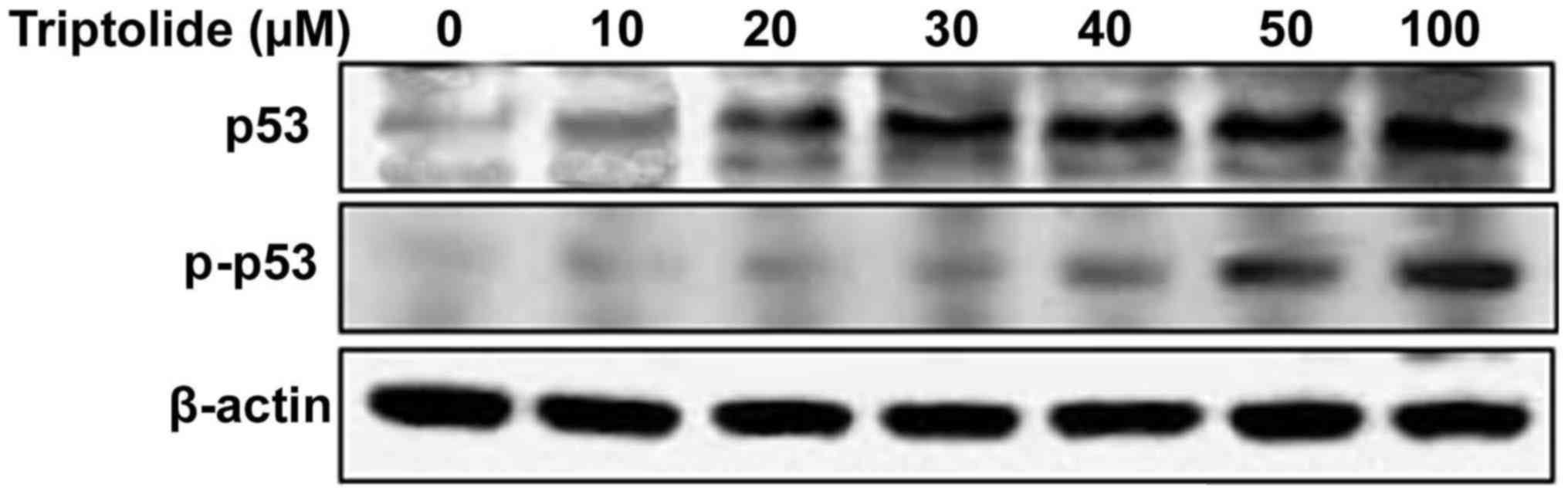
Triptolide increases tumor suppressor p53
phosphorylation
Treatment of HepG2 cells with triptolide at various
concentrations induced phosphorylation of p53 at serine-15 residue.
The phosphorylation of p53 was significant at 2 h of treatment with
50 µM concentration of triptolide in HepG2 cells (Fig. 3).
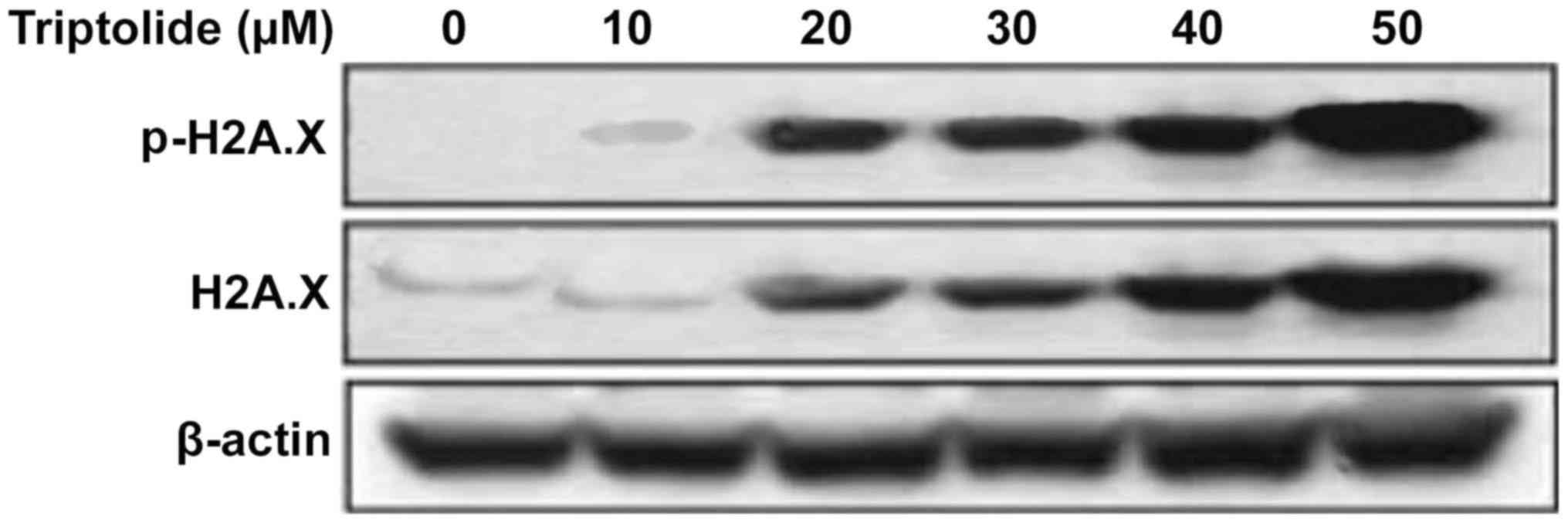
Phosphorylation at serine-15 of p53 in
HepG2 cells by triptolide is independent of DNA damage signals
Analysis of the phosphorylation of histone H2A.X at
serine 139, indicator of DNA damage showed a significant increase
by triptolide treatment from 12 h in HepG2 cells. However, no
significant increase in the phosphorylation of histone H2A.X was
observed at 1 h of triptolide treatment (Fig. 4).
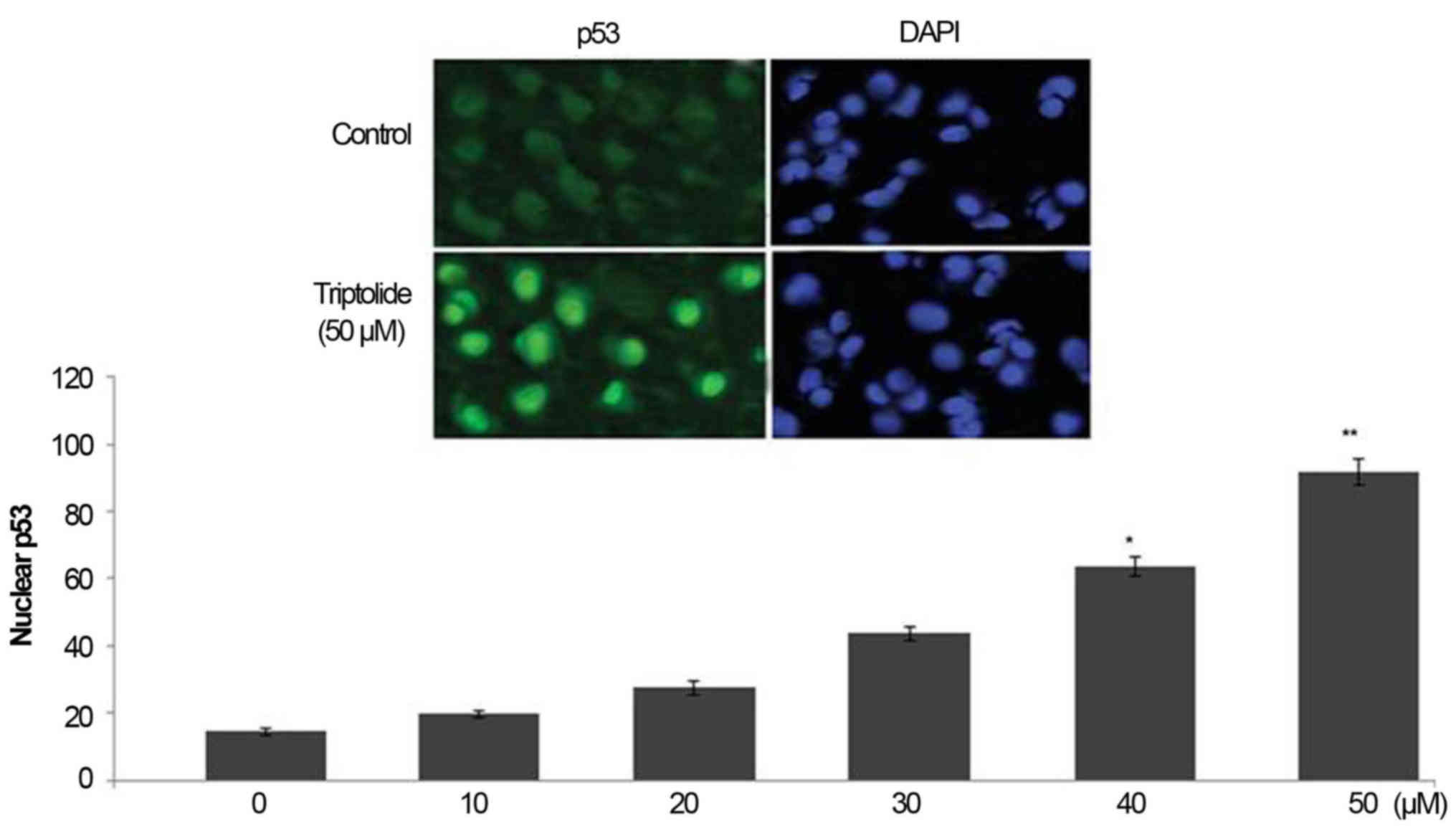
Triptolide treatment increases p53 level
in HepG2 cell nuclei
Treatment of HepG2 cells with triptolide caused a
significant increase in the level of p53 in HepG2 cell nuclei
(Fig. 5). In 6 h-treatment with 0,
10, 20, 30, 40 and 50 µM concentration of triptolide the
level of nuclear p53 was 15.3, 19.6, 28.5, 43.7, 63.8 and 91.5%,
respectively (Fig. 5).
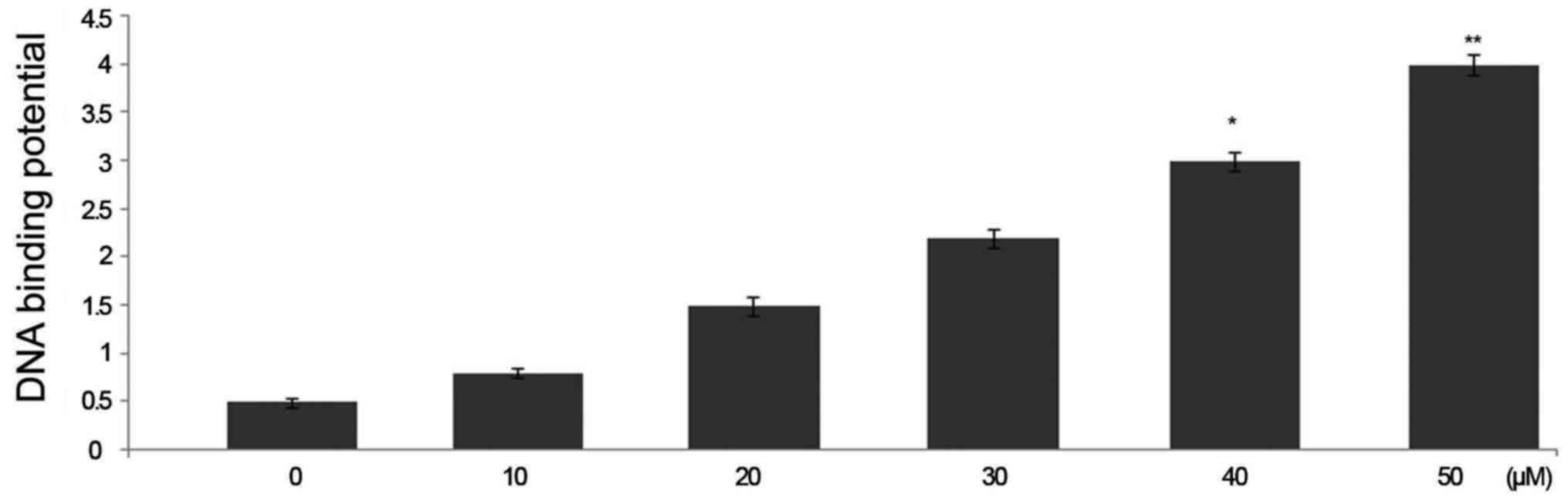
Triptolide treatment increases nuclear
p53 DNA-binding activity in HepG2 cells
Treatment of HepG2 cells with triptolide at 50
µM concentration caused a significant increase in the
binding potential of p53 to DNA. The binding potential of p53 to
DNA was significantly higher in 6 h triptolide treatment (Fig. 6).
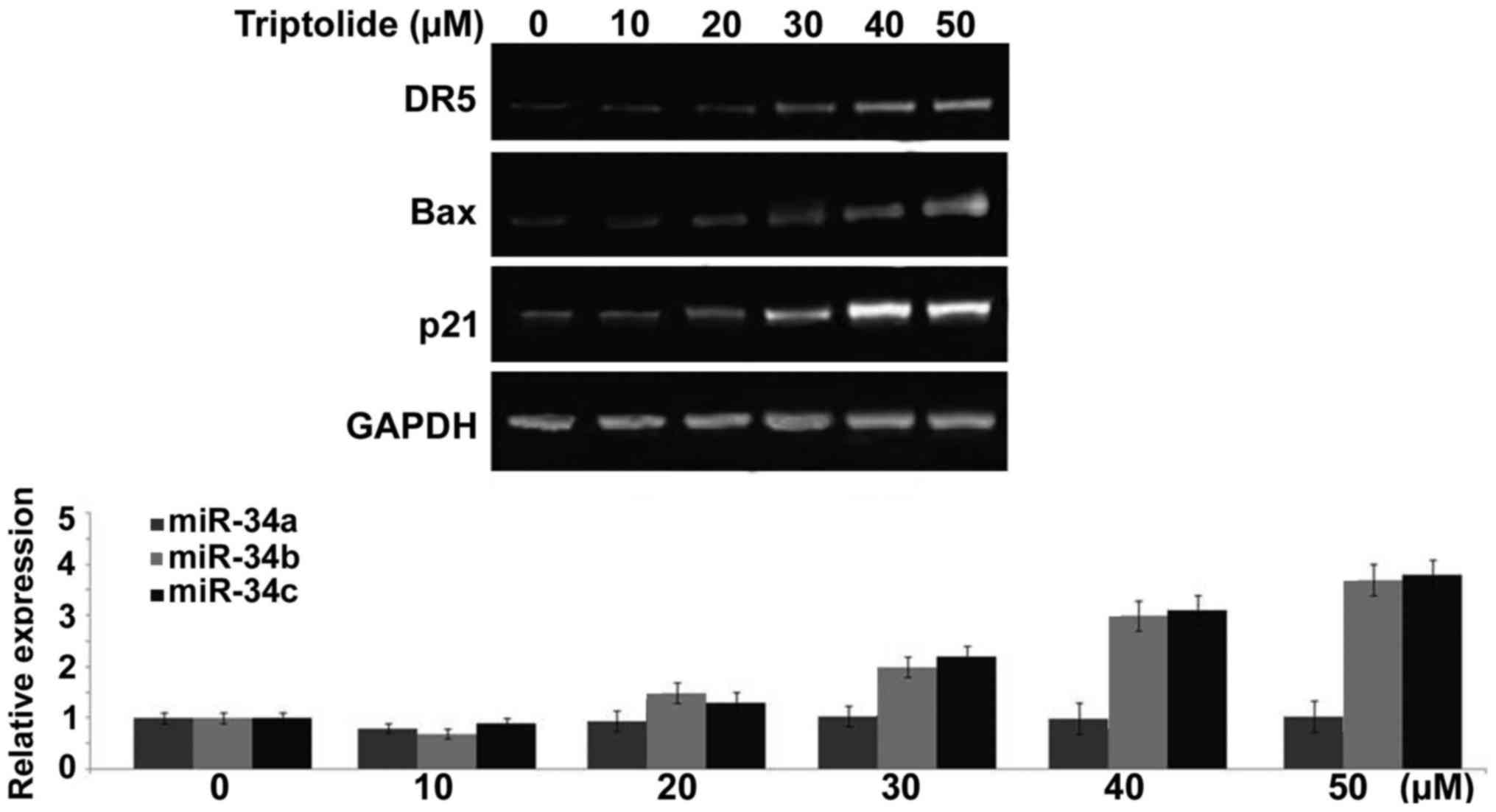
Treatment with triptolide induces rapid
transcription of p53 target genes in liver cancer cells
Triptolide treatment of HepG2 cells caused a
significant increase in the expression of various genes such as
p21, Bax and DR5 at 6 h. Compared to control HepG2 cells the
expression of p21, Bax and DR5 genes was markedly higher on
treatment with 50 µM concentration of triptolide (Fig. 7). In addition, triptolide treatment
also increased the expression of miR-34b and miR-34c in HepG2 cells
markedly (Fig. 7). However, no
increase was observed in the expression levels of miR-34a in HepG2
cells on triptolide treatment (Fig.
7).
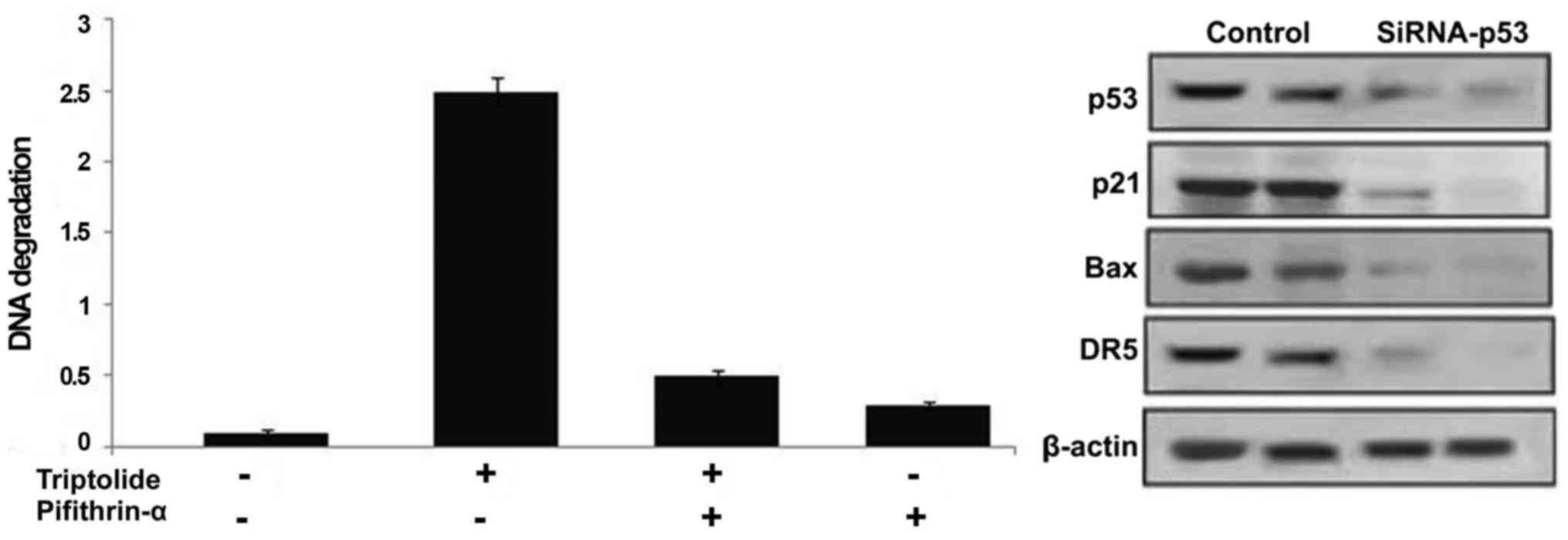
Inhibition of p53 prevents triptolide
induced HepG2 cell apoptosis
Treatment of HepG2 cells with p53 inhibitor,
pifithrin-α prior to incubation with triptolide significantly
prevented induction of cell apoptosis (Fig. 8). Suppression of p53 expression by
siRNA inhibited the expression of p53 as well as its target genes
along with the prevention of apoptosis induction (Fig. 8).
Discussion
In the present study, triptolide treatment reduced
viability and induced apoptosis in HepG2 liver cancer cells. The
mechanistic study revealed that triptolide exhibited its effect
through activation of p53 tumor suppressor gene. Activation of p53
inhibits its interaction and subsequent degradation by MDM2 as well
as increases the expression of nuclear p53 (26,27).
Various kinases have been found to activate p53 by phosphorylating
it at serine-15 during stress (28,29).
Our results revealed that triptolide treatment of HepG2 cells at
various concentrations induced phosphorylation of p53 at serine-15
residue. It is reported that activation of p53 can occur because of
DNA damage signals as well during absence of these signals
(30,31). In the present study, analysis of
the activation of histone H2A.X indicator of DNA damage revealed
its phosphorylation in 6 h-treatment. However, phosphorylation of
p53 was induced only after 2 h of treatment with triptolide in
HepG2 cells. These findings revealed that activation of p53 in
HepG2 cells by triptolide treatment is independent of the signals
produced by DNA damage. Our results showed that triptolide
treatment of HepG2 cells increased expression level of p53 by its
transcription. The expression of activated p53 was found to be more
in nuclear region of triptolide treated HepG2 cells. Following
activation p53 binds with various gene promoters and induces their
expression in the cells. Our results showed that triptolide
treatment of HepG2 cells induced expression levels of p21, Bax and
DR5 mRNAs. The expression level of miR-34b, miR-34c, and microRNAs
in HepG2 cells was also increased on treatment with triptolide. In
various types of cancers it has been found that miR-34a gene is
deleted from its location in the chromosome (32). Our results showed that triptolide
treatment of HepG2 cells induced no alteration in its expression.
Different types of stresses are able to induce cancer cell
apoptosis either in p53-dependent or -independent manner. In the
present study, the role of p53 in induction of apoptosis by
triptolide treatment was examined. The results from p53 suppression
experiments revealed that p53 plays an important role in HepG2
cancer cell apoptosis induction on treatment with triptolide. The
suppression of p53 by siRNA prevented induction of apoptosis in
HepG2 cell cultures by triptolide treatment.
In conclusion, the present study demonstrated that
triptolide inhibits viability and induces apoptosis in liver cancer
cells through activation of tumor suppressor gene p53. Thus,
triptolide can be used for the treatment of liver cancer.
References
|
1
|
Zhen QS, Ye X and Wei ZJ: Recent progress
in research on Tripterygium: A male antifertility plant.
Contraception. 51:121–129. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tengchaisri T, Chawengkirttikul R,
Rachaphaew N, Reutrakul V, Sangsuwan R and Sirisinha S: Antitumor
activity of triptolide against cholangiocarcinoma growth in vitro
and in hamsters. Cancer Lett. 133:169–175. 1998. View Article : Google Scholar
|
|
3
|
Hachida M, Lu H, Zhang X, Saito S,
Furutani Y, Matsuoka R, Hoshi H and Koyanagi H: Inhibitory effect
of triptolide on platelet derived growth factor-A and coronary
arteriosclerosis after heart transplantation. Transplant Proc.
31:2719–2723. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Carter BZ, Mak DH, Schober WD, McQueen T,
Harris D, Estrov Z, Evans RL and Andreeff M: Triptolide induces
caspase-dependent cell death mediated via the mitochondrial pathway
in leukemic cells. Blood. 108:630–637. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cohen JJ: Apoptosis. Immunol Today.
14:126–130. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
White E: Life, death, and the pursuit of
apoptosis. Genes Dev. 10:1–15. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Williams GT and Smith CA: Molecular
regulation of apoptosis: Genetic controls on cell death. Cell.
74:777–779. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wyllie AH, Kerr JF and Currie AR: Cell
death: The significance of apoptosis. Int Rev Cytol. 68:251–306.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma S, Chan KW and Guan XY: In search of
liver cancer stem cells. Stem Cell Rev. 4:179–192. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tomuleasa C, Soritau O, Rus-Ciuca D, Pop
T, Todea D, Mosteanu O, Pintea B, Foris V, Susman S, Kacso G, et
al: Isolation and characterization of hepatic cancer cells with
stem-like properties from hepatocellular carcinoma. J
Gastrointestin Liver Dis. 19:61–67. 2010.PubMed/NCBI
|
|
11
|
Ricci-Vitiani L, Lombardi DG, Pilozzi E,
Biffoni M, Todaro M, Peschle C and De Maria R: Identification and
expansion of human colon-cancer-initiating cells. Nature.
445:111–115. 2007. View Article : Google Scholar
|
|
12
|
Lee TK, Castilho A, Ma S and Ng IO: Liver
cancer stem cells: Implications for a new therapeutic target. Liver
Int. 29:955–965. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma L, Wagner J, Rice JJ, Hu W, Levine AJ
and Stolovitzky GA: A plausible model for the digital response of
p53 to DNA damage. Proc Natl Acad Sci USA. 102:14266–14271. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Harris SL and Levine AJ: The p53 pathway:
Positive and negative feedback loops. Oncogene. 24:2899–2908. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Levine AJ: p53, the cellular gatekeeper
for growth and division. Cell. 88:323–331. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bean LJ and Stark GR: Phosphorylation of
serines 15 and 37 is necessary for efficient accumulation of p53
following irradiation with UV. Oncogene. 20:1076–1084. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
She QB, Chen N and Dong Z: ERKs and p38
kinase phosphorylate p53 protein at serine 15 in response to UV
radiation. J Biol Chem. 275:20444–20449. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vazquez A, Bond EE, Levine AJ and Bond GL:
The genetics of the p53 pathway, apoptosis and cancer therapy. Nat
Rev Drug Discov. 7:979–987. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yin Y, Terauchi Y, Solomon GG, Aizawa S,
Rangarajan PN, Yazaki Y, Kadowaki T and Barrett JC: Involvement of
p85 in p53-dependent apoptotic response to oxidative stress.
Nature. 391:707–710. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lowe SW, Ruley HE, Jacks T and Housman DE:
p53-dependent apoptosis modulates the cytotoxicity of anticancer
agents. Cell. 74:957–967. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brugarolas J, Chandrasekaran C, Gordon JI,
Beach D, Jacks T and Hannon GJ: Radiation-induced cell cycle arrest
compromised by p21 deficiency. Nature. 377:552–557. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Miyashita T and Reed JC: Tumor suppressor
p53 is a direct transcriptional activator of the human bax gene.
Cell. 80:293–299. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Oda E, Ohki R, Murasawa H, Nemoto J,
Shibue T, Yamashita T, Tokino T, Taniguchi T and Tanaka N: Noxa, a
BH3-only member of the Bcl-2 family and candidate mediator of
p53-induced apoptosis. Science. 288:1053–1058. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yu J, Zhang L, Hwang PM, Kinzler KW and
Vogelstein B: PUMA induces the rapid apoptosis of colorectal cancer
cells. Mol Cell. 7:673–682. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shieh SY, Ikeda M, Taya Y and Prives C:
DNA damage-induced phosphorylation of p53 alleviates inhibition by
MDM2. Cell. 91:325–334. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Prives C: Signaling to p53: Breaking the
MDM2-p53 circuit. Cell. 95:5–8. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Brew CT, Aronchik I, Hsu JC, Sheen JH,
Dickson RB, Bjeldanes LF and Firestone GL: Indole-3-carbinol
activates the ATM signaling pathway independent of DNA damage to
stabilize p53 and induce G1 arrest of human mammary epithelial
cells. Int J Cancer. 118:857–868. 2006. View Article : Google Scholar
|
|
29
|
Loehberg CR, Thompson T, Kastan MB,
Maclean KH, Edwards DG, Kittrell FS, Medina D, Conneely OM and
O'Malley BW: Ataxia telangiectasia-mutated and p53 are potential
mediators of chloroquine-induced resistance to mammary
carcinogenesis. Cancer Res. 67:12026–12033. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jiang C, Hu H, Malewicz B, Wang Z and Lü
J: Selenite-induced p53 Ser-15 phosphorylation and caspase-mediated
apoptosis in LNCaP human prostate cancer cells. Mol Cancer Ther.
3:877–884. 2004.PubMed/NCBI
|
|
31
|
Zhao R, Xiang N, Domann FE and Zhong W:
Expression of p53 enhances selenite-induced superoxide production
and apoptosis in human prostate cancer cells. Cancer Res.
66:2296–2304. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chang TC, Wentzel EA, Kent OA,
Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M,
Ferlito M, Lowenstein CJ, et al: Transactivation of miR-34a by p53
broadly influences gene expression and promotes apoptosis. Mol
Cell. 26:745–752. 2007. View Article : Google Scholar : PubMed/NCBI
|