According to central dogma, information flow from
the genome is dictated by the transcription of coding genes to
mRNA, followed by translation to proteins. Multi-faceted omics
information yields high-volume data associated with the
whole-genome sequence, epigenome, methylome, transcriptome,
proteome and metabolome, all of which have been linked to
disease-specific cell phenotypes (1). The metabolome comprises of
physiologically active substances such as nutrients (e.g.,
glucose), lipids, amino acids (e.g., serine and glycine) and
nucleic acids. Importantly, in tumor cells, the processes of cell
growth and proliferation requires construction of building blocks
for new cellular components from substances associated with a redox
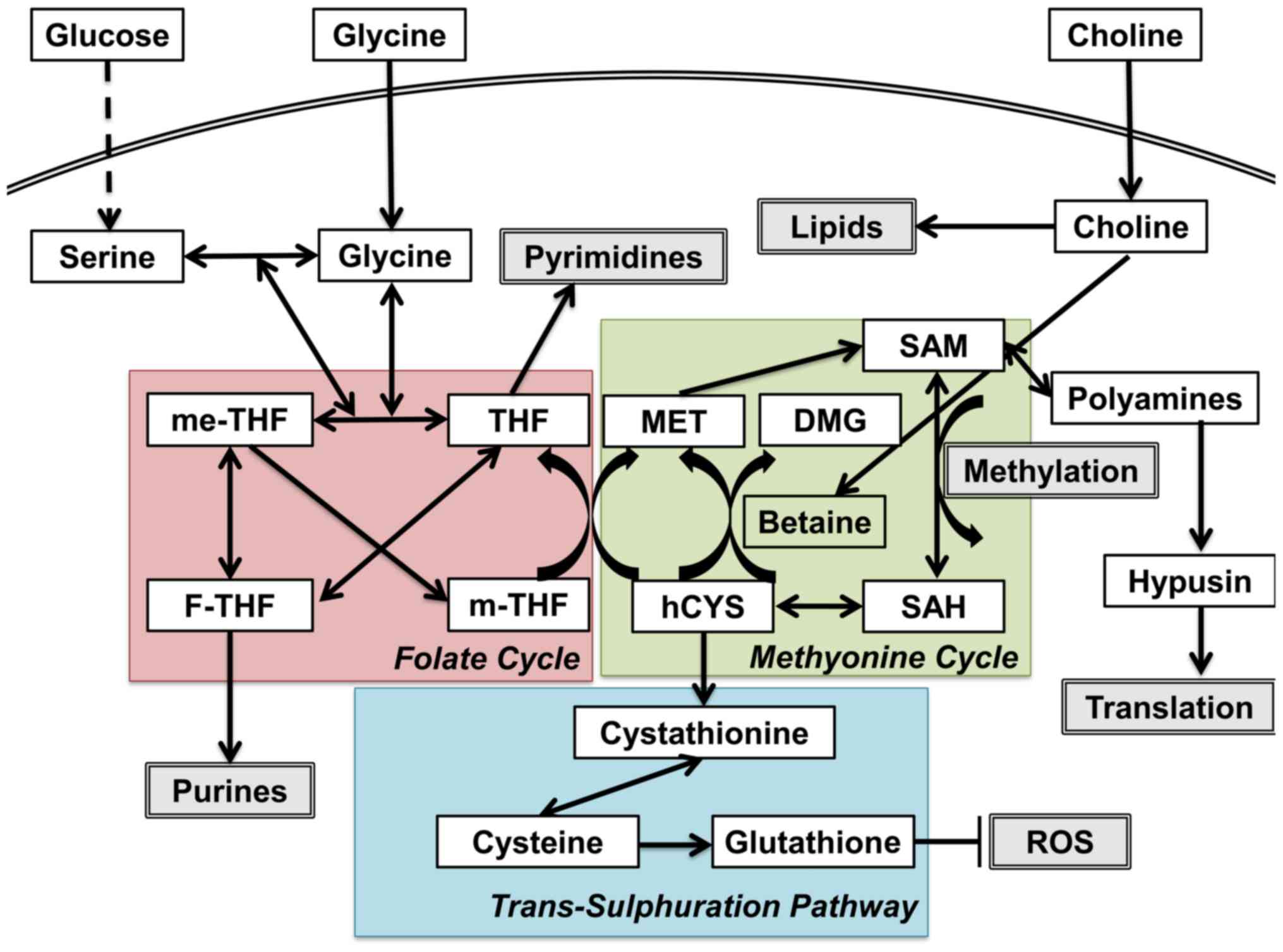
status (Fig. 1) (2). One-carbon (C1) metabolism encompasses
a complex metabolic network based on the chemical reaction of
folate compounds (3). The folate
cycle couples with the methionine cycle to form a bi-cyclic
metabolic pathway that circulates carbon units as part of a process
referred to as the C1 metabolism (3). These two cycles also link with the
trans-sulfuration pathway, which plays a critical role in the
regulation of the redox state by producing glutathione (3). C1 metabolism is critical for the
maintenance of genomic stability through nucleotide metabolism as
well as for the epigenetic control of DNA and histones, altered
expression of which is a characteristic attribute of tumor cells.
Ultimately, these findings should unravel new opportunities for
translational approaches, drug discovery and studies of cancer
pathogenesis. The study and control of C1 metabolism is the
foundation for precision medicine in the context of disease
prevention, identification of biomarkers, diagnosis, and treatment
of various diseases, including cancer (3–5).
High expression of C1 metabolic enzymes such as SHMT2, MTHFD2 and
ALDH1L2 was shown to be independently associated with RFS. These
findings suggest that mitochondrial folate metabolic enzymes could
serve as potential therapeutic targets for treatment of colorectal
cancer (6). The genomic analysis
of clinical samples is an entry point for developments in Precision
Medicine. Here we highlight recent developments in C1 metabolism
research.
Naturally, researchers have considered folate
metabolism as a plausible target for disease control. Antagonism of
folate metabolism has been the principal plank of chemotherapeutic
concept for more than 60 years. Farber and colleagues (7) noted that folic acid could stimulate
proliferation of acute lymphoblastic leukemia (ALL) cells and
wondered whether the intermediates of chemical synthesis could
antagonize cell proliferation. They conducted a pioneering study in
which they used aminopterin, one of the above-mentioned
intermediates, to induce clinical remission in patients with ALL
(8). Thereafter, multiple pathways
downstream of C1 metabolism were identified and targeted by various
cytotoxic chemotherapeutic agents. For example, methotrexate (MTX),
an anti-folate agent that targets dihydrofolate reductase, is used
to treat various cancer and is an effective therapy for rheumatoid
arthritis (RA), despite its associated toxicity (9). The first documented use of
5-fluorouracil (5-FU) was reported by Spears et al (10); it was later approved for the
treatment of colorectal cancer. 5-FU is an analogue of the DNA
base, uracil, and is a potent thymidine synthase inhibitor that
blocks methylation of dUMP to dTMP and disrupts the folate cycle
(11). Similarly, gemcitabine,
another nucleotide metabolism inhibitor in the C1 metabolic
pathway, is used to treat pancreatic cancer (12). A previous study of
gemcitabine-resistant pancreatic cancer cells indicated that
microRNA-1246, which belongs to a class of non-coding RNAs, is
involved in the modulation of chemotherapy resistance and cancer
stem cell properties, which suggests a critical role of nucleotide
metabolism in cancer cell metabolism (12). The conceptual basis of 5-FU has
been used to develop a thymidine analog, trifluorothymidine (TFT),
as discussed below.
Recently, C1 metabolic enzymes were shown to be
novel therapeutic targets for cancer. Pandey et al (13) showed that inhibition of SHMT1 with
targeted siRNAs reduced tumor size in a mouse xenograft model.
Pickman et al (14)
demonstrated inhibition of acute myeloid leukemia cells by MTHFD2
knockdown-induced suppression of TCA in vivo. Small
compounds for inhibition of SHMT1 or MTHFD2 have already been
identified (15–18). These compounds may undergo further
development as novel drugs for cancer therapy in the foreseeable
future.
Regarding nucleotide medicine, microRNAs have been
shown to exert various effects on cells, such as epigenetic
reprogramming via modulation of the methylation pathway (19,20).
Later studies indicated that specific microRNAs, such as
microRNA-302, could induce reprogramming in cancer cells, thus,
identifying these as candidate moieties for treatment of refractory
cancer cells from a nucleotide medicine perspective (21–23).
Furthermore, microRNA-369 was shown to modulate the activity of a
splicing factor of pyruvate kinase (PK), which induces metabolic
reprogramming (24). Taken
together, nucleotide metabolism plays a critical role in C1
metabolism and allows the generation of useful tools for
mechanistic studies and therapeutic tools with which to target
cancer cells.
Control of methylation events might be plausible,
given the significance of epigenetic events with regard to the
malignant phenotype of cancer (25,26).
Previous research has shown that a temporarily distinct
subpopulation of slow-cycling melanoma cells in which the H3K4
demethylase JARID1B (KDM5B/PLU-1/RBP2-H1) play a role is required
for continuous tumor growth (27).
These slow-cycling cells, which exhibit slow DNA replication and
are likely resistant to chemotherapeutic reagents (e.g., genotoxic
agents) and radiation, may be instrumental in tumor relapse and
metastasis. In solid cancers, KDM family members are implicated in
carcinogenesis, and knockdown of associated genes has been shown to
inhibit tumorigenicity and elicit cellular senescence (28,29).
Several reagents, such as dimethyl sulfoxide (DMFO), have been
developed to target methylation donors, ornithine decarboxylation
(ODC), and polyamine metabolism and have been evaluated in clinical
trials (30).
The methionine cycle produces S-adenocyl methionine
(SAM), which acts as a methyl donor in methylation reactions
(40). SAM is involved in the
methylation of histones, DNA and RNA, as well as of lysine and
arginine in general proteins. SAM is coupled with ornithine
metabolic pathway. In a study of PK, which catalyzes the last step
of glycolysis, PKM2 knock-down in the allele contributed to the
generation of SAM in mice (24),
which suggests an important role of PKM2 in the modulation of
cancer phenotypes via SAM-mediated control of methylation. PKM2,
which results from alternative splicing of the PK gene, was
preferentially expressed in tumors relative to PKM1, which is
expressed in differentiated cells. PK contributes to the production
and transportation of pyruvate in the mitochondria and is thus,
associated with folate production in C1 metabolism. This gateway
function of PK is altered in colorectal cancer, wherein the
translocation of PKM2 protein into the nucleus via TGF-β
stimulation has been observed in metastatic cancer cells (41); notably, pyruvate dehydrogenase is
also affected in cancer cells (42).
SAM production is associated with polyamine
metabolism in which ornithine decarboxylation (ODC) functions as a
restricting step in the metabolic flow (43). Studies of an ODC enzyme revealed
the characteristic cancer stem cell properties of fluorescent
cancer cells harboring a GFP-ODC enzyme fusion cassette (44–46).
These GFP-ODC labeled cancer cells exhibited the most aggressive
tumorigenicity in immunodeficient mice, were resistant to
chemotherapy and radiation therapy and exhibited reduced production
of reactive oxygen species (ROS). A trans-omics mathematical
analysis that linked metabolome data with transcriptome data
revealed novel functions of the ornithine metabolic pathway in
cancer stem cells (47). Given
that ornithine is located upstream of polyamine metabolism, the
polyamine flow might play a role in the maintenance of cancer
stemness. Thus, C1 metabolism helps to control treatment-refractory
cancer stem cells.
Although genetic alterations are not the sole
pathogenetic mechanism of carcinogenesis, these factors undoubtedly
play a significant role in disease initiation and progression
(48–50). Studies of hereditary diseases that
are known to predispose to cancer have indicated the involvement of
ectopically activated oncogenes and the inhibition of tumor
suppressor genes (51). In the
1990s, numerous studies suggested that in cancer patients, commonly
deleted genomic regions might contain tumor suppressor genes
(52); accordingly, introduction
of these missing genes to cancer cells might inactivate tumor cell
proliferation and cell cycle progression and thus suppress
tumorigenicity (53). Positional
cloning approaches to the identification of critical genes in the
common fragile sites on chromosome 3p14 led to the identification
of the fragile histidine triad (FHIT) gene, which encodes an
enzyme with dinucleotide hydrolase activity (diadenosine
tri-phosphate hydrolase) and a role in purine metabolism (54). A subsequent biochemical study
indicated the importance of His96 as a catalyst for the hydrolysis
of phosphoanhydrides such as Ap3A (55). More than 50% of human tumors
exhibit focal deletion of this gene (56). Experiments in mice have indicated a
deficiency in FHIT-induced genomic instability and
spontaneous tumor formation, both of which were suppressed by the
introduction of FHIT (3,57).
Mitochondrial quality is known to influence cellular
differentiation. For example, certain mutations in mitochondrial
DNA (mtDNA) affect cellular reprogramming. Reprogramming induction
in fibroblasts harboring mtDNA mutations revealed drastically
reduced reprogramming efficiency of these cells relative to that of
wild-type fibroblast cells (63).
Reduced reprogramming efficiency has also been observed in human
cells that harbor large mtDNA deletions (64), as well as in clonal human
fibroblast cells with very high frequency of mt-tRNA point
mutations. In addition, mtDNA has been suggested to affect
reprogramming efficiency (57,58).
However, the induced pluripotent stem cell lines showed different
pathological mtDNA point mutations (20,25,63–66).
In these cells, no significant difference in reprogramming
efficiency was observed between the normal and mutated lines. Many
studies have associated heteroplasmic mtDNA mutations with specific
segregation patterns during reprogramming. This phenomenon was not
only observed in the induced pluripotent stem cells, but also in
mouse germ cells and during epiblast differentiation in monkey
embryos (11,67).
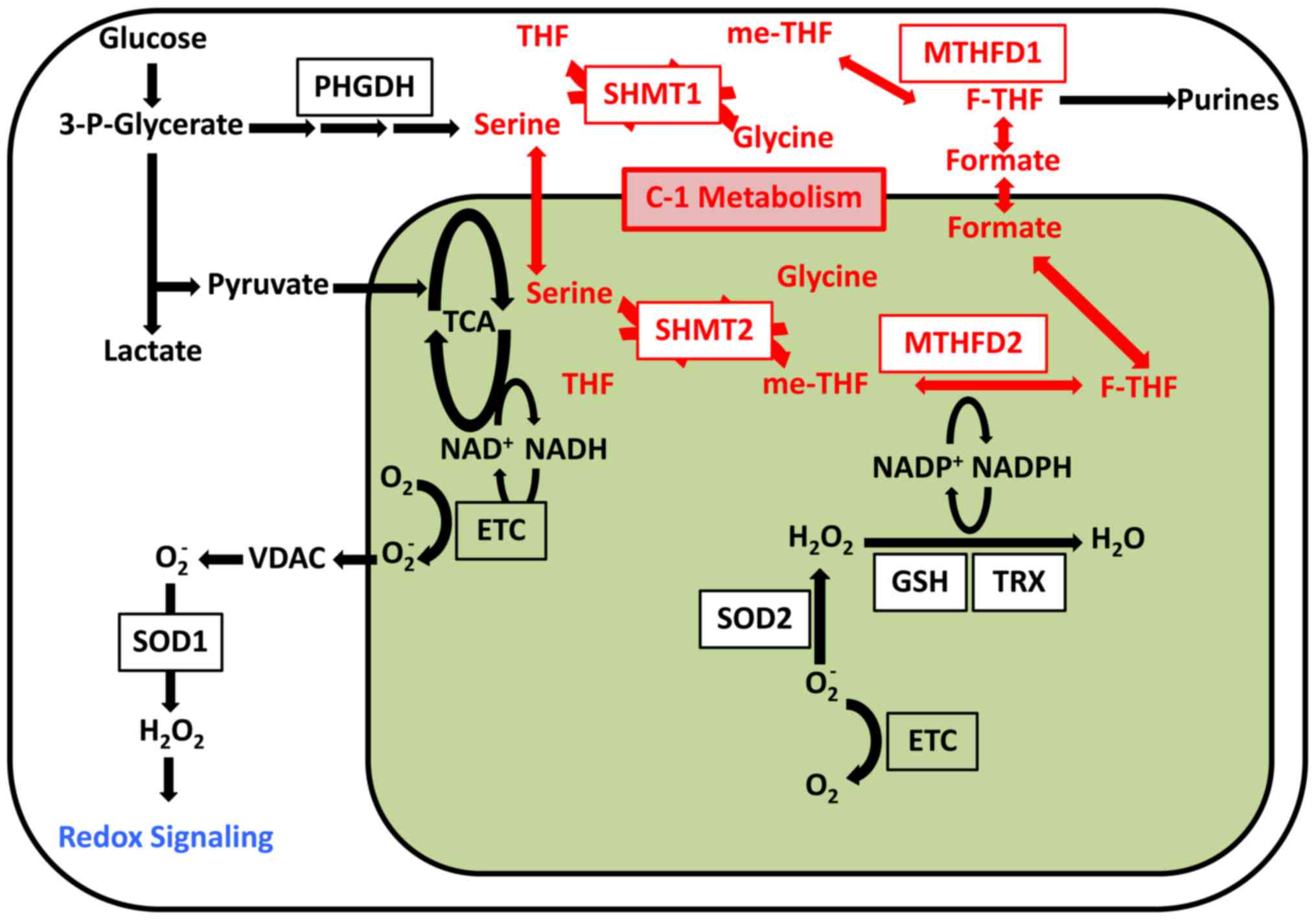
Furthermore, tDNA mutation was found to induce ROS.
ROS signaling determines cell fate. For example, mitochondrial ROS
was shown to induce differentiation of hematopoietic stem cells
(HSCs) (9,30). Therefore, ROS was thought to
mediate signaling and thus affect cell differentiation. Induced
pluripotent stem cells with mtDNA mutations retain high levels of
ROS (63), although this phenotype
can be rescued via treatment with antioxidants such as
n-acetyl-l-cysteine (NAC). Altered ROS signaling is thought to
induce the mtDNA mutation phenotype in stem cells (63). Therefore, the mitochondria is an
organelle involved in signal transduction (Fig. 2).
The present review was supported in part by a
Grant-in-Aid for Scientific Research from the Ministry of
Education, Culture, Sports, Science and Technology; a Grant-in-Aid
from the Third Comprehensive 10-year Strategy for Cancer Control,
Ministry of Health, Labor and Welfare; a grant from the Kobayashi
Cancer Research Foundation; a grant from the Princess Takamatsu
Cancer Research Fund, Japan; a grant from the National Institute of
Biomedical Innovation; and a grant from the Osaka University Drug
Discovery Funds. A.H. is a research fellow of the Japan Society for
the Promotion of Science. Partial support was received from Taiho
Pharmaceutical, Co., Ltd., (H.I., J.K. and M.M.), Chugai, Co.,
Ltd., Yakult Honsha, Co., Ltd., Merck, Co., Ltd., Takeda Science
Foundation and Takeda Medical Research Foundation (M.K., M.M., N.N.
and H.I.) through institutional endowments.
|
1
|
Ghosh D and Poisson LM: 'Omics' data and
levels of evidence for biomarker discovery. Genomics. 93:13–16.
2009. View Article : Google Scholar
|
|
2
|
Zong WX, Rabinowitz JD and White E:
Mitochondria and Cancer. Mol Cell. 61:667–676. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Locasale JW: Serine, glycine and
one-carbon units: Cancer metabolism in full circle. Nat Rev Cancer.
13:572–583. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hanley MP and Rosenberg DW: One-carbon
metabolism and colorectal cancer: Potential mechanisms of
chemoprevention. Curr Pharmacol Rep. 1:197–205. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Padmanabhan N and Watson ED: Lessons from
the one-carbon metabolism: Passing it along to the next generation.
Reprod Biomed Online. 27:637–643. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Miyo M, Konno M, Colvin H, Nishida N,
Koseki J, Kawamoto K, Tsunekuni K, Nishimura J, Hata T, Takemasa I,
et al: The importance of mitochondrial folate enzymes in human
colorectal cancer. Oncol Rep. 37:417–425. 2016.PubMed/NCBI
|
|
7
|
Farber S, Cutler EC, Hawkins JW, Harrison
JH, Peirce EC II and Lenz GG: The action of pteroylglutamic
conjugates on man. Science. 106:619–621. 1947. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Farber S, Diamond LK, Mercer RD, Sylvester
RF Jr and Wolff JA: Temporary remissions in acute leukemia in
children produced by folic acid antagonist, 4-aminopteroyl-glutamic
acid. N Engl J Med. 238:787–793. 1948. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chabner BA and Roberts TG Jr: Timeline:
Chemotherapy and the war on cancer. Nat Rev Cancer. 5:65–72. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Spears CP, Shahinian AH, Moran RG,
Heidelberger C and Corbett TH: In vivo kinetics of thymidylate
synthetase inhibition of 5-fluorouracil-sensitive and -resistant
murine colon adenocarcinomas. Cancer Res. 42:450–456.
1982.PubMed/NCBI
|
|
11
|
Burris HA III, Moore MJ, Andersen J, Green
MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo
AM, Tarassoff P, et al: Improvements in survival and clinical
benefit with gemcitabine as first-line therapy for patients with
advanced pancreas cancer: A randomized trial. J Clin Oncol.
15:2403–2413. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hasegawa S, Eguchi H, Nagano H, Konno M,
Tomimaru Y, Wada H, Hama N, Kawamoto K, Kobayashi S, Nishida N, et
al: MicroRNA-1246 expression associated with CCNG2-mediated
chemoresistance and stemness in pancreatic cancer. Br J Cancer.
111:1572–1580. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pandey S, Garg P, Lee S, Choung HW, Choung
YH, Choung PH and Chung JH: Nucleotide biosynthesis arrest by
silencing SHMT1 function via vitamin B6-coupled vector and effects
on tumor growth inhibition. Biomaterials. 35:9332–9342. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pikman Y, Puissant A, Alexe G, Furman A,
Chen LM, Frumm SM, Ross L, Fenouille N, Bassil CF, Lewis CA, et al:
Targeting MTHFD2 in acute myeloid leukemia. J Med Chem.
213:1285–1306. 2016.
|
|
15
|
Marani M, Paone A, Fiascarelli A, Macone
A, Gargano M, Rinaldo S, Giardina G, Pontecorvi V, Koes D,
McDermott L, et al: A pyrazolopyran derivative preferentially
inhibits the activity of human cytosolic serine
hydroxymethyltransferase and induces cell death in lung cancer
cells. Oncotarget. 7:4570–4583. 2016.
|
|
16
|
Paiardini A, Fiascarelli A, Rinaldo S,
Daidone F, Giardina G, Koes DR, Parroni A, Montini G, Marani M,
Paone A, et al: Screening and in vitro testing of antifolate
inhibitors of human cytosolic serine hydroxymethyltransferase.
ChemMedChem. 10:490–497. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Witschel MC, Rottmann M, Schwab A,
Leartsakulpanich U, Chitnumsub P, Seet M, Tonazzi S, Schwertz G,
Stelzer F, Mietzner T, et al: Inhibitors of plasmodial serine
hydroxymethyltransferase (SHMT): Cocrystal structures of
pyrazolopyrans with potent blood- and liver-stage activities. J Med
Chem. 58:3117–3130. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gustafsson R, Jemth AS, Gustafsson
Sheppard N, Färnegårdh K, Loseva O, Wiita E, Bonagas N, Dahllund L,
Llona-Minguez S and Häggblad M: Crystal structure of the emerging
cancer target MTHFD2 in complex with a substrate-based inhibitor.
Cancer Res. Nov 29–2016.Epub ahead of print. PubMed/NCBI
|
|
19
|
Miyoshi N, Ishii H, Nagano H, Haraguchi N,
Dewi DL, Kano Y, Nishikawa S, Tanemura M, Mimori K, Tanaka F, et
al: Reprogramming of mouse and human cells to pluripotency using
mature microRNAs. Cell Stem Cell. 8:633–638. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Anokye-Danso F, Trivedi CM, Juhr D, Gupta
M, Cui Z, Tian Y, Zhang Y, Yang W, Gruber PJ, Epstein JA, et al:
Highly efficient miRNA-mediated reprogramming of mouse and human
somatic cells to pluripotency. Cell Stem Cell. 8:376–388. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Miyoshi N, Ishii H, Nagai K, Hoshino H,
Mimori K, Tanaka F, Nagano H, Sekimoto M, Doki Y and Mori M:
Defined factors induce reprogramming of gastrointestinal cancer
cells. Proc Natl Acad Sci USA. 107:40–45. 2010. View Article : Google Scholar :
|
|
22
|
Dewi D, Ishii H, Haraguchi N, Nishikawa S,
Kano Y, Fukusumi T, Ohta K, Miyazaki S, Ozaki M, Sakai D, et al:
Reprogramming of gastrointestinal cancer cells. Cancer Sci.
103:393–399. 2012. View Article : Google Scholar
|
|
23
|
Ogawa H, Wu X, Kawamoto K, Nishida N,
Konno M, Koseki J, Matsui H, Noguchi K, Gotoh N, Yamamoto T, et al:
MicroRNAs induce epigenetic reprogramming and suppress malignant
phenotypes of human colon cancer cells. PLoS One. 10:e01271192015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Konno M, Koseki J, Kawamoto K, Nishida N,
Matsui H, Dewi DL, Ozaki M, Noguchi Y, Mimori K, Gotoh N, et al:
Embryonic microRNA-369 controls metabolic splicing factors and
urges cellular reprograming. PLoS One. 10:e01327892015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Avgustinova A and Benitah SA: The
epigenetics of tumour initiation: Cancer stem cells and their
chromatin. Curr Opin Genet Dev. 36:8–15. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rotili D and Mai A: Targeting histone
demethylases: A new avenue for the fight against cancer. Genes
Cancer. 2:663–679. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Roesch A, Fukunaga-Kalabis M, Schmidt EC,
Zabierowski SE, Brafford PA, Vultur A, Basu D, Gimotty P, Vogt T
and Herlyn M: A temporarily distinct subpopulation of slow-cycling
melanoma cells is required for continuous tumor growth. Cell.
141:583–594. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kano Y, Konno M, Ohta K, Haraguchi N,
Nishikawa S, Kagawa Y, Hamabe A, Hasegawa S, Ogawa H, Fukusumi T,
et al: Jumonji/Arid1b (Jarid1b) protein modulates human esophageal
cancer cell growth. Mol Clin Oncol. 1:753–757. 2013.
|
|
29
|
Ohta K, Haraguchi N, Kano Y, Kagawa Y,
Konno M, Nishikawa S, Hamabe A, Hasegawa S, Ogawa H, Fukusumi T, et
al: Depletion of JARID1B induces cellular senescence in human
colorectal cancer. Int J Oncol. 42:1212–1218. 2013.PubMed/NCBI
|
|
30
|
Casero RAJ Jr and Marton LJ: Targeting
polyamine metabolism and function in cancer and other
hyperproliferative diseases. Nat Rev Drug Discov. 6:373–390. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Warren TK, Jordan R, Lo MK, Ray AS,
Mackman RL, Soloveva V, Siegel D, Perron M, Bannister R, Hui HC, et
al: Therapeutic efficacy of the small molecule GS-5734 against
Ebola virus in rhesus monkeys. Nature. 531:381–385. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Longley DB, Harkin DP and Johnston PG:
5-fluorouracil: Mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sakuramoto S, Sasako M, Yamaguchi T,
Kinoshita T, Fujii M, Nashimoto A, Furukawa H, Nakajima T, Ohashi
Y, Imamura H, et al ACTS-GC Group: Adjuvant chemotherapy for
gastric cancer with S-1, an oral fluoropyrimidine. N Engl J Med.
357:1810–1820. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kaufman HE and Heidelberger C: Therapeutic
antiviral Action of 5-trifluoromethyl-2′-deoxyuridine in Herpes
simplex keratitis. Science. 145:585–586. 1964. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mayer RJ, Van Cutsem E, Falcone A, Yoshino
T, Garcia-Carbonero R, Mizunuma N, Yamazaki K, Shimada Y, Tabernero
J, Komatsu Y, et al RECOURSE Study Group: Randomized trial of
TAS-102 for refractory metastatic colorectal cancer. N Engl J Med.
372:1909–1919. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yoshino T, Mizunuma N, Yamazaki K, Nishina
T, Komatsu Y, Baba H, Tsuji A, Yamaguchi K, Muro K, Sugimoto N, et
al: TAS-102 monotherapy for pretreated metastatic colorectal
cancer: A double-blind, randomised, placebo-controlled phase 2
trial. Lancet Oncol. 13:993–1001. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Honma Y, Yamada Y, Terazawa T, Takashima
A, Iwasa S, Kato K, Hamaguchi T, Shimada Y, Ohashi M, Morita S, et
al: Feasibility of neoadjuvant S-1 and oxaliplatin followed by
surgery for resectable advanced gastric adenocarcinoma. Surg Today.
46:1076–1082. 2016. View Article : Google Scholar
|
|
38
|
Uehara K and Nagino M: Neoadjuvant
treatment for locally advanced rectal cancer: A systematic review.
Surg Today. 46:161–168. 2016. View Article : Google Scholar
|
|
39
|
Park IJ, Kim JY, Yu CS, Lee JS, Lim SB,
Lee JL, Yoon YS, Kim CW and Kim JC: Preoperative chemoradiotherapy
for clinically diagnosed T3N0 rectal cancer. Surg Today. 46:90–96.
2016. View Article : Google Scholar
|
|
40
|
Su X, Wellen KE and Rabinowitz JD:
Metabolic control of methylation and acetylation. Curr Opin Chem
Biol. 30:52–60. 2016. View Article : Google Scholar :
|
|
41
|
Hamabe A, Konno M, Tanuma N, Shima H,
Tsunekuni K, Kawamoto K, Nishida N, Koseki J, Mimori K, Gotoh N, et
al: Role of pyruvate kinase M2 in transcriptional regulation
leading to epithelial-mesenchymal transition. Proc Natl Acad Sci
USA. 111:15526–15531. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hamabe A, Yamamoto H, Konno M, Uemura M,
Nishimura J, Hata T, Takemasa I, Mizushima T, Nishida N, Kawamoto
K, et al: Combined evaluation of hexokinase 2 and phosphorylated
pyruvate dehydrogenase-E1α in invasive front lesions of colorectal
tumors predicts cancer metabolism and patient prognosis. Cancer
Sci. 105:1100–1108. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gerner EW and Meyskens FL Jr: Polyamines
and cancer: Old molecules, new understanding. Nat Rev Cancer.
4:781–792. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hayashi K, Tamari K, Ishii H, Konno M,
Nishida N, Kawamoto K, Koseki J, Fukusumi T, Kano Y, Nishikawa S,
et al: Visualization and characterization of cancer stem-like cells
in cervical cancer. Int J Oncol. 45:2468–2474. 2014.PubMed/NCBI
|
|
45
|
Kano Y, Konno M, Kawamoto K, Tamari K,
Hayashi K, Fukusumi T, Satoh T, Tanaka S, Ogawa K, Mori M, et al:
Novel drug discovery system for cancer stem cells in human squamous
cell carcinoma of the esophagus. Oncol Rep. 31:1133–1138.
2014.PubMed/NCBI
|
|
46
|
Tamari K, Hayashi K, Ishii H, Kano Y,
Konno M, Kawamoto K, Nishida N, Koseki J, Fukusumi T, Hasegawa S,
et al: Identification of chemoradiation-resistant osteosarcoma stem
cells using an imaging system for proteasome activity. Int J Oncol.
45:2349–2354. 2014.PubMed/NCBI
|
|
47
|
Koseki J, Matsui H, Konno M, Nishida N,
Kawamoto K, Kano Y, Mori M, Doki Y and Ishii H: A Trans-omics
mathematical analysis reveals novel functions of the ornithine
metabolic pathway in cancer Stem cells. Sci Rep. 6:207262016.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nowell PC: Foundations in cancer research.
Chromosomes and cancer: The evolution of an idea. Adv Cancer Res.
62:1–17. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Nowell PC and Croce CM: Chromosomes,
genes, and cancer. Am J Pathol. 125:7–15. 1986.PubMed/NCBI
|
|
52
|
Weinberg RA: Tumor suppressor genes.
Science. 254:1138–1146. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sherr CJ: Cancer cell cycles. Science.
274:1672–1677. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ohta M, Inoue H, Cotticelli MG, Kastury K,
Baffa R, Palazzo J, Siprashvili Z, Mori M, McCue P, Druck T, et al:
The FHIT gene, spanning the chromosome 3p14.2 fragile site and
renal carcinoma-associated t(3;8) breakpoint, is abnormal in
digestive tract cancers. Cell. 84:587–597. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Huang K and Frey PA: Engineering human
Fhit, a diadenosine triphosphate hydrolase, into an efficient
dinucleoside polyphosphate synthase. J Am Chem Soc. 126:9548–9549.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Huebner K and Croce CM: FRA3B and other
common fragile sites: The weakest links. Nat Rev Cancer. 1:214–221.
2001. View Article : Google Scholar
|
|
57
|
Dumon KR, Ishii H, Fong LY, Zanesi N,
Fidanza V, Mancini R, Vecchione A, Baffa R, Trapasso F, During MJ,
et al: FHIT gene therapy prevents tumor development in
Fhit-deficient mice. Proc Natl Acad Sci USA. 98:3346–3351. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Inoue H, Ishii H, Alder H, Snyder E, Druck
T, Huebner K and Croce CM: Sequence of the FRA3B common fragile
region: Implications for the mechanism of FHIT deletion. Proc Natl
Acad Sci USA. 94:14584–14589. 1997. View Article : Google Scholar
|
|
59
|
Mimori K, Druck T, Inoue H, Alder H, Berk
L, Mori M, Huebner K and Croce CM: Cancer-specific chromosome
alterations in the constitutive fragile region FRA3B. Proc Natl
Acad Sci USA. 96:7456–7461. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ishii H, Mimori K, Inoue H, Inageta T,
Ishikawa K, Semba S, Druck T, Trapasso F, Tani K, Vecchione A, et
al: Fhit modulates the DNA damage checkpoint response. Cancer Res.
66:11287–11292. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Semba S, Trapasso F, Fabbri M, McCorkell
KA, Volinia S, Druck T, Iliopoulos D, Pekarsky Y, Ishii H, Garrison
PN, et al: Fhit modulation of the Akt-survivin pathway in lung
cancer cells: Fhit-tyrosine 114 (Y114) is essential. Oncogene.
25:2860–2872. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Arlt MF, Casper AM and Glover TW: Common
fragile sites. Cytogenet Genome Res. 100:92–100. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Dayem AA, Choi HY, Kim JH and Cho SG: Role
of oxidative stress in stem, cancer, and cancer stem cells. Cancers
(Basel). 2:859–884. 2010. View Article : Google Scholar
|
|
64
|
Al-Hajj M, Wicha MS, Benito-Hernandez A,
Morrison SJ and Clarke MF: Prospective identification of
tumorigenic breast cancer cells. Proc Natl Acad Sci USA.
100:3983–3988. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ambrosone CB: Oxidants and antioxidants in
breast cancer. Antioxid Redox Signal. 2:903–917. 2000. View Article : Google Scholar
|
|
66
|
Barreiro E, Peinado VI, Galdiz JB, Ferrer
E, Marin-Corral J, Sánchez F, Gea J and Barberà JA; ENIGMA in COPD
Project: Cigarette smoke-induced oxidative stress: A role in
chronic obstructive pulmonary disease skeletal muscle dysfunction.
Am J Respir Crit Care Med. 182:477–488. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Cairns RA, Harris IS and Mak TW:
Regulation of cancer cell metabolism. Nat Rev Cancer. 11:85–95.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Kobayashi CI and Suda T: Regulation of
reactive oxygen species in stem cells and cancer stem cells. J Cell
Physiol. 227:421–430. 2012. View Article : Google Scholar
|
|
69
|
Turrens JF: Mitochondrial formation of
reactive oxygen species. J Physiol. 552:335–344. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Dickinson BC and Chang CJ: Chemistry and
biology of reactive oxygen species in signaling or stress
responses. Nat Chem Biol. 7:504–511. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Lee KW, Lee DJ, Lee JY, Kang DH, Kwon J
and Kang SW: Peroxiredoxin II restrains DNA damage-induced death in
cancer cells by positively regulating JNK-dependent DNA repair. J
Biol Chem. 286:8394–8404. 2011. View Article : Google Scholar :
|
|
72
|
Phillips TM, McBride WH and Pajonk F: The
response of CD24−/low/CD44+ breast
cancer-initiating cells to radiation. J Natl Cancer Inst.
98:1777–1785. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Giannoni E, Buricchi F, Raugei G, Ramponi
G and Chiarugi P: Intracellular reactive oxygen species activate
Src tyrosine kinase during cell adhesion and anchorage-dependent
cell growth. Mol Cell Biol. 25:6391–6403. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Hoeijmakers JH: DNA damage, aging, and
cancer. N Engl J Med. 361:1475–1485. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Yee C, Yang W and Hekimi S: The intrinsic
apoptosis pathway mediates the pro-longevity response to
mitochondrial ROS in C. elegans. Cell. 157:897–909. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Fruehauf JP and Meyskens FL Jr: Reactive
oxygen species: A breath of life or death? Clin Cancer Res.
13:789–794. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Szatrowski TP and Nathan CF: Production of
large amounts of hydrogen peroxide by human tumor cells. Cancer
Res. 51:794–798. 1991.PubMed/NCBI
|
|
78
|
Halliwell B: Oxidative stress and cancer:
Have we moved forward? Biochem J. 401:1–11. 2007. View Article : Google Scholar
|
|
79
|
Trachootham D, Alexandre J and Huang P:
Targeting cancer cells by ROS-mediated mechanisms: A radical
therapeutic approach? Nat Rev Drug Discov. 8:579–591. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Chan SM and Majeti R: Role of DNMT3A,
TET2, and IDH1/2 mutations in pre-leukemic stem cells in acute
myeloid leukemia. Int J Hematol. 98:648–657. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Hermann PC, Huber SL, Herrler T, Aicher A,
Ellwart JW, Guba M, Bruns CJ and Heeschen C: Distinct populations
of cancer stem cells determine tumor growth and metastatic activity
in human pancreatic cancer. Cell Stem Cell. 1:313–323. 2007.
View Article : Google Scholar
|
|
82
|
Eyler CE and Rich JN: Survival of the
fittest: Cancer stem cells in therapeutic resistance and
angiogenesis. J Clin Oncol. 26:2839–2845. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Kurtova AV, Xiao J, Mo Q, Pazhanisamy S,
Krasnow R, Lerner SP, Chen F, Roh TT, Lay E, Ho PL, et al: Blocking
PGE2-induced tumour repopulation abrogates bladder
cancer chemoresistance. Nature. 517:209–213. 2015. View Article : Google Scholar
|
|
84
|
Schafer ZT, Grassian AR, Song L, Jiang Z,
Gerhart-Hines Z, Irie HY, Gao S, Puigserver P and Brugge JS:
Antioxidant and oncogene rescue of metabolic defects caused by loss
of matrix attachment. Nature. 461:109–113. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Wang K, Zhang T, Dong Q, Nice EC, Huang C
and Wei Y: Redox homeostasis: The linchpin in stem cell
self-renewal and differentiation. Cell Death Dis. 4:e5372013.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Shi X, Zhang Y, Zheng J and Pan J:
Reactive oxygen species in cancer stem cells. Antioxid Redox
Signal. 16:1215–1228. 2012. View Article : Google Scholar : PubMed/NCBI
|