Introduction
It is well known that the extracellular pH (pHe)
within the microenvironment of tumours is significantly lower (more
acidic) compared with that of normal tissues (1). Although several factors play a role
in the acidification process, it is well accepted that the major
contribution arises from a shift in the ATP generation; according
to the Warburg effect, ATP is produced via aerobic glycolysis even
in presence of oxygen (2). In this
contest, poor vascularisation and increased activity of plasma
membrane ion pumps and transporters (H+-ATPases, the
Na+-H+ exchanger NHE1 and the
monocarboxylate-H+ efflux co-transporters MCT1 and MCT4)
contribute to the extracellular acidification of most, but not all
tumours (3). Many studies on solid
tumours and in particular on breast tumours suggested that a
glycolytic environment is related not only to uncontrolled
proliferation and evasion of apoptosis induction, but also to the
expansion through the extra-cellular matrix, the increase of the
metastatic potential and the promotion of angiogenesis (4). Therefore, novel drugs addressing
specific aspects of the deregulated tumour metabolism have been
proposed for inhibiting tumour growth and survival.
Dichloroacetate (DCA), a mitochondria-targeting
small molecule of 150 Da used to treat lactic acidosis, can reverse
this cancer-specific metabolic remodelling (5). DCA inhibits pyruvate dehydrogenase
kinase (PDK) whose expression is high in many cancers, causing
pyruvate conversion into lactate (6). DCA-mediated inhibition of PDK
re-establishes metabolism of pyruvate into the tricarboxylic acid
cycle and decreases the lactate accumulation and production. Thus,
DCA increases the glucose oxidation therefore promoting apoptosis
and blocking tumour proliferation (7,8).
Several clinical studies tested the DCA antitumour efficacy and its
safety in patients with advanced solid tumours (9). Overall, these studies reported that
oral DCA is well tolerated and safe, it reduces lactate levels and
it can act as an apoptosis sensitizer in combination with cytotoxic
treatments.
Despite the specific mechanism of action of DCA on
tumour glycolysis, tumour response to DCA treatment has
conventionally been assessed through simple measurements of changes
in tumour size by using morphological imaging techniques such as
magnetic resonance imaging (MRI) or computed tomography (CT).
However, the assessment of changes in tumour volume does not
provide indications on tumour acidosis variations induced by
metabolism-targeting drugs. Several MRI methods have been proposed
for non-invasive measurements of in vivo tumour pH (10). In particular,
31P-magnetic resonance spectroscopy (31P-MRS)
allows measuring intracellular and extracellular pH changes from
the chemical shift of endogenous phosphate or of exogenous agents,
but with poor spatial resolution (11). Gd-based contrast agents that
exhibit a pH-dependent relaxivity have been exploited for measuring
tumour pH in a rat glioma model, but a limitation of this approach
is related to the need of injecting two Gd-based contrast agents
for accurate pH measurements (12). Hyperpolarized
13C-bicarbonate can provide pHe map with high
sensitivity (13), while several
hyperpolarised molecules can provide insight into metabolism in
both cells and animals (14–18).
However, this technique is expensive, limited by low spatial
resolution and requires sophisticated instrumentations that are not
easily available in the clinical setting.
Recently, chemical exchange saturation transfer
(CEST) imaging has been proposed as a novel MRI-based technique and
several agents have been considered for assessing tumour metabolism
and pHe (19–23). Among them, clinical approved
radiographic contrast agents and heterocyclic compounds have been
exploited for measuring pH and pathological-induced pH changes
(24–28). In addition, MRI-CEST pH mapping was
demonstrated to be an excellent tool to investigate the
relationship between glycolysis and acidosis at clinical magnetic
field (29).
In this study, we investigated whether DCA-induced
changes in metabolism and tumour acidosis in a murine breast cancer
model can be monitored non-invasively by MRI-CEST pH mapping.
Materials and methods
Cell culture
TS/A cells, derived from a spontaneous BALB/c
mammary tumour, were grown in RPMI-1640 medium supplemented with
10% fetal bovine serum (FBS), 100 U/ml penicillin and 100
μg/ml streptomycin (Pen/Strep) and 2 mM L-glutamine
(30). 4T1 cells, a BALB/c-derived
mouse mammary carcinoma corresponding to stage IV of human breast
cancer, were purchased from American Type Culture Collection (ATCC
LGC Standards, Sesto San Giovanni, Italy) and cultured as TS/A
cells. TUBO cells were derived from a lobular carcinoma arising
spontaneously in a BALB-neuT mouse and were grown in DMEM medium
supplemented with 20% FBS and Pen/Strep (31).
J774 non-tumour cell line (purchased from ATCC) was
grown in DMEM medium supplemented with 10% FBS, Pen/Strep and
L-glutamine. All the cell lines were cultured in a humidified
atmosphere (37°C, 5% CO2).
Cell vitality test
The cytotoxic effect of DCA (Sigma-Aldrich, St.
Louis, MO, USA) on cells was analysed with the CellTiter-Blue Cell
Viability Assay (Promega Corp., Madison, WI, USA). Briefly, TS/A
(5×103) cells were plated in 96-well culture plates and
after 24 h of incubation were treated with DCA (1, 5 and 10 mM) for
24 h. The non-tumour cell line J774 (30×103) was used as
control and treated in the same way. Afterwards, cells were washed
with PBS and then CellTiter-Blue reagent was added to each well.
Fluorescence was measured at 560Ex/590Em using a 96-well plate
reader. Each assay was repeated at least three times.
pH measurement in normoxia and hypoxia
condition
TS/A cells were seeded in 60-mm culture dish with a
final volume of 3 ml at a density of 4×105 cells. After
24 h of incubation in 20% O2 (normoxia) or 1%
O2 (hypoxia) (New Brunswick™ Galaxy® 48 R,
Eppendorf S.r.l., Milan, Italy) cells were treated with a solution
of DCA (1, 5 and 10 mM) and kept in the culture medium for
additional 24 h. Then the culture medium was collected and the pH
was immediately measured using a pH meter (Hamilton®
Slim Trode, GR, Switzerland) previously calibrated. The non-tumour
cell line J774 underwent the same procedure. Each experiment was
performed in triplicate.
FACS analysis
The cell cycle perturbations were analysed by
propidium iodide (PI) DNA staining. TS/A cells (5×105)
were treated with DCA (1, 5 and 10 mM) for 24 h. At the end of each
treatment, cells were collected after a centrifugation (200 × g, 5
min) and then fixed in ethanol (70%, 3 min, 4°C). Ethanol-suspended
cells were diluted with phosphate buffered saline (PBS) and then
centrifuged to remove residual ethanol (471 × g, 5 min). For cell
cycle analysis, the pellets were suspended in 0.1 ml of PBS
containing 50 μg/ml of PI, 100 μg/ml of RNase A and
0.05% of Triton X-100 and incubated at 37°C for 40 minutes. Cell
cycle profiles were studied using a CyanADP flow cytometer and
analysed with Summit 3.4 software (Beckman Coulter, Milano, Italy)
and BD FACSuite software (Becton Dickinson, Milano, Italy). Each
assay was repeated a minimum of three times.
Animal experiments
BALB/c female mice (Charles River Laboratories
Italia S.r.l., Calco, Italy) were maintained in the animal facility
of the Dept. Molecular Biotechnology and Health Sciences,
University of Turin, under specific pathogen-free conditions. All
animal studies were approved by the University Ethics Committee in
accordance with the European guidelines under directive
2010/63.
Mice were inoculated subcutaneously with
2.5×105 TS/A mammary adenocarcinoma cells on both
flanks. When the tumours were approximately 60 mm3, TS/A
tumour-bearing mice were randomly divided in two groups: untreated
group that received drinking water and intraperitoneal injections
of PBS, and DCA-treated group that received DCA by oral
administration of 0.45 g/l (100 mg/kg/day) and also by
intraperitoneal injections of 50 g/l (200 mg/kg/day) (32). DCA or PBS solutions were
administered every day after baseline measurements. All mice were
scanned at day 0 (untreated n=10 mice, treated n=8 mice), 3 days
(untreated n=10 mice, treated n=8 mice) and 15 days (untreated n=7
mice, treated n=5 mice) post-treatment. At each time point
post-treatment, three mice per group were sacrificed and tumour
tissues were excised for lactate level quantification.
For MRI acquisition mice were anesthetized and the
breath rate was monitored by an air pillow placed below the animal
(SA Instruments, Stony Brook, NY, USA). MRI-CEST pH mapping was
performed upon i.v. injection of 4 g I/kg b.w. iopamidol (Bracco
Imaging SpA, Colleretto Giacosa, Italy) into the tail vein through
a 27-gauge needle.
MRICEST pH-mapping acquisition and
analysis
MR images were acquired with a Bruker 7T Avance 300
MRI scanner (Bruker Biospin, Ettlingen, Germany) equipped with a
30-mm 1H birdcage coil before starting the treatment, after 3 and
15 days of treatment.
Anatomical T2w images were acquired with
a Fast Spin Echo (FSE) sequence and the same geometry was used for
the following CEST experiments. CEST images were acquired before
and after iopamidol i.v. injection with a single shot FSE sequence
with centric encoding preceded by a continuous-wave saturation
pulse (power: 3μT, duration: 5 sec) on a central tumour
slice (field-of-view: 3 cm, matrix: 128×128, in-plane resolution:
234 μm, slice thickness: 1.5 mm). CEST images were analysed
using homemade MATLAB scripts (Mathworks, Inc., Natick, MA, USA).
Briefly, saturation transfer effects were calculated upon
irradiating at 4.2 and at 5.5 ppm, respectively, and post-contrast
ST maps were subtracted to pre-contrast ST ones, to obtain the
corresponding ST contrast difference (ΔST) maps. The pixel-by-pixel
extracellular pH (pHe) maps were calculated using the ratio between
the two contrast difference maps at 4.2 and 5.5 ppm according to
the previously described method (24). The calculated pHe maps were
superimposed to the anatomical reference image.
A novel estimate, dubbed acidity score, was
calculated for taking into account the heterogeneity of pHe
distribution values within the tumour region. The tumour pixels
were clustered into three groups: group I for pixels showing pHe
values >7.0, group II for pixels showing pHe values >6.7 and
<7.0, group III for pixels showing pHe values <6.7. The
percentage of pixels of each group was multiplied by a factor
between 1 and 3, to obtain the acidity score, in accordance to the
equation:
The acidity score ranges from 1 (less acidic) to 3 (more acidic),
describing tumour regions with different acidosis levels.
Survival curves
A second cohort of female BALB/c mice was inoculated
with 2.5×105 TS/A cells into the right flank for
long-term effect following DCA treatment. After the tumour reached
1 mm mean diameter, animals were randomized into two groups:
untreated (n=9 mice) and DCA-treated (n=11 mice). DCA-treated group
received DCA by oral administration (100 mg/kg/day) and also by
intraperitoneal injection (200 mg/kg/day) every day. Untreated
group received equal volumes of PBS. Mice were monitored every day
and volumes were measured using a calliper and calculated from
orthogonal measurements of external dimensions as
(width2 × length)/2. Mice were euthanized when tumour
volume reached values around 600 mm3.
Lactate assay
TS/A cells were seeded in 60-mm culture dish with a
final volume of 3 ml and incubated under standard cell culture
condition overnight for 24 h. Thereafter, cells were treated with
DCA for 24 h and culture media was collected and tested for lactate
level following the manufacturer's instructions of Lactate assay
kit (MAK064 Sigma-Aldrich, St. Louis, MO, USA).
After MRI acquisition, three mice per each time
point and group were sacrificed and tumour tissues were excised and
frozen in liquid nitrogen. Tumour tissue was explanted after animal
sacrifice and it was frozen in liquid nitrogen. Tumour was then
homogenized on ice in 4 volumes of lactate assay buffer; the sample
was centrifuged at 13.000 × g for 10 min to remove insoluble
material and deproteinized with a 10 kDa MWCO spin filter to remove
endogenous lactate dehydrogenase. The soluble fraction was then
assayed by Lactate assay kit.
Statistical analysis
Calculations were performed using GraphPad Prism
(GraphPad Software, La Jolla, CA, USA) software package.
Significance between DCA-treated and control groups was determined
by one-way analysis of variance, followed by post-hoc tests with
the Dunnett's multiple comparison test. Correlation analysis was
performed using Pearson's r correlation coefficient. Data are
presented as mean ± SD unless otherwise stated. Statistical
significance was established at P<0.05.
Results
DCA metabolic inhibition impairs TS/A
proliferation but not the cell cycle
In order to investigate the metabolic inhibition
efficiency of DCA, a panel of breast cancer cell lines was treated
with several concentrations of DCA for 24 h under normoxia
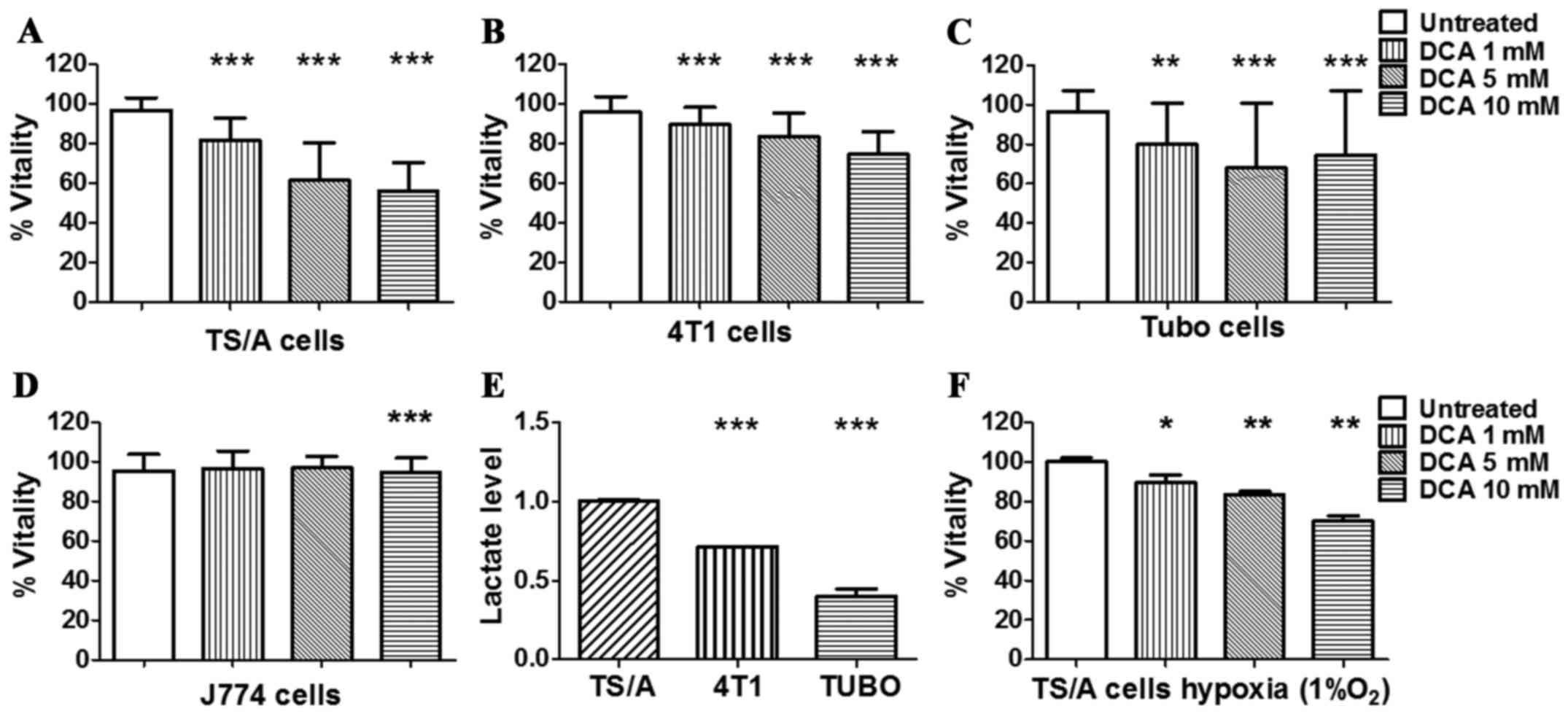
condition. As shown in Fig. 1,
after 24 h of treatment, TS/A, 4T1 and TUBO cancer cell lines
showed a reduction in their metabolic capacity even at low
concentration, whereas the non-tumour J774 control cell line was
not affected (Fig. 1D). The
response of TS/A breast cancer after 24 h of DCA treatment was
dose-dependent (Fig. 1A) and a
significant reduction of cell vitality was already observed at 5 mM
DCA concentration.
Furthermore, compared to the other cell lines,
untreated TS/A cells produced significantly higher level of lactate
(Fig. 1E), hence reflecting a more
glycolytic phenotype that could explain their higher sensitivity to
DCA treatment. On the basis of these observations, TS/A cell line
was selected for subsequent in vitro and in vivo
studies.
DCA effect was also investigated under hypoxia (1%
O2) to mimic in vivo tumour hypoxic conditions. A
significant decrease in TS/A vitality was observed for all the
investigated DCA concentrations, compared to untreated cells
(Fig. 1F).
In order to verify if the effect exerted by DCA on
cancer cell vitality was due to a perturbation of their cell cycle,
as reported for glioblastoma, glioma, non-small cell lung cancer
and other cancer cell lines (33–36),
TS/A cells were incubated with DCA for 24 h, then stained with PI
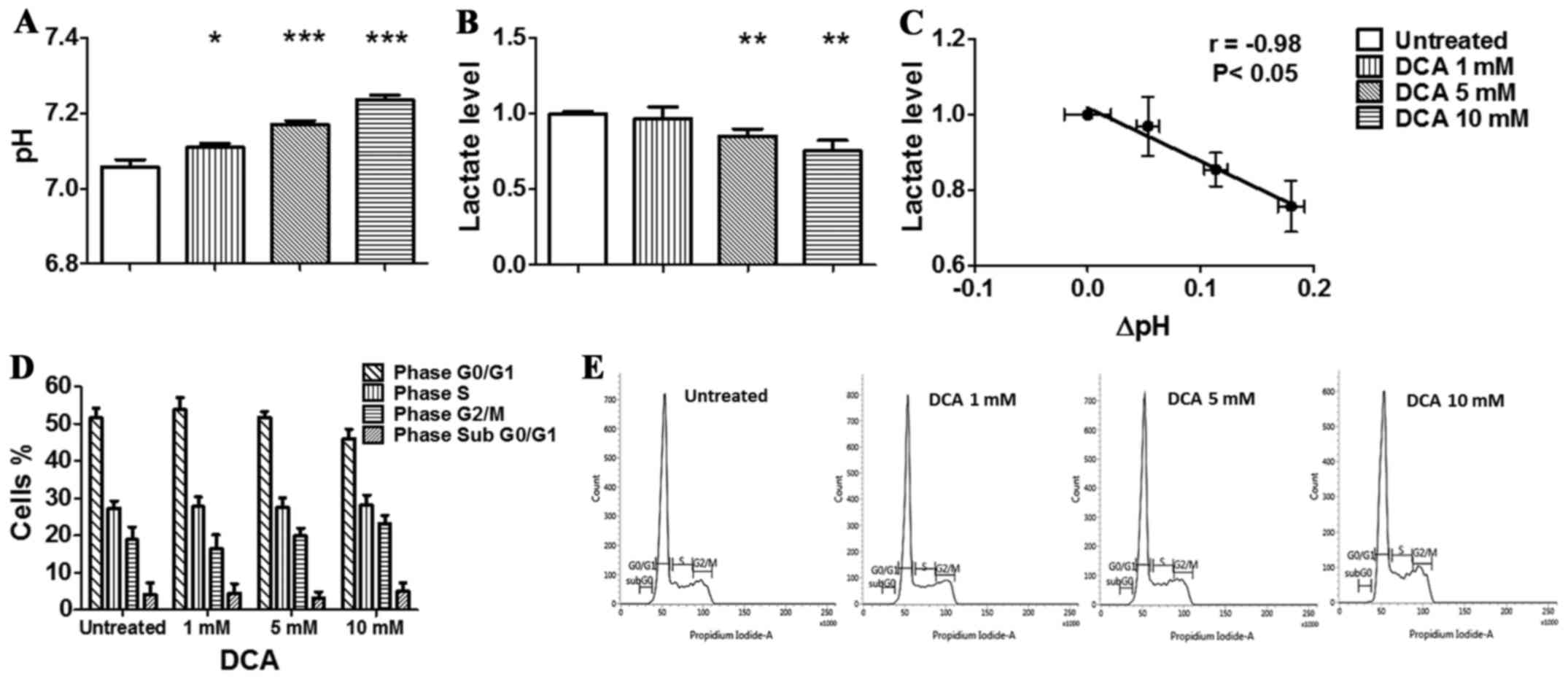
and analysed by flow cytometry. As shown in the graph (Fig. 2D) and in the representative
histograms reported in Fig. 2E,
DCA did not affect the cell cycle of TS/A cells, although the
amount of cells in the G1/G0 phase decreased from 51.6% (untreated
cells) to 45.9% in the presence of 10 mM DCA while cells in G2/M
phase increased from 18.8% (untreated cells) to 23.1%. The amount
of TS/A cells in the S phase did not change between untreated and
treated with 10 mM of DCA. Moreover, the percentage of hypodiploid
cells undergoing apoptosis-induced DNA fragmentation, represented
by the sub-G1/G0 population, was very low in all the conditions
analysed, and was not increased by DCA treatment. These data
suggest that DCA can affect cell vitality through different
mechanisms in different types of tumours, mainly acting as a
metabolic agent.
DCA promotes extracellular pH
alkalinisation in both normoxia and hypoxia conditions
Treatment of TS/A cells for 24 h in normoxic
condition (Fig. 2A) resulted in a
moderate but significant and constant increase of culture medium pH
values (from 7.06±0.02 to 7.23±0.01) with increasing DCA
concentrations. The increase of pHe was significantly more
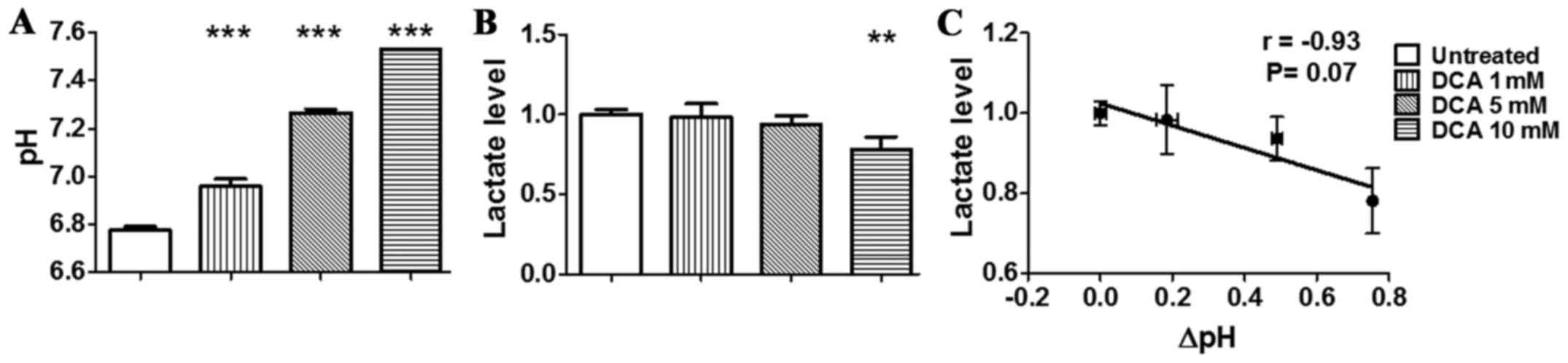
pronounced in the hypoxic condition (Fig. 3A) than in the normoxic condition.
In fact, hypoxic condition resulted in lower pH of the culture
medium for untreated TS/A cells in comparison to normoxic condition
(pHe = 6.78±0.02). Moreover, DCA treatment resulted in an even
higher increase of pHe up to 7.53±0.01 (P<0.001). The measured
increase in extracellular pH displayed by TS/A cells after
treatment is linked to the DCA-induced switch from glycolytic to
more oxidative phenotype as evidenced by decreased lactate levels
measured in the extracellular medium of TS/A cells. In particular,
we observed a significant decrease in lactate production following
DCA treatment in both normoxic and hypoxic conditions (Figs. 2B and 3B). Moreover, the in vitro
anti-glycolytic effect of DCA revealed a strong and significant
correlation between pH and lactate changes in normoxic condition
(r= −0.98, P=<0.05, Fig. 2C),
likely reflecting the impaired glycolytic activity that results in
a reduced lactate production, hence acidification. A strong
correlation was also found for cells treated in hypoxic condition
even though not statistically significant (r= −0.93, P=0.07,
Fig. 3C). Taken together, these
data confirmed the in vitro reversal of the glycolytic
phenotype following DCA treatment.
DCA does not influence tumour growth and
survival
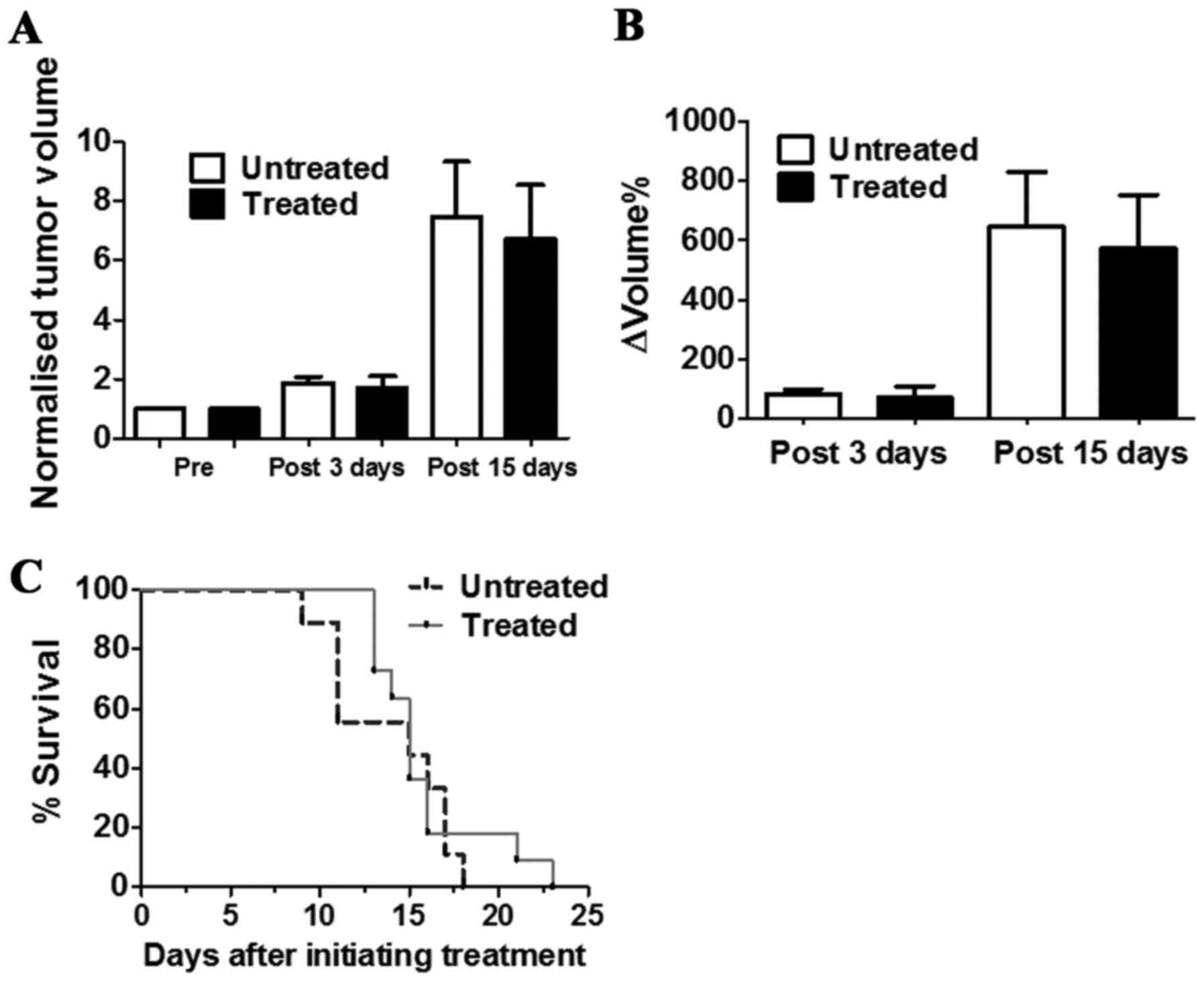
The in vivo antitumour activity of DCA was
evaluated by measuring tumour growth in a group of TS/A
tumour-bearing BALB/c mice that received drinking water and PBS
intraperitoneal injection (untreated) or that were treated with DCA
by oral administration and intraperitoneal injection every day for
3 or 15 consecutive days (Fig.
4A). DCA treatment slightly reduced the growth of TS/A breast
tumours after three days of treatment (ΔVolume% = 70.7±38.6 and
83.1±15.7, for treated and untreated mice, respectively;
P>0.05). This limited growth reduction is maintained up to 15
days of DCA treatment (ΔVolume% = 571.9±180.7 and 646.7±184.0, for
untreated and treated mice, respectively; P>0.05) (Fig. 4B).
The survival study conducted in another group of
mice revealed that DCA-treated mice survived slightly longer than
the untreated mice (Fig. 4C),
despite this difference not being statistically significant. These
data suggest that DCA did not influence tumour growth and did not
improve mouse survival for the TS/A tumour model.
DCA affects in vivo tumour acidosis and
lactate production
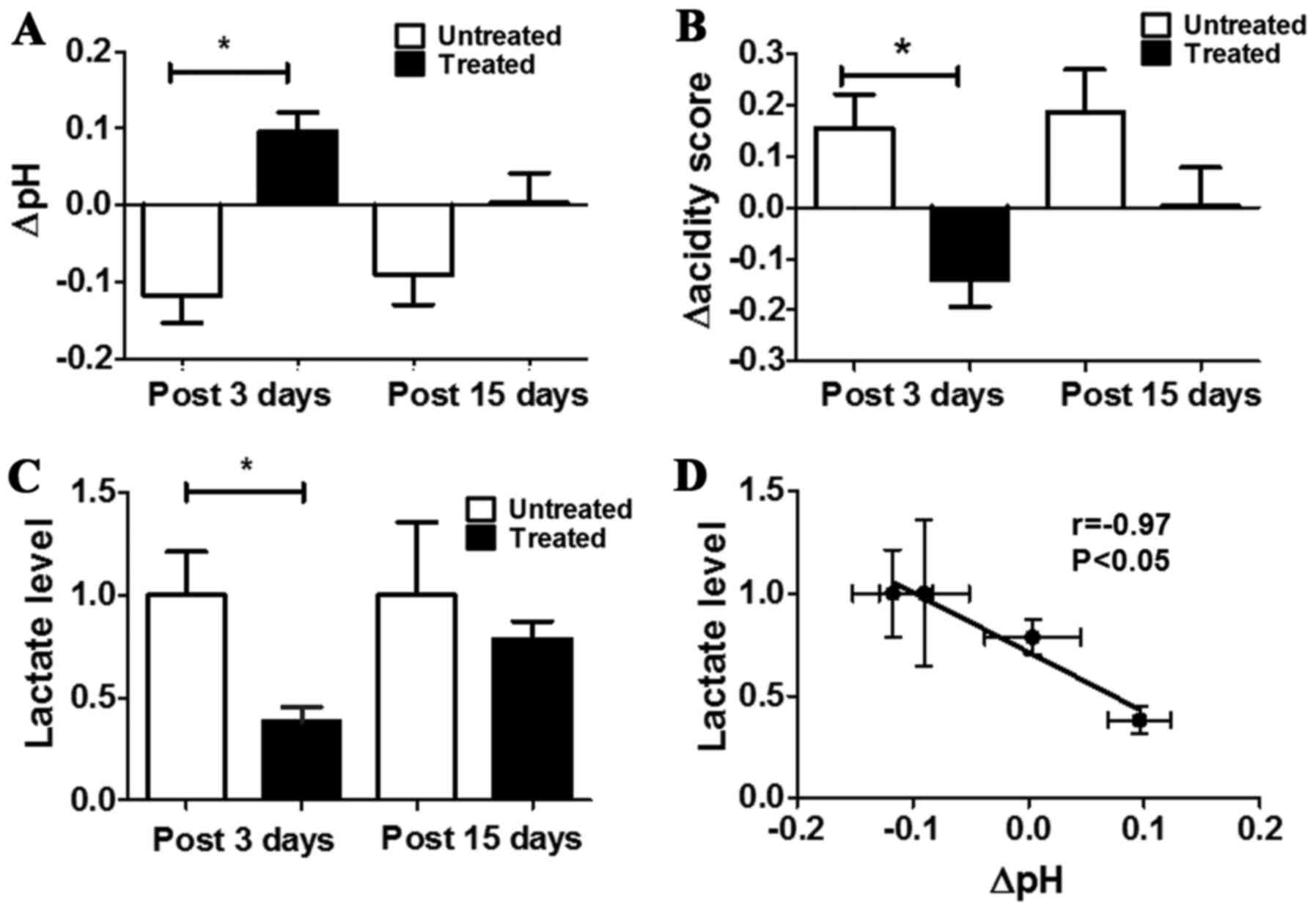
A significant pHe increase was observed for treated
mice in comparison to untreated ones after three days of DCA
treatment (ΔpHe = +0.10±0.03 and −0.12±0.03 for treated and
untreated, respectively, P<0.05, Fig. 5A). The same pHe variations were
maintained also after 15 days of treatment, despite less marked
(ΔpHe= +0.004±0.04 and −0.09±0.04 for treated and untreated,
respectively, Fig. 5A).
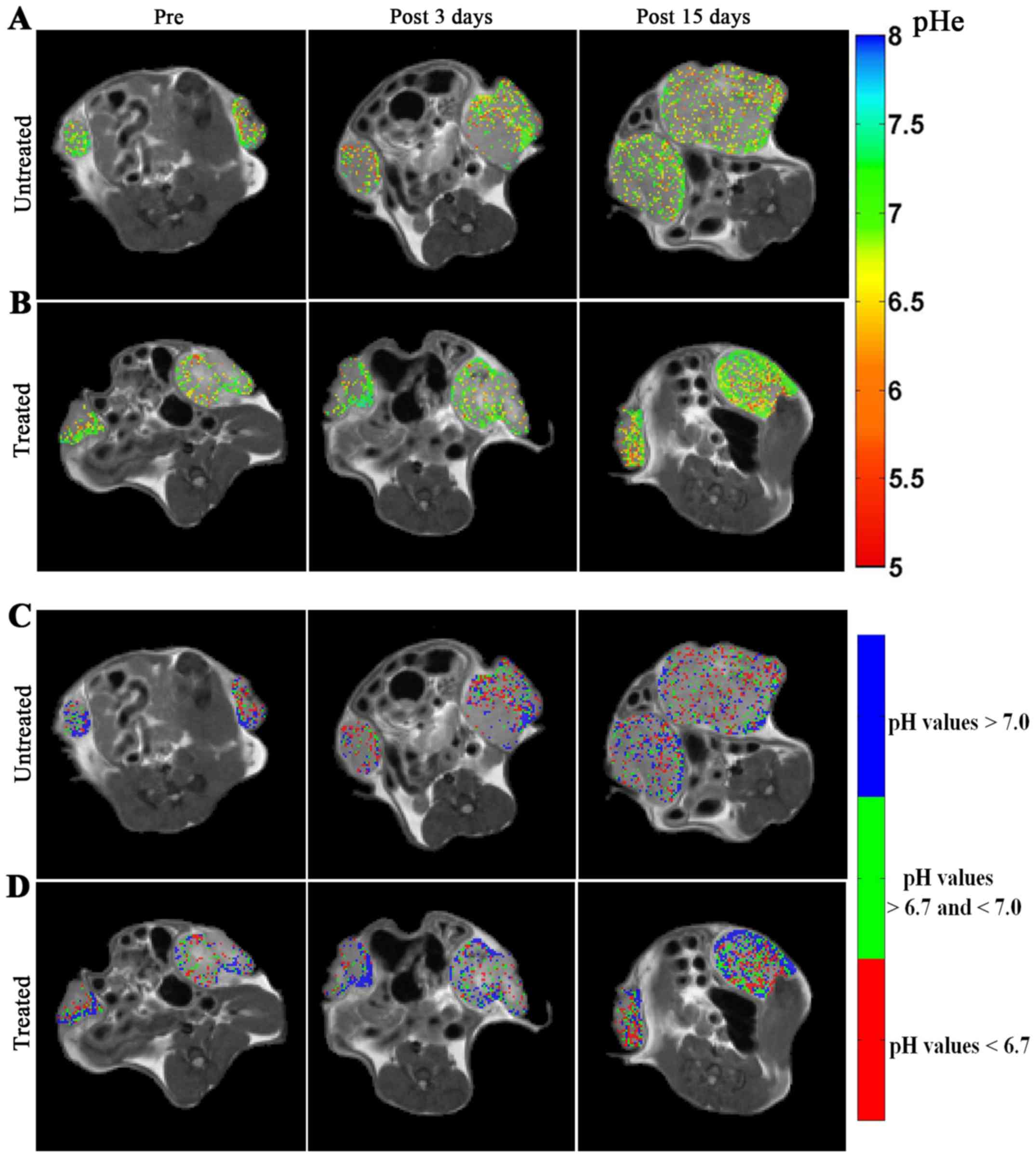
Representative MRI-CEST pHe images over imposed to anatomical
images are shown in Fig. 6 for
untreated (Fig. 6A) and treated
(Fig. 6B) mice. Following
DCA-treatment, an increase of the number of pixels with more
neutral pHe values is visible, in contrast to untreated mice at
both the investigated time points (i.e. after 3 and 15 days of DCA
treatment).
To assess more precisely the heterogeneity of the
extracellular pH distribution inside the tumour region, it was
deemed of interest to calculate the acidity score as an index of
spatial distribution of acidosis. A marked and statistically
significant difference was observed in the changes of the acidity
scores between untreated and treated mice after 3 days of treatment
(Δacidity score = −0.14±0.23 and +0.15±0.34, P<0.05, Fig. 5B). After 15 days a marked
difference in pHe distribution still remains between treated and
untreated mice (Δacidity score = +0.003±0.24 and +0.18±0.35 for
treated and untreated mice, respectively; P>0.05).
Representative acidity score maps are shown in Fig. 6C and D for untreated and treated
mice, respectively. Upon DCA-treatment, a decrease in the number of
pixels clustered at more acidic pH values (colour coded in red) is
well detected after three days of treatment. The MRI-based
measurements of tumour extracellular pH showed that DCA was
effective in inhibiting tumour glycolysis in vivo, resulting
in a marked decreased acidification of the interstitial space, as
observed for the in vitro studies.
Tumour lactate concentration of treated mice was
significantly decreased compared with untreated ones (lactate
levels in treated mice was ca. three times lower than in untreated
mice, P=0.0077). No significant variation in tumour lactate levels
was observed comparing treated to untreated mice after 15 days
(Fig. 5C). A strong and
significant inverse correlation was found between lactate levels
and changes in tumour pHe (r= −0.97, P<0.05, Fig. 5D). These results suggest that DCA
can inhibit the glycolytic activity of TS/A tumours, at least for
early time points and that changes in lactate production and
extracellular pH are highly correlated.
Discussion
High rate of glucose uptake and of lactate
production are two distinctive features of metabolically altered
tumour cells. DCA is able to revert the glycolytic phenotype
through metabolic inhibition of PDK that allows pyruvate to enter
into the tricarboxylic acid cycle thus limiting lactate production
and in turn, decreasing H+ ions pumped out in the
extracellular space. Herein, we investigated the effect of DCA on
tumour pHe in a breast cancer murine model using a non-invasive
MRI-based CEST pH imaging approach.
All the investigated breast cancer cell lines showed
a marked reduction in their metabolic capacity and vitality, but
their response to DCA was dependent on DCA concentration as well as
on the cell line itself, hence indicating different sensitivity.
Previous studies in other breast cancer cell lines showed that they
were sensitive to DCA (32) but
not all to the same extent, indicating a cell line dependency.
Sensitivity to DCA may be dependent on several factors, including
different expression and/or activity of PDK-PDH isoenzymes
(37), or DCA internalization that
is dependent on the ability to reach mitochondria matrix (38).
Moreover, evidence confirmed that the DCA effect on
cell cycle is also cellular-dependent and studies conducted on
glioblastoma, glioma, non-small cell lung cancer and colon rectal
cancer cells revealed that DCA treatment at 20 mM concentration
caused apoptosis and G2 phase cell cycle arrest (33,34).
In TS/A cells, 10 mM DCA induced a small increase in G2/M phase
although not statistically significant. Taken together these data
indicate that DCA is not a cytotoxic drug but it acts as a
metabolic agent.
DCA-induced glycolysis inhibition is expected to
reduce lactate production, hence alter extracellular acidification,
as already demonstrated in previous in vitro studies by
measuring pHe changes (39,40).
Within this study, we observed a marked increase of extracellular
pH that was dependent on DCA concentration; moreover, the
alkalinisation of pHe was even more pronounced when TS/A cells were
cultured in hypoxic conditions (1% O2) than in normoxic
conditions, simulating a poorly-perfused tumour microenvironment.
Our in vivo MRI-based observations reflect similar findings,
since we measured a significant increase in tumour pHe as early as
three days after DCA-treatment.
Other studies in breast and prostate cell lines
showed that lactate production is reduced upon DCA treatment
(41-43). We observed that lactate production
was reduced following DCA treatment both in vitro, just
after 24 h, as well as in vivo, with a marked decrease in
lactate levels after three days of treatment. Notably, these
changes in lactate levels were highly correlated with changes in
pHe in vitro and the same strong correlation was found in
vivo, likely reflecting the intertwined dependence between
glycolysis, lactate levels and tumour acidosis (Figs. 2C, 3C and 5D).
Although other in vivo studies confirmed the
DCA antitumour activity on solid tumours (44), in this study treated mice showed a
limited tumour growth reduction following 15 days of treatment and
only a slight increase of the survival time was observed as
compared to untreated mice. This may be explained considering that
DCA alone has a moderate efficiency as chemotherapeutic drug, and
its antineoplastic pharmacological effect can be augmented when
used in combination with other drugs (41,45,46).
Our results are in agreement with these observations, as similar
lactate levels were observed in untreated and treated mice after 15
days of treatment. These results parallel those obtained by
measuring in vivo tumour pHe, which at 15 days show a
reduced difference between treated and untreated mice in terms of
pHe changes and of acidity scores. These findings confirm the
ability of the proposed non-invasive approach to assess the onset
of resistance to DCA, since tumour acidosis returned to almost
baseline values after 15 days. This behaviour was confirmed by
similar changes in lactate levels. Moreover, the inefficacy of DCA
to halt tumour glycolysis after 15 days, as measured by the
proposed approach, anticipated the lack of difference in terms of
survival times between treated and untreated groups.
In this study, in vivo pH changes were
assessed by MRI-CEST imaging using iopamidol, an MRI-CEST
pH-responsive agent able to map pHe and tumour perfusion in the
microenvironment in which it is distributed (47–49).
Although iopamidol showed a heterogeneous distribution in the
tumour region, the application of the ratiometric pH method allowed
obtaining representative pH maps for the region of interest. To get
more insight into the tumour pHe heterogeneity, we calculated the
acidity score that reports on the distribution of pixels clustered
on the basis of their relative acidity inside the tumour region.
Representative acidity score maps showed an increase in blue pixels
during DCA treatment for treated mice (Fig. 6D), reflecting the DCA-induced
glycolysis inhibition, hence alkalinisation of tumour pHe, although
we cannot exclude that there are other pathways that may contribute
to tumour extracellular acidification (50). A similar decrease of tumour
acidosis was observed in a xenograft model of B-cell lymphoma upon
treatment with metaiodobenzylguanidine by using an analogous
pH-responsive CEST agent (51). Of
note, also endogenous CEST pH mapping allowed monitoring
intracellular acidification following lonidamine or topiramate
treatment in orthotopic glioblastoma tumours (52,53).
All these results confirm the feasibility of MRI-CEST pH mapping to
monitor the response to drugs targeting tumour metabolism.
In conclusion, despite the lack of tumour growth
reduction, this study demonstrated that MRI-CEST pH imaging is able
to detect the early response to DCA by measuring changes in tumour
pHe. These findings correlated well with the observed reduced
lactate levels as a consequence of the reversed glycolytic
phenotype. These results suggest that MRI-CEST pH imaging may serve
as a useful imaging biomarker for monitoring changes in metabolism
following drugs targeting tumour deregulated glycolysis.
Acknowledgments
Financial support from European Community's Seventh
Framework Programme (H2020 GLINT project 667510) is gratefully
acknowledged. L.C. was supported by a fellowship from Fondazione
Umberto Veronesi.
References
|
1
|
Gerweck LE and Seetharaman K: Cellular pH
gradient in tumor versus normal tissue: Potential exploitation for
the treatment of cancer. Cancer Res. 56:1194–1198. 1996.PubMed/NCBI
|
|
2
|
Warburg O, Wind F and Negelein E: The
metabolism of tumors in the body. J Gen Physiol. 8:519–530. 1927.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Webb BA, Chimenti M, Jacobson MP and
Barber DL: Dysregulated pH: A perfect storm for cancer progression.
Nat Rev Cancer. 11:671–677. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hashim AI, Zhang X, Wojtkowiak JW,
Martinez GV and Gillies RJ: Imaging pH and metastasis. NMR Biomed.
24:582–591. 2011.PubMed/NCBI
|
|
5
|
Michelakis ED, Webster L and Mackey JR:
Dichloroacetate (DCA) as a potential metabolic-targeting therapy
for cancer. Br J Cancer. 99:989–994. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McFate T, Mohyeldin A, Lu H, Thakar J,
Henriques J, Halim ND, Wu H, Schell MJ, Tsang TM, Teahan O, et al:
Pyruvate dehydrogenase complex activity controls metabolic and
malignant phenotype in cancer cells. J Biol Chem. 283:22700–22708.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bonnet S, Archer SL, Allalunis-Turner J,
Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta
L, Bonnet S, et al: A mitochondria-K+ channel axis is
suppressed in cancer and its normalization promotes apoptosis and
inhibits cancer growth. Cancer Cell. 11:37–51. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
De Preter G, Neveu MA, Danhier P, Brisson
L, Payen VL, Porporato PE, Jordan BF, Sonveaux P and Gallez B:
Inhibition of the pentose phosphate pathway by dichloroacetate
unravels a missing link between aerobic glycolysis and cancer cell
proliferation. Oncotarget. 7:2910–2920. 2016.
|
|
9
|
Dunbar EM, Coats BS, Shroads AL, Langaee
T, Lew A, Forder JR, Shuster JJ, Wagner DA and Stacpoole PW: Phase
1 trial of dichloroacetate (DCA) in adults with recurrent malignant
brain tumors. Invest New Drugs. 32:452–464. 2014. View Article : Google Scholar
|
|
10
|
Zhang X, Lin Y and Gillies RJ: Tumor pH
and its measurement. J Nucl Med. 51:1167–1170. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gillies RJ, Liu Z and Bhujwalla Z:
31P-MRS measurements of extracellular pH of tumors using
3-aminopropylphosphonate. Am J Physiol. 267:C195–C203.
1994.PubMed/NCBI
|
|
12
|
Garcia-Martin ML, Martinez GV, Raghunand
N, Sherry AD, Zhang S and Gillies RJ: High resolution pH(e) imaging
of rat glioma using pH-dependent relaxivity. Magn Reson Med.
55:309–315. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gallagher FA, Kettunen MI, Day SE, Hu DE,
Ardenkjaer- Larsen JH, Zandt R, Jensen PR, Karlsson M, Golman K,
Lerche MH, et al: Magnetic resonance imaging of pH in vivo using
hyperpolarized 13C-labelled bicarbonate. Nature.
453:940–943. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Serrao EM and Brindle KM: Potential
clinical roles for metabolic imaging with hyperpolarized
[1-13C]pyruvate. Front Oncol. 6:592016. View Article : Google Scholar
|
|
15
|
Reineri F, Daniele V, Cavallari E and Aime
S: Assessing the transport rate of hyperpolarized pyruvate and
lactate from the intra- to the extracellular space. NMR Biomed.
29:1022–1027. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reineri F, Boi T and Aime S: Parahydrogen
induced polarization of 13C carboxylate resonance in
acetate and pyruvate. Nat Commun. 6:58582015. View Article : Google Scholar
|
|
17
|
Viale A, Reineri F, Dastrù W and Aime S:
Hyperpolarized (13) C-pyruvate magnetic resonance imaging in cancer
diagnostics. Expert Opin Med Diagn. 6:335–345. 2012. View Article : Google Scholar
|
|
18
|
Menzel MI, Farrell EV, Janich MA, Khegai
O, Wiesinger F, Nekolla S, Otto AM, Haase A, Schulte RF and
Schwaiger M: Multimodal assessment of in vivo metabolism with
hyperpolarized [1-13C]MR spectroscopy and 18F-FDG PET
imaging in hepatocellular carcinoma tumor-bearing rats. J Nucl Med.
54:1113–1119. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rivlin M and Navon G: Glucosamine and
N-acetyl glucosamine as new CEST MRI agents for molecular imaging
of tumors. Sci Rep. 6:326482016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu X, Chan KW, Knutsson L, Artemov D, Xu
J, Liu G, Kato Y, Lal B, Laterra J, McMahon MT, et al: Dynamic
glucose enhanced (DGE) MRI for combined imaging of blood-brain
barrier break down and increased blood volume in brain cancer. Magn
Reson Med. 74:1556–1563. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rivlin M, Tsarfaty I and Navon G:
Functional molecular imaging of tumors by chemical exchange
saturation transfer MRI of 3-O-Methyl-D-glucose. Magn Reson Med.
72:1375–1380. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Walker-Samuel S, Ramasawmy R, Torrealdea
F, Rega M, Rajkumar V, Johnson SP, Richardson S, Gonçalves M,
Parkes HG, Arstad E, et al: In vivo imaging of glucose uptake and
metabolism in tumors. Nat Med. 19:1067–1072. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hingorani DV, Bernstein AS and Pagel MD: A
review of responsive MRI contrast agents: 2005–2014. Contrast Media
Mol Imaging. 10:245–265. 2015. View Article : Google Scholar
|
|
24
|
Longo DL, Dastrù W, Digilio G, Keupp J,
Langereis S, Lanzardo S, Prestigio S, Steinbach O, Terreno E,
Uggeri F, et al: Iopamidol as a responsive MRI-chemical exchange
saturation transfer contrast agent for pH mapping of kidneys. In
vivo studies in mice at 7. T Magn Reson Med. 65:202–211. 2011.
View Article : Google Scholar
|
|
25
|
Longo DL, Busato A, Lanzardo S, Antico F
and Aime S: Imaging the pH evolution of an acute kidney injury
model by means of iopamidol, a MRI-CEST pH-responsive contrast
agent. Magn Reson Med. 70:859–864. 2013. View Article : Google Scholar
|
|
26
|
Chen LQ, Howison CM, Jeffery JJ, Robey IF,
Kuo PH and Pagel MD: Evaluations of extracellular pH within in vivo
tumors using acidoCEST MRI. Magn Reson Med. 72:1408–1417. 2014.
View Article : Google Scholar :
|
|
27
|
Moon BF, Jones KM, Chen LQ, Liu P, Randtke
EA, Howison CM and Pagel MD: A comparison of iopromide and
iopamidol, two acidoCEST MRI contrast media that measure tumor
extracellular pH. Contrast Media Mol Imaging. 10:446–455. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang X, Song X, Ray Banerjee S, Li Y, Byun
Y, Liu G, Bhujwalla ZM, Pomper MG and McMahon MT: Developing
imidazoles as CEST MRI pH sensors. Contrast Media Mol Imaging.
11:304–312. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Longo DL, Bartoli A, Consolino L, Bardini
P, Arena F, Schwaiger M and Aime S: In vivo imaging of tumor
metabolism and acidosis by combining PET and MRI-CEST pH imaging.
Cancer Res. 76:6463–6470. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nanni P, de Giovanni C, Lollini PL,
Nicoletti G and Prodi G: TS/A: A new metastasizing cell line from a
BALB/c spontaneous mammary adenocarcinoma. Clin Exp Metastasis.
1:373–380. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lanzardo S, Conti L, Rooke R, Ruiu R,
Accart N, Bolli E, Arigoni M, Macagno M, Barrera G, Pizzimenti S,
et al: Immunotargeting of antigen xCT attenuates stem-like cell
behavior and metastatic progression in breast cancer. Cancer Res.
76:62–72. 2016. View Article : Google Scholar
|
|
32
|
Sun RC, Fadia M, Dahlstrom JE, Parish CR,
Board PG and Blackburn AC: Reversal of the glycolytic phenotype by
dichloroacetate inhibits metastatic breast cancer cell growth in
vitro and in vivo. Breast Cancer Res Treat. 120:253–260. 2010.
View Article : Google Scholar
|
|
33
|
Duan Y, Zhao X, Ren W, Wang X, Yu KF, Li
D, Zhang X and Zhang Q: In vitro and in vivo evaluation. Onco
Targets Ther. 6:189–198. 2013.
|
|
34
|
Takahashi M, Watari E and Takahashi H:
Dichloroacetate induces cell cycle arrest in human glioblastoma
cells persistently infected with measles virus: A way for
controlling viral persistent infection. Antiviral Res. 113:107–110.
2015. View Article : Google Scholar
|
|
35
|
Allen KT, Chin-Sinex H, DeLuca T,
Pomerening JR, Sherer J, Watkins JB III, Foley J, Jesseph JM and
Mendonca MS: Dichloroacetate alters Warburg metabolism, inhibits
cell growth, and increases the X-ray sensitivity of human A549 and
H1299 NSC lung cancer cells. Free Radic Biol Med. 89:263–273. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Madhok BM, Yeluri S, Perry SL, Hughes TA
and Jayne DG: Dichloroacetate induces apoptosis and cell-cycle
arrest in colorectal cancer cells. Br J Cancer. 102:1746–1752.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bowker-Kinley MM, Davis WI, Wu P, Harris
RA and Popov KM: Evidence for existence of tissue-specific
regulation of the mammalian pyruvate dehydrogenase complex. Biochem
J. 329:191–196. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Babu E, Ramachandran S, CoothanKandaswamy
V, Elangovan S, Prasad PD, Ganapathy V and Thangaraju M: Role of
SLC5A8, a plasma membrane transporter and a tumor suppressor, in
the antitumor activity of dichloroacetate. Oncogene. 30:4026–4037.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Haugrud AB, Zhuang Y, Coppock JD and
Miskimins WK: Dichloroacetate enhances apoptotic cell death via
oxidative damage and attenuates lactate production in
metformin-treated breast cancer cells. Breast Cancer Res Treat.
147:539–550. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kumar A, Kant S and Singh SM: Novel
molecular mechanisms of antitumor action of dichloroacetate against
T cell lymphoma: Implication of altered glucose metabolism, pH
homeostasis and cell survival regulation. Chem Biol Interact.
199:29–37. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Robey IF and Martin NK: Bicarbonate and
dichloroacetate: Evaluating pH altering therapies in a mouse model
for metastatic breast cancer. BMC Cancer. 11:2352011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xintaropoulou C, Ward C, Wise A, Marston
H, Turnbull A and Langdon SP: A comparative analysis of inhibitors
of the glycolysis pathway in breast and ovarian cancer cell line
models. Oncotarget. 6:25677–25695. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kailavasan M, Rehman I, Reynolds S, Bucur
A, Tozer G and Paley M: NMR-based evaluation of the metabolic
profile and response to dichloroacetate of human prostate cancer
cells. NMR Biomed. 27:610–616. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kinnaird A, Dromparis P, Saleme B, Gurtu
V, Watson K, Paulin R, Zervopoulos S, Stenson T, Sutendra G, Pink
DB, et al: Metabolic modulation of clear-cell renal cell carcinoma
with dichloroacetate, an inhibitor of pyruvate dehydrogenase
kinase. Eur Urol. 69:734–744. 2016. View Article : Google Scholar
|
|
45
|
Sanchez WY, McGee SL, Connor T, Mottram B,
Wilkinson A, Whitehead JP, Vuckovic S and Catley L: Dichloroacetate
inhibits aerobic glycolysis in multiple myeloma cells and increases
sensitivity to bortezomib. Br J Cancer. 108:1624–1633. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Xie J, Wang BS, Yu DH, Lu Q, Ma J, Qi H,
Fang C and Chen HZ: Dichloroacetate shifts the metabolism from
glycolysis to glucose oxidation and exhibits synergistic growth
inhibition with cisplatin in HeLa cells. Int J Oncol. 38:409–417.
2011.
|
|
47
|
Longo DL, Sun PZ, Consolino L, Michelotti
FC, Uggeri F and Aime S: A general MRI-CEST ratiometric approach
for pH imaging: Demonstration of in vivo pH mapping with
iobitridol. J Am Chem Soc. 136:14333–14336. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Longo DL, Michelotti F, Consolino L,
Bardini P, Digilio G, Xiao G, Sun PZ and Aime S: In vitro and in
vivo assessment of nonionic iodinated radiographic molecules as
chemical exchange saturation transfer magnetic resonance imaging
tumor perfusion agents. Invest Radiol. 51:155–162. 2016. View Article : Google Scholar
|
|
49
|
Anemone A, Consolino L and Longo DL:
MRI-CEST assessment of tumour perfusion using X-ray iodinated
agents: Comparison with a conventional Gd-based agent. Eur Radiol.
27:2170–2179. 2017. View Article : Google Scholar
|
|
50
|
Kato Y, Ozawa S, Miyamoto C, Maehata Y,
Suzuki A, Maeda T and Baba Y: Acidic extracellular microenvironment
and cancer. Cancer Cell Int. 13:892013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chen LQ, Howison CM, Spier C, Stopeck AT,
Malm SW, Pagel MD and Baker AF: Assessment of carbonic anhydrase IX
expression and extracellular pH in B-cell lymphoma cell line
models. Leuk Lymphoma. 56:1432–1439. 2015. View Article : Google Scholar
|
|
52
|
Mcvicar N, Li AX, Meakin SO and Bartha R:
Imaging chemical exchange saturation transfer (CEST) effects
following tumor-selective acidification using lonidamine. NMR
Biomed. 28:566–575. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Marathe K, Mcvicar N, Li A, Bellyou M,
Meakin S and Bartha R: Topiramate induces acute intracellular
acidification in glioblastoma. J Neurooncol. 130:465–472. 2016.
View Article : Google Scholar : PubMed/NCBI
|