Introduction
Uveal melanoma (UM), originated from melanocyte
malignant transformation, is the most common intraocular tumor in
adults (1). The yearly incidence
is 5–6 cases per million, and the median age at diagnosis is 62
years (2). Hepatic metastasis is
the leading cause of death in UM patients, and the average survival
time for patients diagnosed with liver metastasis is only 2–14
months (2). Although there are
multiple options available for the treatment of primary UM
patients, such as surgical resection of diseased eye ball, plaque
radiotherapy, transpupillary thermotherapy and chemotherapy
(2–4), the 5-year survival rate displayed no
improvement in past decades, which is attributed to lacking
effective therapy for metastasis (2). Therefore, there is an urgent demand
to develop more efficient agents against both primary and
metastatic UM.
Aberrant activation of NF-κB is widely present in
diverse malignancies (5–9). NF-κB pathway regulates the
transcription of numerous genes which are involved in diverse
cellular functions, including apoptosis, proliferation,
angiogenesis, immune response, cell invasion, and cancer stem-like
cells (CSCs) (10). More
importantly, NF-κB pathway may be a potential molecular target for
cancer therapy (11). Previous
evidence indicates that constitutive activation of NF-κB can
increase UM cellular proliferation and evasion of apoptosis, and is
involved in both primary and metastatic UM (12). Thus, pharmacological inhibition of
NF-κB may be an effective approach to kill UM cells.
Previous studies demonstrated that pristimerin, a
natural product isolated from natural herb plants, is a potent
inhibitor of NF-κB pathway (9,13,14)
and displays antimicrobial, anti-inflammatory, anti-peroxidation
activities (13) and anticancer
effects in various human malignancies (15–18).
Moreover, pristimerin has shown inhibitory effects on
proliferation, survival, angiogenesis, metastasis, and CSCs
characteristics (16,17,19–22).
Thus, we wondered whether this compound also had anticancer
activity in UM.
In the present study, we showed the inhibitory
activity of pristimerin against TNFα-induced NF-κB activation in UM
cells. We also found that the downregulation of survivin on mRNA
level was critical in pristimerin-inducing apoptosis in UM cells.
In addition, pristimerin attenuated the properties of CSCs and the
combination of pristimerin and vinblastine, a frontline therapeutic
agent, showed a synergistic effect against UM cells. This study
suggests that pristimerin is a promising compound for UM
treatment.
Materials and methods
Reagents and antibodies
Pristimerin (Fig.
1A, purity >99%, HPLC) was purchased from Paypaytech Inc.
(Shenzhen, China), and dissolved in dimethyl sulphoxide (DMSO,
Sigma-Aldrich, Shanghai, China) at a stock concentration of 20 mM,
and stored at −20°C. Vinblastine was obtained from Selleck
(Shanghai, China). Antibodies against survivin, Bcl-XL
and PCNA were from Santa Cruz Biotech (Santa Cruz, CA, USA).
Antibodies against p65, IκBα, phospho-IκBα (S32), Tubulin, matrix
metalloproteinase 2 (MMP2), matrix metalloproteinase 9 (MMP9),
Slug, Sox2, Nanog and KLF4 were purchased from Cell Signaling
Technology (Beverly, MA, USA). Antibodies against PARP (clone
4C10–5), caspase-3, active caspase-3 (CM1), cytochrome c
(clone 6H2.B4), X-linked inhibitor of apoptosis protein (XIAP),
Bcl-2, and c-Myc were purchased from BD Biosciences (San Jose, CA,
USA). Anti-mouse and anti-rabbit immunoglobulin G
fluorescent-conjugated secondary antibodies were purchased from
LI-COR Biotechnology (NE, USA). Dual-luciferase assay kit was
provided by Promega (Madison, WI, USA). The Aldeflour kit was from
StemCell Technologies (Vancouver, Canada). Gelatin (G1890-100G) was
obtained from Sigma-Aldrich). Coomassie Brilliant Blue R-250
(44329-7C) was from Farco Chemical Supplies (Hong Kong, China).
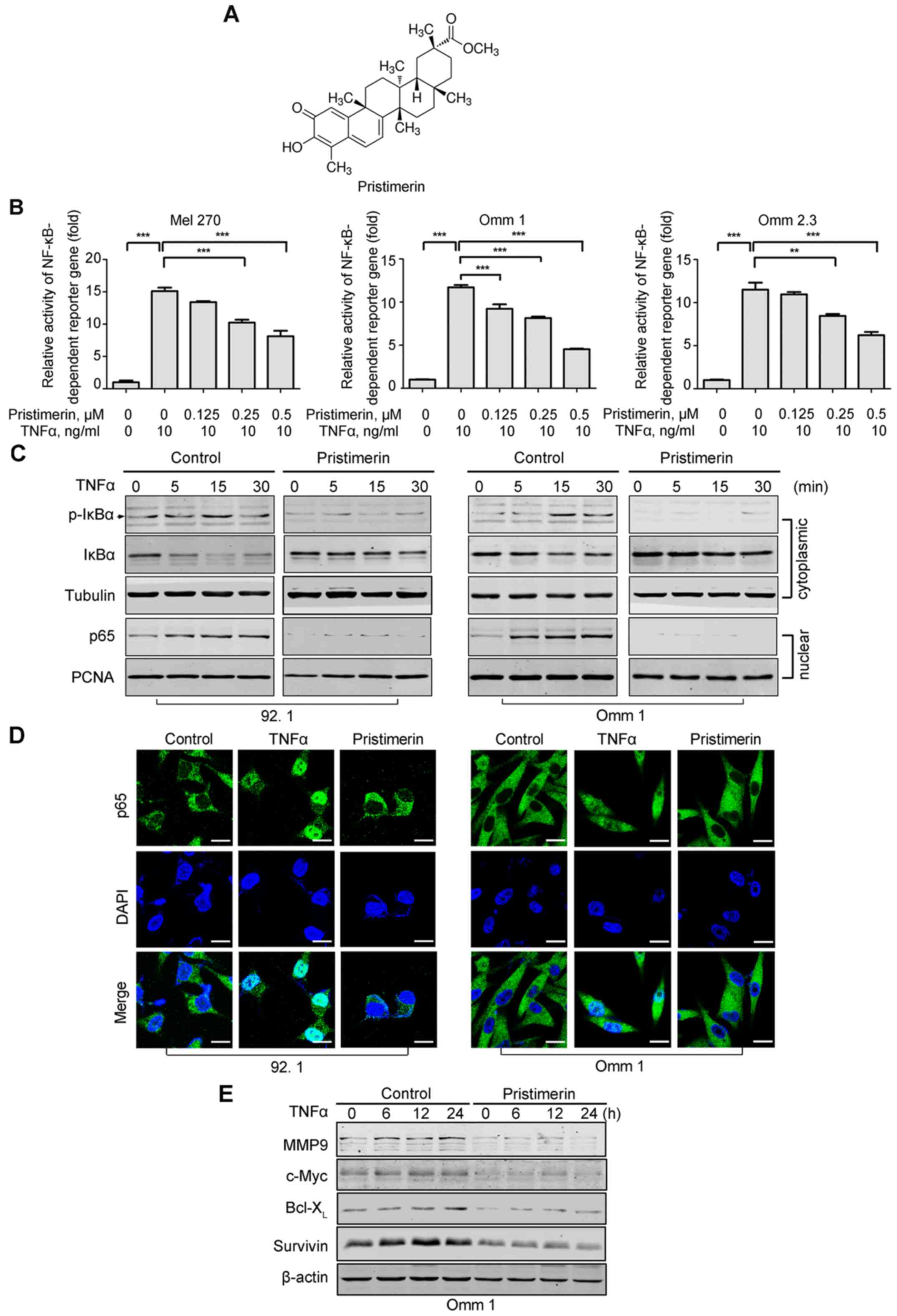 | Figure 1Pristimerin suppresses TNFα-induced
NF-κB activation in uveal melanoma cells. (A) The structure of
pristimerin is shown. (B) Pristimerin inhibited TNFα-induced
NF-κB-dependent reporter gene expression in UM cells. Mel 270, Omm
1 and Omm 2.3 cells co-transfected with NF-κB-TATA-Luc
reporter plasmid and Renilla luciferase reporter plasmid
were treated with various concentrations of pristimerin for 16 h,
and then stimulated with TNFα for 8 h; the luciferase activity of
cells was detected. The levels of firefly luciferase activity were
normalized to Renilla luciferase activity. The results
represented the means ± SE of three independent experiments.
**P<0.01, ***P<0.001, one-way ANOVA,
post hoc intergroup comparisons, Tukey's test. (C and D)
Pristimerin blocked the nuclear translocation of p65 in UM cells
stimulated by TNFα. 92.1 or Omm 1 cells were pretreated with or
without 1.0 µM pristimerin for 12 h, then stimulated with
TNFα (10 ng/ml) at the indicated time; cytosolic and nucleus
fractionations were subjected to western blot analysis. Tubulin was
a cytoplasmic loading control, while PCNA was a nuclear loading
control (C). 92.1 and Omm 1 cells were pretreated with medium
containing DMSO or 0.5 µM pristimerin for 24 h, and then
incubated with TNFα (10 ng/ml) for 15 min, undergoing
immunofluorescence analysis of p65. DAPI was used to stain the
nuclear (D). Scale bar represents 20 µm. (E) Pristimerin
decreased the levels of NF-κB-dependent prosurvival proteins in UM
cells. After pre-incubation with pristimerin for 12 h, Omm 1 cells
were exposed to 0.1 nM TNFα for the indicated durations, and then
western blot analysis was performed. |
Cell culture
The UM cells 92.1, Mel 270, Omm 1 and Omm 2.3 were
cultured in RPMI-1640 supplemented with 10% fetal bovine serum
(FBS), penicillin (100 U/ml), streptomycin (100 µg/ml) and
L-glutamine (2 mM), and incubated at 37°C and 5% CO2, as
described previously (23). 293T
cells were cultured in DMEM supplemented with 10% FBS, penicillin
(100 U/ml), streptomycin (100 µg/ml) and incubated at 37°C
and 5% CO2.
Dual luciferase reporter assay
Omm 1, 92.1 and Omm 2.3 cells seeded in 24-well
plates were co-transfected with NF-κB-TATA-Luc reporter
plasmid (0.5 µg) and Renilla luciferase reporter
plasmid (10 ng), respectively. After 24 h, these UM cells were
exposed to the indicated concentrations of pristimerin for 16 h,
then added with TNFα (10 ng/ml) for 8 h and measured with
dual-luciferase assay kit as described previously (9,24).
The luciferase activities were normalized with NF-κB-dependent
firefly luciferase to Renilla luciferase activity.
Western blot analysis
The prepared cell lysates were quantified by Pierce™
BCA Protein assay kit (Thermo Scientific, USA) according to the
manufacturer's instructions. Protein samples were subjected to
SDS-PAGE and then blotted to nitrocellulose membranes. After
blocked by 5% skim milk, membranes were subsequently incubated with
the primary antibodies overnight at 4°C before incubation with
appropriate secondary antibodies. The immunoblots were recorded
with the Odyssey infrared imaging system (LI-COR).
Immunofluorescence staining
After cultured overnight, 92.1 and Omm 1 cells were
incubated with or without pristimerin for 24 h, and then the cells
were stimulated with TNFα (20 ng/ml) for 15 min before fixed by 4%
paraformaldehyde. After permeabilization by 0.2% (v/v) Triton X-100
at room temperature for 15 min, the samples were washed with 500
µl of PBS for 5 min three times. Subsequently, PBS buffer
containing 5% bovine serum albumin was used to block non-specifical
binding for 1 h and then the cells were incubated with 200
µl of PBS containing the primary antibody for 1 h at room
temperature. After washing with PBS containing 0.1% (v/v) NP-40,
the cells were incubated with 200 µl of PBS containing the
secondary antibody for 1 h at room temperature. Nuclear staining
was performed with DAPI for 20 min. After washing with PBS for 5
min, all samples were added with a half drop of anti-fade regent
and then sealed by nail polish (9).
Preparation of cytoplasmic and nuclear
fractionations
After stimulation with TNFα (10 ng/ml), cells
treated with or without pristimerin were collected by
centrifugation and washed with PBS. Pellets resuspended in 150
µl cold lysis buffer (10 mM HEPES, 10 mM KCl, 0.1 mM EDTA,
0.4% NP-40, 1 mM DTT, 0.5 mM PMSF, 1 mM sodium orthovanadate and
Complete Protease Inhibitor Mix) by pipetting up and down ~10 times
were incubated on ice for 10 min, the lysates were centrifuged at
20,000 g (9,25). The supernatants were transferred to
a fresh tube as cytoplasmic extracts. After rinsed with the lysis
buffer, the pellets were vigorously suspended in nuclear protein
extraction buffer (20 mM HEPES, 0.4 mM NaCl, 1 mM EDTA, 1 mM DTT,
0.5 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM sodium
orthovanadate and Complete Protease Inhibitor Mix) and incubated
for 15 min. After centrifugation with 10 min at 20,000 g, the
supernatant was referred to as nuclear fractions (9,25).
Preparation of whole cell lysates and
cytosolic fractionations
Control or treated cells were pelleted by
centrifugation and then rinsed with PBS. The whole cell lysates
were prepared with RIPA buffer (1X PBS, 0.1% SDS, 1% NP-40, 0.5%
sodium deoxycholate) supplemented with 1X protease inhibitor
cocktail, 10 mM β-glycerophosphate, 1 mM sodium orthovanadate, 10
mM sodium fluoride, and 1 mM PMSF (26). The cytosolic fraction was extracted
with digitonin extraction buffer (10 mM PIPES, 0.015% digitonin,
300 mM sucrose, 100 mM NaCl, 3 mM MgCl2, 5 mM EDTA, and
1 mM PMSF) on ice for 10 min and then centrifuged at 20,000 g for
10 min at 4°C. The supernatants were collected as cytosolic
fractions (27).
Cell viability assay
The UM cells (5,000 cells/well) seeded into 96-well
plates overnight were then exposed to serially diluted pristimerin
for 72 h. Optical intensity was measured by microplate reader after
addition with 20 µl mixture of MTS and PMS (28).
Colony formation assay
The UM cells were treated with various
concentrations of pristimerin for 24 h and rinsed with PBS, then
8,000 cells were seeded to a modified drug-free double layer soft
agar system (29). After 10–14
days, the number of colonies composed of >50 cells was counted
under a microscope.
Cell cycle analysis by flow
cytometry
After pretreated with or without pristimerin for 24
h and washed with PBS, UM cells were fixed with 66% ethanol
overnight. Cells rinsed with PBS were pelleted by centrifugation.
Subsequently, the pellets were resuspended in PBS and stained with
propidium iodide (PI, 50 µg/ml) and RNase (2.5 µg/ml)
for 30 min at room temperature. DNA content was analyzed by flow
cytometry (9).
Apoptosis analysis by flow cytometry
Annexin V-FITC/PI double staining was used to detect
apoptosis by flow cytometry. Cells pre-incubated with or without
pristimerin were collected by centrifugation and washed with PBS,
and then stained with 0.3% Annexin V-FITC in binding buffer for 15
min at room temperature in the dark. After centrifuged at 250 g and
resuspended in 1X binding buffer, the cells were added with PI
before flow cytometry analysis (9,29).
Measurement of mitochondrial
transmembrane potential
UM cells were incubated with 2.0 µM
pristimerin for different durations, and then stained with
MitoTracker probes (CMXRos and MTGreen) for 1 h at 37°C. After
being centrifuged at 250 g for 10 min, the pellets were raised in
PBS and subjected to flow cytometry to detect the changes in
mitochondrial transmembrane potential (Δψ) (25,29).
Lentiviral transfection
Lentiviruses were produced by transient transfection
in 293T cells with control shRNA (Scramble) or specific shRNA
together with the pCMV-dR8.2 packing construct and the pCMV-VSVG
envelope construct. 92.1 and Omm 1 cells (1×105
cells/well) were infected twice with virus-containing supernatants.
The cells were then incubated in the presence of puromycin (1
µg/ml) for ~5 days. Scramble and specific shRNAs were
purchased from Sigma-Aldrich, and the sequences were as follows:
PLKO.1-Non-target shRNA: CCG GGC GCG ATA GCG CTA ATA ATT TCT CGA
GAA ATT ATT AGC GCT ATC GCG CTT TTT; shsurvivin: CCG GGA AGA ATT
AAC CCT TGG TGA ACT CGA GTT CAC CAA GGG TTA ATT CTT CTT TTTG. The
Omm 1 cells overexpressed survivin were established with the same
methods. Constructs bearing full length human survivin cDNA in
pTSB-CMV-MCS-SBP-tRFP-F2A-PuroR and empty vector were provided by
Transheep (Shanghai, China). Human baculoviral inhibitor of
apoptosis repeat-containing 5 (BIRC5, NCBI Reference
Sequence ID: NG_029069.1) was cloned into the
pTSB-CMV-MCS-SBP-tRFP-F2A-PuroR (lentivirus) plasmid using
ClonExpress MultiS One Step Cloning kit, which was provided by
Vazyme (Nanjing, China) and clone sites were EcoRI and
BamHI. The efficiency of knockdown or overexpression was
examined by western blot analysis.
Real-time quantitative PCR
After pre-incubated with various concentrations of
pristimerin, total RNA was extracted by TRIzol reagent, and then
reverse-transcribed into first-strand complementary DNA with maxima
first strand cDNA synthesis kit. GAPDH was used as an endogenous
reference. The PCR primers were as follows: survivin forward,
5′-CAT CTC TAC ATT CAA GAA CTG G-3′; reverse, 5′-GGT TAA TTC TTC
AAA CTG CTT C-3′ (30). GAPDH
forward, 5′-GAT CGA ATT AAA CCT TAT CGT CGT-3′; reverse, 5′-AGC AGC
AGA ACT TCC ACT CGG T-3′.
The qRT-PCR reaction was performed in SYBR Premix EX
Taq with Bio-Rad CFX96 Real-Time Thermocycler according to the
manufacturer's instructions (26,29).
In relative quantification, ΔΔCq method was used, as described
previously (31).
The scratch wound healing assay
92.1 and Omm 1 cells (5×105/well) were
cultured in a 6-well plate supplemented with 10% FBS. The monolayer
was gently and slowly scratched with a 200-µl pipette tip
across the center of the well at ~90% confluence. The cells were
treated with or without pristimerin (0.25 or 0.5 µM), the
same wounded area was recorded under a microscope at different time
periods (29,32).
Migration and invasion assay
After exposed to vehicle, 0.25 or 0.5 µM
pristimerin for 24 h, equal amounts of 92.1 and Omm 1 cells were
cultured in the upper of Transwell chamber covered with or without
Matrigel and contained with FBS-free medium, while to the lower
chamber 20% FBS was added as chemoattractant. After 48 h, the cells
on the surface of chamber were removed by a cotton swab, and the
migrated or invaded cells were fixed with 4% paraformaldehyde and
stained by crystal violet. The cells in three randomly selected low
power fields were counted (29).
Gelatin zymography assay
92.1 and Omm 1 cells were seeded into 6-well culture
plates. When cells grew to ~80% confluency, were washed by
serum-free medium three times. Cells were incubated with serum-free
medium with or without pristimerin (0.25 and 0.5 µM) for 24
h. MDA-MB-231 cells were used as positive control (33). Serum-free medium without cells was
used as negative control. After incubation, the supernatants were
collected and were subjected to SDS-PAGE using 10% acrylamide gels
containing 0.1% gelatin. After electrophoresis, the gels were
washed for 15 min at room temperature in a buffer (50 mM Tris-Cl pH
7.6, 10 mM CaCl2, 20 mM NaCl, 1 µM
ZnCl2 and 2.5% Triton X-100) three times. After
incubation with activation buffer (50 mM Tris-Cl pH 7.6, 10 mM
CaCl2, 20 mM NaCl, 1 µM ZnCl2) for 48
h at 37°C, gels were stained with 0.25% Coomassie Brilliant Blue
R-250 in 40% methanol and 10% acetic acid and then briefly
destained in 10% acetic acid and 30% methanol. The locations of
gelatinolytic enzymes were visualized as clear bands on the
background (34). The experiment
was repeated three times and the optical density of each bands was
quantitated by image pro plus.
Aldehyde dehydrogenase (ALDH) assay
The Aldefluor kit was used to identify a population
of cells with high ALDH enzymatic activity (35). In brief, UM cells (92.1 and Omm 1)
pre-incubated with DMSO or 1.0 µM pristimerin for 24 h, and
then incubated with 5 µl ALDH reagent in the absence or
presence of 5 µl DEAB for 1 h at 37°C. After washed with
ALDH assay buffer, the cells were resuspended in ALDH assay buffer.
The proportion of ALDH+ cells was defined by flow
cytometry (29,35).
Melanosphere culture
92.1 and Omm 1 cells pretreated with DMSO or 1.0
µM pristimerin for 24 h were seeded to 24-well low
attachment plates containing 500 µl DMEM/F12 medium
supplemented with 1X B27, 10 ng/ml basic fibroblast growth factor
(bFGF), 20 ng/ml epidermal growth factor (EGF) each well. After ~14
days, melanospheres with >50 cells were counted as per the
universal standard (29,36). The secondary and tertiary rounds of
melanosphere cultures were implemented in drug-free culture after
collecting the first or second round of tumor sphere culture cells.
Representative images were taken by a microscope.
Statistical analysis
All experiments were performed at least thrice, and
the results are expressed as the means ± standard error (SE),
unless otherwise stated. GraphPad Prism 5.0 software was used for
statistical analysis. Comparison between 2 groups used two-tailed
Student's t-test, and comparison among multiple groups involved
one-way ANOVA with post hoc intergroup comparisons using Tukey's
test. P<0.05 was regarded as statistically significant.
Results
Pristimerin blocks TNFα-induced NF-κB
activation in UM cells
Our previous report indicated that pristimerin
potently inhibits activation of canonical NF-κB pathway in leukemia
cells (9). Here, we tested the
effect of pristimerin in UM cells. We first examined whether
pristimerin exerted the inhibitory effect on TNFα-induced
NF-κB-dependent reporter gene transcription. The results showed
that the NF-κB dependent luciferase activity was increased after
TNFα stimulation in UM cells. This effect was inhibited by
pristimerin in a dose-dependent manner (Fig. 1B). Because the
ubiquitination-dependent degradation of IκBα protein triggers p65
nuclear translocation, which is a critical step in the activation
of the canonical NF-κB pathway (37), we determined the influence of
pristimerin on IκBα and p65. Western blot analysis of cytoplasmic
fractionations showed that the levels of phosphorylated IκBα in the
cytoplasm were appreciably increased after TNFα induction compared
to the untreated control, while the total IκBα was coincidently
decreased (Fig. 1C). However, the
phosphorylation of IκBα was blocked, and the level of total IκBα
remained constant in the cells that were pretreated with
pristimerin (Fig. 1C).
Consistently, the level of p65 in nuclear fractionation was
remarkably escalated after TNFα stimulation (Fig. 1C), which was diminished by the
presence of pristimerin (Fig. 1C).
On the other hand, immunofluorescence staining analysis similarly
revealed that nuclear translocation of p65 was prominently
increased after TNFα stimulation; whereas pristimerin abrogated its
translocation in both 92.1 and Omm 1 cells (Fig. 1D).
We next ascertained the effect of pristimerin on the
expression of encoding products of NF-κB-dependent genes involved
in cell survival by western blotting (10). The expression of MMP9, c-Myc,
Bcl-XL and survivin was increased after stimulation with
TNFα (Fig. 1E). Nevertheless,
pristimerin blocked such an increase (Fig. 1E).
Taken together, our results suggested that
pristimerin represses the activation of the canonical NF-κB
pathway.
Pristimerin suppresses the growth of UM
cells
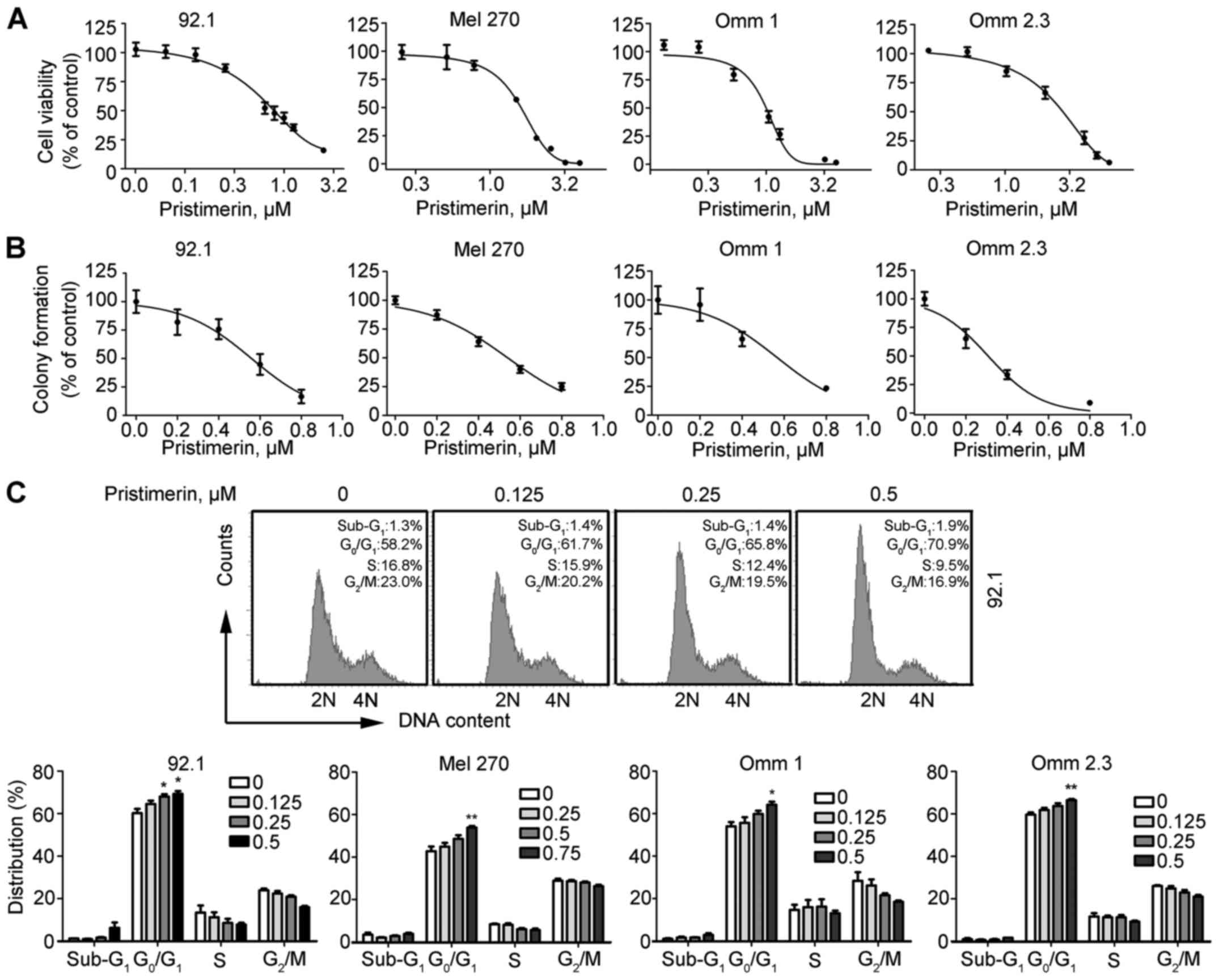
Seventy-two-hour cell-based MTS assay revealed that
pristimerin significantly inhibited the cell viability of 92.1, Mel
270, Omm 1 and Omm 2.3 cells, and the IC50 values were
1.1, 1.7, 2.2 and 2.9 µM, respectively (Fig. 2A). Given the advantages of colony
formation assay in reflecting malignant behavior of tumor cells, we
assessed the impact of pristimerin on anchorage-independent growth
of UM cells in soft agar. The results indicated that pristimerin
significantly inhibited the formation of colony in a dose-dependent
manner (Fig. 2B).
To further study the role of pristimerin in the
growth of UM cells, cell cycle distribution analysis was performed
after UM cells were exposed to different concentrations of
pristimerin for 24 h. The results showed that pristimerin caused
G1-phase arrest in UM cells (Fig. 2C).
Pristimerin exerts induction of apoptosis
in UM cells
We carried out flow cytometry analysis based on
Annexin V-FITC/PI double staining and found that pristimerin
remarkably induced cell death in UM cells in a dose- and
time-dependent manner (Fig. 3A).
Western blot analysis revealed that pristimerin induced cleavage of
PARP and caspase-3 in UM cells (Fig.
3B), which demonstrated the occurrence of apoptosis induced by
pristimerin.
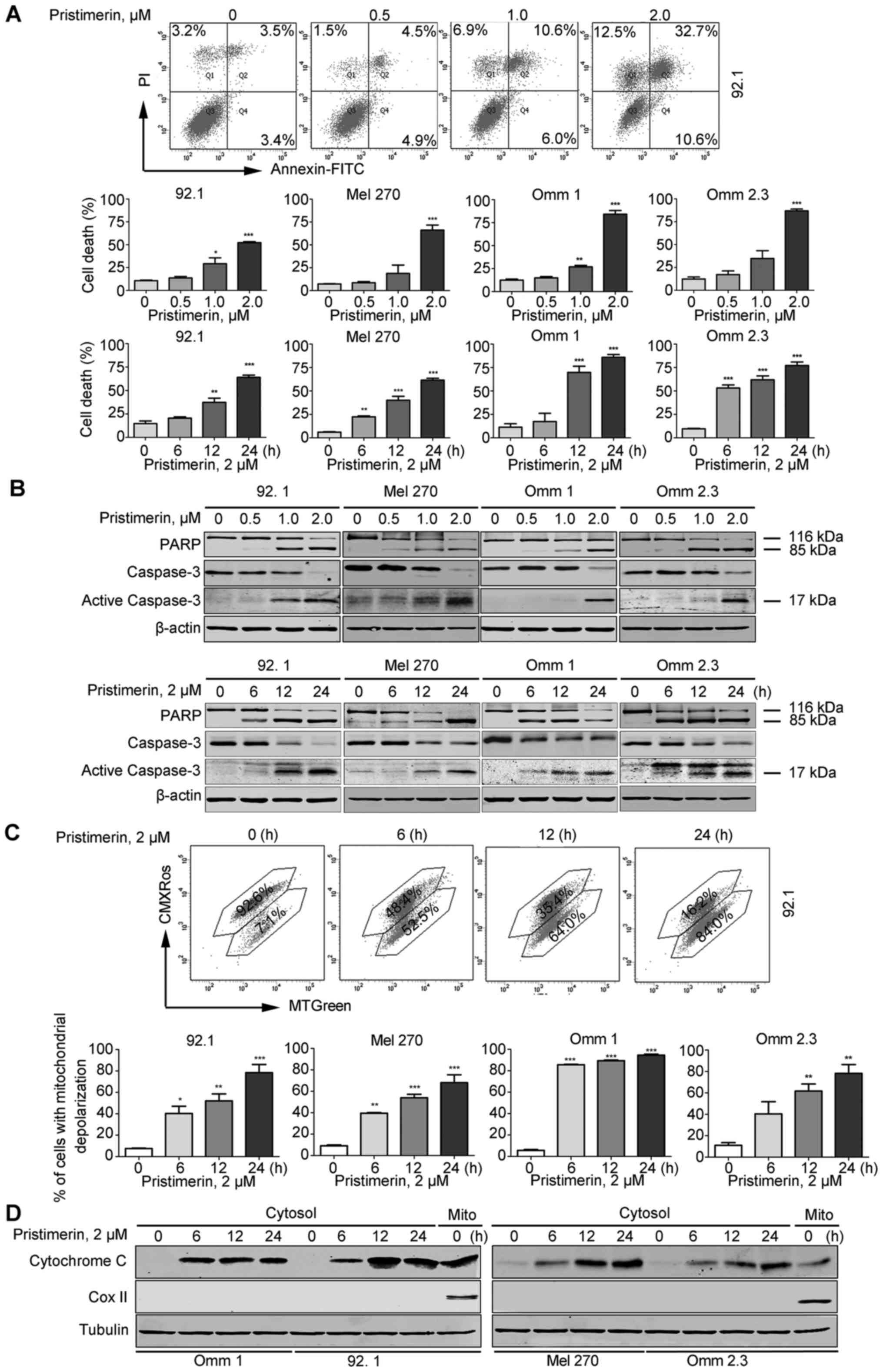 | Figure 3Pristimerin induces intrinsic
apoptosis in UM cells. (A-D) UM cells were treated with different
concentrations of pristimerin for 24 h, or 2.0 µM
pristimerin for the indicated times. (A) Pristimerin induced
apoptosis of UM cells in a dose- and time-dependent manner. The
representative flow cytometry histograms of Annexin V-FITC/PI
double staining are shown (top), statistical chart showed the
quantitative analysis of three independent experiments (bottom).
The results represent the means ± SE of three independent
experiments. *P<0.05, **P<0.01,
***P<0.001, one-way ANOVA, post hoc intergroup
comparisons, Tukey's test. (B) Pristimerin decreased the expression
of apoptosis-indicative proteins in dose- and time-dependent
manner. Western blotting showed the effect of pristimerin on
expression of apoptosis-indicative proteins including PARP,
caspase-3 and active caspase-3. (C) Pristimerin triggered the
mitochondrial depolarization of UM cells. The mitochondrial
depolarization of UM cells was detected by flow cytometry after
treatment with pristimerin. UM cells pre-incubated with 2.0
µM pristimerin in the indicated time periods and then
stained with CMXRos and MTGreen. Mitochondrial membrane potential
(Δψ) was analyzed by flow cytometry. The results represented the
means ± SE of three independent experiments. *P<0.05,
**P<0.01, ***P<0.001, one-way ANOVA,
post hoc intergroup comparisons, Tukey's test. (D) Pristimerin
induced the release of cytochrome c in UM cells. Western
blotting showed the level of cytochrome c in cytoplasm, and
tubulin as a loading control for cytosol. |
Next, we evaluated the effect of pristimerin on
mitochondrial depolarization in UM cells. After being treated with
pristimerin, the cell populations with loss of mitochondrial
transmembrane potential were significantly increased (Fig. 3C). In parallel, western blot
analysis showed that the levels of cytochrome c in the
cytosolic fractionation were increased in a time-dependent manner
(Fig. 3D). These results suggested
that pristimerin might cause damage of mitochondria and trigger the
intrinsic pathway of apoptosis in UM cells.
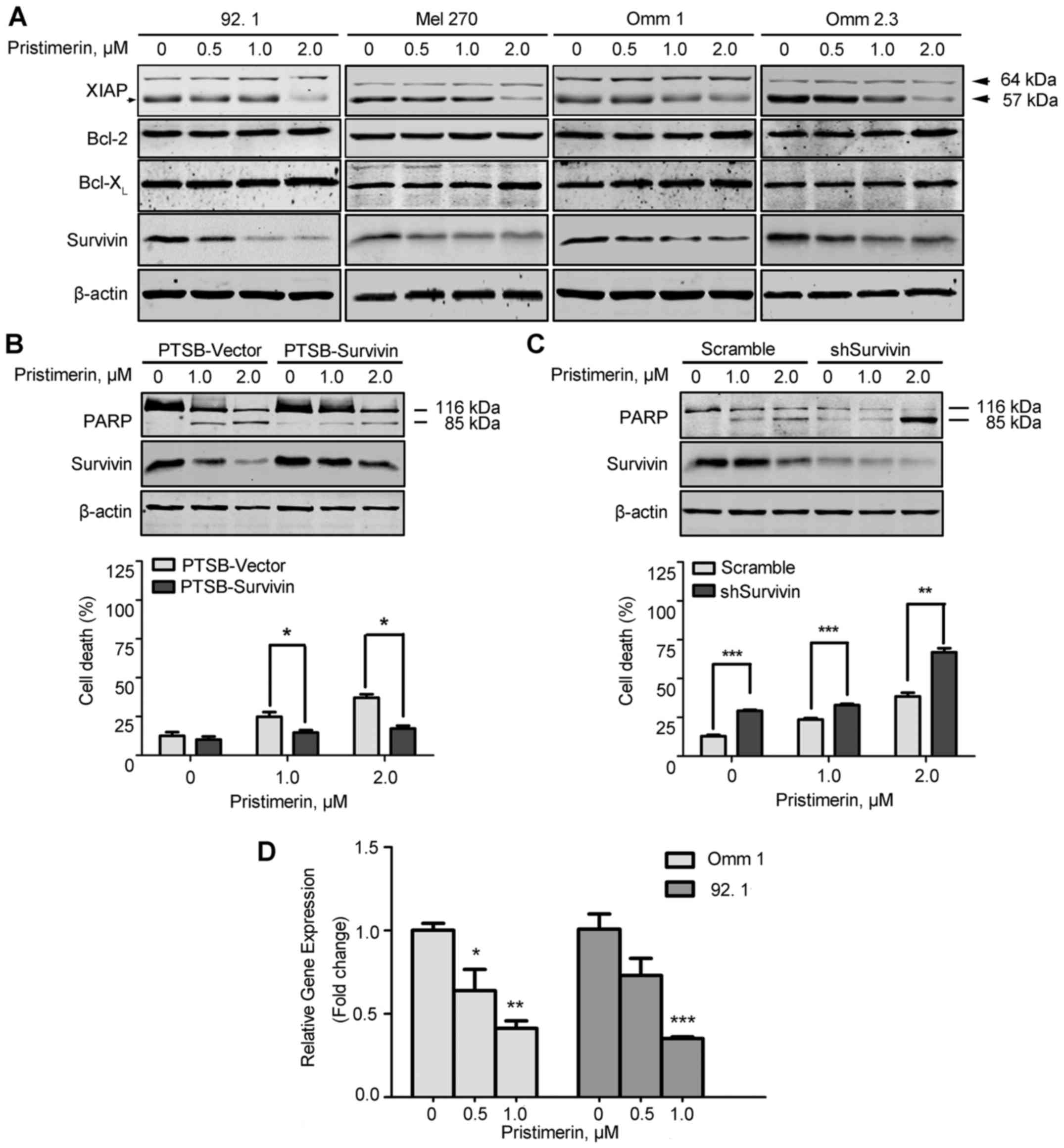
Survivin plays an important role in
pristimerin-induced apoptosis
To dissect the mechanism by which pristimerin
induced apoptosis in UM cells, we next measured apoptosis-related
proteins. The results showed that pristimerin decreased the
expression of XIAP and survivin, but not Bcl-2 and
Bcl-XL (Fig. 4A). Given
that survivin was obviously declined and the anti-apoptotic role of
survivin in pristimerin-inducing apoptosis in prostate cancer cells
(38), we hypothesized that
survivin might be critical in pristimerin-inducing apoptosis in UM
cells. The Omm 1 cell ectopic overexpression of survivin by
lentiviral construct encoding survivin were more resistant to
pristimerin than cells transfected with empty vector (Fig. 4B). On the other hand, 92.1 cells
were transfected with scramble or survivin shRNA, and the results
showed that knockdown of survivin facilitated the sensitivity of UM
cells to pristimerin (Fig. 4C).
These data imply that survivin indeed plays a critical role in
pristimerin-inducing apoptosis.
We next studied the regulation of survivin by
pristimerin. qRT-PCR analysis showed that pristimerin treatment in
92.1 and Omm 1 cells led to a decrease in the mRNA level of BIRC5
(encoding survivin) (Fig. 4D). The
results suggested that downregulation of survivin by pristimerin
occurs at the level of transcription in UM cells.
Pristimerin inhibits migration and
invasion of UM cells
Although multiple therapies are available for
primary UM patients, therapeutic options are limited for the
metastatic patients with no apparent reduced mortality in the past
decades (1,2,4). In
order to investigate the effect of pristimerin on the motility of
UM cells, wound healing ability was tested in 92.1 and Omm 1 cells.
After 72 h of pristimerin treatment, the occlusion of UM cells was
remarkably slowed (Fig. 5A). We
next examined the inhibitory effect of pristimerin on Transwell
migration and invasion and found that pristimerin profoundly
decreased the number of migrated (Fig.
5B) and invaded (Fig. 5C)
cells. Local invasion is an initial step of metastasis with
involvement of matrix metalloproteinases (MMPs) (29,39).
We therefore assumed that pristimerin suppressed the invasion of UM
cells by decreasing the expression or activity of MMP2 and MMP9.
Western blotting results showed that pristimerin decreased the
protein levels of MMP2 and MMP9 in a dose-dependent manner
(Fig. 5D). Moreover, gelatin
zymography assay results indicated that pristimerin inhibited the
activity of MMP9 in a dose-dependent manner (Fig. 5E).
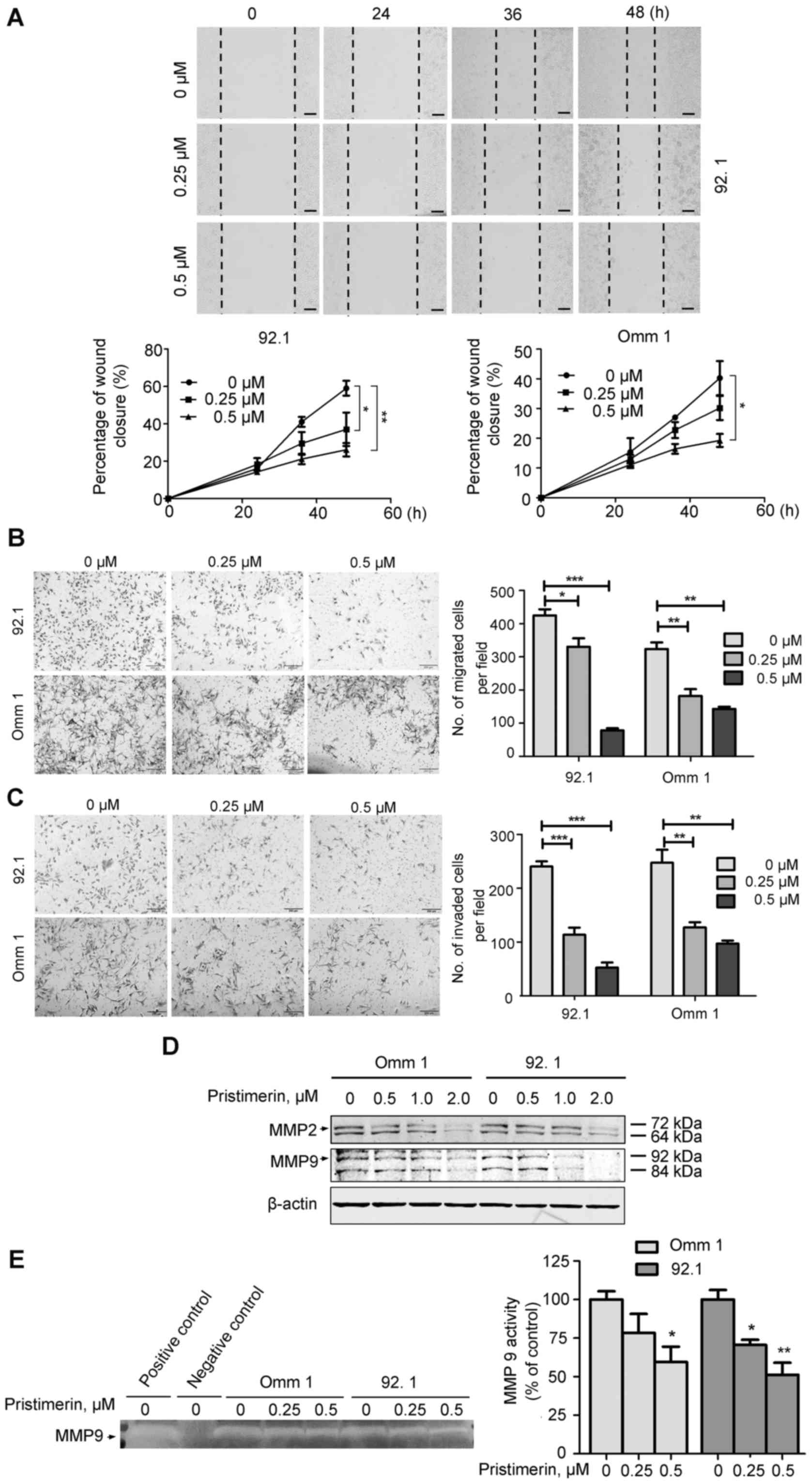 | Figure 5Pristimerin impairs the ability of
migration and invasion in UM cells. (A) Pristimerin reduced the
motility of UM cells. After 92.1 and Omm 1 cells were seeded in
6-well plates overnight, a wound was made by a 200-µl
pipette tip. The cells were exposed to vehicle, 0.25 or 0.5
µM pristimerin for the indicated periods, and the wound
closure was recorded under microscope. The images were the
representative results (top), while the graphs were the
quantitative analysis of the percentage of wound closure at
different time-points (bottom). Data were shown as the means ± SE.
*P<0.05, **P<0.01, one-way ANOVA was
used for statistical analysis. Scale bar represents 200 µm,
original magnification, ×200. (B) Pristimerin inhibited the
Transwell migration ability of UM cells. Left, representative image
of migration assay of 92.1 and Omm 1 cells after exposed to
vehicle, 0.25 or 0.5 µM pristimerin for 24 h. Right,
quantitative analysis of the migration cells was showed. Values
were expressed as mean cell numbers in three random fields of view
(×200) in three independent experiments. Error bars represented the
means ± SE. *P<0.05, **P<0.01,
***P<0.001, one-way ANOVA was used for statistical
analysis. (C) Pristimerin suppressed the invasion ability of UM
cells. The representative images of invaded cells which were
treated with vehicle, 0.25 or 0.5 µM pristimerin for 24 h.
Scale bar represents 200 µm (left), and results of the
invaded cells of per field (three random field) (right). The
results showed as the means ± SE. **P<0.01,
***P<0.001, one-way ANOVA was used for statistical
analysis. (D) Pristimerin reduced the expression of MMP2 and MMP9
in a dose-dependent manner. The whole lysates of cells were used to
detect the protein level of MMP2 and MMP9 by western blot analysis
after Omm 1 and 92.1 cells were treated with pristimerin for 24 h.
(E) Pristimerin inhibited the activity of MMP9 in a dose-dependent
manner. Omm 1 and 92.1 cells were cultured and treated with or
without pristimerin (0.25 and 0.5 µM) for 24 h. The
serum-free medium of cells was used to detect the activity of MMP9
by gelatin zymography assay. The serum-free medium of MDA-MB-231
cells and serum-free medium without cells were used as positive
control and negative control, respectively. The images (left) were
the representative result, while the graphs (right) were the
quantitative analysis of MMP9 activity at different concentration
of pristimerin (n=3). The results showed as the means ± SE.
*P<0.05, **P<0.01, one-way ANOVA was
used for statistical analysis. |
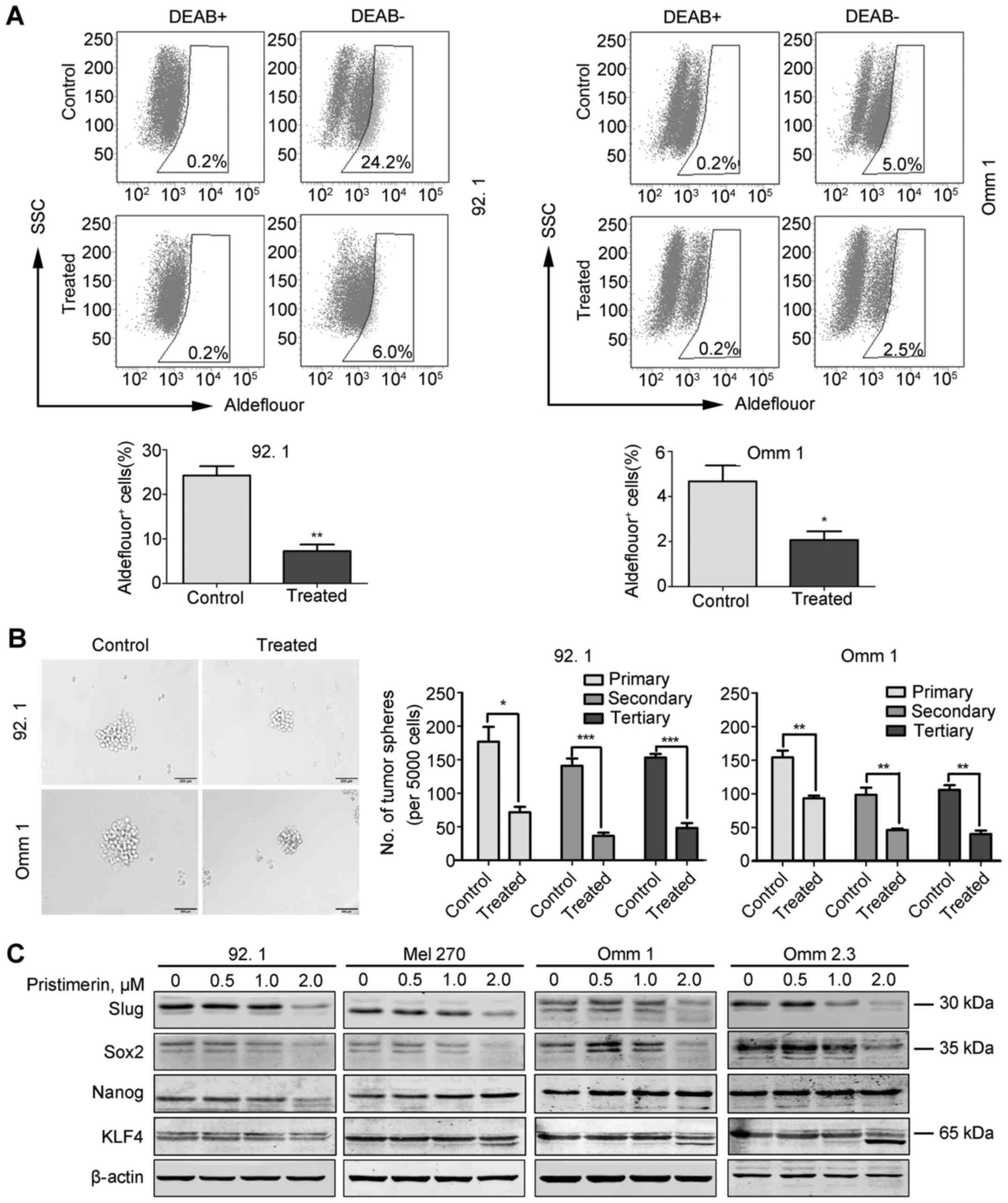
Pristimerin weakened the properties of
cancer stem-like cells in UM cells
CSC theory holds a view that CSC is the main cause
for metastasis and therapeutic resistant (40). Several in vitro assays such
as ALDH+ assay and tumor sphere formation allow for the
study of CSCs (10). Flow
cytometry analysis showed that the percentage of ALDH+
cells was significantly decreased in the cells treated with
pristimerin compared to that in control group (Fig. 6A). On the other hand, three rounds
of serially re-plating culture of melanoshpere showed that the
numbers of melanosphere in the pristimerin-treated cells were
significantly lower than those in control group (Fig. 6B), implying that pristimerin exerts
an inhibitory activity on the self-renewal capacity of CSCs in UM
cells. We also examined the universal stemness-related proteins
which were also regulated by NF-κB pathway such as Slug, Sox2,
Nanog and KLF4 (10), the results
showed that the expression of Slug and Sox2 was decreased in the
cells treated with the highest concentration (2 µM) of
pristimerin (Fig. 6C). The protein
levels of Nanog and KLF4 remained constant (Fig. 6C).
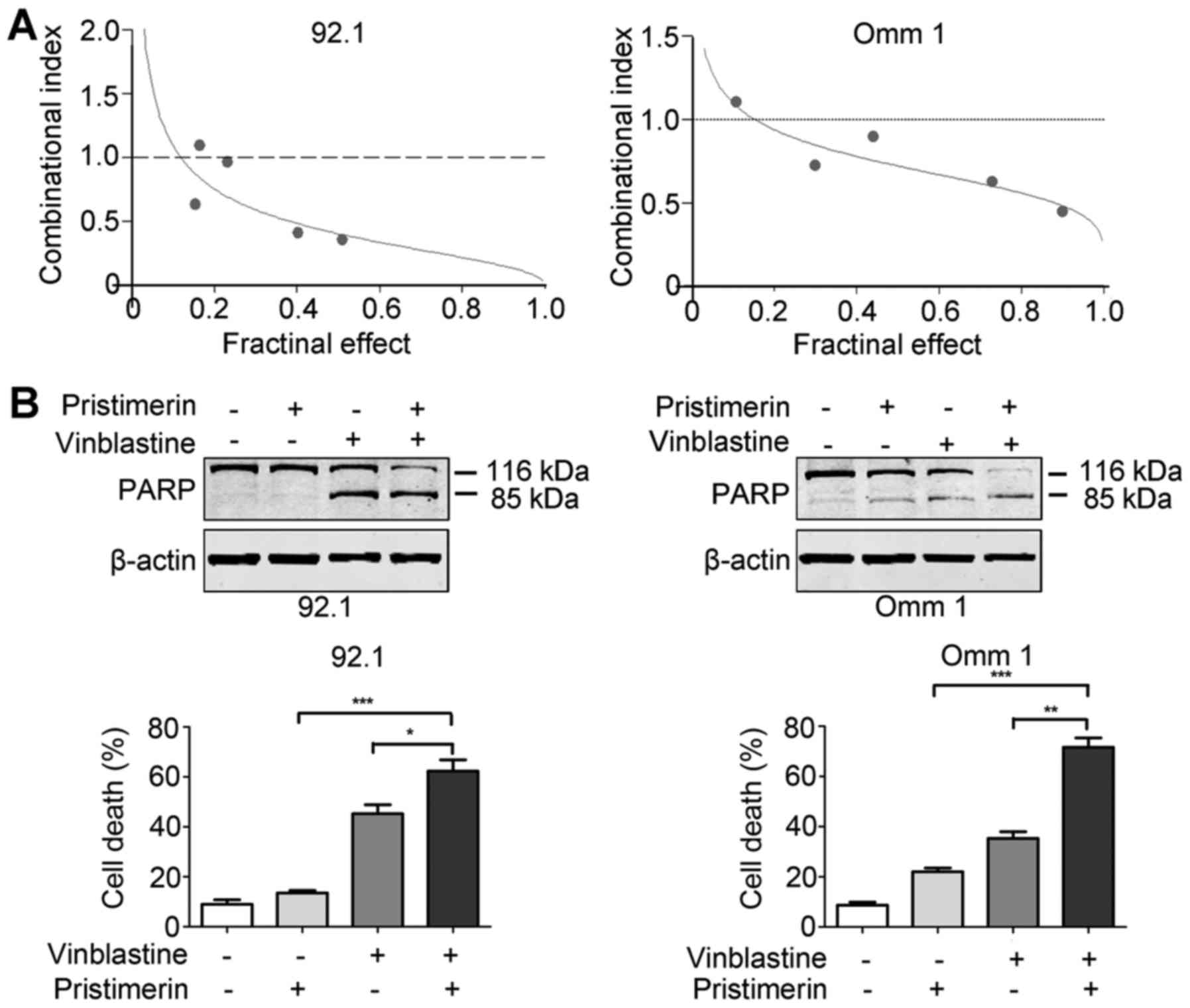
Pristimerin enhances the sensitivity of
UM cells to vinblastine
Vinblastine, a conventional chemotherapeutic agent,
was used in clinical treatment of UM patients (3). We thus exploited whether pristimerin
increased sensitivity of UM cells to vinblastine. The results of 72
h of combinational treatment showed a synergistic effect in 92.1
and Omm 1 cells (combinational index, CI<1) (Fig. 7A). Furthermore, detection of PARP
cleavage and trypan blue exclusion assay confirmed an enhanced
apoptosis-inducing effect in UM cells (Fig. 7B).
Discussion
Although recent technical advances have opened up
many new ways in treating primary UM patients, there are no
effective therapeutic approaches for those who have metastasized.
In this study, our results demonstrated that pristimerin displayed
significant anticancer activities by blocking the NF-κB pathway in
UM cells. Pristimerin suppressed TNFα-induced IκBα phosphorylation
and degradation, translocation of p65, and expression of
NF-κB-dependent genes. Pristimerin effectively inhibited the
malignant phenotypes such as proliferation, resistance to
apoptosis, migration, invasion and CSCs in UM cells. Additionally,
pristimerin decreased the expression of survivin at transcriptional
level which plays a critical role in pristimerin-inducing apoptosis
of UM cells.
It is well-known that cancer-related inflammation is
a hallmaker of cancer (41). In
physiologic condition, eyes are in a state of immune privilege
without existences of immune cells. Once tumor occurs in the eyes,
this state is challenged with infiltration with myeloid and T cells
which constitute an intraocular inflammatory microenvironment
(42). Previous evidence has
demonstrated that cytokines and chemokines (e.g., TNFα, IL-6, IL-8,
IP-10, MIP-1 and IFN-γ) in the vitreous fluid of eyes with UM are
elevated (43). Among them, TNFα
can activate the NF-κB pathway (37). Moreover, hyperactivation of NF-κB
not only contributes to aberrant local tumor cell survival and
growth, but also promotes distant metastasis (12,41,43).
Thus, NF-κB inhibition appears to offer a promising strategy in
cancer therapy in UM.
The CSC theory has emerged as an important landmark
in the understanding of drug resistance and cancer recurrence, and
CSCs are regarded as an essential cause of tumor metastasis,
recurrence and drug tolerance (40). So far, many classic signal pathways
such as Wnt/β-catenin, Hedgehog, and Notch pathway have shown
extensive involvement in the coordination of CSCs (40). Additionally, accumulating evidence
indicated the existence of links between CSCs and NF-κB pathway
(10). Our study showed that
pristimerin inhibited the characteristic of CSCs in UM cells, which
is consistent with a recent report in prostate cancer cells
(20,22). Given the fundamental role of Slug
in CSCs in breast cancer (44),
the role of Slug in UM CSCs need to be defined in the future.
In order to acquire better therapeutic outcomes,
concomitant drugs become a conventional approach in clinical cancer
therapy. Therefore, we chose vinblastine as a representative front
line agent in our research to explore the potential value of
pristimerin in UM clinical combination treatment. This is the first
report that pristimerin has synergistic effect with vinblastine in
UM cells.
Pristimerin is a compound isolated from nature
products and the antitumor activity of this compound is
complicated. It is reported that pristimerin modulates other
molecular targets such as cyclins, apoptosis-related proteins,
proteasome activity, AKT/mTOR and MAPK/ERK pathways contributing to
its antitumor activity (14).
Our results indicated that pristimerin effectively
blocks the NF-κB pathway, which contributes to the inhibition of
malignant phenotypes in UM cells. However, we could not exclude the
possibility of this compound to target other molecular pathways in
UM cells.
In conclusion, pristimerin not only killed the bulk
of tumor cells, but also effectively eliminated CSCs in UM cells.
Pristimerin as a lead compound also exerted favorable anticancer
effects both in single administration and combination with
vinblastine. Thus, further clinical trial of pristimerin against UM
may be warranted.
Acknowledgments
This study was supported by grants from National
Natural Science Funds (nos. U1301226, and 81373434 to J. Pan), the
National Basic Research Program of China (973 program no.
2009CB825506 to J. Pan), the Research Foundation of Education
Bureau of Guangdong Province, China (no. cxzd1103 to J. Pan), and
Natural Science Foundation of Guangdong province (no.
2015A030312014 to J. Pan).
References
|
1
|
Singh AD, Bergman L and Seregard S: Uveal
melanoma: Epidemiologic aspects. Ophthalmol Clin North Am.
18:75–84. viii2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Singh AD, Turell ME and Topham AK: Uveal
melanoma: Trends in incidence, treatment, and survival.
Ophthalmology. 118:1881–1885. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bedikian AY, Johnson MM, Warneke CL,
Papadopoulos NE, Kim KB, Hwu WJ, McIntyre S, Rohlfs M, Homsi J and
Hwu P: Does complete response to systemic therapy in patients with
stage IV melanoma translate into long-term survival? Melanoma Res.
21:84–90. 2011. View Article : Google Scholar
|
|
4
|
Damato B and Heimann H: Personalized
treatment of uveal melanoma. Eye (Lond). 27:172–179. 2013.
View Article : Google Scholar
|
|
5
|
Li B, Li YY, Tsao SW and Cheung AL:
Targeting NF-κB signaling pathway suppresses tumor growth,
angiogenesis, and metastasis of human esophageal cancer. Mol Cancer
Ther. 8:2635–2644. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sakamoto K and Maeda S: Targeting NF-κB
for colorectal cancer. Expert Opin Ther Targets. 14:593–601. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Luedde T and Schwabe RF: NF-κB in the
liver - linking injury, fibrosis and hepatocellular carcinoma. Nat
Rev Gastroenterol Hepatol. 8:108–118. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nogueira L, Ruiz-Ontañon P,
Vazquez-Barquero A, Moris F and Fernandez-Luna JL: The NF-κB
pathway: A therapeutic target in glioblastoma. Oncotarget.
2:646–653. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lu Z, Jin Y, Chen C, Li J, Cao Q and Pan
J: Pristimerin induces apoptosis in imatinib-resistant chronic
myelogenous leukemia cells harboring T315I mutation by blocking
NF-κB signaling and depleting Bcr-Abl. Mol Cancer. 9:1122010.
View Article : Google Scholar
|
|
10
|
Rinkenbaugh AL and Baldwin AS: The NF-κB
pathway and cancer stem cells. Cells. 5:52016. View Article : Google Scholar
|
|
11
|
Lee CH, Jeon YT, Kim SH and Song YS: NF-κB
as a potential molecular target for cancer therapy. Biofactors.
29:19–35. 2007. View Article : Google Scholar
|
|
12
|
Dror R, Lederman M, Umezawa K, Barak V,
Pe'er J and Chowers I: Characterizing the involvement of the
nuclear factor-κB (NF κB) transcription factor in uveal melanoma.
Invest Ophthalmol Vis Sci. 51:1811–1816. 2010. View Article : Google Scholar
|
|
13
|
Dirsch VM, Kiemer AK, Wagner H and Vollmar
AM: The triterpenoid quinonemethide pristimerin inhibits induction
of inducible nitric oxide synthase in murine macrophages. Eur J
Pharmacol. 336:211–217. 1997. View Article : Google Scholar
|
|
14
|
Yousef BA, Hassan HM, Zhang LY and Jiang
ZZ: Anticancer potential and molecular targets of pristimerin: A
mini-review. Curr Cancer Drug Targets. 17:100–108. 2017. View Article : Google Scholar
|
|
15
|
Lee SO, Kim JS, Lee MS and Lee HJ:
Anti-cancer effect of pristimerin by inhibition of HIF-1α involves
the SPHK-1 pathway in hypoxic prostate cancer cells. BMC Cancer.
16:7012016. View Article : Google Scholar
|
|
16
|
Xie G, Yu X, Liang H, Chen J, Tang X, Wu S
and Liao C: Pristimerin overcomes adriamycin resistance in breast
cancer cells through suppressing Akt signaling. Oncol Lett.
11:3111–3116. 2016.PubMed/NCBI
|
|
17
|
Yousef BA, Hassan HM, Guerram M, Hamdi AM,
Wang B, Zhang LY and Jiang ZZ: Pristimerin inhibits proliferation,
migration and invasion, and induces apoptosis in HCT-116 colorectal
cancer cells. Biomed Pharmacother. 79:112–119. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao H, Wang C, Lu B, Zhou Z, Jin Y, Wang
Z, Zheng L, Liu K, Luo T, Zhu D, et al: Pristimerin triggers
AIF-dependent programmed necrosis in glioma cells via activation of
JNK. Cancer Lett. 374:136–148. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Deng Q, Bai S, Gao W and Tong L:
Pristimerin inhibits angiogenesis in adjuvant-induced arthritic
rats by suppressing VEGFR2 signaling pathways. Int Immunopharmacol.
29:302–313. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang S, He P, Peng X, Li J, Xu D and Tang
Y: Pristimerin inhibits prostate cancer bone metastasis by
targeting PC-3 stem cell characteristics and VEGF-induced
vasculogenesis of BM-EPCs. Cell Physiol Biochem. 37:253–268. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yousef BA, Guerram M, Hassan HM, Hamdi AM,
Zhang LY and Jiang ZZ: Pristimerin demonstrates anticancer
potential in colorectal cancer cells by inducing G1 phase arrest
and apoptosis and suppressing various pro-survival signaling
proteins. Oncol Rep. 35:1091–1100. 2016.PubMed/NCBI
|
|
22
|
Zuo J, Guo Y, Peng X, Tang Y, Zhang X, He
P, Li S, Wa Q, Li J, Huang S, et al: Inhibitory action of
pristimerin on hypoxia-mediated metastasis involves stem cell
characteristics and EMT in PC-3 prostate cancer cells. Oncol Rep.
33:1388–1394. 2015.PubMed/NCBI
|
|
23
|
Ma YW, Liu YZ and Pan JX: Verteporfin
induces apoptosis and eliminates cancer stem-like cells in uveal
melanoma in the absence of light activation. Am J Cancer Res.
6:2816–2830. 2016.
|
|
24
|
Jin B, Ding K and Pan J: Ponatinib induces
apoptosis in imatinib-resistant human mast cells by
dephosphorylating mutant D816V KIT and silencing β-catenin
signaling. Mol Cancer Ther. 13:1217–1230. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jin Y, Lu Z, Ding K, Li J, Du X, Chen C,
Sun X, Wu Y, Zhou J and Pan J: Antineoplastic mechanisms of
niclosamide in acute myelogenous leukemia stem cells: Inactivation
of the NF-κB pathway and generation of reactive oxygen species.
Cancer Res. 70:2516–2527. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jin Y, Zhou J, Xu F, Jin B, Cui L, Wang Y,
Du X, Li J, Li P, Ren R, et al: Targeting methyltransferase PRMT5
eliminates leukemia stem cells in chronic myelogenous leukemia. J
Clin Invest. 126:3961–3980. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pan J, Quintás-Cardama A, Kantarjian HM,
Akin C, Manshouri T, Lamb P, Cortes JE, Tefferi A, Giles FJ and
Verstovsek S: EXEL-0862, a novel tyrosine kinase inhibitor, induces
apoptosis in vitro and ex vivo in human mast cells expressing the
KIT D816V mutation. Blood. 109:315–322. 2007. View Article : Google Scholar
|
|
28
|
Jin B, Wang C, Li J, Du X, Ding K and Pan
J: Anthelmintic niclosamide disrupts the interplay of p65 and
FOXM1/β-catenin and eradicates leukemia stem cells in chronic
myelogenous leukemia. Clin Cancer Res. 23:789–803. 2017. View Article : Google Scholar
|
|
29
|
Dai W, Zhou J, Jin B and Pan J: Class
III-specific HDAC inhibitor Tenovin-6 induces apoptosis, suppresses
migration and eliminates cancer stem cells in uveal melanoma. Sci
Rep. 6:226222016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Morrison DJ, Hogan LE, Condos G, Bhatla T,
Germino N, Moskowitz NP, Lee L, Bhojwani D, Horton TM,
Belitskaya-Levy I, et al: Endogenous knockdown of survivin improves
chemotherapeutic response in ALL models. Leukemia. 26:271–279.
2012. View Article : Google Scholar
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
32
|
Yarrow JC, Perlman ZE, Westwood NJ and
Mitchison TJ: A high-throughput cell migration assay using scratch
wound healing, a comparison of image-based readout methods. BMC
Biotechnol. 4:212004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dinicola S, Pasqualato A, Cucina A,
Coluccia P, Ferranti F, Canipari R, Catizone A, Proietti S,
D'Anselmi F, Ricci G, et al: Grape seed extract suppresses
MDA-MB231 breast cancer cell migration and invasion. Eur J Nutr.
53:421–431. 2014. View Article : Google Scholar
|
|
34
|
Toth M and Fridman R: Assessment of
gelatinases (MMP-2 and MMP-9 by gelatin zymography. Methods Mol
Med. 57:163–174. 2001.PubMed/NCBI
|
|
35
|
Ginestier C, Hur MH, Charafe-Jauffret E,
Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG,
Liu S, et al: ALDH1 is a marker of normal and malignant human
mammary stem cells and a predictor of poor clinical outcome. Cell
Stem Cell. 1:555–567. 2007. View Article : Google Scholar
|
|
36
|
Nonaka M, Yawata T, Takemura M, Higashi Y,
Nakai E, Shimizu K and Ueba T: Elevated cell invasion in a tumor
sphere culture of RSV-M mouse glioma cells. Neurol Med Chir
(Tokyo). 55:60–70. 2015. View Article : Google Scholar
|
|
37
|
Chen LF and Greene WC: Shaping the nuclear
action of NF-κB. Nat Rev Mol Cell Biol. 5:392–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu YB, Gao X, Deeb D, Brigolin C, Zhang
Y, Shaw J, Pindolia K and Gautam SC: Ubiquitin-proteasomal
degradation of antiapop-totic survivin facilitates induction of
apoptosis in prostate cancer cells by pristimerin. Int J Oncol.
45:1735–1741. 2014.PubMed/NCBI
|
|
39
|
Huang Q, Lan F, Wang X, Yu Y, Ouyang X,
Zheng F, Han J, Lin Y, Xie Y, Xie F, et al: IL-1β-induced
activation of p38 promotes metastasis in gastric adenocarcinoma via
upregulation of AP-1/c-fos, MMP2 and MMP9. Mol Cancer. 13:182014.
View Article : Google Scholar
|
|
40
|
Clarke MF, Dick JE, Dirks PB, Eaves CJ,
Jamieson CH, Jones DL, Visvader J, Weissman IL and Wahl GM: Cancer
stem cells-perspectives on current status and future directions:
AACR Workshop on cancer stem cells. Cancer Res. 66:9339–9344. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bronkhorst IH and Jager MJ: Inflammation
in uveal melanoma. Eye (Lond). 27:217–223. 2013. View Article : Google Scholar
|
|
43
|
Nagarkatti-Gude N, Bronkhorst IH, van
Duinen SG, Luyten GP and Jager MJ: Cytokines and chemokines in the
vitreous fluid of eyes with uveal melanoma. Invest Ophthalmol Vis
Sci. 53:6748–6755. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Storci G, Sansone P, Mari S, D'Uva G,
Tavolari S, Guarnieri T, Taffurelli M, Ceccarelli C, Santini D,
Chieco P, et al: TNFalpha up-regulates SLUG via the
NF-kappaB/HIF1alpha axis, which imparts breast cancer cells with a
stem cell-like phenotype. J Cell Physiol. 225:682–691. 2010.
View Article : Google Scholar : PubMed/NCBI
|