Introduction
Globally, prostate cancer (PCa) is the most commonly
diagnosed type of cancer in males (1). In 2015, the incidence of PCa in
Chinese males was estimated to be 9 per 100,000 individuals and the
mortality rate was approximately 4.5 per 100,000 individuals. From
2005 to 2011, the average percentage increase in the incidence of
PCa in China was 4.7%, representing the fastest growth rate in all
cancer types in males (2). These
data indicate a high disease burden by PCa in China
The majority of cases of PCa are diagnosed at an
early stage and treatments include surgery and radiotherapy, with
or without androgen deprivation (1,3,4).
Depending on the tumor stage, between 55 and 90% of all PCa cases
can be permanently controlled by radiotherapy (4–6).
However, approximately 50% of patients with PCa receiving
radiotherapy will relapse within 5 years of treatment (7,8).
Relapse following radiotherapy is multifactorial. It can result
from an intrinsic radioresistance of a subpopulation of clones
within the tumor (9), or from
acquired radioresistance during radiotherapy (10–12).
Therefore, radioresistance is a major challenge in the treatment of
PCa.
Oxygen is one of the most influential factors on the
cytotoxic effects of radiation, due to its high affinity for free
radicals produced by radiation in cellular components. Solid tumors
usually have highly abnormal blood vessels and distended
capillaries with leaky walls and sluggish flow, indicating a
hypoxic environment that reduces the effects of radiation and
promotes resistance to radiotherapy in solid tumors (13,14).
In addition to the lack of oxygen as a chemical mechanism of
resistance to radiation, cellular adaptive responses to hypoxia
mediated by hypoxia-inducible factor-1α (HIF-1α) are involved in
the induction of radioresistance in cancer cells (14,15).
HIF-1α is activated in cancer cells, not only under hypoxic
conditions, but also under normoxic conditions through
cancer-specific genetic alterations and the resulting imbalance in
intermediate metabolites (16).
Indeed, HIF-1α is activated in the presence of decreased
Fe2+ or α-ketoglutarate levels (16). HIF-1α mediates radioresistance
through a number of methods: i) By the reprogramming of glucose
metabolism and the overproduction of antioxidants; ii) nucleic acid
synthesis; iii) cell cycle regulation; iv) the protection of tumor
blood vessels; and v) the repopulation of surviving cells after
radiotherapy (16). Therefore,
HIF-1α is considered to play crucial roles in tumor
radioresistance; however, the molecular mechanisms downstream of
HIF-1α which are responsible for tumor radioresistance remain
unclear.
The β-catenin signaling pathway regulates
embryogenesis and tumor progression. Existing studies have
demonstrated that the inhibition of β-catenin can lead to an
enhanced radio-sensitivity of the PCa cell line, PC3, and of the
significantly radioresistant cancer cell line, AMC-HN-9 (17,18).
The over-expression of HIF-1α promotes the invasive potential of
human PCa cells through epithelial-mesenchymal transition (EMT) and
β-catenin plays a vital role in this process (19,20).
The knockdown of β-catenin induced by HIF-1α causes the reversal of
EMT and metastatic phenotypic changes (19). This suggests a potential mechanism
involving β-catenin through which HIF-1α controls radioresistance
in PCa.
The present study aimed to investigate the molecular
mechanisms in relation to β-catenin that are downstream of HIF-1α,
which may contribute to the radioresistance of prostate tumors. In
addition, we utilized both PCa cell lines and animal models to
examine our hypothesis through in vitro and in vivo
interventions. Furthermore, we investigated protein markers for
cell proliferation, cell invasion, cell cycle distribution, cell
death and DNA repair in order to provide a comprehensive
understanding of biological functional changes under the activation
or inhibition of β-catenin with or without radiation treatment.
Materials and methods
Cell lines
The human PCa cell lines, LNCaP and C4-2B, were
generous gifts from Dr Likun Li (MD Anderson Cancer Center). These
two cell lines were validated by short tandem repeat DNA
fingerprinting with the AmpFLSTR Identifiler kit (Applied
Biosystems, Foster City, CA, USA) at the MD Anderson's
Characterized Cell Line Core Facility. Both cell lines were
cultured in DMEM containing 1 mM sodium pyruvate, 2.5 mM glutamine,
10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin at
37°C in a humidified incubator containing 5% CO2. The
LNCaP or C4-2B cells were divided into 3 experimental groups as
follows: The negative control (no treatment), the HIF-1α
overexpression group [transfected with the PcDNA3.1(−)/HIF-1α
plasmid and screened for HIF-1α high expression clones] and the
β-catenin silenced group (PSuper-β-catenin-shRNA vector used in
HIF-1α-overexpressing cells to inhibit β-catenin expression and
selected for clones with stable silencing of β-catenin). The
negative control and β-catenin silenced groups were incubated under
normoxic conditions (18%oxygen). Cells in the HIF-1α overexpression
group were cultured under hypoxic conditions (1% oxygen;
Biospherix, Ltd., Parish, NY, USA) during clone selection, and
grown under normoxic conditions after the clones were identified
and stabilized.
Plasmid transfection and RNA
interference
In the HIF-1α overexpression group,
PcDNA3.1(−)/HIF-1α recombinant plasmid transfection was conducted
using Lipofectamine 2000 (Invitrogen Inc., Carlsbad, CA, USA) as
described previously (20). During
preliminary experiments (data not shown), to further confirm that
HIF-1α overexpression was due to transfection with the
PcDNA3.1(−)/HIF-1α plasmids, a second negative control using
PcDNA3.1(−) plasmid transfection was conducted in both cell
lines.
A PSuper-enhancement green fluorescent protein 1
(EGFP1) vector was used to construct the PSuper-β-catenin-ShRNA and
PSuper-Scramble-β-catenin-ShRNA plasmids based on a previous study
(21). The β-catenin-shRNA and
scramble-β-catenin-shRNA primer sequences are listed below. In the
β-catenin silenced group, HIF-1α overexpression clones were further
knocked down for β-catenin expression using the β-catenin-shRNA
plasmids using Lipofectamine RNAiMax transfection reagent (Life
Technologies Co., Grand Island, NY, USA). During preliminary
experiments (data not shown), to further that confirm β-catenin
inhibition in the β-catenin silenced group was caused by
β-catenin-shRNA transfection, a second negative control using
scramble-β-catenin-ShRNA transfection was conducted in both cell
lines. β-catenin expression was identified in these clones by a
fluorescence microscope, reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) and western blot analysis. RNA
interference was conducted based on a previously published protocol
(19).
The primer sequences of the shRNAs were as follows:
β-catenin-shRNA forward, 5′-GAT CCC CGC AAC AGT CTT ACC TGG ACT TCA
AGA GAG TCCAGG TAA GAC TGT TGC TTT TTA-3′ and reverse, 3′GGG CGT
TGT CAG AAT GGA CCT GAA GTT CTC TCA GGT CCA TTC TGA CAA CGA AAA ATT
CGA5′; and scramble-β-catenin-shRNA forward, 5′-GAT CCC CAA CGA GTG
TGC CTA CAT CCT TCA AGA GAG GAT GTA GGC ACA CTC GTT TTT TTA-3′ and
reverse, 3′-GGG TTG CTC ACA CGG ATG TAG GAA GTT CTC TCC TAC ATC CGT
GTG AGC AAA AAA ATT CGA-5′.
Immunofluorescence
The cells were fixed in 10% paraformaldehyde for 30
min and blocked with goat serum for 30 min. The cells were
incubated at 37°C for 1 h with mouse anti-human β-catenin
monoclonal antibody (sc7963, Santa Cruz Biotechnology, Santa Cruz,
CA, USA) at a dilution of 1:200. After being washed 3 times with
PBS, the cells were co-incubated with fluorescein isothiocyanate
(FITC)-conjugated goat anti-mouse antibody (sc2010, Santa Cruz
Biotechnology) at 37°C for 1 h. The fluorescence staining intensity
and intracellular localization were then determined by a
fluorescence microscope (Olympus Corporation, Tokyo, Japan).
Western blot analysis
The cells were harvested after being washed with
phosphate-buffered saline (PBS) twice, lysed with RIPA cell lysis
buffer (Thermo Fisher Scientific, Waltham, MA, USA) for 30 min on
ice, and centrifuged at 16,000 x g for 15 min at 4°C. The
concentration of total protein was determined using a BCA protein
assay kit. The clarified protein lysates (50 μg/load) were
then separated on denaturing 10% SDS-PAGE gels and
electro-transferred onto nitrocellulose membranes. The membranes
were initially incubated with 5% non-fat dry milk in TBS for 2 h,
and then probed at 4°C overnight with the following antibodies:
anti-HIF-1α (sc53546, 1:1,000), anti-Glut-1 (sc1605, 1:1,000),
anti-VEGF (sc7269, 1:500) (HIF-1α signaling markers, Santa Cruz
Biotechnology), anti-t-β-catenin (ab16051, 1:1,000),
anti-p-β-catenin (Ser45, ab18824, 1:500), anti-p-β-catenin (Y654,
ab59430, 1:500), anti-t-GSK-3β (ab131356, 1:500), anti-p-GSK-3β
(Ser9, ab131097, 1:500) (β-catenin signaling markers; Abcam,
Cambridge, MA, USA), anti-E-cadherin (sc8426, 1:1,000) and
anti-CK18 (sc70917, 1:1,000) (epithelial markers; Santa Cruz
Biotechnology), anti-vimentin (sc6260, 1:1,000), anti-fibronectin
(sc8422, 1:500), anti-matrix metalloproteinase (MMP)2 (sc13595,
1:500) (mesenchymal markers; Santa Cruz Biotechnology),
anti-vascular endothelial growth factor receptor (VEGFR; ab2349,
1:50) (angiogenesis marker; Abcam), anti-CDK1 (ab18, 1:1,000),
anti-p-CDK1 (Y15, ab47594, 1:1,000), anti-Chk1 (ab47574, 1:1,000),
anti-p-Chk1 (S296, ab79758, 1:500), anti-Chk2 (ab47433, 1:500),
anti-p-Chk2 (T387, ab55319, 1:500), anti-Rb (ab226979, 1:1,000),
anti-p-Rb (S780, ab173289, 1:100), anti-p21 (ab227443, 1:500) (cell
cycle markers; Abcam), anti-caspase-3 (cs9662) (1:1,000),
anti-cleaved-caspase-3 (Asp 175, cs9661, 1:1,000), anti-caspase-7
(cs9492, 1:500), anti-cleaved-caspase-7 (Asp 198, cs9491, 1:500),
anti-cleaved poly(ADP-ribose) polymerase (PARP)-1 (Asp214, cs5625,
1:500), anti-Bax (cs2772, 1:1,000) (apoptosis markers; Cell
Signaling Technology, Danvers, MA, USA), anti-Bcl-2 (cs2872,
1:1,000), and anti-Bcl-xL (cs2762, 1:1,000) (anti-apoptosis
markers; Cell Signaling Technology), anti-γH2AX (cs7631, 1:250)
[DNA double-strand break (DSB) marker; Cell Signaling Technology],
anti-Ku70 (ab53126, 1:500), anti-Ku80 (ab53126, 1:500) and
anti-DNA-dependent protein kinase, catalytic subunit (DNA-PKcs;
ab230, 1:500) [non-homologous end joining (NHEJ) pathway markers;
Abcam] and anti-GAPDH (sc47724, 1:1,000) (internal control, Santa
Cruz Biotechnology) antibodies. The membranes were then hybridized
with an appropriate horseradish peroxidase (HRP)-conjugated
secondary antibody (sc2004/sc2005, Santa Cruz Biotechnology) for 2
h at room temperature. An enhanced chemiluminescence system
(Amresco Inc., Solon, OH, USA) was used to detect the
immunopositive protein bands.
MTT assay
The cells (1×104/well) were placed into
96-well plates. At 24, 48 and 72 h, 50 μl MTT solution (2.5
mg/ml; 50 μl) were added followed by incubation for an
additional 4 sh. Cell-associated MTT crystals were dissolved in
DMSO (150 μl/well). The color intensity was measured at 570
nm using a microplate reader (Bio-Rad, Hercules, CA, USA).
In vitro invasion assay
Polycarbonate filters (8-μm-thick; Millipore
corp., Billerica, MA, USA) were coated with 50
μg/cm2 of reconstituted Matrigel (Sigma, St.
Louis, MO, USA). The cells (50,000 in 300 μl of serum-free
growth medium) were seeded into the upper chamber. The cells were
incubated under normoxic conditions and allowed to migrate toward
the complete growth medium for 24, 48 and 72 h. Non-invading cells
were removed mechanically using cotton swabs. The migrated cells,
located on the lower surface, were fixed with methanol and stained
with Giemsa (G1062, Solarib Biotechnology, Beijing, China). The
number of migrating cells was determined by counting 10 random
fields of view on each membrane and photographed by field at ×400
magnification under an inverted microscope (Olympus Corporation).
Data are presented as the mean number of cells per field. Each
experiment was repeated 3 times.
Flow cytometry
The cells were harvested and stained with propidium
iodide for 30 min at room temperature. Analysis was performed at a
405-nm excitation and emission collected with a 450/50 band-pass
filter using a FACSCanto II Flow Cytometer (BD Biosciences,
Franklin Lakes, NJ, USA). Histograms of DNA content were analyzed
using FlowJo software (Tree Star Inc, Ashland, OR, USA) to
determine cell cycle distribution.
Clonogenic survival assay
The cells were seed in 6-well plates at
2×104/well. After sequential radiation and maintenance,
the cells were fixed with 100% cold methanol for 20 min, washed
once with PBS, and stained with 0.5% crystal violet (G1062, Solarib
Biotechnology) diluted in 20% methanol for 20 min. Colonies were
stained with crystal violet and counted with a computer-assisted
program as described previously. Colonies with >50 cells were
counted under a microscope.
DNA fragmentation assay
A DNA fragmentation assay was performed using a Cell
Death Detection ELISA kit (Roche, Mannheim, Germany) for the
apoptotic evaluation of in vitro radiation to cells with
different β-catenin expression and location, according to
previously published methods (22). DNA fragmentation was quantified by
measuring absorbance at 405 nm with a reference wavelength at 490
nm. Data presented are representative of 3 or more independent
experiments.
In vitro radiation treatment
The cells were seeded onto proper cell-culture
plates 24 h prior to irradiation. The cells were irradiated at room
temperature with a single dose of 6 Gy at a rate of 1 Gy/min using
a Gamma cell 40 Exactor (137Cs γ-ray photon radiation; Nordion,
Ottawa, Canada). Following irradiation, all samples were returned
to a 5% CO2 incubator and maintained 72 h for DNA
fragmentation assay, sub-G1 population detection, clonogenic
survival assay, flow cytometry and western blot analysis, and 14
days for colony formation assay.
Animals
BALB/c nude mice and SCID mice (male, 4 weeks old,
20–25 g) were purchased from Charles River Laboratories (Boston,
MA, USA) and maintained in a specific pathogen-free (SPF) class 100
clean room. Animal studies were conducted according to the
recommendations outlined in the Guide for the Care and Use of
Laboratory Animals in the Weatherall report. Animal experiments
were approved by the Committee on the Ethics of Animal Experiments
of the Capital Medical University, Beijing, China.
Orthotopic LNCaP tumor xenografts
The cells (3×106/animal; LNCaP-luc,
LNCaP-luc/HIF-1α, or LNCaP-luc/HIF-1α + shRNA) were injected
orthotopically into the dorsolateral prostate of 4-week old athymic
nude male mice. Approximately 2–6 weeks later, all mice were
monitored using an IVIS Lumina Imaging System (Perkin-Elmer Life
Sciences, Waltham, MA, USA). Mice with a strong luciferase
bioluminescence signal >5×106 were treated with
radiation as described below. Tumor size was monitored every 5 days
according using the luminescence signal.
Subcutaneous C4-2B tumor xenografts
The cells (C4-2B, C4-2B/HIF-1α and C4-2B/HIF-1α +
shRNA) were re-suspended in serum-free DMEM, mixed 1:1 with
Matrigel (BD Biosciences). The cells (1×106/animal) were
injected subcutaneously into the left flanks of previously
castrated SCID mice (Charles River Laboratories, Wilmington, MA,
USA). When palpable tumors reached a volume of 30–50
mm3, the mice were subjected to radiation as described
below. Tumor size was monitored by measuring two dimensions and the
volume was calculated by calculating length ×
width2/2.
In vivo radiation treatment
The nice were irradiated using an Elekta6-MV photon
linear accelerator. Five fractions of 2 Gy were delivered over 5
consecutive days for a total dose of 10 Gy with a dose rate of 1
Gy/min. After the final irradiation treatment, the mice were
observed for 21 consecutive days. When the 21-day protocol was
completed, all mice were euthanized by carbon dioxide inhalation,
and the tumors were harvested. CO2 was displaced in the
euthanisia chamber at the rate of 10–30% of chamber volume per min.
Mice were also euthanized ahead of protocol if they became severely
weak or if the tumor reached 20 mm.
Immunohistochemistry
At the endpoint of animal protocol, tumors were
harvested intactly and clearly, and then fixed by 4%
paraformaldehyde for at least 24 h. Subsequently, speciments were
successively dehydrated by ethanol and treated by xylene. Finally,
speciments were embedded by paraffin and prepared as
2-μm-thick slices for immunohistochemical staining. BRCA1
and Ki67 immunohistochemistry was carried out on formalin-fixed and
paraffin-embedded tissue sections from the 21-day LNCaP and C4-2B
tumor xenografts following in vivo radiation. After the
tissue sections were de-paraffinized and rehydrated through graded
alcohol, they were heated in a microwave in 0.01 mol/l citrate
buffer at pH 6.0 for 10 min to retrieve antigens. Following a
30-min incubation in Dako protein blockage solution, the tissue
sections were incubated in rabbit monoclonal antibodies against
BRCA1 (1:250, ab16780; Abcam) or Ki67 (1:300, sc15402; Santa Cruz
Biotechnology) for 90 min, followed by incubation in a HRP
polymer-conjugated secondary antibody (k4061; Dako, Glostrup,
Denmark) for 40 min. The immunoreaction was visualized in
DAB/H2O2. The specificity of the
immunoreactions was verified by replacing the primary antibodies
with PBS. Ten high-power fields were selected randomly in slides of
each group using an image analysis system (Eclipse 90i; Nikon
Instrument Inc., Tokyo, Japan), and the numbers of positively
stained cells were counted using the Nikon NIS-Elements version 3.0
software and the percentage of positive cells was calculated.
Statistical analysis
Data are presented as the means ± standard deviation
and analyzed using ANOVA with Tukey's post hoc test. Statistical
analysis was undertaken using SPSS 13.0 software for Windows (SPSS
Inc., Chicago, IL, USA). Two-sided P-values <0.05 were
considered to indicate statistically significant differences.
Results
Transfection of the PCa cell lines, LNCaP
and C4-2B, with HIF-1α overexpression plasmid leads to β-catenin
translocation to the nucleus
In both the LNCaP and C4-2B cells, the expression of
HIF-1α and its downstream regulatory proteins (Glut-1 and VEGF) was
significantly increased 72 h following transfection with the HIF-1α
overexpression plasmid (Fig.
1A–D). In the HIF-1α overexpression group, the total expression
of β-catenin (Fig. 1A and E), the
phosphorylation of Ser45 and Y654 of β-catenin (Fig. 1A, F and G), the total expression of
GSK-3β (Fig. 1A and H) and the
phosphorylation of Ser9 of GSK-3β (Fig. 1A and I) were all significantly
increased compared to the negative control group. By contrast,
β-catenin expression and phosphorylation at Ser45 and Y654 were
successfully inhibited in the β-catenin silenced group in both cell
lines; however, the increased phosphorylation of GSK-3β Ser9 was
not reversed by β-catenin silencing.
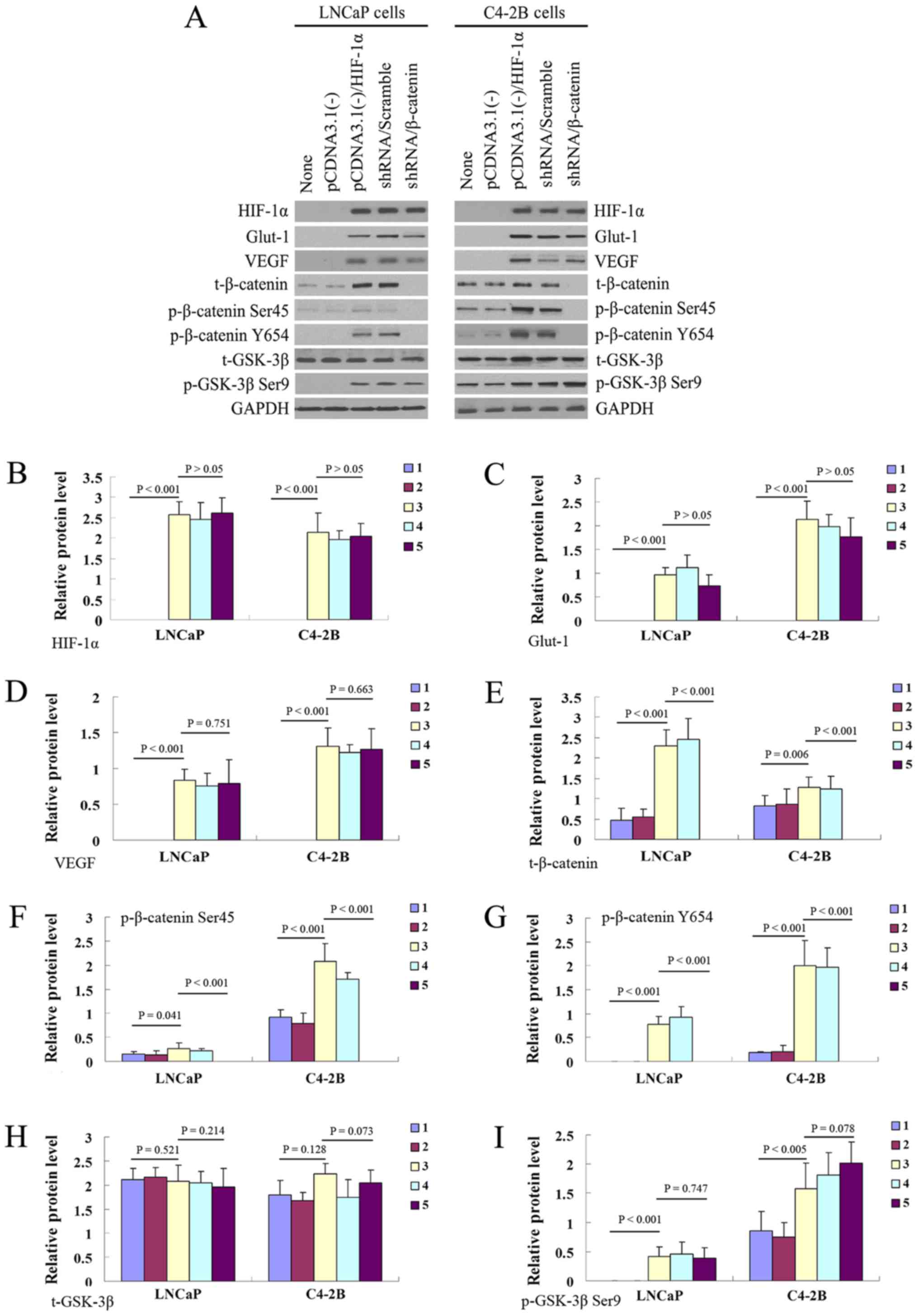 | Figure 1Effect of HIF-1α overexpression on
several functional proteins associated with β-catenin nuclear
translocation in the prostate cancer cell lines, LNCaP and C4-2B.
(A) Western blot analysis of the protein expression of HIF-1α,
Glut-1, VEGF, t-β-catenin, p-β-catenin-Ser45, p-β-catenin-Y654,
t-GSK-3β and p-GSK-3β Ser9 at 72 h in the negative control, HIF-1α
overexpression, β-catenin silenced and scrambled shRNA groups.
(B–I) Statistic analysis of the expression of these functional
proteins. Bar 1 indicates untreated cells, bar 2 indicates
PcDNA3.1(−) control vector-transfected cells, bar 3 indicates
PcDNA3.1(−)/HIF-1α plasmid-transfected cells, bar 4 indicates
HIF-1α-overexpressing cells transfected with
PSuper-Scramble-β-catenin-ShRNA control vector, and bar 5 indicates
HIF-1α-overexpressing cells transfected with PSuper-β-catenin-shRNA
plasmid. |
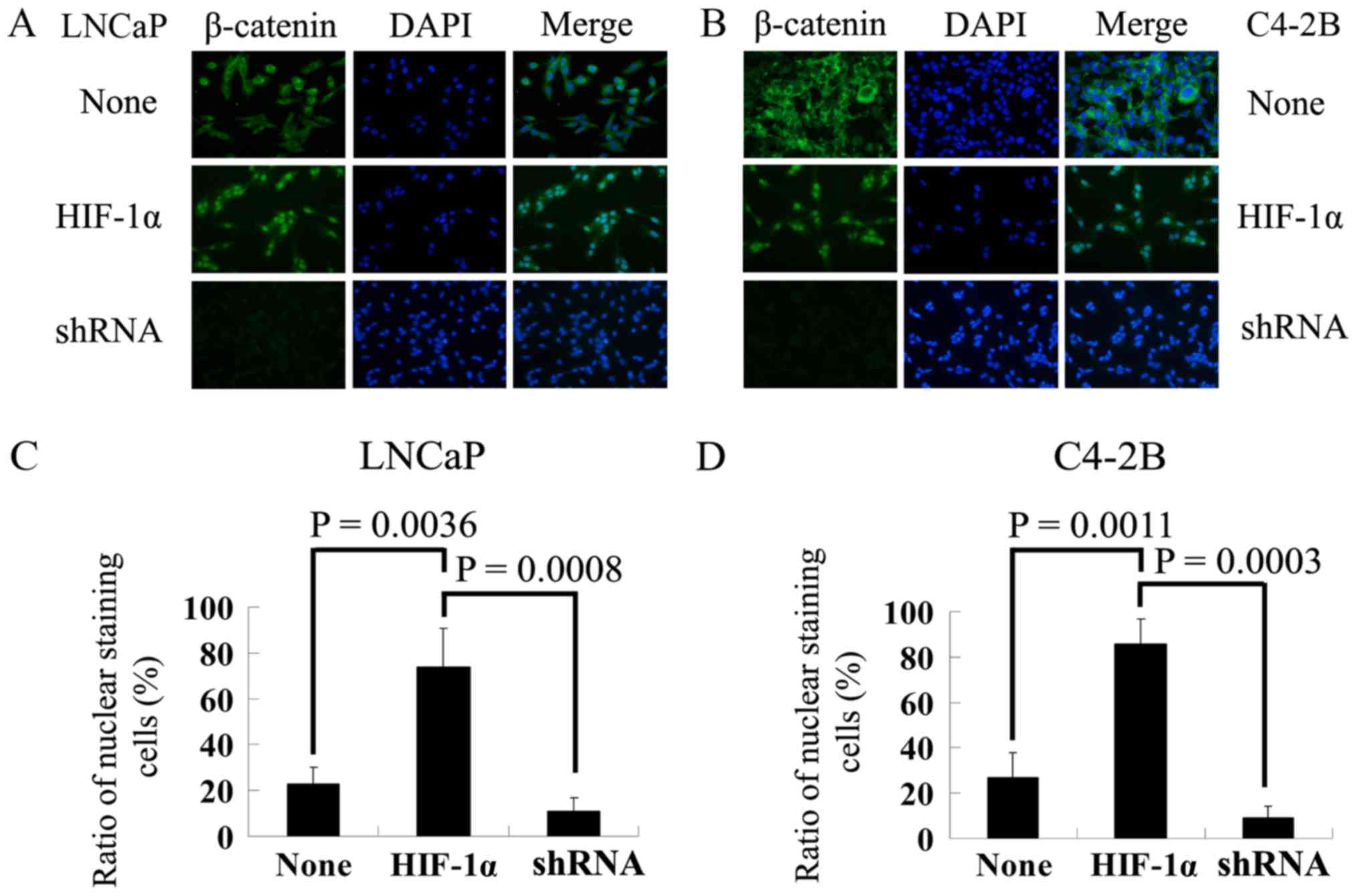
The immunofluorescence staining of β-catenin
suggested that in negative control group, β-catenin expression was
mainly found in the cytoplasm in both the LNCaP and C4-2B cells. In
the HIF-1α overexpression group, β-catenin expression was observed
mainly in the nucleus, and the inhibition of β-catenin in the
β-catenin silenced group led to diminished β-catenin staining
(Fig. 2A and B). In both cell
lines (Fig. 2C and D), the
percentage of stained cells for β-catenin in the nucleus was
significantly higher in the HIF-1α overexpression group than in the
negative control group (LNCaP cells, P=0.0036; C4-2B cells,
P=0.0011) and β-catenin silenced group (LNCaP cells, P=0.0008;
C4-2B cells, P=0.0003).
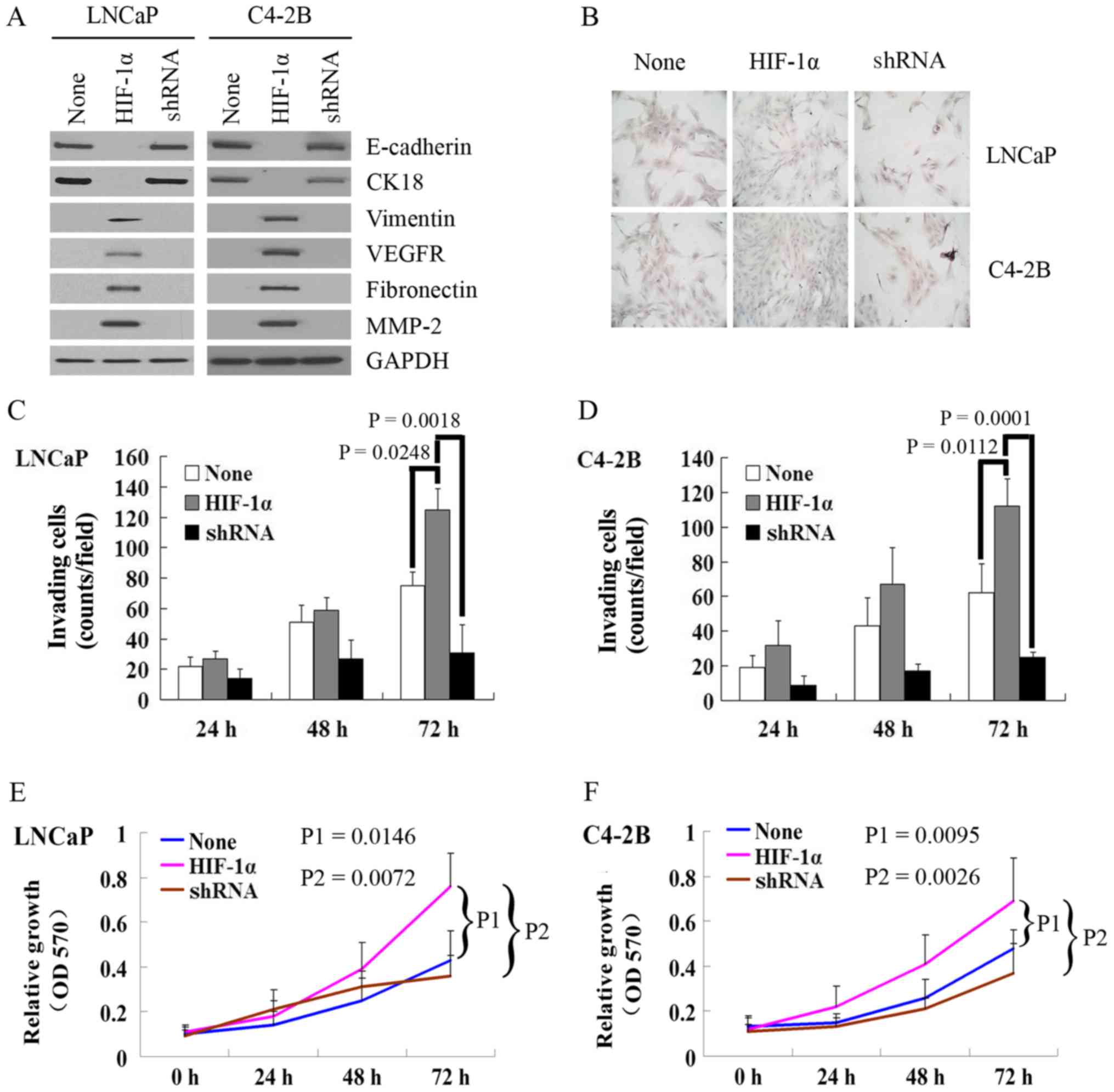
Downstream signals induced by HIF-1α
overexpression trigger β-catenin nuclear translocation
Western blot analysis of both the LNCaP and C4-2B
cells revealed that the HIF-1α overexpression group had a
significantly reduced expression of the transmembrane epithelial
proteins, E-cadherin and CK18, but an increased expression of the
cytoplasmic mesenchymal proteins, vimentin, VEGFR, fibronectin and
MMP-2 (Fig. 3A) compared with the
two other groups. The in vitro invasion assay revealed that
cell invasion was increased in the HIF-1α overexpression group from
24 to 72 h compared with the negative control group (LNCaP cells,
P=0.0248; C4-2B cells, P=0.0112) and the β-catenin silenced group
(LNCaP cells, P=0.0018; C4-2B cells, P=0.0001) at 72 h in both cell
lines (Fig. 3B–D). Furthermore,
the above-mentioned observations were associated with an increased
cell proliferation in the HIF-1α overexpression group from 0 to 72
h compared with the negative control group (LNCaP cells, P=0.0146;
C4-2B cells, P=0.0095) and β-catenin silenced group (LNCaP cells,
P=0.0072; C4-2B cells, P=0.0026) at 72 h, based on the results of
MTT assay (Fig. 3E and F).
Effect of β-catenin nuclear translocation
on cell cycle distribution, apoptosis, and NHEJ repair
Western blot analysis of both the LNCaP and C4-2B
cells revealed that the phosphory-lation of cell cycle regulators
(including CDK1, Chk1, Chk2 and Rb) was significantly enhanced at
72 h in the HIF-1α over-expression group compared with the other 2
groups (P<0.05), while the total expression of each of these
proteins did not differ among the 3 groups (Fig. 4).
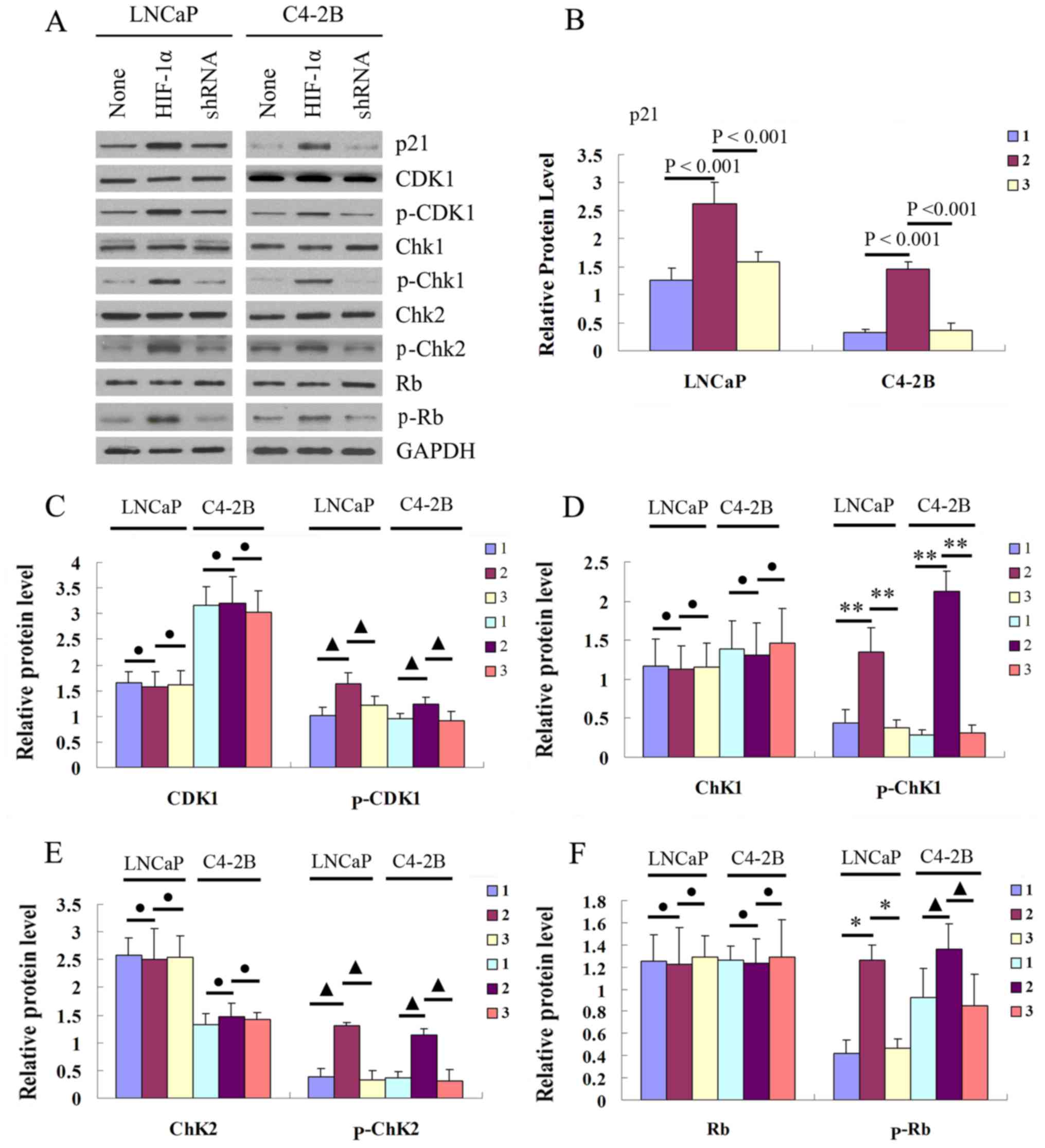 | Figure 4Effect of β-catenin nuclear
translocation on cell cycle distribution in the prostate cancer
cell lines, LNCaP and C4-2B. (A) Western blot analysis of the
protein expression of cell cycle regulatory proteins, including
p21, CDK1, p-CDK1, Chk1, p-Chk1, Chk2, p-Chk2, Rb and p-Rb at 72 h
in the negative control, HIF-1α overexpression and β-catenin
silenced groups of LNCaP and C4-2B cells. (B–F) Statistic analysis
of the expression of cell cycle regulators. Bar 1 indicates
untreated cells, bar 2 indicates PcDNA3.1(−)/HIF-1α plasmid
transfected cells, and bar 3 indicates HIF-1α high expression cells
transfected with PSuper-β-catenin-shRNA plasmid.
●P>0.05; ▲P<0.05;
*P<0.01; **P<0.001. |
Flow cytometric analysis (Fig. 5A and B) revealed that transfection
with the HIF-1α overexpression plasmid decreased the percentage of
cells in the sub-G1 phase (LNCaP cells, P=0.038; C4-2B cells,
P=0.047) and G2/M phase (LNCaP cells, P=0.0411; C4-2B cells,
P=0.0627), and increased the percentage of cells in the G0/G1 phase
(LNCaP cells, P=0.0294; C4-2B cells, P=0.3615) and S phase (LNCaP
cells, P=0.0137; C4-2B cells, P=0.0185), compared with the negative
control. In addition, β-catenin knockdown resulted in a greater
distribution of sub-G1 cells (LNCaP cells, P=0.0016; C4-2B cells,
P=0.0009) and a reduction in the G0/G1 cell population (LNCaP
cells, P=0.0009; C4-2B cells, P=0.0003) compared with the HIF-1α
overexpression group in both the LNCaP and C4-2B cells.
 | Figure 5Effect of β-catenin nuclear
translocation on apoptosis and non-homologous end joining (NHEJ)
repairing potential in the prostate cancer cell lines, LNCaP and
C4-2B. (A and B) Flow cytometric analysis of the cell cycle
distribution (G0/G1, S and G2/M phase) at 72 h in the negative
control, HIF-1α over-expression and β-catenin silenced groups. (C)
Western blot analysis of the protein expression of apoptotic
regulators (including caspase-3, cleaved-caspase-3, caspase-7,
cleaved-caspase-7, cleaved-PARP-1 and Bax), anti-apoptotic proteins
(Bcl-2 and Bcl-xL), the DNA double strand breaks (DSB) marker,
γH2AX, and NHEJ repair proteins (Ku70, Ku80 and DNA-PKcs) at 72 h
in the negative control, HIF-1α overexpression and β-catenin
silenced groups. |
Western blot analysis of both the LNCaP and C4-2B
cells revealed that the protein expression of apoptotic regulators
(including caspase-3, cleaved-caspase-3, caspase-7,
cleaved-caspase-7, cleaved-PARP-1 and Bax) and the DSB marker
(γH2AX) was decreased at 72 h in the HIF-1α overexpression group
compared with the other 2 groups. By contrast, the protein
expression levels of anti-apoptotic proteins (including Bcl-2 and
Bcl-xL) and NHEJ repair proteins (including Ku70, Ku80 and
DNA-PKcs) was increased in the HIF-1α overexpression group compared
with the other 2 groups (Fig. 5C).
In addition, the expression of the above-mentioned apoptotic
regulators, anti-apoptotic proteins, and NHEJ repair proteins was
similar between the β-catenin silenced and negative control
groups.
Effect of β-catenin nuclear translocation
on cell cycle distribution, apoptosis and NHEJ repair following
irradiation
The results of flow cytometric assay (Fig. 6A–D) revealed that in the both LNCaP
and C4-2B cells following irradiation treatment, the HIF-1α
overexpression group had a significantly greater S phase cell
population (LNCaP cells, P=0.0092; C4-2B cells, P=0.0027) and a
decreased sub-G1 cell population (LNCaP cells, P=0.0045; C4-2B
cells, P=0.0039) compared with the negative control group.
β-catenin silencing markedly reversed the effects of HIF-1α
transfection and led to a decrease in the S phase cell population
(LNCaP cells, P=0.0045; C4-2B cells, P=0.0006) and an increase in
the sub-G1 cell population (LNCaP cells, P=0.0014; C4-2B cells,
P=0.0003).
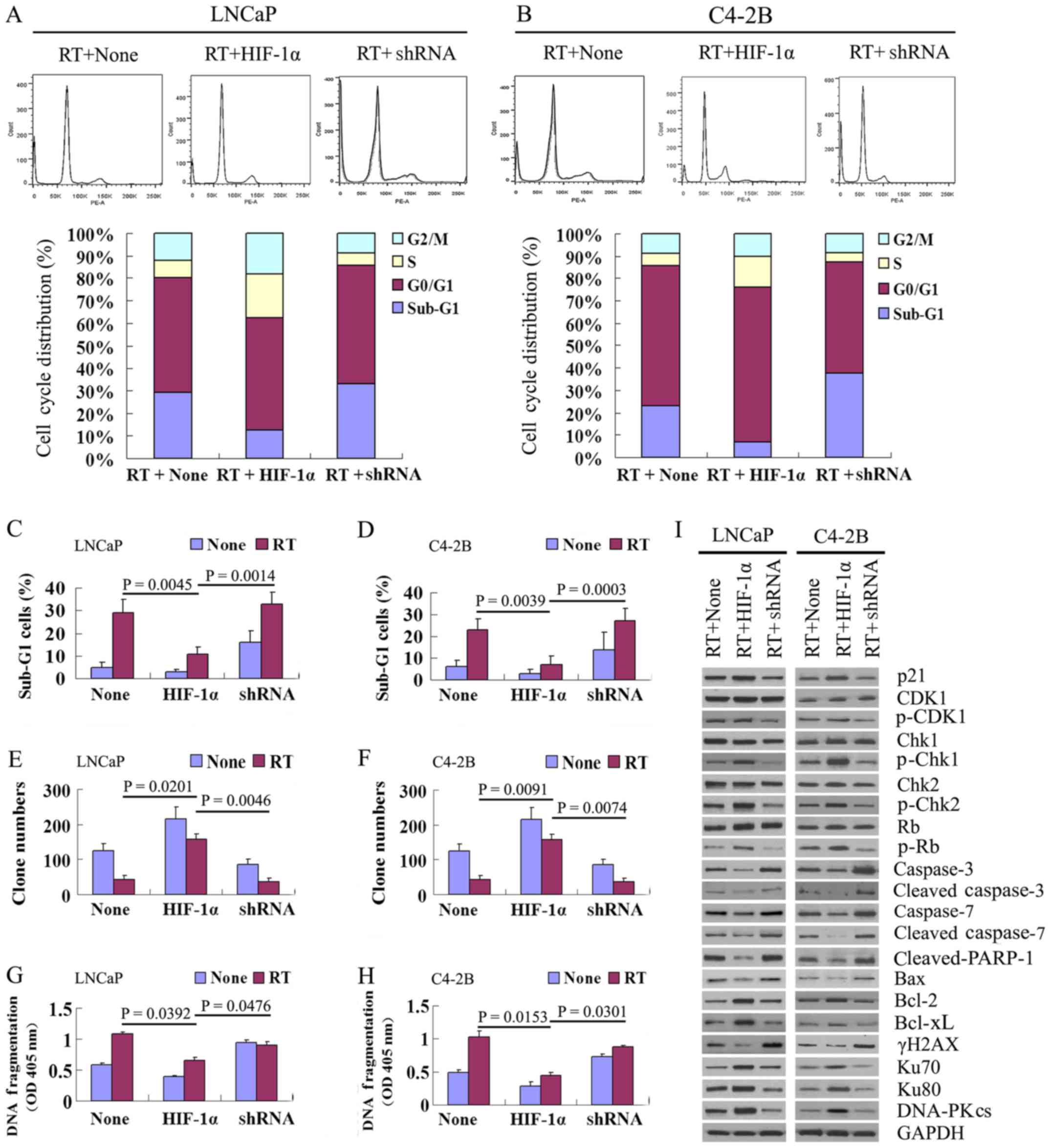 | Figure 6Effect of β-catenin nuclear
translocation on cell cycle distribution, apoptosis and
non-homologous end joining (NHEJ) repair following irradiation.
(A–D) Results of flow cytometry of the cell cycle distribution and
sub-G1 cell ratio at 72 h in the negative control, HIF-1α
overexpression and β-catenin silenced groups of LNCaP and C4-2B
cells following in vitro irradiation. (E and F) Colony
formation assay showing colony-forming capability at 72 h in the
negative control, HIF-1α overexpression and β-catenin silenced
groups of LNCaP and C4-2B cells following in vitro
irradiation. (G and H) ELISA results at 72 h showing cell death
(DNA fragmentation) in the negative control, HIF-1α overexpression
and β-catenin silenced groups of LNCaP and C4-2B cells following
in vitro irradiation. (I) Western blot analysis of the
protein expression of cell cycle regulators (p21, CDK1, p-CDK1,
Chk1, p-Chk1, Chk2, p-Chk2, Rb and p-Rb), apoptotic proteins
(including caspase-3, cleaved-caspase-3, caspase-7,
cleaved-caspase-7, cleaved-PARP-1 and Bax), anti-apoptotic proteins
(Bcl-2 and Bcl-xL), the DNA double strand breaks (DSB) marker,
γH2AX, and non-homologous end joining (NHEJ) repair proteins (Ku70,
Ku80 and DNA-PKCs) at 72 h in the negative control, HIF-1α
overexpression and β-catenin silenced groups of LNCaP and C4-2B
cells following in vitro irradiation. |
In addition, the results of colony formation assay
revealed an approximately 4-fold greater number of clones in the
HIF-1α overexpression group than in the negative control group
(LNCaP cells, P=0.0201; C4-2B cells, P=0.0091) and β-catenin
silenced group (LNCaP cells, P=0.0046; C4-2B cells, P=0.0074) at 72
h, while no significant differences were observed between the
β-catenin silenced and negative control groups (Fig. 6E and F).
DNA fragmentation was significantly lower in the
HIF-1α overexpression group than in the negative control group
(LNCaP cells, P=0.0392; C4-2B cells, P=0.0153) and β-catenin
silenced group (LNCaP cells, P=0.0476; C4-2B cells, P=0.0301) at 72
h in both the LNCaP and C4-2B cells following irradiation treatment
(Fig. 6G and H).
Similar to the above-mentioned experiments without
radiation in both the LNCaP and C4-2B cells, the post-irradiation
expression of cell cycle regulators (including p21, p-CDK1, p-Chk1,
p-Chk2 and p-Rb) was significantly enhanced at 72 h in the HIF-1α
group overexpression compared with the β-catenin silenced and
negative control groups, while the total expression of each of
these proteins did not differ among the 3 groups. Additionally, the
post-irradiation protein expression of apoptotic regulators
(including caspase-3, cleaved-caspase-3, caspase-7,
cleaved-caspase-7, cleaved-PARP-1 and Bax) and the DNA DSB marker
(γH2AX) was decreased at 72 h in the HIF-1α overexpression group
compared with the other 2 groups. By contrast, the protein
expression of anti-apoptotic proteins (including Bcl-2 and Bcl-xL),
and NHEJ repair proteins (including Ku70, Ku80 and DNA-PKcs) was
increased in the HIF-1α overexpression group compared with the
β-catenin silenced and negative control groups (Fig. 6I). In addition, the expression of
the above apoptotic regulators, anti-apoptotic proteins, and NHEJ
repair proteins was similar between the β-catenin silencing and
negative control groups.
Effect of β-catenin nuclear translocation
on tumor growth following in vivo irradiation in mice
The in vivo imaging of orthotopic models
indicated that HIF-1α-overexpressing LNCaP cells produced tumors
with a 3-fold greater volume (P=0.0031) and a 2-fold greater wet
weight (P=0.0394) than those produced by the negative control cells
on day 21. The cells in which β-catenin was silenced produced
tumors with a reduced tumor volume (P=0.0003) and tumor wet weight
(P=0.0175) than the HIF-1α-overexpressing cells (Fig. 7A–C). In addition, the C4-2B
subcutaneous models exhibited similar tumor-promoting effects
induced by HIF-1α-overexpressing cells (tumor volume, P=0.0001; and
wet weight, P=0.0473); in addition, the cells in which β-catenin
was silenced also produced tumors with a lower volume and wet
weight (tumor volume, P<0.0001; and wet weight, P=0.0221),
similarly as in the LNCaP orthotopic tumor xenograft model with
regard to tumor volume and tumor wet weight (Fig. 7D–F).
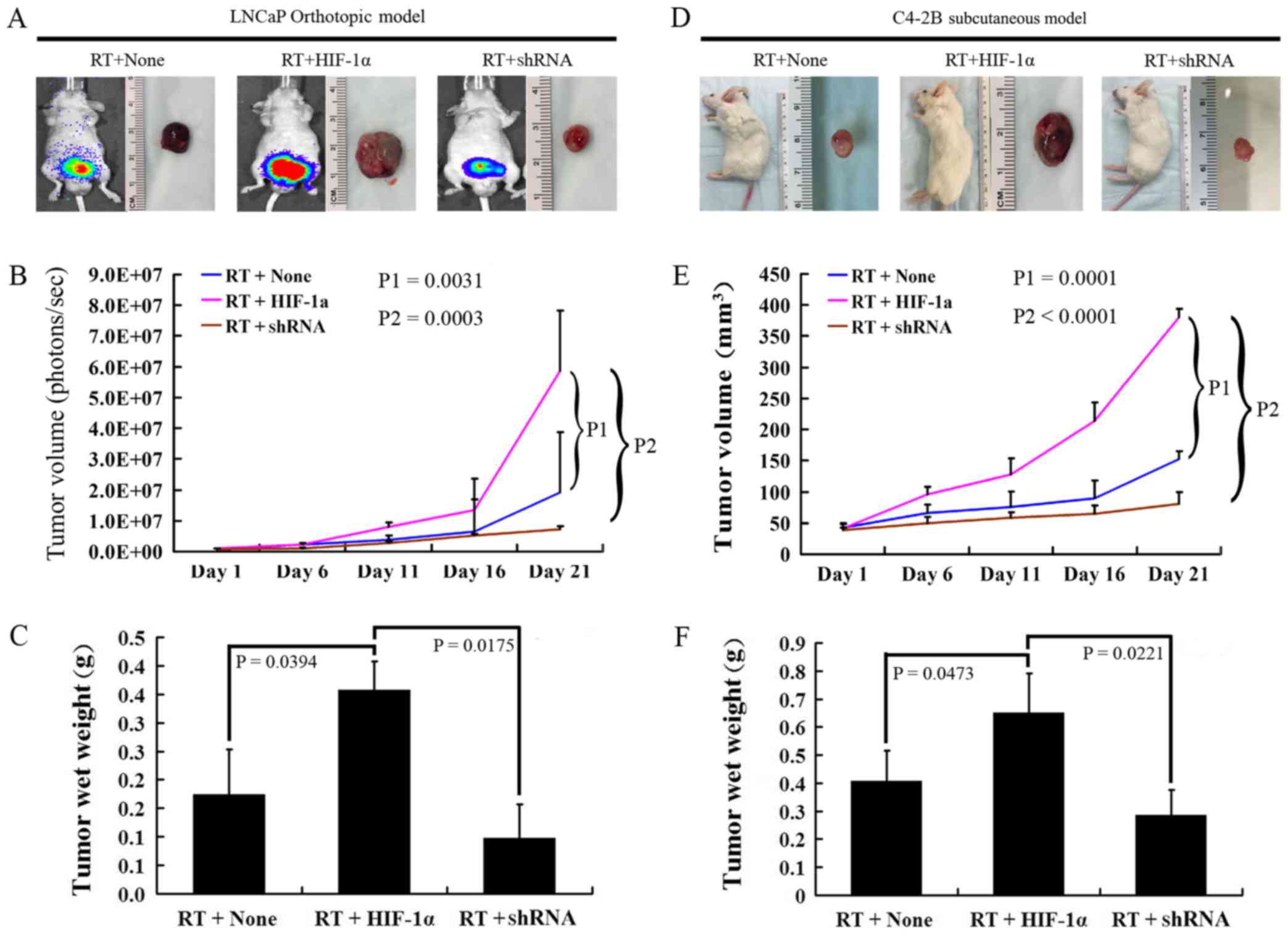 | Figure 7Effect of β-catenin nuclear
translocation on tumor growth following in vivo irradiation
in mice. (A and B) Luminescence signal intensity representing tumor
volume measured at 1, 6, 11, 16 and 21 days in the negative
control, HIF-1α overexpression, and β-catenin silenced groups of
the LNCaP orthotopic model using BALB/c-nu mice following
irradiation. (C) Tumor wet weight on day 21 in the negative
control, HIF-1α overexpression and β-catenin silenced groups of the
LNCaP orthotopic model using BALB/c-nu mice following irradiation.
(D and E) Vital imaging of tumor and tumor volume at 1, 6, 11, 16
and 21 days in the negative control, HIF-1α overexpression and
β-catenin silenced groups of the C4-2B subcutaneous model using
SCID mice following irradiation. (F) Tumor wet weight on day 21 in
the negative control, HIF-1α overexpression and β-catenin silenced
groups of the C4-2B subcutaneous model using SCID mice following
irradiation (n=15/group). |
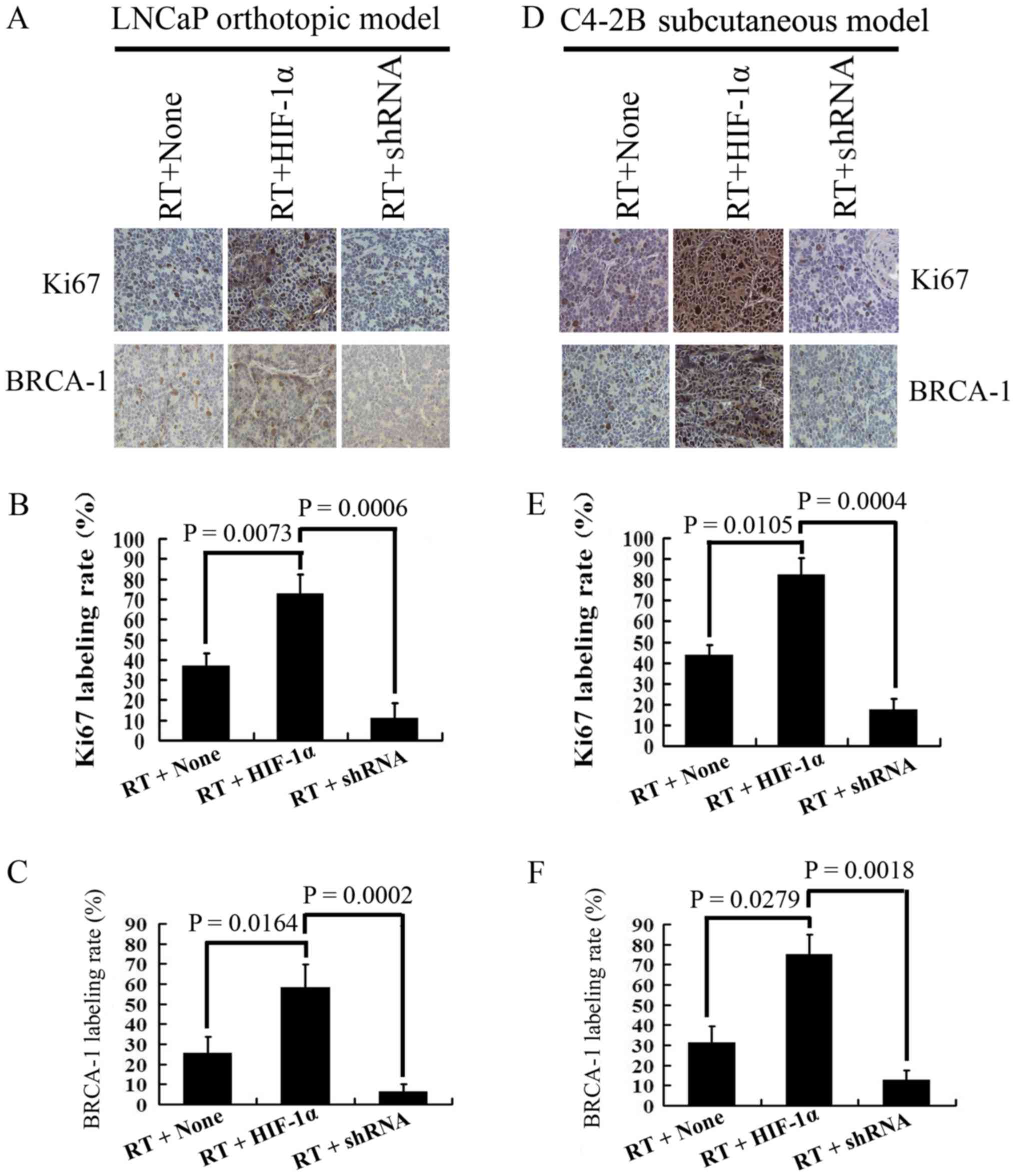
Effect of β-catenin nuclear translocation
on tumor proliferation following in vivo irradiation in mice
The results of immunohistochemistry revealed that
the expression of Ki67 and BRCA-1 was significantly increased in
the HIF-1α overexpression group compared with the negative control
group (Ki67: LNCaP cells, P=0.0073; C4-2B cells, P=0.0105; BRCA-1:
LNCaP cells, P=0.0164; C4-2B cells, P=0.0279) on day 21 in both the
LNCaP orthotopic and C4-2B subcutaneous in vivo irradiation
models. After β-catenin was silenced, the expression levels of Ki67
and BRCA-1 were both decreased (Ki67: LNCaP cells, P=0.0006; C4-2B
cells, P=0.0004; BRCA-1: LNCaP cells, P=0.0002; C4-2B cells,
P=0.0018) compared with the HIF-1α overexpression group (Fig. 8).
Discussion
Although recent studies have suggested that the
activation of the FGFR/PLCγ signaling pathway (23) induced by HIF-1α or the functional
interplay of the ERKs/DNA-PKcs signaling pathway (24) with HIF-1α exerts an independent
effect on radioresistance in malignant tumors, the molecular
mechanisms responsible for the radioresistance promoted by HIF-1α
remain unclear.
β-catenin is involved in the PCa metastatic cascade.
Therefore, this study aimed to investigate the role of HIF-1α and
β-catenin in radioresistance in PCa. The results revealed that
HIF-1α overexpression led to β-catenin activation and nuclear
translocation in the PCa cell lines, LNCaP and C4-2B, and in turn
this upregulated cell proliferation and cell invasion, increased
cell cycle distribution at the G0/G1 phase, reduced apoptosis/DNA
fragmentation, and enhanced DNA NHEJ repairing activity. By
contrast, β-catenin silencing reversed these changes. Following
irradiation, β-catenin activation reduced DNA fragmentation and
enhanced colony formation, which was reversed by β-catenin
silencing. Orthotopic and subcutaneous tumor models further
confirmed that tumor growth was the highest in the HIF-1α
overexpression tumor models and the lowest in the β-catenin
silenced tumor models. Taken together, these results strongly
suggest that β-catenin nuclear translocation is a key process in
the radioresistance of PCa.
Cojoc et al found that acquired
radioresistance was associated with the loss of E-cadherin
expression and the increased expression of β-catenin (18). The present study also demonstrated
that HIF-1α overexpression led to the loss of E-cadherin, while it
enhanced β-catenin nuclear translocation, which is of importance,
as E-cadherin binding to β-catenin keeps β-catenin in the cytoplasm
and prevents its nuclear translocation, while HIF-1α can suppress
the expression of E-cadherin by upregulating Snail (25,26).
Therefore, the expression of E-cadherin and β-catenin is both
regulated by HIF-1α. In other type of cancer (such as lung cancer
and rectal adenocarcinoma), β-catenin has been found to regulate
tumor radioresistance (27,28).
Wang et al were able to use the overexpression (>50% of
positive tumor cells) of nuclear β-catenin to predict
radioresistance in patients with rectal adenocarcinoma and achieved
83% accuracy, 65% sensitivity and 88% specificity (27). The present study using both PCa
cell lines and animal prostate tumor xenograft models produced
similar results in terms of enhanced radioresistance post-β-catenin
nuclear translocation.
β-catenin, through its nuclear translocation,
achieves transcriptional activity by binding to the transcriptional
factor, LEF/TCF (29). In the
study by Zhao et al, the overexpres-sion of HIF-1α
stimulated the invasive potential of human PCa cells through the
EMT pathway, and the Wnt/β-catenin signaling pathway played a vital
role in this process (19). This
is due to the loss of epithelial features and the acquisition of
mesenchymal properties through the EMT pathway leads to migration
of individual cells (30). In the
present study, the enhanced expression of cytoplasmic proteins over
transmembrane proteins, as well as enhanced cell invasion
post-β-catenin nuclear translocation were induced by HIF-1α
overexpression, in line with the findings of previous studies
mentioned above.
Previous studies have suggested that radioresistance
is a result of intrinsic adaptations; for instance, the activation
of the radiation-induced DNA damage response, enhanced DNA repair
capability, increased intracellular defense against ROS, and the
activation of the survival pathways (31,32).
Cellular response to DNA damage is coordinated primarily by two
signaling cascades, the ATM-Chk2 and ATR-Chk1 pathways. Following
DNA damage by ionizing irradiation, Chk1/2 becomes phosphorylated
by ATR/ATM and arrests cell proliferation to allow DNA repair
(18). In the present study,
radiation treatment led to β-catenin activation and nuclear
translocation, as well as to the activation of the Chk1 and Chk2
pathways, reduced apoptotic markers and DNA fragmentation, enhanced
cell proliferation, and cell cycle arrest at G0 (increased Ki67
expression) that allows sufficient DNA repair before cell
division.
Based on the studies mentioned above and the current
findings, a radioresistance mechanism in prostate tumors can be
summarized as follows: First, the hypoxic environment in prostate
tumors activates the expression of HIF-1α, which in turn suppresses
E-cadherin expression and promotes the entry of free β-catenin into
the nucleus. In addition, hypoxia induces Src kinase-dependent
β-catenin phosphorylation at Y654 (33). Y654-β-catenin phosphorylation
disrupts the association between β-catenin and E-cadherin, favoring
its transcriptional activity (34). Once β-catenin achieves nuclear
translocation, it regulates the transcription of genes involved in
the EMT pathway and regulates cell cycle, DNA repair, apoptosis and
cell proliferation. Subsequently, β-catenin nuclear translocation
leads to radioresistance of cancer cells. In the present study, in
the HIF-1α overexpression group, the increased phosphorylation of
GSK-3β at Ser9 was not reversed by β-catenin silencing, indicating
that the phosphorylation of GSK-3β is likely regulated by hypoxia
or HIF-1α independent of β-catenin pathway.
In conclusion, this study firstly described a
comprehensive molecular mechanism that may contribute to the
development to radioresistance of PCa under HIF-1α overexpression
using both PCa cell lines and animal prostate tumor xenograft
models. The results of our findings shed light into the selection
of molecular targets for improving the radiosensitivity of prostate
tumors and hence the effectiveness of radiation treatment.
Abbreviations:
|
PCa
|
prostate cancer
|
|
HIF-1α
|
hypoxia-inducible factor-1α
|
|
NHEJ
|
non-homologous end joining
|
|
DSB
|
DNA double-strand breaks
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
|
EGFP1
|
enhanced green fluorescent protein
1
|
|
FITC
|
fluorescein isothiocyanate
|
|
SPF
|
specific pathogen-free
|
Acknowledgments
Not applicable.
Funding
This study was supported by grants from the National
Natural Science Foundation of China (nos. 30700968 and 81372858)
and High Level Backbone Doctor Training Project of Beijing Health
System (no. 2015-3-054). The funders had no role in the study
design, data collection and analysis, decision to publish, or
preparation of the manuscript.
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
YLu, SPB, JZ and LL were responsible for the study
design, original article drafting and editing, data acquisition and
data analysis. YLu, SPB, JZhang and LL were responsible for article
revision. YLu, ML, XZ, JZhao, YH, YLi, YW performed the
experiments. YLu, XZ, SPB, JZhang, YJ and LL were responsible for
data analysis. ML, XZ, JZhao, YH, YLi and YW were responsible for
data acquisition. YLu, XZ, SPB, JZhang and LL were responsible for
data interpretation and methodology. JZhang, YJ and LL were
responsible for supervision. YLu, XZ, SPB, JZhang and JZhao revised
the manuscript. LL was responsible for funding acquisition and
supervision. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Animal studies were conducted according to the
recommendations outlined in the Guide for the Care and Use of
Laboratory Animals in the Weatherall report. Animal experiments
were approved by the Committee on the Ethics of Animal Experiments
of the Capital Medical University, Beijing, China.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Horwich A, Parker C and de Reijke T:
Prostate cancer: ESMO Clinical Practice Guidelines for diagnosis,
treatment and follow-up. Ann Oncol. 24(Suppl 6): vi106–vi114. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mohler JL, Kantoff PW, Armstrong AJ,
Bahnson RR, Cohen M, D'Amico AV, Eastham JA, Enke CA, Farrington
TA, Higano CS, et al National Comprehensive Cancer Network:
Prostate cancer, version 2.2014. J Natl Compr Canc Netw.
12:686–718. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zietman AL, Bae K, Slater JD, Shipley WU,
Efstathiou JA, Coen JJ, Bush DA, Lunt M, Spiegel DY, Skowronski R,
et al: Randomized trial comparing conventional-dose with high-dose
conformal radiation therapy in early-stage adenocarcinoma of the
prostate: Long-term results from proton radiation oncology
group/american college of radiology 95-09. J Clin Oncol.
28:1106–1111. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baumann M, Krause M and Hill R: Exploring
the role of cancer stem cells in radioresistance. Nat Rev Cancer.
8:545–554. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Johansson S, Aström L, Sandin F, Isacsson
U, Montelius A and Turesson I: Hypofractionated proton boost
combined with external beam radiotherapy for treatment of localized
prostate cancer. Prostate Cancer. 2012:6548612012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Khuntia D, Reddy CA, Mahadevan A, Klein EA
and Kupelian PA: Recurrence-free survival rates after external-beam
radiotherapy for patients with clinical T1-T3 prostate carcinoma in
the prostate-specific antigen era: What should we expect? Cancer.
100:1283–1292. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cooper BT and Sanfilippo NJ: Concurrent
chemoradiation for high-risk prostate cancer. World J Clin Oncol.
6:35–42. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
West CM, Davidson SE, Elyan SA, Swindell
R, Roberts SA, Orton CJ, Coyle CA, Valentine H, Wilks DP, Hunter
RD, et al: The intrinsic radiosensitivity of normal and tumour
cells. Int J Radiat Biol. 73:409–413. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Balmukhanov SB, Yefimov ML and Kleinbock
TS: Acquired radioresistance of tumour cells. Nature. 216:709–711.
1967. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wei K, Kodym R and Jin C: Radioresistant
cell strain of human fibrosarcoma cells obtained after long-term
exposure to x-rays. Radiat Environ Biophys. 37:133–137. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
McDermott N, Meunier A, Mooney B, Nortey
G, Hernandez C, Hurley S, Lynam-Lennon N, Barsoom SH, Bowman KJ,
Marples B, et al: Fractionated radiation exposure amplifies the
radioresistant nature of prostate cancer cells. Sci Rep.
6:347962016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Brown JM and Giaccia AJ: The unique
physiology of solid tumors: Opportunities (and problems) for cancer
therapy. Cancer Res. 58:1408–1416. 1998.PubMed/NCBI
|
|
14
|
Brown JM and Wilson WR: Exploiting tumour
hypoxia in cancer treatment. Nat Rev Cancer. 4:437–447. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Harada H, Kizaka-Kondoh S, Li G, Itasaka
S, Shibuya K, Inoue M and Hiraoka M: Significance of HIF-1-active
cells in angiogenesis and radioresistance. Oncogene. 26:7508–7516.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Harada H: Hypoxia-inducible factor
1-mediated characteristic features of cancer cells for tumor
radioresistance. J Radiat Res (Tokyo). 57(Suppl 1): i99–i105. 2016.
View Article : Google Scholar
|
|
17
|
Chang HW, Nam HY, Kim HJ, Moon SY, Kim MR,
Lee M, Kim GC, Kim SW and Kim SY: Effect of β-catenin silencing in
overcoming radioresistance of head and neck cancer cells by
antagonizing the effects of AMPK on Ku70/Ku80. Head Neck. 38(Suppl
1): E1909–E1917. 2016. View Article : Google Scholar
|
|
18
|
Cojoc M, Peitzsch C, Kurth I, Trautmann F,
Kunz-Schughart LA, Telegeev GD, Stakhovsky EA, Walker JR, Simin K,
Lyle S, et al: Aldehyde dehydrogenase is regulated by β-catenin/TCF
and promotes radioresistance in prostate cancer progenitor cells.
Cancer Res. 75:1482–1494. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao JH, Luo Y, Jiang YG, He DL and Wu CT:
Knockdown of β-catenin through shRNA cause a reversal of EMT and
metastatic phenotypes induced by HIF-1α. Cancer Invest. 29:377–382.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Luo Y, Lan L, Jiang YG, Zhao JH, Li MC,
Wei NB and Lin YH: Epithelial-mesenchymal transition and migration
of prostate cancer stem cells is driven by cancer-associated
fibroblasts in an HIF-1α/β-catenin-dependent pathway. Mol Cells.
36:138–144. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang J, Zhou D, He X, Wang Y, Hu W, Jiang
L and Dou J: Effect of downregulated β-catenin on cell
proliferative activity, the sensitivity to chemotherapy drug and
tumorigenicity of ovarian cancer cells. Cell Mol Biol
(Noisy-le-grand). 57(Suppl): OL1606–OL1613. 2011.
|
|
22
|
Rahbar Saadat Y, Saeidi N, Zununi Vahed S,
Barzegari A and Barar J: An update to DNA ladder assay for
apoptosis detection. Bioimpacts. 5:25–28. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gouazé-Andersson V, Delmas C, Taurand M,
Martinez-Gala J, Evrard S, Mazoyer S, Toulas C and
Cohen-Jonathan-Moyal E: FGFR1 induces glioblastoma radioresistance
through the PLCγ/Hif1α pathway. Cancer Res. 76:3036–3044. 2016.
View Article : Google Scholar
|
|
24
|
Marampon F, Gravina GL, Zani BM, Popov VM,
Fratticci A, Cerasani M, Di Genova D, Mancini M, Ciccarelli C,
Ficorella C, et al: Hypoxia sustains glioblastoma radioresistance
through ERKs/DNA-PKcs/HIF-1α functional interplay. Int J Oncol.
44:2121–2131. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Orsulic S, Huber O, Aberle H, Arnold S and
Kemler R: E-cadherin binding prevents beta-catenin nuclear
localization and beta-catenin/LEF-1-mediated transactivation. J
Cell Sci. 112:1237–1245. 1999.PubMed/NCBI
|
|
26
|
Zhang Y, Fan N and Yang J: Expression and
clinical significance of hypoxia-inducible factor 1α, Snail and
E-cadherin in human ovarian cancer cell lines. Mol Med Rep.
12:3393–3399. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang L, Zhang XM, Li Z, Liu XJ, Chai J,
Zhang GY and Cheng YF: Overexpression of nuclear β-catenin in
rectal adenocarcinoma is associated with radioresistance. World J
Gastroenterol. 19:6876–6882. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang Q, Gao M, Luo G, Han X, Bao W, Cheng
Y, Tian W, Yan M, Yang G and An J: Enhancement of radiation
sensitivity in lung cancer cells by a novel small molecule
inhibitor that targets the β-catenin/Tcf4 interaction. PLoS One.
11:e01524072016. View Article : Google Scholar
|
|
29
|
Nelson WJ and Nusse R: Convergence of Wnt,
beta-catenin, and cadherin pathways. Science. 303:1483–1487. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guarino M: Epithelial-mesenchymal
transition and tumour invasion. Int J Biochem Cell Biol.
39:2153–2160. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bütof R, Dubrovska A and Baumann M:
Clinical perspectives of cancer stem cell research in radiation
oncology. Radiother Oncol. 108:388–396. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Peitzsch C, Kurth I, Kunz-Schughart L,
Baumann M and Dubrovska A: Discovery of the cancer stem cell
related determinants of radioresistance. Radiother Oncol.
108:378–387. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xi Y, Wei Y, Sennino B, Ulsamer A, Kwan I,
Brumwell AN, Tan K, Aghi MK, McDonald DM, Jablons DM, et al:
Identification of pY654-β-catenin as a critical co-factor in
hypoxia-inducible factor-1α signaling and tumor responses to
hypoxia. Oncogene. 32:5048–5057. 2013. View Article : Google Scholar
|
|
34
|
Roura S, Miravet S, Piedra J, García de
Herreros A and Duñach M: Regulation of E-cadherin/Catenin
association by tyrosine phosphorylation. J Biol Chem.
274:36734–36740. 1999. View Article : Google Scholar : PubMed/NCBI
|