Introduction
Excessive alcohol consumption triggers a variety of
liver disorders, ranging from simple steatosis to severe forms of
liver injury, including fatty liver, steatohepatitis, liver
fibrosis, cirrhosis and even liver cancer (1). Alcoholic liver disease (ALD) is a
major healthcare concern, which inflicts individuals, and society
as a whole, with damaging consequences and is a significant cause
of economic burden worldwide (2,3). The
elucidation of the detailed mechanisms responsible for the
development of ALD is important in order to determine an effective
treatment. Mounting evidence indicates that oxidative stress plays
a key role in ALD. Ethanol-induced oxidative stress directly
influences the elevated production of reactive oxygen species (ROS)
and increases lipid peroxidation and damage to the antioxidant
system, which leads to cell apoptosis and necrosis (4,5). It
was recently reported that hepatocyte cell death via apoptosis and
necrosis may be a critical process in ALD (6). An increasing number of studies have
noted that oxidative stress and superfluous intracellular ROS
production induced by ethanol and its metabolites exert a pivotal
effect on ethanol-induced cellular apoptosis (7,8), and
suggested that apoptosis is mainly induced via the Fas- and
mitochondria-mediated pathway (9).
NADPH oxidase 4 (NOX4), is expressed particularly in
hepatocytes and hepatic stellate cells (HSCs), and is therefore an
important source of ROS in signal transduction, playing a vital
role in the physiological and pathological processes of ALD
(5,10). It is a reasonable hypothesis that
ROS derived from NOX4 on the membrane may be associated with Fas
activation (11). Relatively high
levels of intracellular ROS induce redox imbalance, causing cell
apoptosis via the mitogen-activated protein kinase (MAPK) signaling
pathway (12).
It is known that MAPK determines the fate of various
cells, and that p38 MAPK and JNK may positively influence the
mitochondrial pathways that lead to the apoptosis of
ethanol-exposed SK-Hep1 cells, suggesting an interaction between
apoptosis and MAPK signaling systems (13). L-02 is a new cell line established
by the Shanghai Biochemical Institute of Chinese Academy of
Sciences and was selected for use in a previous study (14); this cell line was selected for the
present study as these cells have alcohol dehydrogenase (ADH)
activity (15). Therefore, the
results may be more exact, compared with cancerous hepatocytes,
such as HepG2 or Hep3B.
Methyl ferulic acid (MFA) is a monomer extracted and
purified from Securidaca inappendiculata Hassk. which has
been traditionally used in the treatment of acute or chronic
hepatitis and which has been shown to exert some inhibitory effects
against HBsAg (16,17). However, its anti-apoptotic effects
and its distinct mechanisms of action have yet to be elucidated.
Thus, in the present study, we aimed to investigate the possible
mechanisms responsible for the anti-apoptotic effects of MFA
against the ethanol-induced apoptosis of L-02 cells.
Materials and methods
Drug and reagents
MFA (3,4-dimethoxycinnamic acid) at >98% purity
and all other reagents (unless otherwise indicated) were obtained
from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). Ethanol was
purchased from JiuYi Chemical Factory (Shanghai, China).
Cells and cell culture
L-02, a normal human hepatic cell line (Chinese
Academy of Sciences, Shanghai, China), was cultured in DMEM
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
containing 10% (v/v) fetal bovine serum and antibiotics (100 U/ml
penicillin and 100 µg/ml streptomycin) (both from HyClone;
GE Healthcare Life Sciences, Little Chalfont, UK) and maintained in
a humidified atmosphere of 5% CO2 and 95% air at
37°C.
Cells between generations 3 and 5 were used in the
experiments. The L-02 cells were treated under the following
conditions unless otherwise stated: Following 12 h of
pre-incubation in serum-free DMEM, MFA (25, 50 and 100 µM)
was added to the cultures for pretreatment. Following incubation
with MFA for 1 h, ethanol (400 mM) was added to DMEM and the cells
incubated for a further 24 h.
Cell viability assay
Cell viability was ascertained by MTT assay. First,
the cells were seeded into 96-well plates (5×104
cells/well) 24 h prior to treatment. The cells were then treated
with MFA at various concentrations (25, 50, 100, 250, 500, 1,000
and 2,000 µM for 24, 48 or 72 h, respectively) prior to
being incubated with a working solution of MTT (5 mg/ml) at 37°C
for 4 h. Following treatment with 150 µl of DMSO to dissolve
the crystals, the cells were placed in a microplate reader
(Mutiskan FC; Thermo Fisher Scientific, Inc.) to measure the
absorbance at 570 nm. All experiments were conducted with strict
sterile precautions and repeated ≥3 times.
Apoptosis detection by flow
cytometry
To further examined whether MFA could L-02 cell
apoptosis induced by ethanol, apoptosis was quantified using an
Annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection kit
(Roche Diagnostics GmbH, Mannheim, Germany) in accordance with the
manufacturer's instructions. Briefly, following treatment with MFA
for 24 h at various concentrations as mentioned above, the cells
were harvested and washed twice with cold PBS. Following
centrifugation (500 × g, 5 min, 25°C), the cells were stained with
Annexin V-FITC and PI, and then analyzed by flow cytometry (flow
cytometer; BD Biosciences, San Jose, CA, USA).
Measurement of aspartate aminotransferase
(AST), glutathione peroxidase (GSH-Px), superoxide dismutase (SOD)
and catalase (CAT) levels
Firstly 3×106 cells were cultured in a
dish (diameter, 10 cm) for 24 h, and the DMEM was then discarded.
Secondly, the MFA (100, 50 and 25 µm) group was pre-cultured
with MFA for 1 h, and the cells in the model group and MAF group
were then supplemented with 400 mM ethanol and culture for 24 h.
Thirdly, the supernatant from the cultured cells was collected and
used immediately for the assay of AST using the aspartate
aminotransferase assay kit (C010-2; Nanjing Jiancheng Bio Co.,
Nanjing, China) according to the manufacturer's instructions. The
cells were then split and the active protein was extracted using
the One Step Animal Cell Active Protein Extraction kit (C500022;
Sangon Biotech Co., Ltd., Shanghai, China). The antioxidant enzyme
(SOD and CAT) and GSH-Px activities were then evaluated using
different detection kits respectively according to the
manufacturer's instructions as follows: The superoxide dismutase
assay kit (A001-3), catalase assay kit (A007-2) and glutathione
peroxidase (GSH-PX) assay kit (A005) (all from Nanjing Jiancheng
Bio Co.).
Measurement of ROS generation
ROS generation was evaluated with an intracellular
ROS Assay kit in accordance to the manufacturer's instructions
(Cell Biolabs Inc., San Diego, CA, USA). DCFH-DA itself does not
fluoresce, but can freely cross the cell membrane and enter the
cell to be hydrolyzed to DCFH by esterase in the cell. DCFH cannot
permeate the cell membrane, and thus the probe is easily loaded
into the cell. ROS in the cell can oxidize the non-fluorescent DCFH
to produce the fluorescent DCF, and the amount of DCF fluorescence
can be used to identify the level of ROS in the cells. The ROS
level was verified with a Tecan Model infinite M200 PRO Microplate
reader (Tecan Group, Ltd., Mannedorf, Switzerland) at an emission
wavelength and excitation wavelength of 523 and 502 nm,
respectively.
RNA extraction and reverse transcription
(RT)-PCR
Following the manufacturer's instructions, total RNA
was isolated from the cells with the total RNA isolation kit
(Tripure Reagent; Mai Bio Co., Shanghai, China), and then reverse
transcribed into cDNA with a cDNA synthesis kit (TIANScript cDNA;
Tiangen Biotech Co., Ltd., Beijing, China) in accordance with the
manufacturer's instructions. Target genes were amplified by the MJ
ptc-200 PCR amplification system (MJ Research, Inc., Waltham, MA,
USA) with a RT-PCR kit (2X Taq PCR Master Mix; Aidlab
Biotechnologies Co., Ltd., Beijing, China) according to the
manufacturer's instructions. The specific primers for the target
genes, GAPDH and β-actin, were synthesized by Sangon Biotech Co.,
Ltd. and are listed in Table I.
β-actin was used as an internal control. The parameters of the
reaction were based on those in our previous study (17). The PCR products were identified
using 1.5% agarose gel electrophoresis, and the optical density of
the target gene bands in each sample was calculated using the
ChemiDoc imaging system with adjustment through β-actin correction
to finally obtain the relative expression of the target genes in
each sample.
 | Table ISequences of primers used for the
determination of NOX4, p22phox, Bax, Bcl-2 and β-actin
gene expression. |
Table I
Sequences of primers used for the
determination of NOX4, p22phox, Bax, Bcl-2 and β-actin
gene expression.
| Gene | Oligonucleotide
primer sequence | Annealing
temperature | Number of
cycles |
|---|
| NOX4 (136 bp) | Forward:
5′-TGTGCCGAACACTCTTGGC-3′
Reverse: 5′-ATATGCACGCCTGAGAAAATA-3′ | 58°C | 35 |
| p22phox
(103 bp) | Forward:
5′-TATTGTTGCAGGAGTGCTCA-3′
Reverse: 5′-CACAGCGGTCAGGTACTTCT-3′ | 58°C | 35 |
| Bax (155 bp) | Forward:
5′-CCCGAGAGGTCTTTTTCCGAG-3′
Reverse: 5′-CCAGCCCATGATGGTTCTGAT-3′ | 53°C | 35 |
| Bcl-2 (89 bp) | Forward:
5′-GGTGGGGTCATGTGTGTGG-3′
Reverse: 5′-CGGTTCAGGTACTCAGTCATCC-3′ | 55°C | 35 |
| β-actin (199
bp) | Forward:
5′-GGACTCCTATGTGGGTGACGA-3′
Reverse: 5′-ACGGTTGGCCTTAGGGTTCA-3′ | 56°C | 35 |
Western blot analysis
Total proteins were extracted from the cells and the
concentration was analyzed with a BCA protein concentration assay
kit (Beyotime Institute of Biotechnology, Haimen, China). Sample
proteins were separated by electrophoresis by a 12% SDS-PAGE
separating gel with a Bio-Rad electrophoresis system (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Equivalent amounts (50
µg) of protein were then transferred onto Pure
Nitrocellulose Blotting membranes and blocked with 5% fat-free
milk, the proteins were then incubated with the primary antibodies
[anti-NOX4 (1:1,000; ab13303), Bcl-2 (1:1,000; ab59348) (both from
Abcam, Cambridge, UK), caspase-3 (1:1,000; 9662S; Cell Signaling
Technology Inc., Danvers, MA, USA), cleaved caspase-3 (1:500;
D260009; Sangon Biotech Co., Ltd.), Bax (1:1,000; ab53154), JNK1/2
(1:1,000; ab124956), p-JNK1/2 (1:1,000; ab207477) (all from Abcam),
anti-p22phox rabbit antibody (1:500; Bioworld
Technology, Inc., Nanjing, China), anti-p-p38 MAPK (1:1,000;
ab4822) and anti-p38 MAPK antibodies (1:1,000; ab170099) (both from
Abcam)] overnight at 4°C. The membranes were then incubated with
the corresponding horseradish peroxidase-conjugated secondary
antibodies (1:50,000; TA140003; goat anti-rabbit IgG; OriGene
Technologies, Inc., Beijing, China) at room temperature for 1 h.
The qualitative and quantitative analysis of the blots were
estimated with Molecular Imager Chemi Doc XRS (Bio-Rad
Laboratories, Inc.) by enhanced chemiluminescence (E002-100; 7Sea
Pharmatech Co., Ltd., Shanghai, China) and the JS-780 automatic gel
imaging analysis system. β-actin (1:1,000; C640018; Sangon Biotech
Co., Ltd.) was used as the internal control.
NOX4 overexpression
The L-02 cells were electroporated with the P5
primary cell Nucleofector™ kit (Lonza, Basel, Switzerland). For
each transfection with NOX4-cDNA (provided by Shanghai Genechem
Co., Ltd., Shanghai, China), 18 µl supplemented P5 primary
cell solution were added to the prepared mixed solution of 3
µg of NOX4-cDNA dissolved with 82 µl Nucleofector.
Approximately 107–108 cells were resuspended
in 100 µl cDNA plus P5 primary cell solution and
electroporation conducted in a 4D-Nucleofector X kit L cuvette. The
cells were then transferred to 6-well plates containing 2.5 ml warm
complete medium in a humidified atmosphere of 5% CO2 at
37°C. The medium was changed after 24 h of incubation.
Transfection of small interfering
(si)RNA
siRNA directed against NOX4 (NOX4-siRNA) and
negative control siRNA (NC-siRNA) were obtained from Gene Pharma
(Shanghai, China). The NOX4- or NC-siRNA was transfected into the
L02 cells using Lipofectamine® (Invitrogen; Thermo
Fisher Scientific, Inc.) in accordance with the manufacturer's
instructions. Briefly, the cells were seeded in 6-well plates at a
density of 2×105 cells/well in 2 ml of complete DMEM.
When the cells grew to ~70% confluence transfection with siRNA
commenced as follows: A total of 100 pmol of siRNA was mixed with 2
µl of Lipofectamine® in each well, and 500
µl of antibiotic- and serum-free DMEM medium were then
added. The cells were incubated with the transfection mixture for 6
h. At the final stage of incubation, 1.5 ml of antibiotic-free
complete medium was replenished and the cells were incubated for a
further 18 h. Following exposure to ethanol for 24 h, the cells
were harvested and the NOX4 protein and mRNA expression levels were
evaluated by western blot analysis and RT-PCR, respectively.
Statistical analysis
The results are expressed as the means ± standard
deviation. All experimental data were analyzed using SPSS software
version 17.0 (SPSS, Inc., Chicago, IL, USA). One-way analysis of
variance was used to determine significant differences among groups
with the Student-Newman-Keuls (SNK-q) post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Effect of MFA on the viability of L-02
cells
The cytotoxic effects of MFA on the cells were
evaluated by MTT assay. The cells were treated with various
concentrations of MFA (25, 50, 100, 250, 500, 1,000 and 2,000
µM) for 24, 48 or 72 h, respectively. The percentages of
cell growth inhibition for each treatment group were calculated by
adjusting the untreated control group to 100%. Only 15% cell growth
inhibition was observed at a MFA concentration <250 µM,
even after 72 h of treatment (Fig.
1A).
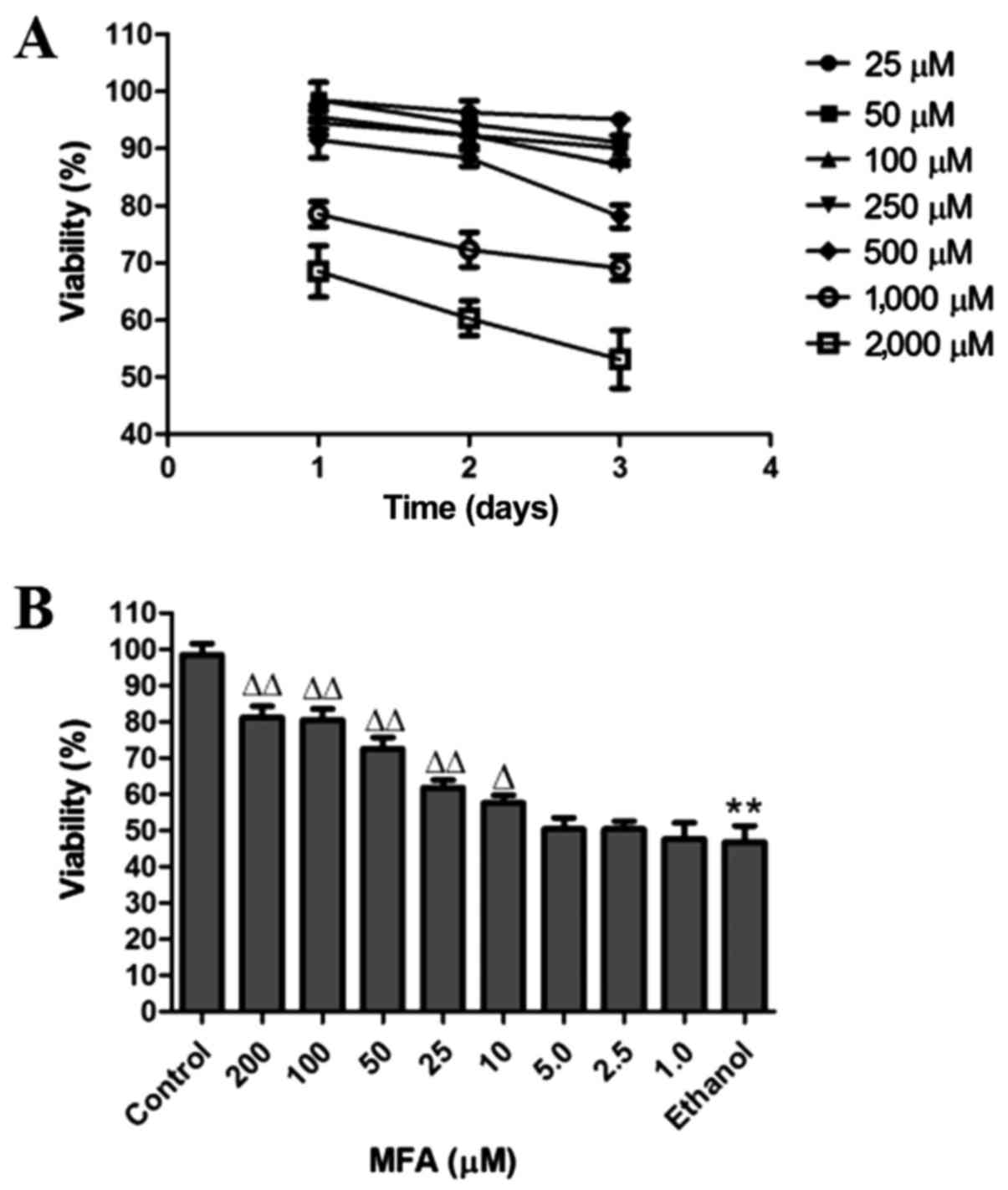 | Figure 1Effects of methyl ferulic acid (MFA)
on ethanol-induced L-02 cell viability. L-02 cell viability was
examined by MTT assay following treatment with (A) MFA (25, 50,
100, 250, 500, 1,000 and 2,000 µM); and (B) MFA (1.0, 2.5,
5, 10, 25, 50, 100 and 200 µM) with or without ethanol (400
mM) as described in the Materials and methods. Data are expressed
as the means ± SD, n=6. **P<0.01 vs. the control
group; △P<0.05 and △△P<0.01 vs. the
ethanol group. |
To examine the effects of ethanol on the viability
of cells, the L-02 cells were cultured in DMEM with or without (as
a control) ethanol. A pre-experimental experiment was conducted in
which viability for 3, 6, 12, 24 and 48 h was estimated at various
concentrations of ethanol (100, 200, 300, 400, 500, 600 and 700
mM). The results demonstrated that the cell inhibition rate of 400
mM ethanol was close to 50% at 24 h (data not shown). It was thus
determined that 400 mM ethanol was the appropriate concentration
for inducing L-02 cell apoptosis.
In order to obtain the optimal experimental
conditions, the concentrations of 1, 2.5, 5, 10, 25, 50, 100 and
200 µM of MFA and 400 mM ethanol were used for the
preliminary experiment (Fig. 1B).
The concentrations of 100, 50 and 25 µM of MFA and 400 mM of
ethanol were selected for use in further experiments as they
produced significant changes in the viability of the cells compared
with the control.
MFA attenuates the ethanol-induced
apoptosis of ethanol-exposed L-02 cells
In order to determine whether MFA mediates the
ethanol-induced apoptosis or necrosis of L-02 cells, Annexin V-PI
flow cytometric analysis, which can differentiate between apoptotic
and necrotic cells, was performed. MFA treatment caused a
significant decrease in the number of Annexin V-positive L-02 cells
in a concentration-dependent manner. The results demonstrated that
the apoptotic rate in the group exposed to ethanol was
significantly increased by 89% (P<0.01) and the apoptotic rate
in the MFA group was significantly lower compared with that in the
ethanol group (P<0.01). The level of apoptosis of L-02 cells
decreased by 29.1, 20.7 and 15.3% following treatment with MFA at
100, 50 and 25 µM for 24 h, respectively. These results
suggested that MFA attenuates the ethanol-induced apoptosis of L-02
cells (Fig. 2A).
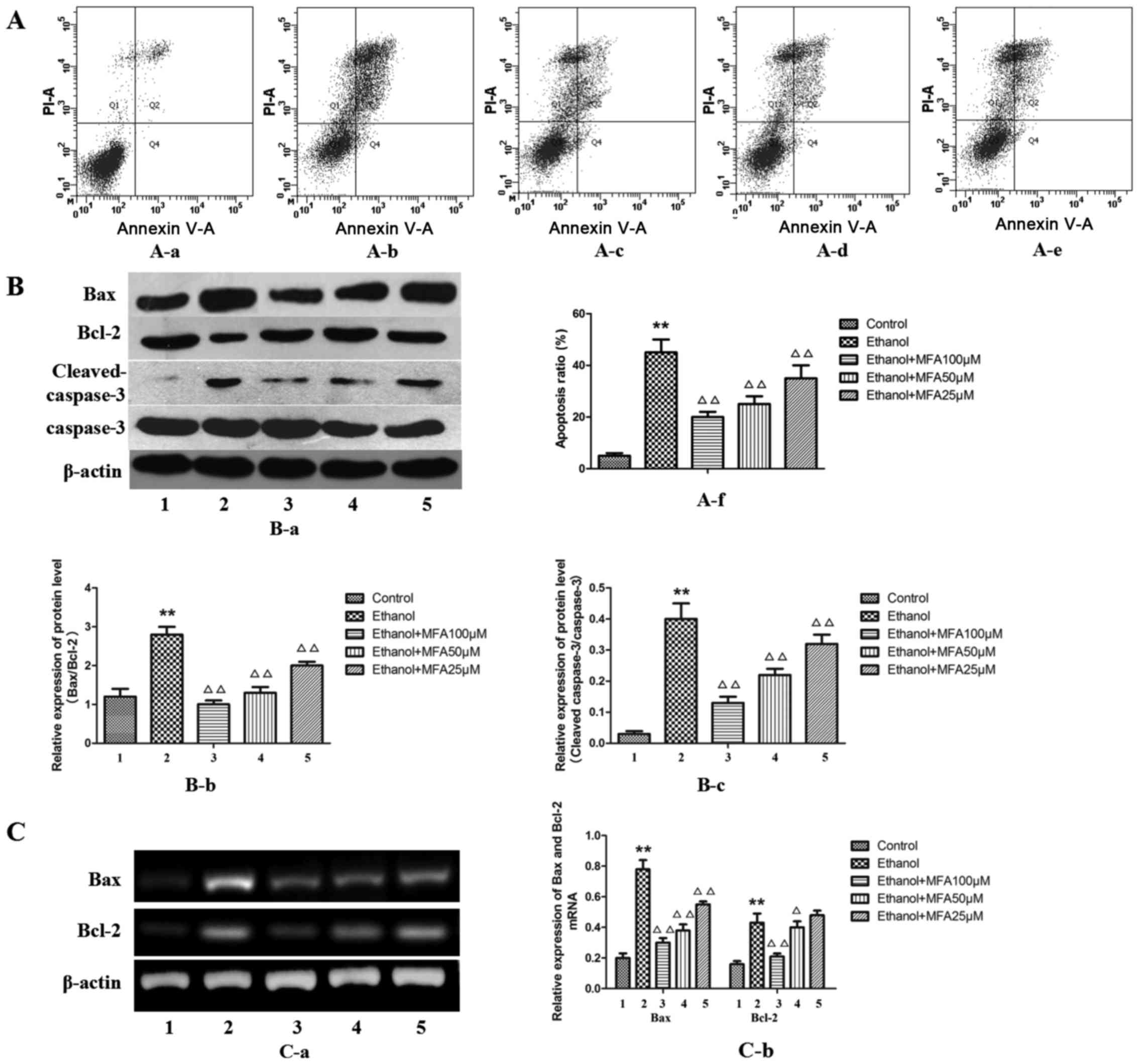 | Figure 2Effects of methyl ferulic acid (MFA)
on ethanol-induced L-02 cell apoptosis. (A) Cell apoptosis was
influenced by pre-incubation with MFA (100, 50 and 25 µM) in
ethanol-exposed L-02 cells; (B) Protein expression levels of Bax,
Bcl-2, cleaved caspase-3, caspase-3 and β-actin were detected by
western blot analysis after the L-02 cells were treated with MFA
(100, 50 and 25 µM) in the absence/presence of ethanol (400
mM) for 24 h; (C) The mRNA levels of Bax and Bcl-2 ere normalized
to β-actin. Data are presented as the means ± SD, n=3.
**P<0.01 vs. the control group; △P<0.05
and △△P<0.01 vs. the ethanol group. Lanes and bars
are numbered as follows: 1, control; 2, ethanol; 3, ethanol + MFA
100 µM; 4, ethanol + MFA 50 µM; 5, ethanol + MFA 25
µM. |
To further evaluate the anti-apoptotic effects of
MFA, the effects of MFA on apoptosis-associated proteins that serve
as biomarkers of cell apoptosis (Bax, Bcl-2, cleaved caspase-3 and
caspase-3) were also examined in vitro. The results from
western blot analysis (Fig. 2B)
demonstrated that ethanol exposure mainly increased Bax, caspase-3
and cleaved caspase-3 protein expression, but decreased Bcl-2
protein expression compared with the control. Although at the mRNA
level, ethanol was also found to increase both the level of Bax and
Bcl-2 expression compared with the control (Fig. 2C), the protein level of Bcl-2
decreased with ethanol treatment compared to that of the control.
Western blot analysis also revealed that the alcohol-exposed L-02
cells exhibited an elevated Bax/Bcl-2 protein expression
(P<0.01). In addition, the protein level of anti-apoptotic Bcl-2
was increased following treatment with MFA in the L-02 cells
compared with the control group (P<0.01), while the levels of
the apoptotic proteins, caspase-3 and Bax, were decreased
(P<0.01). Taken together, these findings suggest that MFA
attenuates the ethanol-induced apoptosis of L-02 cells in a
dose-dependent manner by regulating the levels of apoptotic
proteins. The effect of 100 µM MFA dose is particularly
prominent.
MFA affects the generation of AST,
GSH-Px, SOD and CAT in ethanol-exposed L-02 cells
It has been reported that ethanol-induced cell death
is partly mediated by oxidative stress (13). To alleviate the cumulative burden
of oxidative stress, cells generally utilize antioxidant defense
systems to eliminate ROS. SOD, GSH-Px and CAT are the first line of
defense against oxidative stress and can block free radical
formation and prevent the cells from oxidative damage by ROS
(19). SOD is able to convert the
superoxide radical into H2O2, which can be
broken down to O2 by CAT and GSH-Px (20). In the present study, to determine
whether the protective effects of MFA against ethanol-induced
injury are mediated via antioxidant enzymes, the activities of
antioxidant enzymes in L-02 cells with or without pretreatment with
MFA were investigated. First the L-02 cells were treated with MFA
in the presence or absence of ethanol for 24 h, and the activities
of AST in the culture medium and the levels of GSH-Px, CAT and SOD
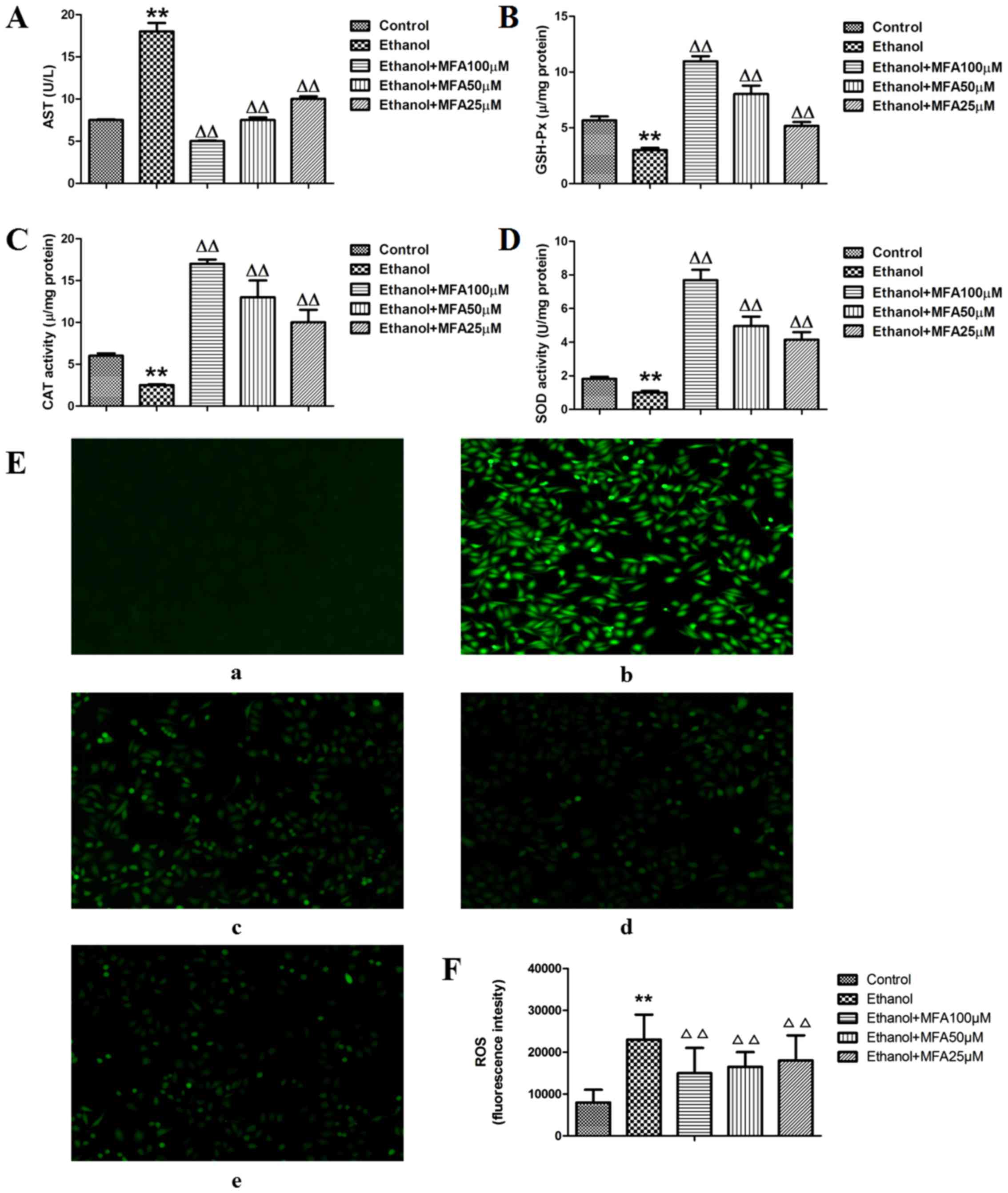
in the cell lysates were then detected. As shown in Fig. 3A, the activities of AST in the
ethanol-exposed group were significantly increased by 125% compared
with those of the control group (P<0.01). However, treatment
with MFA (100, 50 and 25 µM) significantly inhibited the
ethanol-induced elevation of AST activities by 65.5, 52.8 and
39.7%, respectively. In addition, MFA significantly increased the
levels of GSH-Px, CAT and SOD levels compared with those in the
ethanol-exposed cells (Fig. 3B–D).
MFA (100, 50 and 25 µM) increased the GSH-Px levels by 230,
170 and 110% compared with the ethanol-exposed cells, respectively.
MFA (100, 50 and 25 µM) also increased the CAT levels by
556, 412.1 and 223.1%, respectively compared with those in the
ethanol-exposed cells, and MFA (100, 50 and 25 µM) also
elevated the SOD levels by 631.2, 432.3 and 337.0%, respectively
compared with those in the ethanol-exposed cells. Notably,
treatment with MFA significantly reduced ethanol-induced oxidative
stress, as demonstrated by the reduction in the ROS levels when
compared with the ethanol-exposed cells (Fig. 3E and F).
The anti-apoptotic effects of MFA are
mediated via ROS generation
In recent years, a number of studies have
demonstrated that oxidative stress can cause cellular apoptosis
(6,13). ROS production increases in
ethanol-stimulated L-02 cells and leads to apoptosis (14). Therefore, the present study
investigated whether the inhibitory effects of MFA on the
ethanol-induced apoptosis of L-02 cells were associated with the
accumulation of ROS. First, the ROS levels in ethanol-exposed L-02
cells were estimated by determining the oxidative conversion of
non-fluorescent DCFH-DA to fluorescent DCF. Consistent with the
findings of a previous study (17), the exposure of L-02 cells to
ethanol triggered the generation of ROS and apoptosis. The results
of the present study demonstrated that ethanol triggered ROS
production in the L-02 cells; the level of ROS was significantly
increased by about 2 times compared with that of the control group
(P<0.01) (Fig. 3E and F).
Following treatment with MFA (100, 50 and 25 µM), the levels
of ROS generation levels were partly blocked; compared with the
ethanol group, the levels of ROS were reduced in all the 3 MFA
treatment (all 3 concentrations) (P<0.01).
As mentioned above, the exposure of the L-02 cells
to ethanol induced the production of ROS and apoptosis. MFA
treatment successfully reduced ethanol-induced ROS generation
(Fig. 3E and F) and attenuated the
ethanol-induced apoptosis of L-02 cells (Fig. 2A). These results suggested that ROS
generation acts upstream of apoptosis in ethanol-exposed L-02
cells. A similar association was also confirmed in two other
hepatoma carcinoma cell lines (SK-HEP-1 and HepG2; data not shown).
Collectively, these findings suggested that the signaling pathway
through which ethanol induces the apoptosis of L-02 cells involves
ROS as a second messenger. MFA reverses ethanol-induced ROS
production and apoptosis.
MFA inhibits the expression levels of
Nox4 and p22phox in L-02 cells
Recently, studies have emphasized NADPH oxidases
(NOX) as a key source of ROS. NOX4 and its subunit
p22phox are highly expressed in hepatocytes and HSCs
(7,18). In this study, to further understand
the mechanisms underlying the MFA-induced reduction in ROS
generation, the effects of MFA on NOX4, one of the major sources of
ROS generation found in cells, were assessed. NOX4 appears to be
the most abundant isoform of NOXs expressed in the liver (18); therefore, the expression levels of
NOX4, as well as its regulatory subunit, p22phox, were
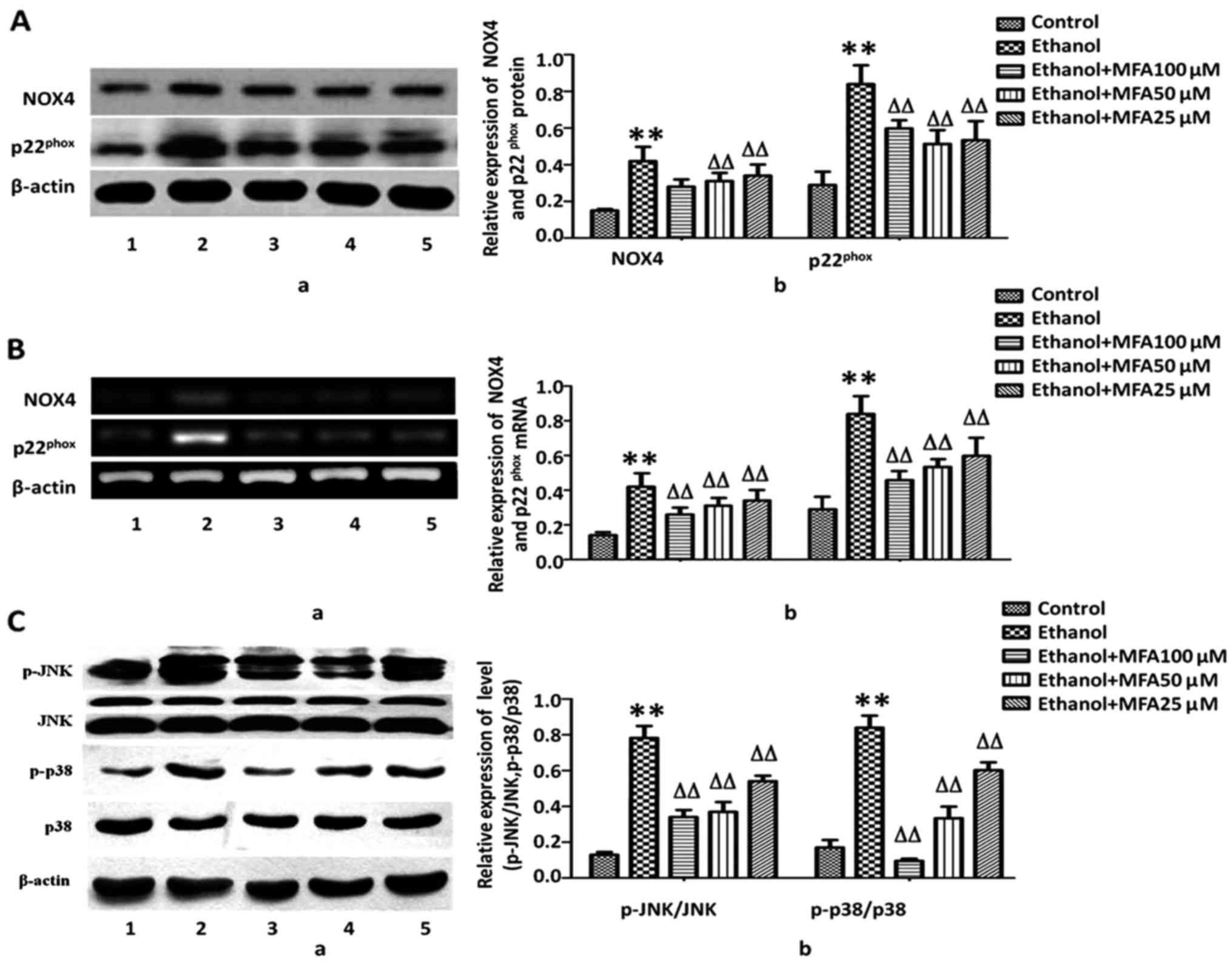
measured. It was identified that the protein expression of NOX4 and
p22phox was significantly increased (75.3 and 120.5%,
respectively) compared with the control group in response to
ethanol. By contrast, treatment with MFA significantly decreased
the expression of NOX4 by 56.2% and the expression of
p22phox by 25.6%, even at the low concentration of 25
µM (P<0.01). A dose-dependent pattern was identified
(Fig. 4A). To further determine
whether the downregulation of NOX4 and p22phox protein
expression induced by MFA was due to alterations in mRNA
transcription, RT-PCR was performed to analyze the mRNA levels of
NOX4 and p22phox. Following exposure to ethanol, the
mRNA levels of NOX4 were increased by 256.2% and those of
p22phox were increased by 320.5% compared with the
control group. Concomitant with the reduction in the protein
expression induced by MFA, treatment with MFA significantly
decreased the mRNA expression levels of NOX4 and
p22phox. MFA (100, 50 and 25 µM) decreased the
mRNA levels of NOX4 by 68.8, 56.2 and 40.6% (P<0.01),
respectively, compared with those of the control group. MFA (100,
50 and 25 µM) decreased mRNA levels of p22phox by
75.3, 53.4 and 50.7% (P<0.01), respectively, compared with those
of the control group (Fig.
4B).
MFA treatment attenuates ethanol-induced
MAPK phosphorylation in L-02 cells
ROS has been regarded as a potent regulator of MAPK
family members and subsequent cell death. To activate p38 MAPK
pathway would accelerate cell apoptosis, JNKs have been considered
to be involved in stimulating apoptotic signaling. Oxidative stress
can switch on JNK to bring about apoptosis by receptor-initiated
extrinsic and mitochondrial intrinsic apoptotic pathways. JNKs also
serve an essential role in modulating the functions of pro- and
anti-apoptotic proteins located in the mitochondria (12).
MAPK cascades are pivotal signaling pathways
involved in the regulation of normal cell proliferation and
survival. Hence, the level of phosphorylated MAPKs in the presence
and/or absence of MFA was measured in this study. As demonstrated
in Fig. 4C, compared with the
control, the phosphorylation levels of p38 MAPK and JNK were
markedly increased in the L-02 cells following exposure to ethanol,
and the levels of p-p38 and p-JNK increased by 50.3 and 76.2%,
respectively compared with the control. By contrast, compared with
the ethanol-exposed group, treatment with MFA (100, 50 and 25
µM) effectively decreased the level of p-p38 by 70.3, 46.0
and 11.1%, and the level of p-JNK by 59.3, 45.0 and 25.1%,
respectively, thereby exhibiting its promising activation of the
p38 MAPK and JNK pathways. MFA decreased MAPK activation in a
concentration-dependent manner, suggesting that MAPK inhibition by
MFA can result in increased survival rates of L-02 cells (Fig. 4C).
Effects of MFA on cell viability and ROS
generation in ethanol-exposed L-02 cells following transfection
with NOX4 overexpression and NOX4 siRNA
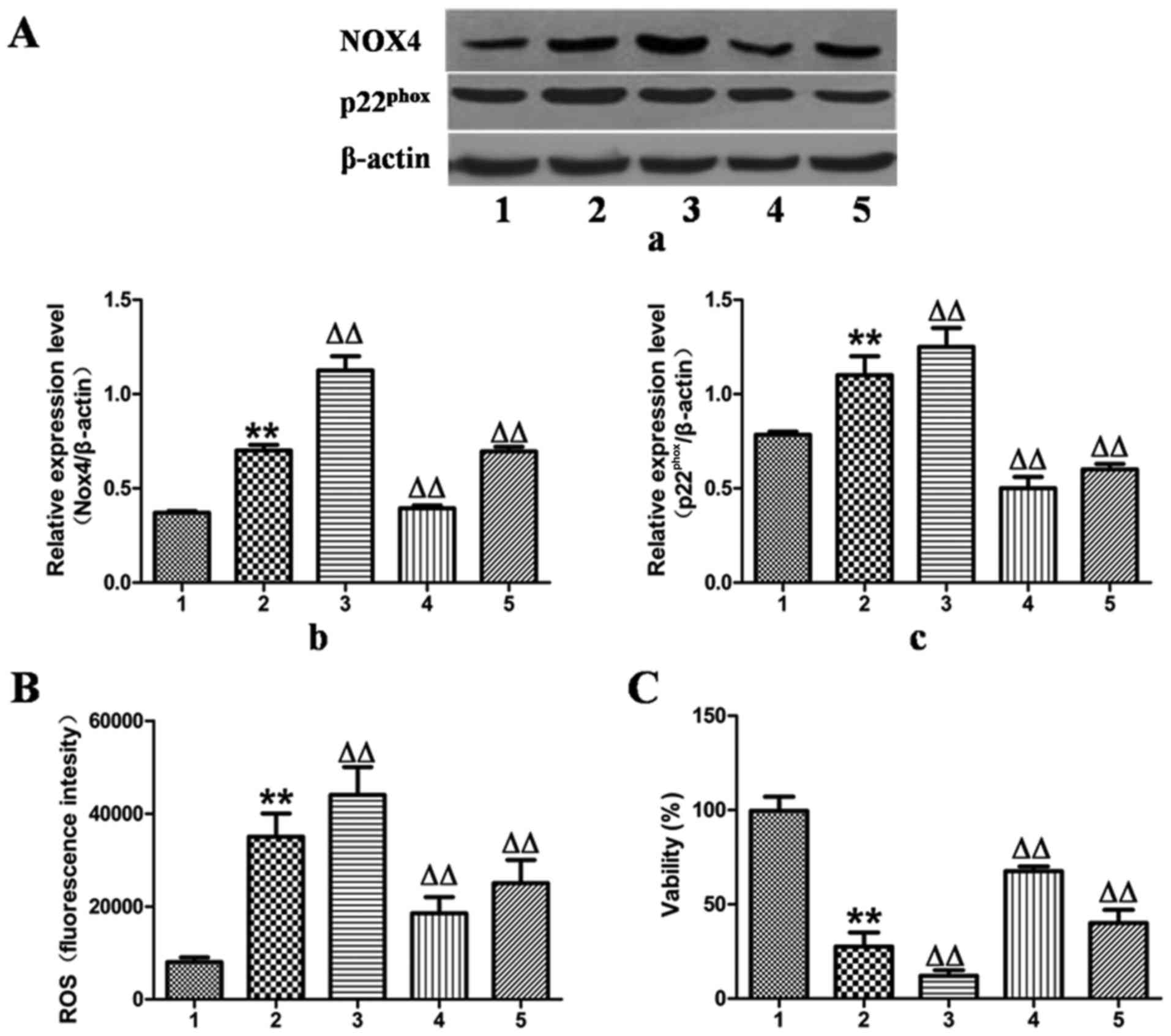
To validate the hypothesis that MFA decreased
ethanol-induced ROS generation by blocking the NOX4 signaling
pathway, the expression of NOX4 was upregulated by transfection of
the L-02 cells with NOX4 overexpression cDNA for 24 h, which
increased the protein expression levels of NOX4 and
p22phox by 55 and 18%, as verified by western blot
analysis (Fig. 5A).
Correspondingly, ROS production increased by 25% and the cell
activity decreased by 65% compared with the control group (Fig. 5B and C). However, these responses
were effectively reversed by treatment with MFA.
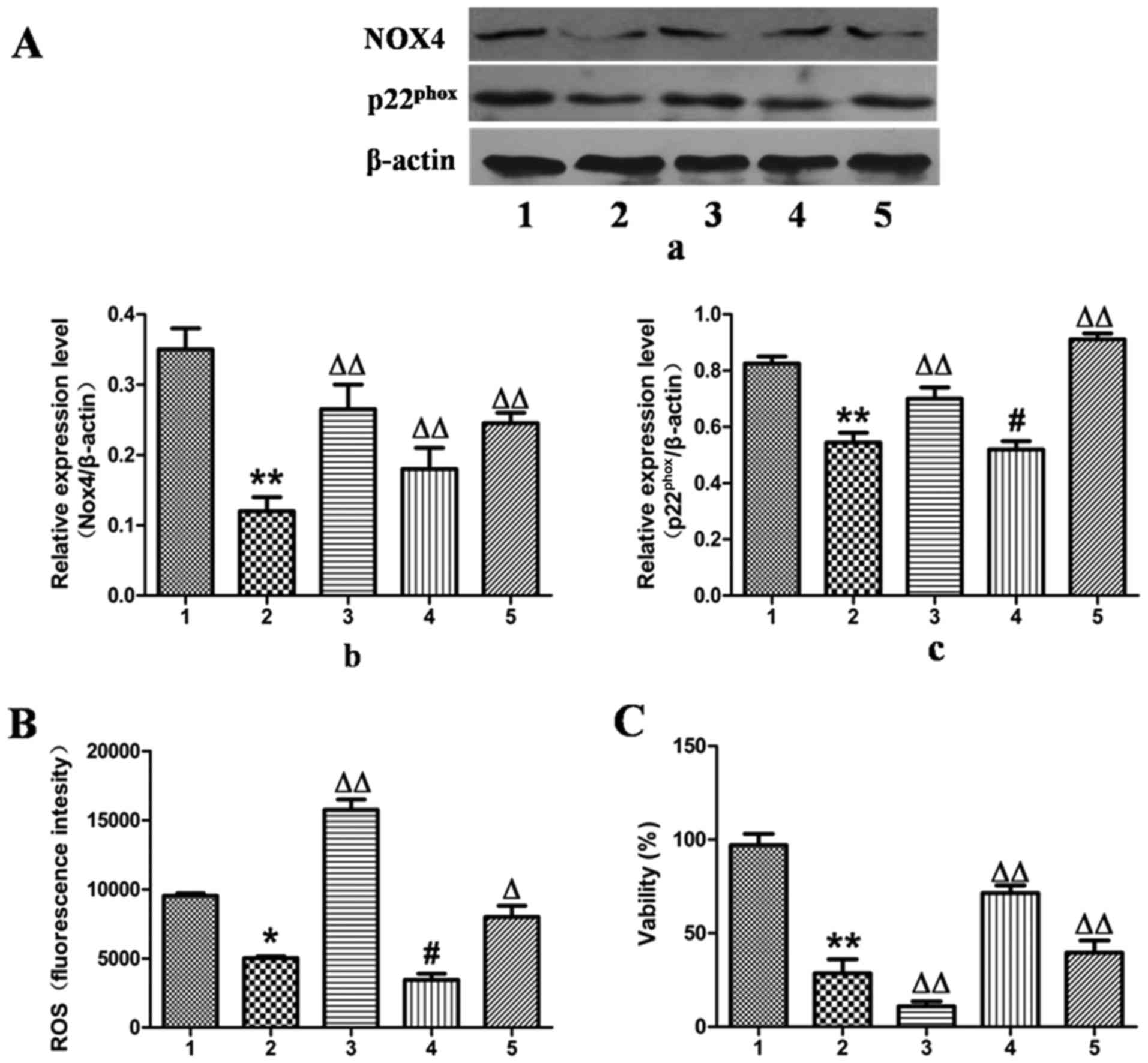
In contrast to the results observed for NOX4
overexpression, the expression of NOX4 was downregulated by
transfection of the L-02 cells with NOX4-siRNA for 24 h, which
reduced the protein expression levels of NOX4 and
p22phox by 67 and 34%, as determined by western blot
analysis (Fig. 6A).
Correspondingly, ROS production was decreased by 49% and cell
viability was increased by 65% compared with the ethanol-exposed
group (Fig. 6B and C).
Additionally, when treatment with MFA was applied to
the NOX4-overexpressing or NOX4-siRNA-transfected L-02 cells,
analysis by cell flow cytometry revealed that treatment with MFA
further blocked ROS elevation triggered by ethanol (Figs. 5B and 6B). MTT assay demonstrated that when the
L-02 cells were exposed to ethanol following treatment with MFA,
the decrease in cellular viability induced by ethanol was
suppressed, indicating that MFA protected the L-02 cells from
ethanol-induced cell toxicity (Figs.
5C and 6C). Taken together,
these results suggested that signaling driven by
NOX4/p22phox was the major mechanism underlying the
anti-oxidative stress activity of MFA against the generation of ROS
triggered by ethanol in L-02 cells. These results suggested that
MFA protected hepatocytes from ethanol-induced apoptosis through
the inactivation of the NOX4/ROS/p38/JNK pathway.
Discussion
As the pharmacological options available for the
therapy of liver diseases are limited, proof of effective hepatic
protective agents from natural sources is important. Therefore, it
is significant to evaluate plant extracts that can help to restore
liver function. MFA is a monomer isolated from Securidaca
inappendiculata Hassk., which possesses positive antiviral
activity via the specific combination with GP120 to prevent the
virus reverse transcriptase from interaction with the lymphocytes
(21). The present study surveyed
the hepatoprotective activity of MFA using an ethanol-exposed L-02
cell model. The results demonstrated that treatment with MFA
significantly reduce elevated ROS levels and reversed
ethanol-induced L-02 cell apoptosis, indicating that MFA was
responsible for a hepatoprotective effect.
The present study explored the possible effects of
MFA on the viability and apoptosis induced by ethanol. The results
from in vitro experiments revealed that MFA attenuated the
ethanol-induced inhibition of the viability and apoptosis of L-02
cells. Previous studies have reported the use of MFA in the therapy
of acute or chronic hepatitis and that it demonstrated some
inhibitory effect on HBsAg (16,17).
Its analog improved antioxidant activity and anti-lipid
peroxidation that protects cells against oxidative stress. Ferulic
acid (FA) alleviated the oxidative stress and decreased cell
apoptosis induced by high glucose in hepatocytes (22). Whether MFA inhibited the apoptosis
induced by ethanol in L-02 cells remained to be elucidated. In this
study, the results from flow cytometry demonstrated that MFA
inhibited the ethanol-induced apoptosis of L-02 cells, suggesting
that its antioxidant properties may contribute to its
anti-apoptotic activity.
Additionally, the present study identified that
culturing the L-02 cells with MFA (25 µM) reduced apoptosis
and ROS production (Figs. 2A and
5B). Maruf et al (23) examined FA and demonstrated that it
can protect isolated rat hepatocytes against glyoxal- or
methylglyoxal-induced cytotoxicity and oxidative stress. FA
attenuated hepatocyte apoptosis which induced ischemia/reperfusion
(I/R) via the inhibition of JNK activation (24). In addition, the results of the
study by Urias-Lugo et al (25) demonstrated that culturing HepG2
cells and primary hepatocytes with phenolic acids exerted an
anti-proliferative effect. FA may cause cell cycle arrest in PC-3
cells and leads to the apoptosis of LNCaP cells (26). MFA treatment at a concentration of
25 µM inhibited the proliferation induced by TGF-β in
HSC-LX-2 cells (27). These
various results indicate that MFA may exert differential effects in
different cell types; that is, MFA can be either cytoprotective or
cytotoxic depending on the cell type.
In this study, when the L-02 cells were incubated
with ethanol at the concentration of 400 mM, ROS generation was
increased with the time of incubation. As one of 6 homologues of
transmembrane NADPH oxidase, NOX4 has been identified to be
involved in ROS generation and highly expressed in the liver
(28). In the liver, NOX4 is
expressed in hepatocytes and is upregulated by ethanol or TGF-β
in vitro as well as in vivo. It is activated in HSCs
and, to a small extent, in sinusoidal endothelial cells, although
Chuffer cells do not express it (29,30).
Furthermore, NOX4 and p22phox are upregulated in
patients exposed to ethanol (31).
In this study, the pronounced expression of NOX4 was identified by
western blot analysis and was confirmed by RT-PCR in L-02 cells.
The results of the present study demonstrated that the mRNA levels
of NOX4 and p22phox were increased when the L-02 cells
were incubated with ethanol (Fig.
4B), demonstrating that NOX4 and p22phox are
required when activating NOX4 on the membrane. The data from
western blot analysis of NOX4 further substantiated this at the
protein level.
In the NOX4 overexpression and NOX4 siRNA knockdown
experiments, it was identified that NOX4 upregulation caused the
upregulation of its regulatory subunit, p22phox and,
simultaneously, NOX4 downregulation caused the downregulation of
p22phox in L-02 cells exposed to ethanol. The results of
the present study support those of previous findings in that
SK-Hep1 cells and alveolar macrophage mRNA levels of NOX4 and
p22phox increased when the cells were incubated with
ethanol (13), which also
demonstrated that both are required to activate NOX4 on the
membrane. However, the mechanisms behind the interaction between
NOX4 and p22phox remain to be elucidated; further
studies are warranted to elucidate these mechanisms.
Based on the results of the present study, the
release of ROS was, at least in part, mediated by NOX4 in the
ethanol-exposed L-02 cells. The overproduction of ROS is one of the
main causes of increasing oxidative stress and triggering apoptosis
(32,33). The results of the present study
suggested that ROS generation mediated by NOX4 on the hepatocyte
membrane was a trigger of apoptosis in ethanol-exposed L-02
cells.
Compared with the ethanol-exposed group, MFA
significantly decreased the elevated NOX4 mRNA and protein
expression levels induced by ethanol in varying degrees (Figs. 4Figure 5–6). This result was consistent with the
results obtained from the ROS and apoptosis experiments, in that
ROS generation and the apoptosis of L-02 cells corresponded with
the changes in NOX4 expression. Further investigations into the
underlying mechanisms of the anti-apoptotic effects of MFA are
required.
Recently, various studies have identified that ROS
may play a critical role in the induction of apoptosis (23,34).
Oxidative stress can be induced by abnormal ROS release or their
constant generation and this is related to apoptosis and other
biological events (35). ROS are
one of the pivotal regulators of cell signal transduction and are
associated with apoptosis, senescence and proliferation (28). A number of drug candidates fulfil a
cytotoxic role through the production of ROS as a critical
regulator. Studies have demonstrated that the metabolism of ethanol
in liver cells can induce ROS production (32,36).
It has been demonstrated that ethanol-mediated ROS production can
cause alterations in cellular morphology and functions and/or
eventually lead to apoptosis (35,36).
For example, the dysfunction of the mitochondria induced by
excessive ROS production results in apoptosis (37). ROS are also known to be activators
of the MAPK signaling pathway (38). The results of the present study
demonstrated that the sustained phosphorylation of p38 MAPK and JNK
was caused by ROS production following exposure to ethanol
(Fig. 4A and B). The suppression
of ROS generation by MFA treatment alleviated the effects of
ethanol on JNK and p38 MAPK phosphorylation, suggesting that
ethanol induced the generation of ROS, which consequently
transformed phosphorylated p38 MAPK and JNK, and caused the
activation of p38 MAPK and JNK, finally leading to the
translocation of Bax to the mitochondria.
Ethanol induced the inhibition of cell growth and
triggered the apoptosis of L-02 cells; however, this process was
effectively blocked by MFA treatment. Ethanol-induced apoptosis
occurs via a decline in Bcl-2 protein synthesis and the transfer of
Bax to the mitochondria (39). The
present study demonstrated that ROS, JNK, p38 MAPK and Bax
participated in the ethanol-triggered apoptotic pathway. In line
with this finding, ROS may serve as an upstream signal mediator of
the p38 MAPK and JNK signaling pathways in L-02 cells treated with
ethanol (Fig. 3E). There is
evidence from a previous study to indicate that ROS can aggravate
apoptosis induced by a variety of stimuli (40).
One of the important factors for the apoptosis
induced by the mitochondrial pathway is the collapse of
mitochondrial membrane potential, which leads to the release of
cytochrome c and the activation of caspase-9. This event is
mediated by anti-apoptotic proteins of the Bcl-2 family.
Particularly, the transfer of Bax to the mitochondria leads to the
alteration of mitochondrial membrane potential and Bax plays a
critical role in triggering apoptosis in response to various
stimuli (41). Although Bax is
mainly located in the cytoplasm, when stimulated, it can move
closer to the mitochondrial membrane (42). When Bax converges onto the
mitochondria, combining with other pro-apoptotic Bcl-2 family
members, it induces the release of cytochrome c through the
pore channels formed in the outer membrane of the mitochondria by
oligomerization or by other channels (43–45).
Phosphorylation of JNK and p38 MAPK activates Bax, either alone or
in combination (46). In the
present study, MFA inhibited induction of caspase-3 activation by
ethanol in L-02 cells. The increased ratio of Bax and Bcl-2 can
alter the mitochondrial membrane potential and lead to the release
of cytochrome c, and then further activate caspase-3 to
trigger apoptosis (47,48). As shown in Fig. 2B and C, exposure to ethanol
upregulated the mRNA expression of Bax and Bcl-2, but also
increased the ratio of Bax to Bcl-2 both at the mRNA and protein
level; these effects of ethanol were antagonized by MFA. The
results indicated that apoptosis plays an important role in
ethanol-induced L-02 cell injury and its effect on the
mitochondrial pathway. The results of the present study seem to
suggest that exposure to ethanol leads to mitochondrial damage and
the caspase-dependent apoptosis of L-02 cells.
Although originally identified as an antioxidant,
MFA is currently believed to act through different mechanisms in
different biological responses. However, the mechanisms through
which MFA inhibits ethanol-induced L-02 cell apoptosis are unclear.
The present study demonstrated that MFA decreased apoptosis by
inhibiting the activation of the p38 MAPK and JNK pathways in L-02
cells. The data identified that MFA treatment caused the persistent
inactivation of p38 MAPK and JNK in L-02 cells (Fig. 4C). The signaling proteins in the
MAPK family promote a variety of biological reactions in cells and
p38 MAPK and JNK play a key role in the signal transduction of
apoptosis (49). Since the release
of cytochrome c from damaged mitochondria is a key step in
the activation of caspases, the finding that JNK and p38 MAPK
activities are essential for caspase activation indicates that JNK
and p38 MAPK can regulate some of the other mitochondrial-related
factors (e.g., Bax). In L-02 cells, MFA treatment caused the
downregulation of JNK and p-38 phosphorylation (Fig. 4C), which is in part similar to the
results obtained in I/R-induced hepatocytes treated with FA
(24). Briefly, MFA attenuated the
apoptosis of L-02 cells induced by ethanol via the inhibition of
the ROS-dependent JNK/p38 MAPK signaling pathway. However, further
studies are required to elucidate the mechanisms through which MFA
inhibits NOX4 to reduce ROS generation.
In brief, the results of the present study
demonstrated that MFA suppressed ethanol-triggered oxidative stress
in L-02 cells by inhibiting ROS production and the upregulation of
GSH-PX, CAT and SOD. The role of MFA in modulating MAPK
phosphorylation, and its established antioxidant effect, suggest
that this compound may prove to be a good candidate for use in the
treatment of ALD.
In conclusion, the present study demonstrated for
the first time, to the best of the authors' knowledge, that MFA
exerts beneficial effects against the ethanol-induced lack of
viability and apoptosis by inhibiting ROS-dependent JNK, the p38
MAPK signaling pathway and the Bcl-2/Bax signaling pathway in L-02
cells. MFA treatment led to the upregulation of p-JNK, p-p38 MAPK
and Bax, and the downregulation of Bcl-2 and cleaved caspase-3. A
new understanding of the role of MFA was developed in that it
attenuated the apoptosis of L-02 cells induced by ethanol by
inhibiting the activation of the MAPK signaling pathway. In the
present study, the L-02 cells used in the in vitro
experiments were normal human hepatocytes. Whether MFA treatment
can inhibit the apoptosis of hepatocytes in vivo requires
further investigation. The present study provides a novel
theoretical basis for the possible use of MFA in the treatment of
liver injury.
Acknowledgments
Not applicable.
Funding
This study was funded by The National Natural
Science Foundation of China (grant nos. 81360497 and 81760669).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
LL and YZ wrote manuscript, performed experiments
and analyzed data; CY, CL, LC, HW and MZR performed experiments and
analyzed data. ZM, CY, LL, HW and YL analyzed the data. ZM and YL
contributed to the discussions and critically edited the
manuscript. All authors have reviewed and approved the manuscript.
All authors are responsible for the integrity of the data.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Duncan C: Rethinking excessive habits and
addictive behaviors. Alcohol Alcohol. 52:128–129. 2017. View Article : Google Scholar
|
|
2
|
Owens RE, Snyder HS, Twilla JD and
Satapathy SK: Pharmacologic treatment of alcoholic hepatitis:
Examining outcomes based on disease severity stratification. J Clin
Exp Hepatol. 6:275–281. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim JW, Yang H, Kim HW, Kim HP and Sung
SH: Lignans from Opuntia ficus-indica seeds protect rat primary
hepatocytes and HepG2 cells against ethanol-induced oxidative
stress. Biosci Biotechnol Biochem. 81:181–183. 2017. View Article : Google Scholar
|
|
4
|
Sugimoto K and Takei Y: Pathogenesis of
alcoholic liver disease. Hepatol Res. 47:70–79. 2017. View Article : Google Scholar
|
|
5
|
Magdaleno F, Blajszczak CC and Nieto N:
Key events participating in the pathogenesis of alcoholic liver
disease. Biomolecules. 7:E92017. View Article : Google Scholar
|
|
6
|
Sinha K, Das J, Pal PB and Sil PC:
Oxidative stress: The mitochondria-dependent and
mitochondria-independent pathways of apoptosis. Arch Toxicol.
87:1157–1180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Simplicio JA, do Vale GT, Gonzaga NA,
Leite LN, Hipólito UV, Pereira CA, Tostes RC and Tirapelli CR:
Reactive oxygen species derived from NAD(P)H oxidase play a role on
ethanol-induced hypertension and endothelial dysfunction in rat
resistance arteries. J Physiol Biochem. 73:5–16. 2017. View Article : Google Scholar
|
|
8
|
Zhu H, Jia Z, Misra H and Li YR: Oxidative
stress and redox signaling mechanisms of alcoholic liver disease:
Updated experimental and clinical evidence. J Dig Dis. 13:133–142.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zwolak A, Surdacka A and Daniluk J: Bcl-2
and Fas expression in peripheral blood leukocytes of patients with
alcoholic and autoimmune liver disorders. Hum Exp Toxicol.
35:799–807. 2016. View Article : Google Scholar
|
|
10
|
Paik YH, Kim J, Aoyama T, De Minicis S,
Bataller R and Brenner DA: Role of NADPH oxidases in liver
fibrosis. Antioxid Redox Signal. 20:2854–2872. 2014. View Article : Google Scholar :
|
|
11
|
Pan JH, Lim Y, Kim JH, Heo W, Lee KY, Shin
HJ, Kim JK, Lee JH and Kim YJ: Root bark of Ulmus davidiana var.
japonica restrains acute alcohol-induced hepatic steatosis onset in
mice by inhibiting ROS accumulation. PLoS One. 12:e01883812017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Venugopal SK, Chen J, Zhang Y, Clemens D,
Follenzi A and Zern MA: Role of MAPK phosphatase-1 in sustained
activation of JNK during ethanol-induced apoptosis in
hepatocyte-like VL-17A cells. J Biol Chem. 282:31900–31908. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Morio Y, Tsuji M, Inagaki M, Nakagawa M,
Asaka Y, Oyamada H, Furuya K and Oguchi K: Ethanol-induced
apoptosis in human liver adenocarcinoma cells (SK-Hep1): Fas- and
mitochondria-mediated pathways and interaction with MAPK signaling
system. Toxicol In Vitro. 27:1820–1829. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yao X, Bai Q, Yan D, Li G, Lü C and Xu H:
Solanesol protects human hepatic L02 cells from ethanol-induced
oxidative injury via upregulation of HO-1 and Hsp70. Toxicol In
Vitro. 29:600–608. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Guo X, Cui R, Zhao J, Mo R, Peng L and Yan
M: Corosolic acid protects hepatocytes against ethanol-induced
damage by modulating mitogen-activated protein kinases and
activating autophagy. Eur J Pharmacol. 791:578–588. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li L, Li YW and Tang AC: The inhibitory
effect of methyl ferulic acid on HBsAg and HBeAg in HepG2.2.15
cell. Pharmacol Clin Chin Materia Med. 27:14–16. 2011.
|
|
17
|
Zheng MS and Li W: Inhibitory effect of
400 kinds of Chinese herbal medicine on HBsAg. Chin J Integr Tradit
West Med Liver Diseas. 6:30–31. 1991.
|
|
18
|
Li C, Li L, Yang CF, Zhong YJ, Wu D, Shi
L, Chen L and Li YW: Hepatoprotective effects of Methyl ferulic
acid on alcohol-induced liver oxidative injury in mice by
inhibiting the NOX4/ROS-MAPK pathway. Biochem Biophys Res Commun.
493:277–285. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Müller TE, Nunes SZ, Silveira A, Loro VL
and Rosemberg DB: Repeated ethanol exposure alters social behavior
and oxidative stress parameters of zebrafish. Prog
Neuropsychopharmacol Biol Psychiatry. 79:105–111. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Grasselli E, Compalati AD, Voci A,
Vecchione G, Ragazzoni M, Gallo G, Borro P, Sumberaz A, Testino G
and Vergani L: Altered oxidative stress/antioxidant status in blood
of alcoholic subjects is associated with alcoholic liver disease.
Drug Alcohol Depend. 143:112–119. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang XD, Xu LZ and Yang SL: Advances in
studies on medicinal plants of Securidaca. Chin Tradit Herbal
Drugs. 31:392–393. 2000.
|
|
22
|
Song Y, Wen L, Sun J, Bai W, Jiao R, Hu Y,
Peng X, He Y and Ou S: Cytoprotective mechanism of ferulic acid
against high glucose-induced oxidative stress in cardiomyocytes and
hepatocytes. Food Nutr Res. 60:30323–30331. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maruf AA, Lip H, Wong H and O'Brien PJ:
Protective effects of ferulic acid and related polyphenols against
glyoxal- or methylglyoxal-induced cytotoxicity and oxidative stress
in isolated rat hepatocytes. Chem Biol Interact. 234:96–104. 2015.
View Article : Google Scholar
|
|
24
|
Kim HY and Lee SM: Ferulic acid attenuates
ischemia/reperfusion-induced hepatocyte apoptosis via inhibition of
JNK activation. Eur J Pharm Sci. 45:708–715. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Urias-Lugo DA, Heredia JB, Muy-Rangel MD,
Valdez-Torres JB, Serna-Saldívar SO and Gutiérrez-Uribe JA:
Anthocyanins and phenolic acids of hybrid and native blue maize
(Zea mays L.) extracts and their antiproliferative activity in
mammary (MCF7), liver (HepG2), colon (Caco2 and HT29) and prostate
(PC3) cancer cells. Plant Foods Hum Nutr. 70:193–199. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Eroğlu C, Seçme M, Bağcı G and Dodurga Y:
Assessment of the anticancer mechanism of ferulic acid via cell
cycle and apoptotic pathways in human prostate cancer cell lines.
Tumour Biol. 36:9437–9446. 2015. View Article : Google Scholar
|
|
27
|
Xiong M, LI Y, LI L, Yang C and Zhong Y:
Inhibitory effect of methy-ferulic acid on proliferation and
activation of TGF-β1-induced human hepatic stellate cells. Shandong
Pharmaceuticals. 56:1–4. 2016.
|
|
28
|
Kleniewska P, Piechota A, Skibska B and
Gorąca A: The NADPH oxidase family and its inhibitors. Arch Immunol
Ther Exp (Warsz). 60:277–294. 2012. View Article : Google Scholar
|
|
29
|
Ceni E, Mello T and Galli A: Pathogenesis
of alcoholic liver disease: Role of oxidative metabolism. World J
Gastroenterol. 20:17756–17772. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bettaieb A, Jiang JX, Sasaki Y, Chao TI,
Kiss Z, Chen X, Tian J, Katsuyama M, Yabe-Nishimura C, Xi Y, et al:
Hepatocyte nicotinamide adenine dinucleotide phosphate reduced
oxidase 4 regulates stress signaling, fibrosis, and insulin
sensitivity during development of steatohepatitis in mice.
Gastroenterology. 149:468–80.e10. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Colmenero J, Bataller R, Sancho-Bru P,
Bellot P, Miquel R, Moreno M, Jares P, Bosch J, Arroyo V,
Caballería J, et al: Hepatic expression of candidate genes in
patients with alcoholic hepatitis: Correlation with disease
severity. Gastroenterology. 132:687–697. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yeligar SM, Harris FL, Hart CM and Brown
LA: Glutathione attenuates ethanol-induced alveolar macrophage
oxidative stress and dysfunction by downregulating NADPH oxidases.
Am J Physiol Lung Cell Mol Physiol. 306:L429–L441. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Valero T: Mitochondrial biogenesis:
Pharmacological approaches. Curr Pharm Des. 20:5507–5509. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhu Y, Jiang Y, Shi L, Du L, Xu X, Wang E,
Sun Y, Guo X, Zou B, Wang H, et al: 7-O-Geranylquercetin induces
apoptosis in gastric cancer cells via ROS-MAPK mediated
mitochondrial signaling pathway activation. Biomed Pharmacother.
87:527–538. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ma L, Dong JX, Wu C, Li XY, Chen J, Zhang
H and Liu Y: Spectroscopic, polarographic, and microcalorimetric
studies on mitochondrial dysfunction induced by ethanol. J Membr
Biol. 250:195–204. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen LY, Chen Q, Zhu XJ, Kong DS, Wu L,
Shao JJ and Zheng SZ: Diallyl trisulfide protects against
ethanol-induced oxidative stress and apoptosis via a hydrogen
sulfide-mediated mechanism. Int Immunopharmacol. 36:23–30. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hoyt LR, Randall MJ, Ather JL, DePuccio
DP, Landry CC, Qian X, Janssen-Heininger YM, van der Vliet A, Dixon
AE, Amiel E, et al: Mitochondrial ROS induced by chronic ethanol
exposure promote hyper-activation of the NLRP3 inflammasome. Redox
Biol. 12:883–896. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang C, Jia X, Bao J, Chen S, Wang K,
Zhang Y, Li P, Wan JB, Su H, Wang Y, et al: Polyphyllin VII induces
apoptosis in HepG2 cells through ROS-mediated mitochondrial
dysfunction and MAPK pathways. BMC Complement Altern Med. 16:58–69.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bonet-Ponce L, Saez-Atienzar S, da Casa C,
Flores-Bellver M, Barcia JM, Sancho-Pelluz J, Romero FJ, Jordan J
and Galindo MF: On the mechanism underlying ethanol-induced
mitochondrial dynamic disruption and autophagy response. Biochim
Biophys Acta. 1852:1400–1409. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang L, Wu L, Du S, Hu Y, Fan Y and Ma J:
1,25(OH)2D3 inhibits high glucose-induced apoptosis and ROS
production in human peritoneal mesothelial cells via the MAPK/P38
pathway. Mol Med Rep. 14:839–844. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Andreu-Fernández V, Sancho M, Genovés A,
Lucendo E, Todt F, Lauterwasser J, Funk K, Jahreis G, Pérez-Payá E,
Mingarro I, et al: Bax transmembrane domain interacts with
prosurvival Bcl-2 proteins in biological membranes. Proc Natl Acad
Sci USA. 114:310–315. 2017. View Article : Google Scholar :
|
|
42
|
Kim JA, Kim JC, Min JS, Kang I, Oh J and
Ahn JK: HSV-1 ICP27 induces apoptosis by promoting Bax
translocation to mitochondria through interacting with 14-3-3θ. BMB
Rep. 50:257–262. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Reshi L, Wang HV, Hui CF, Su YC and Hong
JR: Anti-apoptotic genes Bcl-2 and Bcl-xL overexpression can block
iridovirus serine/threonine kinase-induced
Bax/mitochondria-mediated cell death in GF-1 cells. Fish Shellfish
Immunol. 61:120–129. 2017. View Article : Google Scholar
|
|
44
|
Salvador-Gallego R, Mund M, Cosentino K,
Schneider J, Unsay J, Schraermeyer U, Engelhardt J, Ries J and
García-Sáez AJ: Bax assembly into rings and arcs in apoptotic
mitochondria is linked to membrane pores. EMBO J. 35:389–401. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gómez-Crisóstomo NP, López-Marure R,
Zapata E, Zazueta C and Martínez-Abundis E: Bax induces cytochrome
c release by multiple mechanisms in mitochondria from MCF7 cells. J
Bioenerg Biomembr. 45:441–448. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tsai MH, Liu JF, Chiang YC, Hu SC, Hsu LF,
Lin YC, Lin ZC, Lee HC, Chen MC, Huang CL, et al: Artocarpin, an
isoprenyl flavonoid, induces p53-dependent or independent apoptosis
via ROS-mediated MAPKs and Akt activation in non-small cell lung
cancer cells. Oncotarget. 8:28342–28358. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yang Y, Zong M, Xu W, Zhang Y, Wang B,
Yang M and Tao L: Natural pyrethrins induces apoptosis in human
hepatocyte cells via Bax- and Bcl-2-mediated mitochondrial pathway.
Chem Biol Interact. 262:38–45. 2017. View Article : Google Scholar
|
|
48
|
Yan X, Jiang Z, Bi L, Yang Y and Chen W:
Salvianolic acid A attenuates TNF-α- and D-GalN-induced ER
stress-mediated and mitochondrial-dependent apoptosis by modulating
Bax/Bcl-2 ratio and calcium release in hepatocyte LO2 cells. Naunyn
Schmiedebergs Arch Pharmacol. 388:817–830. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chuang WL, Lin PY, Lin HC and Chen YL: The
Apoptotic effect of ursolic acid on SK-Hep-1 cells is regulated by
the PI3K/Akt, p38 and JNK MAPK signaling pathways. Molecules.
21:460–470. 2016. View Article : Google Scholar
|