Introduction
Polymyositis (PM) is a systemic autoimmune disease,
which is characterized by progressive muscular atrophy as a result
of chronic muscular inflammation (1). PM has been reported to be associated
with a high risk of carcinogenesis (2,3);
however, the exact mechanisms underlying the etiology and pathology
of PM remain to be fully understood. The combined action of natural
immunity, acquired immunity and nonimmune processes may lead to
skeletal muscle injury in patients with PM (4). Various immune cells, cytokines and
chemokines, as well as endoplasmic reticulum stress and autophagy
participate in the pathogenesis of PM (5–9).
Identifying the pathogenesis and key immune regulatory pathway of
PM may improve understanding of the progression of PM, and may
result in the identification of potential diagnostic and drug
treatment targets that enable the future targeting of PM and
PM-associated cancers.
High-mobility group box 1 (HMGB1) is a conserved
non-histone nuclear protein that is widely identified in the lymph,
brain, liver, lung, heart, kidney and other tissues. HMGB1 controls
the stability of nucleosomes, DNA recombination, replication,
repair and transcription in nucleus (10). HMGB1 is secreted to the
extracellular environment where it induces cytokine production, and
is involved in the pathological processes of sepsis, cancer and
arthritis (11,12). HMGB1 may also be released by
cellular necrosis, and it functions critically in natural and
acquired immunity (13–15). It has previously been reported that
activation of HMGB1 is closely associated with the development of
PM (16). Therefore, the present
study aimed to investigate whether HMGB1 is a potential therapeutic
target for PM.
Cluster of differentiation (CD)163, which is a
member of the class B scavenger receptor-cysteine-rich family, is
exclusively expressed in the monocyte-macrophage system. The
positive expression of CD163 has been detected in inflamed tissues,
and CD163 serves an important role in the anti-inflammatory
response (17). IL-17 is a
proinflammatory cytokine released by T-helper cells, which induces
the expression of IL-1β, IL-6, TNF-α and granulocyte-colony
stimulating factor (G-CSF) (18).
Numerous studies have focused on IL-17, most of which concern
common autoimmune diseases, including rheumatoid arthritis
(19,20), systemic lupus erythematosus
(21,22), multiple sclerosis (23,24)
and atheroclerosis (25,26). Intercellular adhesion molecule
(ICAM)-1 is expressed on the surface of numerous classes of cells,
including endothelial cells and immune cells. In addition, ICAM-1
is induced by cell factors, including IL-1, TNF-α and bacterial
lipopolysacharide (27). A
previous study confirmed that the synergistic effects of IL-17 and
TNF-α enhance ICAM-1 expression in several inflammatory diseases
(28).
MicroRNAs (miRNAs/miRs) are a class of non-coding,
single-stranded small RNA molecules, which regulate the expression
of target genes (29). miRNAs
possess numerous biological functions in cellular development,
multiplication, variation and apoptosis (30–33).
The dysregulation of miR-381 has been detected in various diseases,
including acquired immunodeficiency syndrome (34), gastric cancer (35) and chondropathy (36). Therefore, it was hypothesized that
miR-381 may play an important role in PM. The present study aimed
to explore the role of miR-381 in PM and to investigate the
mechanism underlying the effects of miR-381 on inflammation and
macrophage infiltration.
Materials and methods
Ethics statement
The present study was approved by the ethics
committee of the First Affiliated Hospital of Zhengzhou University
(Zhengzhou, China).
Clinical samples
A total of 25 patients with PM (male:female, 12:13;
age range, 40–80 years; average age, 60 years) were recruited from
the First Affiliated Hospital of Zhengzhou University between June
2014 and May 2016. All enrolled patients were treated with
high-dose dexamethasone, and individual patients were treated with
tacrolimus or mercaptopurine. Muscle tissue samples were obtained
from each patient: One sample was obtained prior to treatment and
the other was obtained after treatment. Four men and six women were
included in the healthy control group; these individuals had no
clinical signs of muscle disease (mean age, 60 years). The clinical
features of the selected patients and healthy control individuals
are listed in Table I.
 | Table IClinical data of patients with PM and
healthy controls. |
Table I
Clinical data of patients with PM and
healthy controls.
| No. | Age (years) | Sex | Diagnosis | Duration
(months) | Treatment |
|---|
| 1 | 45 | F | PM | 8 | DXMS, TAC |
| 2 | 69 | F | PM | 4 | DXMS |
| 3 | 55 | M | PM | 9 | DXMS |
| 4 | 78 | F | PM | 5 | DXMS, TAC |
| 5 | 56 | F | PM | 14 | DXMS |
| 6 | 63 | M | PM | 3 | DXMS |
| 7 | 52 | F | PM | 5 | DXMS |
| 8 | 59 | M | PM | 13 | DXMS, MER |
| 9 | 48 | M | PM | 18 | DXMS, TAC |
| 10 | 61 | M | PM | 6 | DXMS |
| 11 | 75 | F | PM | 11 | DXMS, MER |
| 12 | 54 | M | PM | 4 | DXMS |
| 13 | 55 | F | PM | 8 | DXMS |
| 14 | 68 | F | PM | 10 | DXMS |
| 15 | 43 | M | PM | 7 | DXMS |
| 16 | 49 | F | PM | 8 | DXMS, MER |
| 17 | 57 | M | PM | 3 | DXMS |
| 18 | 62 | F | PM | 3 | DXMS |
| 19 | 64 | F | PM | 5 | DXMS, TAC |
| 20 | 58 | M | PM | 12 | DXMS, MER |
| 21 | 73 | M | PM | 14 | DXMS |
| 22 | 79 | M | PM | 8 | DXMS, MER |
| 23 | 54 | F | PM | 3 | DXMS |
| 24 | 62 | M | PM | 6 | DXMS |
| 25 | 74 | F | PM | 8 | DXMS, TAC |
| 26 | 52 | F | – | – | – |
| 27 | 64 | F | – | – | – |
| 28 | 62 | M | – | – | – |
| 29 | 76 | M | – | – | – |
| 30 | 54 | F | – | – | – |
| 31 | 80 | M | – | – | – |
| 32 | 46 | M | – | – | – |
| 33 | 55 | F | – | – | – |
| 34 | 48 | F | – | – | – |
| 35 | 79 | F | – | – | – |
Sample collection
Serum samples were collected from the 25 patients
with PM pretreatment and post-treatment. Serum samples were also
collected from the healthy controls. Briefly, the blood samples
were collected into anticoagulant tubes and centrifuged at 1,000 ×
g for 10 min at 4°C. The serum samples were then maintained at
−80°C for subsequent analyses. Written informed consent was
obtained from the participants.
Animal model
A mouse model of experimental autoimmune myositis
(EAM) was generated in the present study. BALB/c mice were obtained
from Guangdong Medical Laboratory Animal Center (Foshan, China).
The mice were housed under the following conditions: Temperature,
21°C; humidity, 40–60%; 12-h light/dark cycle and free access to
food and water. Briefly, 1.5 mg rabbit myosin (M1636) and an equal
volume of complete Freund's adjuvant (CFA, 1 mg/ml, F5881) (both
from Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) were mixed,
after which 1 ml CFA mixed with 5 mg Mycobacterium tuberculosis
(cat. no. 231141; BD Biosciences, Franklin Lakes, NY, USA) was
added to the mixture. The obtained reagent was initially injected
into the left hind leg of mice. After 1 week, it was injected into
the tail root of mice. Furthermore, all of the mice were
intraperitoneally injected with pertussis toxin (PT, cat. no.
P2980; Sigma-Aldrich; Merck KGaA; 200 µl normal saline mixed
with 500 ng PT) alongside each of the two previous injections.
Healthy adult female BALB/c mice (weight, 15–18 g; age, 5–6 weeks)
were randomly divided into the following groups (n=5 mice/group):
Control group, in which mice were untreated; model group, in which
mice underwent EAM induction; anti-IL-17 group, in which EAM mice
were injected daily with anti-IL-17 antibody (100 µg/mice,
cat. no. 13838; Cell Signaling Technology, Inc., Danvers, MA, USA)
14 days post-EAM induction; and anti-HMGB1 group, in which EAM mice
were intraperitoneally injected with anti-HMGB1 antibody daily (100
µg/mice, cat. no. 6893; Cell Signaling Technology, Inc.) 14
days post-EAM induction. All animals were euthanized by rapid
cervical dislocation 4 weeks after antibody injection. The animal
study was approved by the Ethics Committee of the First Affiliated
Hospital of Zhengzhou University.
Hematoxylin and eosin staining assay
The mouse muscle tissues were fixed with 4%
paraformaldehyde for 6 h at room temperature. After dehydration
with ethanol (from a low concentration to a high concentration),
the tissues were embedded in paraffin. Paraffin-embedded mouse
muscle tissues were then sliced into 4–6 µm sections and the
sections were mounted onto the slides. The slides were placed into
hematoxylin (Beijing Solarbio Science & Technology Co., Ltd.,
Beijing, China) for 10 min at room temperature. After being washed
with tap water for 1–2 min, the slides were then placed in 10%
glacial acetic acid for 10 sec and placed in 1% ammonia water until
the sections turned blue. Subsequently, after being washed with tap
water for 1–2 min, the slides were placed in eosin (Beijing
Solarbio Science & Technology Co., Ltd.) for 10 sec. After
dehydration in a gradual series of ethanol (70, 90, 95 and 100%),
the slides were placed in xylene for 2 min; this step was repeated
once. Finally, the slides were sealed with neutral gum. The slides
were observed under an inverted microscope, in order to count the
percentage of inflammatory cells in the whole field. The
histopathologic scores were evaluated as follows: 0 score, no
inflammatory cells; 1 score, <25% inflammatory cells; 2 score,
25–50% inflammatory cells; 3 score, >75% inflammatory cells.
Immunohistochemistry
Muscle tissues obtained from the patients or mice,
were fixed in 4% paraformaldehyde and 30% sucrose solution at 4°C
for 73 h. The tissue samples were embedded in paraffin and then
dissected into sections (4–6 µm). Following treatment with
3% hydrogen peroxide in methanol for 30 min at room temperature,
the slides were incubated with primary antibodies against CD163
(1:500 dilution, cat. no. ab182422; Abcam, Cambridge, MA, USA) at
4°C overnight. The tissue samples were then incubated with
horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G
(IgG) secondary antibodies (1:1,000 dilution, cat. no. ab6721;
Abcam) at room temperature for 2 h. Finally, the tissue samples
were treated with DAB (Sigma-Aldrich; Merck KGaA). Cells in the
negative group were incubated with nonspecific IgG (1:500 dilution,
cat. no. ab108337; Abcam).
Isolation of peritoneal macrophages
As previously described (37), isolation of peritoneal macrophages
was conducted. Briefly, the normal mice were euthanatized by rapid
cervical dislocation and were disinfected in 70% ethanol for 5 min.
Under sterile conditions, a small incision was made along the
midline of the mice with sterile scissors. Subsequently,
pre-chilled saline (4°C) was injected into the peritoneal cavity;
fluid from the peritoneum was aspirated and was transferred into a
50-ml conical polypropylene centrifuge tube on ice. The peritoneal
exudate cells were centrifuged at 400 × g for 10 min at 4°C.
Following removal of the supernatant, the cells were resuspended in
cold Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and were gently pipetted up and
down. The cells were then seeded at a density of
1×106/ml in RPMI-1640 medium (Thermo Fisher Scientific,
Inc.) and incubated at 37°C in an incubator containing 5%
CO2. Following 2 h incubation, the macrophages were
grouped as follows: Control group, which consisted of untreated
cells; negative control (NC)-mimics group, in which cells were
transfected with NC mimics; mimics group, in which cells were
transfected with miR-381 mimics; NC-inhibitors group, in which
cells were transfected with NC inhibitors negative control;
inhibitors group, in which cells were transfected with miR-381
inhibitors; NC-small interfering (si)RNA, in which cells were
transfected with NC siRNA (no specific sequence); and inhibitors +
siRNA-HMGB1group, in which cells were co-transfected with miR-381
inhibitors and siRNA-HMGB1.
Cell transfection
The cells were seeded into a 6-well plate at a
density of 3×104/well. Once cell confluence reached 70%,
the cells were transfected with miRNA (50 nM) and/or siRNA (50
pmol) using Lipofectamine® 3000 (Invitrogen; Thermo
Fisher Scientific, Inc.) at 37°C. After 48 h, the cells underwent
further experimentation. The miRNAs and siRNA used were as follows:
miR-381 mimics (miR10017081-1-5); miRNA negative control
(miR01201-1-5); miR-381 inhibitor (miR20017081-1-5); miRNA
inhibitor control (miR02201-1-5) (all from Guangzhou RiboBio Co.,
Ltd., Guangzhou, China); si-HMGB1 (MBS8222378) and siRNA negative
control (NC) (MBS8241404) (both from MyBioSource, San Diego, CA,
USA).
ELISA
The secreted levels of IL-17 (mouse, M1700; human,
D1700) and ICAM-1 (mouse, MIC100; human, Dy720) were detected using
ELISA kits (R&D Systems, Inc., Minneapolis, MN, USA). The mouse
serum creatine kinase (s-CK) ELISA kit (CSB-E14407m) was purchased
from Cusabio Technology LLC (Houston, TX, USA). The human s-CK
ELISA kit (NR-R10105) and the HMGB1 ELISA kits (mouse, NB-S11134;
human BG-HUM11133) were purchased from Novatein Biosciences, Inc.,
(Woburn, MA, USA). All experiments were performed according to the
manufacturer's protocols. Finally, the optical density at 450 nm
was measured on a microplate reader (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
The mouse tissues were ground in liquid nitrogen;
total RNA from tissues and cultured cells was extracted by
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.).
Subsequently, 2 µg RNA was used for cDNA synthesis using
first strand cDNA kit (Sigma-Aldrich; Merck KGaA), according to the
manufacturer's protocol. RT-qPCR was performed using the SYBR Green
Premix Reagent (Takara Bio, Inc., Otsu, Japan) on an ABI 7500
Thermocycler (Applied Biosystems; Thermo Fisher Scientific, Inc.).
The qPCR thermocycling conditions were as follows: 94°C for 5 min;
followed by 30 cycles at 94°C for 30 sec and 60°C for 30 sec; and a
final extension step at 72°C for 10 min. GAPDH/U6 was used as an
internal control. The sequences of the primers (Invitrogen; Thermo
Fisher Scientific, Inc.) used for PCR amplification are indicated
in Table II.
| Table IIPrimer sequences used in the reverse
transcription-quantitative polymerase chain reaction analysis. |
Table II
Primer sequences used in the reverse
transcription-quantitative polymerase chain reaction analysis.
| Gene | Sequence
(5′-3′) |
|---|
| HMGB1 | F:
AAGAAGTGCTCAGAGAGGTGGAAG |
| R:
CTAGTTTCTTCGCAACATCACCA |
| IL-17 | F:
TACCTCAACCGTTCCACTTCACCC |
| R:
GGCACTTCTCAGGCTCCCTCTTC |
| ICAM-1 | F:
GGAGACTAACTGGATGAAAGACGA |
| R:
TCCCACGGAGCAGCACTACT |
| GAPDH | F:
TGTCATATTTCTCGTGGTTCA |
| R:
TGTCATATTTCTCGTGGTTCA |
| miR-381 | F:
AAAGCGAGGTTGCCCTTTGT |
| R:
TACTCACAGAGAGCTTGCCC |
| U6 | F:
CTCGCTTCGGCAGCACA |
| R:
AACGCTTCACGAATTTGCGT |
| s-CK | F:
CTCACCCCCACCATCTATGC |
| R:
GATGCATCCAGGTCCGTAGG |
Western blotting
The collected tissues were ground in liquid nitrogen
and proteins were isolated using the total extraction sample kit
(Sigma-Aldrich; Merck KGaA). Cultured cells were lysed on ice in
radioimmunoprecipitation assay lysis buffer (pH 7.4; 1 mM
MgCl2, 1% Triton X-100, 10 mM Tris-HCl and 0.1% SDS).
Protein concentration was determined using the bicinchoninic acid
protein assay kit (Bio-Rad Laboratories, Inc.). An equal quantity
(50 µg) of protein from each sample was separated by 8%
SDS-PAGE and transferred onto nitrocellulose membranes (EMD
Millipore, Billerica, MA, USA). Nonspecific antigens were blocked
by immersing the membranes in 5% non-fat milk at room temperature
for 2 h. Subsequently, the membranes were incubated overnight at
4°C with the following primary antibodies: Anti-HMGB1 (1:500
dilution, cat. no. ab18256), anti-IL-17 (1:5,000 dilution, cat. no.
ab9565), anti-ICAM-1 (1:1,000 dilution, cat. no. ab2213) and
anti-GAPDH (1:2,500 dilution, cat. no. ab9485) (all from Abcam).
The membranes were then incubated with the corresponding
horseradish peroxidase-conjugated secondary antibodies (1:2,000;
cat. nos. ab6789 and ab6721; Abcam) at room temperature for 1 h.
The signals were detected using an enhanced chemiluminescence
reagent (EMD Millipore). The blots were analyzed using Quantity One
software version 4.62 (Bio-Rad Laboratories).
Luciferase activity assay
Bioinformatics prediction was performed using the
online databases TargetScan (http://www.targetscan.org/) and miRDB (http://www.mirdb.org/). The HMGB1-3′ untranslated
region (3′UTR) was synthesized by Invitrogen; Thermo Fisher
Scientific, Inc. The HMGB-3′UTR was amplified to the downstream
site of pGL4 luciferase vectors (Promega Corporation, Madison, WI,
USA). The HMGB1-3′UTR mutant (mut) was generated using a rapid
site-directed mutagenesis kit (Thermo Fisher Scientific, Inc.).
Cells were seeded at 3×104/well in a 24-well plate;
after 24 h, the cells were transfected with 1 µg HMGB1-3′UTR
(wild-type or mut)-luciferase plasmid, 50 nM miR-381 mimics/miR-NC
and 150 ng Renilla luciferase plasmid using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.). The cells were then incubated at 37°C for 36 h.
Luciferase activity was detected using a dual-luciferase reporter
assay kit (Promega Corporation), according to the manufacturer's
protocol. All data were normalized to Renilla luciferase
activity. The relative luciferase activities were expressed as
relative fluorescence units.
Transwell assay
Cells (~5×104 cells/ml) were
serum-starved for 24 h in serum-free DMEM at 37°C. The cells were
then plated into a 24-well Transwell plate (Corning Incorporated,
Corning, NY, USA). DMEM supplemented with 15% fetal bovine serum
(Gibco; Thermo Fisher Scientific, Inc.) was placed into the lower
chamber. After 24 h, cells that penetrated into the lower chamber
were fixed with 95% ethyl alcohol and were stained with 0.1%
crystal violet at room temperature for 20 min. Finally, the cells
were observed under an inverted microscope and the number of
migrated cells was counted.
Statistical analysis
All experimental results are presented as the means
± standard deviation and experiments were repeated at least three
times. Kaplan-Meier analysis was used for survival analysis; log
rank test was used to compare the differences. The receiver
operating characteristic analysis was performed to distinguish the
patients with high HMGB1 expression from the patients with low
HMGB1 expression. GraphPad Prism 6.0 (GraphPad, Inc., La Jolla, CA,
USA) was used to perform data analysis. All assays were analyzed by
one-way analysis of variance followed by Turkey's test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Expression levels of s-CK, HMGB1, IL-7
and miR-381 in healthy controls and patients with PM
Compared with in the healthy control individuals,
the serum concentrations of s-CK (434±70 vs. 97±11 pg/ml), HMGB1
(348±63 vs. 102±19 pg/ml) and IL-17 (297±38 vs. 89±14 pg/ml) were
markedly increased in patients with PM. However, following
treatment with a glucocorticoid or other immunosuppressive drugs,
s-CK (212±35 vs. 434±70 pg/ml), HMGB1 (220±38 vs. 348±63 pg/ml) and
IL-17 (158±21 vs. 297±38 pg/ml) expression levels were decreased,
as detected by ELISA (P<0.05; Fig.
1A–C). Furthermore, in patients with PM, the expression levels
of HMGB1 were significantly increased, whereas the expression
levels of miR-381 were markedly decreased compared with in healthy
control individuals. Conversely, the expression levels of HMGB1 and
miR-381 were reversed in the post-treatment group (P<0.05;
Fig. 1D and E). As shown in
Fig. 1F, patients with PM with a
high expression of HMGB1 had a worse prognosis (P=0.0204). A
receiver operating characteristic curve analysis was conducted to
distinguish patients with PM with high HMGB1 expression from
patients with PM with low HMGB1 expression. The cutoff value was
14.2 ng/ml. The area under the curve was 0.733 (Fig. 1G).
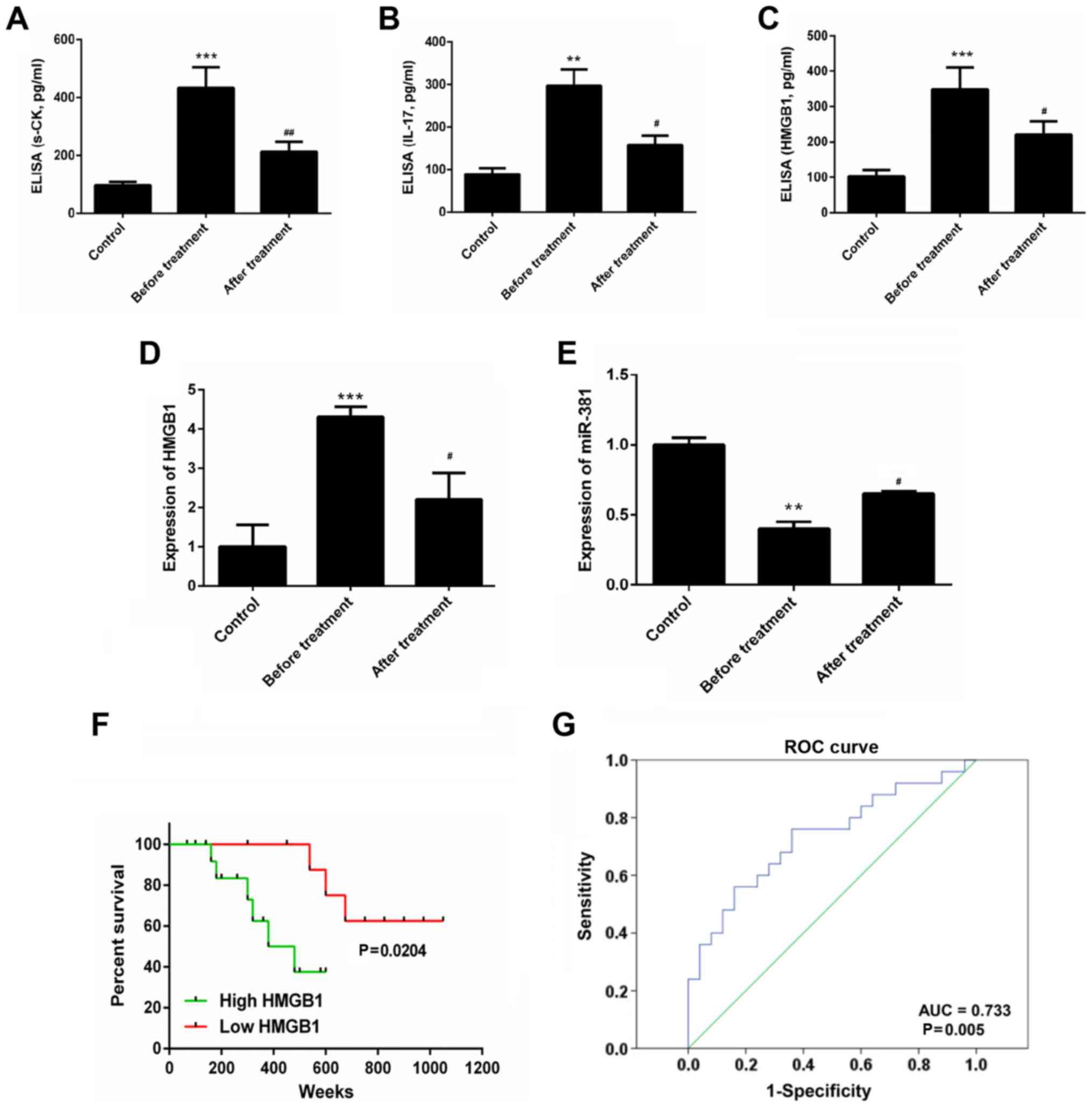 | Figure 1ELISA was performed to assess (A)
s-CK, (B) IL-17 and (C) HMGB1 expression in serum samples. Reverse
transcription-quantitative polymerase chain reaction analysis was
conducted to detect the expression levels of (D) HMGB1 and (E)
miR-381 in serum samples. (F) Overall survival rates of patients
with PM based on serum HMGB1 levels, as measured by Kaplan-Meier
survival analysis. P-values were calculated by the log-rank test.
(G) ROC curve analysis was performed to distinguish patients with
PM with high HMGB1 expression from patients with PM with low HMGB1
expression; cutoff value, 14.2 ng/ml. **P<0.01,
***P<0.01 vs. the control group.
#P<0.05, ##P<0.01 vs. the before
treatment group. AUC, area under the curve; HMGB1, high mobility
group box protein 1; IL-17, interleukin-17; miR-381, microRNA-381;
PM, polymyositis; ROC, receiver operating characteristic. |
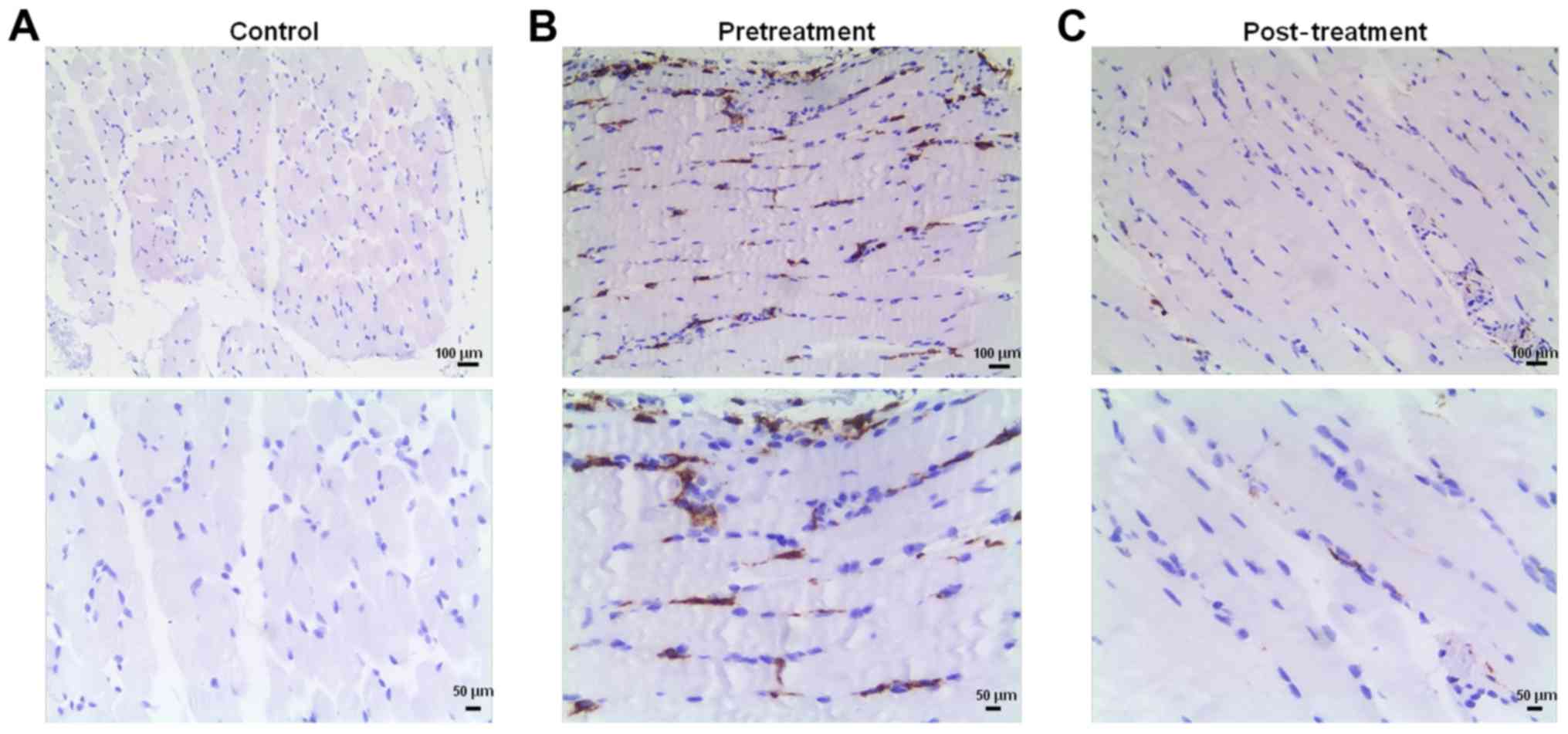
CD163 expression in muscle samples from
patients with PM
Muscle samples from control, pretreatment and
post-treatment groups were stained with anti-CD163. The expression
of CD163 was almost negative in the control group (Fig. 2A). Conversely, strong CD163
staining was identified in the pretreatment group (Fig. 2B), whereas CD163 expression was
markedly reduced in the post-treatment group (Fig. 2C).
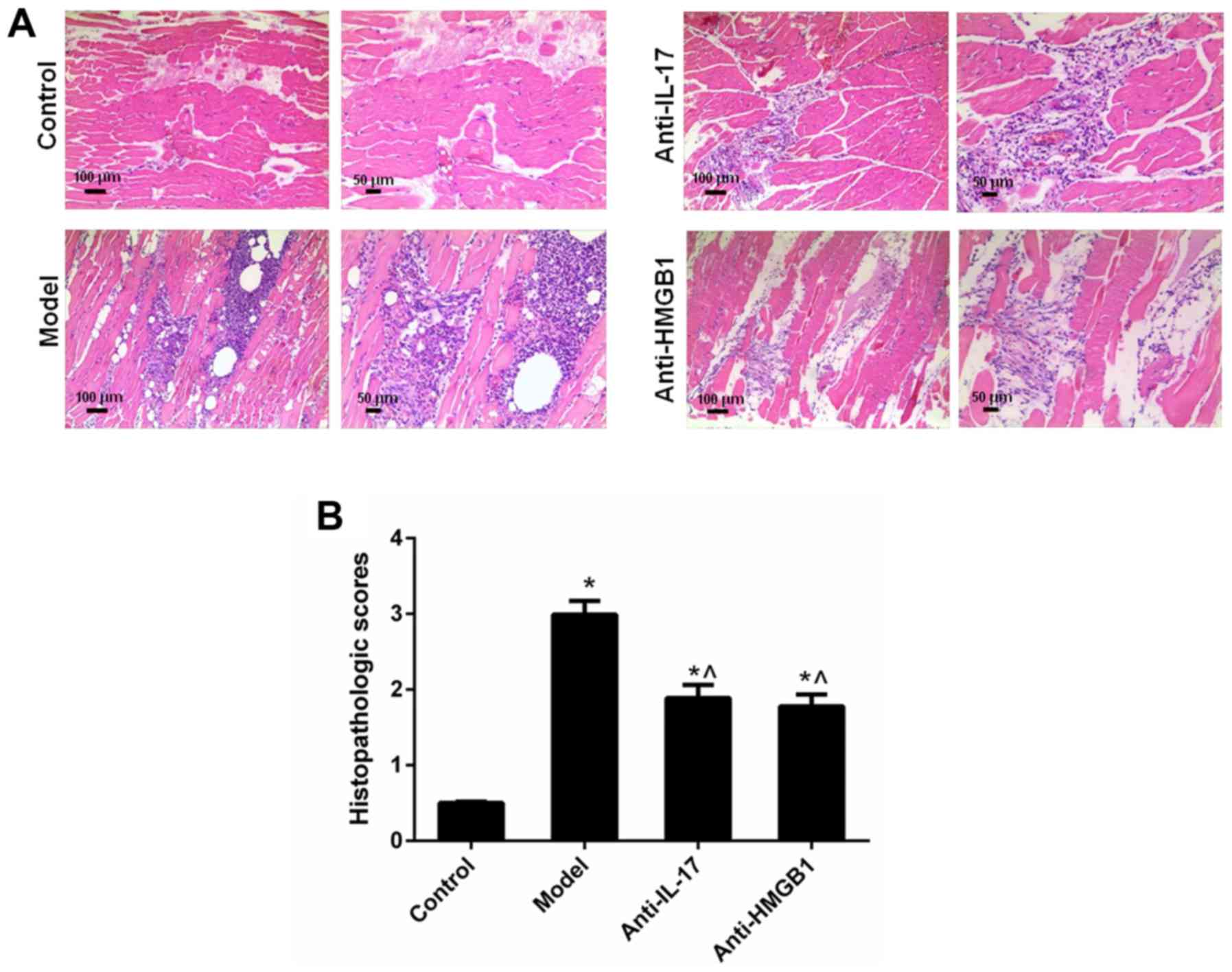
Inflammation of muscle tissues in
mice
In the mouse control group, muscle fibers were
polygonal in shape and were roughly the same size. A large amount
of inflammation and macrophage infiltration was detected, and
muscle fibers with varied sizes were observed in the EAM model
group. Conversely, inflammation and macrophage infiltration was
inhibited in the anti-IL-17 and anti-HMGB1 groups, compared with in
the model group (Fig. 3A).
Similarly, the histopathologic score in the model group was
elevated, whereas treatment with anti-IL-17 and anti-HMGB1 resulted
in a reduction in histopathologic score (Fig. 3B; Table III).
| Table IIIHistopathologic scores of the muscle
samples from the experimental autoimmune myositis model rats (n=5;
mean ± standard deviation). |
Table III
Histopathologic scores of the muscle
samples from the experimental autoimmune myositis model rats (n=5;
mean ± standard deviation).
| Group | Histopathologic
scores |
|---|
| Control | 0 |
| Model | 2.99±0.182a |
| Anti-IL-17 | 1.89±0.173a,b |
| Anti-HMGB1 | 1.78±0.156a,b |
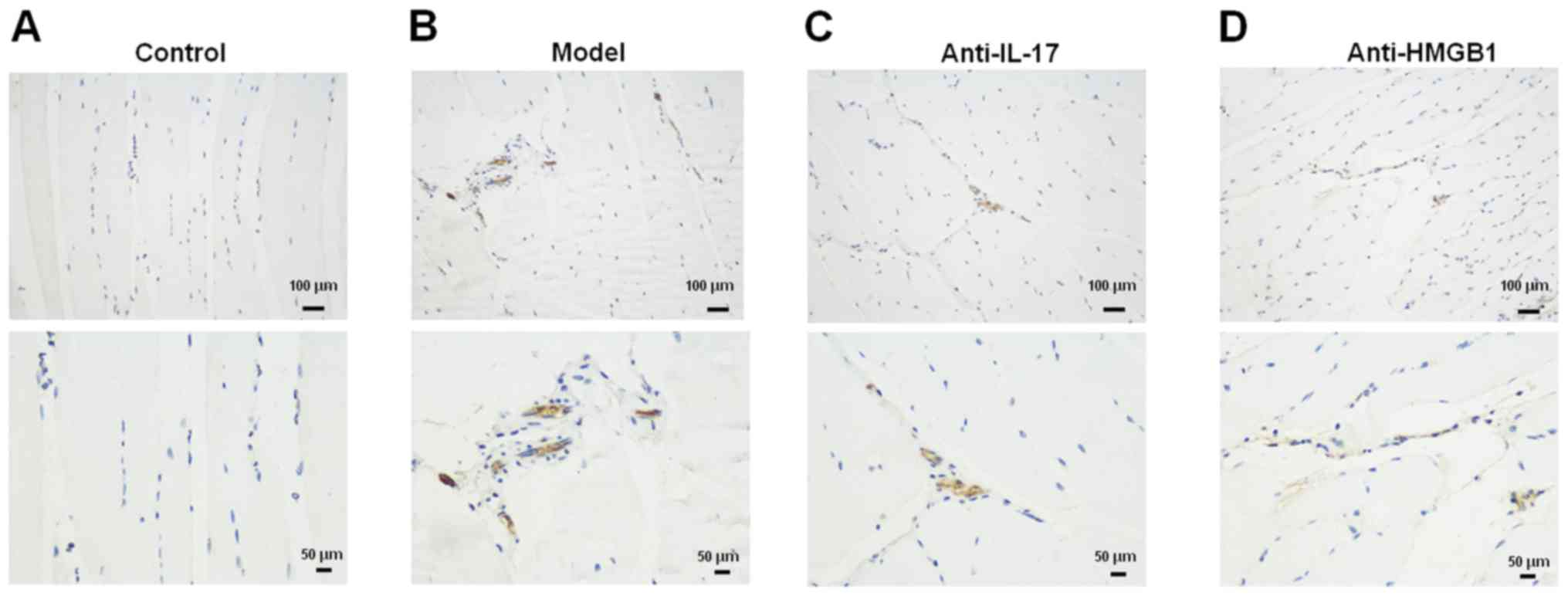
Expression of CD163 in muscle samples
from mice
CD163 expression was increased in the model group
compared with in the control group (Fig. 4A and B). Conversely, following
treatment with anti-IL-17 and anti-HMGB1, CD163 expression was
markedly reduced (Fig. 4C and
D).
Expression of s-CK, HMGB1, IL-7, ICAM-1
and miR-381 in mice
The results of an ELISA indicated that, compared
with in the control group, the levels of s-CK, HMGB1 and IL-17 were
significantly increased in the model group. However, the expression
levels of s-CK, HMGB1 and IL-17 were reduced following anti-IL-17
and anti-HMGB1 treatment (Fig.
5A–C). Furthermore, the results of RT-qPCR and western blotting
revealed that the expression levels of HMGB1, ICAM-1 and IL-17
exhibited a consistent trend with the results of the ELISA.
Furthermore, the expression levels of miR-381 were reduced in the
model group, whereas anti-IL-17 and anti-HMGB1 did not restore the
expression of miR-381 (Fig.
5D–F).
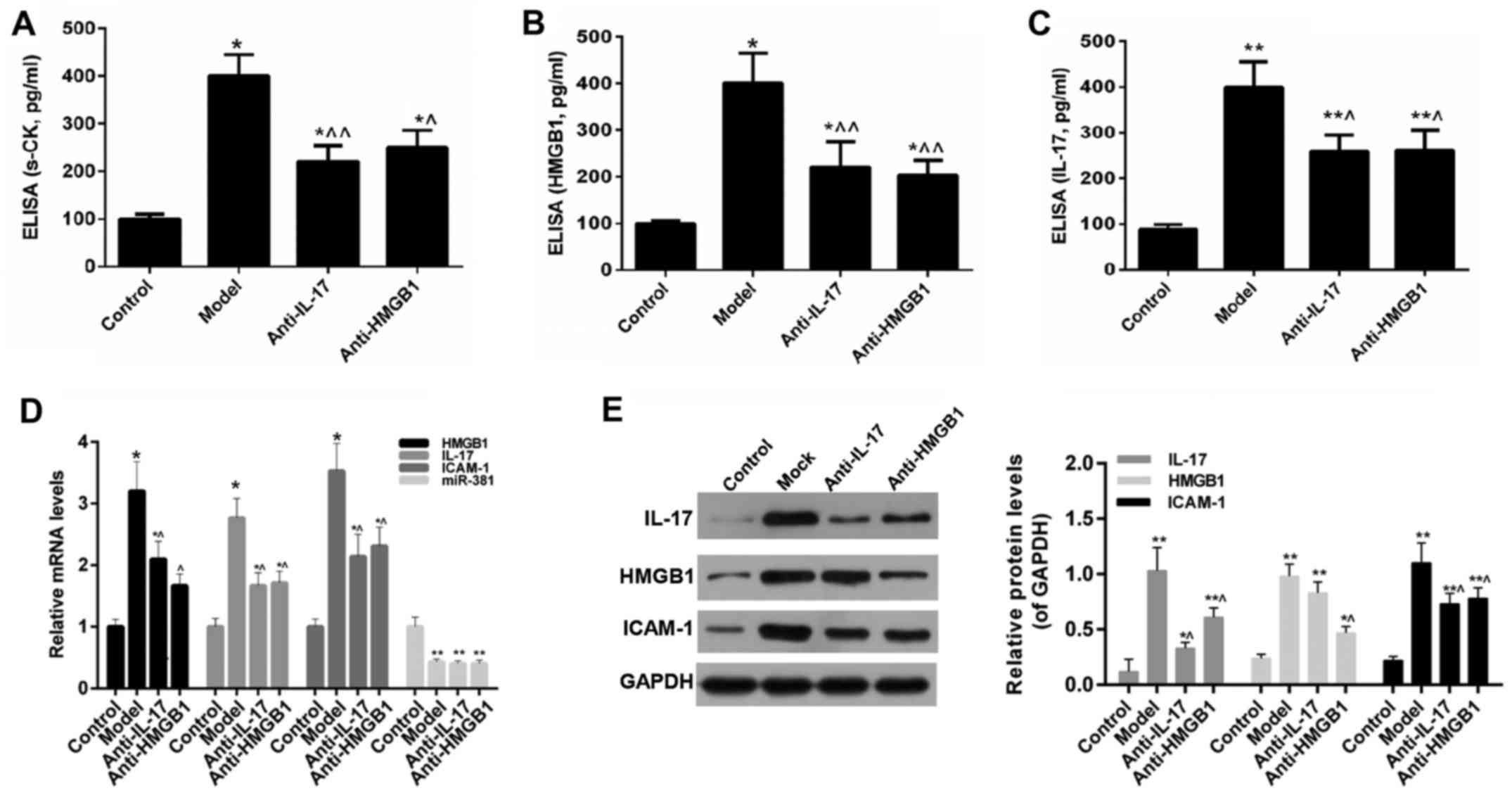 | Figure 5ELISA was performed to detect (A)
s-CK, (B) HMGB1 and (C) IL-17 expression in mice. (D) Reverse
transcription-quantitative polymerase chain reaction analysis was
carried out to evaluate the expression levels of HMGB1, IL-17,
ICAM-1 and miR-381 in mice. (E) Western blot analysis was performed
to detectIL-17, HMGB1 and ICAM-1 expression in mice.
*P<0.05, **P<0.01 vs. the control
group; ^P<0.05, ^^P<0.01 vs. the model
group. HMGB1, high mobility group box protein 1; ICAM-1,
intercellular adhesion molecule 1; IL-17, interleukin-17; miR-381,
microRNA-381; s-CK, serum creatine kinase. |
HMGB1 may be a target gene of
miR-381
Bioinformatics analysis identified a possible
binding site for miR-381 in the 3′UTR of HMGB1 (Fig. 6A). The results of a luciferase
assay in cells indicated that the luciferase activity of
HMGB1-3′UTR was decreased in the presence of miR-381 (Fig. 6B, P<0.05).
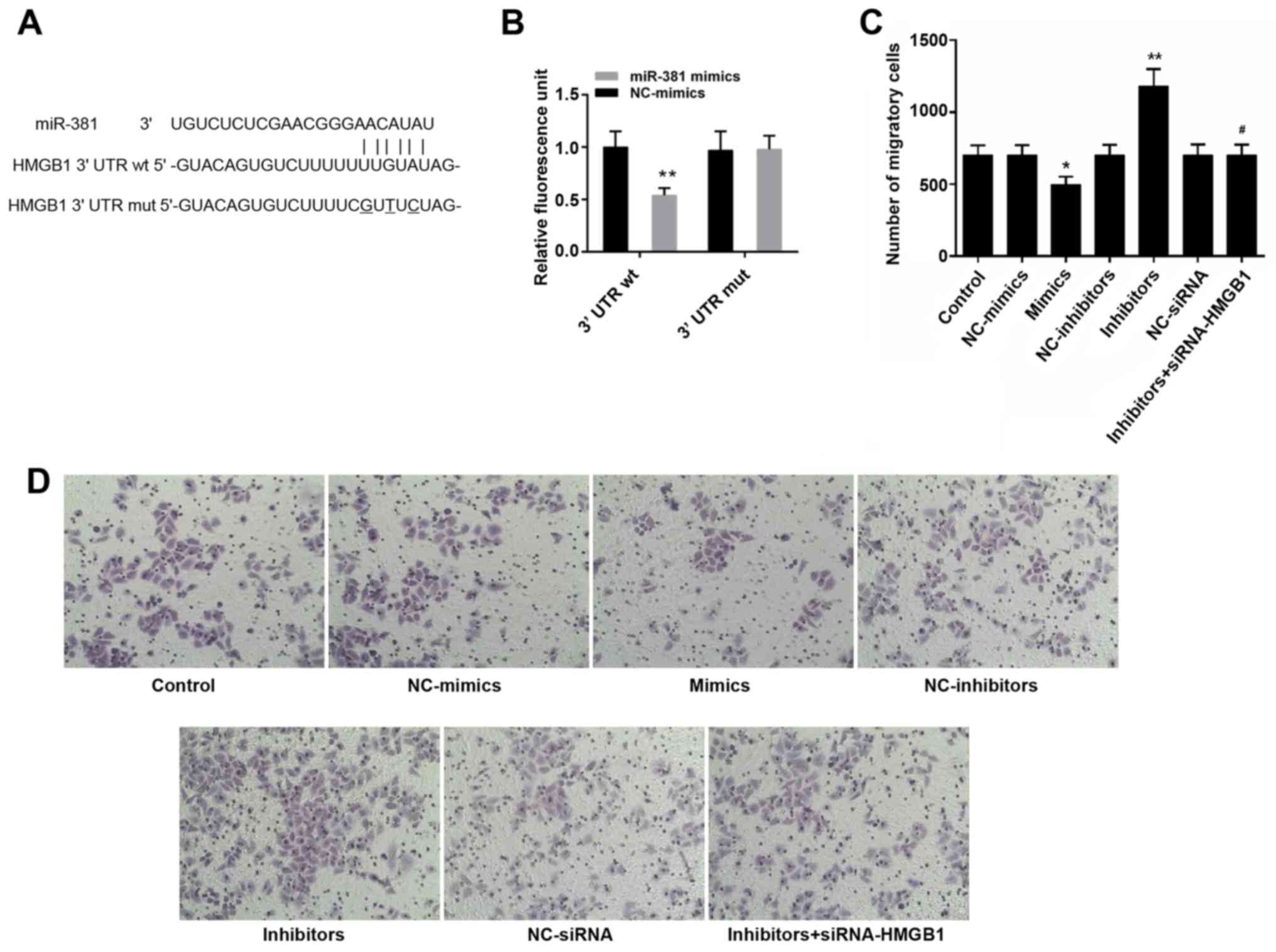 | Figure 6(A) Bioinformatics prediction of the
binding between miR-381 and HMGB1-3'UTR. (B) Luciferase activity
assay was conducted to assess the binding of miR-381 and
HMGB1-3'UTR. (C) Number of migratory cells. (D) Images of cells
that penetrated through the membrane in the control, NC-mimics,
mimics, NC-inhibitors, inhibitors, NC-siRNA and inhibitors +
siRNA-HMGB1 groups. Magnification, ×100. *P<0.05,
**P<0.01 vs. the control group; #P<0.05
vs. the inhibitors group. 3'UTR, 3' untranslated region; HMGB1,
high mobility group box protein 1; miR-381, microRNA-381; mut,
mutant; NC, negative control; siRNA, small interfering RNA; wt,
wild-type. |
miR-381 reduces the migration of
macrophages
As shown in Fig. 6C and
D, an obvious decrease in the number of migratory cells was
observed following transfection with miR-381 mimics (P<0.05).
The number of migratory cells in the miR-381 inhibitors group was
higher than in the other groups (P<0.05). Conversely, the
effects of miR-381 inhibitors were blocked by siRNA-HMGB1.
Effects of miR-381 and HMGB1 on the
expression levels of s-CK, ICAM-1 and IL-17
As shown in Fig.
7A, miR-381 expression in the miR-381 mimics group was
significantly increased compared with in the other groups
(P<0.05). Furthermore, an obvious decrease in miR-381 expression
was observed in the miR-381 inhibitors group (P<0.05). The
expression levels of HMGB1, IL-17 and ICAM-1 in macrophages were
decreased following transfection with miR-381 mimics, whereas
miR-381 inhibitors increased the expression levels of HMGB1, IL-17
and ICAM-1 (Fig. 7B–E).
Conversely, the effects of miR-381 inhibitors were inhibited by
siRNA-HMGB1.
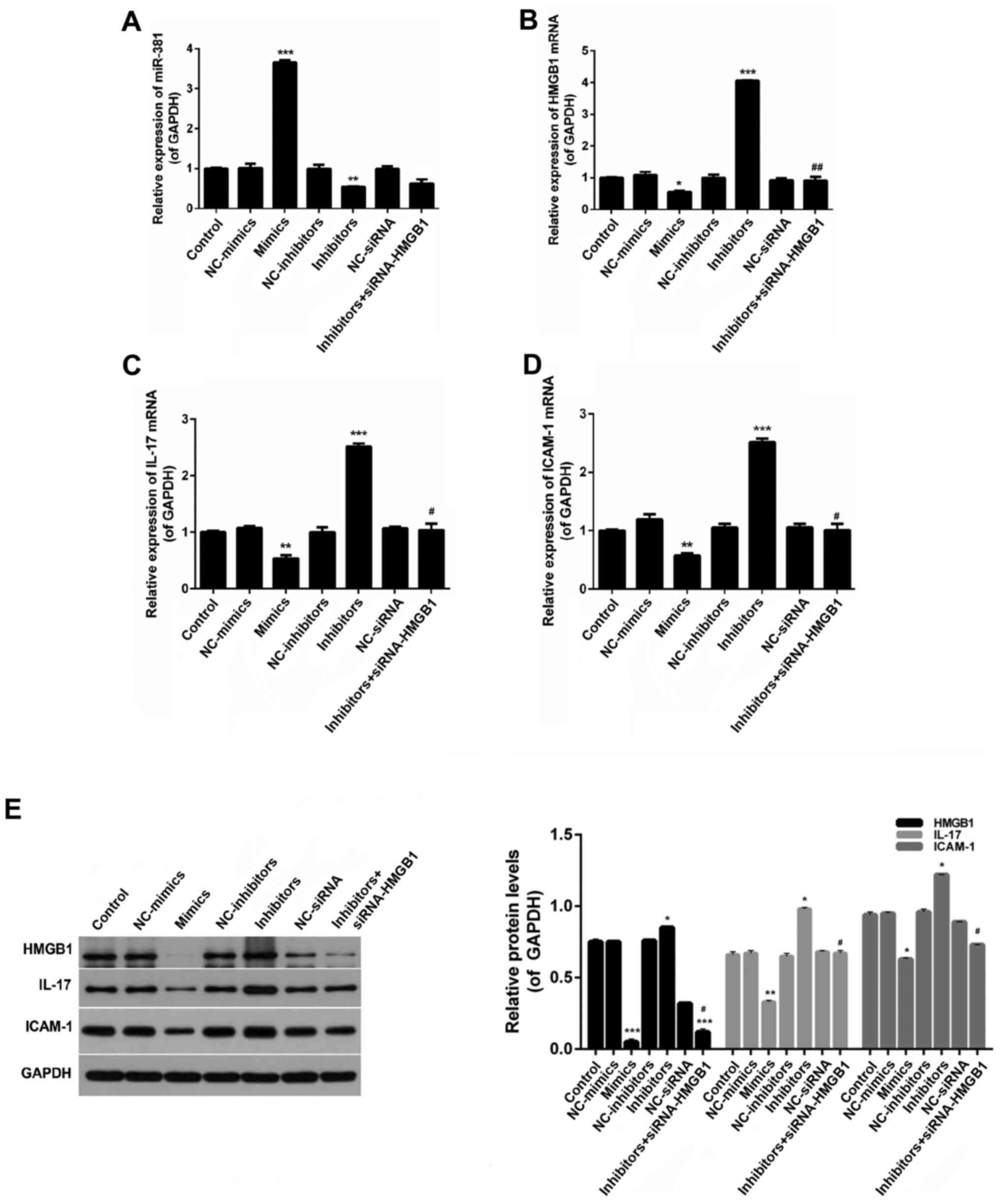 | Figure 7Reverse transcription-quantitative
polymerase chain reaction analysis was performed to evaluate the
expression levels of (A) miR-381, (B) HMGB1, (C) IL-17 and (D)
ICAM-1 in macrophages. (E) Western blot analysis was performed to
detectHMGB1, IL-17, and ICAM-1 expression in macrophages.
*P<0.05, **P<0.01,
***P<0.01 vs. the control group;
#P<0.05, ##P<0.05 vs. the inhibitors
group. HMGB1, high mobility group box protein 1; ICAM-1,
intercellular adhesion molecule 1; IL-17, interleukin-17; miR-381,
microRNA-381; NC, negative control; siRNA, small interfering
RNA. |
Discussion
PM is characterized by inflammation in muscle
tissues and chronic muscle weakness, and can lead to physical
disability and a shortened lifespan. The necrotic muscle tissues of
patients with PM are mainly characterized by inflammatory cell
infiltration, including the entry of macrophages into skeletal
muscles (38–40). Treatment with high doses of
glucocorticoids and supplementation with additional
immunosuppressive drugs is a widely adopted method to treat
patients with PM (41). However,
the exact pathogenesis of PM is currently unclear. A recent study
confirmed that cellular inflammation serves a significant role in
the development of PM (42).
Nevertheless, little is currently known about the explicit effects
of these pathways on the progression of PM. Therefore, studies
regarding the exact mechanisms underlying PM development are
required.
In the present study, the expression levels of s-CK
were markedly increased in patients with PM. Following treatment
with high doses of glucocorticoids or immunosuppressive drugs, s-CK
expression was markedly reduced. In addition, HMGB1 expression was
significantly higher in patients with PM compared with in the
control individuals, and a marked decrease was identified in the
post-treatment group. It has previously been reported that
activation of HMGB1 may be associated with the development of PM
(16). Furthermore, IL-17 levels
were significantly higher in patients with PM compared with in the
control group, whereas IL-17 levels declined in the post-treatment
group. A previous study regarding IL-17 in PM revealed that IL-17
levels are increased in PM compared with in a control group
(43). The strong effects of IL-17
have been reported on the pathogenesis of numerous autoimmune
diseases, including multiple sclerosis (44), rheumatoid arthritis (45) and psoriasis (46). An association between the
IL-17/ICAM-1 pathway and PM has also been reported (47–49).
The present study also detected the expression levels of miR-381 in
healthy controls and patients with PM. The results demonstrated
that the expression levels of miR-381 were markedly reduced in the
PM group compared with in the control group, whereas after
treatment with drugs, miR-381 expression was distinctly higher than
in the pretreatment group. The results of a survival analysis
further revealed that survival time was longer in patients with PM
with low HMGB1 expression. Based on these results, it may be
hypothesized that HMGB1 is a critical factor in the mechanism
underlying the development of PM.
The present results demonstrated that, in PM, the
expression levels of HMGB1 and miR-381 exhibited opposing trends.
Therefore, it was hypothesized that miR-381 might serve a critical
role in the pathogenesis of PM. An EAM animal experimental model
was established, in order to verify this assumption. The results
revealed that inflammation of muscle tissues was reduced by
anti-IL-17 and anti-HMGB1, compared with in the model group.
Furthermore, CD163 expression was also decreased by anti-IL-17 and
anti-HMGB1. It has previously been reported that IL-17 is a
proinflammatory cytokine released by T-helper cells, which may
induce the expression of IL-1β, IL-6, TNF-α and G-CSF (18). Therefore, the present study
confirmed that downregulation of HMGB1 may alleviate inflammatory
responses. To further verify this conclusion, the expression levels
of HMGB1, IL-17 and ICAM-1 were estimated. The results revealed
that the increased levels of HMGB1, IL-17 and ICAM-1 could be
decreased by anti-IL-17 and anti-HMGB1. This finding was consistent
with the results of a previous study (50); in this previous study, it was
revealed that the expression of ICAM-1 is reduced by anti-IL-17.
Similarly, another study suggested that HMGB1 can stimulate the
expression of IL-17 (51). These
results indicated that the HMGB1-IL-17-ICAM1 pathway has a critical
role in the inflammatory response of PM. Furthermore, the
expression levels of miR-381 were significantly decreased in the
EAM model mice; however, treatment with anti-IL-17 and anti-HMGB1
did not increase the expression levels of miR-138, thus suggesting
that miR-138 was not downstream of IL-17 or HMGB1.
To illustrate the underlying mechanism in the
regulation of HMGB1, a bioinformatics analysis was conducted using
available online databases. The results demonstrated that a
potential binding site for miR-381 was present in the 3′UTR of
HMGB1; this was confirmed by a dual luciferase assay. These
findings suggested that HMGB1 may be a downstream target of
miR-381; therefore, the association between HMGB1 and miR-381 was
determined. The results demonstrated that transfection with miR-381
mimics inhibited the migration of macrophages, and that the
expression levels of HMGB1, IL-17 and ICAM-1 were decreased.
Conversely, miR-381 inhibitors exerted the opposite effects
compared with miR-381 mimics, thus suggesting that miR-381 may
reduce the inflammatory responses in EAM mice. Notably,
transfection with si-HMGB1 suppressed the effects of miR-381
inhibitors. Taken together, these findings suggested that HMGB1 may
be a downstream target of miR-381, and that miR-381 may inhibit
inflammation and macrophage infiltration via downregulating the
expression of HMGB1.
In conclusion, the present study indicated that
miR-381 downregulated HMGB1, so as to reduce inflammation and
macrophage infiltration and the expression of IL-17/ICAM-1. The
present study provided a novel molecular clue to aid understanding
of the progression of PM and a potential target to combat PM.
Acknowledgments
Not applicable.
References
|
1
|
Maoz CR, Langevitz P, Livneh A, Blumstein
Z, Sadeh M, Bank I, Gur H and Ehrenfeld M: High incidence of
malignancies in patients with dermatomyositis and polymyositis: An
11-year analysis. Semin Arthritis Rheum. 27:319–324. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang J, Guo G, Chen G, Wu B, Lu L and Bao
L: Meta-analysis of the association of dermatomyositis and
polymyositis with cancer. Br J Dermatol. 169:838–847. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hill CL, Zhang Y, Sigurgeirsson B, Pukkala
E, Mellemkjaer L, Airio A, Evans SR and Felson DT: Frequency of
specific cancer types in dermatomyositis and polymyositis: A
population-based study. Lancet. 357:96–100. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rayavarapu S, Coley W and Nagaraju K: An
update on pathogenic mechanisms of inflammatory myopathies. Curr
Opin Rheumatol. 23:579–584. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fasth AE, Dastmalchi M, Rahbar A,
Salomonsson S, Pandya JM, Lindroos E, Nennesmo I, Malmberg KJ,
Soderberg-Naucler C, Trollmo C, et al: T cell infiltrates in the
muscles of patients with dermatomyositis and polymyositis are
dominated by CD28null T cells. Journal of immunology (Baltimore, Md
: 1950). 183:4792–4799. 2009. View Article : Google Scholar
|
|
6
|
Henriques-Pons A and Nagaraju K: Nonimmune
mechanisms of muscle damage in myositis: Role of the endoplasmic
reticulum stress response and autophagy in the disease
pathogenesis. Curr Opin Rheumatol. 21:581–587. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Salajegheh M, Kong SW, Pinkus JL, Walsh
RJ, Liao A, Nazareno R, Amato AA, Krastins B, Morehouse C, Higgs
BW, et al: Interferon-stimulated gene 15 (ISG15) conjugates
proteins in dermatomyositis muscle with perifascicular atrophy. Ann
Neurol. 67:53–63. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Walsh RJ, Kong SW, Yao Y, Jallal B, Kiener
PA, Pinkus JL, Beggs AH, Amato AA and Greenberg SA: Type I
interferon-inducible gene expression in blood is present and
reflects disease activity in dermatomyositis and polymyositis.
Arthritis Rheum. 56:3784–3792. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wenzel J, Scheler M, Bieber T and Tüting
T: Evidence for a role of type I interferons in the pathogenesis of
dermatomyositis. Br J Dermatol. 153:462–463; author reply 463–464.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Scovell WM: High mobility group protein 1:
A collaborator in nucleosome dynamics and estrogen-responsive gene
expression. World J Biol Chem. 7:206–222. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Musumeci D, Roviello GN and Montesarchio
D: An overview on HMGB1 inhibitors as potential therapeutic agents
in HMGB1-related pathologies. Pharmacol Ther. 141:347–357. 2014.
View Article : Google Scholar
|
|
12
|
Yang Q, Liu X, Yao Z, Mao S, Wei Q and
Chang Y: Penehyclidine hydrochloride inhibits the release of
high-mobility group box 1 in lipopolysaccharide-activated RAW264.7
cells and cecal ligation and puncture-induced septic mice. J Surg
Res. 186:310–317. 2014. View Article : Google Scholar
|
|
13
|
Harris HE, Andersson U and Pisetsky DS:
HMGB1: A multifunctional alarmin driving autoimmune and
inflammatory disease. Nat Rev Rheumatol. 8:195–202. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lotze MT and Tracey KJ: High-mobility
group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal.
Nat Rev Immunol. 5:331–342. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pisetsky DS, Erlandsson-Harris H and
Andersson U: High-mobility group box protein 1 (HMGB1): An alarmin
mediating the pathogenesis of rheumatic disease. Arthritis Res
Ther. 10:2092008. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Müller S, Scaffidi P, Degryse B, Bonaldi
T, Ronfani L, Agresti A, Beltrame M and Bianchi ME: New EMBO
members' review: The double life of HMGB1 chromatin protein:
architectural factor and extracellular signal. EMBO J.
20:4337–4340. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moestrup SK and Møller HJ: CD163: A
regulated hemoglobin scavenger receptor with a role in the
anti-inflammatory response. Ann Med. 36:347–354. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Niimoto T, Nakasa T, Ishikawa M, Okuhara
A, Izumi B, Deie M, Suzuki O, Adachi N and Ochi M: MicroRNA-146a
expresses in interleukin-17 producing T cells in rheumatoid
arthritis patients. BMC Musculoskelet Disord. 11:2092010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hirota K, Hashimoto M, Yoshitomi H, Tanaka
S, Nomura T, Yamaguchi T, Iwakura Y, Sakaguchi N and Sakaguchi S: T
cell self-reactivity forms a cytokine milieu for spontaneous
development of IL-17+ Th cells that cause autoimmune
arthritis. J Exp Med. 204:41–47. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee YK, Mukasa R, Hatton RD and Weaver CT:
Developmental plasticity of Th17 and Treg cells. Curr Opin Immunol.
21:274–280. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Crispin JC, Oukka M, Bayliss G, Cohen RA,
Van Beek CA, Stillman IE, Kyttaris VC, Juang YT and Tsokos GC:
Expanded double negative T cells in patients with systemic lupus
erythe-matosus produce IL-17 and infiltrate the kidneys. J Immunol.
181:8761–8766. 2008. View Article : Google Scholar
|
|
22
|
Pickens SR, Volin MV, Mandelin AM 2nd,
Kolls JK, Pope RM and Shahrara S: IL-17 contributes to angiogenesis
in rheumatoid arthritis. J Immunol. 184:3233–3241. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Langrish CL, Chen Y, Blumenschein WM,
Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA and
Cua DJ: IL-23 drives a pathogenic T cell population that induces
autoimmune inflammation. J Exp Med. 201:233–240. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tzartos JS, Friese MA, Craner MJ, Palace
J, Newcombe J, Esiri MM and Fugger L: Interleukin-17 production in
central nervous system-infiltrating T cells and glial cells is
associated with active disease in multiple sclerosis. Am J Pathol.
172:146–155. 2008. View Article : Google Scholar :
|
|
25
|
Gao Q, Jiang Y, Ma T, Zhu F, Gao F, Zhang
P, Guo C, Wang Q, Wang X, Ma C, et al: A critical function of Th17
proinflam-matory cells in the development of atherosclerotic plaque
in mice. J Immunol. 185:5820–5827. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Smith E, Prasad KM, Butcher M, Dobrian A,
Kolls JK, Ley K and Galkina E: Blockade of interleukin-17A results
in reduced atherosclerosis in apolipoprotein E-deficient mice.
Circulation. 121:1746–1755. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Othumpangat S, Noti JD, McMillen CM and
Beezhold DH: ICAM-1 regulates the survival of influenza virus in
lung epithelial cells during the early stages of infection.
Virology. 487:85–94. 2016. View Article : Google Scholar :
|
|
28
|
Gabr MA, Jing L, Helbling AR, Sinclair SM,
Allen KD, Shamji MF, Richardson WJ, Fitch RD, Setton LA and Chen J:
Interleukin-17 synergizes with IFNgamma or TNFalpha to promote
inflammatory mediator release and intercellular adhesion molecule-1
(ICAM-1) expression in human interver-tebral disc cells. J Orthop
Res. 29:1–7. 2011. View Article : Google Scholar
|
|
29
|
Plaisance-Bonstaff K and Renne R: Viral
miRNAs. Methods Mol Biol. 721:43–66. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang J, Zhang SY, Gao YM, Liu YF, Liu YB,
Zhao ZG and Yang K: MicroRNAs as oncogenes or tumour suppressors in
oesophageal cancer: Potential biomarkers and therapeutic targets.
Cell Prolif. 47:277–286. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li J, You T and Jing J: MiR-125b inhibits
cell biological progression of Ewing's sarcoma by suppressing the
PI3K/Akt signalling pathway. Cell Prolif. 47:152–160. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li M, Yu M, Liu C, Zhu H, He X, Peng S and
Hua J: miR-34c works downstream of p53 leading to dairy goat male
germline stem-cell (mGSCs) apoptosis. Cell Prolif. 46:223–231.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li Z, Yu X, Shen J and Jiang Y: MicroRNA
dysregulation in uveal melanoma: A new player enters the game.
Oncotarget. 6:4562–4568. 2015.PubMed/NCBI
|
|
34
|
Xu Z, Asahchop EL, Branton WG, Gelman BB,
Power C and Hobman TC: MicroRNAs upregulated during HIV infection
target peroxisome biogenesis factors: Implications for virus
biology, disease mechanisms and neuropathology. PLoS Pathog.
13:e10063602017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cao Q, Liu F, Ji K, Liu N, He Y, Zhang W
and Wang L: MicroRNA-381 inhibits the metastasis of gastric cancer
by targeting TMEM16A expression. J Exp Clin Cancer Res. 36:292017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hou C, Meng F, Zhang Z, Kang Y, Chen W,
Huang G, Fu M, Sheng P, Zhang Z and Liao W: The role of
MicroRNA-381 in chondrogenesis and interleukin-1-beta induced
chondrocyte responses. Cell Physiol Biochem. 36:1753–1766. 2015.
View Article : Google Scholar
|
|
37
|
Zhang X, Goncalves R and Mosser DM: The
isolation and characterization of murine macrophages. Curr Protoc
Immunol Chapter 14. Unit. 14:12008.
|
|
38
|
Arahata K and Engel AG: Monoclonal
antibody analysis of mono-nuclear cells in myopathies. I:
Quantitation of subsets according to diagnosis and sites of
accumulation and demonstration and counts of muscle fibers invaded
by T cells. Ann Neurol. 16:193–208. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Engel AG and Arahata K: Monoclonal
antibody analysis of mononuclear cells in myopathies. II:
Phenotypes of autoinvasive cells in polymyositis and inclusion body
myositis. Ann Neurol. 16:209–215. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Goebels N, Michaelis D, Engelhardt M,
Huber S, Bender A, Pongratz D, Johnson MA, Wekerle H, Tschopp J,
Jenne D, et al: Differential expression of perforin in
muscle-infiltrating T cells in polymyositis and dermatomyositis. J
Clin Invest. 97:2905–2910. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zong M and Lundberg IE: Pathogenesis,
classification and treatment of inflammatory myopathies. Nat Rev
Rheumatol. 7:297–306. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Notarnicola A, Lapadula G, Natuzzi D,
Lundberg IE and Iannone F: Correlation between serum levels of
IL-15 and IL-17 in patients with idiopathic inflammatory
myopathies. Scand J Rheumatol. 44:224–228. 2015. View Article : Google Scholar
|
|
43
|
Yin Y, Li F, Shi J, Li S, Cai J and Jiang
Y: MiR-146a regulates inflammatory infiltration by macrophages in
polymyositis/derma-tomyositis by targeting TRAF6 and affecting
IL-17/ICAM-1 pathway. Cell Physiol Biochem. 40:486–498. 2016.
View Article : Google Scholar
|
|
44
|
Miossec P: Interleukin-17 in fashion, at
last: Ten years after its description, its cellular source has been
identified. Arthritis Rheum. 56:2111–2115. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kenna TJ and Brown MA: The role of
IL-17-secreting mast cells in inflammatory joint disease. Nat Rev
Rheumatol. 9:375–379. 2013. View Article : Google Scholar
|
|
46
|
Baeten DL and Kuchroo VK: How Cytokine
networks fuel inflammation: Interleukin-17 and a tale of two
autoimmune diseases. Nat Med. 19:824–825. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sallum AM, Kiss MH, Silva CA, Wakamatsu A,
Vianna MA, Sachetti S and Marie SK: Difference in adhesion molecule
expression (ICAM-1 and VCAM-1) in juvenile and adult
dermatomyositis, polymyositis and inclusion body myositis.
Autoimmun Rev. 5:93–100. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Szodoray P, Alex P, Knowlton N, Centola M,
Dozmorov I, Csipo I, Nagy AT, Constantin T, Ponyi A, Nakken B, et
al: Idiopathic inflammatory myopathies, signified by distinctive
peripheral cytokines, chemokines and the TNF family members B-cell
activating factor and a proliferation inducing ligand. Rheumatology
(Oxford). 49:1867–1877. 2010. View Article : Google Scholar
|
|
49
|
Tournadre A, Porcherot M, Chérin P, Marie
I, Hachulla E and Miossec P: Th1 and Th17 balance in inflammatory
myopathies: Interaction with dendritic cells and possible link with
response to high-dose immunoglobulins. Cytokine. 46:297–301. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yin Y, Li F, Shi J, Li S, Cai J and Jiang
Y: MiR-146a regulates inflammatory infiltration by macrophages in
polymyositis/derma-tomyositis by targeting TRAF6 and affecting
IL-17/ICAM-1 Pathway. Cell Physiol Biochem. 40:486–498. 2016.
View Article : Google Scholar
|
|
51
|
Tang Q, Li J, Zhu H, Li P, Zou Z and Xiao
Y: Hmgb1-IL-23-IL-17-IL-6-Stat3 axis promotes tumor growth in
murine models of melanoma. Mediators Inflamm. 2013:7138592013.
View Article : Google Scholar
|