Introduction
The American Cancer Society estimates that 164,690
new cases of prostate cancer (PC) will be diagnosed in 2018. PC is
the third leading cause of cancer-associated mortality among
American men, following lung and colorectal cancer. It is estimated
that ~1/7 men are diagnosed with the disease during their lifetime,
with 1/39 succumbing to the disease (1).
The most common treatments for PC include radiation
therapy, chemotherapy and hormone therapy. Radiation therapy is not
always ideal as healthy living cells are damaged by the treatment,
causing numerous side effects in patients. Hormone therapy requires
surgical castration and the use of anti-androgens, which also have
side effects and affect the patient’s lifestyle (2). In addition to the side effects that
follow hormone therapy, patients often become castration-resistant,
meaning the cancer is no longer affected by the treatment (2). Once this occurs, chemotherapeutics,
including docetaxel, are used. Docetaxel only increases a patient’s
survival rate by ~3 months. This is largely due to resistance that
develops within the cancer cells (3).
The protein kinase C (PKC) family of isozymes
transduce signals and control other proteins through
phosphorylation (4). The PKC
family comprises 14 known isozymes, which are found in varying
ratios in the cytoplasmic and membrane fraction of cells depending
on the type of tissue and its physiological state (5). PKC isozymes can be classified into
three groups. Group I includes Ca2+-dependent isozymes:
cPKC-α, cPKC-βl, cPKC-βll, and cPKC-γ. Isozymes in Group II
(nPKC-δ, nPKC-ε, nPKC-η and PKC-θ) are Ca2+-independent.
Group III includes the atypical (a)PKCs, including aPKC-ι (6), aPKC-ζ, aPKC-ζll (7) aPKC-µ (protein kinase D) and aPKC-ν
(8), which are insensitive to
diacylglycerol and calcium and do not bind to or become activated
by phorbol esters. PKC regulates cellular functions, metabolism and
proliferation by phosphorylating proteins in response to
transmembrane signals from hormones, growth factors,
neuro-transmitters, and pharmacological agents. The activation of
PKC by various agonists, including radiation, results in the
altered transcription of a considerable number of genes. PKCs are
involved in the day-to-day functioning of normal cells, however,
they can cause several adverse effects when disrupted. PKC is the
major receptor for tumor-promoting phorbol esters, however, the
extent to which PKC is involved in cellular malignancy remains to
be fully elucidated. Various studies have indicated that increased
tumorigenicity results from either the dysregulation of PKC
activity, changes in PKC concentration, or both (9-13).
PKC isozymes have been implicated in carcinogenesis,
however, investigation of the functional significance of these
enzymes in human cancer has been limited so far. PKC isozymes α, β,
δ, ε, γ, η, ζ, ι, and µ are expressed in the normal and cancerous
prostate (14). LNCaP cells are a
widely used model for investigation of PC. Besides classical PKC-α,
the novel PKC-δ is overexpressed in LNCaP cells and provokes
phorbol ester-induced apoptosis. In PC-3 and DU-145 cells, PKC-δ is
involved in the cell motility and invasion of prostate tumor cells
(15,16). In addition, PKC-ε alongside Akt
promotes matrix adhesion-containing actin-filaments and β-intergins
in recurrent PC cells (15). In
DU-145 cells, the downregulation of PKC-ε prevents apoptosis
(16). Therefore, PKC-ε may be
targeted for prostate therapy. Finally, the PKC-µ and PKC-ζ
mammalian target of rapamycin (mTOR)/p70 S6 kinase pathway is
associated with the progression of androgen- dependent PC to
androgen-independent PC (17-19).
Therefore, understanding PKC isoforms in PC may contribute to
possible novel diagnostic and therapeutic strategies.
PKCs is involved in tumor growth and formation when
the levels of PKC are markedly altered (5). PKC-ι is a member of the PKC family
located in chromosome 3 at 3q26.2, and is a human oncogene. PKCs
can be overexpressed in various types of cancer, including ovarian,
lung, head and neck cancer, and PC (20-22).
Elevated levels of PKC-ι have been correlated with poorer
prognosis. It was found that patients with lung cancer who had
elevated levels of PKC-ι during the early stages were 10 times more
likely to succumb to mortality from the disease, compared with
those who had low levels of PKC-ι. PKC-ι is also involved in
several oncogenic signaling path- ways (23). For these reasons, the in
vitro effects of two novel aPKC inhibitors,
5-amino-1-(1R,2S,3S,4R)-2,3-dihydroxy-4-methylcyclopentyl)-1H-imidazole-4-carboxamide
(ICA-1) and 2-acetyl-1,3-cyclopentanedione (ACPD), on the normal
RWPE-1 cell line, and the DU-145 and PC-3 PC cell lines, were
investigated in the present study. ICA-1 has been shown to target
PKC-ι, whereas ACPD has been shown to target PKC-ι and PKC-ζ
(24,25).
The nuclear factor (NF)-κB signaling pathway is
involved in cancer propagation and dissemination in several types
of cancer, however, its involvement in PC remains to be fully
elucidated. The inhibitor of NF-κB kinase (IKK) complex is
comprised of IKKα and IKKβ, both of which are necessary for the
activation of NF-κB. In the present study, it was hypothesized that
PKC-ι acts on IKKα/β, causing the release and translocation of
NF-κB. Inhibition of this pathway following treatment with ICA-1 is
expected allow normal apoptosis to take place with minimal effect
on RWPE-1 cells, but with more marked effects in DU-145 and PC-3
cells.
The results of the present study showed a
correlation between the presence of PKC-ι and PC. It also revealed
the efficacy of ACPD and ICA-1 on PKC-ι and indicated the role of
PKC-ι in the survival of PC. Cumulatively, the results led to the
conclusion that the detection of PKC-ι may be used as a biomarker
of prostate carcinogenesis and that PKC-ι inhibition may be an
alternative therapy in patients with PC.
Materials and methods
ICA-1 was synthesized by Therachem
Research Medilab (Jaipur, India) and ACPD was purchased from
Sigma-Aldrich; EMD Millipore (Billerica, MA, USA)
The inhibitors were dissolved in sterile distilled
water prior to use. Dulbecco’s phosphate-buffered saline without
Mg2+ and Ca2+ (DPBS) was purchased from the
American Type Culture Collection (Manassas, VA, USA).
Trypsin-ethylenediaminetetraacetic acid (EDTA) solution was
purchased from Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
Polyclonal primary antibodies were purchased from the following
companies: Anti-PKC-ι mouse monoclonal (cat. no. 610176) and B-cell
lymphoma 2 (Bcl-2; cat. no. 610538) from BD Transduction Laboratory
(Lexington, KY, USA). PKC-ζ (cat. no. sc-17781), NF-κB p65 (cat.
no. sc-372-G), inhibitor of NF-κBα (IκBα; cat. no. sc-1643),
phosphorylated (phospho) IκBα (cat. no. sc-8404) β-actin (cat. no.
sc-1616) goat polyclonal, PKC-α (cat. no. sc-8393) mouse
monoclonal, cytochrome c (cat. no. sc-13156), survivin (cat.
no. sc-17779) and caspase-3 (cat. no. sc-7272) from Santa Cruz
Biotechnology Co., Ltd. (Santa Cruz, CA, USA), phosphorylated
phosphatase and tensin homolog (PTEN; S380; cat. no. 9551),
phosphorylated AKT (S473; cat. no. 4059S), phosphorylated IKKα/β
(S176/180; cat. no. 2697), poly (ADP-ribose) polymerase (PARP; cat.
no. 9532) and cleaved-PARP (cat. no. 9185) from Cell Signaling
Technology Inc. (Danvers, MA, USA). β-catenin (cat. no. ab16051)
from Abcam (Cambridge, MA, USA). Secondary antibodies were
purchased from the following companies: Horseradish peroxidase
(HRP) goat x mouse IgG (cat. no. JGM035146), HRP goat x rabbit IgG
(cat. no. JGZ035144) from Accurate (Westbury, NY, USA); HRP bovine
anti-goat IgG (cat. no. sc-2350 from Santa Cruz Biotechnology,
Inc.. The RWPE-1 (ATCC® CRL-11609™) epithelial cells and
DU-145 (ATCC® HTB-81™) human prostate carcinoma cells
were purchased from the American Type Culture Collection. The PC-3
cells were acquired from Moffitt Cancer Center (Tampa, FL,
USA).
Prostate tissue analysis
The protein for western blot analysis was extracted
from human biopsy-derived benign prostate hyperplasia (BPH) tissues
obtained from the Cooperative Human Tissue Network (Southern
Division) at the University of Alabama (Birmingham, AL, USA). For
the purposes of the present study, BPH was defined as a
non-cancerous enlargement of the prostate gland. The BPH tissue
samples were obtained from men of varying ages (57-80 years old)
with a mean age of 67.6 years. Protein extraction from the
fresh-frozen radical prostatectomy samples of patients with PC were
obtained during surgery performed at the James A. Haley Veterans
Hospital (Tampa, FL, USA) between May and August 2007. All patients
provided informed consent as part of a clinical protocol approved
by the Institutional Review Board (IRB no. 103023) of the
University of South Florida (Tampa, FL, USA). The samples were
placed on ice immediately following prostatectomy and were frozen
in liquid nitrogen within 30-60 min post-prostatectomy. The
prostate tissues (0.5-1 g) were re-suspended and sonicated in 2 ml
homogenization buffer containing 50 mM HEPES (pH 7.5), 150 mM NaCl,
0.1% Tween-20, 1 mM ethylenediaminetetraacetic acid, 2 mM ethylene
glycol bis(β-aminoethyl ether)-N,N,N′,N′,-tetraacetic acid, 0.1 mM
orthovanadate, 1 mM NaF, 2 mM phenyl-methylsulfonly fluoride, 2.5
µg/ml leupeptin, 1 mM dithiothreitol and 0.15 U/ml
aprotinin. The suspension was sonicated for three 15 sec cycles on
ice. The prostate tissue suspensions or cell lysates were
centrifuged at 40,000 g for 30 min at 4°C to obtain cell extracts.
The proteins were quantified according to the Bradford method
(26). Tissue extracts containing
equal quantities of protein in each lane were run on 10% SDS-PAGE
gels according to Laemmli (27)
and the proteins were transblotted on nitrocellulose membrane
according to Towbin et al (28). The tissue lysates (100 µg)
were used for western blot analysis with antibodies against PKC-α
(10 µg, 1:500 dilution), PKC-ι (5 µg, 1:4,000
dilution) and β-actin (10 µg, 1:500 dilution) and incubated
for a duration of 1 h at room temperature. The secondary
antibodies, obtained from Accurate (cat. nos. JGM035146 and
JG2035744) were used at a dilution of 1:15,000 (1 µg) and
incubated for 2 h at room temperaure. The blots were subjected to
chemiluminescence and results were captured on X-ray film and
developed by machine. Observable bands were scanned and quantified
by densitometry analysis. The specimens used were from 10 patients
with PC (one core with adenocarcinoma from each patient), eight
patients with high grade prostate intraepithelial neoplasma (HGPIN;
1 core with HGPIN from each patient) and nine patients with BPH (1
core with from each patient), comprising a total of 27 samples.
Samples obtained from cell cultures and examined by western blot
analysis and were visualized digitally by the Protein Simple (San
Jose, CA, USA) FlourChem system and analyzed with the accompanying
alpha viewer software.
Immunohistochemical staining
For immunohistochemistry (IHC), de-identified,
archival, unstained sections of full-core prostate needle biopsies
(PNBs) were used, which were obtained for diagnostics in the
systematic PC screening program at the James A. Haley Veterans
Administration Medical Center. The PNB specimens examined were from
10 patients with PC (1 core with adenocarcinoma from each patient),
eight patients with HGPIN (1 core with HGPIN from each patient) and
nine patients with BPH (1 core with from each patient), comprising
a total of 27 samples. Following the deparaffinization of the
sections, and citrate microwave antigen retrieval, blocking was
performed. For the detection of PKC-α, the sections were incubated
with monoclonal anti-PKC-α antibody (1:100 dilution; BD
Transduction Laboratories) for 60 min at room temperature, followed
by washing and detection with the ‘EnVision’ detection system using
mouse IgG polymer and DAB chromogen. For the detection of PKC-ι,
separate sections were subsequently incubated overnight with
purified mouse monoclonal anti-PKC-ι (1:200 dilution; BD
Transduction Laboratories), followed by washing and a subsequent
30-min incubation at room temperature with 1:200 rat anti-mouse
IgG2b. Final detection was performed using DAB chromogen. All
sections were examined for PKC-α and PKC-ι and scored using the
Allred semi-quantitative scoring system (29). The Allred score is a composite of
the percentage of cells stained and the intensity of their
staining. The percentage of cells stained, termed the proportion
score, is classified between 0 and 5. The intensity of the cells
stained is termed the intensity score and it is rated as 1, 2 or 3;
thus, composite scores range between 0 and 8, with 0 being the
lowest and 8 being the maximum score. The adenocarcinoma glands,
glands with HGPIN, BPH glands, and stromal cells were scored
separately using a microscope (Olympus, Tokyo, Japan).
Cell culture
The cells were grown as a monolayer in a T25 tissue
culture flask with 5 ml of growth medium and were maintained in a
37°C incubator with 5% CO2. The E-MEM and F-12 growth
media were obtained from American Type Culture Collection. The
medium was supplemented with 10% fetal bovine serum (FBS) and a mix
of the antibiotics penicillin (10,000 IU) and streptomycin (10,000
µg/ml) in a 100X concentration purchased from Corning Life
Sciences (Tewksbury, MA, USA).
Cell proliferation assay
The RWPE-1, DU-145 and PC-3 cells were cultured in
T25 cell culture flasks with 20,000 cells seeded into each. The
flasks were treated with either ICA-1 or ACPD at doses of 1, 5, and
10 µM, in addition to an untreated control set for each. The
treatment was repeated over the course of 72 h and samples were
taken at 24-h intervals. The cells were trypsinized and placed into
a 1.5 ml microcentrifuge tube at intervals of 24, 48, and 72 h. The
tubes were inverted gently to maintain solution homogeneity and
cell numbers were measured using a Cell Scepter counter (EMD
Millipore).
4-[3-(4-iodophenyl)-2-(4-nitropheny))-2H-5-tetrazolio]-1,3- benzene
disulfonate (WST-1) assay for cell viability and cytotoxicity
The WST-1 assay (in vitro) was performed by
culturing ~4×103 cells/well (RWPE-1, DU-145 and PC-3
cells) in a 96-well plate. After 24 h, fresh media were supplied
(200 µl/well) and the cells were treated with either an
equal volume of sterile water (vehicle control) or with 10
µM of ICA-1 or 10 µM of ACPD, which was identified as
the optimal working concentration for the PC cell lines for the two
inhibitors. Additional doses were supplied every 24 h during a
3-day incubation period. At the end of day 3, media were removed
and fresh media (100 µl) were added with WST-1 reagent (10
µl) to each well. The absorbance was measured at 450 nm
every 1 h up to 8 h using the Synergy HT microplate reader from
Biotek Instruments, Inc., (Winooski, VT, USA). The detailed
procedure was performed as described by Ratnayake et al
(30).
Cell sub-fractionation
The DU-145 and PC-3 cells were cultured in a T25
cell culture flasks (20,000 cells seeded into each). The flasks
were treated with either ICA-1 or ACPD at a dose 10 µM, with
an untreated control set for each. The treatment was repeated over
the course of 72 h and samples were taken at 24-h intervals. At 30
min prior to harvesting, the cells were exposed to 10 nM of tumor
necrosis factor (TNF)-α. The NE-PER Nuclear and Cytoplasmic
Extraction kit from Thermo Fisher Scientific, Inc. was used to
harvest the proteins and separate the nuclear protein from the
cytoplasmic protein.
Flow cytometry for the analysis of
apoptosis
The DU-145 and PC-3 cells (40,000 cells/flask) were
cultured in a T25 cell culture flasks. The flasks were treated with
either ICA-1 or ACPD at a dose 10 µM, with an untreated
control set for each. After 24 h, the media were removed and dead
cells were collected by centrifugation using 350 × g for 4 min at
4°C. The cells were washed twice with ice-cold DPBS (1X) prior to
lifting with trypsin and trypsin was neutralized with an equal
volume of media. The cells were then combined with the collected
dead cells and washed twice with ice- cold DPBS (1X). A PE Annexin
V apoptosis detection kit (BD Biosciences, Pharmingen, San Jose,
CA, USA) was used to detect the apoptosis according to the detailed
protocol provided by BD Pharmingen. The BD Accuri™ C6 Plus personal
flow cytometer instrument was used to analyze the samples. Three
replicate experiments were performed.
Statistical analysis
All values are presented as the mean ± standard
deviation (n=3, where n is a single trial and each trial contained
9-24 replicates). Statistical analysis was performed with Student’s
t-test or one-way analysis of variance followed by Tukey’s multiple
comparison tests using the ‘VassarStats’ web tool (vassarstats.net) for statistical analysis. P≤0.05 was
considered to indicate a statistically significant difference.
Results
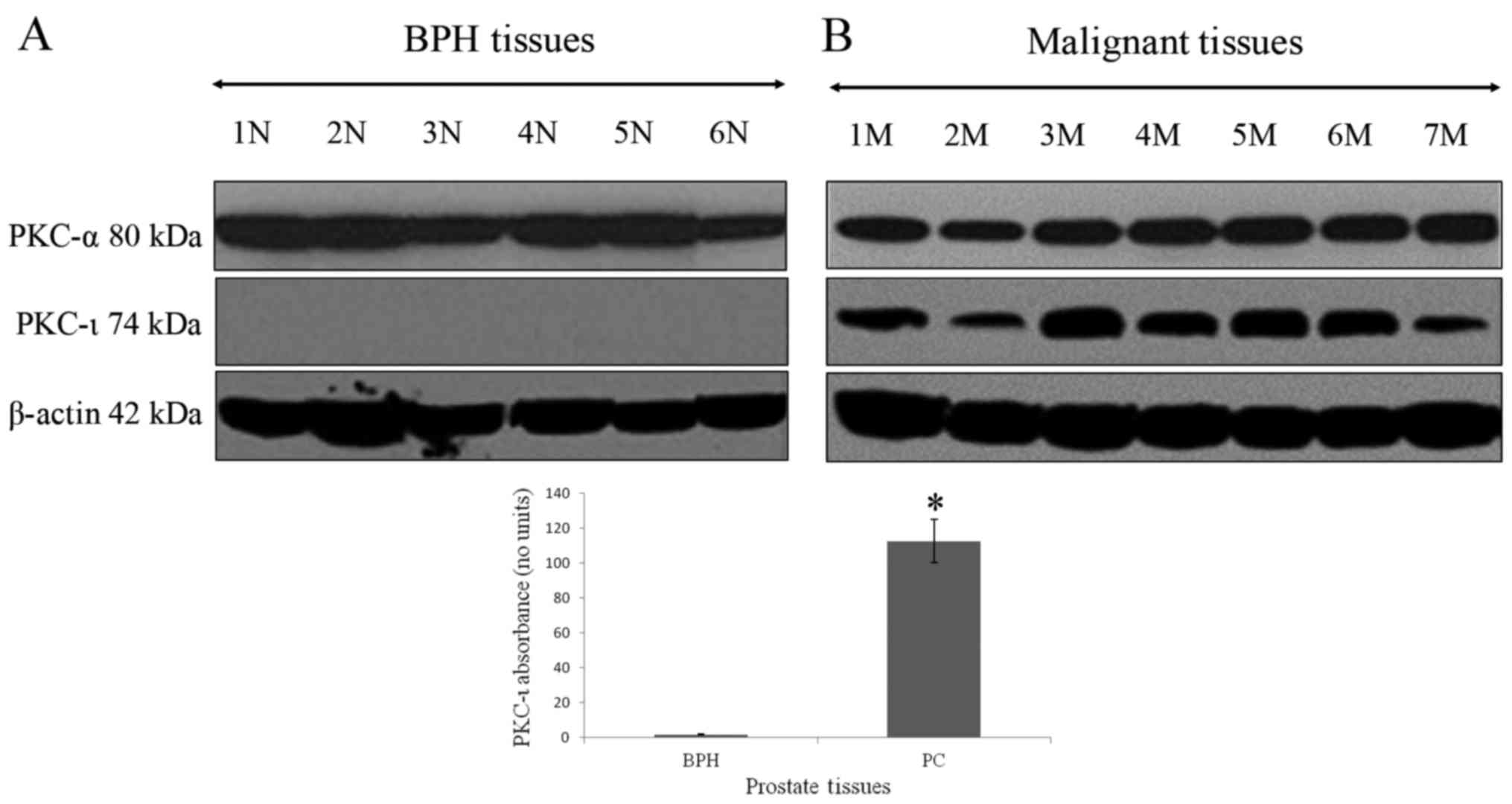
The association between the absence of PKC-ι in BPH
tissue and its robust presence in PC is shown in Fig. 1. PKC-ι was abundant in all PC
tissues. PKC-α (positive control PKC) and PKC-ι were identified in
western blot analysis by bands with molecular weights of 80 and 74
kDa, respectively, which corresponded to the immunoreactive signal
obtained from U-373MG glioma cells, which contain PKC-ι and PKC-α
(data not shown). The western blot controls for PKC-α did not show
a pattern of expression specific to BPH, or malignant prostate
tumors. β-actin was used as the internal control to ensure equal
quantities of protein loaded in to each well of the SDS-PAGE gel. A
100-fold increase in PKC-ι immunoreactivity was detected in PC
tissue when compared with BPH tissue (Fig. 1A and B). The level of PKC-ι in the
BPH tissue differed significantly from that in the PC tissue
(P=0.00048). This demonstrated that PKC-ι was significantly
overexpressed in PC tissues compared with BPH tissues.
Subsequently, IHC was performed to investigate
comparatively the tissue distribution and intracellular
localization of PKC-ι and PKC-α. The IHC data showed that PKC-α was
expressed in the stromal cells but showed minimal to no expression
in the majority of the BPH (Fig.
2A), HGPIN (Fig. 2B) or PC
(Fig. 2C) glands. In only one case
did the PC glands show moderate expression of PKC-α. In that case
the PC glands expressing PKC-α were Gleason pattern 4 (Table I), which may explain the increase
of PKC-α.
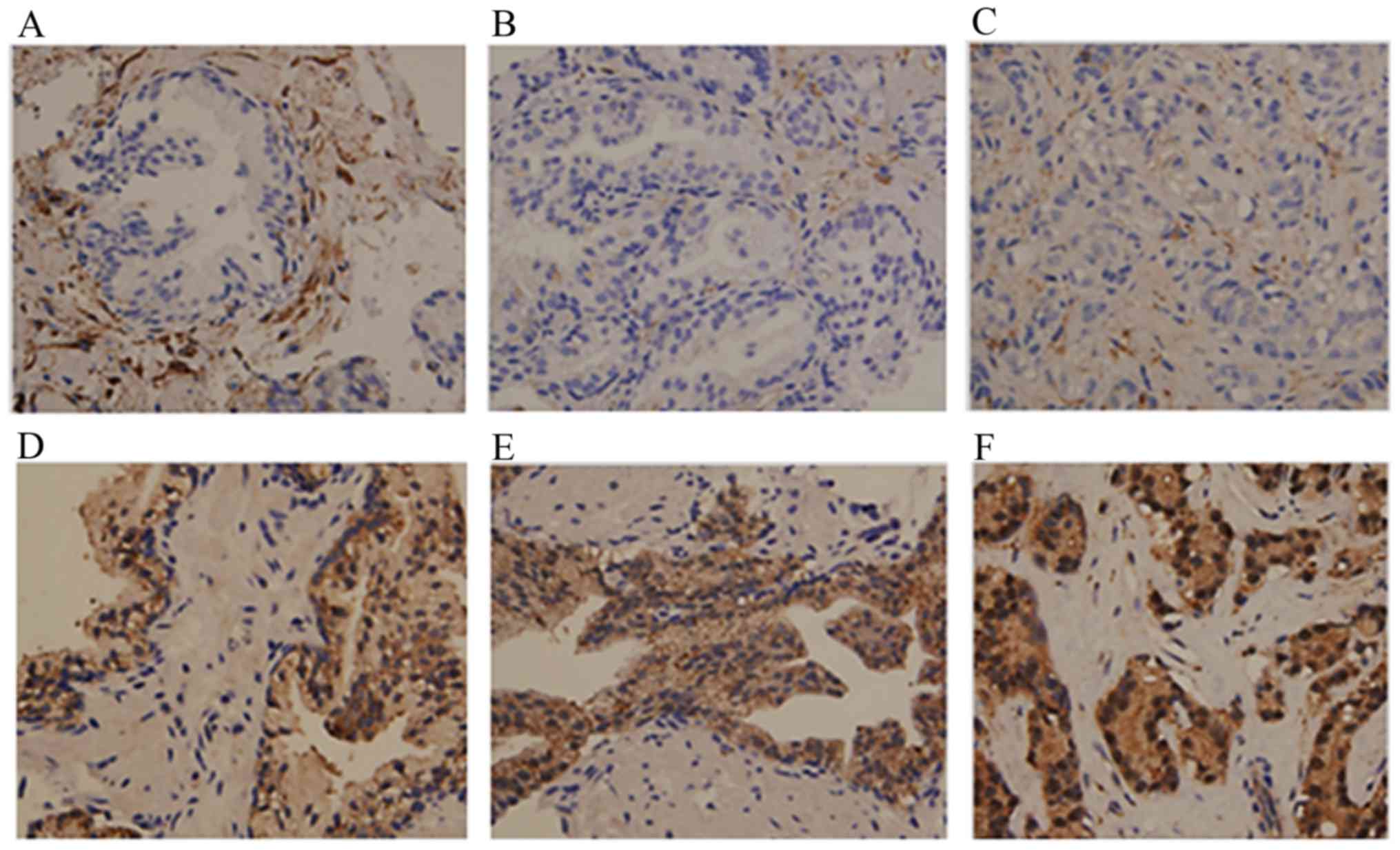 | Figure 2Immunohistochemical staining of PKC-α
and PKC-ι in BPH, HGPIN and PC. Tissue was stained with PKC-α
antibody as a control for PKC-ι staining. Results show PKC-ι
staining in (A) BPH, (B) HGPIN, and (C) PC tissue. PKC-α staining
is shown in (D) BPH glands, (E) glands with HGPIN, and (F) PC
glands. Tissues examined comprised BPH (n=9), HGPIN (n=8) and PC
tissues (n=10). Magnification for all micrographs was x400. Three
experiments were performed in triplicate. BPH, benign prostate
hyperplasia; HGPIN, high grade prostate intraepithelial neoplasma;
PC, prostate adenocarcinoma; PKC, protein kinase C. |
 | Table IPKC-α and PKC-ι staining of glands
and stroma of patients with BPH, HGPIN and prostate
adenocarcinoma. |
Table I
PKC-α and PKC-ι staining of glands
and stroma of patients with BPH, HGPIN and prostate
adenocarcinoma.
| Total cases | PKC-α | Staining (No. of
cases)a
|
|---|
| 8+ | 7+ | 6+ | 5+ | 4+ | 3+ | 2+ | 1+ | 0 |
|---|
| BPH | Glands | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 4 |
| 9 | Stroma | 0 | 1 | 2 | 5 | 1 | 0 | 0 | 0 | 0 |
| HGPIN | Glands | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 5 |
| 8 | Stroma | 0 | 1 | 3 | 4 | 0 | 0 | 0 | 0 | 0 |
| Adenocarcinoma | Glands | 0 | 0 | 1 | 0 | 1 | 0 | 4 | 0 | 4 |
| 10 | Stroma | 0 | 1 | 4 | 2 | 1 | 0 | 2 | 0 | 0 |
|
| Total cases | PKC-ι | Staining (No. of
cases)a
|
| 8+ | 7+ | 6+ | 5+ | 4+ | 3+ | 2+ | 1+ | 0 |
|
| BPH | Glands | 0 | 2 | 7 | 0 | 0 | 0 | 0 | 0 | 0 |
| 9 | Stroma | 0 | 0 | 0 | 2 | 4 | 2 | 1 | 0 | 0 |
| HGPIN | Glands | 1 | 3 | 3 | 1 | 0 | 0 | 0 | 0 | 0 |
| 8 | Stroma | 0 | 0 | 0 | 0 | 3 | 4 | 1 | 0 | 0 |
| Adenocarcinoma | Glands | 9 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 10 | Stroma | 0 | 0 | 0 | 3 | 5 | 2 | 0 | 0 | 0 |
By contrast, the expression of PKC-ι was noted in
all glands, with a proportion score of 5 in the majority of cases
(Table I and Fig. 2D-F), however, the intensity of
staining was more marked in the PC glands with Allred scores of +8
and +7 (Table I and Fig. 2F), compared with that in the BPH
glands (Fig. 2D) and glands with
HGPIN (Table I and Fig. 2E). The BPH glands showed similar
weak staining in samples obtained from patients with and without
prostatic adenocarcinoma. The stromal cells showed weak to moderate
staining in samples with and without adenocarcinoma. PKC-ι was
present at low levels in a comparative number of the BPH samples,
however, the intensity was markedly increased in the adenocarcinoma
samples, resulting in higher composite scores. Overall, the IHC
data from the PNB specimens confirmed the higher expression of
PKC-ι in PC detected in the western blot analysis from fresh frozen
excisional clinical prostate tissue specimens.
To establish whether ACPD or ICA-1 affected PKC-ι
and PKC-ζ, their protein levels were determined by western blot
analysis. In the malignant cells (DU-145 and PC-3), there was an
abundance of PKC-ι (Fig. 3)
compared with the normal cells (RWPE-1). There was a marginal
difference in the RWPE cells, however, neither drug reduced the
levels of PKC-ι significantly (P=0.077). The effects were more
marked in the DU-145 cells. ICA-1 inhibited PKC-ι and, following 3
days of treatment, there was a decrease of 64% (P=0.01) in PKC-ι.
Following 3 days of treatment with ACPD, which inhibits PKC-ι and
PKC-ζ, there was an average decrease of 51% (P=0.018) in PKC-ι. In
the PC-3 cells, there was a 40% (P=0.028) decrease in PKC-ι
following treatment with ICA-1 and a 37% (P=0.036) decrease in
PKC-ι following treatment with ACPD. ICA-1 had no significant
effect on the levels of PKC-ζ in any of the cell lines, whereas
ACPD had a significant effect, reducing the levels of PKC-ζ by 23%
(P≤0.05), in the PC-3 cells only.
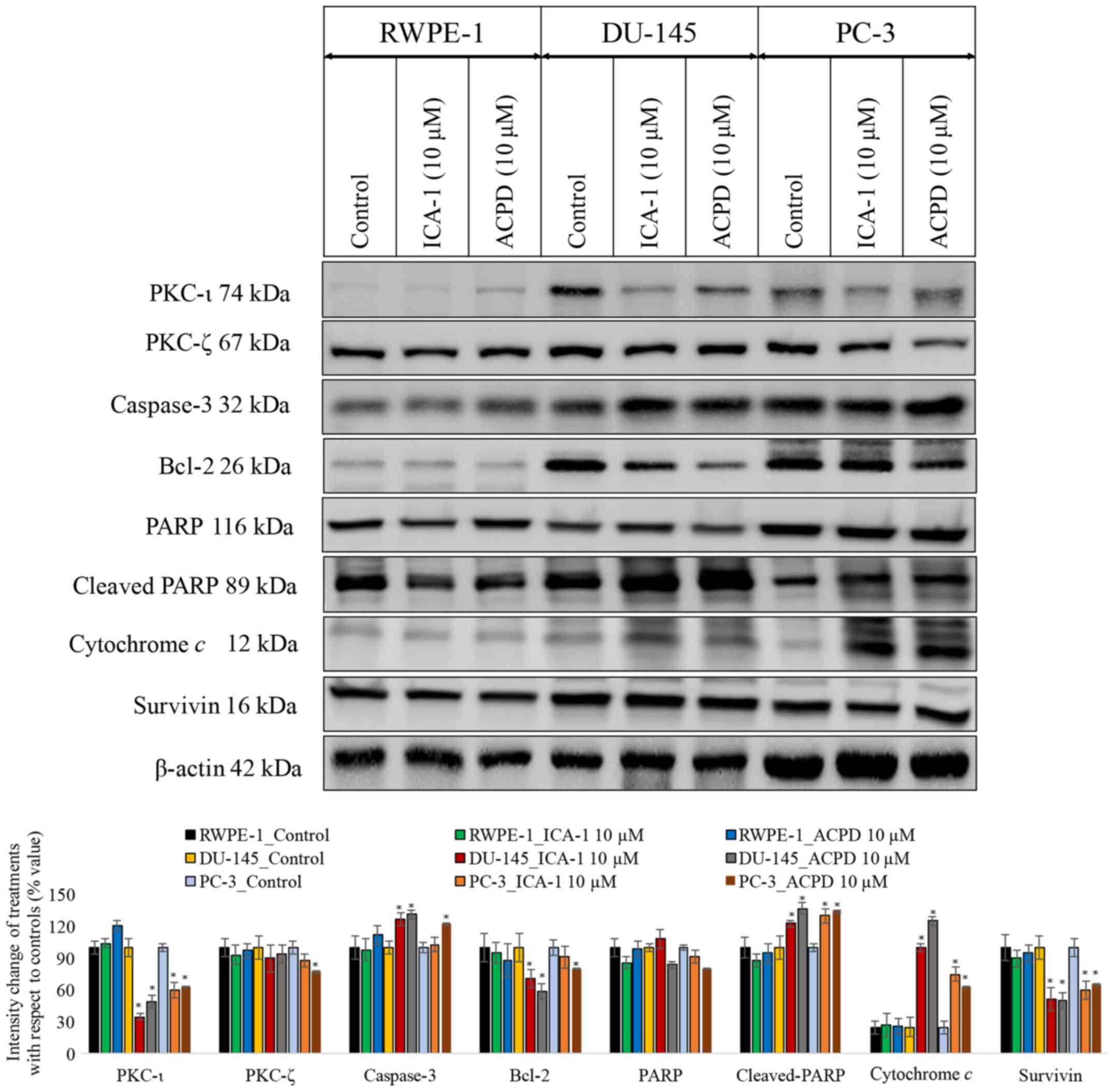 | Figure 3Effects on PKC-ι regulation and
apoptosis. Effects on PKC-ι and apoptosis were examined following
inhibition of PKC-ι with either ICA-1 (10 µM) or ACPD (10 µM) for
72 h with respect to their controls. Protein expression of PKC-ι,
PKC-ζ, caspase 3, Bcl-2, PARP, cytochrome c, and survivin in
the RWPE-1 cell line and the DU-145 and PC-3 malignant cell lines.
A total of 35 µg protein was loaded into each well and results were
normalized by β-actin. Three trials were performed in triplicate.
Densitometry bar graphs show the percentage change of the treated
sample with respect to their controls (mean ± standard devia-
tion). *P≤0.05, vs. control. PKC, protein kinase C;
Bcl-2, B-cell lymphoma 2; PARP, poly (ADP-ribose); ACPD,
polymerase; 2-acetyl-1,3-cyclopentanedione; ICA-1,
5-amino-1-(1R,2S,3S,4R)-2,3-dihydroxy-4-methylcyclopentyl)-1H-imidazole-4-carboxamide. |
To determine whether ICA-1 or ACPD induced
apoptosis, the apoptotic markers caspase-3, Bcl-2, PARP, cleaved
PARP, survivin and cytochrome c were used to detect ongoing
apoptosis in the cell lines (Fig.
3). In all three cell lines treated with ICA-1 and ACPD, there
was a measurable increase in apoptosis. Following ICA-1 treatment
for 72 h, the RWPE-1 control line showed no significant change in
either cytochrome c or survivin. Following treatment with
ACPD for 72 h, the RWPE-1 cells showed no significant change in
cytochrome c or in survivin. By contrast, the DU-145 cell
line showed a more substantial change, with an almost 3-fold
increase in cytochrome c and a reduction in survivin by 50%
following treatment of ICA-1. Following treatment with ACPD for 72,
the DU-145 cells showed ~400% (P<0.001) increase in cytochrome
c and a 50% (P=0.015) decrease in survivin. In the PC-3
cells treated with ICA-1, there was a 2-fold increase in cytochrome
c (P=0.011) and a 40% decrease in survivin (P=0.028). The
PC-3 cells treated with ACPD showed a 150% (P=0.013) increase in
cytochrome c and a 35% decrease in survivin (P=0.042). The
levels of caspase-3 and cleaved-PARP were significantly increased
and those of Bcl-2 and total PARP were significantly reduced upon
inhibitor treatment in the PC cell lines. All cell lines were also
probed with β-actin to ensure equal loading between lanes.
The effects of ICA-1 (10 µM) and ACPD (10
µM) on cell proliferation were also determined (Fig. 4A-C). There was a noticeable but
statistically insignificant effect of the inhibitors ICA-1 and ACPD
on the RWPE-1 cell line. Both inhibitors retained >90% of the
cell population present in the untreated RWPE-1 cells (Fig. 4A). By contrast, there was a
substantial decrease in the population of DU-145 cells treated with
the inhibitors ICA-1 (P≤0.01) and ACPD (P≤0.01) (Fig. 4B). The PC-3 cells showed a similar
trend of population decline following treatment with ICA-1 (P=0.01)
and ACPD (P=0.0052) (Fig. 4C).
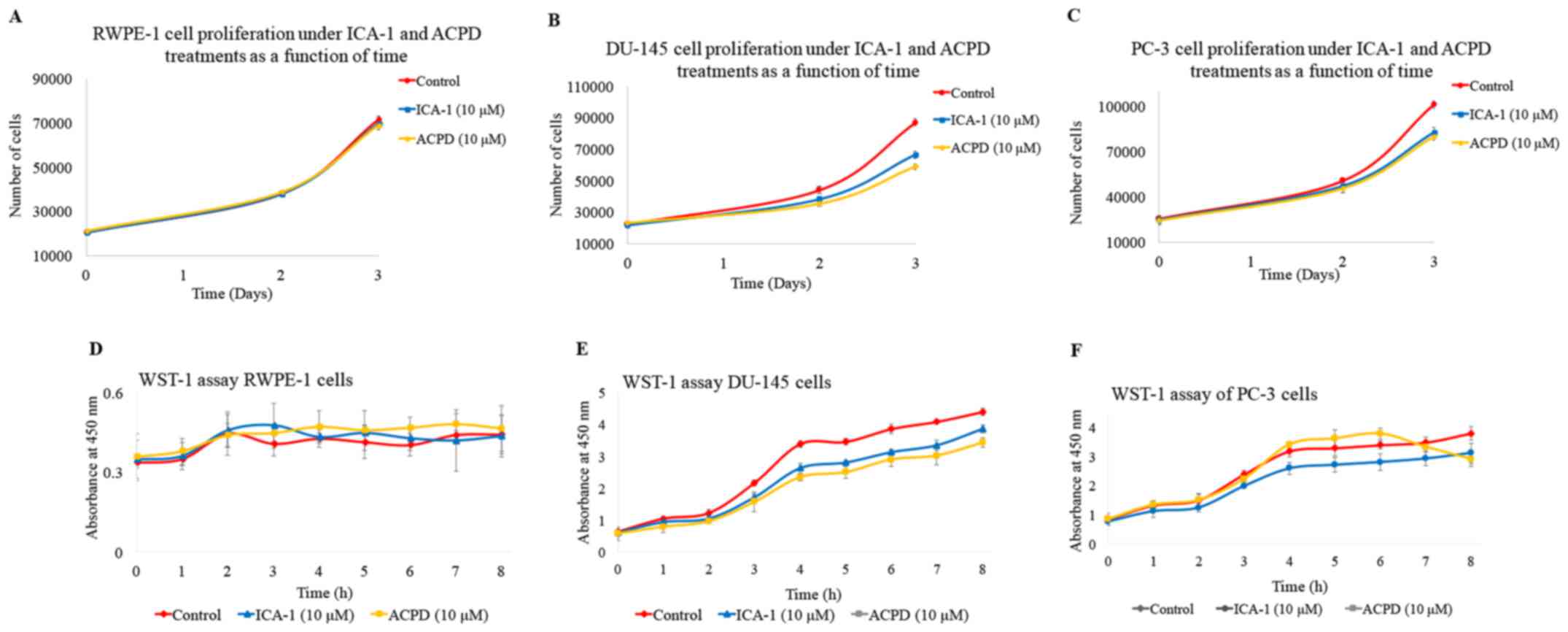 | Figure 4Effects of ICA-1 or ACPD on cell
populations in a 72-h period. Populations of cells under normal
conditions (red) and under treatment with either 10 µM ICA-1 (blue)
or 10 µM ACPD (yellow) in (A) RWPE-1, (B) DU-145, and (C) PC-3 cell
lines. Effects of ICA-1 and ACPD on in vitro cytotoxicity
over 8 h were measured by absorbance in nm under normal conditions
(red) and under the effects of either 10 µM ICA-1 (blue) or 10 µM
ACPD (yellow), in (D) RWPE-1, (E) DU-145, and (F) PC-3 cell lines.
There were 24 replicates per sample performed over three trials.
ACPD, polymerase; 2-acetyl-1,3-cyclopentanedione; ICA-1,
5-amino-1-(1R,2S,3S,4R)-2,3-dihydroxy-4-methylcyclopentyl)-1H-imidazole-4-carboxamide. |
To determine the in vitro cytotoxicity of
ICA-1 and ACPD on normal and malignant cell lines, a WST-1 assay
was performed. ICA-1 and ACPD demonstrated no significant
cytotoxicity towards the normal prostate epithelial RWPE-1 cell
line (Fig. 4D). As shown in
Fig. 4E and F, ICA-1 exhibited
significant cytotoxicity (P≤0.05) on the two metastatic prostate
cancer cell lines (DU-145 and PC-3). These results indicated that
ICA-1 and ACPD appeared to be cytostatic more than cytotoxic in the
time range assessed, thereby retarding PC cell growth and
proliferation.
As shown in Fig. 5,
flow cytometric analysis of apoptosis showed that PC-3 viability
decreased from 91.8% (Fig. 5A) to
60.8% following ICA-1 treatment (Fig.
5B) and 60.9% following ACPD treatment (Fig. 5C). Early apoptosis increased from
6.8% (Fig. 5A) to 37.3 and 36.3%
following ICA1 and ACPD treatments, respectively. By contrast, the
DU-145 cells showed a marginally lower response, in which viability
decreased from 97.9% (Fig. 5D) to
89% following ICA-1 treatment (Fig.
5E) and 85.5% following ACPD treatment (Fig. 5F). Early apoptosis increased from
0.9% (Fig. 5D) to 3.4 and 2.6%
following ICA-1 and ACPD treatments, respectively (Fig. 5E and F). The late stage of
apoptosis significantly increased from 1.2% (Fig. 5D) to 7.6 and 12% for ICA-1 and ACPD
treatments, respectively, in the DU-145 cell line (Fig. 5E and F).
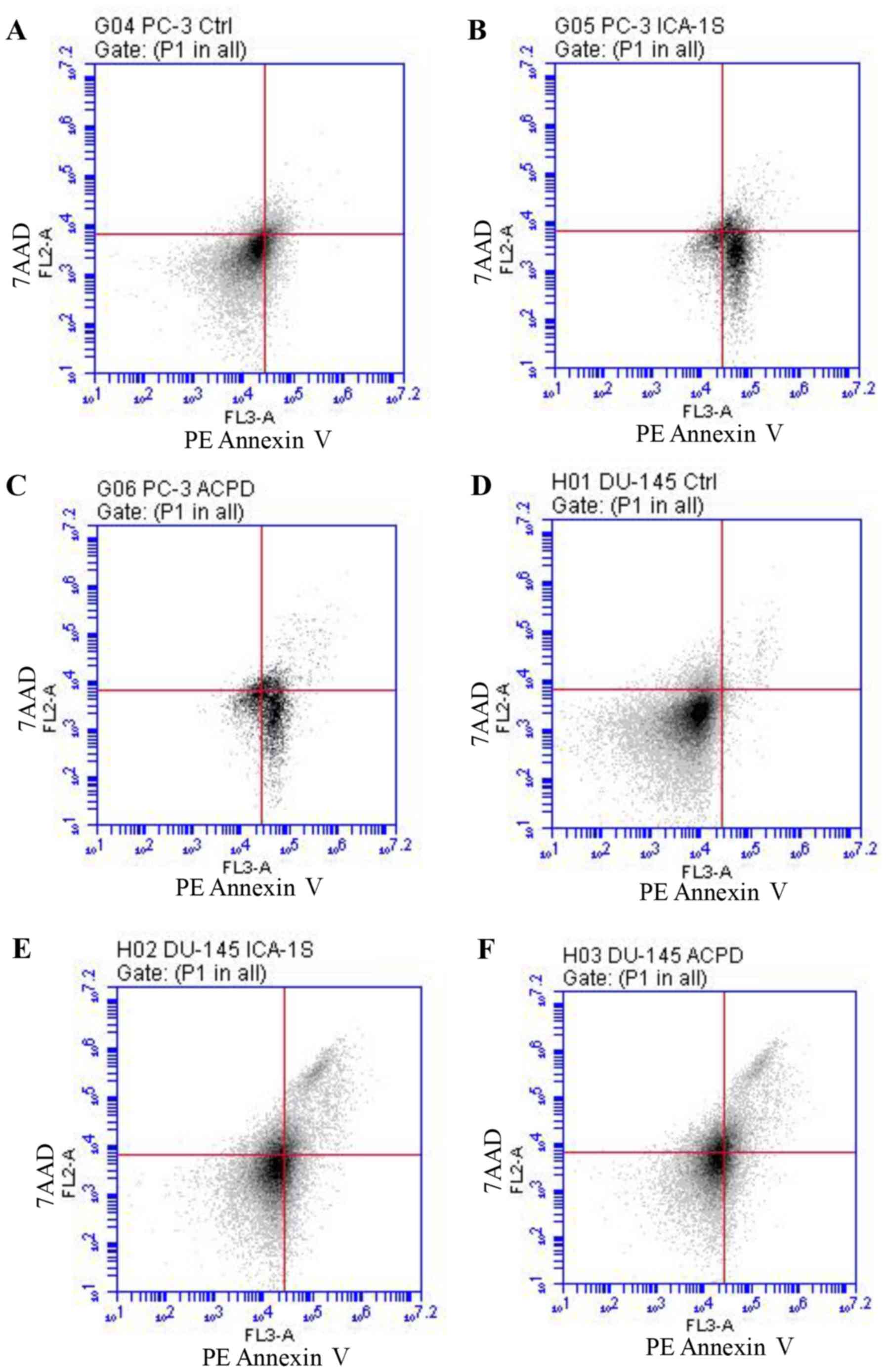 | Figure 5Flow cytometric analysis of PE
Annexin V staining for prostate cancer cells. Graphs show
fluorescent emission of PC-3 cells in the (A) control, (B), ICA-1-
and (C) ACPD-treated groups, and DU-145 cells in the (D) control,
(E) ICA-1- and (F) ACPD-treated groups. Cells were incubated with
PE Annexin V (x-axis) against 7AAD (y-axis). Cells were treated
with ICA-1 and ACPD with respective IC50 concentrations
for 24 h and 50,000 events were analyzed and recorded to obtain
FL3-A (PE-Annexin V), vs. FL-2A (7-AAD). Three experiments were
performed with 24 replicates and representative plots are shown.
ACPD, polymerase; 2-acetyl-1,3-cyclopentanedione; ICA-1,
5-amino-1-(1R,2S,3S,4R)-2,3-dihydroxy-4-methylcyclopentyl)-1H-imidazole-
4-carboxamide; 7AAD, 7-amino-actinomycin; Ctrl, control. |
Subsequently, the present study determined whether
ICA-1 or ACPD affected cell survival by inhibiting the
translocation of NF-κB from the cytoplasm to the nucleus (Fig. 6). Following exposure to TNF-α, the
protein levels of NF-κB were measured in the nucleus and the
cytosol of DU-145 and PC-3 cells. Following treatment with ICA-1,
there was a 5-fold decrease in nucleic NF-κB in the DU-145 cells
(P=0.02) and a 3-fold decrease of nucleic NF-κB in the PC-3 cells
(P=0.025), compared with the untreated samples. With ACPD, there
was a 1.5-fold decrease in nucleic NF-κB (P=0.046) in the DU-145
cells and a 40% decrease in NF-κB (P=0.028) in the PC-3 cells,
compared with the untreated samples (Fig. 6). In addition, the phosphorylation
of IKKαβ (S176/180) and IκBα (S32) were examined, both of which
reduced significantly (P≤0.05) following inhibitor treatments in
the two PC cell lines, which confirmed a reduction of NF-κB
translocation to the nuclei. The phosphorylation of PTEN (S380) and
AKT (S473) also decreased significantly (P≤0.05) in the two PC cell
lines, indicating that the AKT pathway also slowed down upon
inhibitor treatment.
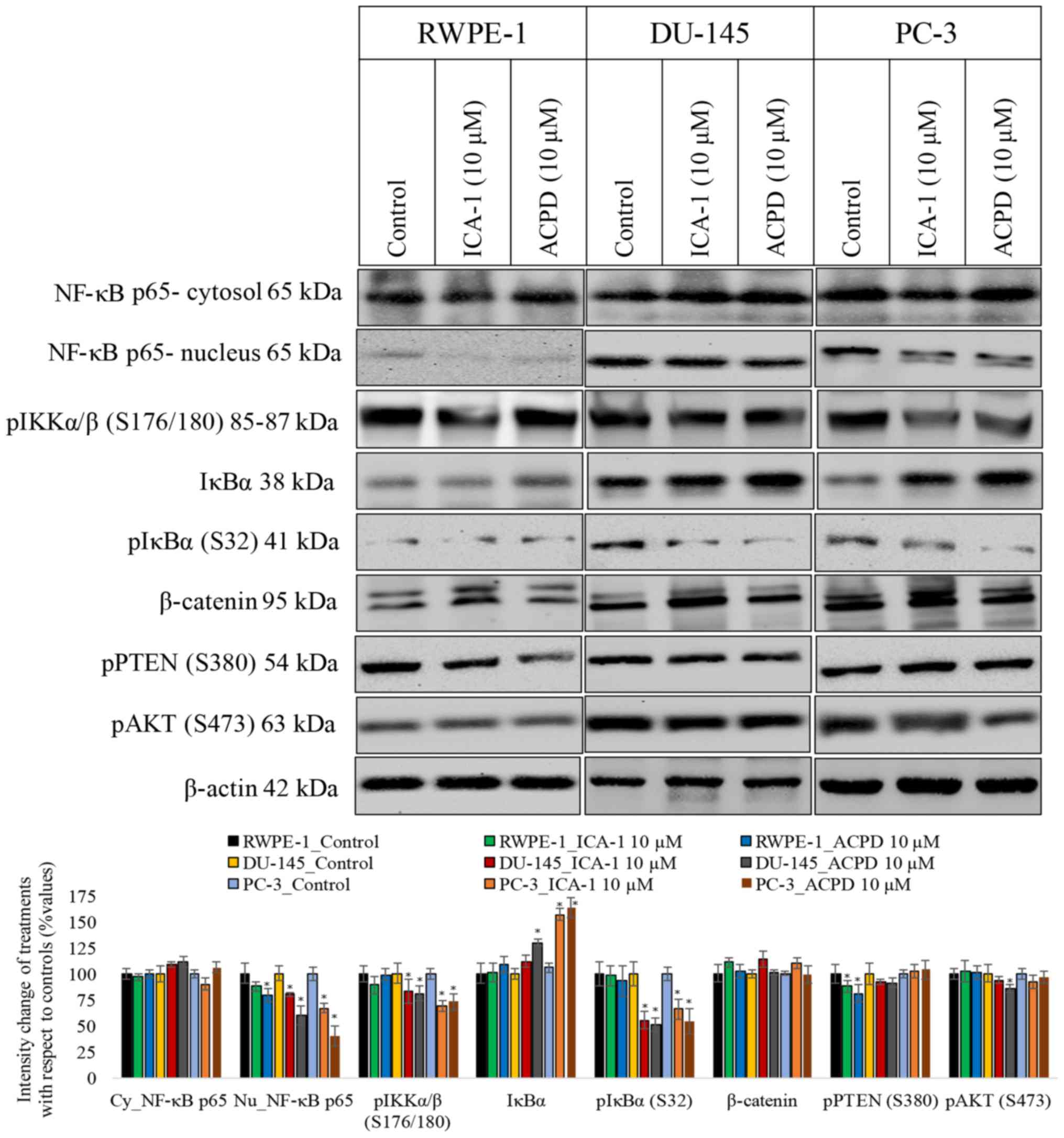 | Figure 6Effects of 10 µM ICA-1 and 10 µM ACPD
on NF-κB translocation in RWPE-1, DU-145, and PC-3 cells. Protein
expression of NF-κB p65 is shown in nuclear and cytoplasmic cell
fractions following 72 h of treatment. Cells were exposed to 10 nM
of TNF-α 30 min prior to harvest to induce NF-κB transloca- tion.
Other targets in the proposed pathway and targets known to be
affected by PKC-ι activation were assessed, including pIKKα/β,
IKKα/β, pIκBα, IκBα, β-catenin, pPTEN, and pAKT. A total of 50 µg
protein was loaded into each well and the results were normalized
by β-actin. Three trials were performed in triplicate. Densitometry
bar graphs are shown as the percentage change of the treated sample
with respect to their controls (mean ± standard deviation).
*P≤0.05, vs. control. NF-κB, nuclear factor-κB; IκBα,
inhibitor of NF-κBα; IKK, IκBα kinase; PTEN, phosphatase and tensin
homolog; p, phosphorylated; ACPD, polymerase;
2-acetyl-1,3-cyclopentanedione; ICA-1,
5-amino-1-(1R,2S,3S,4R)-2,3-dihydroxy-4-methylcyclopentyl)-1H-imidazole-4-carboxamide. |
Discussion
There has been increased interest in the status of
aPKC-ι, which does not contain a Ca2+-binding region,
has one zinc finger-like motif, and is the human homolog of the
mouse PKC-λ (31). Eder et
al (32) provided evidence for
the role of PKC-ι in cell proliferation by demonstrating that
increased protein levels of PKC-ι were associated with increased
protein expression of cyclin E and proliferation in ovarian cancer.
Eder et al (32)
demonstrated in non-serous ovarian cancer that increased protein
levels of PKC-ι markedly decreased overall survival rate. PKC-ι is
critical for non-small cell lung cancer proliferation in
vivo via activation of the Ras-related C3 botulinum toxin
substrate 1 (Rac1)/Rac1-activated kinase/mitogen-activated protein
kinase kinase 1,2/extracellular signal-regulated kinase 1,2
signaling pathway, which has been implicated in tumor cell
proliferation, indicating PKC-ι as an oncogene in human non- small
cell lung cancer (20,23). Additionally, the importance of
PKC-ι is further recognized due to its exclusive association with
transformed phenotype of human melanomas in vivo and in
vitro (33), its
overexpression in human non-small lung cancer cell lines and
cholangiocarcinoma (34), and its
presence in the transformed growth of the human lung adenocarcinoma
A549 cell line in vitro and tumorigenicity in vivo
(20). Our previous study also
highlighted the exclusive association of PKC-ι with the transformed
phenotype of glioma and meningioma (35). However, the linkage between the PKC
family of proteins and PC remains to be fully elucidated.
The results of the western blot analysis
demonstrated that the PKC-ι protein was overexpressed in all PC
tissues, but not in BPH tissues (P=0.00048). Furthermore, the IHC
results demonstrated increased PKC-ι staining intensity in PC
glands compared with BPH glands and glands with HGPIN. The BHP
glands showed weak staining of PKC-ι, similar to samples obtained
from patients with and without prostatic adenocarcinoma. The
stromal cells showed weak to moderate staining of PKC-ι in samples
with and without adenocarcinoma. It is possible that the reduced
categorical expression of PKC-ι observed in the comparison between
malignant and BPH tissues may be associated with the antigen
retrieval methodology of the IHC process in formalin-fixed,
archival specimens. The observation of marked and consistent
expression of PKC-ι in all PC specimens and certain HGPIN specimens
is significant, as it may allow the prediction of the proportion of
HGPIN patients, estimated to be up to 50%, who progress to clinical
PC. The fact that benign prostatic acini in patients with PC do not
significantly express PKC-ι further underscores the
histopathological dichotomy of the BPH and PC pathways. Finally, if
PKC-ι is demonstrated to have be significant in prostate
carcinogenesis, it may offer an opportunity to target PKC-ι in
HGPIN patients for the prevention of PC. The results of the present
study suggested that PC showed a higher dependency on PKC-ι
compared with non-cancerous prostate tissue. The increase in the
expression of PKC-ι may be due to gene amplification or genetic
duplication of PKC-ι. Previous studies have shown that chromosome
3q26, where PKC-ι is located, is an amplicon and that gene
amplification of PKC-ι is associated with non-small cell lung
cancer and esophageal squamous cell carcinoma (23,36).
Studies have also shown that PKC-ι is co-amplified with neighboring
gene SRY-box 2 (37). These
findings have clinical ramifications as the increased expression of
PKC-ι has been shown to correlate with poor prognosis in other
types of cancer (32,34). The results of the present study
suggest that prostate cancer behaves similarly and that increased
expression of PKC-ι indicates a less favorable prognosis.
Although PKC-ι has been previously shown to be
involved in the survival of the normal RWPE-1 cell line, these
results were observed following serum starvation (38). As PKC-ι is activated under stress,
for example, following the addition of TNF-α or serum starvation,
and the results from the tissues were so different, the present
study examined the reliance of cultured cell lines on PKC-ι without
serum starvation. It was found that PKC-ι had a significant effect
in PC cells, but had minimal effect in normal cells. This provided
evidence of the dependence of PC on PKC-ι and assists in explaining
its overexpression in PC cells. As PKC-ι can be a predictor for PC
(37) and is also an
anti-apoptotic factor (39),
resistance to apoptosis is decreased by inhibiting PKC-ι. The
results of the present study showed that inhibitors, including
ICA-1, may not be directly toxic to cancer cells; however, by
inhibiting PKC-ι, carcinoma cells that overexpress and rely on
PKC-ι are profoundly affected. The WST-1 results demonstrated that
the inhibitors had a cytostatic effect on the PC cells thereby
inhibiting their growth, differentiation and proliferation prior to
the induction of apoptosis.
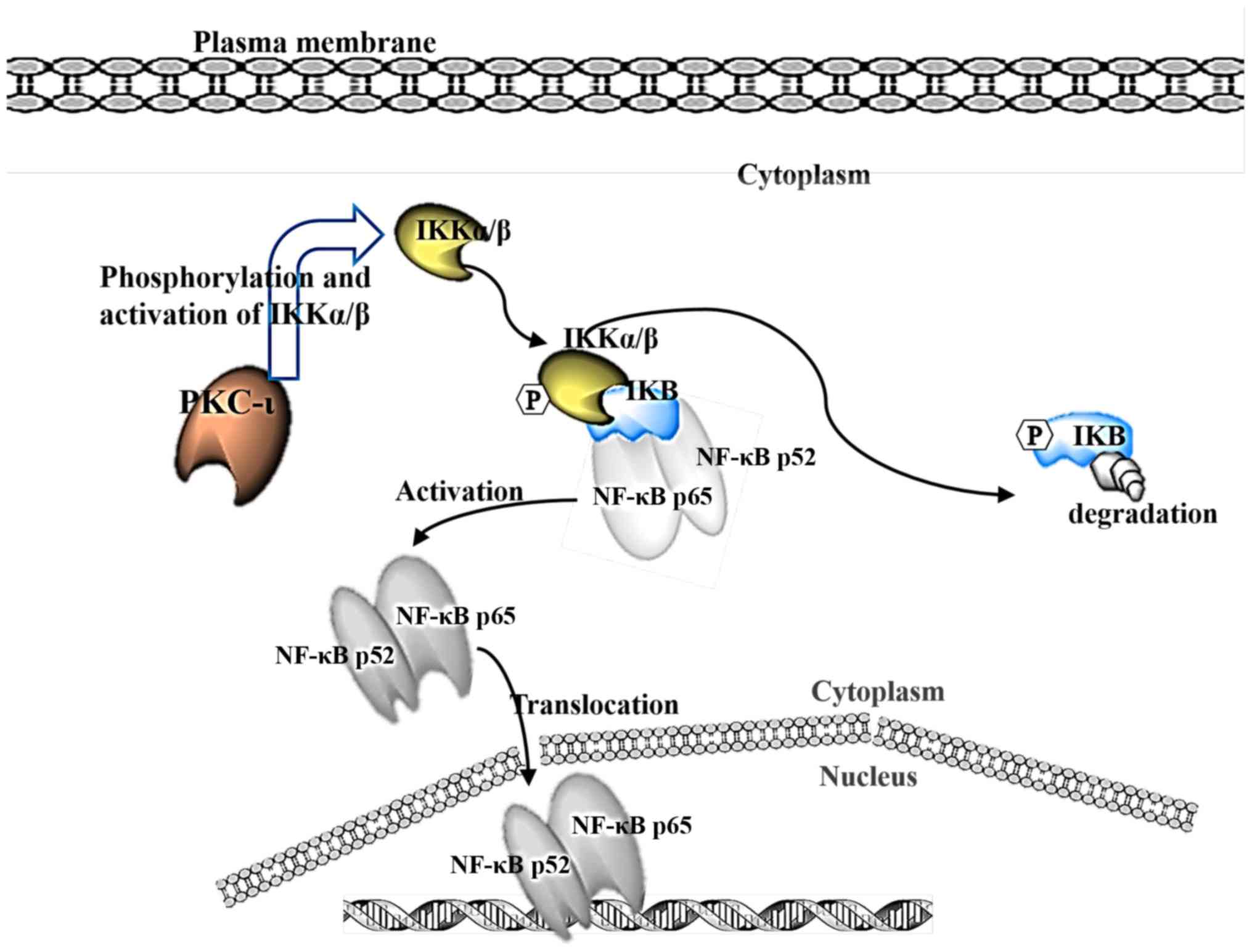
It is suggested that PKC-ι phosphorylates IKK, which
in turn releases the inhibitor IκBα from NF-κB, allowing NF-κB to
translocate to the nucleus as a transcription factor known to
propagate cancer (Fig. 7). A
previous study linked the activation of IKK through the action of
PKC (38). The present study
showed that the phosphorylation of IKKα/β decreased upon inhibition
of PKC-ι, which decreased the phosphorylation of IκBα and prevented
its degradation by the release of activated NF-κB, allowing it to
translocate into the nuclei. These data suggest that, by inhibiting
PKC-ι, the transcription factor NF-κB remained inhibited, and the
chain of events was not set in motion. NF-κB is generally released
during periods of stress and is not the only pro-survival factor
affecting the cell. However, this pathway inhibition may explain
the decrease in cell growth rates for the two cell lines treated
with PKC-ι inhibitors. In neuroblastoma cells, ICA-1 has been shown
to be a specific inhibitor to PKC-ι, and its inhibition has been
shown to induce apoptosis (24).
In the present study, the DU-145 cells also showed signs of
increased apoptotic markers and retarded growth rate. The PC-3
cells showed a similar decrease in growth rate and were subject to
apoptotic factors despite their aggressive nature. One limitation
of the present study was the sample size of patient tissues used. A
larger sample combined with the in vivo assessment of the
inhibitors is recommended.
In conclusion, the results in the present study
suggested that aPKCs are major components responsible for inducing
cell growth, differentiation and survival in human PC cells. In
addition, PKC-ι was found to be involved in the activation and
nuclear translocation of NF-κB. The results also suggested that
ICA-and ACPD were effective aPKC inhibitors in PC cells and did not
affect normal prostate epithelial cells at the optimal working
concentrations for PC cells used. These inhibitors reduced cell
proliferation and induced the apoptosis of PC cells. Taken
together, these results indicate that the detection of PKC-ι may be
used as a predictor of prostate carcinogenesis and suggest that
patients with PC may benefit from anti-PKC-ι therapy.
Funding
This study was funded by the generous support of The
Leo and Anne Albert Charitable Trust (USFF 42-1042), The Sapphire
Foundation for Prostate Cancer (USFF 42-0044), The Daniel Tanner
Foundation (USFF 42-044) and The Frederick H. Leonhardt Foundation
(USFF 42-0044).
Availability of data and materials
All data generated or analyzed during this study are
included within the manuscript.
Authors’ contributions
AHA and MAD conceived and planned the experiments.
Cell culturing was performed by AHA, WSR, HWP, CAA and TS. Prostate
tissue collection and analysis was performed by RS and HWP.
Immunohistochemical staining and analysis was performed by LK and
HWP. Western blot analysis was performed by AHA, WSR and HWP, and
TS carried out the experiments. Cell proliferation assays were
performed by AHA, WSR and CAA. Cell subfractionation was performed
by AHA and WSR. Flow cytometry was performed by RH and WSR. AHA,
MAD, RH, RS, WSR, HWP and LK contributed to the interpretation of
the results. AHA took the lead in writing the manuscript. All
authors provided critical feedback and helped shape the research,
analysis and manuscript.
Ethics approval and consent to
participate
All patients provided informed consent as part of a
clinical protocol approved by the Institutional Review Board (IRB
no. 103023) of the University of South Florida.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Dr Hercules
Apostolatos and Ms. Roberta Blanchard of Tampa Bay Technology
Incubator at USF for their assistance with equipment and cell
culturing, and Ms. Gina Bladuell and Ms. Kayla Batista of USF
College of Medicine and Mr. Sloan Breedy of USF for their
assistance with editing.
References
|
1
|
American Cancer Society: Key statistics
for prostate cancer. Prostate Cancer Facts. https://www.cancer.org/cancer/prostate-cancer/about/key-statistics.html
|
|
2
|
Kume H, Kawai T, Nagata M, Azuma T,
Miyazaki H, Suzuki M, Fujimura T, Nakagawa T, Fukuhara H and Homma
Y: Intermittent docetaxel chemotherapy is feasible for
castration-resistant prostate cancer. Mol Clin Oncol. 3:303–307.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kharaziha P, Chioureas D, Rutishauser D,
Baltatzis G, Lennartsson L, Fonseca P, Azimi A, Hultenby K, Zubarev
R, Ullén A, et al: Molecular profiling of prostate cancer derived
exosomes may reveal a predictive signature for response to
docetaxel. Oncotarget. 6:21740–21754. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Newton AC: Protein kinase C: Poised to
signal. Am J Physiol Endocrinol Metab. 298:E395–E402. 2010.
View Article : Google Scholar :
|
|
5
|
Nishizuka Y: Intracellular signaling by
hydrolysis of phospholipids and activation of protein kinase C.
Science. 258:607–614. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Selbie LA, Schmitz-Peiffer C, Sheng Y and
Biden TJ: Molecular cloning and characterization of PKC iota, an
atypical isoform of protein kinase C derived from insulin-secreting
cells. J Biol Chem. 268:24296–24302. 1993.PubMed/NCBI
|
|
7
|
Hirai T, Niino YS and Chida K: PKC zeta
II, a small molecule of protein kinase C zeta, specifically
expressed in the mouse brain. Neurosci Lett. 348:151–154. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hayashi A, Seki N, Hattori A, Kozuma S and
Saito T: PKCnu, a new member of the protein kinase C family,
composes a fourth subfamily with PKCmu. Biochim Biophys Acta.
1450:99–106. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Persons DA, Wilkison WO, Bell RM and Finn
OJ: Altered growth regulation and enhanced tumorigenicity of NIH
3T3 fibroblasts transfected with protein kinase C-I cDNA. Cell.
52:447–458. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Housey GM, Johnson MD, Hsiao WL, O’Brian
CA, Murphy JP, Kirschmeier P and Weinstein IB: Overproduction of
protein kinase C causes disordered growth control in rat
fibroblasts. Cell. 52:343–354. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kamata T, Sullivan NF and Wooten MW:
Reduced protein kinase C activity in a ras-resistant cell line
derived from Ki-MSV transformed cells. Oncogene. 1:37–46.
1987.PubMed/NCBI
|
|
12
|
Weyman CM, Taparowsky EJ, Wolfson M and
Ashendel CL: Partial down-regulation of protein kinase C in C3H 10T
1/2 mouse fibroblasts transfected with the human Ha-ras oncogene.
Cancer Res. 48:6535–6541. 1988.PubMed/NCBI
|
|
13
|
Mizuguchi J, Nakabayashi H, Yoshida Y,
Huang KP, Uchida T, Sasaki T, Ohno S and Suzuki K: Increased
degradation of protein kinase C without diminution of mRNA level
after treatment of WEHI-231 B lymphoma cells with phorbol esters.
Biochem Biophys Res Commun. 155:1311–1317. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hudes GR: Signaling inhibitors in the
treatment of prostate cancer. Invest New Drugs. 20:159–172. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu D, Thakore CU, Wescott GG, McCubrey JA
and Terrian DM: Integrin signaling links protein kinase Cepsilon to
the protein kinase B/Akt survival pathway in recurrent prostate
cancer cells. Oncogene. 23:8659–8672. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rusnak JM and Lazo JS: Downregulation of
protein kinase C suppresses induction of apoptosis in human
prostatic carcinoma cells. Exp Cell Res. 224:189–199. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jaggi M, Rao PS, Smith DJ, Hemstreet GP
and Balaji KC: Protein kinase C mu is down-regulated in
androgen-independent prostate cancer. Biochem Biophys Res Commun.
307:254–260. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rao PS, Jaggi M, Smith DJ, Hemstreet GP
and Balaji KC: Metallothionein 2A interacts with the kinase domain
of PKCmu in prostate cancer. Biochem Biophys Res Commun.
310:1032–1038. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Inoue T, Yoshida T, Shimizu Y, Kobayashi
T, Yamasaki T, Toda Y, Segawa T, Kamoto T, Nakamura E and Ogawa O:
Requirement of androgen-dependent activation of protein kinase
Czeta for androgen-dependent cell proliferation in LNCaP Cells and
its roles in transition to androgen-independent cells. Mol
Endocrinol. 20:3053–3069. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Regala RP, Weems C, Jamieson L, Copland
JA, Thompson EA and Fields AP: Atypical protein kinase Ciota plays
a critical role in human lung cancer cell growth and
tumorigenicity. J Biol Chem. 280:31109–31115. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Weichert W, Gekeler V, Denkert C, Dietel M
and Hauptmann S: Protein kinase C isoform expression in ovarian
carcinoma correlates with indicators of poor prognosis. Int J
Oncol. 23:633–639. 2003.PubMed/NCBI
|
|
22
|
Win HY and Acevedo-Duncan M: Atypical
protein kinase C phosphorylates IKKalphabeta in transformed
non-malignant and malignant prostate cell survival. Cancer Lett.
270:302–311. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Regala RP, Weems C, Jamieson L, Khoor A,
Edell ES, Lohse CM and Fields AP: Atypical protein kinase C iota is
an oncogene in human non-small cell lung cancer. Cancer Res.
65:8905–8911. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Desai S, Pillai P, Win-Piazza H and
Acevedo-Duncan M: PKC-ι promotes glioblastoma cell survival by
phosphorylating and inhibiting BAD through a phosphatidylinositol
3-kinase pathway. Biochim Biophys Acta. 1813.1190–1197. 2011.
|
|
25
|
Pillai P, Desai S, Patel R, Sajan M,
Farese R, Ostrov D and Acevedo-Duncan M: A novel PKC-ι inhibitor
abrogates cell proliferation and induces apoptosis in
neuroblastoma. Int J Biochem Cell Biol. 43:784–794. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Laemmli UK: Cleavage of structural
proteins during the assembly of the head of bacteriophage T4.
Nature. 227:680–685. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Towbin H, Staehelin T and Gordon J:
Electrophoretic transfer of proteins from polyacrylamide gels to
nitrocellulose sheets: Procedure and some applications.
1979.Biotechnology. 24:145–149. 1992.
|
|
29
|
Allred DC, Harvey JM, Berardo M and Clark
GM: Prognostic and predictive factors in breast cancer by
immunohistochemical analysis. Mod Pathol. 11:155–168.
1998.PubMed/NCBI
|
|
30
|
Ratnayake WS, Apostolatos AH, Ostrov DA
and Acevedo- Duncan M: Two novel atypical PKC inhibitors; ACPD and
DNDA effectively mitigate cell proliferation and epithelial to
mesenchymal transition of metastatic melanoma while inducing
apoptosis. Int J Oncol. 51:1370–1382. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Diaz-Meco MT, Municio MM, Sanchez P,
Lozano J and Moscat J: Lambda-interacting protein, a novel protein
that specifically interacts with the zinc finger domain of the
atypical protein kinase C isotype lambda/iota and stimulates its
kinase activity in vitro and in vivo. Mol Cell Biol. 16:105–114.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Eder AM, Sui X, Rosen DG, Nolden LK, Cheng
KW, Lahad JP, Kango-Singh M, Lu KH, Warneke CL, Atkinson EN, et al:
Atypical PKCiota contributes to poor prognosis through loss of
apical-basal polarity and cyclin E overexpression in ovarian
cancer. Proc Natl Acad Sci USA. 102:12519–12524. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Selzer E, Okamoto I, Lucas T, Kodym R,
Pehamberger H and Jansen B: Protein kinase C isoforms in normal and
transformed cells of the melanocytic lineage. Melanoma Res.
12:201–209. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li Q, Wang J-M, Liu C, Xiao B-L, Lu J-X
and Zou S-Q: Correlation of aPKC-iota and E-cadherin expression
with invasion and prognosis of cholangiocarcinoma. Hepatobiliary
Pancreat Dis Int. 7:70–75. 2008.PubMed/NCBI
|
|
35
|
Patel R, Win H, Desai S, Patel K, Matthews
JA and Acevedo- Duncan M: Involvement of PKC-iota in glioma
proliferation. Cell Prolif. 41:122–135. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang Y-L, Chu J-Y, Luo M-L, Wu YP, Zhang
Y, Feng YB, Shi ZZ, Xu X, Han YL, Cai Y, et al: Amplification of
PRKCI, located in 3q26, is associated with lymph node metastasis in
esophageal squamous cell carcinoma. Genes Chromosomes Cancer.
47:127–136. 2008. View Article : Google Scholar
|
|
37
|
Justilien V, Walsh MP, Ali SA, Thompson
EA, Murray NR and Fields AP: The PRKCI and SOX2 oncogenes are
coamplified and cooperate to activate Hedgehog signaling in lung
squamous cell carcinoma. Cancer Cell. 25:139–151. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kim S-W, Schifano M, Oleksyn D, Jordan CT,
Ryan D, Insel R, Zhao J and Chen L: Protein kinase C-associated
kinase regulates NF-κB activation through inducing IKK activation.
Int J Oncol. 45:1707–1714. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xie J, Guo Q, Zhu H, Wooten MW and Mattson
MP: Protein kinase C iota protects neural cells against apoptosis
induced by amyloid beta-peptide. Brain Res Mol Brain Res.
82:107–113. 2000. View Article : Google Scholar : PubMed/NCBI
|