Introduction
Gastric cancer ranks fourth among the most common
cancer types of cancer and is the third leading cause of
cancer-related mortality worldwide (1). Recent advances in the diagnosis and
treatment of the disease have increased the early detection and
have decreased the mortality of patients with gastric cancer over
the past decades; however, there are still an estimated 28,000 new
cases of gastric cancer and 10,960 related deaths in the US in 2017
(2). Gastric cancer is difficult
to cure primarily as the majority of patients present with advanced
disease, and even a large number of patients who undergo surgical
resection succumb to the disease due to recurrence. A variety of
oncogenes and tumor suppressor genes are involved in the
development of gastric cancer; however, the precise mechanisms
underlying the progression of gastric cancer remain to be
determined. Therefore, there is an urgent to identify more specific
biomarkers for the early diagnosis of and therapy targets for
gastric cancer.
MicroRNAs (miRNAs or miRs) are small non-coding RNAs
(~22 nucleotides in length) that can regulate the expression of
their target genes by binding to the complementary sites in the
3′-untranslated regions (3′-UTRs) of their targeting mRNAs and
further regulate either translational repression or mRNA
degradation (3,4). miRNAs have been reported to play
important roles in cellular processes, such as cell
differentiation, cell growth and proliferation, migration,
apoptosis, metabolism and defense (5,6).
Mounting evidence indicates that the aberrant expression of miRNAs
or miRNA mutations are associated with diverse human malignancies,
suggesting that miRNAs can function as tumor suppressors or
oncogenes (7). miRNA expression
profiles, determined using miRNA micro-arrays, reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
next-generation sequencing (NGS) approaches, can be used to
establish sample specificity and to identify cancer type (8). Tsai et al summarized a variety
of aberrantly expressed miRNAs as potential useful biomarkers for
gastric cancer screening, diagnosis, prognosis, disease monitoring,
as well as therapeutic targets (8). However, the exact role of specific
miRNAs in regulating the tumorigenesis and progression of gastric
cancer remains largely unknown.
The miR-17-92 cluster, also known as
oncomiR-1, is one of the most well-known oncogenic miRNAs
(9). Human miR-17-92 is
located within intron 3 of the C13orf25 gene at 13q31.3, and
is frequently amplified in a variety of malignant tumors (10). The miR-17-92 cluster has 6
mature miRNAs which are classified into 3 separate miRNAs families,
including the miR-17 family (miR-17, miR-18
and miR-20), the miR-19 family (miR-19a and
miR-19b) and the miR-92 family (miR-92).
Previous studies have demonstrated that the expression of the
miR-17-92 cluster is upregulated in a variety of tumors
originating from the lungs (11),
breast (12), kidneys (13), liver (14), colon 15) and stomach (16). Generally, the miR-17-92
cluster promotes tumorigenesis by negatively regulating tumor
suppressors or genes, which are associated with a large variety of
biological behaviors including cell proliferation, cell cycle, cell
apoptosis, migration and metastasis (17).
The role of the miR-17-92 cluster in gastric
cancer is unclear. The majority of the published studies indicate
that the miR-17-92 cluster plays oncogenic roles in gastric
cancer progression. In patients with gastric cancer, the levels of
miR-17-92 members in serum have been shown to be
over-expressed, and are thus considered as potential biomarkers for
the early detection of gastric cancer (16). Moreover, the miR-17-92
cluster has been shown to be upregulated in gastric cancer tissues
and serves as an independent prognostic marker in gastric cancer
patients (18). miR-92a is
used as a predictive prognostic marker in gastric cancer (19). Another study demonstrated that
circulating miR-18a can be a useful biomarker for the
screening of gastric cancer and monitoring cancer dynamics
(20). The overexpression of
miR-17-5p has been shown to increase the proliferation and
growth of gastric cancer cells in vitro and in vivo
by repressing suppressors of cytokine signaling 6
(SOCS6) (21). The
overexpression of miR-17 in gastric cancer cells is associated with
certain proliferation-associated genes amplification, including
myelocytomatosis (MYC), cyclin E1
(CCNE1), V-Erb-B2 avian erythroblastic leukemia viral
oncogene homolog 2 (ERBB2) and fibroblast growth
factor receptor 2 (FGFR2) (22). miR-18a acts as an oncogene and
plays an important role in gastric cancer by negatively regulating
protein inhibitor of activated signal transducer and activator
of transcription 3 (PIAS3) (23). miR-19a/b has been shown to
facilitate the migration and invasion of gastric cancer cells by
targeting the antagonist of c-myelocytomatosis-mitotic arrest
deficient protein 1 (c-Myc-MXD1) (24). Moreover, miR-19a/b has been
demonstrated to regulate multidrug resistance in gastric cancer
cells by targeting phosphate and tension homology deleted on
chromsome ten (PTEN) (25). miR-19b, miR-20a and
miR-92a have been reported to play important roles in the
development of gastric cancer stem cells (26). However, the critical role of the
miR-17-92 cluster in regulating gastric cancer progression
remains unclear.
In this study, we systematically investigated the
biological functions and the underlying mechanisms of action of the
miR-17-92 cluster in gastric cancer. The overexpression of
the miR-17-92 cluster in MGC-803 human gastric cancer cells
affected a variety of biological functions, including cell growth,
proliferation, apoptosis, migration and invasion. Activated protein
kinase B (AKT), extracellular-signal-regulated kinase (ERK) and
nuclear factor kappa-light-chain-enhancer of activated B cells
(NF-κB) signaling pathways were involved in these processes.
Importantly, for the first time, at least to the best of our
knowledge, we identified that tumor necrosis factor receptor
associated factor 3 (TRAF3) was a direct and functional
target of the miR-17-92 cluster in MGC-803 gastric cancer
cells. The loss-of-function of TRAF3 led to the acquirement of
phenotypes, similar to what had been observed in the MGC-803 cells
overexpressing the miR-17-92 cluster. Survival analyses
revealed that TRAF3 served as an important prognostic indicator in
patients with gastric cancer. Herein, we demonstrate that the
miR-17-92 cluster functions as an oncogene in gastric cancer
by directly targeting TRAF3. Targeting the
miR-17-92/TRAF3/NF-κB axis may thus prove to be a potent
therapeutic approach in human gastric cancer.
Materials and methods
Cell culture
The human gastric cancer cell lines, AGS, SGC-7901,
BGC-823, MKN-45 and MGC-803, were obtained from the Key Laboratory
of Medicine and Clinical Immunology of Jiangsu Province, the First
Affiliated Hospital of Soochow University. The 293T cells (provided
by Professor Yong Zhao, Institute of Zoology, Chinese Academy of
Sciences, Beijing, China) were maintained in our central
laboratory. All the cells were cultured in RPMI-1640 medium
supplemented with 10% fetal bovine serum (FBS), 100 U/ml
penicillin, 100 µg/ml streptomycin and 2 mM glutamine. All the
cells were cultured at 37°C in a humidified atmosphere containing
5% CO2.
Transfection
The miR-17-92 cluster overexpression vector
was constructed on the MSCV vector containing green fluorescent
protein (GFP; from Professor Yong Zhao, Institute of Zoology,
Chinese Academy of Sciences, Beijing, China). The
MSCV-GFP-miR-17-92 or MSCV-GFP control plasmid was
transfected into the Phoenix A packaging cells (provided by
Professor Yong Zhao, Institute of Zoology, Chinese Academy of
Sciences) using FuGENE HD transfection reagent (cat. no.
04709705001; Roche, Shanghai, China). Viral supernatants were
collected and used to infect the MGC-803 cells. For obtaining
stably expressing cell lines, the cells were selected in the
presence of 5 µg/ml puromycin (cat. no. J593; Amresco, Beijing,
China). A shRNA-carrying sequence targeting the TRAF3 gene
(506-524: 5′-GATAAGGTGTTTAAGGATA-3′) was designed and synthesized
by Invitrogen (Beijing, China). The shRNA-TRAF3 was subcloned into
the pSilencer3.1-H1-neo plasmid (cat no. 5770; Thermo Fisher
Scientific™, Shanghai, China). The recombinant pSilencer3.1-siTRAF3
plasmid or the pSilencer3.1-H1-neo control plasmid were then
trans-fected into the MGC-803 cells using Lipofectamine 2000 (cat
no. 12566014; Thermo Scientific™). To obtain stably transfected
cell lines, the cells were selected in the presence of 600 ng/µl
neomycin (cat. no. A1720-5G; Sigma, Beijing, China).
RT-qPCR
Total RNA (2 µg) was reverse transcribed with
SuperScript M-MLV (cat. no. 28025013; Promega, Shanghai, China)
according to the manufacturer’s instructions. All RT-qPCR reactions
were performed with a LightCycler 480 system (Roche) in triplicate.
Primers were designed using Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/;
NCBI; PubMed) and synthesized from Invitrogen (Beijing, China).
β-actin was used as an internal control. cDNA was amplified
using 2× LC480 SYBR-Green Master Mix (cat. no. 04887352001; Roche).
Data were analyzed with the Pfaffl method (27,28).
The primer sequences were as follows: BIM forward,
5′-ACAGAGCCACAAGACAGGAGCCC-3′ and reverse,
5′-CGCCgGCAACTCTTGGGCGAT-3′; cyclin D1 (CCND1) forward,
5′-CACTTTCAGTCCAATAGGTGTAG-3′ and reverse,
5′-TTCTTCTTGACTGGCACG-3′; PTEN forward,
5′-TGGGGAAGTAAGGACCAGAGA-3′ and reverse,
5′-TGAGGATTGCAAGTTCCGCC-3′; Von Hippel-Lindau (VHL) forward,
5′-GCAGGCGTCGAAGAGTACG-3′ and reverse, 5′-CGGACTGCGATTGCAGAAGA-3′;
PH domain and leucine rich repeat protein phosphatase 2
(PHLPP2) forward, 5′-GTACGCAAGGGAAAGACCCA-3′ and reverse,
5′-AGCAAGGGAGTATTGCCGTC-3′; TRAF3 forward,
5′-TGTAAATACCGGGAAGCCACA-3′ and reverse,
5′-TGCACTCAACTCGCTCCTCA-3′; β-actin reverse,
5′-GCTACGAGCTGCCTGACGG-3′ and reverse,
5′-TGTTGGCGTACAGGTCTTTGC-3′.
RT-qPCR analysis of mature miRNA
expression
Total RNA (2 µg) was polyadenylated with ATP by
E. coli poly (A) polymerase (PAP; cat. no. M0276L; New
England Biolabs, Ltd., Beijing, China). Following phenol-chloroform
extraction and ethanol precipitation, the polyadenylated RNA was
reverse transcribed with M-MLV and universal RT primer sequence.
The cDNA (1:5 dilution) was then amplified using 2× LC480
SYBR-Green Master Mix (Roche). Each sample was determined in
triplicate. U6 expression was considered as an internal
control. Data were analyzed using the Pfaffl method (27,29).
The primer sequences were as follows: hsa-miR-17
5′-GCAAAGTGCTTACAGTGCAGGTAG-3′; hsa-miR-18,
5′-CCGGTAAGGTGCATCTAGT-3′; hsa-miR-19a,
5′-TGTGCAAATCTATGCAAAACTG-3′; hsa-miR-19b,
5′-GCATCCCAGTGTGCAAATCC-3′; hsa-miR-20,
5′-GGTAAAGTGCTTATAGTGCAGGTAG-3′; hsa-miR-92.
5′-ATTGCACTTGTCCCGGCCTGT-3′; RT primer,
5′-AACGAGACGACGACAGACTTTTTTTTTTTTTTTV-3′; U6,
5′-GCAAATTCGTGAAGCGTTCCATA-3′.
Western blot analysis
Whole-cell extracts (WCE), cytoplasmic extracts (CE)
and nuclear extracts (NE) were prepared using suspension buffer (10
mM Tris-HCl, 0.1 M NaCl, 1 mM EDTA) and proteinase inhibitor (cat.
no. 11697498001; Roche Diagnostics, Mannheim, Germany) according to
standard procedures. The total protein was measured with a micro
BCA protein assay. Proteins (50 µg) were fractionated on an 10%
SDS-PAGE gel and then transferred onto nitrocellulose membranes.
After blocking with 5% milk in PBS for 1 h at room temperature, the
membranes were incubated with primary antibodies (Abs). After
washing, the membranes were incubated with secondary Abs. Finally,
proteins were detected using the Odyssey system (LI-COR
Biosciences, Lincoln, NE, USA). Abs (1:200 dilution) against RelA
(sc-372X), RelB (sc-226X), c-Rel (sc-70), p105/p50 (sc-7178X),
p100/p52 (sc-298), inhibitor of kappa B (IκB-α; sc-1643), p-IκB-α
(Ser32/36, sc-101713), cyclin D1 (sc-20044) and Lamin A/C
(sc-20681) were purchased from Santa Cruz Biotechnology (Santa
Cruz, CA, USA). Abs (1:1,000 dilution) against AKT (#4691), p-AKT
(Ser473, #4060), p-AKT (Thr308, #2965), p-c-Raf (#9421), p-glycogen
synthase kinase (GSK)-3β (#5558), p-PTEN (#9551), phosphorylated
phosphoinositide-dependent protein kinase-1 (p-PDK1; #3438), ERK1/2
(#4695), p-ERK1/2 (#4370S), BIM (#2933), PTEN (#9559), VHL (#2738),
TRAF3 (#4729), E-cadherin (#3195), claudin1 (#13255), epithelial
cellular adhesion molecule (EpCAM; #14452), CK8/18 (#4546),
N-cadherin (#13116) and Vimentin (#5741) were obtained from Cell
Signaling Technology. Abs (1:1,000 dilution) against α-tubulin
(AJ1034a) and β-actin (AO1215a) were purchased from Abgent (Suzhou,
China). Secondary Abs (1:10,000 dilution) against IRDye 680CW
(926-32222) and IRDye 800CW (926-32210) were obtained from LI-COR
Biosciences (Lincoln, NE, USA).
NF-κB activity assay
NE preparation and TransAM assay were performed as
previously described (30). The
activity of individual NF-κB subunits was detected by an
ELISA-based NF-κB family transcription factor assay kit (cat. no.
43296; Active Motif, Carlsbad, CA, USA). Briefly, nuclear extracts
(2 µg) were incubated in a 96-well plate, which were immobilized
NF-κB consensus oligonucleotides. The captured complexes were
incubated with specific NF-κB primary Abs and subsequently detected
with HRP-conjugated secondary Abs (included with the kit). Finally,
the optical density (OD) value at 450 nm was measured by
spectrophotometry (ELx800; BioTek Instruments, Winooski, VT,
USA).
Cell proliferation assay (CCK-8)
The cells were harvested and seeded in a 96-well
plate at a density of 5×103 cells per well with 100 µl
of complete culture medium, and incubated overnight at 37°C in a 5%
CO2 humidified incubator. Cell growth rate was assessed
using a cell counting kit-8 (cat. no. CK04; Dojindo, Kumamoto,
Japan) at different time points (0, 24, 48 or 72 h). Briefly, CCK-8
(10 µl) was added to each well. Following incubation for 2 h at
37°C, the absorbance at 450 nm was detected using a plate reader
(ELx800; BioTek Instruments). Cell growth was measured by the
relative absorbance with the absorbency of culture medium
deducted.
Cell viability assay
The cells were harvested and seeded in a 96-well
plate at a density of 5×103 cells per well with 100 µl
of complete culture medium, and incubated overnight at 37°C in a 5%
CO2 humidified incubator. Cell viability assay was
performed using CellTiter-Glo (cat. no. G7570; Promega, Shanghai,
China) according to the manufacturer’s instructions. Briefly,
CellTiter-Glo (100 µl) was added to each well. Following incubation
for 10 min at room temperature, the luciferase activities were
detected using a plate reader (ELx800; BioTek Instruments)
CellTiter-Glo measurements were taken at individual time points (0,
24, 48 or 72 h) to track cell proliferation.
Terminal nucleotidyl transferase-mediated
nick-end labeling (TUNEL) assay
The cells were cultured on cover slides for 24, 48
and 72 h at 37°C overnight. TUNEL assay was performed according to
the manufacturer’s instructions of the TUNEL system kit (Roche). In
brief, the slides were fixed by 4% para-formaldehyde for 30 min and
blocked with 3% H2O2 methanol solution for 10
min. The slides were then treated with 1% Triton PBS solution for 2
min. Subsequently, 50 µl of TUNEL reaction solution were added to
incubate the cells on slides for 1 h at 37°C. Finally, the TUNEL
signals were converted using peroxidase (POD) for 30 min at 37°C,
and the sections were treated with DAB for 3 min. The results were
observed under alight system microscope IX71 (Olympus, Tokyo,
Japan).
Scratch wound healing assay
For the scratch healing assays, THE cells were
treated with 10 mg/ml mitomycin C (Sigma, Beijing, China) for 3 h.
Subsequently, the cells were wounded using a 200 µl sterile pipette
tip, washed 3 times with PBS, and RPMI-1640 with 10% FBS was added.
The wound closure was imaged continuously for 24, 48 and 72 h under
a magnification of x10 using the light System Microscope IX71
(Olympus).
Transwell migration and invasion
assay
Transwell chambers (cat. no. 3422, 8 µm, 24-well
insert; Corning, Lowell, MA, USA) were used for the migration and
invasion assay. In brief, 600 µl of 10% FBS-containing medium was
added to the lower chamber and 1×105 cells in 200 µl
serum-free medium were added to the upper chamber. The cells were
incubated for 24 h at 37°C, and the non-invading cells were
removed. Finally, the insert membranes were fixed with 4%
paraformaldehyde, stained with 0.1% crystal violet (C8470;
Solarbio, Beijing, China) for 15 min at room temperature, and
photographed under an inverted microscope (SystemMicroscope IX71;
Olympus, Tokyo, Japan). For the invasion assay, the insert
membranes were coated with diluted Matrigel (cat. no. 354234; BD
Biosciences, San Jose, CA, USA). Subsequently, 1×105
cells were added to the upper chamber. At least 3 randomly selected
fields were observed, and the average cell number was
calculated.
Gelatinase zymography assay
For the gelatinase zymography assay, the activities
of matrix metalloproteinase (MMP)-9 and -2 were examined. Culture
medium was loaded on an 8% SDS-PAGE gel in the presence of 0.1%
gelatin under non-reducing conditions. Culture medium samples were
not denatured prior to electrophoresis. Following electrophoresis,
the gels were washed twice in 2.5% Triton X-100 for 30 min. The
gels were then incubated in substrate buffer (50 mM Tris-HCl and 10
mM CaCl2, pH 8.0) at 37°C overnight, and stained with
0.5% Coomassie blue R250 (50% methanol and 10% glacial acetic acid)
for 30 min, and then de-stained. Upon renaturation of the enzyme,
the gelatinases digested the gelatin in the gel to produce clear
bands against an intensely stained background.
Luciferase reporter assay
The 3′-untranslated region (UTR) of TRAF3
mRNA was amplified by PCR from genomic DNA of the MGC-803 human
gastric cancer cell line and ligated into the pmirGLO
dual-luciferase miRNA target expression vector (#E1330; Promega,
Madison, WI, USA). The primer sequences of the 3′-UTR of
TRAF3 mRNA were as follows: 5′-CTGGACATGTCAGCATGTTAAGT-3′
and 5′-CGAGGCTCCGTTCAGAATTG-3′. The mutant constructs were
generated using a Site-Directed Mutagenesis kit (#SDM-15, Beijing
SBS Genetech Co., Ltd., Beijing, China). The 293T cells were seeded
in 24-well plates at a density of 5×104 cells per well
with 500 µl of complete culture medium, and incubated overnight at
37°C in a 5% CO2 humidified incubator. The
pmirGLO-TRAF3-3′-UTR-wt plasmid or pmirGLO-TRAF3-3′-UTR-mut plasmid
was co-transfected into the 293T cells with the miR-17-92
plasmid or control plasmid using Lipofectamine 2000 reagent
(#11668-027; Invitrogen/Thermo Fisher Scientific, Waltham, MA,
USA), respectively. Luciferase and Renilla activities were
evaluated 48 h following transfection using the
Dual-GLO® Luciferase Assay System (#E2920; Promega,
Madison, WI, USA) according to the manufacturer’s instructions.
In vivo tumorigenesis
Four-week-old male BALB/c-nude mice were purchased
from Shanghai Slac Laboratory Animal Co. Ltd., Shanghai, China. The
animals were randomly classified into 2 groups as follows: the
MGC-803-control group and the MGC-803-miR-17-92 group, and each
group had 5 mice. A total of 5×106 cells were
resuspended in 200 µl PBS and then injected subcutaneously into the
right flanks of the nude mice. Tumor volume was measured every 4
days using a digital caliper according to the following formula: TV
(mm3) = 0.5 × length × width2. All the mice
were sacrificed at 32 days after the injection. The tumors were
excised, photographed, measured, weighted and fixed in 10%
neutralized formalin overnight. The tumor tissues were dehydrated,
fixed in paraffin, and cut into 5-µm-thick sections for
histopathological examination and immunohistochemistry (IHC). The
sections were stained for TRAF3 (dilution 1:50) expression using
standard protocols. The sections were developed with
3,3-diaminobenzine (DAB) and counterstained with hematoxylin. All
procedures and animal experiments were approved by the Animal Care
and Use Committee of Soochow University.
Clinical samples
The commercial human gastric cancer tissue
microarray was purchased from Shanghai Outdo Biotech Company from
the National Human Genetic Resources Sharing Service Platform
(2005DKA21300). It consists of 100 paired gastric cancer and
adjacent normal tissues, and was used to evaluate the protein
expression of TRAF3. Patient follow-up information was obtained
from 2007 to 2014. The information was obtained from the microarray
company.
IHC
The IHC assay of tissue microarray was performed
using a standard peroxidase-based staining method. Tissue sections
(5-µm-thick) were incubated in a dry oven at 60°C for 30 min and
then deparaffinized in xylene for 10 min in triplicate, rehydrated
with graded ethanol in 100, 95, 90, 80 and 70% ethanol for 5 min
each. Antigen retrieval was then performed using 0.01 M citrate
buffer (pH 6.0). Subsequently, the slides were treated with 3%
hydrogen peroxide (H2O2) to block endogenous
peroxidase. The slides were then blocked with 5% bovine serum
albumin (BSA; Boster Bioengineering, Wuhan, China), incubated with
mouse anti-TRAF3 antibody (#4729; Cell Signaling Technology, 1:50
dilution) overnight at 4°C. Subsequently, the slides were incubated
with diluted secondary antibody (#7076; Cell Signaling Technology,
1:500 dilution) for 45 min at 37°C. Finally, the slides were
treated with DAB and counterstained with hematoxylin for
microscopic examination. The results were observed by 2 independent
investigators under an Olympus microscope BX 51 (Olympus) and
scored as follows: 0, No staining; 1+, light; 2+, moderate; 3+,
strong, according to the intensity of the staining. The percentage
of positively stained cells was scored as follows: 0, No staining;
1, <25% staining; 2, 26-50% staining; 3, 51-75% staining; and 4,
>75% staining. The product of the intensity and extent grades ≥4
of positive cells was considered high expression, and the score of
0-3 of positive cells was regarded as low expression.
Statistical analysis
All the experiments were repeated at least 3 times.
All quantitative data are presented as the means ± SD and analyzed
using the Student’s t-test. The association between TRAF3
expression and the clinicopathological factors was estimated using
the Chi-square test. Overall survival (OS) curves were plotted on
the basis of the Kaplan-Meier method and analyzed using the
log-rank test. All statistical analyses were performed using
GraphPad Prism version 5.0. A P-value ≤0.05 was considered to
indicate a statistically significant difference, and P-values ≤0.01
and ≤0.001 were considered to indicate highly significant
differences.
Results
Distinct miR-17-92 expression patterns in
human gastric cancer cell lines
The expression of the miR-17-92 cluster,
including miR-17, miR-18, miR-19a,
miR-19b, miR-20 and miR-92, was detected by
RT-qPCR analyses of human gastric cancer cell lines, including AGS,
SGC-7901, BGC-823, MKN-45 and MGC-803. As shown in Fig. 1, all of the 6 members of the
miR-17-92 cluster could be clearly detected in individual
human gastric cancer cell lines, albeit with different levels. The
expression levels of the individual miR-17-92 members in the
AGS cells were comparable to those in the SGC-7901 and BGC-823
cells. However, the expression levels of the miR-17-92
cluster in the MKN-45 and MGC-803 cells were significantly lower
than those in the AGS, SGC-7901 and BGC-823 cells. The expression
levels of miR-17 (P=0.0003), miR-18 (P=0.0041),
miR-19a (P=0.0114), miR-19b (P=0.0008), miR-20
(P=0.0086) and miR-92 (P=0.0348) were significantly
decreased approximately 2- to 4-fold in the MGC-803 cells, as
compared with those in the AGS cells. It was indicated that the
miR-17-92 expression levels in the MGC-803 cells were the
lowest among the 5 human gastric cancer cell lines. Thus, the
MGC-803 cell line was selected to perform the following experiments
to explore the function of the miR-17-92 cluster in gastric
cancer progression.
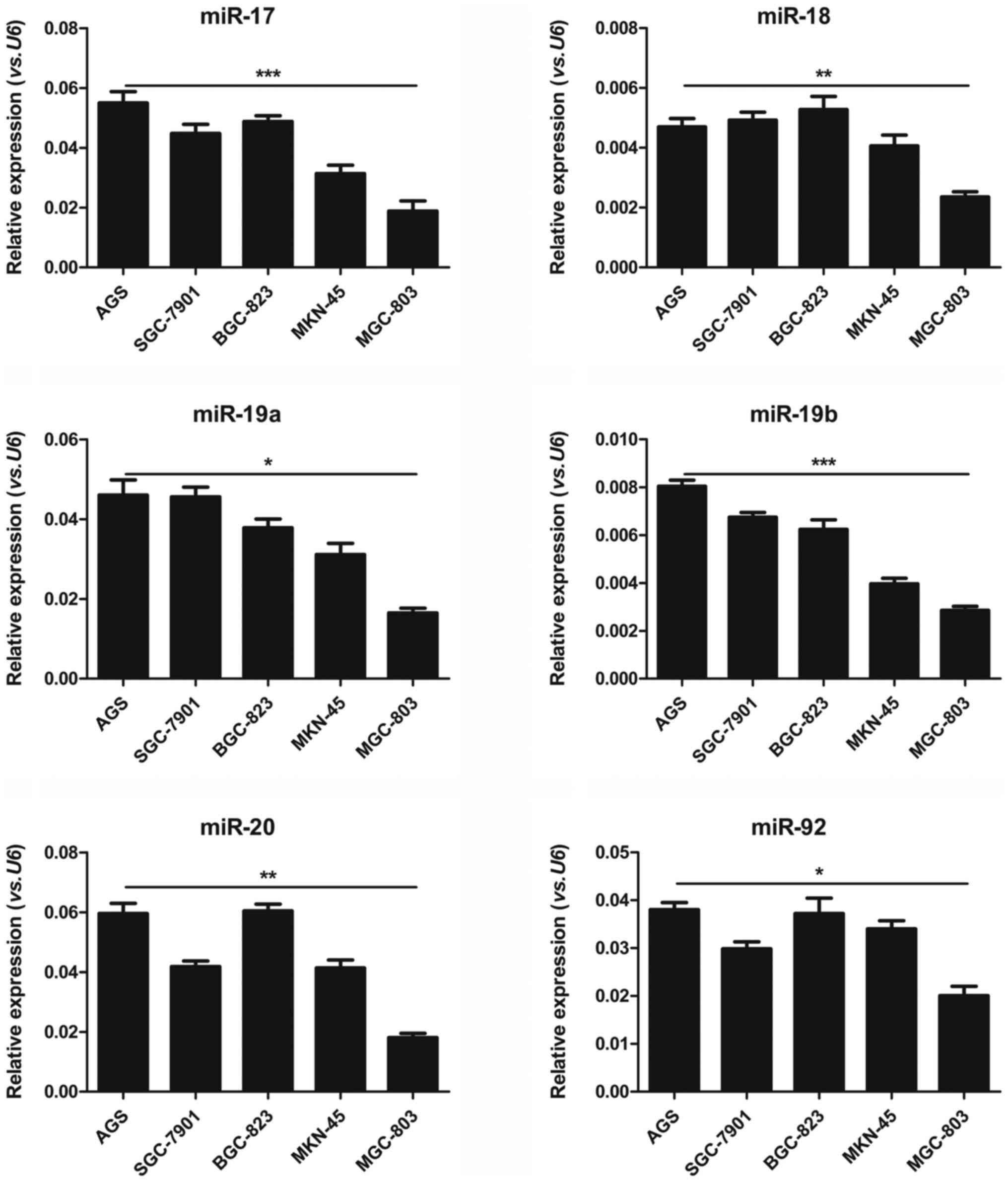 | Figure 1Distinct miR-17-92 expression
patterns in human gastric cancer cell lines. RT-qPCR analysis of
the expression of the 6 miR-17-92 family members
(miR-17, miR-18, miR-19a, miR-19b,
miR-20 and miR-92) in AGS, SGC-7901, BGC-823, MKN-45
and MGC-803 human gastric cancer cell lines. Gene expression was
normalized to U6, and measured in triplicate (Student’s
t-test, *P<0.05, **P<0.01,
***P<0.001). |
Establishment of a
miR-17-92-overexpressing MGC-803 cell line
To explore the effects of the miR-17-92
cluster on the biological behaviors of the MGC-803 cells, the
constructed miR-17-92 overexpression plasmid was transfected
into the MGC-803 cells. The positive monoclones were selected upon
exposure to puromycin (5 ng/µl) for 2 weeks. The formed monoclones
were further verified for miR-17-92 expression by RT-qPCR
analyses. The expression levels of each miR-17-92 family
member were significantly increased in the MGC-803-miR-17-92 cells
compared to those in the MGC-803-control cells, 4-fold for
miR-17 (P=0.0109), 3-fold for miR-18 (P=0.0415),
6-fold for miR-19a (P=0.0111), 5-fold for miR-19b
(P=0.0104), 4-fold for miR-20 (P=0.0058) and 2-fold for
miR-92 (P=0.0169), indicating a successful introduction of
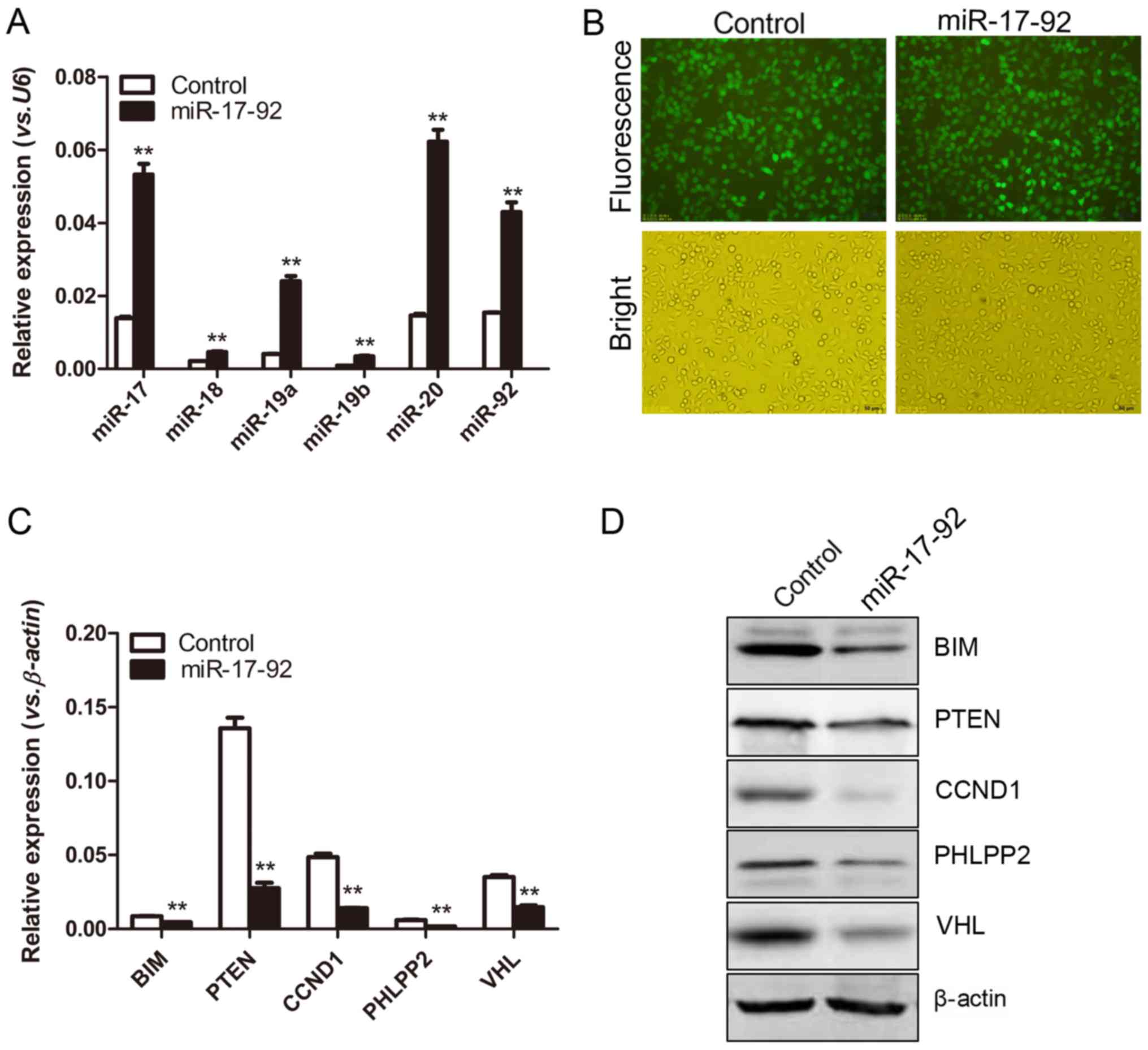
the miR-17-92 cluster into the MGC-803 cells (Fig. 2A). Moreover, GFP was used as a
marker for the transfection of the plasmids. As shown in Fig. 2B, both the established
MGC-803-miR-17-92 and the MGC-803-control cells presented strong
GFP signals observed under a fluorescence microscope (Fig. 2B). The GFP signals of the
MGC-803-miR-17-92 and the MGC-803-control cells were measured by
flow cytometry. The percentages of GFP-positive cells in the 2
established cell lines were 95.71 and 96.88%, respectively (data
not shown), indicating the successful transfection of the plasmids.
Moreover, the expression levels of certain well-known target genes
of the miR-17-92 cluster, including BIM, PTEN,
CCND1, PHLPP2 and VHL were significantly
decreased in the MGC-803-miR-17-92 cells compared to those in the
MGC-803-control cells (Fig. 2C).
Consistently, the expression levels of BIM, PTEN, cyclin D1, PHLPP2
and VHL at the protein level were decreased, as detected by western
blot analysis (Fig. 2D). Taken
together, it was indicated that the MGC-803 gastric cancer cell
line stably overexpressing the miR-17-92 cluster was
successfully established.
miR-17-92 overexpression promotes
proliferation and reduces apoptosis in vitro and accelerates tumor
xenograft growth in vivo
To determine whether the overexpression of the
miR-17-92 cluster modulates the proliferative activity of
the MGC-803 cells, a CCK-8 assay was performed. As shown in
Fig. 3A, the OD values at 450 nm
in the miR-17-92 overexpression group (0.76±0.10 and
1.26±0.11) were significantly higher than those in the control
group (0.37±0.06 and 0.61±0.08) at 48 and 72 h, respectively (both
P<0.01). To further characterize the effects of miR-17-92
on the viability of the MGC-803 cells, a CellTiter-Glo cell
viability assay was carried out. It was shown that the luciferase
activities of the MGC-803-control cells were 37,098±6,833,
45,846±7,009 and 63,883±8,471 at 24, 48 and 72 h, respectively;
while those in the MGC-803-miR-17-92 cells were 54,545±8,151,
86,085±5,774 and 102,027±11,564 (**P<0.01,
***P<0.001, Fig.
3B). To investigate whether the overexpression of
miR-17-92 affected the apoptotic capability of the MGC-803
cells, a TUNEL assay was performed. As shown in Fig. 3C, both the MGC-803-control and
MGC-803-miR-17-92 cells underwent apoptosis in a time-dependent
manner. The apoptotic rates of the MGC-803-control cells were
14.67±1.28, 18.28±1.75 and 20.17±1.76%, while those of the
MGC-803-miR-17-92 cells were 5.33±1.28, 7.11±1.07 and 7.97±1.25% at
24, 48 and 72 h, respectively (all P<0.01). Moreover, the cell
cycle was analyzed by flow cytometry, and the results revealed no
significant differences between the MGC-803-control and
MGC-803-miR-17-92 cells (data not shown).
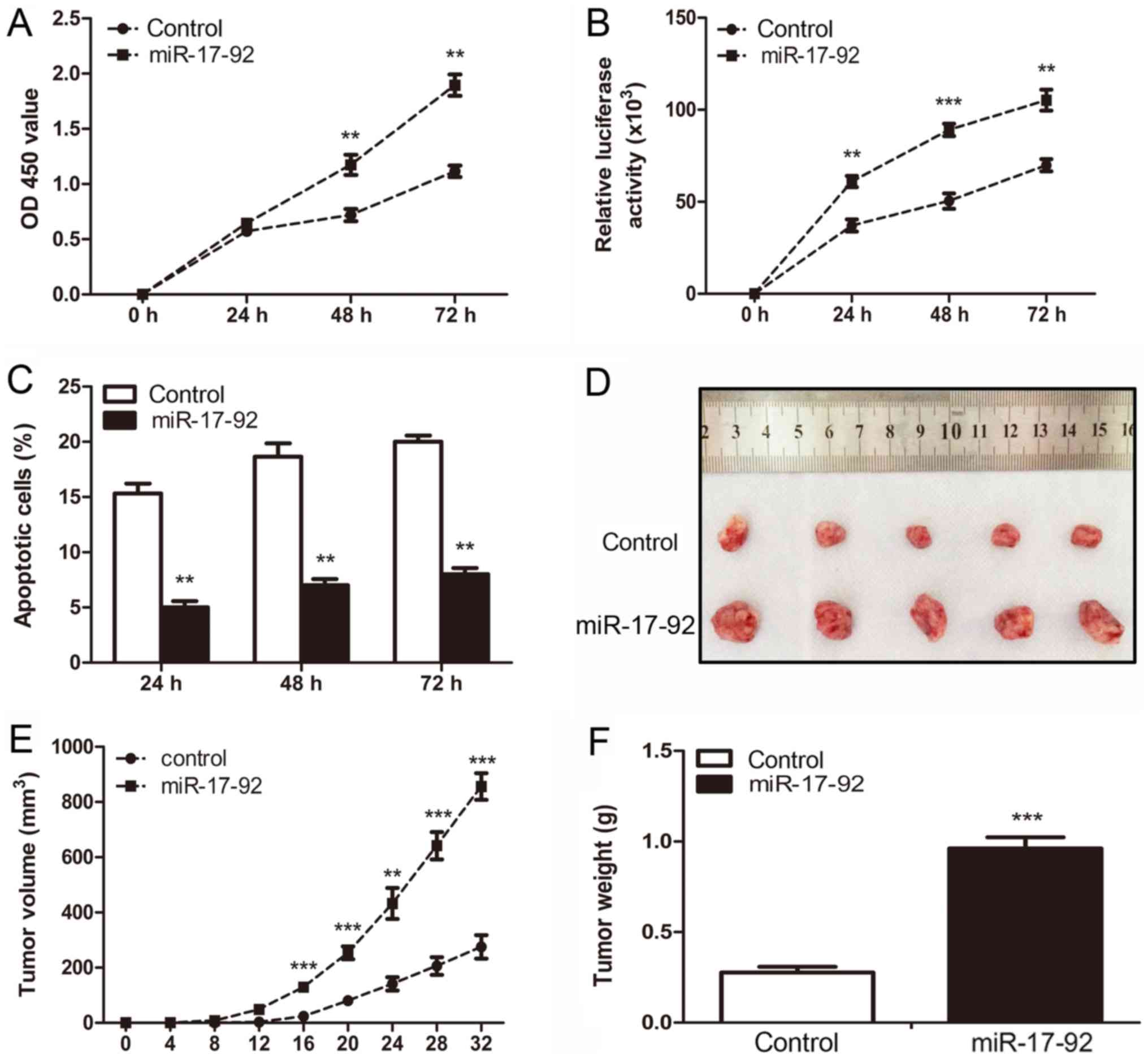 | Figure 3miR-17-92 overexpression
promotes proliferation and reduces apoptosis in vitro and
accelerates xenograft tumor growth in vivo. (A) Cell
proliferation activity between the MGC-803-control and
MGC-803-miR-17-92 cells was analyzed by CCK-8 assay at 24, 48 and
72 h (Student’s t-test, **P<0.01). (B) Cell viability
assay was carried out to detect the viability of the 2 established
cell lines at 24, 48 and 72 h. The relative luciferase activity was
detected among the 2 established cell lines. Significant
differences are indicated (Student’s t-test,
**P<0.01, ***P<0.001). (C) TUNEL assay
was carried out to quantitatively evaluate the apoptotic cells
between the 2 established cell lines at 24, 48 and 72 h.
Significant differences are indicated (Student’s t-test,
**P<0.01). (D) Representative images of tumor growth
in the mouse xenograft model. The MGC-803-control or
MGC-803-miR-17-92 cells were injected subcutaneously into nude
mice. Each group had 5 nude mice. (E) At 32 days after the
injection, the mice were sacrificed and tumor volume was measured
every 4 days after inoculation. Significant differences are
indicated (Student’s t-test, **P<0.01,
***P<0.001). (F) Tumors in individual mice were
weighed. Significant differences are indicated (Student’s t-test,
***P<0.001). |
To further investigate whether miR-17-92
overexpression can promote cell growth in vivo, BALB/c-nude mice
were injected subcutaneously into the right flanks with the
MGC-803-control or MGC-803-miR-17-92 cells to establish a
heterotopic xenograft tumor mouse model. A representative image of
tumors from both the control and the miR-17-92
overexpression group is shown in Fig.
3D. The mean tumor volume in the miR-17-92
overexpression group reached >800 mm3, but was <400
mm3 in the control group at 32 days following
implantation (Fig. 3E). The mean
tumor weight in the miR-17-92 overexpression group
(0.94±0.16 g) was much greater than that in the control group
(0.26±0.08 g, P<0.001, Fig.
3F). Taken together, these results indicated that
miR-17-92 overexpression in the MGC-803 cells increased the
prolifera-tive activity and decreased the apoptosis of the gastric
cancer cells in vitro, and promoted tumor xenograft growth
in vivo.
miR-17-92 overexpression modulates the
AKT, ERK and NF-kB signaling pathways
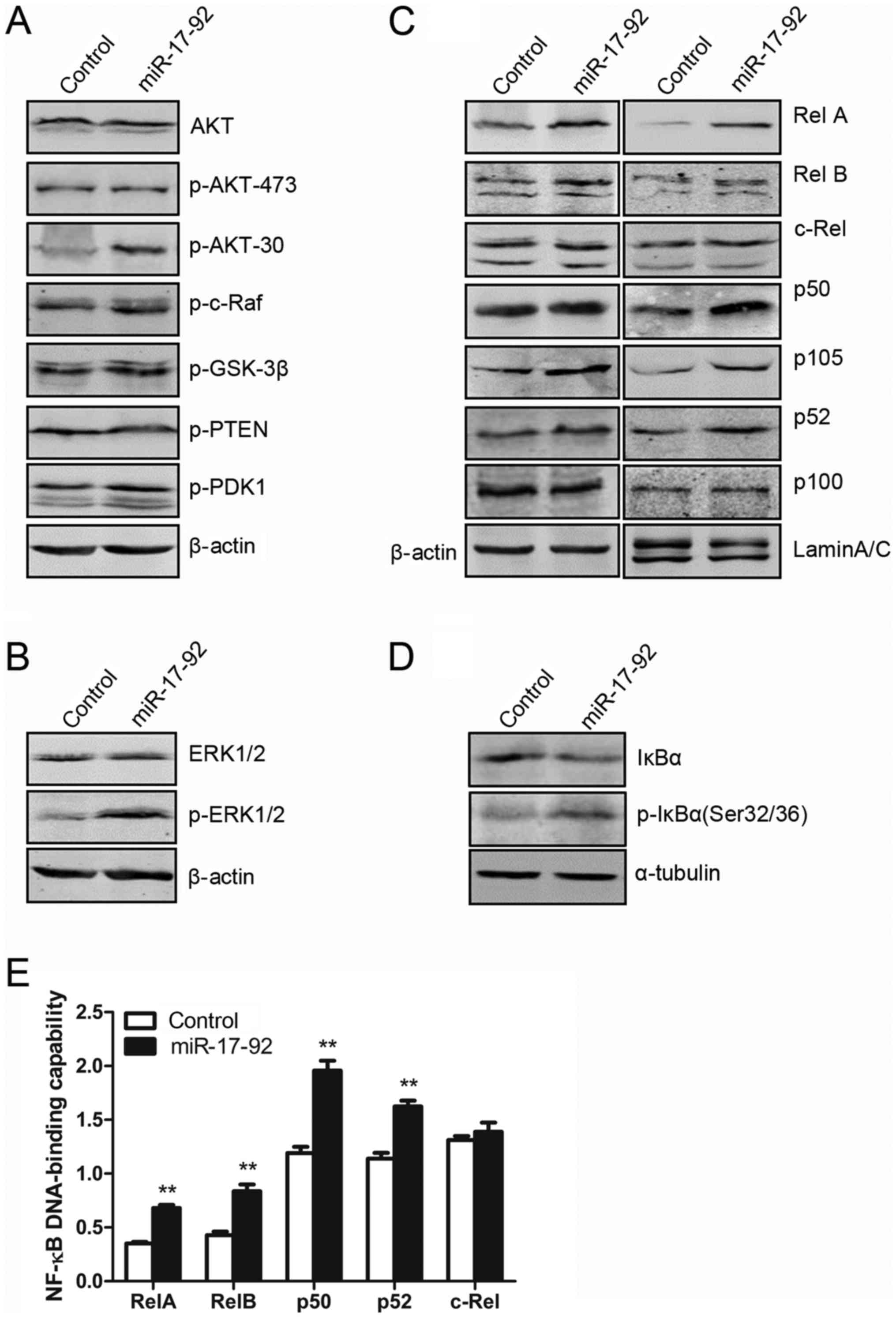
As shown in Fig.
4A, the protein expression of AKT signaling molecules,
including AKT, p-AKT-473, p-AKT-308, p-c-Raf, p-GSK-3β, p-PTEN and
p-PDK1, were measured by western blot analyses. Total AKT
expression was similar between the MGC-803-control and
MGC-803-miR-17-92 cells. However, miR-17-92 over-expression
induced the phosphorylation of AKT at Thr308, but not at Ser473
(Fig. 4A), which was due to the
inhibition of PTEN expression (Fig.
2D). No significant changes were observed in the other AKT
signaling molecules (Fig. 4A). As
shown in Fig. 4B, the protein
expression level of total ERK1/2 in the MGC-803-miR-17-92 cells was
comparable to that in the MGC-803-control cells. However, the
overexpression of the miR-17-92 cluster in the MGC-803 cells
induced the phosphorylation of ERK1/2 (p-ERK1/2). Thus,
miR-17-92 overexpression in the MGC-803 cells induced
p-ERK1/2 expression without affecting total ERK1/2 expression.
Subsequently, to investigate whether
miR-17-92 over-expression affects the activity of NF-κB
signaling, western blot analysis was performed. As shown in
Fig. 4C, the expression of
RelA/p65 and p50, representing canonical NF-κB activity, was
clearly increased in both the CE and NE fractions in the
MGC-803-miR-17-92 cells compared to those in the MGC-803-control
cells. The phosphorylation of IκB-α at Ser32/36 was induced by
miR-17-92 overexpression in the MGC-803 cells (Fig. 4D), leading to the subsequent
degradation of IκB-α. The expression of IκB-α, an important
upstream regulator of the canonical NF-κB signaling, was markedly
decreased in the MGC-803-miR-17-92 cells. Moreover, the expression
of RelB and p52, representing non-canonical NF-κB activity, was
slightly increased in the NE of the MGC-803-miR-17-92 cells.
However, the expression of c-Rel in the MGC-803-miR-17-92 cells was
similar to that in the MGC-803-control cells (Fig. 4C). To determine the exact
contribution of the miR-17-92 gene cluster to the NF-κB
DNA-binding capabilities in the MGC-803 cells, an ELISA-based NF-κB
activity assay was performed. As shown in Fig. 4E, the DNA-binding capability of
each NF-κB subunit including RelA, p50, RelB and p52 in the NE of
the MGC-803-miR-17-92 cells was significantly increased as compared
with that in the MGC-803-control cells. However, the DNA-binding
capability of c-Rel was comparable between the 2 cell lines. Taken
together, these results indicated that the introduction of the
miR-17-92 cluster into the MGC-803 cells activated the AKT,
ERK and NF-κB signaling pathways, likely contributing to the
increased cellular proliferation.
miR-17-92 overexpression enhances the
migratory and invasive abilities of the MGC-803 cells by regulating
epithelial-mesenchymal transition (EMT)
As shown in Fig.
5A, the number of migrated cells in the miR-17-92
overexpression group was increased approximately 3-fold as compared
with that of the control group (P<0.01). Cell motility was also
evaluated by scratch wound healing assay. As shown in Fig. 5B, a scratched cell monolayer was
formed and images were captured at 24, 48 and 72 h. It was shown
that the MGC-803-miR-17-92 cells migrated from the edge towards the
scratch center more rapidly than the MGC-803-control cells,
indicating an enhanced migratory ability. Transwell invasion assay
was performed to further examine the role of miR-17-92 in
the invasive ability of the MGC-803 cells. As shown in Fig. 5C, the number of cells that invaded
the Matrigel layer from the MGC-803-miR-17-92 group was increased
approximately 4-fold compared with that of the MGC-803-control
group (P<0.01). The results of the gelatin zymography assay
revealed that MMP-9 activity was not detected in either of the
established cell lines; however, MMP-2 activity was markedly
increased in the MGC-803-miR-17-92 cells (Fig. 5D). The expression levels of key
molecules involved in EMT, including E-cadherin, claudin1, EpCAM,
CK8/18, N-cadherin and Vimentin, were examined by western blot
analysis. The expression levels of claudin1 and EpCAM, representing
epithelial markers, were decreased in the MGC-803-miR-17-92 cells
compared to the MGC-803-control cells, while the expression levels
of other epithelial markers, such as E-cadherin and CK8/18 were
unaffected. The expression levels of N-cadherin and Vimentin,
mesenchymal markers, were increased in the MGC-803-miR-17-92 cells
compared with the MGC-803-control cells (Fig. 5E). Thus, EMT occurred when the
miR-17-92 cluster was overexpressed in the MGC-803 cells.
Collectively, these results indicated that the enforced
introduction of the miR-17-92 cluster into the MGC-803 cells
enhanced the migratory and invasive abilities of the cells, which
was owing to the occurrence of EMT.
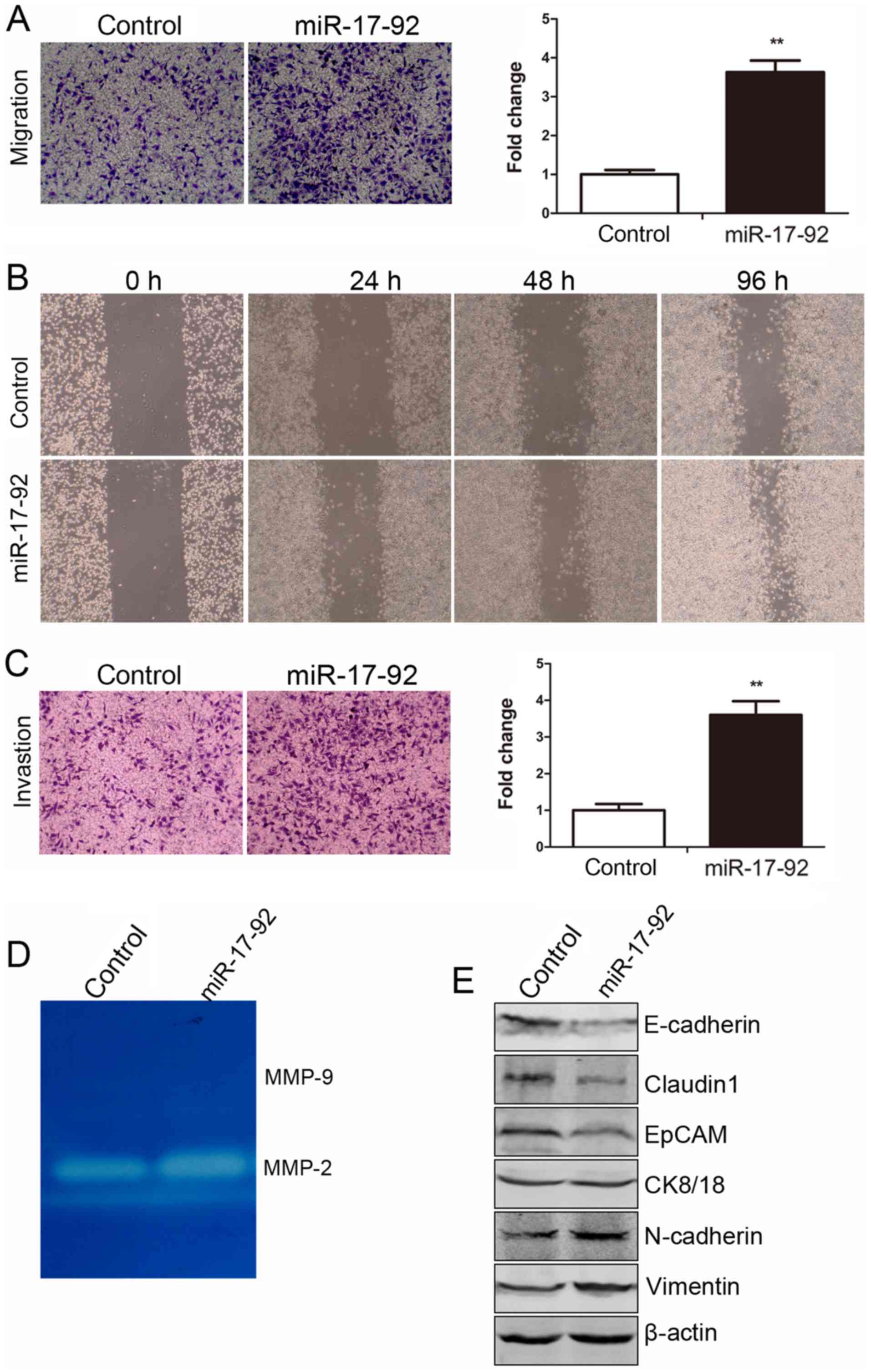 | Figure 5miR-17-92 overexpression
enhances the migratory and invasive abilities of the MGC-803 cells
by regulating epithelial-mesenchymal transition (EMT). (A) The
migratory ability of the MGC-803-control and MGC-803-miR-17-92
cells was detected by Transwell migration assay. Left panel,
representative images of migrated cells between the 2 established
cell lines were photographed under an inverted microscope (x20
magnification). Right panel, the number of migrated cells was
quantified (Student’s t-test, **P<0.01). (B) The
migratory ability of the cells was detected by a scratch wound
healing assay at 24, 48 and 72 h. (C) The invasive ability of the
MGC-803-control and MGC-803-miR-17-92 cells was detected by
Transwell invasion assay. Left panel, representative images of
invaded cells between the 2 established cell lines were
photographed under an inverted microscope (x20 magnification).
Right panel, the number of invaded cells was quantified (Student’s
t-test, **P<0.01). (D) The activity of matrix
metalloproteinase (MMP)-2 and MMP-9 was evaluated by the gelatin
zymography assay. (E) The protein expression levels of E-cadherin,
claudin1, EpCAM, CK8/18, N-cadherin and Vimentin were examined by
western blot analysis in the 2 established cell lines. The level of
each protein was normalized against β-actin. |
TRAF3 is a direct target of the miR-17-92
cluster in the MGC-803 cells
Of note, we focused on the TRAF3 gene, as it was
found to be one of the predicted target genes of the
miR-17-92 cluster using TargetScan Release 5.1 online
software (http://www.targetscan.org/,
Whitehead Institute for Biomedical Research, Cambridge, MA, USA)
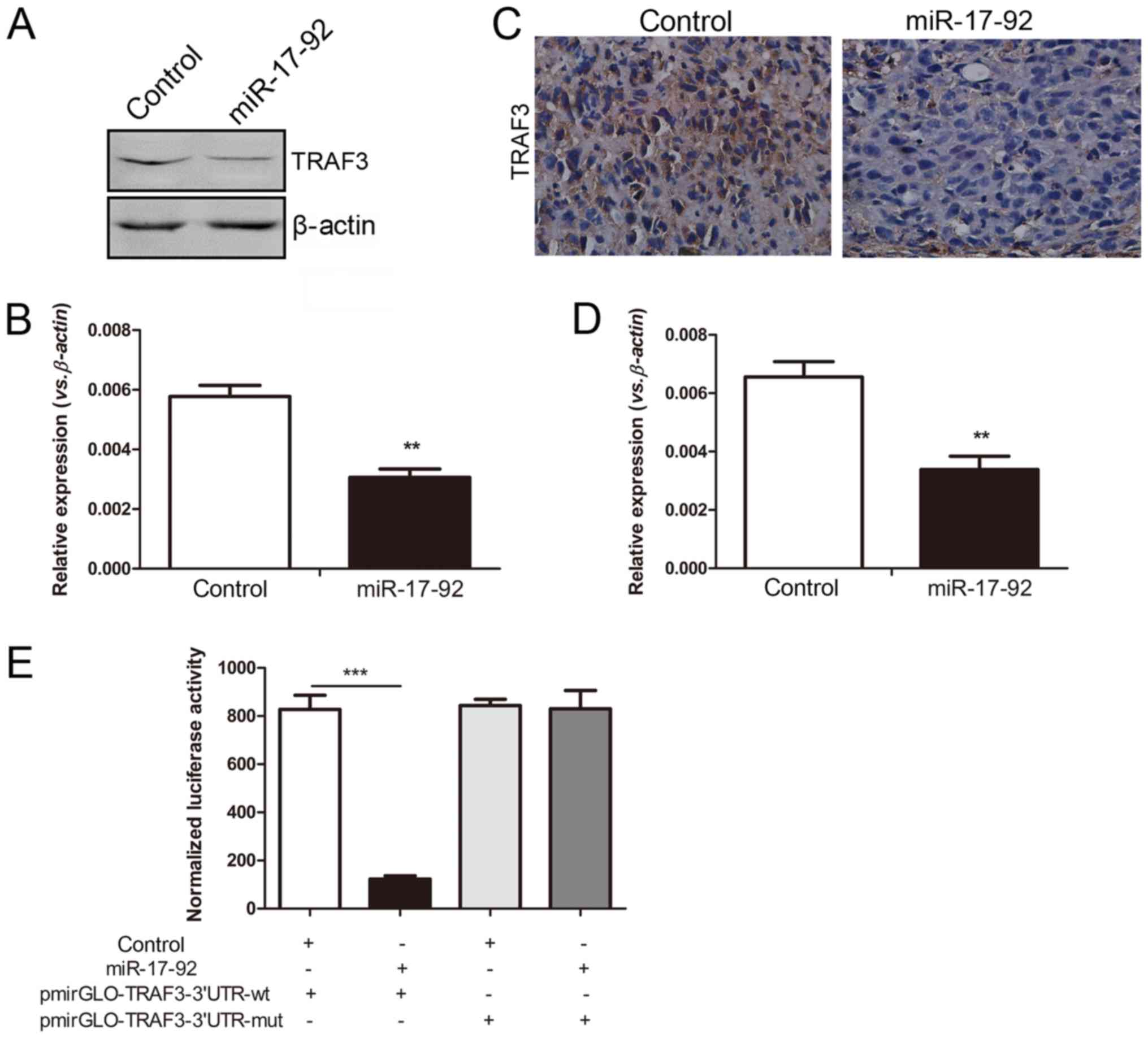
(data not shown). The protein expression of TRAF3 was significantly
decreased in the MGC-803 cells overexpressing the miR-17-92
cluster, which was in line with the decreased mRNA level detected
by RT-qPCR analysis (Fig. 6A and
B). The deregulated NF-κB family members were observed in the cells
overexpressing the miR-17-92 cluster. Compared with the
control group, both the mRNA and protein expression levels of TRAF3
were decreased in the xenograft tumors in the miR-17-92
overex-pression group (Fig. 6C and
D), which was consistent with the in vitro results.
To verify whether TRAF3 is truly a direct
target of the miR-17-92 cluster, a luciferase assay was
performed. The pmirGLO-TRAF3-3′-UTR-wild-type or
pmirGLO-TRAF3-3′-UTR-mutant vector were co-transfected into 293T
cells with the control or miR-17-92 plasmid. As shown in
Fig. 6E, the atopic expression of
the miR-17-92 cluster significantly reduced the relative
luciferase activity of the wild-type 3′-UTR of TRAF3
(P<0.001), but not that of the mutant 3′-UTR of TRAF3.
Taken together, it was confirmed that TRAF3 was a direct target of
miR-17-92, which could downregulate TRAF3 expression by
directly binding to the 3′-UTR of TRAF3.
TRAF3-silencing promotes cellular
proliferation and enhances migration and invasion abilities of
MGC-803 cells in vitro
We further investigated whether TRAF3 functions as
a target gene of the miR-17-92 cluster in the MGC-803 cells.
The MGC-803 cells were transfected with shRNA-TRAF3 or
shRNA-control plasmid, respectively. As shown in Fig. 7A, both the mRNA and protein levels
of TRAF3 were markedly lower in the MGC-803-siTRAF3 cells as
compared with the MGC-803-sictrl cells, indicating a successful RNA
interference (RNAi) of the TRAF3 gene. The expression levels
of RelA and p50 were clearly increased in the NE of the
MGC-803-siTRAF3 cells compared to that in the MGC-803-sictrl cells.
Moreover, the expression levels of RelB and p52 were upregulated in
both the CE and NE in the MGC-803-siTRAF3 cells compared to that in
the control cells (Fig. 7B). To
further determine the exact contribution of TRAF3 silencing to the
NF-κB DNA-binding capabilities in the MGC-803 cells, an ELISA-based
NF-κB activity assay was performed. In line with the results of
western blot analysis, the average DNA-binding capability of RelA,
p50, RelB and p52 in the NE of the MGC-803-siTRAF3 cells were
significantly increased compared to that in the MGC-803-sictrl
cells, while the DNA-binding capability of c-Rel remained unaltered
(Fig. 7C). Therefore, TRAF3
silencing significantly activated not only canonical NF-κB
activity, but also non-canonical NF-κB activity in the MGC-803
cells.
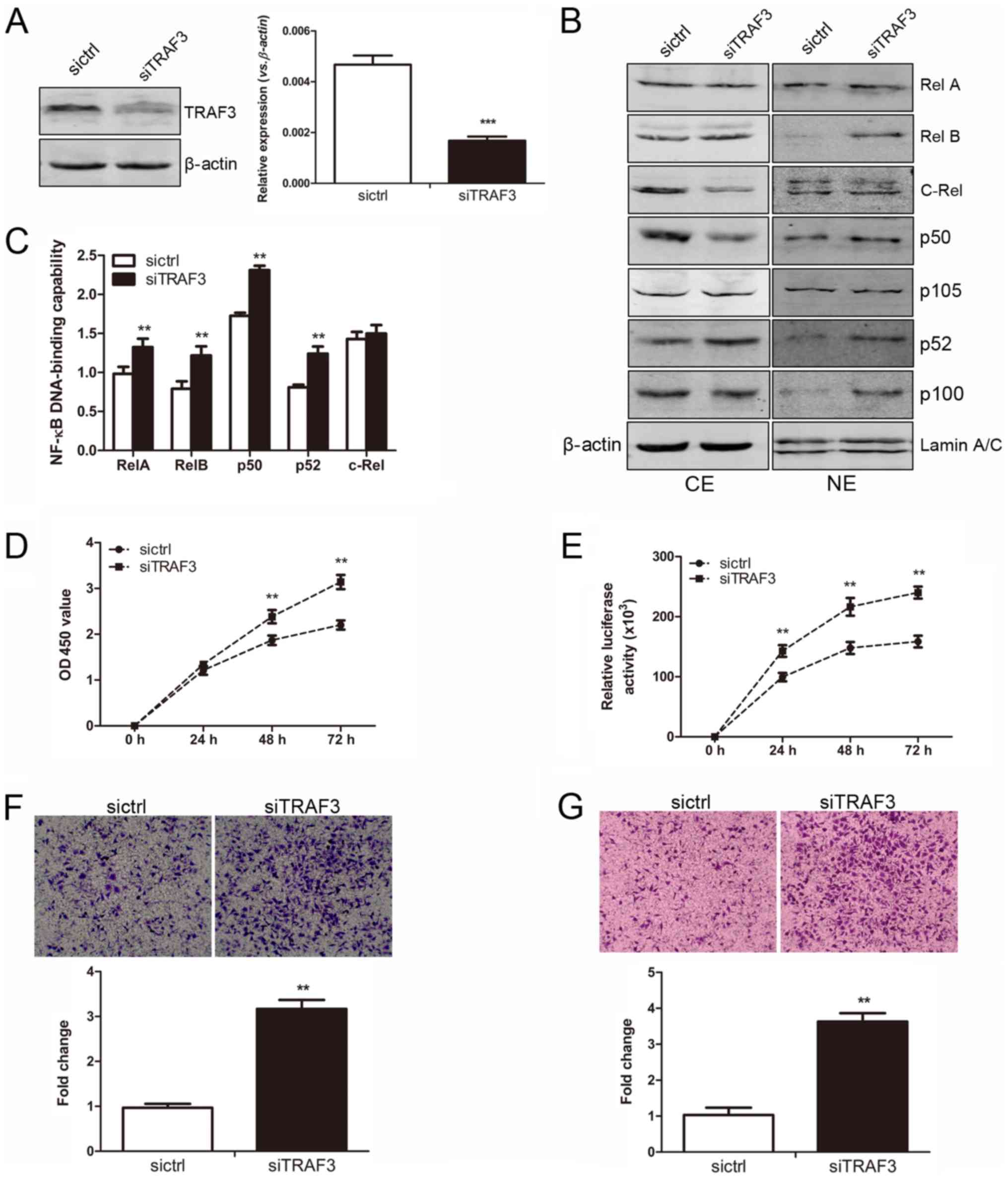 | Figure 7Tumor necrosis factor receptor
associated factor 3 (TRAF3) silencing promotes cellular
proliferation and enhances the migratory and invasive abilities of
the MGC-803 cells in vitro. (A) The expression of TRAF3 was
downregulated in the MGC-803 cells in which TRAF3 was
silenced. Left panel, the protein expression of TRAF3 in the
MGC-803-sictrl and MGC-803-siTRAF3 cells was determined by western
blot analysis. The level of each protein was normalized against
β-actin. Right panel, the mRNA expression of TRAF3 between
the 2 established cell lines was determined by RT-qPCR analysis
Gene expression was normalized to β-actin, and measured in
triplicate. Significant differences are indicated (Student’s
t-test, ***P<0.001). (B) The protein expression of
nuclear factor (NF)-κB subunits between the 2 established cell
lines was examined by western blot analysis. Protein expression in
the cytoplasmic extract (CE) and nuclear extract (NE) was
normalized against β-actin and LaminA/C, respectively. (C) Nuclear
extracts of cells were prepared and tested for the DNA-binding
activities using the TransAM™ NF-κB family transcription factor
assay kit (Student’s t-test, **P<0.01). (D) The cell
proliferative activity between the MGC-803-sictrl and
MGC-803-siTRAF3 cells was examined by CCK-8 assay at 24, 48 and 72
h (Student’s t-test, **P<0.01). (E) A cell viability
assay was carried out to detect the viability of the 2 established
cell lines at 24, 48 and 72 h. The relative luciferase activity was
detected among the 2 established cell lines. Significant
differences are indicated (Student’s
t-test,**P<0.01). (F) The migratory ability of the
MGC-803-sictrl and MGC-803-siTRAF3 cells was detected by Transwell
migration assay. Upper panel, representative images of migrated
cells between the 2 established cell lines were photographed under
an inverted microscope (x20 magnification). Bottom panel, the
number of migrated cells was quantified (Student’s t-test,
**P<0.01). (G) The invasive ability of the
MGC-803-sictrl and MGC-803-siTRAF3 cells was detected by Transwell
invasion assay. Upper panel, representative images of invaded cells
between the 2 established cell lines were photographed under an
inverted microscope (x20 magnification). Bottom panel, the number
of invaded cells was quantified (Student’s t-test,
**P<0.01). |
As shown by CCK-8 assay, the OD values at 450 nm in
the MGC-803-siTRAF3 group (0.72±0.10, 1.71±0.20 and 2.14±0.17) were
significantly higher than those in the MGC-803-sictrl group
(0.64±0.13, 1.39±0.16 and 1.63±0.15) at 24, 48 and 72 h,
respectively (all P<0.01, Fig.
7D). Moreover, a CellTiter-Glo cell viability assay was carried
out. The relative luciferase activities of the MGC-803-sictrl cells
were 110,886±17,428, 165,340±28,222 and 183,612±16,171 at 24, 48
and 72 h, respectively; while those in the MGC-803-siTRAF3 cells
were 150,425±30,618, 224,387±31,175 and 241,320±24,341 at 24, 48
and 72 h, respectively (all P<0.01, Fig. 7E). Transwell migration and invasion
assays were also performed. TRAF3 silencing markedly increased the
number of migrated cells approximately 3-fold in the
MGC-803-siTRAF3 group as compared with the control group
(P<0.01, Fig. 7F). Moreover,
the number of cells that invaded the Matrigel layer from the
MGC-803-siTRAF3 group was increased approximately ut 3-fold
compared with that of the corresponding control group (P<0.01,
Fig. 7G). Taken together, these
results indicated that TRAF3 silencing promoted the proliferation,
and enhanced the migratory and invasive abilities of the MGC-803
cells in vitro.
TRAF3 functions as an important prognosis
factor in patients with human gastric cancer
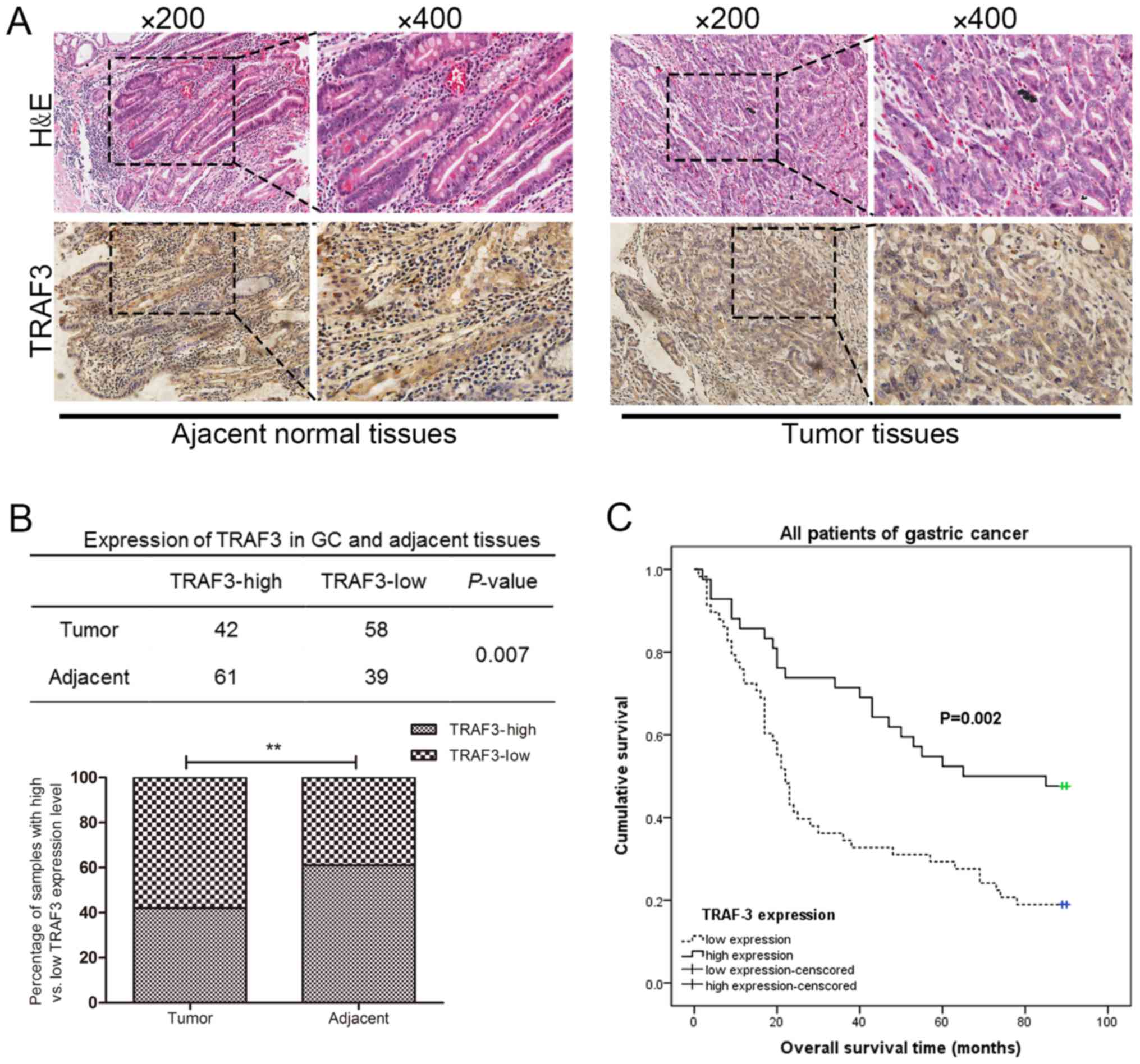
The protein expression levels of TRAF3 in human
gastric cancer tissues and adjacent normal tissues were detected by
IHC analyses. As shown in Fig. 8A,
the expression of TRAF3 could be clearly detected in both the
nuclear and the cytoplasmic portions of the adjacent normal
tissues. By contrast, the expression of TRAF3 was predominantly
detected in the cytoplasm of the gastric tumor tissues. Moreover,
the expression of TRAF3 in the gastric tumor tissues was
significantly lower than that in the adjacent normal gastric
mucosa, and there was a statistically significance (P=0.007)
between the gastric tumor tissues and the adjacent normal tissues
(Fig. 8B). As shown in Table I, the association between TRAF3
expression and the clinicopathological characteristics in the 100
paired gastric cancer patients was evaluated. The expression of
TRAF3 was negatively associated with tumor diameter and the TNM
stage of patients with gastric cancer (P<0.05). The expression
of TRAF3 was not associated with the other clinicopathological
characteristics examined, including age, sex, site of tumor,
histological types and tumor differentiation (P>0.05).
 | Table IAssociation between TRAF3 expression
and clinicopathological characteristics of patients with gastric
cancer. |
Table I
Association between TRAF3 expression
and clinicopathological characteristics of patients with gastric
cancer.
|
Characteristics | No. of patients
(n=100) | TRAF3 expression
| |
|---|
| Low (n=58) | High (n=42) | P-value |
|---|
| Age (years) | | | | |
| Median | 65 (32-81) | | | |
| ≤65 | 51 | 29 | 22 | 0.814 |
| >65 | 49 | 29 | 20 | |
| Sex | | | | |
| Male | 64 | 37 | 27 | 0.960 |
| Female | 36 | 21 | 15 | |
| Tumor diameter | | | | |
| ≤5 cm | 52 | 25 | 27 | 0.036 |
| >5 cm | 48 | 33 | 15 | |
| Site of tumor | | | | |
| Cardia | 13 | 5 | 8 | 0.325 |
| Body | 28 | 18 | 10 | |
| Antrum | 51 | 29 | 22 | |
| Unknown | 8 | 6 | 2 | |
| Histological
type | | | | |
| Tubular
adenocarcinoma | 66 | 42 | 24 | 0.228 |
| Mucinous
adenocarcinoma | 10 | 4 | 6 | |
| Signet ring cell
adenocarcinoma | 11 | 7 | 4 | |
| Undifferentiated
carcinoma | 13 | 5 | 8 | |
| Tumor
differentiation | | | | |
| Well | 15 | 6 | 9 | 0.219 |
| Moderate | 74 | 44 | 30 | |
| Poor | 11 | 8 | 3 | |
| TNM stage | | | | |
| I-II | 42 | 18 | 24 | 0.009 |
| III-IV | 58 | 40 | 18 | |
OS curves were also plotted according to TRAF3
expression using the Kaplan-Meier method. Survival analysis
indicated that the low TRAF3 expression group had a significantly
shorter OS rate than that of the high TRAF3 expression group, and
there was a statistically significance (P=0.002) between the low
TRAF3 expression group and the high TRAF3 expression group
(Fig. 8C). Taken together, these
results indicated that TRAF3 expression was downregulated in
gastric cancer tissues and that TRAF3 functions as an important
prognosis factor in patients with gastric cancer.
Discussion
In this study, the role of the miR-17-92
cluster in gastric cancer progression was systematically examined.
The introduction of the miR-17-92 overexpression plasmid
into the MGC-803 human gastric cancer cells significantly promoted
cell growth both in vitro and in vivo, which was
predominantly due to the activation of the AKT, ERK and NF-κB
signaling pathways. The migratory and invasive abilities of the
cells were also promoted by the overexpression of the
miR-17-92 cluster, which was contributed to the occurrence
of EMT. To the best of our knowledge, in this study, for the first
time, TRAF3, a negative regulator of the NF-κB signaling pathway,
was demonstrated to be a direct target of the miR-17-92
cluster in the MGC-803 human gastric cancer cells. Of note, TRAF3
expression was indicative of a favorable prognosis factor of human
gastric cancer patients.
In this study, we observed a significant and rapid
growth rate of the MGC-803 cells due to the introduction of the
miR-17-92 cluster both in vitro and in vivo.
Similar to the finding of previous studies, we demonstrated that
miR-17-92 overexpression significantly promoted cell growth
due to increased cellular proliferation and decreased cellular
apoptosis. As previously demonstrated, the increased expression of
all the members of the miR-17-92 cluster induced by the
overexpression of E2F transcription factor 1 (E2F1) significantly
promoted the proliferation of mouse palatal mesenchymal cells
(31). The activation of the
miR-17-92 pathway is involved in the growth of small cell
lung cancer due to the driving factor of the friend leukemia virus
integration 1 (FLI1) gene (32). In vivo, the conditional
deletion of miR-17-92 in mouse pancreatic β-cells has been
shown to significantly reduce the proliferative ability of
pancreatic β-cells (33).
miR-17-5p has been demonstrated to promote the proliferation
of gastric cancer cells by targeting SOCS6 (21). Gain- and loss-of-function assays
have also demonstrated that miR-20a affects the
proliferative ability of uveal melanoma cells in vitro
(34). The upregulation of
miR-18a has been shown to promote cell proliferation by
increasing Cyclin D1 expression by regulating PTEN-PI3K-AKT-mTOR
signaling in esophageal squamous cell carcinoma cells (35). The over-expression of miR-17
in gastric cancer cells is associated with several
proliferation-associated oncogenes amplification, including
MYC, CCNE1, ERBB2 and FGFR2. The
knockdown of miR-17 suppresses the proliferative potential
of KATO-III gastric cancer cells accompanied by the downregulation
of p-ERK1/2 expression (22).
Similarly, in this study, the expression of PTEN, which functions
as a negative regulator of AKT signaling, was decreased upon the
overexpression of the miR-17-92 cluster in the MGC-803
cells. Correspondingly, we observed that the expression of p-AKT at
Thr308 was induced without the level of total AKT being affected.
The induction of p-ERK1/2 was also observed. These data confirm the
function of the miR-17-92 cluster in MGC-803 gastric cancer
cells with respect to cell proliferation, and the activation of the
AKT and ERK signaling pathways are implicated in these processes.
Furthermore, we found that the overexpression of the miR-17-92
cluster significantly promoted tumor growth in vivo.
Therefore, we hypothesized that the activation of the AKT and ERK
signaling pathways is important for the rapid tumor growth. Further
studies are required to examine this hypothesis by analyzing these
pathways in tumor tissues using assays, such as western blot
analysis.
Furthermore, a number of studies have revealed the
oncogenic activity of the miR-17-92 cluster by regulating
apoptotic genes. The overexpression of the miR-17-92 cluster
in DU145 prostate cancer cells has been shown to significantly
decrease cellular apoptosis (29).
In vivo, the overexpression of miR-17-92 has been
shown to block the induction of BIM-mediated apoptosis in multiple
transgenic mouse models of acute lymphoblastic leukemia (36). In this study, we observed that
miR-17-92 overexpression in the MGC-803 cells resulted in
the clear inhibition of cellular apoptosis due to the suppressed
expression of BIM, which was a direct target of miR-17-92.
As a pro-apoptotic protein, BIM regulates cell death by
antagonizing anti-apoptotic proteins, such as Bcl-2 (37). The downregulation of PTEN
expression can also contribute to the resistance to apoptosis. In
myeloma cells, the overexpression of miR-19a has been shown
to significantly inhibit cellular apop-tosis and to function as an
oncogenic miRNA by targeting the PTEN-AKT pathway (38). In response to various extracellular
signals, the PI3K/AKT pathway participates in diverse cellular
responses to promote cell survival, proliferation and cell growth.
The NF-κB signaling pathway can be regulated by the
miR-17-92 cluster and regulates the balance between cellular
proliferation and apoptosis (39,40).
For example, miR-17-92 contributes to chronic myeloid
leukemia (CML) leukemo-genesis by targeting A20 and activates NF-κB
signaling (39). miR-17-92
has been demonstrated to drive lymphomagenesis by suppressing the
expression of multiple negative regulators of the PI3K and NF-κB
pathways, and by inhibiting the mitochondrial apoptotic pathway
(41). In this study, in the
MGC-803 cells, both the canonical and non-canonical NF-κB signaling
pathways were activated due to the overexpression of the
miR-17-92 cluster, which contributed to the regulation of
multiple cellular functions, including cell proliferation and
apoptosis.
Furthermore, in this study, we demonstrated that
miR-17-92 overexpression markedly promoted the migratory and
invasive abilities of the MGC-803 cells. Our results are consistent
with those of previous studies that the miR-17-92 cluster
plays an important role in cancer invasion and metastasis (42). The overexpression of the
miR-17-92 cluster in DU145 prostate cancer cells has been
shown to promote cellular migration and invasion in vitro
(29). miR-17 facilitates
the cell motility of melanoma cells by targeting ETV1 (43). miR-19 plays an important
role in the invasion and migration of lung cancer cells by
modulating the EMT process (44).
miR-19a/b promotes gastric cancer cell migration and
invasion by targeting the antagonist of c-Myc-MXD1 (24). By contrast, Bahari et al
reported that miR-17-92 was downregulated in gastric cancer
and that its expression was negatively associated with metastasis
(45). In this study, we found
that miR-17-92 overexpression markedly enhanced the
migratory and invasive abilities of the MGC-803 cells. The
activation of NF-κB signaling in the MGC-803 cells overexpressing
the miR-17-92 cluster increased the expression of MMP-2,
which is a downstream gene of the NF-κB signaling pathway. Of note,
we demonstrated that there was a decrease in E-cadherin, EpCAM and
Claudin1 expression levels coupled with an increase in N-cadherin
and Vimentin expression levels in the MGC-803-miR-17-92 cells,
indicating that EMT had occurred. EMT, which is characterized by
the loss of epithelial characteristics and the acquisition of a
mesen-chymal phenotype, plays a critical role in allowing cancer
cells to invade adjacent tissue and migrate to distant sites, where
these cancer cells continue to proliferate and generate new tumors
(46). Mounting evidence has
indicatd that in epithelial cancers, including gastric cancer, the
induction of EMT is a major event that provides mobility to cancer
cells in order to generate metastasis (45). miR-19 has been reported to
trigger EMT in lung cancer cells, and thus enhances the migratory
and invasive abilities of A549 and HCC827 cells (44). In this study, we demonstrated that
miR-17-92 overexpression promoted the migratory and invasive
abilities of the MGC-803 gastric cancer cells, and the increased
MMP-2 expression and the occurrence of EMT were implicated in these
processes.
In mammals, the transcription factor family of
NF-κB includes RelA/p65, NF-κB1/p50, RelB, NF-κB2/p52 and c-Rel.
NF-κB subunits play important roles in cellular survival,
inflammation, proliferation, apoptosis and tumori-genesis (47). TRAF3 is one of the most enigmatic
members of the TRAF family with distinct cell and context-specific
roles (48). TRAF3 is
characterized to be an ubiquitin E3 ligase, and is mainly composed
of a signature TRAF domain at the carboxyl terminus and a typical
C3HC4 RING finger domain at the N-terminus (49). TRAF3 functions as an upstream
regulator of NF-κB signaling, and suppresses NF-κB activity by
constantly mediating the degradation of NF-κB-inducing kinase (NIK)
(50). The downregulation of TRAF3
in NOD1 ligand-stimulated cells leads to enhanced NF-κB reporter
activity, while it increases TRAF3 expression and suppresses NF-κB
activity (51). In this study, we
found that both the canonical and non-canonical NF-κB activities
were activated in the MGC-803 gastric cancer cells due to the
introduction of the miR-17-92 cluster. We further observed
that the overexpression of the miR-17-92 cluster decreased
TRAF3 expression at both the mRNA and protein level. Therefore, one
possibility is that TRAF3 may be regulated by the miR-17-92
cluster directly in MGC-803 cells. As a matter of fact, TRAF3 can
be regulated by various miRNAs. For example, TRAF3 has been
demonstrated to be targeted by miR-32 to affect
non-canonical NF-κB signaling and consequently, NIK stabi-lization
(52). TRAF3 is also directly
targeted by miR-214-3p to promote osteoclast- and
bone-resorbing activity (53). In
murine macrophages, Burkholderia pseudomallei-derived
miR-3473 has been shown to enhance NF-κB expression by
targeting TRAF3 and is associated with different inflammatory
responses compared to Burkholderia thailandensis (54). TRAF3 has also been demonstrated to
be directly targeted by miR-3178 (55), miR-322 (56) and miR-576-3p (57). In this study, we used a luciferase
reporter gene containing the 3′-UTR of the TRAF3 gene to
testify the direct repression of TRAF3 by the
miR-17-92 cluster. Therefore, miR-17-92 was able to
bind to the complementary sites in the 3′-UTR of the TRAF3
mRNA and regulate either its mRNA degradation or translational
repression. In this study, we demonstrate that TRAF3 is a direct
target of the miR-17-92 cluster. Accordingly, the
overexpression of the miR-17-92 cluster in the MGC-803 cells
caused a reduction in TRAF3-NF-κB signaling, which further
influenced multiple cellular functions. To the best of our
knowledge, this is the first study to demonstrate that TRAF3
is regarded as a novel target of the miR-17-92 cluster.
TRAF3 is ubiquitously expressed in the majority of
tissues, including brain, heart, lung, liver and spleen. The
expression pattern of TRAF3 in human gastric cancer tissue remains
undefined. Zou et al demonstrated that the expression of
TRAF3 was upregulated in Helicobacter pylori-positive
gastric tissues but not in Helicobacter pylori-negative
gastric tissues, and the expression of TRAF3 was also positive in
both the intestinal and diffuse type gastric cancer samples
(55). The clinicopathological
analyses of 100 paired gastric cancer tissues from a tissue
microarray was performed and the results suggested that TRAF3
expression was negatively associated with tumor diameter and TNM
stage. Moreover, Kaplan-Meier survival analysis also revealed that
a low expression of TRAF3 was associated with a shorter OS rate of
patients with gastric cancer. Therefore, TRAF3 expression may be
considered as a novel biomarker for the prognosis of patients with
gastric cancer.
In conclusion, the findings of this study provide a
key tumor-promoting role of the miR-17-92 cluster in the
development and progression of gastric cancer. The miR-17-92
cluster plays an important role in cellular biological functions,
including cell growth, proliferation, apoptosis, migration and
invasion of MGC-803 gastric cancer cells. To the best of our
knowledge, for the first time, we provide evidence that the
miR-17-92 cluster plays a pivotal role in regulating the
NF-κB signaling pathway by directly targeting TRAF3. The
miR-17-92/TRAF3/NF-κB axis may thus be a novel target for human
gastric cancer therapy and may be further considered as a potential
prognostic factor in the future.
Funding
This study was supported by grants from the
National Natural Science Foundation of China (WC, grant no.
81272737), China Scholarship Council, Jiangsu Provincial
Postgraduate Research and Practice Innovation Program (FL, grant
no. KYZZ16_0089), Jiangsu Provincial Science and Technology
Department Clinical Special Project (WC, grant no. BL2014016),
Jiangsu Provincial Natural Science Foundation of China (FG, grant
no. BK20151211) and Jiangsu Provincial Medical Youth Talent (JX,
grant no. QNRC2016725).
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors’ contributions
WC and FG conceived and designed the study.
Experimental procedures were conducted by FL, LC and JX. Data
analysis was performed by LC. The manuscript was prepared by FL,
and JX critically revised the manuscript. Financial support was
obtained by WC, FG and JX. All authors have read and approved the
final manuscript.
Ethics approval and consent to
participate
The human tissues examined in this study were from
a tissue microarray. All procedures involving animals and animal
experiments were approved by the Animal Care and Use Committee of
Soochow University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors greatly acknowledge Professor Yong Zhao
for the gift of the miR-17-92 plasmids.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
Statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang C: Novel functions for small RNA
molecules. Curr Opin Mol Ther. 11:641–651. 2009.
|
|
6
|
Lujambio A and Lowe SW: The microcosmos of
cancer. Nature. 482:347–355. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Esquela-Kerscher A and Slack FJ: Oncomirs
- microRNAs with a role in cancer. Nat Rev Cancer. 6:259–269. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tsai MM, Wang CS, Tsai CY, Huang HW, Chi
HC, Lin YH, Lu PH and Lin KH: Potential diagnostic, prognostic and
therapeutic targets of MicroRNAs in human gastric cancer. Int J Mol
Sci. 17:172016. View Article : Google Scholar
|
|
9
|
He L, Thomson JM, Hemann MT,
Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe
SW, Hannon GJ, et al: A microRNA polycistron as a potential human
oncogene. Nature. 435:828–833. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ota A, Tagawa H, Karnan S, Tsuzuki S,
Karpas A, Kira S, Yoshida Y and Seto M: Identification and
characterization of a novel gene, C13orf25, as a target for
13q31-q32 amplification in malignant lymphoma. Cancer Res.
64:3087–3095. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hayashita Y, Osada H, Tatematsu Y, Yamada
H, Yanagisawa K, Tomida S, Yatabe Y, Kawahara K, Sekido Y and
Takahashi T: A polycistronic microRNA cluster, miR-17-92 is
overexpressed in human lung cancers and enhances cell
proliferation. Cancer Res. 65:9628–9632. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Farazi TA, Horlings HM, Ten Hoeve JJ,
Mihailovic A, Halfwerk H, Morozov P, Brown M, Hafner M, Reyal F,
van Kouwenhove M, et al: MicroRNA sequence and expression analysis
in breast tumors by deep sequencing. Cancer Res. 71:4443–4453.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chow TF, Mankaruos M, Scorilas A, Youssef
Y, Girgis A, Mossad S, Metias S, Rofael Y, Honey RJ, Stewart R, et
al: The miR-17-92 cluster is over expressed in and has an oncogenic
effect on renal cell carcinoma. J Urol. 183:743–751. 2010.
View Article : Google Scholar
|
|
14
|
Zhu H, Han C and Wu T: MiR-17-92 cluster
promotes hepatocarcinogenesis. Carcinogenesis. 36:1213–1222. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu G, Tang JQ, Tian ML, Li H, Wang X, Wu
T, Zhu J, Huang SJ and Wan YL: Prognostic values of the miR-17-92
cluster and its paralogs in colon cancer. J Surg Oncol.
106:232–237. 2012. View Article : Google Scholar
|
|
16
|
Li H, Wu Q, Li T, Liu C, Xue L, Ding J,
Shi Y and Fan D: The miR-17-92 cluster as a potential biomarker for
the early diagnosis of gastric cancer: Evidence and literature
review. Oncotarget. 8:45060–45071. 2017.PubMed/NCBI
|
|
17
|
Mogilyansky E and Rigoutsos I: The
miR-17/92 cluster: A comprehensive update on its genomics,
genetics, functions and increasingly important and numerous roles
in health and disease. Cell Death Differ. 20:1603–1614. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Valladares-Ayerbes M, Blanco M, Haz M,
Medina V, Iglesias-Díaz P, Lorenzo-Patiño MJ, Reboredo M,
Santamarina I, Figueroa A, Antón-Aparicio LM, et al: Prognostic
impact of disseminated tumor cells and microRNA-17-92 cluster
deregulation in gastrointestinal cancer. Int J Oncol. 39:1253–1264.
2011.PubMed/NCBI
|
|
19
|
Ren C, Wang W, Han C, Chen H, Fu D, Luo Y,
Yao H, Wang D, Ma L, Zhou L, et al: Expression and prognostic value
of miR-92a in patients with gastric cancer. Tumour Biol.
37:9483–9491. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tsujiura M, Komatsu S, Ichikawa D,
Shiozaki A, Konishi H, Takeshita H, Moriumura R, Nagata H,
Kawaguchi T, Hirajima S, et al: Circulating miR-18a in plasma
contributes to cancer detection and monitoring in patients with
gastric cancer. Gastric Cancer. 18:271–279. 2015. View Article : Google Scholar
|
|
21
|
Wu Q, Luo G, Yang Z, Zhu F, An Y, Shi Y
and Fan D: miR-17-5p promotes proliferation by targeting SOCS6 in
gastric cancer cells. FEBS Lett. 588:2055–2062. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Park D, Lee SC, Park JW, Cho SY and Kim
HK: Overexpression of miR-17 in gastric cancer is correlated with
proliferation-associated oncogene amplification. Pathol Int.
64:309–314. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu W, Takanashi M, Borjigin N, Ohno SI,
Fujita K, Hoshino S, Osaka Y, Tsuchida A and Kuroda M: MicroRNA-18a
modulates STAT3 activity through negative regulation of PIAS3
during gastric adenocarcinogenesis. Br J Cancer. 108:653–661. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu Q, Yang Z, An Y, Hu H, Yin J, Zhang P,
Nie Y, Wu K, Shi Y and Fan D: MiR-19a/b modulate the metastasis of
gastric cancer cells by targeting the tumour suppressor MXD1. Cell
Death Dis. 5:e11442014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang F, Li T, Zhang B, Li H, Wu Q, Yang L,
Nie Y, Wu K, Shi Y and Fan D: MicroRNA-19a/b regulates multidrug
resistance in human gastric cancer cells by targeting PTEN. Biochem
Biophys Res Commun. 434:688–694. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu Q, Yang Z, Wang F, Hu S, Yang L, Shi Y
and Fan D: MiR-19b/20a/92a regulates the self-renewal and
proliferation of gastric cancer stem cells. J Cell Sci.
126:4220–4229. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pfaffl MW: A new mathematical model for
relative quantification in real-time RT-PCR. Nucleic Acids Res.
29:e452001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu F, Zhou J, Zhou P, Chen W and Guo F:
The ubiquitin ligase CHIP inactivates NF-κB signaling and impairs
the ability of migration and invasion in gastric cancer cells. Int
J Oncol. 46:2096–2106. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou P, Ma L, Zhou J, Jiang M, Rao E, Zhao
Y and Guo F: miR-17-92 plays an oncogenic role and conveys
chemo-resistance to cisplatin in human prostate cancer cells. Int J
Oncol. 48:1737–1748. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu J, Zhou P, Wang W, Sun A and Guo F:
RelB, together with RelA, sustains cell survival and confers
proteasome inhibitor sensitivity of chronic lymphocytic leukemia
cells from bone marrow. J Mol Med (Berl). 92:77–92. 2014.
View Article : Google Scholar
|
|
31
|
Li L, Shi B, Chen J, Li C, Wang S, Wang Z
and Zhu G: An E2F1/MiR-17-92 Negative Feedback Loop mediates
proliferation of Mouse Palatal Mesenchymal Cells. Sci Rep.
7:51482017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li L, Song W, Yan X, Li A, Zhang X, Li W,
Wen X, Zhou L, Yu D, Hu JF, et al: Friend leukemia virus
integration 1 promotes tumorigenesis of small cell lung cancer
cells by activating the miR-17-92 pathway. Oncotarget.
8:41975–41987. 2017.PubMed/NCBI
|
|
33
|
Chen Y, Tian L, Wan S, Xie Y, Chen X, Ji
X, Zhao Q, Wang C, Zhang K, Hock JM, et al: MicroRNA-17-92 cluster
regulates pancreatic beta-cell proliferation and adaptation. Mol
Cell Endocrinol. 437:213–223. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhou J, Jiang J, Wang S and Xia X:
Oncogenic role of microRNA 20a in human uveal melanoma. Mol Med
Rep. 14:1560–1566. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang W, Lei C, Fan J and Wang J: miR-18a
promotes cell proliferation of esophageal squamous cell carcinoma
cells by increasing cylin D1 via regulating PTEN-PI3K-AKT-mTOR
signaling axis. Biochem Biophys Res Commun. 477:144–149. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li Y, Deutzmann A, Choi PS, Fan AC and
Felsher DW: BIM mediates oncogene inactivation-induced apoptosis in
multiple transgenic mouse models of acute lymphoblastic leukemia.
Oncotarget. 7:26926–26934. 2016.PubMed/NCBI
|
|
37
|
Gupta S, Read DE, Deepti A, Cawley K,
Gupta A, Oommen D, Verfaillie T, Matus S, Smith MA, Mott JL, et al:
Perk-dependent repression of miR-106b-25 cluster is required for ER
stress-induced apoptosis. Cell Death Dis. 3:e3332012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang X, Chen Y, Zhao P, Zang L, Zhang Z
and Wang X: MicroRNA-19a functions as an oncogene by regulating
PTEN/ AKT/pAKT pathway in myeloma. Leuk Lymphoma. 58:932–940. 2017.
View Article : Google Scholar
|
|
39
|
Jia Q, Sun H, Xiao F, Sai Y, Li Q, Zhang
X, Yang S, Wang H, Wang H, Yang Y, et al: miR-17-92 promotes
leukemogenesis in chronic myeloid leukemia via targeting A20 and
activation of NF-κB signaling. Biochem Biophys Res Commun.
487:868–874. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Trenkmann M, Brock M, Gay RE, Michel BA,
Gay S and Huber LC: Tumor necrosis factor α-induced microRNA-18a
activates rheumatoid arthritis synovial fibroblasts through a
feedback loop in NF-κB signaling. Arthritis Rheum. 65:916–927.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jin HY, Oda H, Lai M, Skalsky RL, Bethel
K, Shepherd J, Kang SG, Liu WH, Sabouri-Ghomi M, Cullen BR, et al:
MicroRNA-17~92 plays a causative role in lymphomagenesis by
coordinating multiple oncogenic pathways. EMBO J. 32:2377–2391.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Quattrochi B, Gulvady A, Driscoll DR, Sano
M, Klimstra DS, Turner CE and Lewis BC: MicroRNAs of the mir-17~92
cluster regulate multiple aspects of pancreatic tumor development
and progression. Oncotarget. 8:35902–35918. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cohen R, Greenberg E, Nemlich Y, Schachter
J and Markel G: miR-17 regulates melanoma cell motility by
inhibiting the translation of ETV1. Oncotarget. 6:19006–19016.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li J, Yang S, Yan W, Yang J, Qin YJ, Lin
XL, Xie RY, Wang SC, Jin W, Gao F, et al: MicroRNA-19 triggers
epithelial-mesenchymal transition of lung cancer cells accompanied
by growth inhibition. Lab Invest. 95:1056–1070. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bahari F, Emadi-Baygi M and Nikpour P:
miR-17-92 host gene, uderexpressed in gastric cancer and its
expression was negatively correlated with the metastasis. Indian J
Cancer. 52:22–25. 2015. View Article : Google Scholar
|
|
46
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gilmore TD: Introduction to NF-kappaB:
Players, pathways, perspectives. Oncogene. 25:6680–6684. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
He JQ, Oganesyan G, Saha SK, Zarnegar B
and Cheng G: TRAF3 and its biological function. Adv Exp Med Biol.
597:48–59. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Arch RH, Gedrich RW and Thompson CB: Tumor
necrosis factor receptor-associated factors (TRAFs)--a family of
adapter proteins that regulates life and death. Genes Dev.
12:2821–2830. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liao G, Zhang M, Harhaj EW and Sun SC:
Regulation of the NF-kappaB-inducing kinase by tumor necrosis
factor receptor-associated factor 3-induced degradation. J Biol
Chem. 279:26243–26250. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Asano N, Imatani A, Watanabe T, Fushiya J,
Kondo Y, Jin X, Ara N, Uno K, Iijima K, Koike T, et al: Cdx2
expression and intestinal metaplasia induced by H. pylori infection
of gastric cells is regulated by NOD1-mediatedinnate immune
responses. Cancer Res. 76:1135–1145. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Malcomson FC, Willis ND, McCallum I, Xie
L, Lagerwaard B, Kelly S, Bradburn DM, Belshaw NJ, Johnson IT and
Mathers JC: Non-digestible carbohydrates supplementation increases
miR-32 expression in the healthy human colorectal epithelium: A
randomized controlled trial. Mol Carcinog. 56:2104–2111. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liu J, Li D, Dang L, Liang C, Guo B, Lu C,
He X, Cheung HY, He B, Liu B, et al: Osteoclastic miR-214 targets
TRAF3 to contribute to osteolytic bone metastasis of breast cancer.
Sci Rep. 7:404872017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Fang Y, Chen H, Hu Y, Li Q, Hu Z, Ma T and
Mao X: Burkholderia pseudomallei-derived miR-3473 enhances NF-κB
via targeting TRAF3 and is associated with different inflammatory
responses compared to Burkholderia thailandensis in murine
macrophages. BMC Microbiol. 16:2832016. View Article : Google Scholar
|
|
55
|
Zou M, Wang F, Jiang A, Xia A, Kong S,
Gong C, Zhu M, Zhou X, Zhu J, Zhu W, et al: MicroRNA-3178
ameliorates inflammation and gastric carcinogenesis promoted by
Helicobacter pylori new toxin, Tip-α, by targeting TRAF3.
Helicobacter. 22:222017. View Article : Google Scholar
|
|
56
|
Gu H, Yu J, Dong D, Zhou Q, Wang JY and
Yang P: The miR-322-TRAF3 circuit mediates the pro-apoptotic effect
of high glucose on neural stem cells. Toxicol Sci. 144:186–196.
2015. View Article : Google Scholar :
|
|
57
|
Yarbrough ML, Zhang K, Sakthivel R, Forst
CV, Posner BA, Barber GN, White MA and Fontoura BM:
Primate-specific miR-576-3p sets host defense signalling threshold.
Nat Commun. 5:49632014. View Article : Google Scholar : PubMed/NCBI
|