Introduction
As one of the most common types of gastrointestinal
malignancies, colorectal cancer (CRC) remains a major public health
problem worldwide (1,2). In China, CRC ranks as the fifth
leading cause of cancer-associated mortality nationwide, and its
incidence is increasing annually (3). At present, surgical resection is
still the most effective treatment for patients with CRC (4). The clinical outcomes of CRC have
gradually improved due to numerous developments in screening,
detection and adjuvant therapy; however, the prognosis of patients
with advanced CRC remains poor due to tumor metastasis and
recurrence (5,6). Recent studies have revealed that
certain genetic and epigenetic abnormalities are strongly
associated with the clinical outcomes of CRC (7,8).
Therefore, identifying the molecular mechanisms involved in the
onset and progression of CRC is required to improve the therapeutic
effectiveness of treatments for CRC.
The kinesin superfamily proteins (KIFs) are
essential microtubule-dependent molecular motors that transport a
variety of cargo, including organelles, transcripts and proteins in
an ATP-dependent manner (9,10).
At present, >45 KIFs have been identified in mammals and are
classified into 14 subfamilies (11). According to the position of the
motor domain, KIF members are divided into N-kinesin, C-kinesin and
M-kinesin sub-groups (11).
Kinesin family member 14 (KIF14), located on chromosome 1q32.1, is
an N-type kinesin and belongs to the kinesin-3 superfamily
(12,13). KIF14 has been reported to contain
four conserved functional domains, including an N-terminal
extension domain that binds with protein regulator of cytokinesis 1
(14), a motor domain required for
movement along the microtubule via the hydrolysis of ATP (15), a forkhead-associated domain which
regulates phosphorylation (15,16),
and a stalk and tail domain for the interaction with citron kinase
(16,17). The functions of KIF14 have been
associated with a variety of biological processes (18,19).
Previous studies have demonstrated that the depletion of KIF14 may
induce the incompletion of cytokinesis and lead to multi-
nucleation (19,20). Additionally, the homozygous
mutation of KIF14 may result in autosomal recessive primary
microcephaly (21,22).
Aberrant overexpression of KIF14 has been reported
in numerous types of cancer (23-31);
the oncogenic effects of KIF14 are also diverse. Overexpressed
KIF14 promotes cell proliferation in malignancies, including
glioblastoma, lung and ovarian cancer (23,24,30,31),
affects cell cycle progression and cytokinesis in hepatocellular
carcinoma (25) and interferes
with chemosensitivity in triple-negative breast cancer (27,29).
The genomic gain of 1q32 has been detected in several cancer types,
including CRC (32); however, the
role of KIF14 in colorectal carcinogenesis remains poorly
understood. Aside from its functional role, the regulation of KIF14
has not been investigated in CRC.
In the present study, it was demonstrated that KIF14
was upregulated in CRC tissues and promoted tumor cell
proliferation. Abnormally overexpressed KIF14 was proposed to
partly contribute to genomic amplification and transcriptional
activation. Additionally, the present study revealed that microRNA
(miRNA/miR)-200c, a well-reported tumor suppressor (33,34),
regulated the expression of KIF14 at the post-transcriptional
level. To the best of our knowledge, the present study is the first
to comprehensively demonstrate the expression profile and
functional roles of KIF14 in CRC, and reveal that downregulated
miR-200c expression may contribute to increased KIF14
expression.
Materials and methods
Ethics statement
All methods conducted in the present study and all
the experimental protocols were approved by the Research Ethics
Committee of Peking University Cancer Hospital and Institute
(Beijing, China).
Patients and specimens
A total of 36 pairs of CRC specimens accompanied
with adjacent non-cancerous tissues were obtained from patients who
underwent colorectal resection at Peking University Cancer Hospital
and Institute from February 2004 to November 2007. All patients
enrolled in the present study were pathologically confirmed as
primary colorectal adenocarcinoma by endoscopic biopsy, and had not
received adjuvant chemotherapy, radiotherapy, or immunotherapy
prior to surgery. Tissue samples were frozen in liquid nitrogen.
The MKI67 immunostaining scores of the specimens were obtained from
the pathological reports and the clinicopathological features of
patients are presented in Table I.
Prior to surgery, written informed consent was obtained from each
patient.
 | Table IAssociation between the expression of
KIF14 and clinicopathological features in 36 patients with CRC. |
Table I
Association between the expression of
KIF14 and clinicopathological features in 36 patients with CRC.
|
Characteristics | No. of patients
(%) | KIF14a | P-valueb |
|---|
| Sex | | | 0.828 |
| Male | 27 (75.0) | 1.048
(0.553-4.368) | |
| Female | 9 (25.0) | 1.341
(0.533-4.177) | |
| Age (years) | | | 0.249 |
| <60 | 18 (50.0) | 0.791
(0.431-5.137) | |
| ≥60 | 18 (50.0) | 1.435
(0.818-4.118) | |
| Tumor size
(cm) | | | 0.008d |
| <4 | 12 (33.3) | 0.696
(0.338-1.240) | |
| ≥4 | 24 (66.7) | 1.746
(0.786-7.164) | |
| Invasion | | | 0.726 |
| T2+T3 | 12 (33.3) | 1.197
(0.639-5.330) | |
| T4 | 24 (66.7) | 1.194
(0.456-3.480) | |
| Lymphatic
metastasis | | | 0.949 |
| Negative | 14 (38.9) | 1.051
(0.442-8.798) | |
| Positive | 22 (61.1) | 1.320
(0.573-3.797) | |
| Distal
metastasis | | | 0.807 |
| Negative | 18 (50.0) | 1.197
(0.537-6.606) | |
| Positive | 18 (50.0) | 1.116
(0.545-3.417) | |
| Stage (AJCC) | | | 0.949 |
| Stage II | 14 (38.9) | 1.051
(0.442-8.798) | |
| Stage III/IV | 22 (61.1) | 1.320
(0.573-3.797) | |
| MKI67
immunostaining score | | | 0.016c |
| <50% | 17 (47.2) | 0.611
(0.409-1.079) | |
| ≥50% | 19 (52.8) | 1.743
(0.780-4.368) | |
Cell culture and transfection
The human CRC cell lines (SW620, SW480, HCT116, RKO
and LoVo) and 293T cells were purchased from the American Type
Culture Collection (ATCC; Manassas, VA, USA). Cells were maintained
in either Dulbecco’s modified Eagle’s medium or RPMI-1640 medium
(HyClone Laboratories; GE Healthcare Life Sciences, Logan, UT, USA)
supplemented with 10% fetal bovine serum (FBS) in a humidified
atmosphere with 5% CO2 at 37°C.
To overexpress KIF14, the KIF14 coding sequence was
amplified from cDNA template of RKO cells and cloned into the
pcDNA3.1 vector (P3.1-KIF14 CDS for short), which was verified by
DNA sequencing. The miR-200c-binding sequence of the KIF14
3′-untranslated region (UTR) was inserted into the pGL3-Control
construct (Promega Corp., Madison, WI, USA) and was denoted as
pGL3-KIF14 wild-type (WT) 3′UTR. A Takara MutanBEST kit (Takara
Bio, Inc., Otsu, Japan) was applied to generate mutant (MUT)
constructs with a single (MUT1/MUT2) or double (DOU MUT) miR-200c
core binding site deletion; mutants were denoted as pGL3-MUT1-
KIF14, pGL3-MUT2-KIF14 or pGL3-Double-MUT-KIF14, respectively. The
designed primers are summarized in Table II.
| Table IIPrimers and sequences. |
Table II
Primers and sequences.
A, Primers for
amplification for expression vectors
|
|---|
| Name | Sequence |
|---|
| pCDNA3.1-KIF14 CDS
clone | Forward:
5′-CGCGGATCCATGTCATTACACAGTACTCA-3′ |
| Reverse:
5′-CCGCTCGAGTCACACCCACTGAATCCTACT-3′ |
| pGL3-WT-KIF14 3′UTR
luciferase reporter | Forward:
5′-CGTCTAGAACCTGATCCTTTCATTTGCCCT-3′ |
| Reverse:
5′-CGTCTAGAGTCAGTGCACATAATTCCAATAGC-3′ |
| pGL3-MUT1-KIF14
3′UTR luciferase reporter | Forward:
5′-GTGAACCTTGTGTGGTGTTTTCAAGCTCT-3′ |
| Reverse:
5′-ATTTACTTACAATCTCTTCCTAACAAGGA-3′ |
| pGL3-MUT2-KIF14
3′UTR luciferase reporter | Forward:
5′-AATCTTCAGTTTGTGCTTTGTAAACTC-3′ |
| Reverse:
5′-AACTACTATACAGGTACTTTAGGAAGTTCT-3′ |
B, RT-qPCR primers
|
| Name | Sequence |
|
| GAPDH | Forward:
5′-TGCACCACCACCTGCTTAGC-3′ |
| Reverse:
5′-GGCATGGACTGTGGTCATGAG-3′ |
| KIF14 | Forward:
5′-CCGACATTACAGATGCACCA-3′ |
| Reverse:
5′-CTTCATTCCTAAGCCTACACC-3′ |
C, KIF14 siRNAs
|
| Name | Sequence |
|
| KIF14 siRNA 1 | Sense:
5′-CAGCGGUGAUAUUCUUGAUTT-3′ |
| Antisense:
5′-AUCAAGAAUAUCACCGCUGTT-3′ |
| KIF14 siRNA 2 | Sense:
5′-CUCAGAGCAAGUUGGAUAUTT-3′ |
| Antisense:
5′-AUAUCCAACUUGCUCUGAGTT-3′ |
| KIF14 siRNA 3 | Sense:
5′-GCCCGUUUAAUAGUCAACATT-3′ |
| Antisense:
5′-UGUUGACUAUUAAACGGGCTT-3′ |
| siRNA NC | Sense:
5′-UUCUCCGAACGUGUCACGUTT-3′ |
| Antisense:
5′-ACGUGACACGUUCGGAGAATT-3′ |
D, KIF14 shRNAs
|
| Name | Sequence |
|
| KIF14 shRNA
T459 |
5′-CTCAGAGCAAGTTGGATAT-3′ |
| KIF14 shRNA
T460 |
5′-CAGCGGTGATATTCTTGAT-3′ |
| shRNA NC |
5′-TTCTCCGAACGTGTCACGT-3′ |
miR-200c mimics (assay ID MC11714) or inhibitor
(assay ID MH11714), and a control oligo (mi-NC) (cat. no. 4464058)
were obtained from Invitrogen (Thermo Fisher Scientific, Inc.
Waltham, MA, USA). KIF14 small interfering (si)RNAs and
lentiviruses expressing KIF14 short hairpin (sh)RNAs, together with
their negative controls were purchased from Shanghai GenePharma
Co., Ltd. (Shanghai, China).
Plasmids, siRNAs, miR-200c mimics or inhibitor, and
their corresponding controls were transiently transfected into
cells by using Lipofectamine® 3000 (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer’s
instructions.
MTT assay
Cells were seeded into 96-well plates at the
concentration of 5×103 cells/well. At the indicated
time-points, 20 μl MTT was added to each well. Following
incubation, dimethyl sulfoxide was used to solubilize the crystals.
The absorbance was recorded at 570 nm using a Model 680 microplate
reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Plate colony formation assay
At 24 or 48 h after transfection, cells were counted
and seeded into 6-well plates at the concentration of 200
cells/well, and then maintained in complete medium, which was
refreshed every 3 days. Following incubation for 2 weeks, colonies
were notably visible. Subsequently, the cells were fixed with 100%
cold methanol for 15 min, stained with 1% crystal violet for 20 min
at room temperature and washed with PBS at least three times. The
colony numbers were then counted.
Cell cycle
At 48 h after transfection, cells were harvested and
fixed with 70% cold ethanol at −20°C. Then, 1×106 cells
were stained with 0.5 ml propidium iodide (PI)/RNase staining
buffer (BD Pharmingen; BD Biosciences San Jose, CA, USA) for 15 min
at room temperature, and the cell cycle distribution was examined
by flow cytometry. The extent of PI staining was detected by a BD
FACSCalibur Flow Cytometer (BD Biosciences). Additionally, DNA
histograms of the cell cycle distribution were processed using
FlowJo Analysis Software v.8.7.3 (FlowJo LLC, Ashland, OR,
USA).
5-Ethynyl-2-deoxyuridine (EdU)
incorporation assay
The effect of KIF14 on cell proliferation was also
determined via an EdU incorporation assay using the Cell-Light™ EdU
In Vitro Imaging kit (Guangzhou RiboBio Co., Ltd., Guangzhou,
China). Cells (1×104 cells/well) were seeded into a
96-well plate and cultured for 24 h, the indicated cells were
incubated with 50 μM EdU for 2 h at 37°C after 24 h
post-transfection. Following several washes, the cells were fixed
with 4% form- aldehyde for 30 min and treated with 0.5% Triton
X-100 for an additional 20 min at room temperature. Then,
permeabilized cells were incubated with 1X Apollo reaction cocktail
for 30 min at room temperature in the dark. Then, the cells were
stained with Hoechst 33342 (5 µg/ml) for 30 min away from
light. The proliferative activity of treated cells was visualized
under a Zeiss confocal microscope LSM 700 (Carl Zeiss AG,
Oberkochen, Germany). Three replicates were performed for each
group, and five fields per well were selected for counting. The
rate of EdU incorporation was calculated as the ratio of
EdU-positive cells (green cells) to the total number of Hoechst
33342 positive cells (blue cells).
Western blotting
Transfected cells were harvested and lysed in
radioimmunoprecipitation assay buffer with a protease inhibitor
cocktail (Roche Applied Science, Penzberg, Germany). Then, the
concentration of protein samples was determined with a Pierce BCA
Protein assay kit (Pierce; Thermo Fisher Scientific, Inc.). Equal
amounts of protein (30 µg) were separated by 10% SDS-PAGE
and then transferred to nitrocellulose membranes (EMD Millipore,
Billerica, MA, USA). Following blocking with 5% non-fat milk for 1
h at room temperature, the membranes were incubated overnight at
4°C with primary antibodies against KIF14 (polyclonal, rabbit
anti-human IgG; 1:1,000; cat. no. ab3746; Abcam, Cambridge, MA,
USA), protein kinase B (Akt; polyclonal, rabbit anti-human IgG;
1:1,000; cat. no. 9272s; Cell Signaling Technology, Inc., Danvers,
MA, USA), phosphorylated-Akt (polyclonal, rabbit anti-human IgG;
1:200; cat. no. 9271s; Cell Signaling Technology, Inc.) or β-actin
(monoclonal, mouse anti-human IgG; 1:3,000; cat. no. AC-15; Sigma-
Aldrich; Merck KGaA, Darmstadt, Germany). Following washing with 1X
PBS containing 0.05% Tween-20 for three times, the membranes were
incubated at room temperature for 1 h with either horseradish
peroxidase-conjugated goat anti-rabbit (1:2,000; cat. no. ZB-2301;
OriGene Technologies, Inc., Beijing, China) or goat anti-mouse
(1:2,000; cat. no. ZB-2305; OriGene Technologies, Inc.) secondary
antibodies. An enhanced chemiluminescence detection kit (Amersham;
GE Healthcare, Chicago, IL, USA) was applied to detect the signal.
Relative protein expression levels were normalized to β-actin and
quantified with ImageJ software (National Institutes of Health,
Bethesda, MD, USA).
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA from freshly cultured cells or tumor
samples ground in liquid nitrogen was extracted using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). Subsequently,
RNA samples were quantified with a NanoDrop spectrophotometer
(ND-1000, NanoDrop; Thermo Fisher Scientific, Inc.) and 1 µg
of total RNA was reverse transcribed into cDNA using a Reverse
Transcription kit (Promega Corp.). cDNA templates were then mixed
with SYBR Green PCR Master Mix (Toyobo Life Science, Osaka, Japan)
and the corresponding primers; qPCR was subsequently performed on
the ABI 7500 Real-time PCR System (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The thermocycling conditions were set to
95°C for 5 min, followed by 40 cycles of 95°C for 10 sec and 60°C
for 30 sec. The subsequent dissociation stage was set to 95°C for
15 sec, 60°C for 30 sec, and 95°C for 15 sec. The primer sets for
the detected genes are listed in Table II. miRNAs were reverse transcribed
by using TaqMan MicroRNA reverse transcription kit (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer’s
instructions. TaqMan MicroRNA Assays and the TaqMan Universal PCR
Master Mix (Invitrogen; Thermo Fisher Scientific, Inc.) were used
to examine miRNA expression, with the thermocycling conditions as
10 min at 95°C followed by 40 cycles of 95°C for 15 sec and 60°C
for 30 sec. Data analysis was conducted by using the
2−ΔΔCq method (35),
and all RT-qPCR reactions were performed in triplicate.
Dual-Luciferase reporter assay
293T cells (~70% confluence) were co-transfected
with luciferase reporter plasmid (200 ng), miR-200c mimics/mi-NC
(100 nM), and the internal control pRL-SV40 (10 ng). Cells were
harvested and lysed; the luciferase activity was determined by the
Dual-Luciferase Reporter Assay System (Promega Corp.) and
quantified using a luminometer. The Renilla luciferase
activity was used as an internal reference to adjust the deviation
caused by the varying cell numbers plated and transfection
efficiency.
Xenograft tumor model
All experiments were approved by the Animal Ethics
Committee of Peking University Cancer Hospital & Institute and
performed in full compliance with the Experimental Animal
Management Ordinance produced by the Peking University Cancer
Hospital & Institute. A total of 10 female BALB/c nude mice (20
g) were purchased from Hua-Fu-Kang Corp. (Beijing, China) and
randomly divided into two groups. Each mouse was subcutaneously
inoculated with 2×106 of the indicated cells into the
right forelimb armpit. When the tumor mass became palpable, the
tumor volume was measured by a caliper every three days and
calculated using the formula: Volume = length ×
width2/2. At day 32 following inoculation, the mice were
sacrificed, and the xenografts were extracted, imaged and processed
for further analysis.
Bioinformatics analysis
The differential expression of KIF14 mRNA between
cancerous and noncancerous CRC tissues was analyzed by using two
integrated data-mining platforms, Gene Expression across Normal and
Tumor tissue (GENT) (36) and
Oncomine (37), which contain a
collection of rearranged substantial microarray data for cancer
profiling. Data from three datasets GSE8671, GSE21510 and GSE20916
were derived from GENT. To reduce the false discovery rate,
P<0.01 was set as a threshold for Oncomine data mining.
Gene set enrichment analysis (GSEA) was conducted by
using the GSEA-2.2.3 software package (Broad Institute, Cambridge,
MA, USA) (38,39) to identify the genes biologically
associated with CRC. The cBioPortal for Cancer Genomics (40) was used to investigate the genetic
alterations of KIF14. Information regarding KIF14 gene mutations,
copy- number alterations (CNAs), and abnormal mRNA expression
levels (RNA-Seq V2 RSEM) was obtained from the provisional The
Cancer Genome Atlas (TCGA) CRC dataset with 629 samples available
at present. Transcript values were transformed into log2 values,
and CNAs were determined with GISTIC 2.0 (41). miRNA target prediction algorithms,
including microRNA.org (http://www.microrna.org) (42) and TargetScan (http://www.targetscan.org) (43), were applied to predict the
potential miRNAs that target the KIF14 3′UTR.
Statistical analysis
All experiments were conducted no less than three
times with each sample tested in triplicate. Data are presented as
the mean ± standard deviation. A two-tailed Student’s t-test or a
Mann-Whitney U-test was employed to evaluate the difference between
two groups where appropriate. One-way analysis of variance followed
by a post hoc Dunnett’s test was used to determine statistical
differences for multiple comparisons. Pearson correlation analysis
was conducted to assess the correlation between two genes of
interest. Statistical analysis was performed using SPSS software
20.0 (IBM Corp, Armonk, NY, USA) and GraphPad Prism 5 software
(GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
KIF14 is significantly upregulated in
CRC
To assess whether KIF14 was ectopically
overexpressed in CRC, integrated microarray data from GENT
(36) and Oncomine (37) were analyzed in the present study. A
total of three Gene Expression Omnibus (GEO) datasets, including
GSE8671, GSE21510 and GSE20916 were profiled with Affymetrix U133
Plus 2.0 platforms; the gene expression values of the CRC and
corresponding normal tissues were selected from GENT. In each
dataset, KIF14 was significantly overexpressed in CRC specimens
compared with in the corresponding normal samples (Fig. 1A). In addition, KIF14 expression
levels were significantly correlated with the mRNA levels of
proliferation-related Ki-67 antigen (MKI67), a well-established
proliferation marker of CRC (44),
in all examined datasets (Fig.
1B). Consistent with the findings of analysis with GENT,
elevated KIF14 expression levels were also observed in CRC samples
compared with the benign tissues from 14 analyses with the Oncomine
database (Fig. 1C). Furthermore,
KIF14 expression was detected in 36 pairs of fresh frozen CRC
specimens by RT-qPCR. In addition to the results from
bioinformatics analyses, 63.9% (23/36) of cases exhibited
significant upregulation of KIF14 in CRC compared with in adjacent
noncancerous tissues (Fig. 1D).
Increased KIF14 expression levels were significantly associated
with tumor size (P=0.008) and MKI67 immunostaining scores (P=0.016;
Table I). In addition, the mRNA
and protein expression levels of KIF14 were detected in the five
CRC cell lines by RT-qPCR and western blotting (Fig. 1E and F). The results revealed that
KIF14 was upregulated in SW620, SW480 and RKO cells, and
downregulated in HCT116 and LoVo cells.
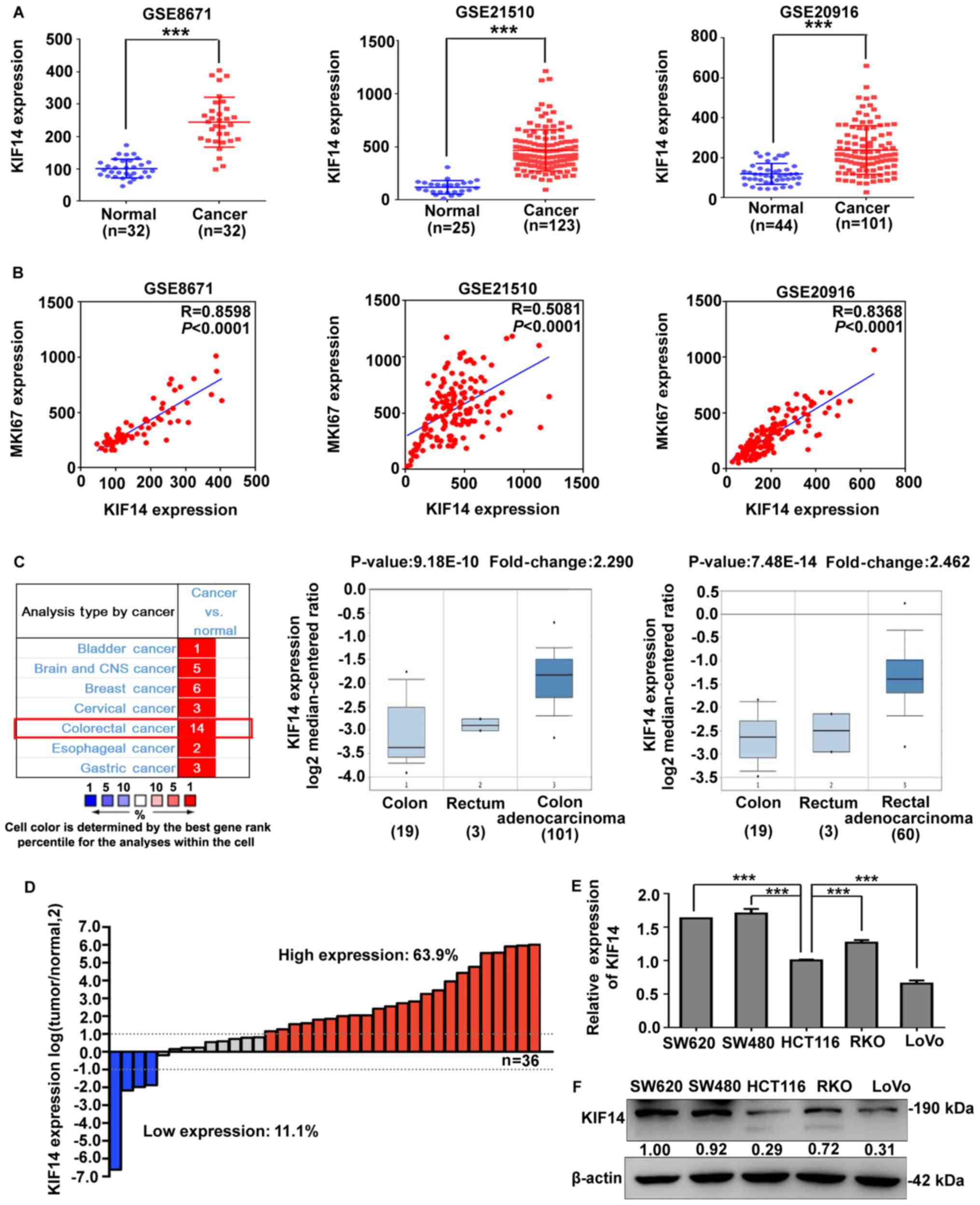 | Figure 1Expression of KIF14 in CRC specimens
and cell lines. (A) KIF14 expression was analyzed in the GSE8671,
GSE21510 and GSE20916 CRC datasets from the Gene Expression across
Normal and Tumor tissue database. (B) Correlation between KIF14 and
MKI67 was investigated via Pearson’s correlation analysis. (C) Left
panel, summarization of KIF14 expression in various cancer types
from the Oncomine database. Middle and right panels, KIF14 mRNA was
overexpressed in colon and rectal adenocarcinomas compared with
normal epithelia. (D) KIF14 mRNA expression in 36 pairs of fresh
frozen CRC tissues was determined by RT-qPCR. Different colors of
the bars indicate the proportions of CRC samples with upregulated
(red, >2-fold), downregulated (blue, <2-fold) and unchanged
(gray) expression of KIF14. (E and F) mRNA and protein expression
levels of KIF14 were evaluated by RT-qPCR and western blotting in
five CRC cell lines. The results were presented as the mean ±
standard deviation from three independent experiments,
***P<0.001. KIF14, kinesin family member 14; CRC,
colorectal cancer; MKI67, proliferation-related Ki-67 antigen;
RT-qPCR, reverse transcription-quantitative polymerase chain
reaction. |
KIF14 accelerates CRC cell proliferation
by modulating progression of the cell cycle
To investigate the function of KIF14 in the
progression of CRC, specific siRNA oligos targeting KIF14 were
introduced into SW480 and RKO cells. Additionally, a recombinant
expression plasmid encoding KIF14 was constructed and transfected
into LoVo cells to observe alterations in phenotype associated with
KIF14. The transfection efficiency of KIF14 siRNA or its
overexpression vector was confirmed by RT-qPCR and western blotting
in SW480 or LoVo cells (Fig. 2A and
B). By using an MTT assay, the present study demonstrated that
decreased KIF14 expression resulted in markedly reduced cell
viability of SW480 and RKO cells, and upregulated KIF14 promoted
the proliferation of LoVo cells (Fig.
2C). In addition, the colony formation assay produced similar
results to that of the MTT assay (Fig.
2D). Furthermore, the DNA synthesis of treated CRC cells was
examined by an EdU incorporation assay. As presented in Fig. 2E, transfection of KIF14 siRNA
attenuated the ratio of EdU-positive SW480 and RKO cells, whereas
overexpressed KIF14 significantly accelerated the DNA synthesis of
LoVo cells.
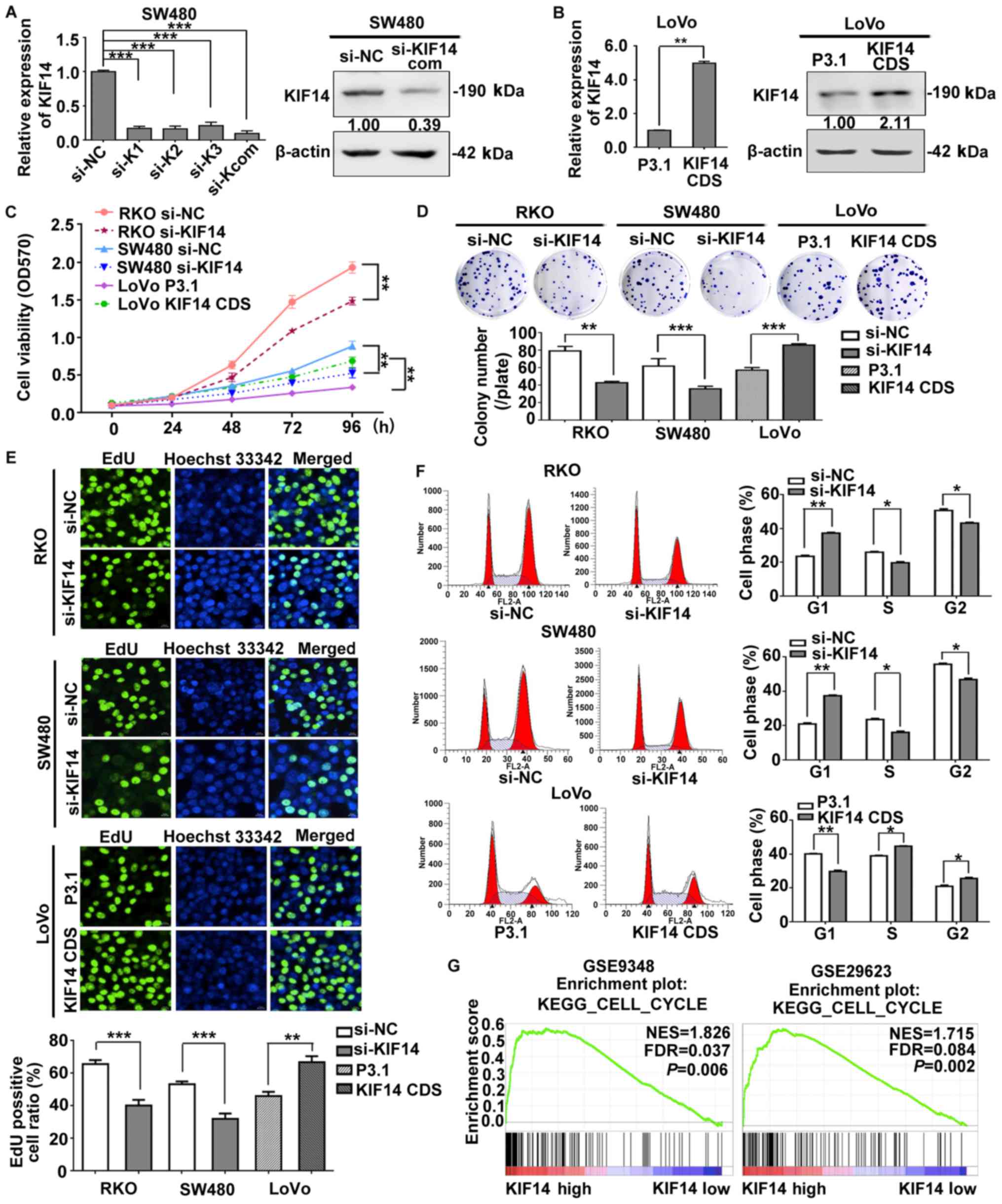 | Figure 2KIF14 promotes CRC cell proliferation
and accelerates the cell cycle. (A) SW480 or (B) LoVo cells were
transfected with three KIF14 siRNA oligos or the siRNA complex and
the P3.1-KIF14 CDS plasmid, respectively, and their corresponding
negative controls; the mRNA and protein expression levels of KIF14
were examined by reverse transcription-quantitative polymerase
chain reaction and western blotting. (C) Cell proliferation of
SW480 and RKO cells transfected with KIF14 siRNA or NC siRNA, and
LoVo cells transfected with P3.1-KIF14 CDS or the control vector,
was detected via a 96 h MTT assay. (D) Cell viability of CRC cells,
which were treated as aforementioned, was determined via a colony
formation assay. (E) DNA synthesis was analyzed in CRC cells
transfected with the corresponding siRNA or plasmids via an EdU
incorporation assay (scale bar, 20 µm). (F) Distribution of
the cell cycle was examined by flow cytometry in silenced KIF14
SW480 and RKO cells, or LoVo cells exhibiting KIF14 overexpression,
in addition to the corresponding controls. The distribution of the
cell population of different treatment groups was presented as a
bar graph. (G) Enrichment plots of gene expression signatures
associated with the cell cycle in the GSE9348 and GSE29623 datasets
according to KIF14 mRNA expression levels. All results were
presented as the mean ± standard deviation (n=3),
*P<0.05, **P<0.01,
***P<0.001. CRC, colorectal cancer; KIF14, kinesin
family member 14; si, small interfering RNA; NC, negative control;
EdU, 5-ethynyl-2-deoxyuridine; FDR, false detection rate; NES,
normalized enrichment score; |
To understand the potential mechanism by which KIF14
promotes CRC cell proliferation, flow cytometry was conducted to
investigate the effects of KIF14 on the cell cycle of CRC.
Depletion of KIF14 increased the G1 phase population of SW480 and
RKO cells, and reduced that of the S and G2 phase population.
Additionally, overexpressed KIF14 promoted the transition of LoVo
cells in G1 to enter into S and G2 phase compared with the control
(Fig. 2F). In addition, the
results from GSEA analysis (38)
with two GEO datasets of CRC revealed that the cell
cycle-associated gene set was significantly enriched in KIF14
overexpressed cases, supporting that KIF14 participates in the
regulation of the cell cycle (Fig.
2G).
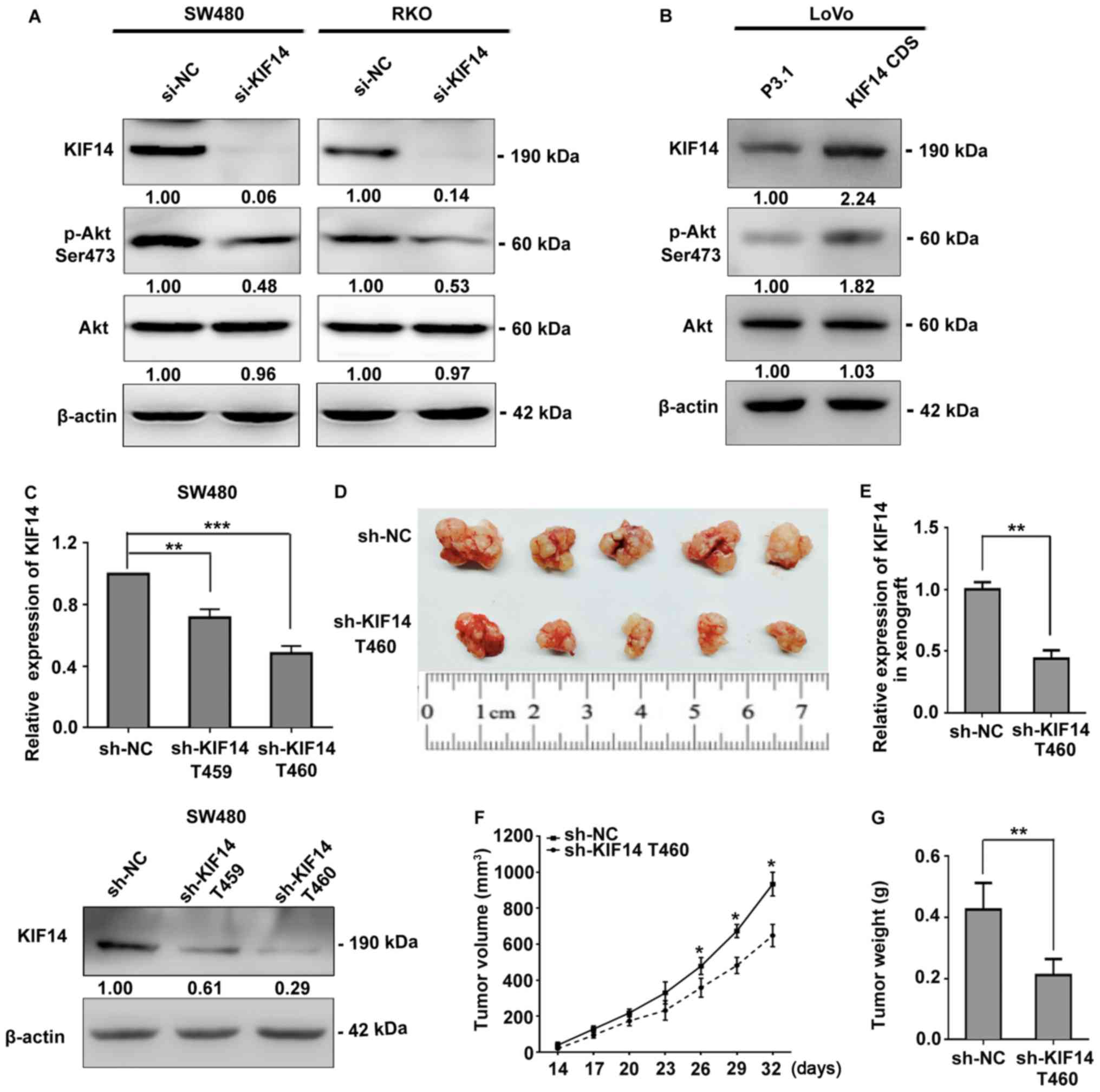
KIF14 activates Akt signaling and
promotes CRC tumorigenesis in vivo
It has been reported that Akt activation is
necessary for cell cycle progression (45,46).
To determine whether KIF14 activated Akt in CRC, thus accelerating
the progression of the cell cycle, western blotting was performed.
As presented in Fig. 3A, silencing
of KIF14 in SW480 and RKO cells resulted in notably decreased
phosphorylation of Akt. Furthermore, upregulation of KIF14 in LoVo
cells led to markedly increased phosphorylation of Akt (Fig. 3B).
To determine whether KIF14 promoted CRC tumor
progression in vivo as observed in vitro, a
tumorigenicity assay with nude mice was performed. Two recombinant
lentiviruses containing shRNA targeting KIF14 (KIF14 shRNA T459 and
T460) or a control were transduced into SW480 cells, respectively.
Subsequently, the cells were selected with puromycin at 2
µg/ml for a week. Following detection via RT-qPCR and
western blotting, the subgroup of cells stably expressing KIF14
shRNA T460 was selected for further analysis due to its higher
efficiency knockdown of KIF14 compared with those without stable
expression (Fig. 3C). Thereafter,
SW480 cells containing KIF14 shRNA T460 or the control shRNA were
subcutaneously inoculated into the right forelimb armpit of nude
mice. The tumor volume was measured every 3 days, starting at day
14 after injection. The mice were sacrificed on day 32 for tumor
weight analysis. The tumor size of the nude mice injected with
KIF14 shRNA T460-containing cells was notably smaller than that of
the control group (Fig. 3D).
Additionally, KIF14 expression was markedly decreased in the
experimental group as determined by RT-qPCR (Fig. 3E). The tumor volume was
significantly decreased in the KIF14 knockdown group for at least
three measurements compared with in the control (Fig. 3F). In accordance with this, the
tumor weight was significantly lower in the KIF14 shRNA
T460-transfected cell group compared with in the control group
(Fig. 3G).
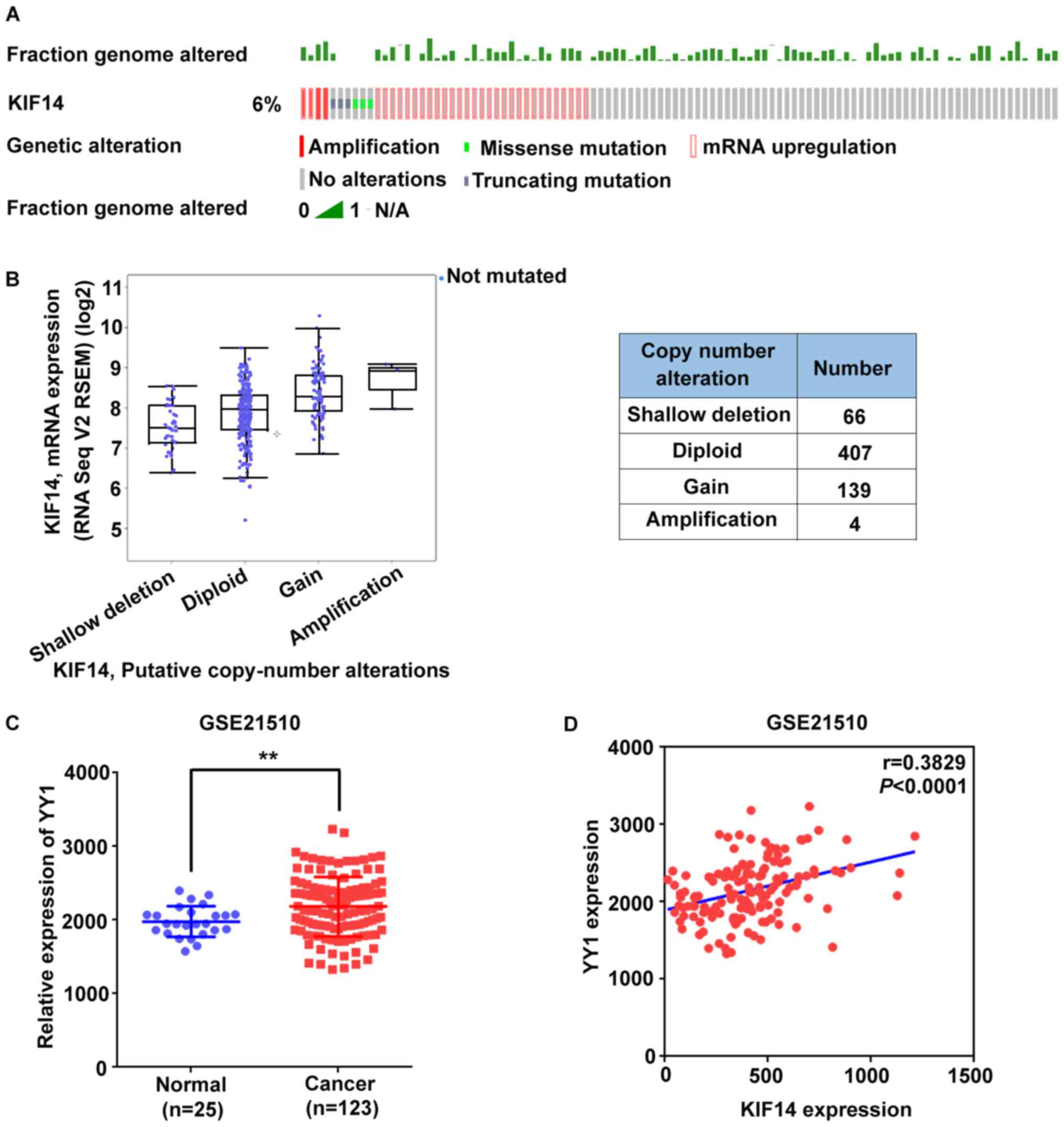
Genomic amplification and transcriptional
regulation may lead to KIF14 overexpression in CRC
To investigate the mechanism underlying the
overexpression of KIF14 in CRC at the genetic level, analysis was
performed using the cBio- Portal for Cancer Genomics (40). A previous study reported that
certain patients with CRC exhibited a chromosomal gain of 1q31-1q32
at the location of the KIF14 coding gene (32), and in silico screening of
KIF14 in TCGA provisional CRC dataset further supported this
finding. As demonstrated in Fig.
4A, 6% cases (39/629) presented at least one genetic alteration
of KIF14. Additionally, KIF14 mRNA expression levels gradually
increased, along with putative KIF14 CNAs (in ascending order as
shallow deletion, diploid, gain and amplification), indicating that
genomic amplifications may partially contribute to abnormally
overexpressed KIF14 in CRC (Fig.
4B). Additionally, the phenomenon that certainpatients with
upregulated KIF14 present a notable unchanged fraction of genomic
alterations (Fig. 4A) suggested
that KIF14 overexpression may not be limited to the genetic level.
In addition, the expression of transcription factor YY1, which
regulates KIF14 (47), was
increased in CRC samples and was also positively correlated with
the expression of KIF14 in the GSE21510 dataset (Fig 4C and D). This indicated that
upregulated expression levels of YY1 may activate KIF14
transcription, leading to KIF14 overexpression.
KIF14 is a direct target of miR-200c
Whether KIF14 is regulated at the
post-transcriptional level in CRC remains unknown. To identify the
potential miRNAs targeting KIF14, two online bioinformatics tools,
microRNA.org (42)
and TargetScan (43), were used,
and the prediction results were comprehensively analyzed. The
candidate miRNAs were sorted and ranked according to the total
mirSVR scores. Analysis revealed that three mature miRNAs from the
miR-200 family, including miR-200c, miR-200b and miR-429, exhibited
the greatest potential to regulate KIF14. To validate the findings
of the present study, the endogenous expression of miR-200c, miR-
200b and miR-429 was detected in the CRC cell lines (Fig. 5A). To identify the specific miRNA
that regulated KIF14 with the highest potency, the expression of
these three miR-200 family members were investigated in
KIF14-silenced CRC cells. As presented in Fig. 5B, knockdown of KIF14 led to notably
increased miR-200c expression levels in SW480 and RKO cells;
miR-200b and miR-429 were upregulated in RKO cells following KIF14
knockdown. These observations indicated that miR-200c bound to and
modulated KIF14 expression in both SW480 and RKO cells; reduced
KIF14 mRNA released more miR-200c and induced elevated expression
levels of miR-200c. While the regulation of KIF14 by miR-200b and
miR-429 may be cell type-specific. The present study found that
miR-200c regulated KIF14 most effectively.
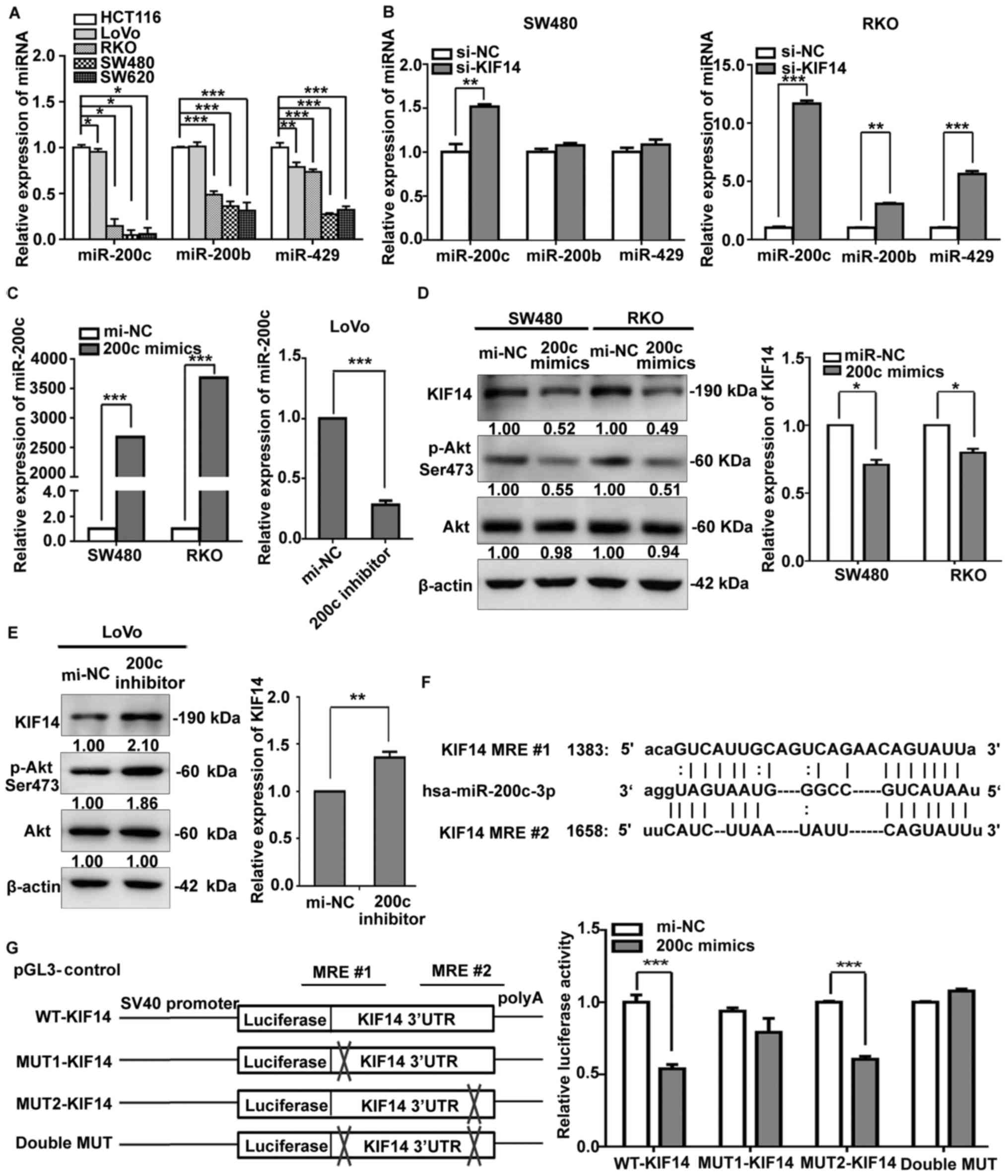 | Figure 5miR-200c modulates KIF14 expression
most potently. (A) miR-200c, miR-200b and miR-429 expression levels
were detected in five colorectal cancer cell lines by RT-qPCR. (B)
Expression of miR-200c, miR-200b and miR-429 was measured by
RT-qPCR in SW480 and RKO cells transfected with KIF14 siRNA or the
control. (C) miR-200c mimics or inhibitor was transfected into
SW480, RKO or LoVo cells respectively, and the transfection
efficiency was determined by RT-qPCR. Effects of miR-200c (D)
mimics or (E) inhibitor on KIF14 protein expression levels and the
phosphorylation levels of Akt were examined by western blotting.
β-actin served as an internal control. RT-qPCR was used to detect
the alteration of KIF14 mRNA expression. (F) Schematic diagram of
miR-200c binding sites in the 3′UTR of KIF14 from microRNA.org. (G)
Left panel, construction of KIF14 luciferase reporter plasmids
harboring the wild-type, single mutated or double mutated miR-200c
response element. Right panel, the relative luciferase activity was
measured in HEK293 cells co-transfected with reporter plasmids
containing the wild-type or mutant KIF14 3′UTR sequences and
miR-200c mimics/mi-NC. All data were expressed as the mean ±
standard deviation (n=3). *P<0.05,
**P<0.01, ***P<0.001. RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; UTR,
untranslated region; miR, microRNA; si, small interfering RNA; NC,
negative control; KIF14, kinesin family member 14; mi-NC, negative
control microRNA; MRE, microRNA response element; WT, wild-type;
MUT, mutant. |
To validate the findings of the present study,
miR-200c mimics transfected into SW480 and RKO, and inhibitor was
transfected into LoVo cells, in addition to a control. Following
confirmation of the transfection efficiency (Fig. 5C), western blotting and RT-qPCR
were conducted to examine whether KIF14 was modulated by miR-200c.
The results demonstrated that upregulated miR-200c led to decreased
expression of KIF14 at the protein and mRNA levels in SW480 and RKO
cells, and also inhibited Akt activity (Fig. 5D). Additionally, miR-200c inhibitor
elevated the protein expression level of KIF14 and the Ser473
phosphorylation of Akt in LoVo cells (Fig. 5E).
According to the bioinformatics analysis, two
miR-200c response elements were observed to be located at 1,402-
1,409 bp and 1,672-1,678 bp of the KIF14 3′UTR (Fig. 5F). To determine whether miR-200c
directly targeted KIF14 via these two binding sites, WT KIF14 3′UTR
fragment or mutated sequences with a MUT1/MUT2 or DOU MUT miR-200c
response element deletion were inserted following the coding
sequence of the pGL3-Control reporter construct (Fig. 5G). 293T cells were co-transfected
with either WT or MUT KIF14 3′UTR reporter plasmids, along with
miR-200c mimics or the corresponding control, respectively.
miR-200c directly reduced the luciferase activity of pGL3-WT KIF14
3′UTR compared with in the control (Fig. 5G). Similarly, attenuated luciferase
activities of pGL3-MUT2 KIF14 3′UTR were detected when cells were
co-transfected with miR-200c mimics other than the control oligo
(Fig. 5G). However, the KIF14
3′UTR reporter constructs with the first miR-200c binding site
(MUT-1) or both miR-200c binding sites (DOU MUT) deleted were
unaffected by miR-200c mimics (Fig.
5G). Therefore, KIF14 was proposed as a direct target of
miR-200c; the 1,402-1,409 bp locus of KIF14 may be the essential
binding site for miR-200c.
miR-200c is involved in KIF14-mediated
CRC cell proliferation and is negatively correlated with KIF14 in
vivo
To confirm whether KIF14 was a functional target of
miR-200c, rescue experiments were performed in RKO cells. The
growth-inhibitory effect caused by KIF14 knockdown was partly
reversed via miR-200c silencing as demonstrated by MTT and colony
formation assays (Fig. 6A and B).
Cell cycle analysis revealed that RKO cells accumulated in G1 phase
due to downregulated KIF14 partially transitioned into S/G2 phase
following co-transfection with miR-200c inhibitor (Fig. 6C). Furthermore, co-transfection of
miR-200c inhibitor restored the decreased protein expression levels
of KIF14 mediated by siRNA silencing, and elevated the reduced
phosphorylation levels of Akt (Fig.
6D). These findings suggested that miR-200c modulated the
function of KIF14, and could influence CRC cell survival by
regulating KIF14.
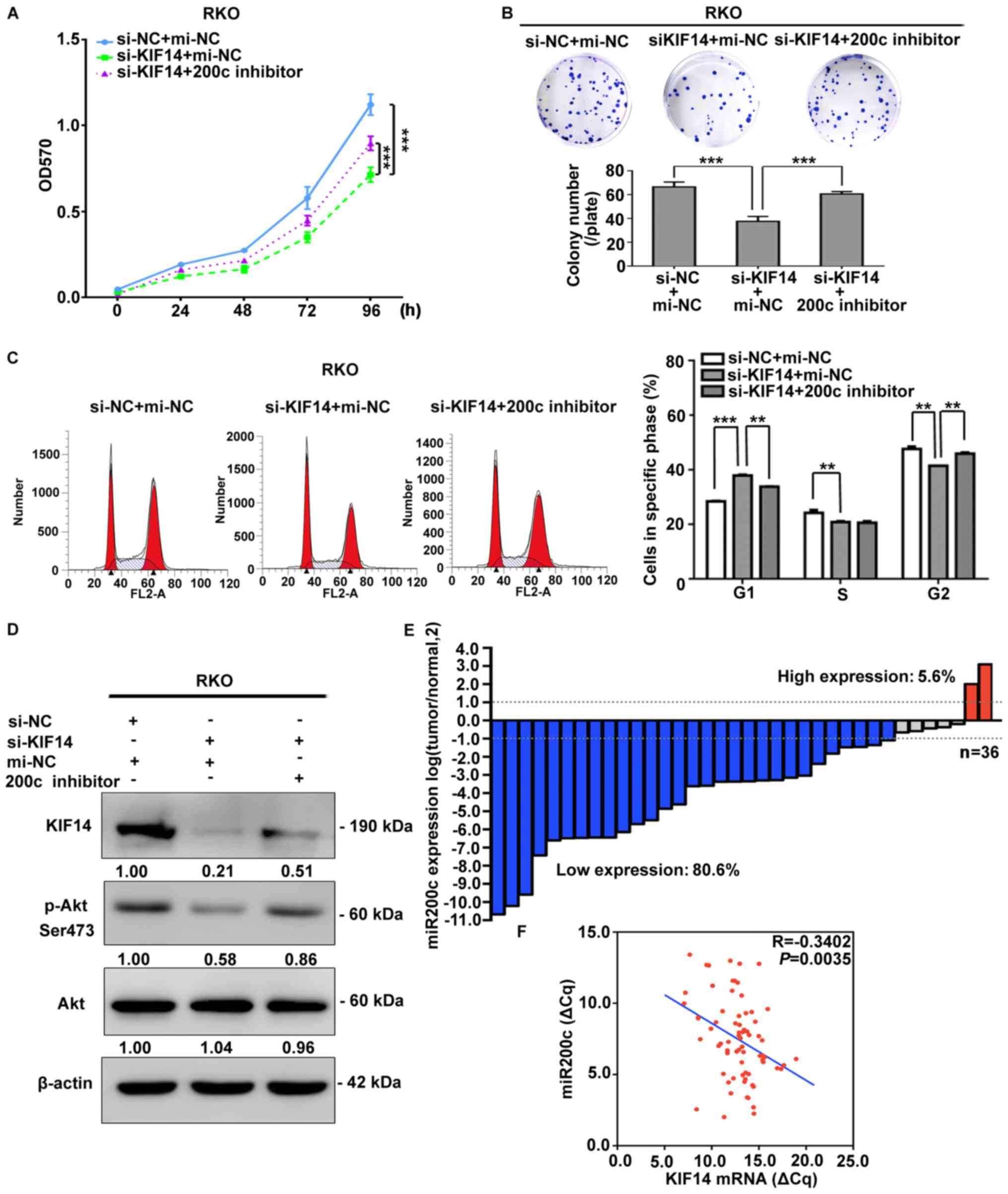 | Figure 6miR-200c participates in the
regulation of CRC cell proliferation by targeting KIF14 and is
negatively correlated with KIF14 in vivo. Reduced cell
viability of RKO cells due to KIF14 siRNA transfection was partly
restored following the silencing of miR-200c, which was detected by
(A) MTT and (B) colony formation assays. (C) Cell cycle was
analyzed by flow cytometry when RKO cells were co-transfected with
KIF14 siRNA and miR-200c inhibitor, or their corresponding
controls. (D) Inhibition of miR-200c elevated the protein
expression levels of KIF14 and the phosphorylation levels of Akt,
which was downregulated by KIF14 siRNA previously. (E) miR-200c
expression was evaluated in 36 paired primary CRC and adjacent
normal tissues by reverse transcription-quantitative polymerase
chain reaction. The various colors of the bars indicated the
proportions of CRC samples with upregulated (red, >2-fold),
downregulated (blue, <2-fold) and unchanged (gray) expression of
miR-200c. (F) Correlation between miR-200c and KIF14 in these
specimens (DCq values were normalized to GAPDH) was determined via
Pearson’s correlation analysis. All data were expressed as the mean
± standard deviation (n=3), **P<0.01,
***P<0.001. CRC, colorectal cancer; miR, microRNA;
si, small interfering RNA; mi-NC, negative control microRNA; KIF14,
kinesin family member 14; p-, phospho-; Akt, protein kinase B. |
In addition, the expression of miR-200c in 36 pairs
of CRC specimens was determined by RT-qPCR. The majority of the CRC
specimens (80.6%) presented significantly decreased expression
levels of miR-200c compared with in the adjacent normal tissues
(Fig. 6E). Additionally, a
significant inverse correlation was observed between the expression
of miR-200c and KIF14 (Pearson R=-0.3402; P=0.0035) (Fig. 6F), which suggested that KIF14 was
regulated by miR-200c in vivo.
Discussion
Accumulating evidence has demonstrated that KIF14 is
involved in the initiation and progression of a variety of human
cancers (23-31); however, the biological function and
clinical significance of KIF14 in colorectal tumorigenesis remain
poorly understood. In the present study, KIF14 was observed to be
abundantly overexpressed in CRC tissues and associated with
proliferative indicators. Functional investigations demonstrated
that KIF14 promoted tumor proliferation via regulation of the cell
cycle and activation of Akt signaling. In addition, the present
study reported that downregulated miR-200c may partly contribute to
KIF14 overexpression and facilitate KIF14 to exert its oncogenic
role in CRC.
Previous studies have revealed that KIF14 is a
robust prognostic indicator in numerous cancers (24,28,31).
Significant upregulation of KIF14 was observed in CRC samples;
however, KIF14 expression was only correlated with the indicators
associated with proliferation, which may be due to the limited
sample size in the present study. To further validate the
correlation between KIF14 and the proliferative index MKI67, the
expression levels of KIF14 and MKI67 were investigated using
publicly available datasets. A strong correlation was observed
between KIF14 and MKI67 mRNA expression levels in the datasets
employed, which further demonstrated the pro-proliferative
characteristics of KIF14. These findings suggested that KIF14 may
be a potential novel marker of proliferation and therapeutic target
for CRC.
In vitro and in vivo experiments
confirmed the correlation between KIF14 overexpression and CRC cell
proliferation. The altered ratio of EdU incorporation along with
altered KIF14 expression levels further indicated that KIF14 was
involved in the cell cycle progression of CRC. Consistently, the
results from flow cytometric analysis revealed that depletion of
KIF14 may induce G1 arrest in RKO and SW480 cells, while
overexpressed KIF14 accelerated the cell cycle of LoVo cells.
Additionally, GSEA analysis demonstrated the promoting effect of
KIF14 on cell cycle.
As a mitotic kinesin, KIF14 has been reported to be
crucial for midbody formation and the completion of cytokinesis
during telophase (19). Carleton
et al (19) documented that
notable reduction in the expression levels of KIF14 prevented
midbody cleavage and induced cytokinesis failure; less efficacious
KIF14 knockdown resulted in acute apoptosis at certain points in
the cell cycle (19). Based on the
aforementioned findings, the KIF14 siRNAs employed in the present
study were suggested to lead to cell division failure and
multinucleation; thus the progression of the cell cycle of
non-viable daughter cells may be inhibited and cells will not
transition into S phase. Apart from interfering with cytokinesis,
downregulated KIF14 may also inhibit the cell cycle by suppressing
the ubiquitination of cyclin-dependent kinase inhibitor 1B in
hepatocellular carcinoma (25),
which further demonstrates the complex functions of KIF14 in a
variety of tumors. In addition, Ser473 phosphorylation of Akt
induced by the overexpression of KIF14 in the present study
suggested that KIF14 promoted cell survival via activation of the
Akt signaling pathway, which was crucial for the progression of the
cell cycle (45,46).
A previous study reported that KIF14 was
overexpressed in ovarian cancer via transcriptional and epigenetic
regulation (47). In the present
study, bioinformatics analysis revealed that abnormally
overexpressed KIF14 in CRC was partially due to genomic
amplification and transcriptional activation. The relatively lower
proportion of KIF14 genetic alterations and the median correlation
coefficient between KIF14 and YY1 may not explain the
overexpression of KIF14 in CRC entirely. Therefore, the present
study investigated the possible mechanism underlying the modulation
of KIF14 at the post- transcriptional level to further improve
understanding of the KIF14 regulatory mechanism.
Following in silico screening and validation
experiments, miR-200c was reported to directly regulate the
expression of KIF14. Rescue experiments were conducted to further
validate whether KIF14 was a functional target of miR-200c in the
present study. In addition, decreased miR-200c expression levels
were inversely correlated with KIF14 expression levels within CRC
tissues. A previous study revealed that miR-200c exerted antitumor
effects by inhibiting metastasis and improving chemosensitivity in
CRC (48); other studies also
reported that the expression of miR-200c was significantly
decreased in CRC compared with in normal tissues (49,50).
However, several studies have demonstrated that miR-200c was
overexpressed in primary CRC lesions and gradually decreased with
increasing tumor stage (51,52).
The factors contributing to the regulation of miR-200c expression
are also complex. Hypomethylation of the miR-200c promoter regions
induces miR-200c upregulation (53), while the aberrantly overexpressed
transcriptional factors achaete-schute complex homolog 2 and Nanog
suppresses the transcription of miR-200c in CRC (49,54),
indicating that expression of miR-200c is regulated by epigenetic
alterations and transcriptional regulation. Therefore, we
hypothesized that within CRC tissues exhibiting unaltered
methylation of miR-200c promoter regions, decreased miR-200c
expression levels were associated with KIF14 overexpression and
enhanced tumor progression.
In summary, the findings of the present study, which
determined the role of KIF14 and its regulatory mechanisms in CRC,
may improve the current understanding of colorectal tumorigenesis
and provide insight for the development of novel therapeutic agents
for the treatment of CRC.
Funding
The present study was supported by the grants from
Beijing Municipal Natural Science Foundation (grant nos. 7172042
and 7162039), the National Natural Science Foundation of China
(grant nos. 81402346, 81672439, 81272766, 81450028, 81470129 and
81502137), the Capital’s Funds for Health Improvement and Research
(grant no. CFH 2018-2-2153), Science Foundation of Peking
University Cancer Hospital (grant no. 2017-14), Beijing Municipal
Administration of Hospitals Clinical Medicine Development of
Special Funding Support (grant nos. XM201309 and ZYLX201701),
Beijing Health System High-level Health Technical Personnel
Training Program (grant no. 2015-3-074) and the Excellent
Overseas-Returnee Scholar Program (Ministry of Education).
Availability of data and materials
The datasets generated and analyzed in the present
study are available at GENT (http://genome.kobic.re.kr/GENT/), Oncomine (http://www.oncomine.org), and cBioPortal for Cancer
Genomics (http://www.cbioportal.org).
Authors’ contributions
ZW, JY and XS conceived the experiments and drafted
the manuscript. ZW and JY conducted the experiments. BJ, JD, PG and
LP helped to analyze and interpret the data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Written informed consent from each patient enrolled
in the study was obtained and ethical approval for the use of human
tissues and for the animal study was obtained from the Ethics
Committee of Peking University Cancer Hospital and Institute
(Beijing, China).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Favoriti P, Carbone G, Greco M, Pirozzi F,
Pirozzi RE and Corcione F: Worldwide burden of colorectal cancer: A
review. Updates Surg. 68:7–11. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen W, Zheng R, Zuo T, Zeng H, Zhang S
and He J: National cancer incidence and mortality in China, 2012.
Chin J Cancer Res. 28:1–11. 2016.PubMed/NCBI
|
|
4
|
DeSantis CE, Lin CC, Mariotto AB, Siegel
RL, Stein KD, Kramer JL, Alteri R, Robbins AS and Jemal A: Cancer
treatment and survivorship statistics, 2014. CA Cancer J Clin.
64:252–271. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Anaya DA, Becker NS and Abraham NS: Global
graying, colorectal cancer and liver metastasis: New implications
for surgical management. Crit Rev Oncol Hematol. 77:100–108. 2011.
View Article : Google Scholar
|
|
6
|
Stein U and Schlag PM: Clinical,
biological, and molecular aspects of metastasis in colorectal
cancer. Recent Results Cancer Res. 176:61–80. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cancer Genome Atlas, N; Cancer Genome and
Atlas Network: Comprehensive molecular characterization of human
colon and rectal cancer. Nature. 487:330–337. 2012. View Article : Google Scholar
|
|
8
|
Uchi R, Takahashi Y, Niida A, Shimamura T,
Hirata H, Sugimachi K, Sawada G, Iwaya T, Kurashige J, Shinden Y,
et al: Integrated multiregional analysis proposing a new model of
colorectal cancer evolution. PLoS Genet. 12:pp. e10057782016,
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hirokawa N, Noda Y, Tanaka Y and Niwa S:
Kinesin superfamily motor proteins and intracellular transport. Nat
Rev Mol Cell Biol. 10:682–696. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Miki H, Setou M, Kaneshiro K and Hirokawa
N: All kinesin superfamily protein, KIF, genes in mouse and human.
Proc Natl Acad Sci USA. 98:7004–7011. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hirokawa N and Tanaka Y: Kinesin
superfamily proteins (KIFs): Various functions and their relevance
for important phenomena in life and diseases. Exp Cell Res.
334:16–25. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lawrence CJ, Dawe RK, Christie KR,
Cleveland DW, Dawson SC, Endow SA, Goldstein LS, Goodson HV,
Hirokawa N, Howard J, et al: A standardized kinesin nomenclature. J
Cell Biol. 167:19–22. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nakagawa T, Tanaka Y, Matsuoka E, Kondo S,
Okada Y, Noda Y, Kanai Y and Hirokawa N: Identification and
classification of 16 new kinesin superfamily (KIF) proteins in
mouse genome. Proc Natl Acad Sci USA. 94:9654–9659. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bassi ZI, Audusseau M, Riparbelli MG,
Callaini G and D’Avino PP: Citron kinase controls a molecular
network required for midbody formation in cytokinesis. Proc Natl
Acad Sci USA. 110:9782–9787. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Arora K, Talje L, Asenjo AB, Andersen P,
Atchia K, Joshi M, Sosa H, Allingham JS and Kwok BH: KIF14 binds
tightly to microtubules and adopts a rigor-like conformation. J Mol
Biol. 426:2997–3015. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gruneberg U, Neef R, Li X, Chan EH,
Chalamalasetty RB, Nigg EA and Barr FA: KIF14 and citron kinase act
together to promote efficient cytokinesis. J Cell Biol.
172:363–372. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Watanabe S, De Zan T, Ishizaki T and
Narumiya S: Citron kinase mediates transition from constriction to
abscission through its coiled-coil domain. J Cell Sci.
126:1773–1784. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Molina I, Baars S, Brill JA, Hales KG,
Fuller MT and Ripoll P: A chromatin-associated kinesin-related
protein required for normal mitotic chromosome segregation in
Drosophila. J Cell Biol. 139:1361–1371. 1997. View Article : Google Scholar
|
|
19
|
Carleton M, Mao M, Biery M, Warrener P,
Kim S, Buser C, Marshall CG, Fernandes C, Annis J and Linsley PS:
RNA interference-mediated silencing of mitotic kinesin KIF14
disrupts cell cycle progression and induces cytokinesis failure.
Mol Cell Biol. 26:3853–3863. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu C, Zhao J, Bibikova M, Leverson JD,
Bossy-Wetzel E, Fan JB, Abraham RT and Jiang W: Functional analysis
of human microtubule-based motor proteins, the kinesins and
dyneins, in mitosis/cytokinesis using RNA interference. Mol Biol
Cell. 16:3187–3199. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Moawia A, Shaheen R, Rasool S, Waseem SS,
Ewida N, Budde B, Kawalia A, Motameny S, Khan K, Fatima A, et al:
Mutations of KIF14 cause primary microcephaly by impairing
cytokinesis. Ann Neurol. 82:562–577. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Makrythanasis P, Maroofian R,
Stray-Pedersen A, Musaev D, Zaki MS, Mahmoud IG, Selim L, Elbadawy
A, Jhangiani SN, Coban Akdemir ZH, et al: Biallelic variants in
KIF14 cause intellectual disability with microcephaly. Eur J Hum
Genet. 26:330–339. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang W, Wang J, Zhang D, Chen W, Hou L,
Wu X and Lu Y: Inhibition of KIF14 suppresses tumor cell growth and
promotes apoptosis in human glioblastoma. Cell Physiol Biochem.
37:1659–1670. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Corson TW, Zhu CQ, Lau SK, Shepherd FA,
Tsao MS and Gallie BL: KIF14 messenger RNA expression is
independently prognostic for outcome in lung cancer. Clin Cancer
Res. 13:3229–3234. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu H, Choe C, Shin SH, Park SW, Kim HS,
Jung SH, Yim SH, Kim TM and Chung YJ: Silencing of KIF14 interferes
with cell cycle progression and cytokinesis by blocking the
p27(Kip1) ubiquitination pathway in hepatocellular carcinoma. Exp
Mol Med. 46:pp. e972014, View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Abiatari I, DeOliveira T, Kerkadze V,
Schwager C, Esposito I, Giese NA, Huber P, Bergman F, Abdollahi A,
Friess H, et al: Consensus transcriptome signature of perineural
invasion in pancreatic carcinoma. Mol Cancer Ther. 8:1494–1504.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Singel SM, Cornelius C, Zaganjor E, Batten
K, Sarode VR, Buckley DL, Peng Y, John GB, Li HC, Sadeghi N, et al:
KIF14 promotes AKT phosphorylation and contributes to
chemoresistance in triple-negative breast cancer. Neoplasia. 16:pp.
247–256. pp. 256.e2422014, https://doi.org/10.1016/j.neo.2014.03.008.
10.1016/j.neo.2014.03.008.
|
|
28
|
Corson TW and Gallie BL: KIF14 mRNA
expression is a predictor of grade and outcome in breast cancer.
Int J Cancer. 119:1088–1094. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Singel SM, Cornelius C, Batten K, Fasciani
G, Wright WE, Lum L and Shay JW: A targeted RNAi screen of the
breast cancer genome identifies KIF14 and TLN1 as genes that
modulate docetaxel chemosensitivity in triple-negative breast
cancer. Clin Cancer Res. 19:2061–2070. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Thériault BL, Cybulska P, Shaw PA, Gallie
BL and Bernardini MQ: The role of KIF14 in patient-derived primary
cultures of high-grade serous ovarian cancer cells. J Ovarian Res.
7:1232014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Thériault BL, Pajovic S, Bernardini MQ,
Shaw PA and Gallie BL: Kinesin family member 14: An independent
prognostic marker and potential therapeutic target for ovarian
cancer. Int J Cancer. 130:1844–1854. 2012. View Article : Google Scholar
|
|
32
|
Corson TW, Huang A, Tsao MS and Gallie BL:
KIF14 is a candidate oncogene in the 1q minimal region of genomic
gain in multiple cancers. Oncogene. 24:4741–4753. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kumar S, Nag A and Mandal CC: A
Comprehensive Review on miR-200c, a promising cancer biomarker with
therapeutic potential. Curr Drug Targets. 16:1381–1403. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mutlu M, Raza U, Saatci Ö, Eyüpoğlu E,
Yurdusev E and Şahin Ö: miR-200c: A versatile watchdog in cancer
progression, EMT, and drug resistance. J Mol Med (Berl).
94:629–644. 2016. View Article : Google Scholar
|
|
35
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
36
|
Shin G, Kang TW, Yang S, Baek SJ, Jeong YS
and Kim SY: GENT: Gene expression database of normal and tumor
tissues. Cancer Inform. 10:149–157. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rhodes DR, Yu J, Shanker K, Deshpande N,
Varambally R, Ghosh D, Barrette T, Pandey A and Chinnaiyan AM:
ONCOMINE: A cancer microarray database and integrated data-mining
platform. Neoplasia. 6:1–6. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES, et al: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mootha VK, Lindgren CM, Eriksson KF,
Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E,
Ridderstråle M, Laurila E, et al: PGC-1alpha-responsive genes
involved in oxidative phosphorylation are coordinately
downregulated in human diabetes. Nat Genet. 34:267–273. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multi- dimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mermel CH, Schumacher SE, Hill B, Meyerson
ML, Beroukhim R and Getz G: GISTIC2.0 facilitates sensitive and
confident localization of the targets of focal somatic copy-number
alteration in human cancers. Genome Biol. 12:R412011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Betel D, Wilson M, Gabow A, Marks DS and
Sander C: The http://microRNA.orgurisimplemicroRNA.org resource:
Targets and expression. Nucleic Acids Res. 36:Database. pp.
D149–D153. 2008, View Article : Google Scholar
|
|
43
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li LT, Jiang G, Chen Q and Zheng JN: Ki67
is a promising molecular target in the diagnosis of cancer
(Review). Mol Med Rep. 11:1566–1572. 2015. View Article : Google Scholar
|
|
45
|
Liu P, Begley M, Michowski W, Inuzuka H,
Ginzberg M, Gao D, Tsou P, Gan W, Papa A, Kim BM, et al:
Cell-cycle-regulated activation of Akt kinase by phosphorylation at
its carboxyl terminus. Nature. 508:541–545. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liang J and Slingerland JM: Multiple roles
of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell
Cycle. 2:339–345. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Thériault BL, Basavarajappa HD, Lim H,
Pajovic S, Gallie BL and Corson TW: Transcriptional and epigenetic
regulation of KIF14 overexpression in ovarian cancer. PLoS One.
9:pp. e915402014, View Article : Google Scholar : PubMed/NCBI
|
|
48
|
O’Brien SJ, Carter JV, Burton JF, Oxford
BG, Schmidt MN, Hallion JC and Galandiuk S: The role of the miR-200
family in epithelial-mesenchymal transition in colorectal cancer: A
systematic review. Int J Cancer. 142:2501–2511. 2018. View Article : Google Scholar
|
|
49
|
Tian Y, Pan Q, Shang Y, Zhu R, Ye J, Liu
Y, Zhong X, Li S, He Y, Chen L, et al: MicroRNA-200 (miR-200)
cluster regulation by achaete scute-like 2 (Ascl2): Impact on the
epithelial- mesenchymal transition in colon cancer cells. J Biol
Chem. 289:36101–36115. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lu YX, Yuan L, Xue XL, Zhou M, Liu Y,
Zhang C, Li JP, Zheng L, Hong M and Li XN: Regulation of colorectal
carcinoma stemness, growth, and metastasis by an
miR-200c-Sox2-negative feedback loop mechanism. Clin Cancer Res.
20:2631–2642. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Toiyama Y, Hur K, Tanaka K, Inoue Y,
Kusunoki M, Boland CR and Goel A: Serum miR-200c is a novel
prognostic and metastasis-predictive biomarker in patients with
colorectal cancer. Ann Surg. 259:735–743. 2014. View Article : Google Scholar :
|
|
52
|
Wang M, Zhang P, Li Y, Liu G, Zhou B, Zhan
L, Zhou Z and Sun X: The quantitative analysis by stem-loop
real-time PCR revealed the microRNA-34a, microRNA-155 and microRNA-
200c overexpression in human colorectal cancer. Med Oncol.
29:3113–3118. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hur K, Toiyama Y, Takahashi M, Balaguer F,
Nagasaka T, Koike J, Hemmi H, Koi M, Boland CR and Goel A:
MicroRNA-200c modulates epithelial-to-mesenchymal transition (EMT)
in human colorectal cancer metastasis. Gut. 62:1315–1326. 2013.
View Article : Google Scholar :
|
|
54
|
Pan Q, Meng L, Ye J, Wei X, Shang Y, Tian
Y, He Y, Peng Z, Chen L, Chen W, et al: Transcriptional repression
of miR-200 family members by Nanog in colon cancer cells induces
epithelial-mesenchymal transition (EMT). Cancer Lett. 392:26–38.
2017. View Article : Google Scholar : PubMed/NCBI
|