Introduction
Tamoxifen is an estrogen receptor (ER) antagonist
used in the treatment of ER-positive breast cancer (1,2). It
is composed of triphenylethylene with an alkylamine side chain, and
cytochrome P450 converts tamoxifen to its active form,
4-hydroxytamoxifen (3), which
strongly binds to ER-α to antagonize the action of estrogen and
exert antitumor effects (4).
The clinical effectiveness of tamoxifen treatment in
ER-positive breast cancer patients is well-established (5,6),
while some clinical studies demonstrated that tamoxifen
administration may also be effective in ER-negative cancer patients
(7). The rates of tamoxifen
responders with complete/partial response or stable disease among
patients with ER-positive and -negative advanced breast cancer were
62 and 27%, respectively (7).
Tamoxifen was also found to be effective for patients with
pancreatic cancer (8,9) or recurrent malignant glioma (10). A number of preclinical studies
indicated that tamoxifen exerted antitumor effects on ER-negative
cancer cells. Tamoxifen inhibited cholangiocarcinoma growth
accompanied by suppression of Akt phosphorylation in a mouse
xenograft model, and induced apoptosis via activating caspase-3, -8
and -9 in vitro (11).
Tamoxifen also effectively inhibited invasion and growth of
follicular thyroid cancer cells in vivo and in vitro
(12). The combination of
tamoxifen and epigallocatechin gallate exerted synergic
antiproliferative effects against ER-negative breast cancer
xenografts, and inhibited the expression of epidermal growth factor
receptor, Akt, mitogen-activated protein kinase
(MAPK)/extracellular signal-regulated kinase kinase (MEK) and
nuclear factor (NF)-κB in the xenografts (13). Tamoxifen administration with cell
division cycle 42 (Cdc42) inhibitor ML141 treatment suppressed
ER-negative breast cancer growth in vivo (14). In vitro studies demonstrated
that tamoxifen decreased glioma (15), leukemia (16), hepatocellular carcinoma (17) and prostate cancer (18) cell viability.
The abovementioned evidence indicates that tamoxifen
is effective against not only ER-positive but also ER-negative
cancer cells, although the mechanisms underlying the anti-tumor
effect of tamoxifen against ER-positive and -negative cancer cells
are quite different. Although tamoxifen is expected to be effective
against various ER-negative cancers, its effect on non-melanoma
skin cancer cells, such as squamous cell carcinoma (SCC) cells,
remains unknown.
The aim of the present study was to elucidate the
antiproliferative effect of tamoxifen on human skin SCC cells and
investigate the involvement of intracellular calcium levels in this
effect.
Materials and methods
Materials
Tamoxifen, verapamil, nifedipine, MEK inhibitor
PD98059 and calcium ionophore A23187 were purchased from Cayman
Chemical, Co. (Ann Arbor, MI, USA). Phloretin was purchased from
Calbiochem, Inc. (San Diego, CA, USA). Cell Counting Kit-8 and Fluo
4-AM was purchased from Dojindo Laboratories Co., Ltd. (Kumamoto,
Japan). Flufenamic acid was purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA).
Cell culture
The human skin SCC cells A431 and DJM-1, and human
acute T-cell leukemia Jurkat cells were obtained from RIKEN
BioResource Center (Tsukuba, Japan). Human skin SCC HSC-1 cells,
human mammary carcinoma MCF-7 cells and human endometrial
adenocarcinoma SNG-II cells were obtained from the Japanese
Collection of Research Bioresources Cell Bank (Osaka, Japan). A431
and HSC-1 cells were cultured in Dulbecco’s modified Eagle’s
medium. Jurkat and MCF-7 cells were cultured in RPMI-1640 medium.
DJM-1 and SNG-II cells were cultured with Eagle’s minimal essential
medium and Ham’s F12 medium, respectively. All media were
supplemented with antibiotics-antimycotics, gentamicin (10
μg/ml, Thermo Fisher Scientific, Inc., Waltham, MA, USA) and
10% heat-inactivated fetal bovine serum. The cells were maintained
at 37°C in a humidified atmosphere of 95% air and 5%
CO2.
Cell proliferation assay
A431, DJM-1 and HSC-1 cells were seeded in 96-well
plates (1,000 cells/well) and incubated for 24 h at 37°C. The cells
were then incubated with tamoxifen (2.5–50 μM) for 48 h, and
the cell numbers were calculated using the WST-8 reagent (Cell
Counting Kit-8, Dojindo Laboratories).
Reverse transcription-polymerase chain
reaction (RT-PCR) analysis
Total RNA was extracted from A431, DJM-1, HSC-1,
MCF-7, Jurkat and SNG-II cells with the RNeasy Mini kit (Qiagen
GmbH, Hilden, Germany). The Superscript III First-Strand Synthesis
System (Thermo Fisher Scientific, Inc.) was then applied. Up to the
synthesis of cDNAs, all experiments were conducted under RNase-free
conditions. First-strand cDNAs were then used as templates in PCR.
The oligonucleotide primers synthesized were 5′-CAGAGAGATGA
CTTGGAAGG-3′ and 5′-GACTTCAAGGTGCTGGATAG-3′ for human ER-α; and
5′-TCTACAGTCCTGCTGTGATG-3′ and 5′-GTGTACCTGCTCGCTAGAAC-3′ for human
ER-β. The experimental conditions for PCR were as follows: 94°C for
5 min (1 cycle), 94°C for 30 sec, 60°C for 30 sec, 72°C for 1 min
(30 cycles), 72°C for 7 min (1 cycle) for human ER-α; and 94°C for
5 min (1 cycle), 94°C for 30 sec, 60°C for 30 sec, 72°C for 1 min
(35 cycles), 72°C for 7 min (1 cycle) for human ER-β. The PCR
products were electrophoresed on 1.5% agarose gel and stained with
GelRed (Biotium, Inc., Fremont, CA, USA).
Measurement of intracellular calcium
concentration
A431, DJM-1 and HSC-1 cells were seeded in 96-well
plates (40,000 cells/well) and incubated for 24 h at 37°C. The
cells were then incubated with Fluo 4-AM (5 μM) for 1 h at
37°C. After residual Fluo 4 removal, tamoxifen (10-50 μM)
was added to the cells, and the fluorescence emission was measured
at 530 nm in response to excitation at 485 nm
(485F530) by a fluorescence plate
reader (CytoFluor Multi-Well Plate Reader Series 4000; PerSeptive
Biosystems, Framingham, MA, USA). Intracellular calcium
concentration was calculated using the equation of Grynkiewicz
et al (19), where
Kd = 345 nM.
Flow cytometry
A431, DJM-1 and HSC-1 cells were seeded in 6-well
plates (1.2×106 cells/well) and incubated for 24 h at
37°C. The cells were then incubated with Fluo 4-AM (5 μM)
for 1 h at 37°C. After residual Fluo 4 removal, tamoxifen (100
μM) was added to the cells, and fluorescence was measured by
FACSCanto II flow cytometer (BD Biosciences, San Jose, CA,
USA).
Western blot analysis
A431, DJM-1 and HSC-1 cells were seeded in 60-mm
dishes (3×106 cells/dish) and incubated for 24 h at
37°C. The cells were then incubated with tamoxifen (20 or 30
μM) for 0, 10, 20, 30 or 60 min. After extensive washing
with phosphate-buffered saline, the cells were lysed with cell
lysis buffer (0.01 M phosphate, 0.15 M NaCl, 5 mM EDTA, 1% Nonidet
P-40, 0.1% sodium dodecyl sulfate and 0.1% sodium deoxycholate).
The protein concentration of the cell lysates was measured with the
Protein Assay Bicinchoninate Kit (Nacalai Tesque, Inc., Kyoto,
Japan).
Cell lysates (90 μg) were electrophoresed on
a polyacrylamide gel under non-reducing conditions, and resolved
proteins were electrophoretically transferred to a PVDF membrane.
The membrane was incubated for 1 h in a blocking solution (5%
skimmed milk in TBS containing 0.05% Tween-20) and probed with
anti-human phospho-protein kinase C (PKC) (pan) rabbit polyclonal
antibody (1:2,000 diluted in TBS containing 1% skimmed milk and
0.05% Tween-20; Cell Signaling Technology, Inc., Danvers, MA, USA),
anti-human PKCα rabbit polyclonal antibody (1:2,000, Cell Signaling
Technology), anti-human PKCδ rabbit monoclonal antibody (D10E2,
1:2,000, Cell Signaling Technology), anti-human phospho-p44/42 MAPK
rabbit monoclonal antibody (D13.14.4E, 1:2,000, Cell Signaling
Technology), anti-human p44/42 MAPK rabbit monoclonal antibody
(1:2,000, Cell Signaling Technology) or anti-human
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) rabbit monoclonal
antibody (14C10, 1:1,000, Cell Signaling Technology) overnight at
4°C. After extensive washing with TBS containing 0.05% Tween-20,
the membrane was further incubated with a horseradish
peroxidase-conjugated goat anti-rabbit IgG antibody (1:4,000
dilution; Cell Signaling Technology), and the bands of antigens
were detected by chemiluminescence of the product of peroxidase
reaction using Luminata Forte Western HRP Substrate (EMD Millipore
Corp., Darmstadt, Germany).
Data analysis
All data are expressed as mean ± standard deviation.
The significance of the difference between two groups was analyzed
by Student’s t-tests and among more than three groups by one-way
and two-way analysis of variance followed by multiple comparison
tests, Dunnett’s test or Tukey’s test. A P-value of <0.05 was
considered to indicate a statistically significant difference. Data
analysis was performed using JMP 9 software (SAS Institute, Inc.,
Cary, NC, USA).
Results
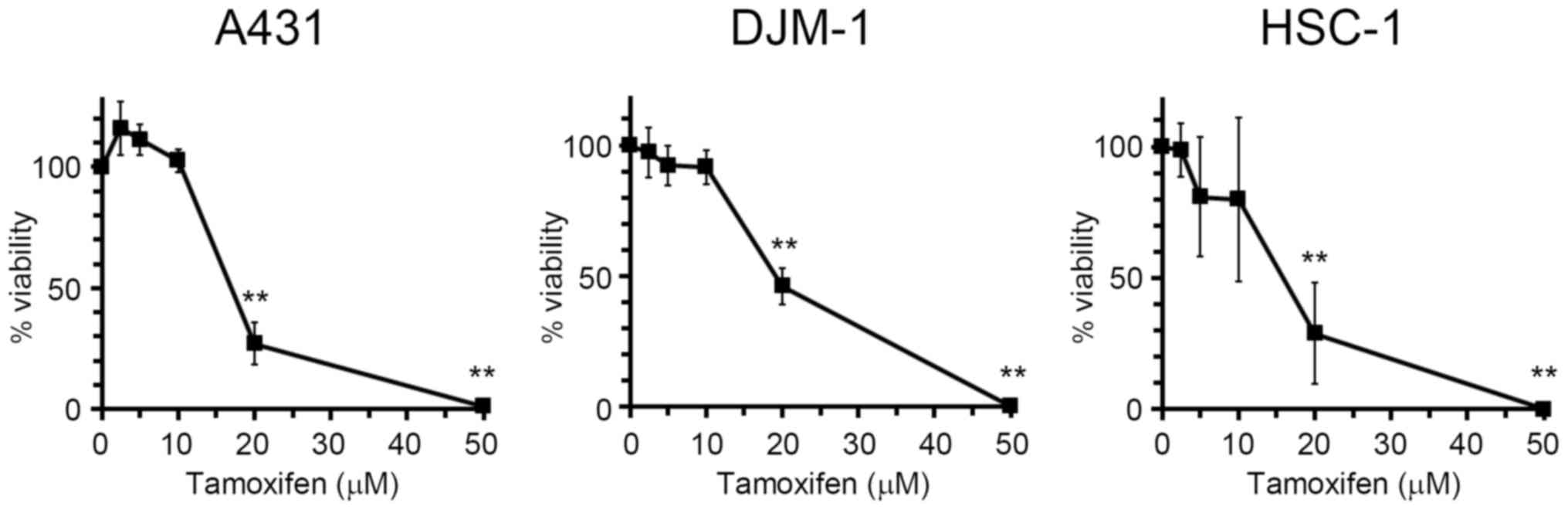
Tamoxifen exerts antiproliferative
effects on human skin SCC cells
Tamoxifen is known to inhibit cell proliferation in
several types of cancer, such as breast cancer (20). In order to evaluate the
antiproliferative effect of tamoxifen on human skin SCC cells,
A431, DJM-1 and HSC-1 cells were incubated with tamoxifen, and the
cell numbers were then estimated by the WST assay (Fig. 1). Tamoxifen inhibited the
proliferation of skin SCC cells dose-dependently at concentrations
of 2.5–50 μM. The GI50 (50% growth inhibition)
values of tamoxifen for A431, DJM-1 and HSC-1 cells were 17.6, 19.4
and 15.6 μM, respectively.
Tamoxifen has been shown to antagonize ER function,
which is a well-known mechanism mediating the antiproliferative
effects of tamoxifen against ER-positive cancer cells. Thus, the
expression of ERs in skin SCC cells was investigated by RT-PCR
(Fig. 2). A431 cells did not
express either ER-α or -β, DJM-1 cells expressed both ER-α and -β,
whereas HSC-1 cells slightly expressed ER-α and -β. It has been
reported that tamoxifen may inhibit the proliferation of both
ER-positive and -negative cells (21). These results indicate that
tamoxifen may exert its antiproliferative effect on certain types
of skin SCC cells via a non-ER-mediated pathway.
 | Figure 2Expression of ER-α and -β in skin SCC
cells. The expression of ER-α and -β in A431, DJM-1 and HSC-1 cells
was evaluated by RT-PCR. The data shown are from one experiment
representative of at least three separate experiments. n.c.,
negative control (Jurkat for ER-α, SNG-II for ER-β); T, target cell
(A431, DJM-1 or HSC-1), p.c., positive control (MCF-7). ER,
estrogen receptor; SCC, squamous cell carcinoma; RT-PCR, reverse
transcription-polymerase chain reaction. |
Tamoxifen increases intracellular calcium
concentration in skin SCC cells
It was reported that tamoxifen-induced apoptosis is
mediated by intracellular calcium signals in human hepatoblastoma
HepG2 cells (17). To elucidate
the mechanism underlying the tamoxifen-induced antiproliferative
effect on skin SCC cells, the changes in intracellular calcium
concentration were investigated. As shown in Fig. 3A, tamoxifen markedly increased
intracellular calcium concentration in A431, DJM-1 and HSC-1 cells
in a dose-dependent manner. The increase in intracellular calcium
levels in A431, DJM-1 and HSC-1 cells induced by tamoxifen was also
evaluated by flow cytometry (Fig.
3B). The data suggest that tamoxifen induces calcium influx in
skin SCC cells.
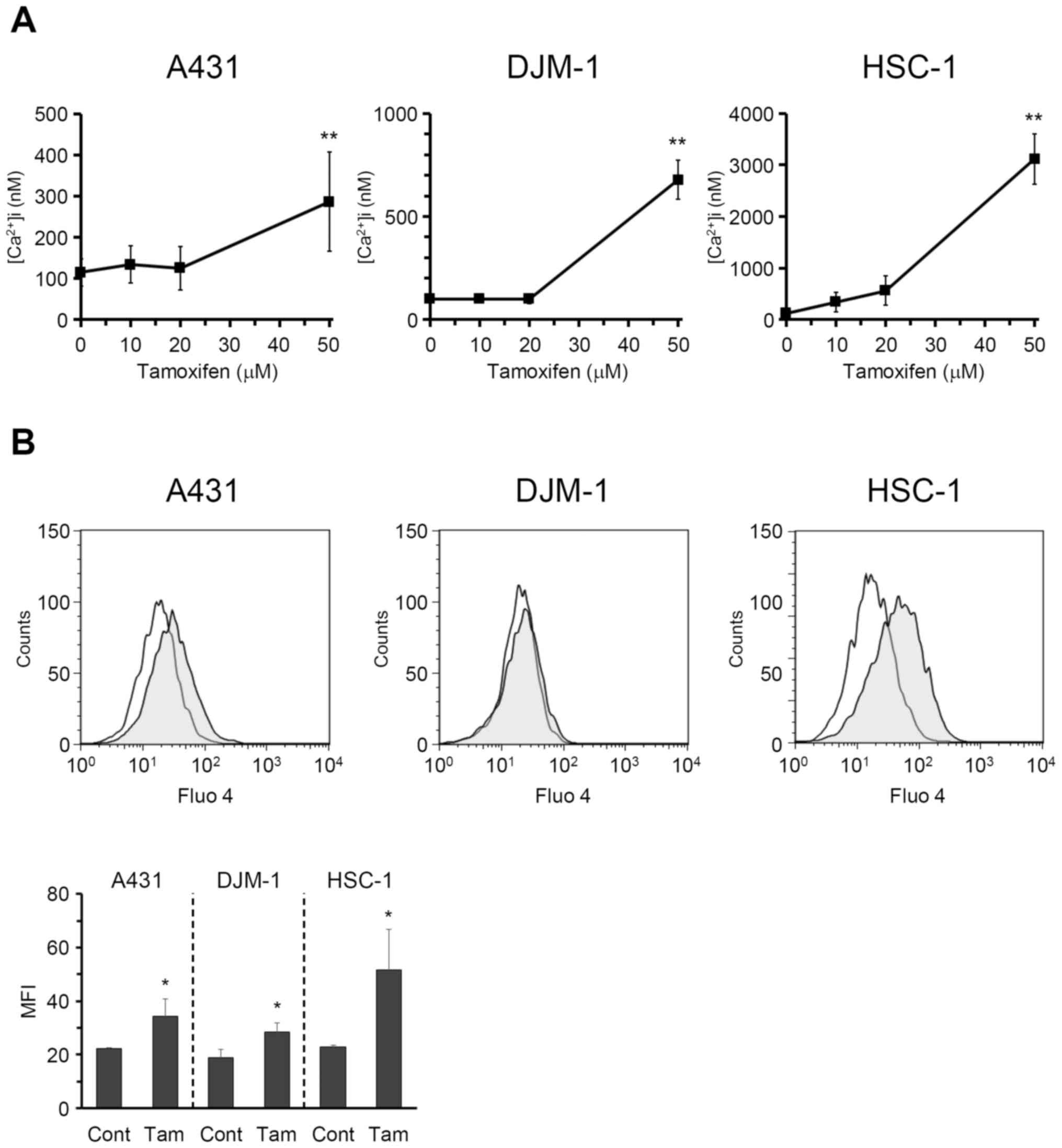 | Figure 3Tamoxifen increased intracellular
calcium concentration of skin SCC cells. (A) A431, DJM-1 and HSC-1
cells were incubated with Fluo 4-AM (5 μM) for 1 h, and then
tamoxifen was added at various concentrations. Fluorescence derived
from Fluo 4 was monitored by a fluorescence plate reader, and
intracellular calcium concentration was calculated.
**P<0.01 (vs. tamoxifen 0 μM, Dunnett’s test).
(B) The increase of intracellular calcium concentration induced by
tamoxifen (100 μM, shaded area) in A431, DJM-1 and HSC-1
cells was also evaluated by FACS. Open areas represent control. The
data shown are from one experiment representative of at least three
separate experiments. The mean fluorescence intensity (MFI) data
generated by the results of flow cytometry are also shown. Cont,
control; Tam, tamoxifen (100 μM). *P<0.05 (vs.
Cont, Student’s t-test). SCC, squamous cell carcinoma. |
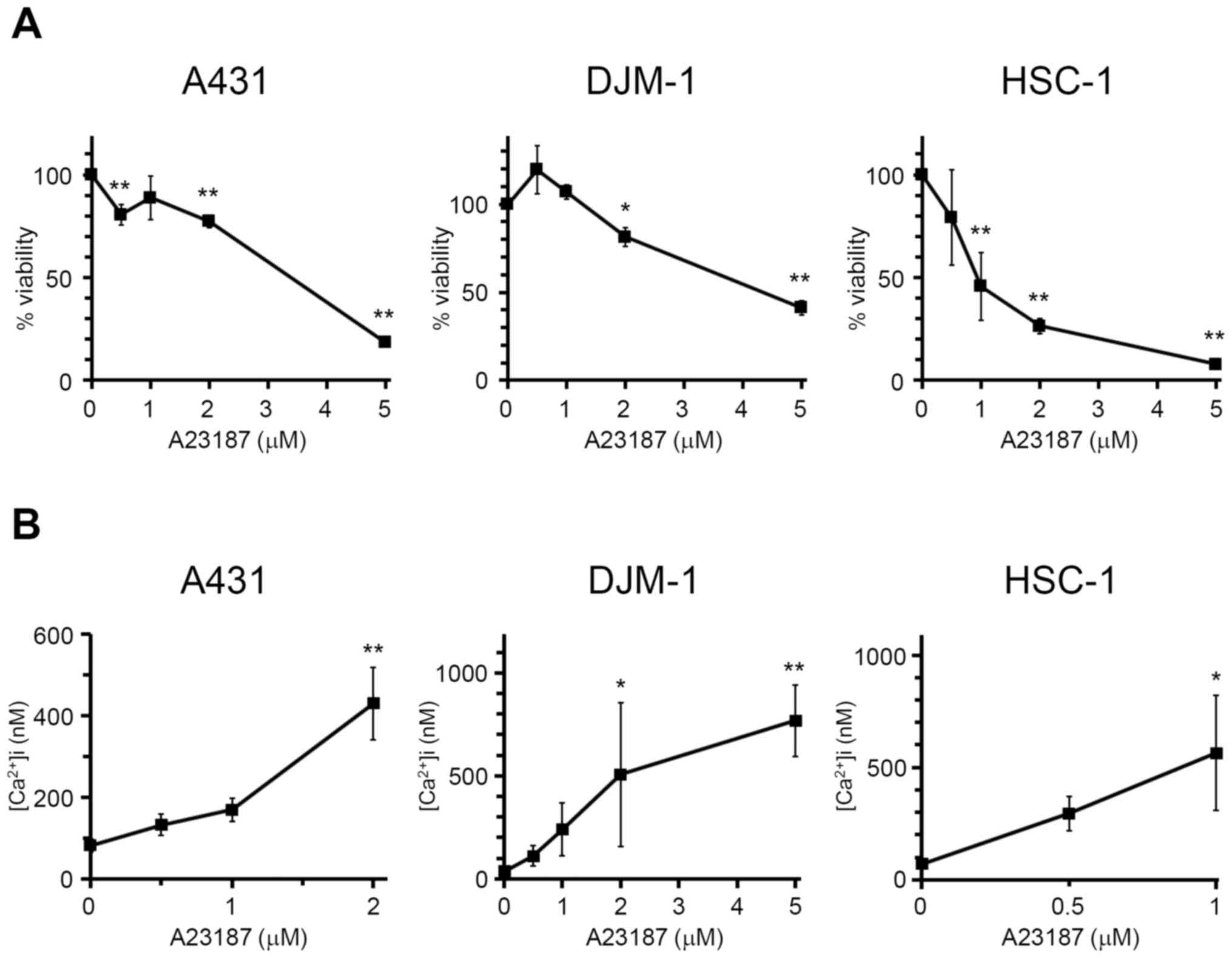
Increase in intracellular calcium
concentration suppresses the proliferation of skin SCC cells
In order to confirm the association between the
increase in intracellular calcium concentration and growth
inhibition of skin SCC cells, A431, DJM-1 and HSC-1 cells were
incubated with calcium ionophore A23187 at various concentrations
(0.5-5 μM), and the cell numbers were then calculated
(Fig. 4A). A23187 significantly
inhibited the growth of skin SCC cells, indicating that the
increase in intracellular calcium concentration caused growth
inhibition of skin SCC cells. It was confirmed that A23187 (0.5–5
μM) increased the intracellular calcium levels in A431,
DJM-1 and HSC-1 cells (Fig. 4B).
Following addition of A23187 at >2 μM for A431 cells and
>1 μM for HSC-1 cells, intracellular calcium
concentration could not be measured, due to the excess fluorescence
over the detection limits. These results indicate that tamoxifen
inhibited the proliferation of human skin SCC cells via increasing
intracellular calcium concentration.
Voltage-gated calcium channels as well as
non-selective cation channels are involved in the increase in
intracellular calcium concentration in skin SCC cells induced by
tamoxifen
Two major pathways are involved in the increase in
intracellular cytosolic calcium concentration: Release from
endoplasmic reticulum calcium stores to the cytosol, and
extracellular calcium influx via plasma membrane calcium channels
(22). To elucidate which pathway
is implicated in the increase in intracellular cytosolic calcium
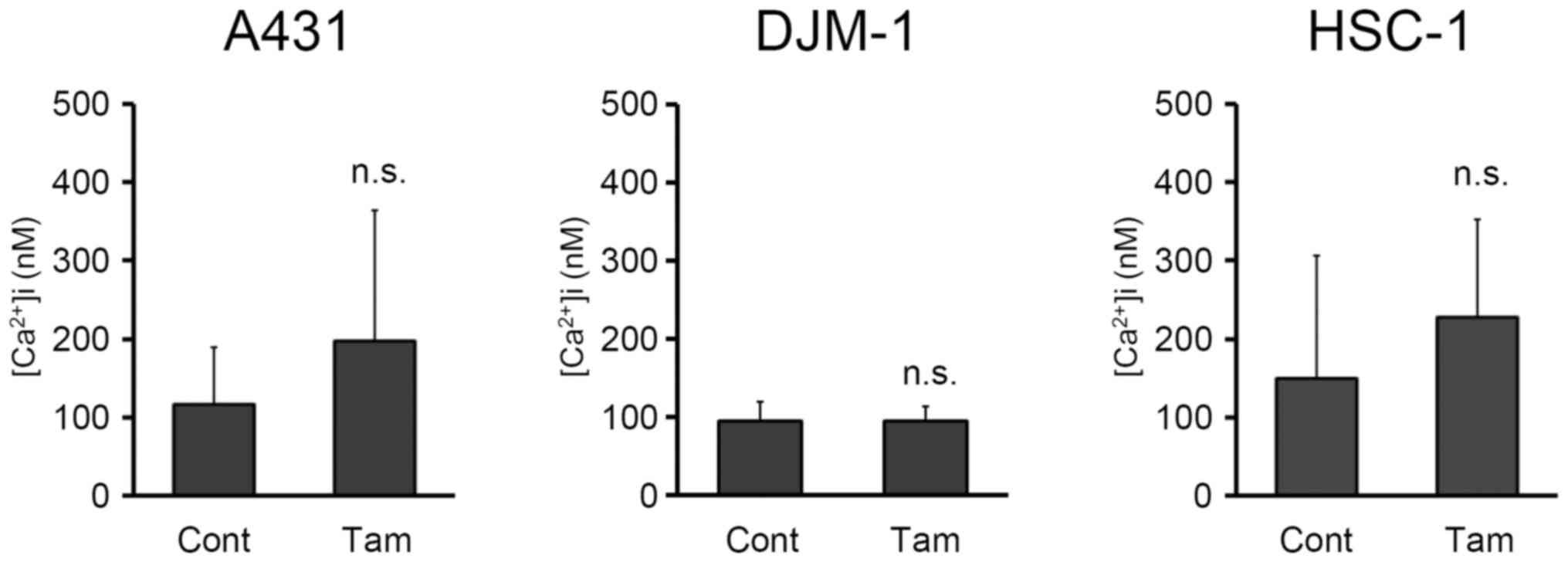
concentration in skin SCC cells induced by tamoxifen, it was first
investigated whether tamoxifen induced calcium release from
intracellular stores. A431, DJM-1 and HSC-1 cells were incubated
with Fluo 4-AM in HEPES buffer solution without calcium, followed
by the addition of tamoxifen. The fluorescence derived from Fluo 4
was monitored and intracellular calcium concentration was
calculated. As shown in Fig. 5,
addition of tamoxifen did not increase the intracellular cytosolic
calcium concentration in skin SCC cells when using calcium-depleted
buffer solution. However, thapsigargin (23), which discharged intracellular
calcium stores by potently inhibiting endoplasmic reticulum calcium
ATPase, increased the intracellular cytosolic calcium concentration
in skin SCC cells. Addition of thapsigargin increased the
intracellular calcium concentration in A431, DJM-1 and HSC-1 cells
up to 231.8±57.8, 299.2±124.9 and 438.1±252.4 μM,
respectively, whereas the intracellular calcium concentrations in
A431, DJM-1 and HSC-1 cells without thapsigargin were 116.0±73.9,
95.2±24.4 and 149.3±157.3 μM, respectively. These data
suggest that the tamoxifen-induced increase in intracellular
cytosolic calcium concentration was not due to calcium release from
the endoplasmic reticulum stores.
It was next investigated whether extracellular
calcium influx via plasma membrane calcium channels was involved in
the increase in intracellular calcium concentration in skin SCCs
induced by tamoxifen. The tamoxifen-induced increase in
intracellular cytosolic calcium concentration was significantly
inhibited by nifedipine, a voltage-gated calcium channel inhibitor,
and flufenamic acid, a non-selective cation channel blocker
(Fig. 6A). Verapamil, another
voltage-gated calcium channel inhibitor, significantly inhibited
the increase in intracellular cytosolic calcium concentration in
DJM-1 cells. From these data, both voltage-gated calcium channels
and non-selective cation channels appear to be involved in the
increase in intracellular cytosolic calcium concentration in skin
SCC cells induced by tamoxifen. As shown in Fig. 6B, blockage of calcium influx by
voltage-gated calcium channel and non-selective cation channel
inhibitors attenuated the tamoxifen-induced toxicity on skin SCC
cells, indicating that tamoxifen-induced cytotoxicity is mediated
by calcium influx.
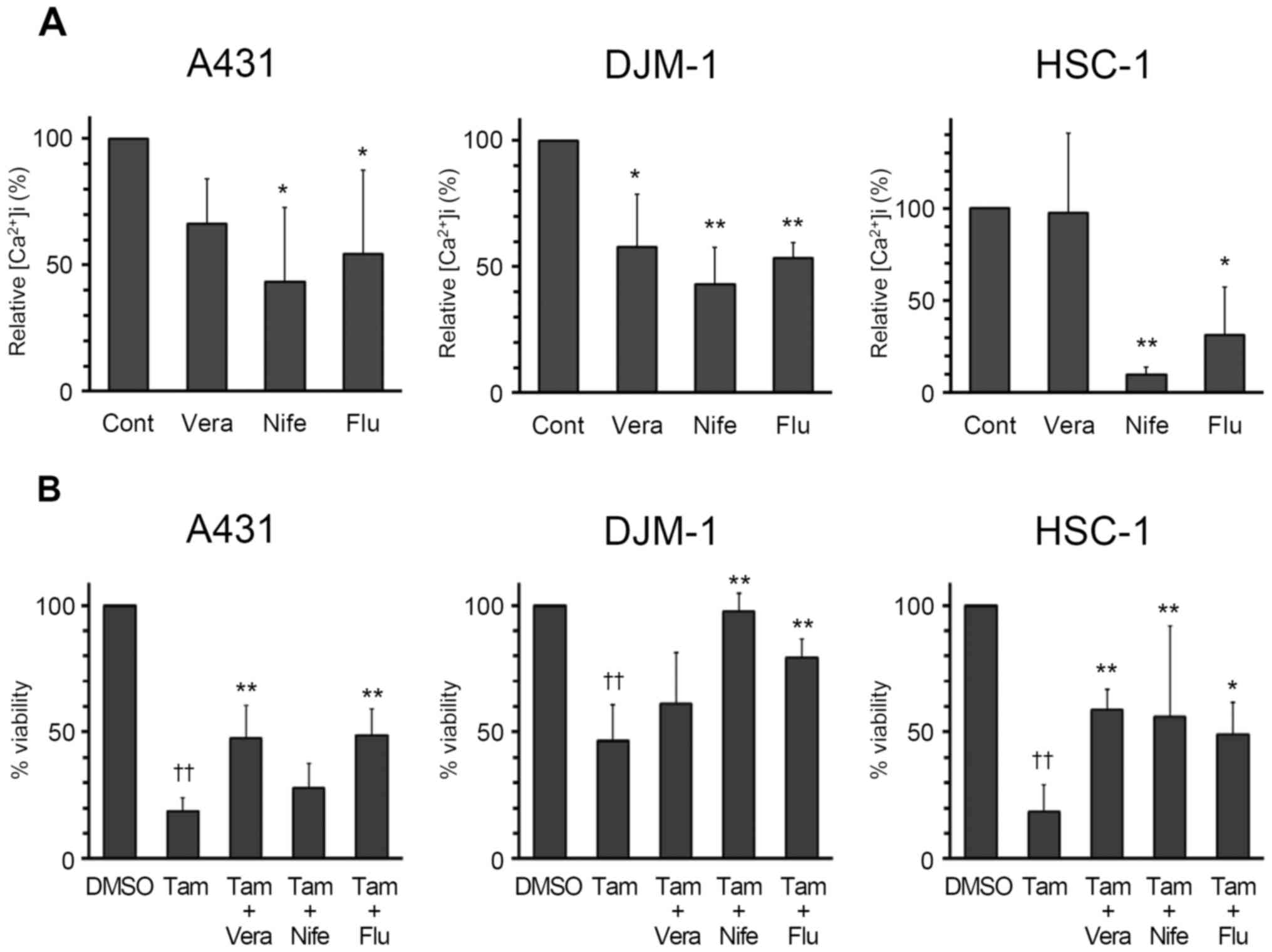 | Figure 6Inhibitory effects of calcium channel
blockers on tamoxifen-induced calcium influx in skin SCC cells. (A)
A431, DJM-1 and HSC-1 cells were pre-incubated with verapamil (100
μM, Vera), nifedipine (100 μM, Nife), flufenamic acid
(100 μM, Flu) or DMSO (Cont) for 1 h, and then incubated
with Fluo 4-AM (5 μM) for 1 h. Calcium influx derived from
the addition of 50 μM tamoxifen (Tam) was monitored, and the
relative intracellular calcium concentration was calculated.
*P<0.05, **P<0.01 (vs. Cont, Dunnett’s
test). (B) A431, DJM-1 and HSC-1 cells were pre-incubated with 100
μM Vera, 100 μM Nife, 100 μM Flu or dimethyl
sulfoxide (DMSO) for 1 h, and then incubated with Tam (20 μM
for A431 and DJM-1 cells, 25 μM for HSC-1 cells) for 48 h,
and the cell numbers were calculated by the WST assay. The number
of cells in the control group (DMSO) was set as 100% and the cell
counts for all treatment groups are expressed as a percentage
against the control. ††P<0.01 (vs. DMSO),
*P<0.05, **P<0.01 (vs. Tam, Tukey’s
test). SCC, squamous cell carcinoma. |
PKC is involved in the antiproliferative
effect of tamoxifen on skin SCC cells
The results in Fig.
3 indicate that tamoxifen increased the intracellular cytosolic
calcium concentration of skin SCC cells. Calcium is a well-known
second messenger that is involved in a number of intracellular
signal transduction pathways, such as the PKC pathway (24). In order to confirm the involvement
of PKC in tamoxifen-induced growth inhibition of skin SCC cells,
A431, DJM-1 and HSC-1 cells were incubated with tamoxifen in the
presence of the broad-spectrum PKC inhibitor phloretin, and the
cell numbers were then calculated by the WST assay (Fig. 7A). Tamoxifen-induced growth
inhibition of skin SCC cells was markedly recovered by phloretin.
The inhibition ratios of phloretin against tamoxifen-induced growth
inhibition were 33.7, 24.0 and 44.2% for A431, DJM-1 and HSC-1
cells, respectively. These data demonstrated that PKC is involved
in the antiproliferative effect of tamoxifen on skin SCC cells.
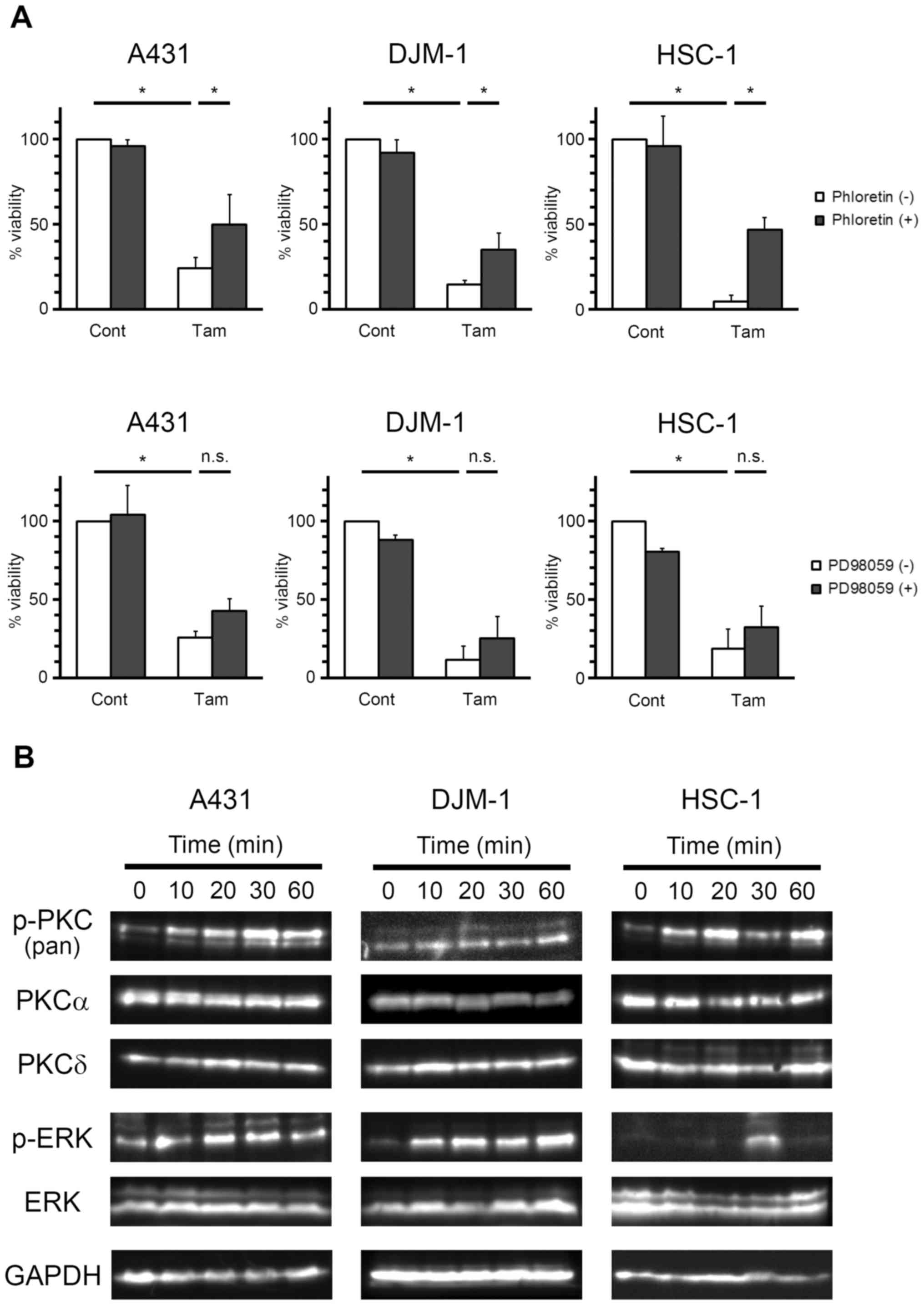 | Figure 7Effects of PKC and MEK inhibitors on
the antiproliferative activity of tamoxifen on skin SCC cells. (A)
A431, DJM-1 and HSC-1 cells were pre-incubated with (+) or without
(−) phloretin (500 μM, upper row) or PD98059 (25 μM,
lower row) for 1 h, and then incubated with tamoxifen (Tam; 20
μM for A431 and HSC-1 cells, 30 μM for DJM-1 cells)
for 48 h. The cell numbers were calculated by the WST assay. The
number of cells in the control group (Cont) was set as 100% and the
cell counts for all treatment groups are expressed as a percentage
against the control. *P<0.05 (Tukey’s test), n.s.,
not significant. (B) A431, DJM-1 and HSC-1 cells were incubated
with Tam (20 μM for A431 and HSC-1 cells, 30 μM for
DJM-1 cells) for various times, and the expression of phospho-PKC
[p-PKC (pan)], PKCα, PKCδ, phospho-ERK (p-ERK), ERK and GAPDH were
evaluated by western blotting. The data shown are from one
experiment representative of at least three separate experiments.
PKC, protein kinase C; ERK, extracellular signal-regulated kinase;
MEK, mitogen-activated protein kinase/extracellular
signal-regulated kinase kinase; SCC, squamous cell carcinoma. |
Calcium is also involved in the MAPK pathway
(25); thus, the involvement of
MAPK/MEK in tamoxifen-induced growth inhibition of skin SCC cells
was investigated. A431, DJM-1 and HSC-1 cells were incubated with
tamoxifen in the presence of the selective MEK inhibitor PD98059,
and the cell numbers were then calculated by the WST assay
(Fig. 7A). Although the effect was
not statistically significant, PD98059 tended to recover the
viability of skin SCC cells against tamoxifen-induced growth
inhibition. The inhibition ratios of PD98059 against
tamoxifen-induced growth inhibition were 23.0, 15.5 and 16.5% for
A431, DJM-1 and HSC-1 cells, respectively. The results from western
blot analysis demonstrated that tamoxifen induced the
phosphorylation of PKC and extracellular signal-regulated kinase1/2
(ERK1/2) in A431, DJM-1 and HSC-1 cells (Fig. 7B). Taken together, these data
indicate that the PKC and MAPK pathways may be involved in the
antiproliferative effects of tamoxifen on skin SCC cells; the
contribution of the MAPK pathway, however, is smaller compared with
that of the PKC pathway.
Discussion
One of the major functions of tamoxifen is to
inhibit the proliferation of cancer cells by antagonizing
ER-induced signaling pathways. The effectiveness of tamoxifen
against several types of ER-negative cancer cells (e.g., glioma and
hepatocellular carcinoma) is also widely reported (26); however, the effect of tamoxifen on
non-melanoma skin cancer remained unknown. In the present study, it
was demonstrated that tamoxifen inhibited the proliferation of
human skin SCC cells via a non-ER-mediated pathway. Tamoxifen
increased the intracellular calcium concentration in skin SCC cells
by influx of extracellular calcium through voltage-gated calcium
channels and non-selective cation channels, resulting in the
activation of PKC.
Tamoxifen inhibited the proliferation of A431, DJM-1
and HSC-1 cells dose-dependently at concentrations of 2.5–50
μM (Fig. 1). The
GI50 values for A431, DJM-1 and HSC-1 cells were 17.6,
19.4 and 15.6 μM, respectively. ER-positive cancer cells are
usually more sensitive to tamoxifen compared with ER-negative
cells. For example, the GI50 values for the ER-positive
breast cancer cell lines MCF-7 and T47D were reported to be <3
μM, whereas the GI50 values for the ER-negative
breast cancer cell lines MDA-MB-231 and HCC1937 was >3 μM
(27). Guo et al (4) reported that the mean value of
GI50 of tamoxifen over all 39 cancer cell lines
(MG-MID), including both ER-positive and -negative cell lines, was
7.41 μM. Although the GI50 values of tamoxifen
for A431, DJM-1 and HSC-1 cells were higher compared with the
MG-MID of tamoxifen, the antiproliferative effect of tamoxifen on
skin SCC cells was notable. A431 cells do not express either ER-α
or -β (Fig. 2), suggesting that
the growth inhibitory effects of tamoxifen on skin SCC cells were
mediated via a non-ER pathway.
In order to elucidate the mechanism underlying the
antiproliferative effects of tamoxifen on skin SCC cells, the
change in intracellular calcium concentration during tamoxifen
treatment was investigated. As shown in Fig. 3A, tamoxifen significantly increased
intracellular calcium concentration in A431, DJM-1 and HSC-1 cells,
thereby inhibiting the growth of skin SCC cells (Fig. 4A). Thus, it was strongly suggested
that the tamoxifen-induced growth inhibition of skin SCC cells was
caused by an increase in the intracellular calcium
concentration.
Tamoxifen at a dose of 20 μM reduced the
viability of skin SCC cells to 50–70% (Fig. 1), although the same concentration
of tamoxifen did not cause a marked elevation in intracellular
calcium concentration (Fig. 3A).
This discrepancy may be explained by the differences in the
incubation times of skin SCC cells with tamoxifen in these assays.
To evaluate the effect of tamoxifen on cell viability, skin SCC
cells were incubated with tamoxifen for 48 h. However,
intracellular calcium concentration was measured immediately
following the addition of tamoxifen to skin SCC cells, as mentioned
in ‘Materials and methods’. These differences in incubation times
of skin SCC cells with tamoxifen in each assay were the cause of
the variations in the effective tamoxifen concentration. Although
the addition of 20 μM tamoxifen did not immediately induce a
marked increase in intracellular calcium concentration, it may have
induced a moderate calcium influx, resulting in SCC cell death.
Additionally, these differences in incubation time
of skin SCC cells in each assay exclude the possibility of calcium
influx through a weakened cell membrane by tamoxifen. As mentioned
above, the intracellular calcium concentration was measured
immediately following the addition of tamoxifen to skin SCC cells.
Thus, it is strongly suggested that the plasma membrane of skin SCC
cells was intact when intracellular calcium concentration was
measured. Therefore, calcium influx may not be triggered in dying
cells when the plasma membrane is likely not intact.
To identify the mechanism underlying the
tamoxifen-induced increase of cytosolic calcium concentration in
skin SCC cells, it was investigated whether tamoxifen induced
calcium release from the endoplasmic reticulum stores. It has been
reported that thapsigargin discharged intracellular calcium stores
by potently inhibiting endoplasmic reticulum calcium ATPase,
followed by an increase of the intracellular cytosolic calcium
concentration (23). Although
thapsigargin increased intracellular cytosolic calcium
concentration in skin SCC cells, tamoxifen did not (Fig. 5), indicating that tamoxifen did not
induce the efflux of calcium from endoplasmic reticulum stores to
the cytosol in skin SCC cells.
Extrinsic stimuli may cause extracellular calcium
influx into the cell via calcium-permeable channels, such as
voltage-gated calcium channels and non-selective cation channels.
The effects of voltage-gated calcium channel inhibitors, such as
verapamil and nifedipine, and the non-selective cation channel
blocker, flufenamic acid, on tamoxifen-induced calcium influx in
skin SCC cells were examined in the present study. Nifedipine and
flufenamic acid significantly inhibited tamoxifen-induced calcium
influx in A431, DJM-1 and HSC-1 cells (Fig. 6A), indicating that both
voltage-gated calcium channels and non-selective cation channels
were involved in tamoxifen-induced calcium influx. In our
experiments, tamoxifen immediately increased intracellular calcium
concentration in skin SCC cells (Fig.
3). Thus, tamoxifen may not act by increasing the expression
level of calcium channels on the plasma membrane. These findings
suggest that tamoxifen modulates the activity of voltage-gated
calcium channels and non-selective cation channels to induce
extracellular calcium influx. Kim et al (17) reported that tamoxifen-induced
calcium influx in ER-negative human hepatoblastoma HepG2 cells was
significantly inhibited by flufenamic acid, although verapamil or
nifedipine did not affect this tamoxifen-induced calcium influx.
This observation together with our data suggest that the mechanism
of tamoxifen-induced calcium influx into the cytosol in ER-negative
cancer cells may vary among different cancer types. Zhang et
al reported that tamoxifen enhanced and prolonged ATP-induced
intracellular calcium elevation in both ER-positive MCF-7 breast
cancer cells and ER-negative C6 glioma cells (28). It was also reported that tamoxifen
increased intracellular calcium concentration in MG-63 osteosarcoma
cells (29), which express both
ER-α and -β (30). Although an
increase of tamoxifen-induced intracellular calcium concentration
may be accompanied by cytotoxicity (31), the effect of ER on
tamoxifen-induced intracellular calcium increase leading to cell
death and the detailed mechanism underlying tamoxifen-induced
calcium influx will be investigated in future studies.
Verapamil significantly inhibited the increase in
intracellular cytosolic calcium concentration of DJM-1 cells, and
tended to inhibit the increase in intracellular cytosolic calcium
concentration in A431 cells (Fig.
6A). However, verapamil did not inhibit the increase in
intracellular cytosolic calcium concentration in HSC-1 cells,
although nifedipine almost completely inhibited the increase in
intracellular cytosolic calcium concentration in this cell line. It
is reported that the inhibitory mechanisms of action of verapamil
and nifedipine on voltage-gated calcium channels are different
(32). Phenylalkylamines (e.g.,
verapamil) bind to the V-binding site in L-type calcium channels,
while dihydropyridines (e.g., nifedipine) bind to the N-binding
site. Administration of these two types of calcium channel
inhibitors results in different antihypertensive effects in
patients with essential hypertension and in normal subjects
(33). Although the details are
unknown, the mechanism of voltage-gated calcium channel activation
by tamoxifen may differ in skin SCC cells.
Tamoxifen-induced growth inhibition of skin SCC
cells was significantly attenuated by the addition of the
broad-spectrum PKC inhibitor phloretin (Fig. 7A). This observation suggests that
PKC may be involved in the calcium-mediated growth-inhibitory
signaling in skin SCC cells induced by tamoxifen. The
phosphorylation of PKCα increases during tamoxifen-induced
apoptosis in ER-negative human breast cancer MCF-7/ADR cells
(21), and our data suggested that
some isoforms of PKC in skin SCC cells were phosphorylated during
incubation with tamoxifen (Fig.
7B). The role of the PKC isoform involved in tamoxifen-induced
growth inhibition of skin SCC cells remains to be elucidated.
There was a tendency for the MEK inhibitor PD98059
to attenuate tamoxifen-induced growth inhibition of skin SCC cells
(Fig. 7A), suggesting that the
MAPK pathway was also involved in calcium-mediated
growth-inhibitory signaling in skin SCC cells induced by tamoxifen.
It is well-known that intracellular calcium affects the MAPK
pathway via small GTPase Ras activation (34). Tamoxifen increased the
phosphorylation of ERK1/2, downstream kinases of MEK, in A431,
DJM-1 and HSC-1 cells (Fig. 7B).
Xie et al (21) reported
that tamoxifen induced ERK1/2 phosphorylation in both ER-positive
and -negative breast cancer cells. Therefore, the MEK/ERK pathway
may be involved in the tamoxifen-induced growth inhibition of
cancer cells, although its contribution is limited.
Our data indicate that tamoxifen activates the PKC
and MAPK pathways during growth inhibition in skin SCC cells.
However, the downstream effect of PKC and MAPK activation on
tamoxifen-induced growth inhibition of skin SCC cells remains
unclear. It has been reported that PKC and MAPK activation affect
the expression and trafficking of tumor necrosis factor (TGF)-β,
which inhibits cancer cell growth. TGF-β is known to inhibit the
growth of A431 cells (35), and
PKC-ε and -ζ are involved in the trafficking of TGF-β (36). P38 MAPK, ERK and JNK are involved
in the expression of TGF-β in fibroblasts during apoptosis
(37). It was also reported that
tamoxifen induces the expression of TGF-β in ER-positive and
-negative breast cancer cells (38). In addition, it was observed that
tamoxifen increased the transcription level of TGF-β in HSC-1 cells
(data not shown). Taken together, these findings indicate that
tamoxifen may induce TGF-β expression by activating the PKC and
MAPK pathways through an increase in intracellular calcium
concentration, resulting in growth inhibition of skin SCC
cells.
In conclusion, the data of the present study
demonstrated that tamoxifen inhibits the proliferation of skin SCC
cells. To the best of our knowledge, this is the first report to
elucidate the antiproliferative effect of tamoxifen on skin SCC
cells. Tamoxifen induces extracellular calcium influx into the
cytoplasm via voltage-gated calcium channels and non-selective
cation channels, and then activates the PKC and MEK/ERK pathways.
These data suggest that the increase in intracellular calcium
concentration is key to the antiproliferative effects of tamoxifen
on ER-negative cancer cells.
Abbreviations:
|
ER
|
estrogen receptor
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
GI50
|
50% growth inhibition
|
|
JNK
|
c-Jun N-terminal kinase
|
|
MAPK
|
mitogen-activated protein kinase
|
|
MEK
|
mitogen-activated protein
kinase/extracellular signal-regulated kinase kinase
|
|
PKC
|
protein kinase C
|
|
TGF-β
|
tumor necrosis factor-β
|
Acknowledgments
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during this study
are available from the corresponding author on reasonable
request.
Authors’ contributions
GH was a major contributor to designing the study,
performing the experiments, analyzing the data and writing the
manuscript. KA, YN, YY and NH performed the experiments. MS
designed the study and wrote the manuscript. All authors have read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests to disclose.
References
|
1
|
Osborne CK: Tamoxifen in the treatment of
breast cancer. N Engl J Med. 339:1609–1618. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Johnston SR and Dowsett M: Aromatase
inhibitors for breast cancer: Lessons from the laboratory. Nat Rev
Cancer. 3:821–831. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Crewe HK, Notley LM, Wunsch RM, Lennard MS
and Gillam EM: Metabolism of tamoxifen by recombinant human
cytochrome P450 enzymes: Formation of the 4-hydroxy, 4′-hydroxy and
N-desmethyl metabolites and isomerization of
trans-4-hydroxytamoxifen. Drug Metab Dispos. 30:869–874. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guo WZ, Wang Y, Umeda E, Shiina I, Dan S
and Yamori T: Search for novel anti-tumor agents from ridaifens
using JFCR39, a panel of human cancer cell lines. Biol Pharm Bull.
36:1008–1016. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cancer NCCf: 41 Endocrine therapy.
Advanced Breast Cancer: Diagnosis and Treatment. National
Collaborating Centre for Cancer (UK); Cardiff: pp. 17–21. 2009
|
|
6
|
Bonneterre J, Buzdar A, Nabholtz JM,
Robertson JF, Thürlimann B, von Euler M, Sahmoud T, Webster A,
Steinberg M, Committee AW, et al Investigators Committee Members:
Anastrozole is superior to tamoxifen as first-line therapy in
hormone receptor positive advanced breast carcinoma. Cancer.
92:2247–2258. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Patterson J, Furr B, Wakeling A and
Battersby L: The biology and physiology of ‘Nolvadex’ (tamoxifen)
in the treatment of breast cancer. Breast Cancer Res Treat.
2:363–374. 1982. View Article : Google Scholar
|
|
8
|
Bakkevold KE, Pettersen A, Arnesjø B and
Espehaug B: Tamoxifen therapy in unresectable adenocarcinoma of the
pancreas and the papilla of Vater. Br J Surg. 77:725–730. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rosenberg L: Treatment of pancreatic
cancer. Promises and problems of tamoxifen, somatostatin analogs,
and gemcitabine. Int J Pancreatol. 22:81–93. 1997.PubMed/NCBI
|
|
10
|
Couldwell WT, Hinton DR, Surnock AA,
DeGiorgio CM, Weiner LP, Apuzzo ML, Masri L, Law RE and Weiss MH:
Treatment of recurrent malignant gliomas with chronic oral
high-dose tamoxifen. Clin Cancer Res. 2:619–622. 1996.PubMed/NCBI
|
|
11
|
Pawar P, Ma L, Byon CH, Liu H, Ahn EY,
Jhala N, Arnoletti JP, McDonald JM and Chen Y: Molecular mechanisms
of tamoxifen therapy for cholangiocarcinoma: Role of calmodulin.
Clin Cancer Res. 15:1288–1296. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hoelting T, Duh Q-Y, Clark OH and Herfarth
C: Tamoxifen antagonizes proliferation and invasion of estrogen
receptor-negative metastatic follicular thyroid cancer cells via
protein kinase C. Cancer Lett. 100:89–93. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Scandlyn MJ, Stuart EC, Somers-Edgar TJ,
Menzies AR and Rosengren RJ: A new role for tamoxifen in oestrogen
receptor-negative breast cancer when it is combined with
epigallocatechin gallate. Br J Cancer. 99:1056–1063. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen HY, Yang YM, Stevens BM and Noble M:
Inhibition of redox/Fyn/c-Cbl pathway function by Cdc42 controls
tumour initiation capacity and tamoxifen sensitivity in basal-like
breast cancer cells. EMBO Mol Med. 5:723–736. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moodbidri MS and Shirsat NV: Activated JNK
brings about accelerated apoptosis of Bcl-2-overexpressing C6
glioma cells on treatment with tamoxifen. J Neurochem. 92:1–9.
2005. View Article : Google Scholar
|
|
16
|
Maccarrone M, Fantini C, Ranalli M, Melino
G and Agrò AF: Activation of nitric oxide synthase is involved in
tamoxifen-induced apoptosis of human erythroleukemia K562 cells.
FEBS Lett. 434:421–424. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim JA, Kang YS, Jung MW, Lee SH and Lee
YS: Involvement of Ca2+ influx in the mechanism of
tamoxifen-induced apoptosis in HepG2 human hepatoblastoma cells.
Cancer Lett. 147:115–123. 1999. View Article : Google Scholar
|
|
18
|
Shim J-H, Choi C-S, Lee E-C, Kim M-Y and
Chun Y-J: Tamoxifen suppresses clusterin level through Akt
inactivation and proteasome degradation in human prostate cancer
cells. Biomol Ther (Seoul). 17:25–31. 2009. View Article : Google Scholar
|
|
19
|
Grynkiewicz G, Poenie M and Tsien RY: A
new generation of Ca2+ indicators with greatly improved
fluorescence properties. J Biol Chem. 260:3440–3450.
1985.PubMed/NCBI
|
|
20
|
Lippman M, Bolan G and Huff K: The effects
of estrogens and antiestrogens on hormone-responsive human breast
cancer in long-term tissue culture. Cancer Res. 36:4595–4601.
1976.PubMed/NCBI
|
|
21
|
Li Z, Wang N, Fang J, Huang J, Tian F, Li
C and Xie F: Role of PKC-ERK signaling in tamoxifen-induced
apoptosis and tamoxifen resistance in human breast cancer cells.
Oncol Rep. 27:1879–1886. 2012.PubMed/NCBI
|
|
22
|
Berridge MJ, Bootman MD and Roderick HL:
Calcium signalling: Dynamics, homeostasis and remodelling. Nat Rev
Mol Cell Biol. 4:517–529. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Thastrup O, Cullen PJ, Drøbak BK, Hanley
MR and Dawson AP: Thapsigargin, a tumor promoter, discharges
intracellular Ca2+ stores by specific inhibition of the
endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci USA.
87:2466–2470. 1990. View Article : Google Scholar
|
|
24
|
Racay P and Lehotský J: Intracellular and
molecular aspects of Ca(2+)-mediated signal transduction in
neuronal cells. Gen Physiol Biophys. 15:273–289. 1996.PubMed/NCBI
|
|
25
|
White CD and Sacks DB: Regulation of MAP
kinase signaling by calcium. MAP Kinase Signaling Protocols. 2nd.
Seger R: Humana Press; Totowa, NJ: pp. 151–165. 2010, View Article : Google Scholar
|
|
26
|
Radin DP and Patel P: Delineating the
molecular mechanisms of tamoxifen’s oncolytic actions in estrogen
receptor-negative cancers. Eur J Pharmacol. 781:173–180. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Johnson N, Bentley J, Wang LZ, Newell DR,
Robson CN, Shapiro GI and Curtin NJ: Pre-clinical evaluation of
cyclin-dependent kinase 2 and 1 inhibition in
anti-estrogen-sensitive and resistant breast cancer cells. Br J
Cancer. 102:342–350. 2010. View Article : Google Scholar :
|
|
28
|
Zhang W, Couldwell WT, Song H, Takano T,
Lin JH and Nedergaard M: Tamoxifen-induced enhancement of calcium
signaling in glioma and MCF-7 breast cancer cells. Cancer Res.
60:5395–5400. 2000.PubMed/NCBI
|
|
29
|
Lu YC, Jiann BP, Chang HT, Huang JK, Chen
WC, Su W and Jan CR: Effect of the anti-breast cancer drug
tamoxifen on Ca(2+) movement in human osteosarcoma cells. Pharmacol
Toxicol. 91:34–39. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jones PS, Parrott E and White IN:
Activation of transcription by estrogen receptor alpha and beta is
cell type- and promoter-dependent. J Biol Chem. 274:32008–32014.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jan CR, Cheng JS, Chou KJ, Wang SP, Lee
KC, Tang KY, Tseng LL and Chiang HT: Dual effect of tamoxifen, an
anti-breast-cancer drug, on intracellular Ca(2+) and cytotoxicity
in intact cells. Toxicol Appl Pharmacol. 168:58–63. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sandmann S and Unger T: L- and T-type
calcium channel blockade - the efficacy of the calcium channel
antagonist mibefradil. J Clin Basic Cardiol. 2:187–201. 1999.
|
|
33
|
Agabiti-Rosei E, Muiesan ML, Romanelli G,
Castellano M, Beschi M, Corea L and Muiesan G: Similarities and
differences in the antihypertensive effect of two calcium
antagonist drugs, verapamil and nifedipine. J Am Coll Cardiol.
7:916–924. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cullen PJ and Lockyer PJ: Integration of
calcium and Ras signalling. Nat Rev Mol Cell Biol. 3:339–348. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Goldkorn T and Mendelsohn J: Transforming
growth factor beta modulates phosphorylation of the epidermal
growth factor receptor and proliferation of A431 cells. Cell Growth
Differ. 3:101–109. 1992.PubMed/NCBI
|
|
36
|
Zhang F, Dong W, Zeng W, Zhang L, Zhang C,
Qiu Y, Wang L, Yin X, Zhang C and Liang W: Naringenin prevents
TGF-β1 secretion from breast cancer and suppresses pulmonary
metastasis by inhibiting PKC activation. Breast Cancer Res.
18:382016. View Article : Google Scholar
|
|
37
|
Xiao YQ, Freire-de-Lima CG, Schiemann WP,
Bratton DL, Vandivier RW and Henson PM: Transcriptional and
translational regulation of TGF-beta production in response to
apoptotic cells. J Immunol. 181:3575–3585. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Perry RR, Kang Y and Greaves BR:
Relationship between tamoxifen-induced transforming growth factor
beta 1 expression, cytostasis and apoptosis in human breast cancer
cells. Br J Cancer. 72:1441–1446. 1995. View Article : Google Scholar : PubMed/NCBI
|