Introduction
Pancreatic cancer is a highly malignant tumor of the
digestive system with a 5-year survival rate of <7%; the
incidence rate of pancreatic cancer is almost equal to its
mortality rate (1). The majority
of patients with pancreatic cancer lack apparent symptoms until it
has evolved into an advanced stage. Therefore, only ~20% of
patients with pancreatic cancer have the opportunity for radical
resection (2). Even among those
who undergo potentially curative resection, the majority of
patients will eventually relapse, and the 5-year survival rate of
patients who undergo a complete resection is <25% (3). Autopsy cases have revealed that ~90%
of patients with pancreatic cancer also have distant metastases
(4); therefore, it is imperative
to clarify the mechanism underlying its early metastasis and high
recurrence rate, in order to improve therapeutic efficacy.
It has previously been reported that there is a
subpopulation of cells in pancreatic ductal adenocarcinomaå(PDAC),
which is the most common type of pancreatic cancer, that exhibits
stem cell-like traits; these cells are termed pancreatic cancer
stem cells (PCSCs), and have the capacity of self-renewal and
differentiation into heterogeneous cancer cells. PCSCs serve a key
role in tumor initiation, invasion, metastasis and therapeutic
resistance, as well as in local recurrence following curative
resection. Therefore, the elimination of PCSCs from pancreatic
cancer is considered an effective strategy for the treatment of
this highly refractory malignancy (5-7).
Although additional molecular markers of CSCs are still being
discovered, three transcription factors; namely, SRY-box 2 (Sox2),
octamer-binding protein 4 (Oct4) and Nanog, have been reported as
master mediators of pluripotency (8). Epithelial-mesenchymal transition
(EMT) is characterized by the loss of intercellular adhesion and
decreased expression of epithelial markers, including E-cadherin,
and enhanced expression of mesenchymal markers, such as vimentin
(9). EMT has an important role in
tumor invasion, metastasis and therapeutic resistance. Notably, a
direct connection has been reported between EMT and the acquirement
of stem cell-like properties (10), as EMT generates neoplastic stem
cells. In addition, neoplastic stem cells tend to express higher
levels of the molecular markers of EMT during metastasis (10). Furthermore, non-PCSCs can be
converted into PCSCs by introducing ectopic expression of zinc
finger E-box binding homeobox 1, a key transcriptional factor
associated with EMT (11), which
facilitates cell invasion and treatment resistance to enhance the
malignant phenotype (12). This
further highlights the close association between the properties of
CSCs and EMT.
5′ adenosine monophosphate-activated protein kinase
(AMPK) is a heterotrimeric complex that consists of catalytic
α-subunits, and regulatory β- and γ-subunits. It exerts a crucial
role in regulating energy metabolism in cancer as an energy sensor
(13). AMPK activation inhibits
energy consumption under nutrient deprivation, leading to the
suppression of protein synthesis and cellular proliferation
(14). Previous studies have
confirmed that AMPK signaling is also involved in regulating
various pathological aspects of fibrosis, and modulates migration
and invasion (15-19). Alteration of cellular bioenergetics
through the regulation of AMPK signaling may be a prerequisite to
stemness (20). Recently, Sengupta
et al revealed that activation of tumor suppressor-liver
kinase B1 by honokiol subsequently enhances AMPK phosphorylation,
which in turn restricts the recruitment of signal transducer and
activator of transcription 3 (STAT3) to the promoter regions of
Sox2, Oct4 and Nanog, leading to inhibition of the stem-like
phenotype in breast cancer (8).
Similarly, methylisoindigo, which is a natural product of indirubin
and a derivative of isoindigo, is able to kill PCSCs by modulating
cell metabolism via activation of AMPK in PDAC (21). Metformin is an activator of AMPK,
which also serves important roles in targeting PCSCs via regulating
metabolism and microRNA expression (22,23).
Although AMPK signaling is involved in the stemness of pancreatic
cancer, its explicit mechanism has not been completely clarified
and there is currently a lack of effective drugs that
preferentially kill PCSCs via the modulation of AMPK signaling.
Betulinic acid (BA) is a natural pentacyclic
triterpene purified from bark, particularly bark from Betula
sp., which exhibits a wide spectrum of pharmacological and
biological activities. BA has been reported to exert antidepressive
(24), anti-inflammatory (25,26)
and anti-acquired immune deficiency syndrome (AIDS) (27,28)
effects, and possesses hepatoprotective potential (29) and anticancer efficacy (30-32).
It has previously been suggested that the combined use of BA and
mithramycin A may effectively suppress angiogenesis, proliferation
and invasion of pancreatic cancer through downregulation of SP1
(33). A previous study further
verified that lamin B1 is a novel therapeutic target in BA-treated
pancreatic cancer independent of SP1 signaling (34). BA may also effectively ameliorate
non-alcoholic fatty liver disease (NAFLD) via activation of AMPK
and modulation of calcium/calmodulin-dependent protein kinase
kinase-AMPK-mammalian target of rapamycin (mTOR)-sterol regulatory
element-binding protein 1 signaling (35). However, whether BA exerts
anticancer effects on pancreatic cancer and the underlying
mechanism of action remain elusive. Therefore, the present study
aimed to demonstrate whether BA could inhibit the EMT and stemness
of pancreatic cancer cells through regulating the expression of
pluripotency-induced transcription factors (i.e. Sox2, Oct4 and
Nanog) via the activation of AMPK signaling. In addition, the study
aimed to elucidate the contribution of BA to pancreatic cancer
therapy.
Materials and methods
Reagents and antibodies
BA, gemcitabine, 5-aminoimidazole-4-carboxamide
1-β-D-ribofuranoside (AICAR), dimethyl sulfoxide (DMSO) and MTT
were purchased from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany).
BA and AICAR were initially dissolved in dimethyl sulfoxide at
stock concentrations of 50 mM and 2 M, respectively. Working
concentrations for BA and AICAR were diluted immediately in culture
medium prior to use. Human epidermal growth factor (EGF) and
fibroblast growth factor (FGF) were purchased from PeproTech, Inc.
(Rocky Hill, NJ, USA).The antibodies used in this study were as
follows: Rabbit anti-Sox2 (1:1,000 dilution; cat. no. ab97959),
anti-Oct4 (1:1,000 dilution; cat. no. ab18976) and anti-Nanog
(1:1,000 dilution; cat. no. ab80892) (all from Abcam, Cambridge,
UK), mouse anti-β-actin (1:10,000 dilution; cat. no. 19526;
KangChen Bio-Tech, Inc., Shanghai, China), mouse anti-E-cadherin
(1:1,000 dilution; cat. no. sc-71008), rabbit anti-cluster of
differentiation (CD)133 (1:1,000 dilution; cat. no. sc-11406),
mouse anti-aldehyde dehydrogenase (ALDH)1 (1:1,000 dilution; cat.
no. sc-374149) and mouse anti-epithelial cell adhesion molecule
(EpCAM) (1:1,000 dilution; cat. no. sc-66020) (all from Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), and rabbit anti-vimentin
(1:800 dilution; cat. no. 12826), anti-AMPK (1:800 dilution; cat.
no. 5831) and anti-phosphorylated (P)-AMPK (Thr172) (1:800
dilution; cat. no. 2535) (all from Cell Signaling Technology, Inc.,
Danvers, MA, USA).
Cell culture
The Mia PaCa-2 and Panc-1 human pancreatic cancer
cell lines were purchased from the Type Culture Collection of the
Chinese Academy of Sciences (Shanghai, China). Mia PaCa-2 and
Panc-1 cells were cultured in Dulbecco’s modified Eagle’s medium
(DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% heat-inactivated fetal bovine serum (FBS)
(HyClone; GE Healthcare Life Sciences, Logan, UT, USA), 100 U/ml
penicillin and 100 µg/ml streptomycin (Gibco; Thermo Fisher
Scientific, Inc.). All cells were cultured in a humidified
atmosphere at 37°C containing 5% CO2.
Cell viability assay
Mia PaCa-2 and Panc-1 cells were seeded into 96-well
plates at a density of 5×103 cells/well and treated with
various concentrations of BA (0, 12.5, 25, 50, 100 and 200
µM) for 24, 48 and 72 h at 37°C. Subsequently, 10 µl
MTT (Sigma-Aldrich; Merck KGaA) was added and incubated for 4 h at
37°C. The supernatant was then replaced with 100 µl DMSO
(Sigma-Aldrich; Merck KGaA) and absorbance was detected at 490 nm
using a multiwell microplate reader (BioTek Instruments, Inc.,
Winooski, VT, USA).
Colony formation assay
Mia PaCa-2 and Panc-1 cells were digested, counted
and seeded into 6-well plates at a density of 800 cells/well. After
adherence overnight, pancreatic cancer cells were treated with BA
(50 µM), gemcitabine (5 µM), or gemcitabine (5
µM) combined with BA (50 µM) for 24 h at 37°C, after
which the medium was replaced with drug-free medium containing 10%
FBS. The cells were then allowed to grow for 2 weeks to form
colonies. After 2 weeks, the colonies were washed with PBS, fixed
with 4% paraformaldehyde for 30 min at room temperature and stained
for 10 min at room temperature with 0.1% crystal violet solution,
followed by rinsing and imaging. The number of colonies with a
diameter >0.5 mm was counted under a microscope (Nikon Eclipse
Ti-S; Nikon Corporation, Tokyo, Japan).
Apoptosis assay
The apoptosis of pancreatic cancer cells was
assessed using flow cytometry with an Annexin V-fluorescein
isothiocyanate (FITC)/7-aminoactinomycin D (7-AAD) apoptosis
detection kit (BD Biosciences, Franklin Lakes, NJ, USA), according
to the manufacturer’s protocol. The preprocessing of pancreatic
cancer cells was performed as previously described (36). Cells treated with gemcitabine (5
µM), BA (50 µM), or gemcitabine (5 µM)
combined with BA (50 µM) for 24 h at 37°C were washed twice
in cold PBS and resuspended in 1X binding buffer (BD Biosciences)
at a density of 1×106 cells/ml. Cell suspensions
(2.5×105 cells) were added to 1.5 ml Eppendorf tubes, to
which 5 µl allophycocyanin Annexin V and 7-AAD were added,
and were gently vortexed. Subsequently, cells were incubated at
room temperature for 15 min in the dark, and the percentage of
apoptotic cells was quantified by flow cytometry using a
FACSCalibur flow cytometer (BD Biosciences). Data were analyzed
using Winmdi2.9 software (The Scripps Research Institute, San
Diego, CA, USA). The total apoptotic rate was assessed by adding
the rate of Annexin V-FITC+/7-AAD- cells
(early apoptotic cells) and Annexin
V-FITC+/7-AAD+ cells (late apoptotic cells)
together.
Tumorsphere formation assay
Following treatment with BA (50 µM) or AICAR
(2 mM), or with BA (50 µM) combined with siRNA to knockdown
AMPK or siControl for 24 h at 37°C, pancreatic cancer cells were
digested, counted and seeded in 6-well ultra-low attachment plates
(Corning Incorporated, Corning, NY, USA) at a density of 5,000
cells/well in serum-free DMEM/F12 medium (Gibco; Thermo Fisher
Scientific, Inc.) containing 20 ng/ml human FGF, 20 ng/ml human EGF
and 1% B27 (Gibco; Thermo Fisher Scientific, Inc.). Cells were
subsequently cultured at 37°C in an atmosphere containing 5%
CO2 for 1 week to form tumorspheres. After 1 week,
tumorsphere formation was counted and recorded under a light
microscope (Nikon Corporation) at a magnification of ×200.
Wound-scratch assay
The wound-scratch assay was conducted to examine the
migratory capacity of pancreatic cancer cells. Briefly, once Mia
PaCa-2 and Panc-1 cells were cultured to 90-100% confluence, a
10-µl pipette tip was used to generate a wound in the
surface of the cells in a 6-well plate, and then the cells were
treated with or without BA (50 µM) for 48 h at 37°C. Images
of the same fields at the indicated time-points (0 and 48 h) were
captured under a light microscope (Nikon Corporation) at a
magnification of ×100.
Transwell invasion assay
Transwell chamber (EMD Millipore, Billerica, MA,
USA) assays were performed to assess the invasive ability of
pancreatic cancer cells, in accordance with a previously described
protocol (37). Briefly, Mia
PaCa-2 and Panc-1 cells were serum-starved for 6-8 h and were
pretreated with BA (50 µM) or AICAR (2 mM) for 24 h at 37°C.
In addition, the upper chamber was coated with Matrigel
(Sigma-Aldrich; Merck KGaA) and was incubated at 37°C in an
atmosphere containing 5% CO2 for 5 h. Subsequently, Mia
PaCa-2 and Panc-1 cells (1×105) were digested,
resuspended in serum-free medium and seeded into the upper chamber.
The pancreatic cancer cells were allowed to invade into the lower
chamber, which contained medium supplemented with 10% FBS, for 24 h
at 37°C. The non-invading cells on the upper side were scraped off
using a cotton swab, and the membrane was then fixed with 4%
paraformaldehyde for 30 min at room temperature and stained for 10
min at room temperature with 0.1% crystal violet. Subsequently, the
number of cells on each membrane was counted in 10 random fields
and images were captured under a light microscope (Nikon
Corporation) at ×200 magnification. The values reported are the
mean of triplicate experiments.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Following treatment with or without BA (50
µM), with various concentrations of gemcitabine (0, 1 and 5
µM), or with gemcitabine (5 µM) combined with BA (50
µM) for 24 h at 37°C, total RNA was extracted from the cells
using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer’s protocol. The isolated total RNA
was then reverse transcribed into cDNA using a PrimeScript RT
reagent kit (Takara Biotechnology Co., Ltd., Dalian, China),
according to the manufacturer’s protocol. An iQ5 Multicolor
Real-Time PCR Detection system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) and a SYBR Green PCR kit (Takara Biotechnology
Co., Ltd.) were used to conduct RT-qPCR, according to the
manufacturers’ protocols. The RT-qPCR experimental steps were
conducted as previously described (38). The specificity of the amplified PCR
products was evaluated by melting curve analysis, and the
comparative Cq method was used to determine the expression levels
of each target gene, with GAPDH as a normalization control, as
previously described (39). The
primer sequences used for RT-qPCR were as follows: Sox-2, forward
5′-GCCGAGTGGAAACTTTTGTCG-3′, reverse 5′-GGCAGCGTGTACTTATCCTTCT-3′;
Nanog, forward 5′-TTTGTGGGCCTGAATAAGCAG-3′, reverse
5′-AGGGCTGTCCTGAATAAGCAG-3′; Oct4, forward
5′-CTGGGTTGATCCTCGGACCT-3′, reverse 5′-CCATCGGAGTTGCTCTCCA-3′;
CD133, forward 5′-TACAACGCCAAACCACGACTGT-3′, reverse
5′-TCTGAACCAATGGAATTCAAGACCCTTT-3′; ALDH1, forward
5′-TGTTAGCTGATGCCGACTTG-3′, reverse 5′-CTTCTTAGCCCGCTCAACAC-3′;
EpCAM, forward 5′-ATGTTTGGTGATGAAGGCAGAA-3′, reverse
5′-ATCGCAGTCAGGATCATAAAGC-3′; and β-actin, forward
5′-ATCGTGCGTGACATTAAGGAGAAG-3′ and reverse
5′-AGGAAGAAGGCTGGAAGAGTG-3′.
Immunofluorescence staining
Following treatment with or without BA (50
µM) for 24 h at 37°C, pancreatic cancer cells
(1×106) were washed with PBS and fixed in 4%
paraformaldehyde for 30 min at room temperature. Fixed cells were
washed again with PBS and permeabilized in 0.5% Triton X-100
diluted in PBS for 10 min; cells on the slides were subsequently
blocked with 5% bovine serum albumin (Beyotime Institute of
Biotechnology, Guangzhou, China) for 1 h at room temperature and
were then incubated with a Sox2 antibody (1:150 dilution) at 4°C
overnight. Following incubation with a secondary antibody (1:150
dilution; cat. no. 913921; Jackson ImmunoResearch Laboratories,
Inc., West Grove, PA, USA) for 1 h at room temperature, the nuclei
of the cells were stained with DAPI and the cells were sealed on
glass slides. A confocal microscope (Zeiss AG, Oberkochen, Germany)
was used to capture images and record the cells at a wavelength of
488 nm and a magnification of ×400.
Western blotting
Following treatment with various concentrations of
BA (0, 25 and 50 µM), with BA (50 µM) for various
durations (0, 5, 15, 30, 60 and 120 min), various concentrations of
AICAR (0 and 2 mM), various concentrations of gemcitabine (0, 1 and
5 µM), or with BA (50 µM) combined with siRNA to
knockdown AMPK or siControl, or with gemcitabine (5 µM)
combined with BA (50 µM) for 24 h at 37°C, total proteins
were extracted from Mia PaCa-2 and Panc-1 cells (1×106)
grown under experimental conditions using radioimmunoprecipitation
assay lysis buffer (Beyotime Institute of Biotechnology). The
bicinchoninic acid protein assay kit (Pierce; Thermo Fisher
Scientific, Inc.) was used to determine protein concentrations,
according to the manufacturer’s protocol. Western blotting was
conducted as previously described (40). Briefly, proteins (150 µg)
were separated by 10% SDS-PAGE and were transferred onto
polyvinylidene difluoride membranes. Subsequently, the membranes
were blocked with 5% non-fat dry milk in Tris-buffered saline
containing 0.1% Tween (TBST) and were incubated with primary
antibodies overnight at 4°C. After three washes in TBST (10
min/wash), the membranes were incubated with goat anti-rabbit
immunoglobulin G (IgG)-horseradish peroxidase (HRP) (1:10,000
dilution; cat. no. ab6721; Abcam) or goat anti-mouse IgG-HRP
(1:10,000 dilution; cat. no. ab6789; Abcam) secondary antibodies
for 1 h at room temperature, and were washed again. The
immunoreactive bands were visualized using a chemiluminescence
detection system (EMD Millipore) through the peroxidase reaction,
and images of the bands were recorded using the ChemiDoc XRS
imaging system (Bio-Rad Laboratories, Inc.). β-actin was used as an
internal loading control.
Immunohistochemical analysis
The present study was approved by the Ethical
Committee of The First Affiliated Hospital of Xi’an Jiaotong
University (Xi’an, China), and written informed consent was
obtained from the patients. A total of 72 pancreatic cancer tissues
and eight normal pancreatic samples were collected from the
Department of Hepatobiliary Surgery, The First Affiliated Hospital
of Xi’an Jiaotong University. The details of these samples were
provided in our previous study (41). The tissue samples were fixed in 10%
paraformaldehyde and were embedded in paraffin for further
immunohistochemical analysis, as previously described (42). Briefly, pancreatic tissues were cut
into 5-µm sections on glass slides, and the sections were
deparaffinized and rehydrated, after which, the sections underwent
antigen retrieval and endogenous enzyme blocking. The sections were
then incubated with primary antibodies (anti-P-AMPK and anti-Sox2;
1:150 dilution) overnight at 4°C, followed by incubation with
streptavidin peroxidase (Dako LSAB + HRP kit; Dako; Agilent
Technologies, Inc., Santa Clara, CA, USA) for 30 min at room
temperature. The sections were incubated with DAB for 5 min
followed by counterstaining with hematoxylin for 2 min at room
temperature. Finally, the sections were observed under a light
microscope (Nikon Corporation).
Gene silencing by small interfering
(si)RNA
Loss-of-function analysis was conducted using siRNA
to knockdown AMPK, which was purchased from Shanghai GenePharma
Co., Ltd. (Shanghai, China). The sequences of siRNA and
corresponding siControl are provided in our previous study
(41). Each siRNA (100 nM) was
mixed with Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) as a carrier and were transfected into
pancreatic cancer cells (1×106) for 10 h at 37°C,
according to the manufacturer’s protocol. The efficacy of AMPK
knockdown was validated by western blot analysis. A total of 48 h
post-transfection, these cells underwent further experimentation
(Transwell invasion assay and tumorsphere formation assay).
Statistical analysis
Data shown are representative of three independent
experiments, and for each experiment, at least three samples were
analyzed for each treatment group. Data are presented as the means
± standard deviation. Differences were evaluated using Student’s
t-test, or one-way analysis of variance for multiple comparisons
with the Student-Newman-Keuls method as a post hoc test.
Statistical analysis of the human tissue data was performed using
Pearson’s χ2 test. All statistical analyses were
performed using SPSS 20.0 (SPSS, Inc., Chicago, IL, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
BA inhibits the proliferation and
tumorsphere formation of pancreatic cancer cells
To determine whether BA could suppress the viability
of pancreatic cancer cells, the MTT assay was used. Briefly, Mia
PaCa-2 and Panc-1 cells were treated with a series of gradually
increasing concentrations of BA [0 (control), 12.5, 25, 50, 100 and
200 µM] for 24, 48 and 72 h, and absorbance was measured at
490 nm at the designated time-points to analyze cell viability. The
results indicated that BA impaired the growth of Mia PaCa-2 and
Panc-1 cells in time- and dose-dependent manners (Fig. 1A and B). The half maximal
inhibitory concentration of BA was ~50 µM for both Panc-1
and Mia PaCa-2 cells, and it exerted no cytotoxic effects on a
pancreatic duct epithelial cell line (data not shown). This
concentration of BA was in accordance with previously used dosages
(34); therefore, 50 µM BA
was used for subsequent experiments. The present study subsequently
examined the effects of BA on colony formation and tumorsphere
formation in Mia PaCa-2 and Panc-1 cells (Fig. 1C-F). As shown in Fig. 1C and E, compared with in untreated
control cells, the number of colonies in cells treated with 50
µM BA was markedly decreased. Since cancer stemness is
associated with mechanisms underlying cancer metastasis and
occurrence, the present study further explored the role of BA in
the stemness of pancreatic cancer. As shown in Fig. 1D and F, tumorsphere formation was
decreased in Mia PaCa-2 and Panc-1 cells upon BA treatment compared
with in untreated cancer cells.
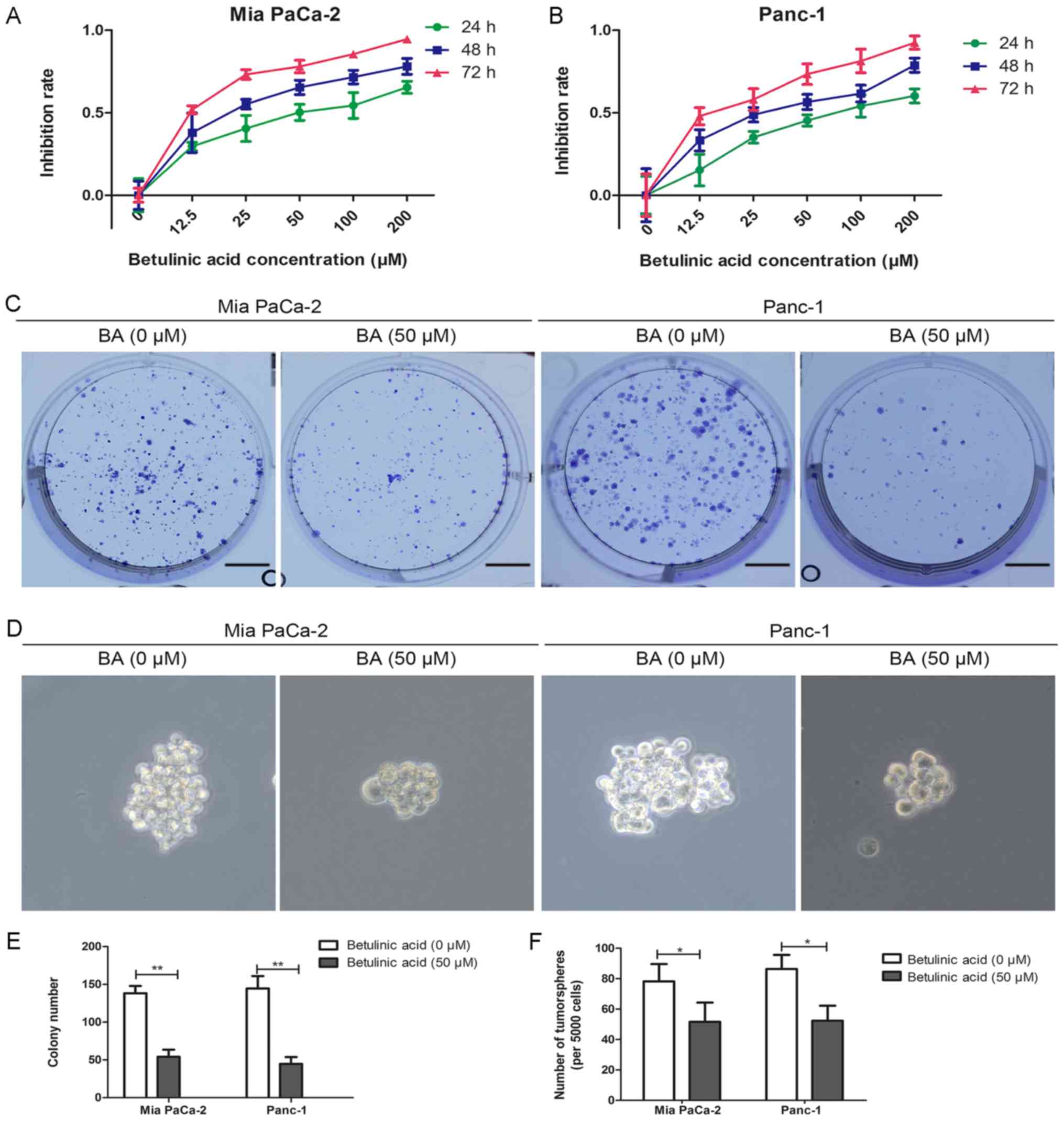 | Figure 1Effects of BA on the proliferation
and tumorsphere formation of pancreatic cancer cells. (A and B) Mia
PaCa-2 and Panc-1 cells were incubated with a series of BA
concentrations (0, 12.5, 25, 50, 100 and 200 µM) for 24, 48
and 72 h, and cell viability was evaluated by MTT assay. (C and E)
Effects of BA on the colony-forming ability of Mia PaCa-2 and
Panc-1 cells were assessed by colony formation assay. Images are
representative of three independent experiments, and the colony
number was counted and plotted. Scale bar, 1 cm. (D and F)
Tumorsphere formation assay of Mia PaCa-2 and Panc-1 cells treated
with or without 50 µM BA. The number of tumorspheres was
counted and plotted. Magnification, ×200. *P<0.05,
**P<0.01. BA, betulinic acid. |
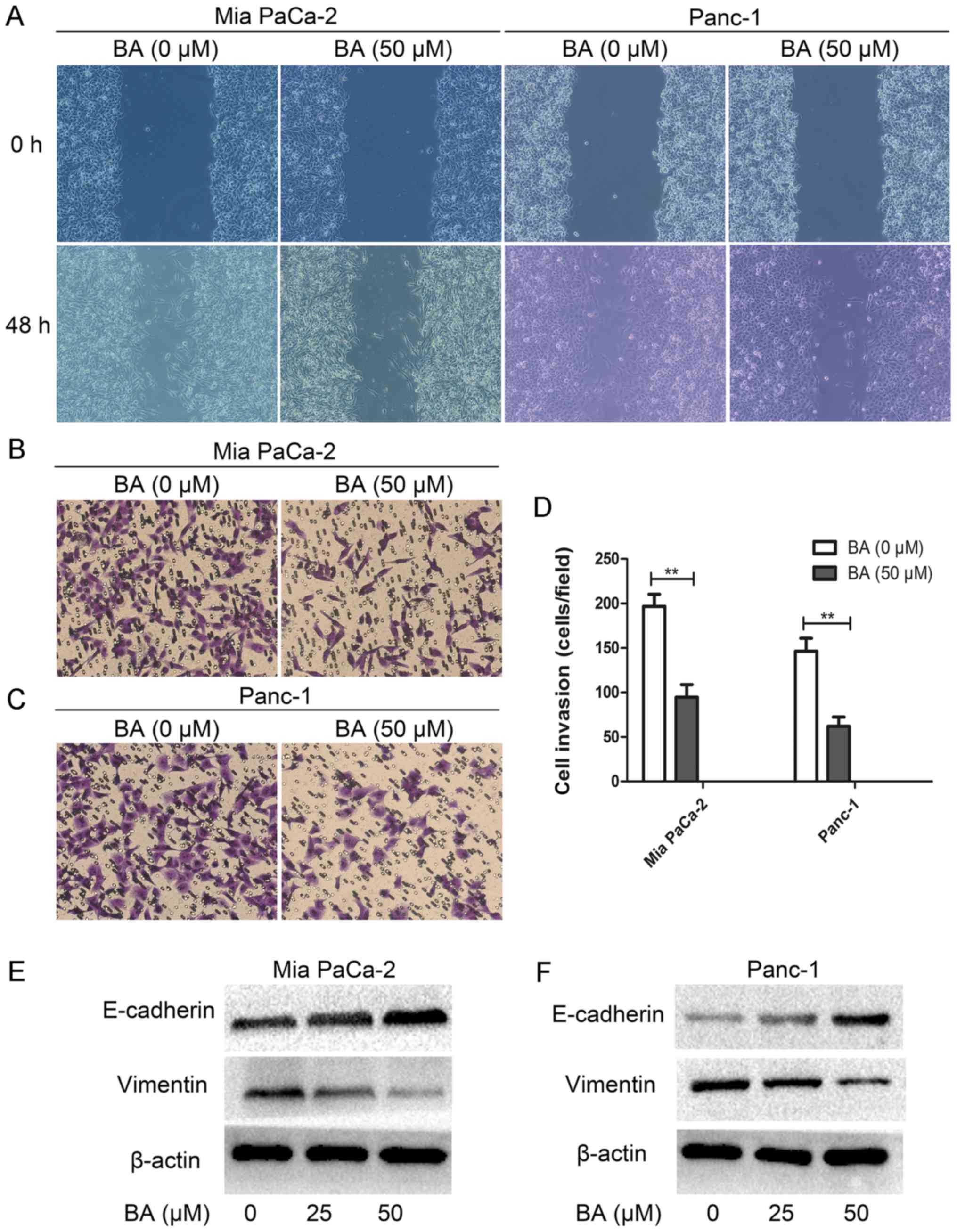
BA suppresses the migration and invasion
of pancreatic cancer cells via inhibiting EMT
It has previously been reported that the EMT process
endows cancer cells with an increased self-renewal capability and
mesenchymal phenotype, which is necessary for tumor metastasis
(43). Therefore the present study
examined the effects of BA on EMT, migration and invasion of
pancreatic cancer cells. Notably, BA-treated Mia PaCa-2 and Panc-1
cells exhibited reduced migration and invasion in vitro compared
with untreated cells (Fig. 2A-D).
In addition, the results of western blotting suggested that the
epithelial marker E-cadherin was elevated, whereas the mesenchymal
marker vimentin was decreased in BA-treated Mia PaCa-2 and Panc-1
cells compared with the control (Fig.
2E and F). Collectively, these data indicated that BA may
inhibit the migration and invasion of pancreatic cancer cells
through targeting EMT.
BA inhibits the stemness of pancreatic
cancer cells and activates AMPK signaling
Although the number of known molecular markers of
the cancer stem-like phenotype is still increasing, three
transcription factors; namely Sox2, Oct4 and Nanog, have been
strongly validated as master regulators in the maintenance of the
cancer stem-like phenotype (8).
The present study examined whether BA treatment affects the
expression of these master mediators of pluripotency (Fig. 3). RT-qPCR analysis revealed that BA
treatment suppressed the mRNA expression levels of Sox2, Oct4 and
Nanog (Fig. 3A and B). In
addition, as shown in Fig. 3C,
western blotting indicated that compared with the group without BA
treatment, treatment of Mia PaCa-2 and Panc-1 cells with 50
µM BA downregulated the expression levels of Sox2, Oct4 and
Nanog, and upregulated P-AMPK expression. The suppressive effects
of BA on other stemness markers (CD133, ALDH1 and EpCAM) were
similar to those on the aforementioned three master regulators
(data not shown). The findings of immunofluorescence analysis also
confirmed that BA could suppress the expression and nuclear
localization of Sox2 in both pancreatic cancer cell lines (Fig. 3F and G). Notably, AMPK signaling
has an important influence on the cancer stem-like phenotype
(20,44). Therefore, to investigate whether BA
could activate AMPK signaling, Mia PaCa-2 and Panc-1 cells were
treated with BA at various time-points (0, 15, 30, 60, 90 and 120
min), and total protein was extracted a to detect the expression
levels of P-AMPK by western blotting. The results revealed that BA
treatment enhanced the expression of P-AMPK in a time-dependent
manner (Fig. 3D and E).
Collectively, these data indicated that BA treatment downregulated
the expression of Sox2, Oct4 and Nanog, and activated AMPK
signaling in pancreatic cancer cells.
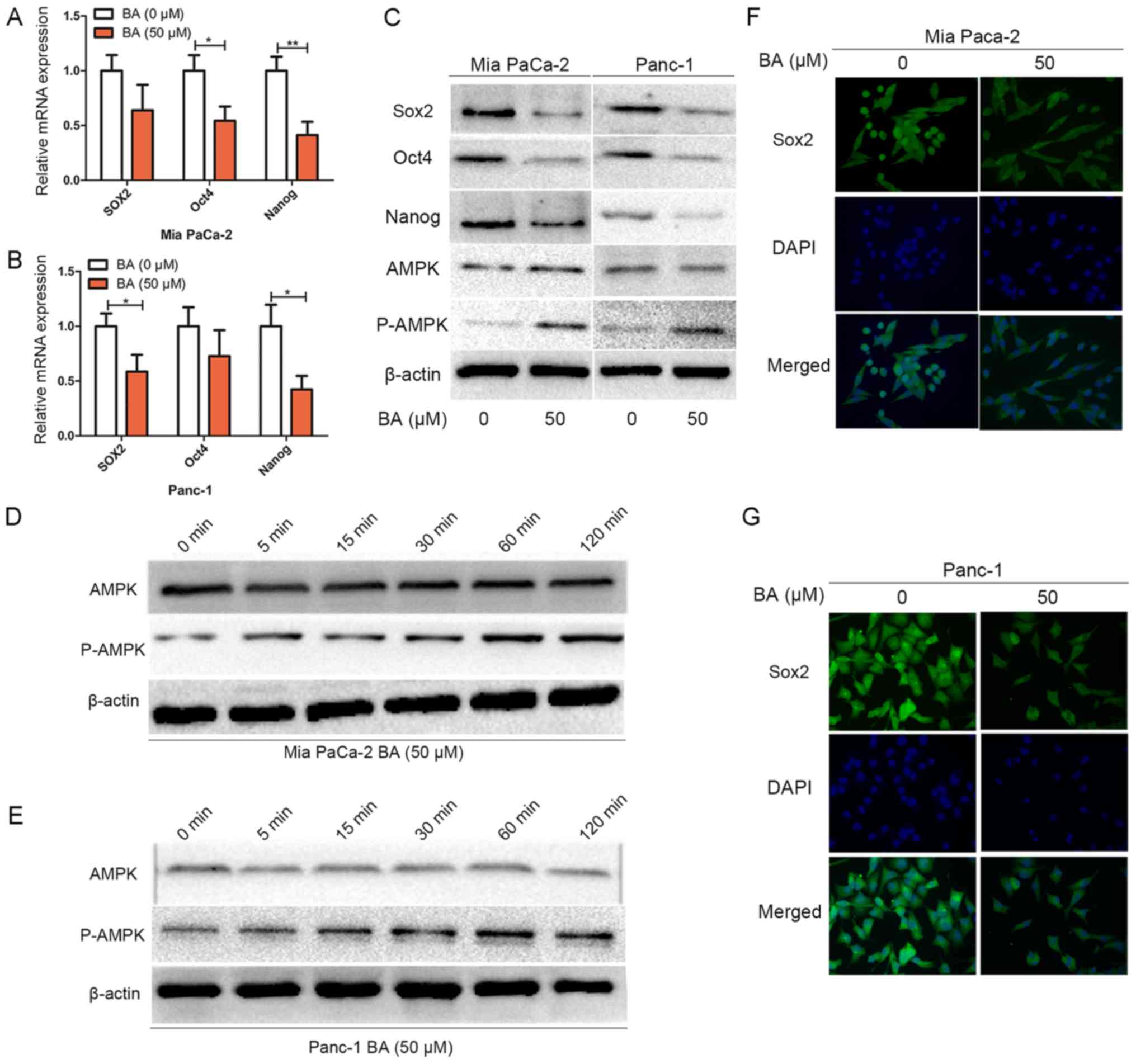 | Figure 3BA inhibits the stemness of
pancreatic cancer cells and activates AMPK signaling. (A and B) Mia
PaCa-2 and Panc-1 cells were pretreated with BA for 24 h, total RNA
was extracted and reverse transcription-quantitative polymerase
chain reaction was conducted to detect the expression levels of
Sox2, Oct4 and Nanog. *P<0.05 and
**P<0.01. (C) Mia PaCa-2 and Panc-1 cells were
pretreated with or without 50 µM BA for 48 h, and western
blot analysis was performed to assess the protein expression levels
of master pluripotency regulators (Sox2, Oct4 and Nanog), total
AMPK and P-AMPK. (D and E) Mia PaCa-2 and Panc-1 cells were treated
with 50 µM BA at various time-points (0, 5, 15, 30, 60 and
120 min), and total protein was extracted to detect the expression
levels of P-AMPK by western blotting. (F and G) Mia PaCa-2 and
Panc-1 cells were pretreated with BA for 24 h, and
immunofluorescence analysis was conducted to assess the expression
and nuclear localization of Sox2 in Mia PaCa-2 and Panc-1 cells.
Magnification, ×400. AMPK, 5′ adenosine monophosphate-activated
protein kinase; BA, betulinic acid; Oct4, octamer-binding protein
4; P, phosphorylated; Sox2, SRY-box 2. |
AMPK activation by AICAR exhibits similar
effects to BA on EMT and stemness of pancreatic cancer cells
To uncover the potential association between AMPK
signaling and cancer stemness, immunohistochemical analysis was
conducted to detect the expression levels of P-AMPK and Sox2 in
normal pancreatic tissues and pancreatic cancer tissues.
Immunohistochemical analysis revealed that, compared with in normal
pancreatic tissues, the expression levels of P-AMPK were decreased
in pancreatic cancer tissues. Conversely, the expression levels of
Sox2 were markedly elevated (Fig.
4A). Furthermore, Sox2 expression was detected in 78.3% (47/60)
of pancreatic cancer tissues with negative P-AMPK expression
(Fig. 4B), thus suggesting an
inverse relationship between P-AMPK signaling and the cancer
stem-like phenotype, and indicating that downregulation of P-AMPK
may lead to increased cancer stemness. In order to further clarify
the effects of AMPK signaling on the maintenance of pancreatic
cancer cell stemness and to determine its role in EMT, an AMPK
activator, AICAR (2 mM), was used to treat Mia PaCa-2 and Panc-1
cells for 24 h. The results suggested that AMPK activation by AICAR
effectively inhibited the invasion and tumorsphere formation of Mia
PaCa-2 and Panc-1 cells compared with the vehicle control (Fig. 4C-F). Furthermore, the results of
western blotting indicated that the expression levels of the
epithelial marker E-cadherin were elevated, whereas those of the
mesenchymal marker vimentin were decreased in AICAR-treated Mia
PaCa-2 and Panc-1 cells compared with the control. In addition, the
expression levels of the master pluripotency inducers (Sox2, Oct4
and Nanog) were decreased (Fig. 4G and
H). Similar effects were observed on other stemness markers
(CD133, ALDH1 and EpCAM; data not shown). These data indicated that
AMPK activation by AICAR may exert similar effects to BA on EMT and
stemness of pancreatic cancer cells.
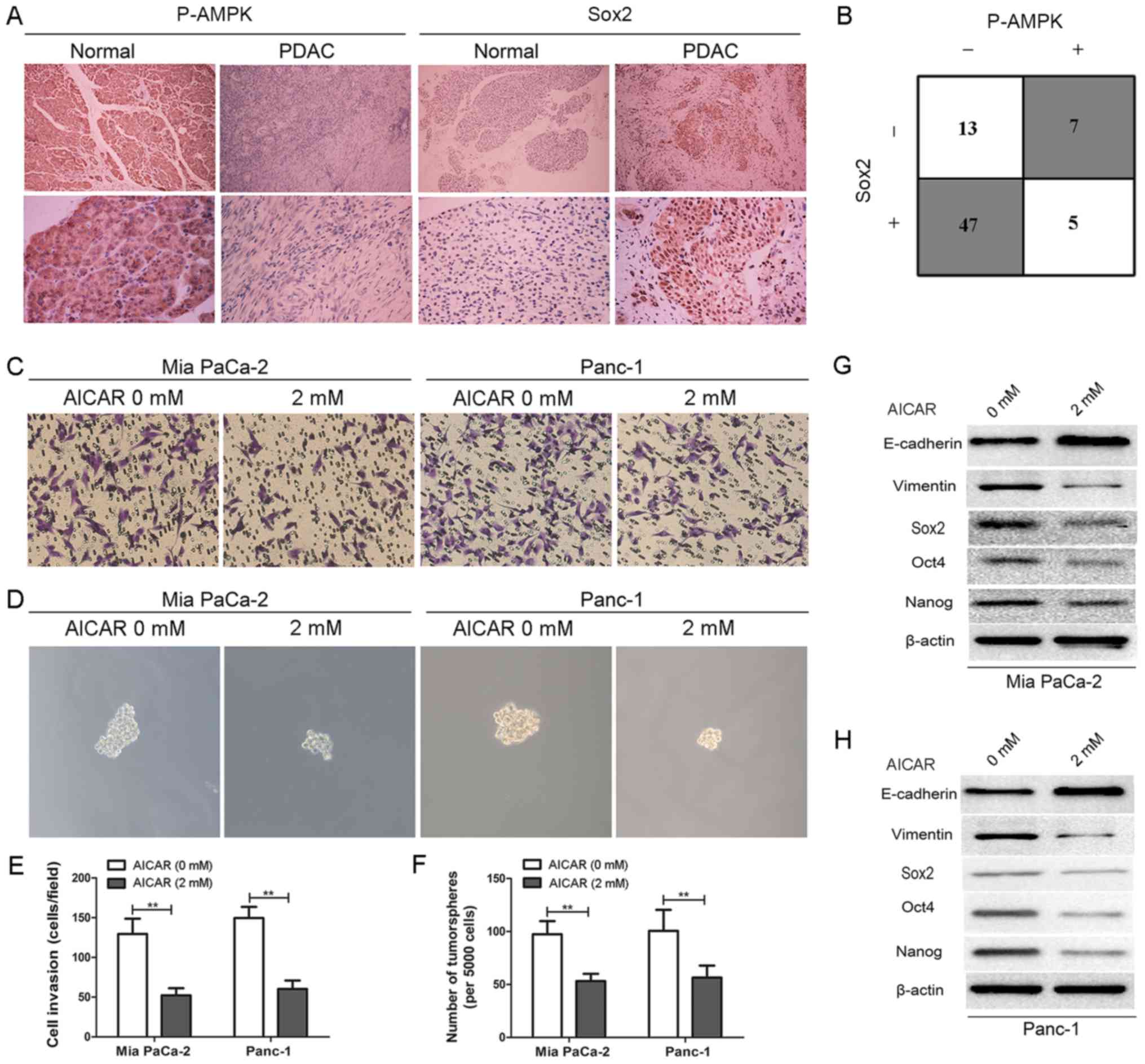 | Figure 4AMPK activation by AICAR exerts
similar effects to betulinic acid on EMT and stemness of pancreatic
cancer cells. (A) Representative images of immunohistochemical
staining of P-AMPK and SOX2 in normal pancreatic tissues and
pancreatic cancer tissues. Magnification, ×100 for the upper images
and ×400 for the lower images. (B) An inverse association between
P-AMPK and SOX2 expression was detected in pancreatic cancer
tissues. P<0.05 by two-tailed χ2 test. (C and E)
Effects of AMPK activation by AICAR (2 mM) on the invasive ability
of Mia PaCa-2 and Panc-1 cells were evaluated by Matrigel invasion
assay. Images are representative of three independent experiments,
and the invasive cells were counted and plotted. Magnification,
×200. **P<0.01. (D and F) Tumorsphere formation assay
of Mia PaCa-2 and Panc-1 cells treated with or without 2 mM AICAR.
The number of tumorspheres was counted and plotted. Magnification,
×200. **P<0.01. (G and H) Mia PaCa-2 and Panc-1 cells
were pretreated with or without 2 mM AICAR for 48 h, and western
blot analysis was performed to assess the expression levels of
master pluripotency regulators (Sox2, Oct4, and Nanog) and EMT
markers (E-cadherin and vimentin). AICAR,
5-aminoimidazole-4-carboxamide 1-β-D-ribofuranoside; AMPK, 5′
adenosine monophosphate-activated protein kinase; EMT,
epithelial-mesenchymal transition; Oct4, octamer-binding protein 4;
P, phosphorylated; PDAC, pancreatic ductal adenocarcinoma; Sox2,
SRY-box 2. |
Knockdown of AMPK rescues BA-induced
suppression of EMT and stemness in pancreatic cancer cells
In order to test the hypothesis that BA inhibits
stemness and EMT of pancreatic cancer through activating AMPK
signaling, AMPK-specific siRNA (si-AMPK) was used to silence AMPK
expression in pancreatic cancer cells in conjunction with BA
treatment. Successful silencing of AMPK expression was confirmed in
Mia PaCa-2 and Panc-1 cells by western blotting (Fig. 5A). Subsequently, AMPK-depleted and
control pancreatic cancer cells (Mia PaCa-2-siAMPK, Mia
PaCa-2-siControl, Panc-1-siAMPK and Panc-1-siControl) were treated
with BA and were subjected to Transwell invasion assay, tumorsphere
formation assay and western blot analysis (Fig. 5). BA could effectively inhibit
invasion (Fig. 5D-F) and
tumorsphere formation (Fig. 5G-I)
in Mia PaCa-2-siControl and Panc-1-siControl cells. Conversely, Mia
PaCa-2-siAMPK and Panc-1-siAMPK cells exhibited a slightly higher
capacity for invasion and tumor-sphere formation, which was not
suppressed by BA treatment. Furthermore, this study analyzed
whether BA could abrogate the expression of master pluripotency
inducers (Sox2, Oct4 and Nanog) and EMT markers (E-cadherin and
vimentin) in the absence of AMPK. As shown in Fig. 5B and C, BA treatment not only
suppressed the expression of Sox2, Oct4, Nanog and vimentin, but
also increased the expression of E-cadherin in Mia PaCa-2-siControl
and Panc-1-siControl cells; however, no alterations were detected
in Mia PaCa-2-siAMPK and Panc-1-siAMPK cells. Similar results were
also determined with regards to other stemness markers (CD133,
ALDH1 and EpCAM; data not shown). Collectively, these results
indicated that BA may inhibit invasion and tumorsphere formation of
pancreatic cancer cells, and regulate the expression levels of
Sox2, Oct4, Nanog and EMT markers in an AMPK-dependent manner.
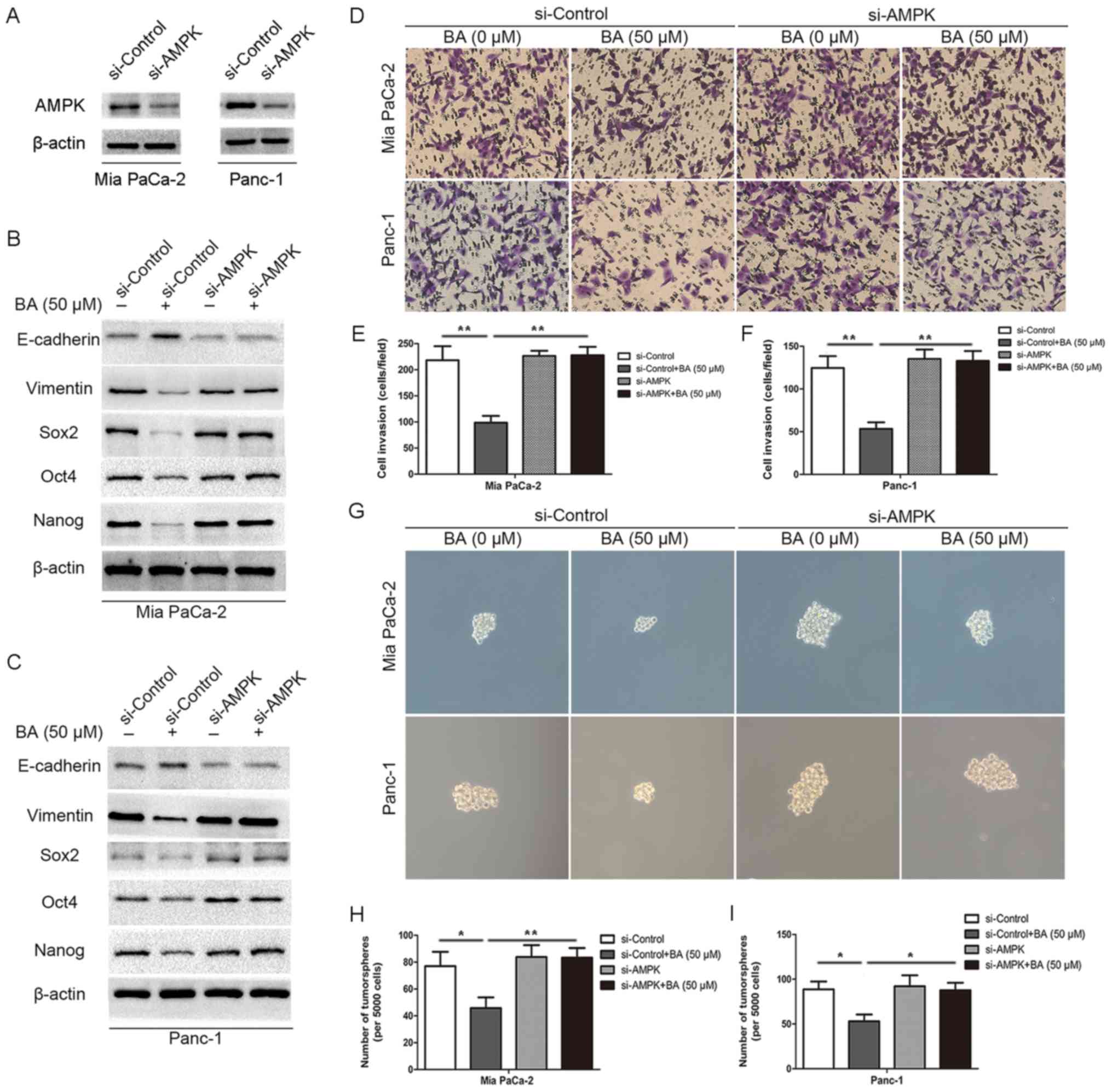 | Figure 5Knockdown of AMPK rescues BA-induced
suppression of EMT and stemness in pancreatic cancer cells. (A)
Western blotting confirmed the successful silencing of AMPK in Mia
PaCa-2 and Panc-1 cells. β-actin was used as an internal loading
control. (B and C) Western blot analysis suggested that silencing
AMPK by siRNA reversed BA-induced inhibition of the expression of
master pluripotency regulators (Sox2, Oct4 and Nanog) and EMT
markers (E-cadherin and vimentin) in Mia PaCa-2 and Panc-1 cells.
β-actin was used as an internal loading control. (D-F) Matrigel
invasion assay revealed that silencing AMPK by siRNA reversed
BA-induced suppression of the invasion of Mia PaCa-2 and Panc-1
cells. Images are representative of three independent experiments,
and the invasive cells were counted and plotted. Magnification,
×200. **P<0.01. (G-I) Tumorsphere formation assay
exhibited that knocking down AMPK by siRNA reversed BA-induced
inhibition of tumorsphere formation in Mia PaCa-2 and Panc-1 cells.
The number of tumorspheres was counted and plotted. Magnification,
×200. *P<0.05, **P<0.01. AMPK, 5′
adenosine monophosphate-activated protein kinase; BA, betulinic
acid; EMT, epithelial-mesenchymal transition; Oct4, octamer-binding
protein 4; si/siRNA, small interfering RNA; Sox2, SRY-box 2. |
BA reverses stemness induced by
gemcitabine and enhances the sensitivity of pancreatic cancer cells
to gemcitabine
Gemcitabine is the major chemotherapeutic agent
currently used in the treatment of pancreatic cancer; however,
chemoresistance is a serious issue that markedly affects the
prognosis of patients. A previous study demonstrated that
gemcitabine treatment induces stemness in pancreatic cancer cells
(45), which may serve a critical
role in resistance to gemcitabine. The present results revealed
that gemcitabine treatment dose-dependently increased the
expression levels of CSC markers, including Sox2, Oct4 and Nanog
(Fig. 6A and B). Similar effects
were observed on other stemness markers (CD133, ALDH1 and EpCAM;
data not shown). Since BA markedly inhibited the stemness of cancer
cells, this study further investigated whether it could attenuate
gemcitabine-induced stemness and facilitate the antitumor effects
of gemcitabine. Notably, BA reversed gemcitabine-induced stemness,
as revealed by decreased Sox2, Oct4 and Nanog expression (Fig. 6C and D). Since Panc-1 cells are
resistant to gemcitabine (46),
Panc-1 cells were selected for further experiments. Notably, BA
combined with gemcitabine was more effective at suppressing
proliferation (Fig. 6E) and
inducing apoptosis (Fig. 6F and G)
of Panc-1 cells. Taken together, these data suggested that BA may
be considered a sensitizer in gemcitabine treatment.
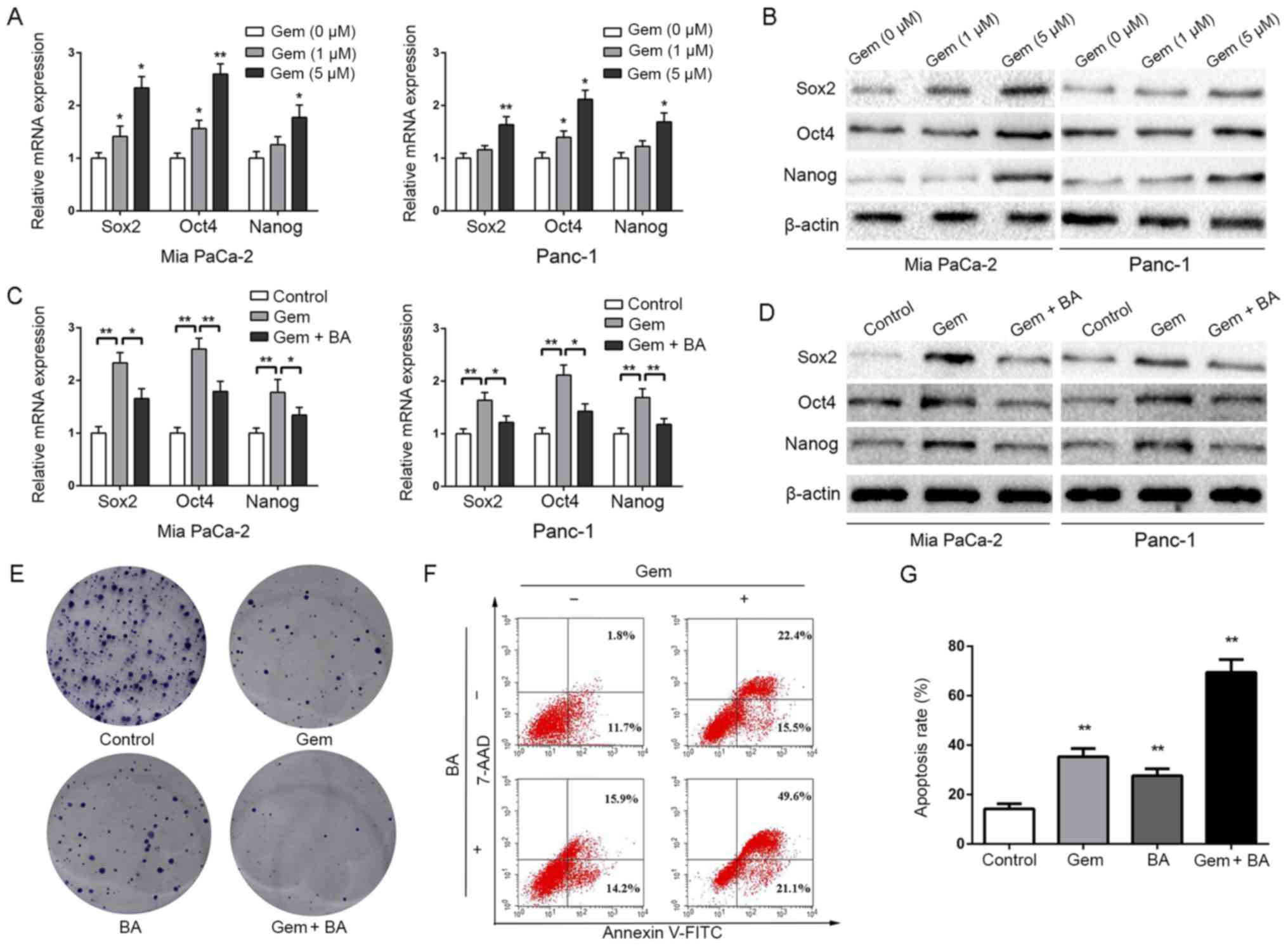 | Figure 6BA reverses gemcitabine-induced
stemness and enhances the sensitivity of pancreatic cancer cells to
gemcitabine. (A) Mia PaCa-2 and Panc-1 cells were pretreated with
various concentrations of gemcitabine (0, 1 and 5 µM) for 24
h, total RNA was extracted and RT-qPCR was conducted to detect the
expression levels of Sox2, Oct4 and Nanog. *P<0.05
and **P<0.01, compared with the control group. (B)
Mia PaCa-2 and Panc-1 cells were pretreated with various
concentrations of gemcitabine (0, 1 and 5 µM) for 24 h, and
western blot analysis was performed to assess the expression levels
of master pluripotency regulators (Sox2, Oct4 and Nanog). (C)
Effects of BA (50 µM) on gemcitabine (5 µM)
treatment-induced stemness were measured by RT-qPCR analysis.
*P<0.05 and **P<0.01. (D) Effects of BA
(50 µM) on gemcitabine (5 µM) treatment-induced
stemness were measured by western blot analysis. (E) Effects of BA
(50 µM) combined with gemcitabine (5 µM) on the
colony-forming ability of Panc-1 cells. (F and G) Effects of BA (50
µM) combined with gemcitabine (5 µM) on the apoptosis
of Panc-1 cells. **P<0.01, compared with the control
group. 7-AAD, 7-aminoactinomycin D; BA, betulinic acid; FITC,
fluorescein isothiocyanate; Oct4, octamer-binding protein 4;
RT-qPCR, reverse transcription-quantitative polymerase chain
reaction; Sox2, SRY-box 2. |
Discussion
The tumor biology of PDAC is conducive to early
metastasis and recurrence, and contributes to chemoradiotherapy
resistance (47). It has
previously been reported that several tumor types, including
pancreatic cancer, exhibit a minority of cells that display a
stem-like phenotype; these cells have an elevated metastatic
potential, thus contributing to tumor recurrence and
chemoresistance (48). The present
study indicated a vital role for PCSCs in metastasis and recurrence
of pancreatic cancer, and suggested that targeting PCSCs may be a
promising strategy for the treatment of this refractory malignancy.
In addition, EMT triggers cancer metastasis, which enhances the
invasion of cancer cells and impels them to disseminate into
secondary tissue sites, forming micrometastatic lesions.
Furthermore, the EMT process confers on these cancer cells acquired
stem-like traits for self-renewal, with an enhanced proliferative
capacity and an ability to form macroscopic metastases from
micrometastatic lesions (43,49).
Therefore, there is a direct link between the EMT process and CSCs.
Notably, the present results revealed that BA effectively inhibited
EMT and stemness of pancreatic cancer cells, thus suggesting that
BA may be an effective compound to suppress metastasis of
pancreatic cancer via targeting PCSCs and EMT.
Although known molecular markers of the cancer
stem-like phenotype are still being discovered, three transcription
factors, namely Sox2, Oct4 and Nanog, have been strongly validated
as master regulators in the maintenance of the cancer stem-like
phenotype (8). Overexpression of
Sox2, Oct4 and Nanog frequently occurs in poorly differentiated
malignancies and overlap with the signatures of embryonic stem
cells (50). It has previously
been reported that inhibition of AMPK signaling results in
activation of the Warburg effect, which further induces stemness
during the reprogramming of somatic cells (51). Furthermore, activation of AMPK
signaling, via a pharmacological strategy, is a metabolic barrier
during the process of somatic cells transforming into stem cells,
which cannot be bypassed even under a p53 deficiency (52), thus indicating the critical role of
AMPK signaling in stemness. It has also been reported that
Sox2-overexpressed breast cancer cells downregulate AMPK signaling
and activate mTOR to maintain their cancer stem-like phenotypes
(44). In the present study, to
explore the relationship between Sox2 and AMPK in pancreatic
cancer, immunohistochemistry was conducted; the results indicated
that there was an inverse association between P-AMPK and the key
stemness regulator, Sox2. Immunohistochemistry also revealed that,
compared with in normal pancreatic tissues, the expression levels
of P-AMPK were significantly decreased in pancreatic cancer
tissues. Conversely, the expression levels of Sox2 were markedly
elevated. Investigating the modulation of AMPK signaling and
suppression of pancreatic cancer stemness, it was revealed that
administration of BA not only enhanced the levels of P-AMPK, but
also inhibited the expression and nuclear localization of Sox2 in
Mia PaCa-2 and Panc-1 cells, in order to restrain the stem-like
phenotype. This study confirmed the finding whereby AMPK signaling
exerts an essential role in modulating EMT and stemness of
pancreatic cancer.
Gemcitabine has been used as a major therapy for the
treatment of advanced pancreatic cancer; however, the majority of
patients develop resistance during the initial period of treatment.
The definite mechanism underlying chemoresistance remains to be
determined. Recent reports have demonstrated that treatment with
gemcitabine fortifies the stemness of cancer cells via the NAPDH
oxidase/reactive oxygen species/nuclear factor-κB/STAT3 signaling
cascade and the long non-coding RNA HOX transcript antisense RNA
(45,53). Similarly, the present data
indicated that gemcitabine treatment promoted upregulation of
stemness markers in MiaPaCa-2 and Panc-1 pancreatic cancer cell
lines. Notably, BA reversed gemcitabine-induced stemness and
facilitated the antitumor effects of gemcitabine in pancreatic
cancer cells; therefore, it may be considered a potential
sensitizer for patients with pancreatic cancer.
BA is known to exert antidepressive (24), anti-inflammatory (25,26)
and anti-AIDs (27,28) effects; in addition, it has
hepatoprotective potential (29)
and can alleviate NAFLD (35).
Furthermore, its potential as a cancer preventive and therapeutic
compound has been verified (30-34).
Of particular interest is its direct and relatively selective
cytotoxic effect on various tumor cells compared with normal or
non-neoplastic cells (54). The
present results revealed that BA may inhibit the proliferation,
migration, invasion and tumorsphere formation of pancreatic cancer
cells, regulate EMT, and alter the expression levels of Sox2, Oct4
and Nanog via activation of AMPK signaling. Furthermore, BA
combined with gemcitabine exerted antitumor effects on pancreatic
cancer cells. However, there are some limitations to the present
study. These findings were based on in vitro experiments, which may
not accurately reflect the physiological conditions in vivo.
Therefore, BA warrants further functional studies and in vivo
investigations, to determine whether it is an effective inhibitor
of the stem-like phenotype in pancreatic cancer cells.
Acknowledgments
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81502528,
81572734, 81672434 and 81702916).
Availability of data and materials
The data and materials used during the present study
are available from the corresponding author upon reasonable
request.
Authors’ contributions
LS, JC, QM and QX designed the experiments. LS and
JC conducted the majority of the experiments. KC analyzed the data.
LC, CZ and BY organized the figures and contributed to data
analysis. LS and JC wrote the manuscript, and QM and QX reviewed
it. WQ, JL, WD, JM, DQ, EW, ZW and QL contributed to the collection
of human specimens and revised the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Ethical Committee of The First Affiliated Hospital of Xi’an
Jiaotong University. Written informed consent was obtained from the
patients.
Patient consent for publication
The authors declare that the patients provided
written informed consent for the publication of any associated data
in the present study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gillen S, Schuster T, Meyer Zum,
Büschenfelde C, Friess H and Kleeff J: Preoperative/neoadjuvant
therapy in pancreatic cancer: A systematic review and meta-analysis
of response and resection percentages. PLoS Med. 7:e10002672010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kamisawa T, Isawa T, Koike M, Tsuruta K
and Okamoto A: Hematogenous metastases of pancreatic ductal
carcinoma. Pancreas. 11:345–349. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gattinoni L, Klebanoff CA and Restifo NP:
Paths to stemness: Building the ultimate antitumour T cell. Nat Rev
Cancer. 12:671–684. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Magee JA, Piskounova E and Morrison SJ:
Cancer stem cells: Impact, heterogeneity, and uncertainty. Cancer
Cell. 21:283–296. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Clevers H: The cancer stem cell: Premises,
promises and challenges. Nat Med. 17:313–319. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sengupta S, Nagalingam A, Muniraj N,
Bonner MY, Mistriotis P, Afthinos A, Kuppusamy P, Lanoue D, Cho S,
Korangath P, et al: Activation of tumor suppressor LKB1 by honokiol
abrogates cancer stem-like phenotype in breast cancer via
inhibition of oncogenic Stat3. Oncogene. 36:5709–5721. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chaffer CL, Marjanovic ND, Lee T, Bell G,
Kleer CG, Reinhardt F, D’Alessio AC, Young RA and Weinberg RA:
Poised chromatin at the ZEB1 promoter enables breast cancer cell
plasticity and enhances tumorigenicity. Cell. 154:61–74. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Abel EV and Simeone DM: Biology and
clinical applications of pancreatic cancer stem cells.
Gastroenterology. 144:1241–1248. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hardie DG: AMP-activated/SNF1 protein
kinases: Conserved guardians of cellular energy. Nat Rev Mol Cell
Biol. 8:774–785. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Young LH, Li J, Baron SJ and Russell RR:
AMP-activated protein kinase: A key stress signaling pathway in the
heart. Trends Cardiovasc Med. 15:110–118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zheng X, Chi J, Zhi J, Zhang H, Yue D,
Zhao J, Li D, Li Y, Gao M and Guo J: Aurora-A-mediated
phosphorylation of LKB1 compromises LKB1/AMPK signaling axis to
facilitate NSCLC growth and migration. Oncogene. 37:502–511. 2018.
View Article : Google Scholar
|
|
16
|
Shi WY, Xiao D, Wang L, Dong LH, Yan ZX,
Shen ZX, Chen SJ, Chen Y and Zhao WL: Therapeutic metformin/AMPK
activation blocked lymphoma cell growth via inhibition of mTOR
pathway and induction of autophagy. Cell Death Dis. 3:e2752012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Handy JA, Saxena NK, Fu P, Lin S, Mells
JE, Gupta NA and Anania FA: Adiponectin activation of AMPK disrupts
leptin-mediated hepatic fibrosis via suppressors of cytokine
signaling (SOCS-3). J Cell Biochem. 110:1195–1207. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
da Silva Morais A, Abarca-Quinones J,
Guigas B, Viollet B, Stärkel P, Horsmans Y and Leclercq IA:
Development of hepatic fibrosis occurs normally in AMPK-deficient
mice. Clin Sci (Lond). 118:411–420. 2009. View Article : Google Scholar
|
|
19
|
Kahn BB, Alquier T, Carling D and Hardie
DG: AMP-activated protein kinase: Ancient energy gauge provides
clues to modern understanding of metabolism. Cell Metab. 1:15–25.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Menendez JA, Joven J, Cufí S,
Corominas-Faja B, Oliveras-Ferraros C, Cuyàs E, Martin-Castillo B,
López-Bonet E, Alarcón T and Vazquez-Martin A: The Warburg effect
version 2.0: Metabolic reprogramming of cancer stem cells. Cell
Cycle. 12:1166–1179. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cheng X, Kim JY, Ghafoory S, Duvaci T,
Rafiee R, Theobald J, Alborzinia H, Holenya P, Fredebohm J, Merz
KH, et al: Methylisoindigo preferentially kills cancer stem cells
by interfering cell metabolism via inhibition of LKB1 and
activation of AMPK in PDACs. Mol Oncol. 10:806–824. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lonardo E, Cioffi M, Sancho P,
Sanchez-Ripoll Y, Trabulo SM, Dorado J, Balic A, Hidalgo M and
Heeschen C: Metformin targets the metabolic achilles heel of human
pancreatic cancer stem cells. PLoS One. 8:e765182013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bao B, Wang Z, Ali S, Ahmad A, Azmi AS,
Sarkar SH, Banerjee S, Kong D, Li Y, Thakur S, et al: Metformin
inhibits cell proliferation, migration and invasion by attenuating
CSC function mediated by deregulating miRNAs in pancreatic cancer
cells. Cancer Prev Res (Phila). 5:355–364. 2012. View Article : Google Scholar
|
|
24
|
Machado DG, Cunha MP, Neis VB, Balen GO,
Colla A, Bettio LE, Oliveira A, Pazini FL, Dalmarco JB, Simionatto
EL, et al: Antidepressant-like effects of fractions, essential oil,
carnosol and betulinic acid isolated from Rosmarinus officinalis L.
Food Chem. 136:999–1005. 2013. View Article : Google Scholar
|
|
25
|
Tsai JC, Peng WH, Chiu TH, Lai SC and Lee
CY: Anti-inflammatory effects of Scoparia dulcis L. and betulinic
acid. Am J Chin Med. 39:943–956. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Viji V, Helen A and Luxmi VR: Betulinic
acid inhibits endotoxin-stimulated phosphorylation cascade and
pro-inflammatory prostaglandin E(2) production in human peripheral
blood mononuclear cells. Br J Pharmacol. 162:1291–1303. 2011.
View Article : Google Scholar :
|
|
27
|
Qian K, Bori ID, Chen CH, Huang L and Lee
KH: Anti-AIDS agents 90. novel C-28 modified bevirimat analogues as
potent HIV maturation inhibitors. J Med Chem. 55:8128–8136. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fujioka T, Kashiwada Y, Kilkuskie RE,
Cosentino LM, Ballas LM, Jiang JB, Janzen WP, Chen IS and Lee KH:
Anti-AIDS agents, 11. Betulinic acid and platanic acid as anti-HIV
principles from Syzigium claviflorum, and the anti-HIV activity of
structurally related triterpenoids. J Nat Prod. 57:243–247. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jain M, Kapadia R, Jadeja RN, Thounaojam
MC, Devkar RV and Mishra SH: Hepatoprotective potential of
Tecomella undulata stem bark is partially due to the presence of
betulinic acid. J Ethnopharmacol. 143:194–200. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang P, Li Q, Li K, Zhang X, Han Z, Wang
J, Gao D and Li J: Betulinic acid exerts immunoregulation and
anti-tumor effect on cervical carcinoma (U14) tumor-bearing mice.
Pharmazie. 67:733–739. 2012.PubMed/NCBI
|
|
31
|
Liu Y and Luo W: Betulinic acid induces
Bax/Bak-independent cytochrome c release in human nasopharyngeal
carcinoma cells. Mol Cells. 33:517–524. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chintharlapalli S, Papineni S, Lei P,
Pathi S and Safe S: Betulinic acid inhibits colon cancer cell and
tumor growth and induces proteasome-dependent and -independent
downregulation of specificity proteins (Sp) transcription factors.
BMC Cancer. 11:3712011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gao Y, Jia Z, Kong X, Li Q, Chang DZ, Wei
D, Le X, Suyun H, Huang S, Wang L, et al: Combining betulinic acid
and mithramycin a effectively suppresses pancreatic cancer by
inhibiting proliferation, invasion, and angiogenesis. Cancer Res.
71:5182–5193. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li L, Du Y, Kong X, Li Z, Jia Z, Cui J,
Gao J, Wang G and Xie K: Lamin B1 is a novel therapeutic target of
betulinic acid in pancreatic cancer. Clinical Cancer Res.
19:4651–4661. 2013. View Article : Google Scholar
|
|
35
|
Quan HY, Kim DY, Kim SJ, Jo HK, Kim GW and
Chung SH: Betulinic acid alleviates non-alcoholic fatty liver by
inhibiting SREBP1 activity via the AMPK-mTOR-SREBP signaling
pathway. Biochem Pharmacol. 85:1330–1340. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ma J, Duan W, Han S, Lei J, Xu Q, Chen X,
Jiang Z, Nan L, Li J, Chen K, et al: Ginkgolic acid suppresses the
development of pancreatic cancer by inhibiting pathways driving
lipogenesis. Oncotarget. 6:20993–21003. 2015.PubMed/NCBI
|
|
37
|
Lei J, Ma J, Ma Q, Li X, Liu H, Xu Q, Duan
W, Sun Q, Xu J, Wu Z, et al: Hedgehog signaling regulates hypoxia
induced epithelial to mesenchymal transition and invasion in
pancreatic cancer cells via a ligand-independent manner. Mol
Cancer. 12:662013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lei J, Huo X, Duan W, Xu Q, Li R, Ma J, Li
X, Han L, Li W, Sun H, et al: α-Mangostin inhibits hypoxia-driven
ROS-induced PSC activation and pancreatic cancer cell invasion.
Cancer Lett. 347:129–138. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Özdemir BC, Pentcheva-Hoang T, Carstens
JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C,
Novitskiy SV, et al: Depletion of carcinoma-associated fibroblasts
and fibrosis induces immunosuppression and accelerates pancreas
cancer with reduced survival. Cancer Cell. 25:719–734. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Duan W, Chen K, Jiang Z, Chen X, Sun L, Li
J, Lei J, Xu Q, Ma J, Li X, et al: Desmoplasia suppression by
metformin-mediated AMPK activation inhibits pancreatic cancer
progression. Cancer Lett. 385:225–233. 2017. View Article : Google Scholar
|
|
42
|
Chen K, Qian W, Li J, Jiang Z, Cheng L,
Yan B, Cao J, Sun L, Zhou C, Lei M, et al: Loss of AMPK activation
promotes the invasion and metastasis of pancreatic cancer through
an HSF1-dependent pathway. Mol Oncol. 11:1475–1492. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Thiery JP: Epithelial-mesenchymal
transitions in development and pathologies. Curr Opin Cell Biol.
15:740–746. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Corominas-Faja B, Cufí S,
Oliveras-Ferraros C, Cuyàs E, López-Bonet E, Lupu R, Alarcón T,
Vellon L, Iglesias JM, Leis O, et al: Nuclear reprogramming of
luminal-like breast cancer cells generates Sox2-overexpressing
cancer stem-like cellular states harboring transcriptional
activation of the mTOR pathway. Cell Cycle. 12:3109–3124. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang Z, Duan Q, Zhao H, Liu T, Wu H, Shen
Q, Wang C and Yin T: Gemcitabine treatment promotes pancreatic
cancer stemness through the Nox/ROS/NF-κB/STAT3 signaling cascade.
Cancer Lett. 382:53–63. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cao J, Yang J, Ramachandran V, Arumugam T,
Deng D, Li Z, Xu L and Logsdon CD: TM4SF1 promotes gemcitabine
resistance of pancreatic cancer in vitro and in vivo. PLoS One.
10:e01449692015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kamisawa T, Wood LD, Itoi T and Takaori K:
Pancreatic cancer. Lancet. 388:73–85. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhao J, Li J, Schlößer HA, Popp F, Popp
MC, Alakus H, Jauch KW, Bruns CJ and Zhao Y: Targeting cancer stem
cells and their Niche: Current therapeutic implications and
challenges in pancreatic cancer. Stem Cells Int.
2017.6012810:2017.
|
|
49
|
Kondo M, Wagers AJ, Manz MG, Prohaska SS,
Scherer DC, Beilhack GF, Shizuru JA and Weissman IL: Biology of
hematopoietic stem cells and progenitors: Implications for clinical
application. Annu Rev Immunol. 21:759–806. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ben-Porath I, Thomson MW, Carey VJ, Ge R,
Bell GW, Regev A and Weinberg RA: An embryonic stem cell-like gene
expression signature in poorly differentiated aggressive human
tumors. Nat Genet. 40:499–507. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
51
|
Faubert B, Boily G, Izreig S, Griss T,
Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, et
al: AMPK is a negative regulator of the Warburg effect and
suppresses tumor growth in vivo. Cell Metab. 17:113–124. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Vazquez-Martin A, Vellon L, Quirós PM,
Cufí S, Ruiz de Galarreta E, Oliveras-Ferraros C, Martin AG,
Martin-Castillo B, López-Otín C and Menendez JA: Activation of
AMP-activated protein kinase (AMPK) provides a metabolic barrier to
reprogramming somatic cells into stem cells. Cell Cycle.
11:974–989. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang L, Dong P, Wang W, Huang M and Tian
B: Gemcitabine treatment causes resistance and malignancy of
pancreatic cancer stem-like cells via induction of lncRNA HOTAIR.
Exp Ther Med. 14:4773–4780. 2017.PubMed/NCBI
|
|
54
|
Pisha E, Chai H, Lee IS, Chagwedera TE,
Farnsworth NR, Cordell GA, Beecher CW, Fong HH, Kinghorn AD, Brown
DM, et al: Discovery of betulinic acid as a selective inhibitor of
human melanoma that functions by induction of apoptosis. Nat Med.
1:1046–1051. 1995. View Article : Google Scholar : PubMed/NCBI
|