Introduction
Inorganic arsenic is ubiquitously found in foods
(i.e., rice and grains) (1,2) and
drinking water (3–5). Chronic arsenic exposure through
contaminated water has been linked to an increased incidence of
several tumors (6,7) and long-term health complications at
levels of exposure below safety limits (10 ppb) (8). Common human exposures to arsenic
include inorganic trivalent arsenite (AsIII) and
pentavalent arsenate (AsV). The AsIII form
has potent estrogen-disrupting activities in connection with its
affinity for the ligand-binding domain of the estrogen receptor-α
(ERα). It also stimulates cell growth and the expression of the
progesterone receptor (PR) (9). As the AsV form is
enzymatically converted to AsIII, it provides a
reservoir for ERα-binding metabolites (10) that may disrupt estrogen signaling
and response to endocrine therapies based on antagonists of the ERα
(11–13).
Approximately 70–80% of diagnosed breast tumors are
ER-positive and they are treated with anti-estrogens, including
tamoxifen (TAM). However, over time, a significant percentage
(~30%) of these tumors become resistant to treatment with
anti-estrogens (14,15). The reasons for this acquired
resistance remain largely unknown. However, the loss of ERα
expression has been linked to a poor response to endocrine therapy
(16–18). The deregulation of ERα signaling
associated with the drinking of water contaminated with arsenic has
been reported both in men and women (19). Arsenic-induced genomic instability
via the Fanconi anemia (FA)/breast cancer (BRCA) pathway disruption
has been shown to directly contribute to arsenic carcinogenic
effects (20). A previous study
using rodent models (e.g., Sprague-Dawley rats) demonstrated that
the in utero exposure to AsIII induced an
increase in the number of mammosphere-forming cells, the branching
of epithelial cells and density in the mammary gland of prepubertal
offspring, and that these changes persisted into adulthood
(21). Other studies using rodent
models concluded that AsIII was a 'complete'
transplacental carcinogen promoting the maternal dose-dependent
induction of tumors in endocrine-related tissues (adrenal gland,
ovary and uterus) in offspring (22,23).
In a spontaneous mammary-tumor model (C3H/St mice), arsenic
exposure was shown to abolish the anticancer effects of selenium
and increase tumor growth rates and multiplicity (24). At the cellular level, in
vitro studies have indicated that chronic exposure to low
levels of arsenic induced the transformation of normal breast
epithelial cells, and accelerated the growth of ERα-positive breast
cancer cells (25,26). Exposure to AsIII has
been shown to inhibit DNA mismatch repair, leading to genomic
instability (27,28). In endocrine-responsive tissue
(e.g., prostate), exposure to AsIII has been reported to
induce the transition to a steroid receptor-independent tumor
phenotype (29). These cumulative
observations have raised the question of whether or not endocrine
disruption associated with AsIII exposure contributes to
breast carcinogenesis.
Epigenetics refers to changes in DNA methylation,
histone post-translational modifications and the expression of
non-coding RNAs (30). Maternal
exposure to arsenic has been shown to alter DNA methylation in
placental tissue (31), and to
increase DNA methylation in children (32). Moreover, preclinical (33,34)
and human (35) studies have
demonstrated that arsenic causes the hypermethylation of tumor
suppressor genes (i.e., p16INK4 and
RASSF1) and a decrease in telomere length associated with
genomic instability (36).
Finally, exposure to AsIII has been found to induce
cancer stem cell-like properties involving the epigenetic silencing
of the let-7c via Ras/NF-κB pathways (37). Based on these observations, the
main objective of this study was to investigate the effects of
AsIII on BRCA1 and ESR1 (ERα) expression
and CpG methylation, and response to TAM in cultured and
xenografted MCF7 breast cancer cells.
Materials and methods
Cells and cell culture
Authenticated breast cancer MCF7 cells (Batch
#62349993) were obtained from the American Type Culture Collection
(ATCC, Manassas, VA, USA) and maintained at 37°C with 5%
CO2 in Dulbecco's modified Eagle's/F12 medium (DMEM)
from Corning Cellgro (Thermo Fisher Scientific, Pittsburgh, PA,
USA) supplemented with 10% fetal calf serum (FCS; HyClone
Laboratories Inc., Logan UT, USA) as previously described (38). Sodium arsenite
(NaAsIII), TAM, genistein (GEN) and 17β-estradiol (E2)
were obtained from Sigma-Aldrich (St. Louis, MO, USA). TAM and E2
were solubilized in stock solutions with ethanol, which was added
to DMEM/F12 as the vehicle control. For cell proliferation
experiments, the MCF7 cells (passage nos. 3–15) were seeded in
6-well plates at a density of 5×105 cells/well in
triplicate overnight, and then switched to phenol-free media
containing 10% charcoal-stripped FCS (HyClone Laboratories Inc.)
for 3 days before the start of each treatment. For proliferation
measurements, the cells were washed with ice-cold PBS and counted
by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT) colorimetric assay (Promega, Madison, WI, USA). This assay is
based on the conversion of the yellow tretrazolium dye MTT to
purple formazan crystals by metabolically active cells. Briefly,
2×104 cells were seeded in 96-well tissue culture plates
and maintained overnight. Six replicates were assigned to each
experimental treatment. Following treatment, 15 µl of MTT
dye solution were added to each well, and the plate was incubated
for 4 h at 37°C. Solubilization/stop solution (100 µl) was
added for 1 h at room temperature and the absorbance at 570/650 nm
was recorded using a Synergy HT plate reader (Bio-Tek Instruments,
Winooski, VT, USA). For flow cytometric analysis, trypsinized cells
were washed in phosphate-buffered saline (PBS), treated with RNAse
and stained with propidium iodide (50 µg/ml). Cell cycle
distribution profiles were determined with a FACscan (BD
Biosciences, Franklin Lakes, NJ, USA), using a CELLQuest program at
the Flow Cytometry Laboratory of the Arizona Cancer Center, and
analyzed with MODFIT.2 software.
Promoter CpG methylation
Quantitative polymerase chain reaction (qPCR)
analysis of human BRCA1 and ESR1 promoter CpG
methylation was performed as previously described (38) with genomic DNA (DNeasy blood and
tissue kit; Qiagen, Hilden, Germany) and bisulfonated with the
Epitect bisulfite kit (Qiagen) using the following unmethylated
(U)- and methylated (M)-specific primers (Sigma-Aldrich):
BRCA1 U-sense, 5′-TTGGTTTTTGTGGTAATGGAAAAGTGT-3′ and
U-antisense, 5′-CAAAAAATCTCAACAAACTCACACCA-3′; M-sense,
5′-TGGTAACGGAAAAGCG-3′ and M-antisense, 5′-ATCTCAACGAACTCACGC-3′;
ESR1 U-sense, 5′-GGATATGGTTTGTATTTTGTTTGT-3′ and
U-antisense, 5′-ACAAACAATTCAAAAACTCCAACT-3′; M-sense,
5′-GGTTTTTGAGTTTTTTGTTTTG-3′ and M-antisense,
5′-AACTTACTACTATCCAAATACACCTC-3′. The qPCR was carried out in a
volume of 10 µl consisting of the following master mix: 5
µl of SYBER-Green mix (Thermo Fisher Scientific), 1
µl each of forward and reverse primers, 2 µl
nuclease-free water, and 1 µl of bisulfonated genomic DNA.
Data from qPCR of bisulfonated DNA were presented as the
fold-change compared to the control of the ratio of CpG M/U, as
previously described (38).
Chromatin immunoprecipitation assay
The Pierce magnetic chromatin immunoprecipitation
(ChIP) kit (Pierce, Rockford, IL, USA) was used to analyze the
occupancy of the BRCA1 promoter by DNA methyltransferase 1
(DNMT1) and RNA polymerase II (PolII) in MCF7 cells according to
instructions provided by the manufacturer. Briefly, the cells were
fixed in 1% paraformaldehyde for 10 min and neutralized with
glycine. After 2 washes with cold PBS and protease inhibitors
cocktail, cells were resuspended in membrane extraction buffer and
prepared for DNA enzymatic digestion. Aliquots of digested
chromatin were immunoprecipitated using antibodies against DNMT1
(Abcam Inc, Cambridge, MA, USA) and PolII (Thermo Fisher
Scientific). qPCR was performed on aliquots of DNA obtained after
the reversal of DNA-protein cross-links and purification through
spin-filtration columns. Briefly, PCR amplification reactions were
done at a final volume of 25 µl consisting of the following:
12.5 µl of SYBR-Green buffer, 1 µl each forward
(5′-CTCCCATCCTCTGATTGTACCTTG AT-3′) and reverse
(5′-CAGGAAGTCTCAGCGAGCTCAC-3′) oligonucleotides flanking exon-1a in
the BRCA1 gene (39); 8.5
µl nuclease free water, and 2 µl DNA purified from
the ChIP assay.
mRNA analyses
Total RNA was purified using RNeasy Mini kit as per
the manufacturer's instructions (Qiagen) (38). The concentrations and quality of
RNA were verified using the Nanodrop 1000 Spectrophotometer (Thermo
Fisher Scientific). Equal amounts of total RNA (500 ng) were
transcribed into cDNA using ISCRIPT supermix kit (Bio-Rad
Laboratories, Hercules, CA, USA). Next, cDNA aliquots were analyzed
by qPCR using the SYBR-Green PCR Reagents kit (Life
Technologies/Thermo Fisher Scientific). Briefly, reactions were run
at a final volume of 25 µl consisting of the following
master mix: 12.5 µl of SYBR-Green mix, 1 µl each of
forward and reverse primers, 9.5 µl nuclease-free water and
1 µl cDNA. The primer (Sigma-Aldrich) sequences were:
ERα sense, 5′-CAAGCCCGCTCATGATCAA-3′ and antisense,
5′-CTGATCATGGAGGGTCAAATCCAC-3′; BRCA1 sense,
5′-AGCTCGCTGAGACTTCCTGGA-3′ and antisense,
5′-CAATTCAATGTAGACAGACGT-3′; cyclin D1 (CCND1 sense,
5′-ACAAACAGATCATCCGCAAACAC-3′ and anti-sense,
5′-TGTTGGGGCTCCTCAGGTTC-3′; folate receptor (FOLR1) sense,
5′-ATTCCTTGGTGCCACTGACC-3′ and antisense,
5′-ATAGAACCTCGCCACCTCCT-3′; methyltenetrahydrofolate reductase
(MTHFR) sense, 5′-AAGCCTCTT CCTTTGTCGCA-3′ and antisense,
5′-AGGACCCTGGCTT TTCGATG-3′; and control glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) sense, 5′-ACCCACTCCTCCACCTTT-and
antisense, 5′-CTCTTGTGCTCTTGCTGGG-3′. Amplification of GAPDH
mRNA was used for the normalization of the transcript levels.
Western blot analysis
Western blot analysis was performed as previously
described (38). Protein lysates
were obtained from cells scraped in triplicates from 6-well plates
and using Pierce RIPA buffer (Thermo Fisher Scientific), with 1%
proteinase inhibitors. The protein concentration was calculated
using a Nanodrop 1000 Spectrophotometer (Thermo Fisher Scientific).
Immunoblotting was carried out with antibodies against BRCA1 (cat.
no. 9010); GAPDH (cat. no. 2118) (both from Cell Signaling
Technology, Beverly, MA, USA); and ERα (cat. no. sc-542) (Santa
Cruz Biotechnology, Dallas, TX, USA). Immunocomplexes were detected
using enhanced chemiluminescence (GE Healthcare Life Sciences,
Little Chalfont, UK). Immunocomplexes for GAPDH were used as an
internal control for the normalization of protein expression.
Western blot analyses were carried out at least twice for each
experiment. The quantification of immunocomplexes was carried out
by densitometry performed using Kodak ID Image Analysis Software
(Eastman Kodak Company, Rochester, NY, USA).
Mouse mammary xenografts
All in vivo mouse xenograft experiments were
performed under the #07–029 protocol approved by the University of
Arizona Institutional Animal Care and Use Committee approved on
02/22/2016. All procedures were performed in compliance with the
standard operating procedures and relevant guidelines of the
University of Arizona Animal Care. MCF7 cells
(7.5–10×106 cells in 50 µl of Matrigel
resuspension) pre-cultured for 4 weeks in control DMEM/F12 media
plus 10% FCS (MCF7 Control) or DMEM/F12 plus 10% FCS with 1
µM NaAsIII (MCFAsIII) were injected
into the left number-4 mammary fat pad of 4-week-old (19–22 g)
ovariectomized (OVX) athymic rTac:NCr-Foxn1 nude female mice
(Taconic Biosciences, Rensselaer, NY, USA) implanted with an
estradiol pellet (0.72 mg, 60 days release; Innovative Research of
America, Sarasota, FL, USA). After 30 days, the mice injected were
with MCF7 control or MCF7NaAsIII cells were implanted
with TAM pellets (5 mg, 60 days release; Innovative Research of
America). Mice (10 animals/group × 4 experimental groups, 40
animals in total) were housed in conventional pathogen-free cages
under a 12 h light/12 h dark cycle, at 20–22°C, and 50–55% humidity
with free access to Teklad Global Rodent Diet (Harlan Laboratories,
Madison, WI, USA) and tap water. The animals were sacrificed at 60
days after the start of TAM treatment. Tumor growth was measured
once/week with a caliper until there were visible signs of tumor
growth, then twice/week until the end of the study. Tumor volume
was estimated using the following formula: [(width)2 ×
length]/2]. Tumor tissue was snap-frozen in liquid nitrogen and
stored at −80°C for further analysis.
Statistical analysis
Data were analyzed by ANOVA as previously described
(38). Post-hoc multiple
comparisons among all means were conducted using Tukey's Test after
main effects and interactions were found to be significant at
P≤0.05. Data are presented as the means ± SEM and statistical
differences highlighted with different letters for multiple
comparisons (a>b>c, etc.) or asterisks when compared to the
control.
Results
NaAsIII reduces the expression
of BRCA1 via CpG hypermethylation in ERα-positive breast cancer
cells
Previously (38–40),
we reported that the expression of BRCA1 was stimulated by E2 in
ER-positive MCF7 breast cancer cells (38). In this study, using western blot
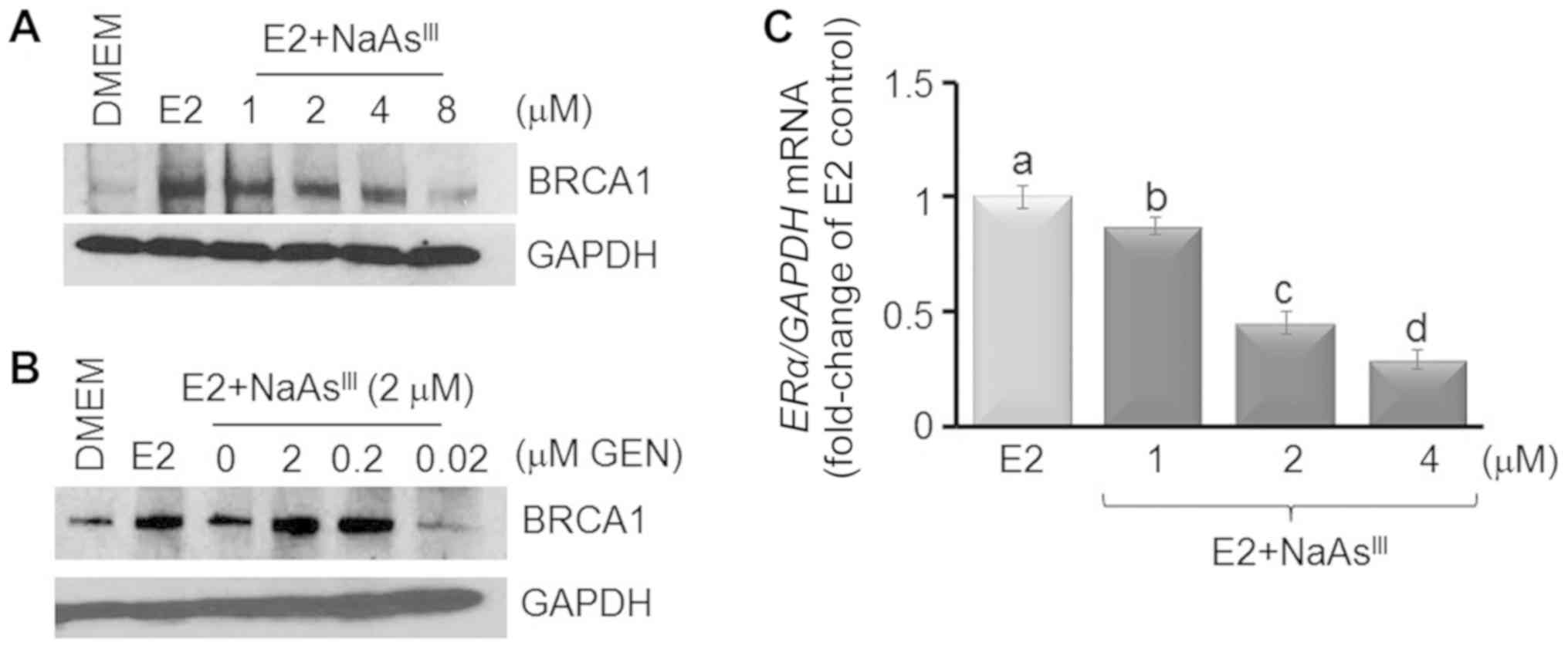
analysis (Fig. 1A), we observed
that E2-induced BRCA1 expression was antagonized by
NaAsIII, starting at the 1 µM concentration, and
to a larger degree upon co-treatment with higher doses of
NaAsIII (2 to 8 µM). As a control, we co-treated
MCF7 cells with NaAsIII (2 µM) plus various doses
(0.02, 0.2 and 2.0 µM) of the isoflavone GEN, which was
found in our previous study to induce BRCA1 expression (38). Co-treatment with 0.2 and 2
µM GEN reversed the repressive effects of NaAsIII
on BRCA1 expression (Fig. 1B).
Based on the information that BRCA1 transcription is
regulated by the ERα (40),
changes in the expression of ERα mRNA were analyzed by qPCR
in MCF7 cells treated for 72 h with various doses of
NaAsIII. Compared to the E2 control, treatment with 1
µM NaAsIII decreased ERα mRNA expression
by ~15%, which was further decreased (55–70%) by higher
concentrations (2 and 4 µM) of NaAsIII (Fig. 1C). Based on these dose-response
results, we examined the long-term effects of exposure to 1
µM NaAsIII, which approximates levels of
AsIII measured in drinking water of populations residing
in the US (41) and other
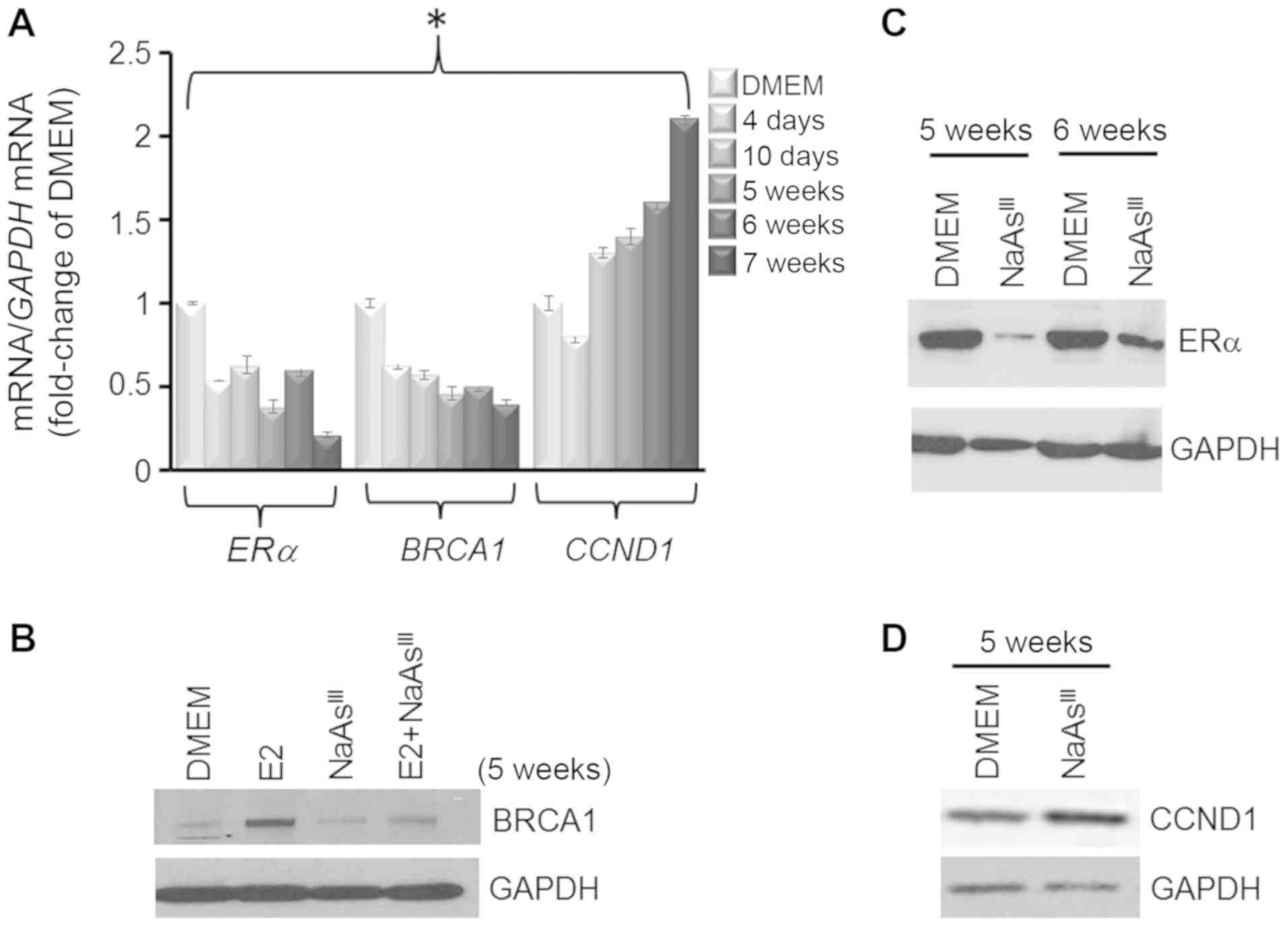
geographical regions (42–44). MCF7 cells were cultured for various
periods of time (4 days to 7 weeks) either as control DMEM cells or
in the presence of 1 µM NaAsIII, which reduced
the expression of ERα and BRCA1 mRNA (Fig. 2A). In parallel, the expression of
CCDN1 was reduced by ~ 25% within 4 days post-treatment with
NaAsIII, whereas the CCND1 levels were enhanced
by longer exposure to NaAsIII. Western blot analysis
confirmed that long-term (5 weeks) exposure to NaAsIII
had repressive effects on E2-induced BRCA1 (Fig. 2B) and ERα (Fig. 2C), but induced the expression of
CCND1.
It has previously been documented (45) that AsIII treatment
decreases the expression of MTHFR, an enzyme involved in one-carbon
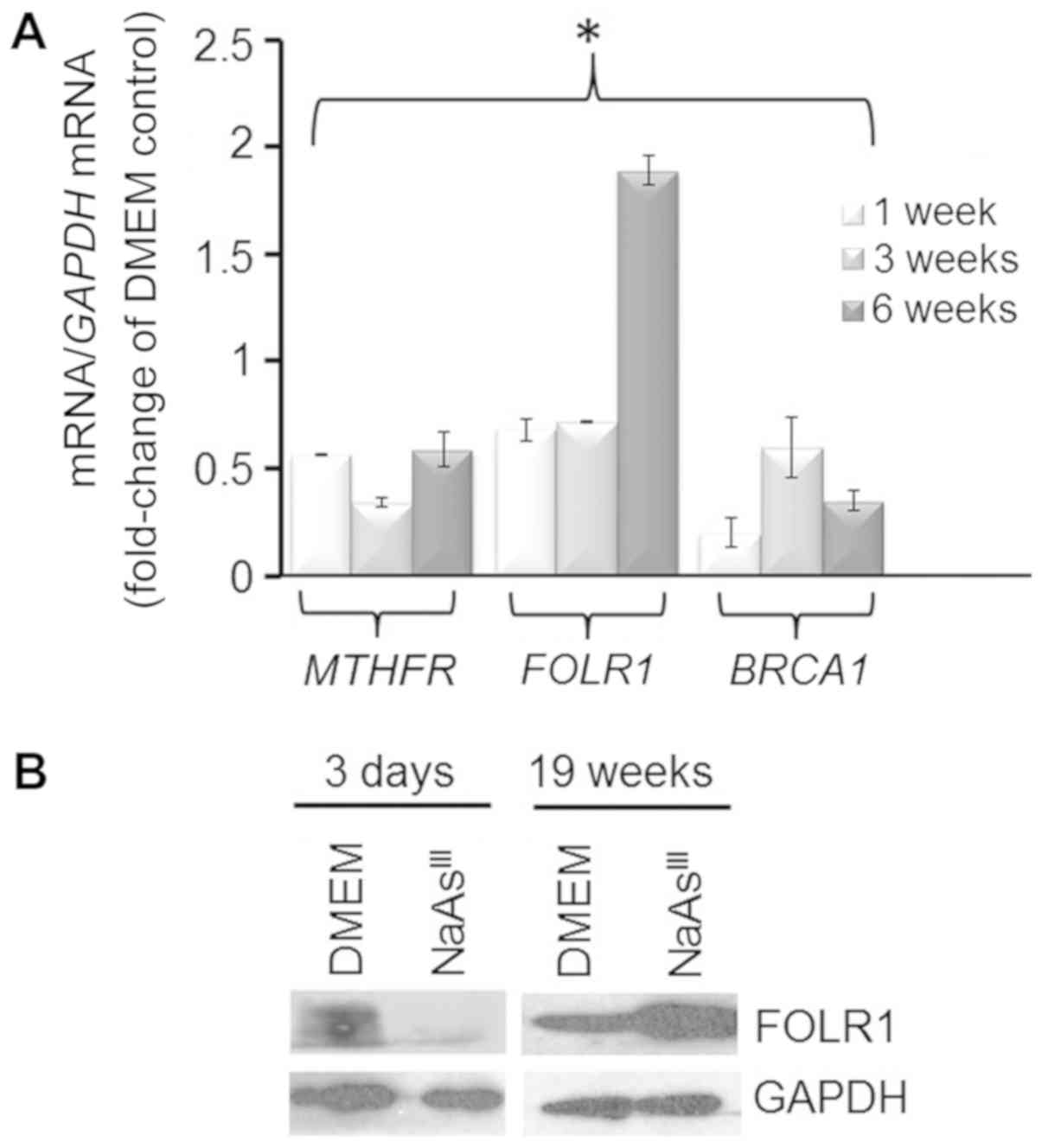
metabolism. Analysis of MTHFR expression by RT-qPCR (Fig. 3A) showed that 1 to 6 weeks exposure
of MCF7 cells to 1 µM NaAsIII reduced markedly
(~50%) MTHFR mRNA. The treatment with NaAsIII had
a biphasic effect on expression of FOLR1 mRNA, which was
reduced at 1 and 3 weeks, but induced at 6 weeks, of exposure.
FOLR1 participates in cellular uptake of 5-methyltetrahydrofolate
into cells, and its overexpression has been linked to poor
prognosis in particular in triple-negative breast cancers (TNBC)
(46). As an additional control,
we confirmed the repressive effects on BRCA1 mRNA expression
by treatment of the MCF7 cells with NaAsIII by RT-qPCR.
As another control, we also examined the expression of FOLR1
protein and found that exposure to NaAsIII reduced its
expression within 3 days, although it had a stimulatory effect
long-term (19 weeks) (Fig.
3B).
One mechanism through which NaAsIII may
lower BRCA1 expression is epigenetic silencing involving DNA
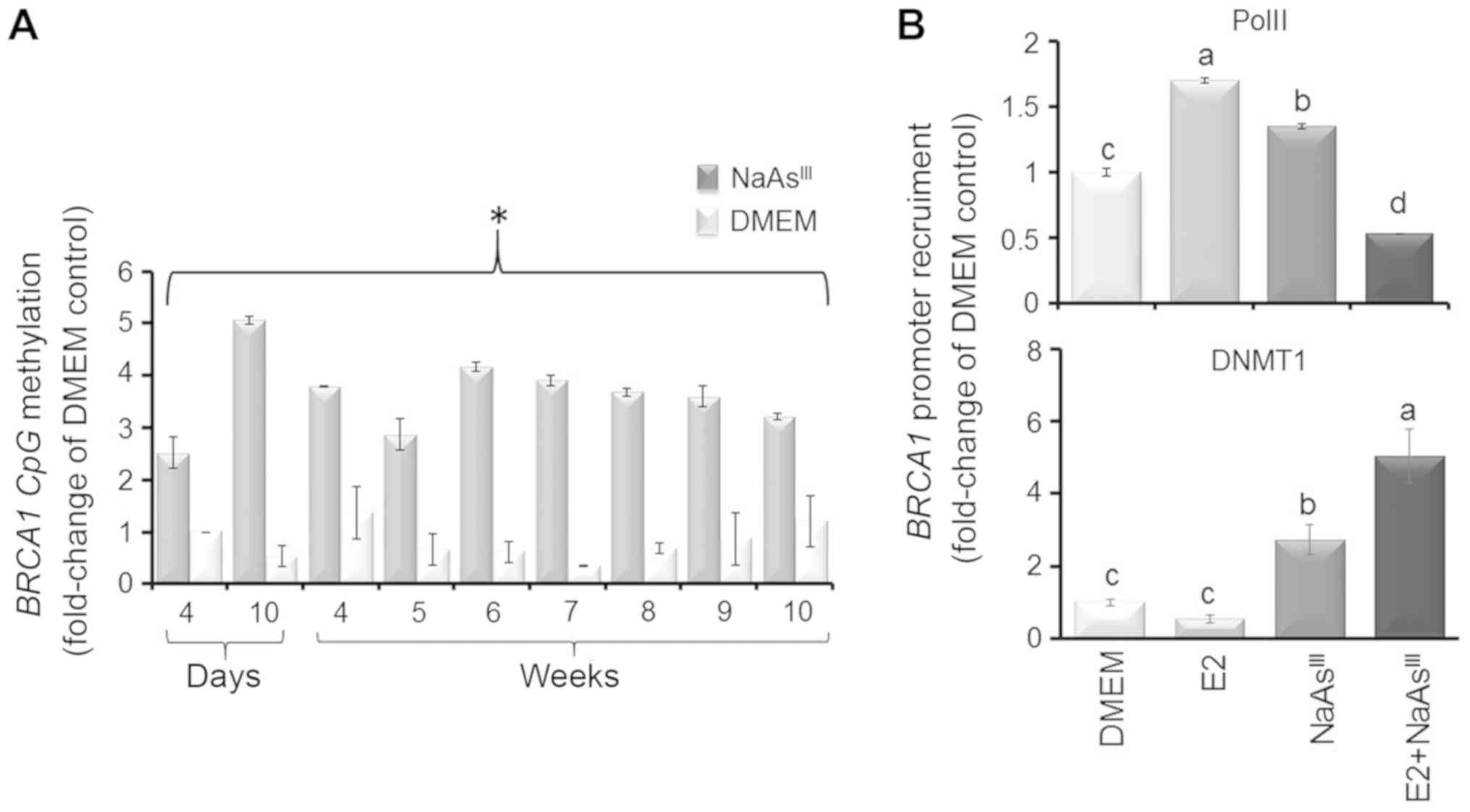
methylation. The analysis of bisulfonated genomic DNA prepared from
the MCF7 cells revealed that exposure to 1 µM
NaAsIII from 4 days to 10 weeks brought about an
increase (2.5- to 5-fold) in BRCA1 CpG methylation (Fig. 4A), which was associated at 6 days
post-treatment with a reduction in the recruitment of PolII to the
BRCA1 promoter and increased occupancy by DNMT1 (Fig. 4B). These results suggested that the
NaAsIII-dependent downregulation of BRCA1 was associated
with the reduced transcription and recruitment of DNA-modifying
enzymes (i.e., DNMT1) to the BRCA1 gene.
NaAsIII disrupts the response
to TAM in MCF7 cells in culture and in mouse mammary tumor
xenografts
The observed reduction in ERα expression depicted in
Figs. 1 and 2 raised the question as to whether
NaAsIII exposure influences E2-induced cell
proliferation and response to TAM. The results presented in
Fig. 5 indicated that treatment of
the MCF7 cells with TAM for 72 h reduced E2-induced cell growth.
Conversely, in the MCF7 cells pre-treated for 6 weeks with 1
µM NaAsIII, treatment with TAM increased cell
proliferation (Fig. 5A). The
results of western blot analysis indicated that pre-treatment with
NaAsIII for 6 weeks antagonized E2-induced BRCA1
expression, while it reduced ERα expression, a known target for TAM
(Fig. 5B). The analysis of cell
cycle distribution by flow cytometry revealed that a larger
percentage of cells co-treated for 6 weeks with NaAsIII
plus TAM or E2 plus TAM were positioned in the S-phase of the cell
cycle compared to the control MCF7 cells (Fig. 5C). These cumulative results
suggested that long-term exposure to environmentally relevant doses
(1 µM) of NaAsIII increased the resistance of
MCF7 cells to TAM through the downregulation of ERα.
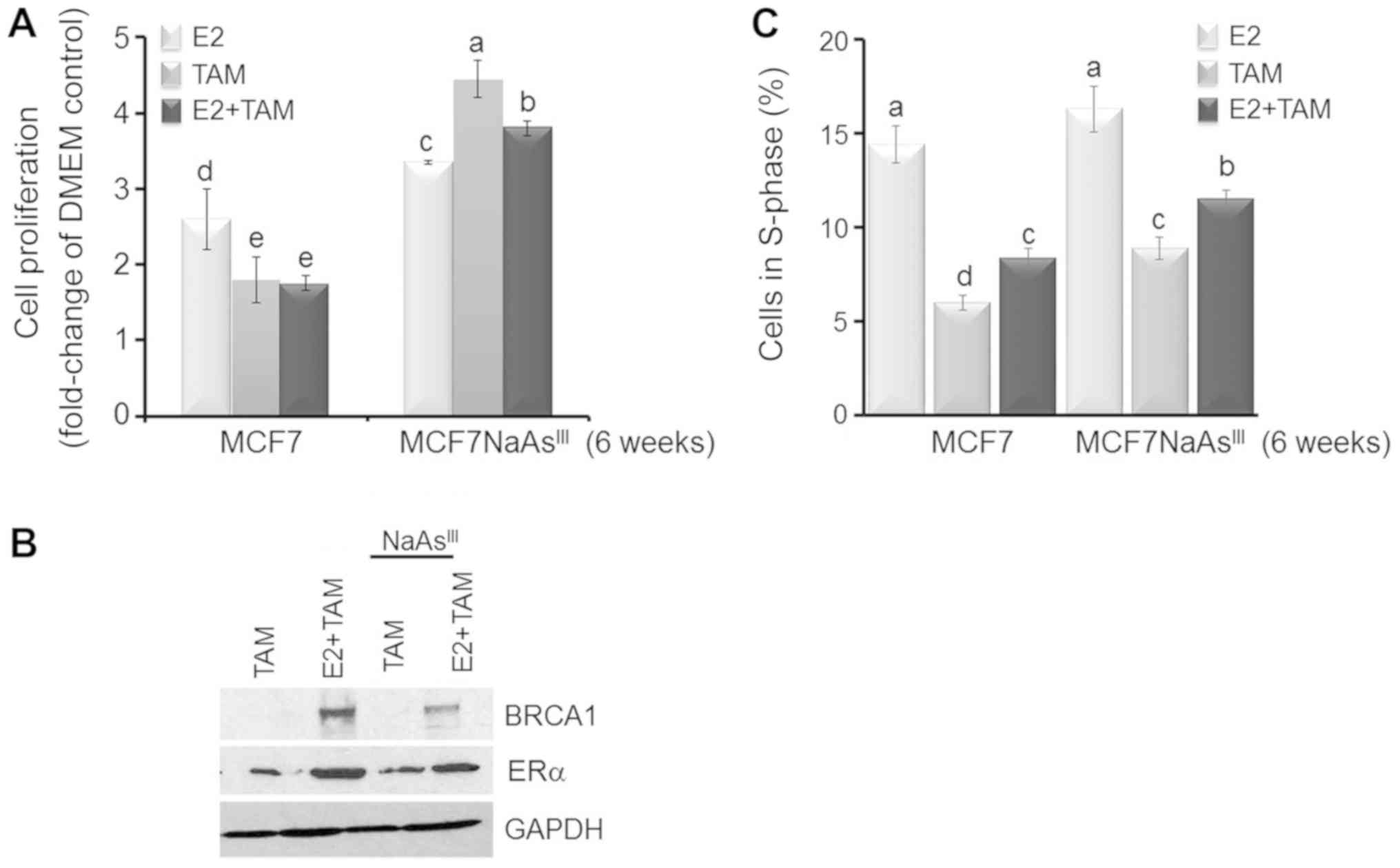 | Figure 5AsIII antagonizes the
TAM-dependent inhibition of proliferation. MCF7 cells and MCF7
cells pre-treated for 6 weeks in the presence of 1 µM
NaAsIII (MCF7NaAsIII) were cultured for 72 h
in control DMEM, or DMEM plus E2 (10 nM), TAM (1 µM), or
their combination. (A) Bars represent the means ± SEM of
quantitation (fold-change of DMEM Control) of proliferation
determined by MTT assay from 2 separate experiments (n=2) with 5
replicates. (B) Bands are representative immunocomplexes for
BRCA-1, ERα and internal standard GAPDH from 2 (n=2) separate
experiments performed in duplicate. (C) Bars represent the means ±
SEM of percentage cells in S-phase measured by flow cytometry from
two separate experiments (n=2) with 5 replicates. In (A) and (C)
different letters represent statistically significant multiple
comparison (a>b>c, etc.) differences (P<0.05).
AsIII, trivalent arsenite; BRCA1, breast cancer 1;
NaAsIII, sodium arsenite; TAM, tamoxifen; E2,
17β-estradiol. |
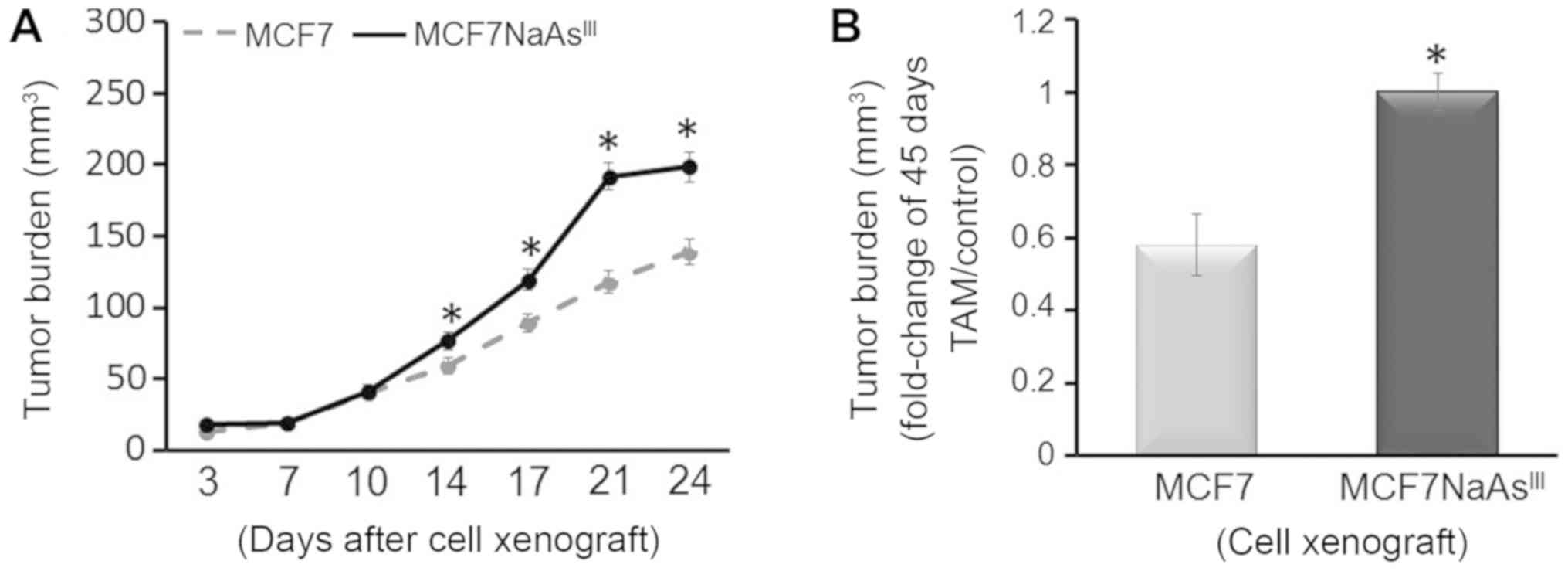
To further investigate the influence of
NaAsIII exposure on tumor development, we injected
control MCF7 cells or MCF7 cells pre-treated with 1 µM
NaAsIII for 4 weeks (MCF7 NaAsIII) into the
cleared mammary fat pad of 4-week-old OVX athymic rTac:NCr-Foxn1
nude female mice also implanted with an E2 pellet. We then
monitored tumor growth for 24 days and noted a higher tumor volume
for mice injected with MCF7 NaAsIII compared to mice
xenografted with control MCF7 cells (Fig. 6A). Subsequently, the xenografted
mice were implanted with a TAM pellet and tumors were allowed to
grow for an additional 45 days. Mammary tumors that originated from
xenografted MCF7 NaAsIII cells were more refractory
(~40%) to TAM treatment compared with tumors that developed from
control MCF7 cells (Fig. 6B). The
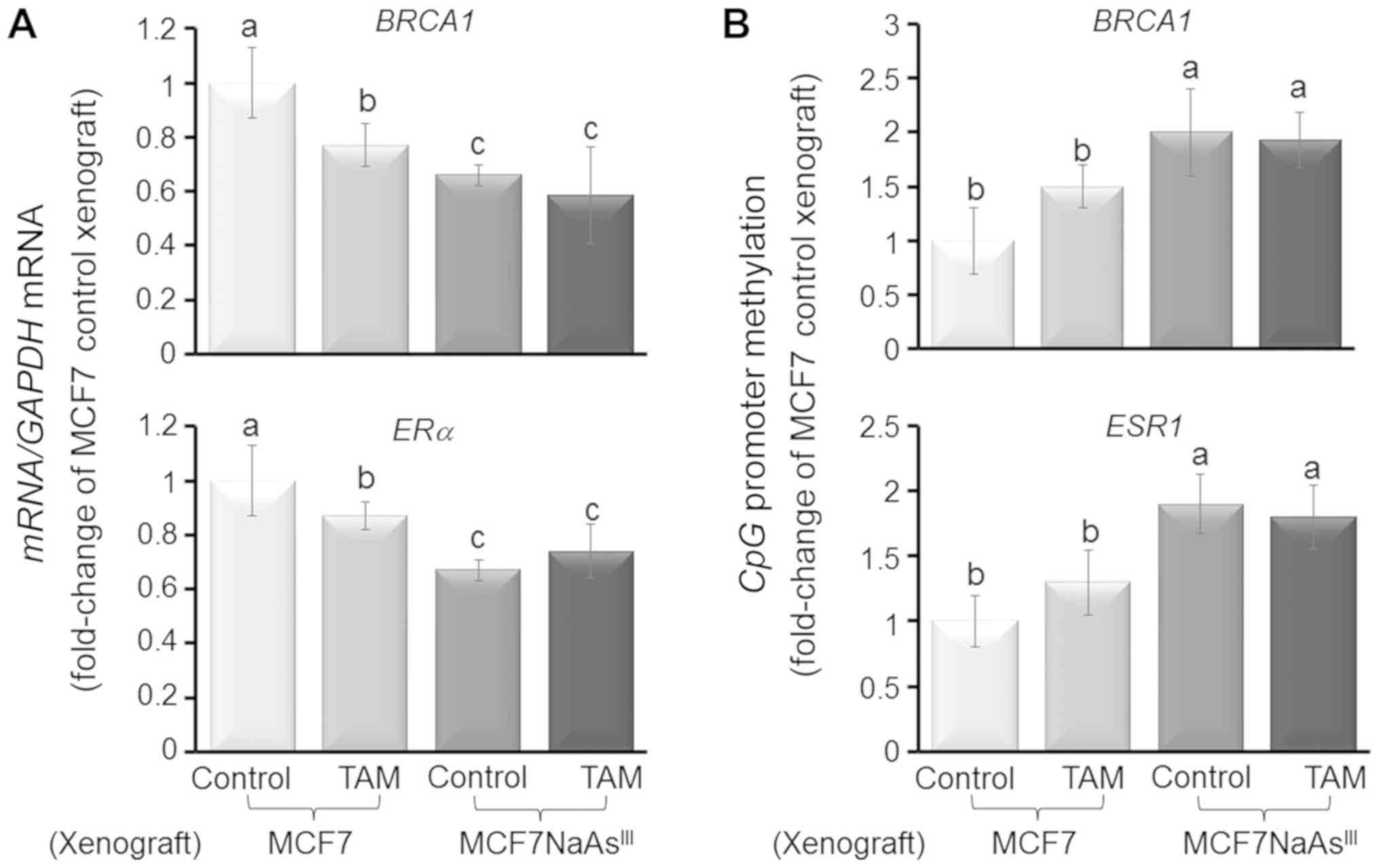
resilience of MCF7 NaAsIII tumors to TAM was coupled
with the reduced expression of BRCA1 and ERα mRNA
(Fig. 7A), and increased CpG
methylation of the respective genes (i.e., BRCA1 and
ESR1) (Fig. 7B). As a
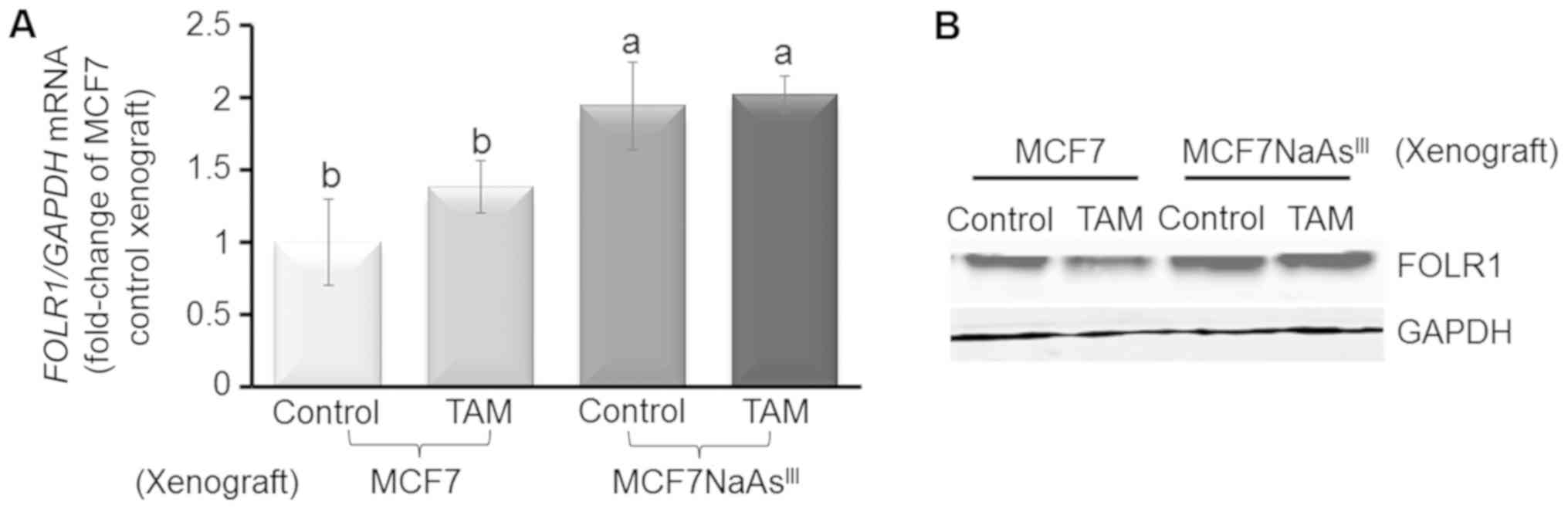
control, we measured the expression of FOLR1 mRNA (Fig. 8A) and FOLR1 protein (Fig. 8B), which were increased (~1.0-fold)
in mammary tumors from xenografted MCF7 NaAsIII cells
compared to tumors that developed from control MCF7 cells. Taken
together, the results of the tumor xenograft experiments indicated
that exposure to NaAsIII conferred the resistance of
mammary tumors to TAM and that this resilience was associated with
the hypermethylation of BRCA1 and ESR1, the reduced
expression of BRCA1 and ERα, and increased levels of
FOLR1 mRNA and tumor burden.
Discussion
The loss of ERα expression has been linked to a poor
response to endocrine therapy (16–18).
Drinking water contaminated with arsenic has been linked to the
disruption of ERα signaling (19)
and arsenic exposure has been shown to contribute to genomic
instability through the disruption of BRCA1-regulated DNA repair
(20). Arsenic may accelerate
cancer growth (24) and confer a
steroid receptor-independent phenotype (29). These cumulative observations
suggest arsenic exposure may interfere with endocrine regulation
and prompted our investigation into whether or not AsIII
contributes to resistance to TAM therapy through the silencing of
BRCA1 and ESR1. In this study, we first examined the
in vitro effects of NaAsIII in ERα-positive
breast cancer cells and found that acute (within 4 days) and
long-term (10 days to 7 weeks) exposure to environmentally relevant
doses of AsIII reduced BRCA1 expression. Furthermore,
NaAsIII compromised ERα expression and the in
vitro response of MCF7 cells to treatment with TAM. In normal
breast epithelial cells, the BRCA1 and ESR1 (encoding
for ERα) genes are regulated through a positive feedback loop in
which ERα induces expression of BRCA1 in the presence of E2
(40). In turn, BRCA1
transcriptionally activates the ESR1 gene (47). This crosstalk between BRCA1 and ERα
is thought to favor DNA repair controlled by BRCA1 before cells
progress through division under the proliferative pressure of
estrogens. Conversely, in BRCA1 mutation and sporadic breast
tumors, the reduced expression of BRCA1, also termed 'BRCAness', is
usually associated with the reduced expression of ERα and
resistance to TAM (48). Our cell
culture data suggested that exposure to NaAsIII may
compromise BRCA1 expression and confer resistance to antagonists of
the ERα such as TAM. The results of this study are in agreement
with those of a previous study (49) showing that environmentally relevant
doses of NaAsIII (~1–5 µM) reduced the expression
of the ERα.
A mechanism that may contribute to the
NaAsIII-dependent loss of BRCA1 is epigenetic silencing
via CpG methylation, which has been documented in sporadic breast
tumors, particularly in those that are more invasive (i.e., TNBC)
compared to lobulo-alveolar breast cancers (50). In this study, we documented that in
MCF7 cells both the short- (4 days) and long- (10 days to 10 weeks)
term in vitro exposure to NaAsIII induced
BRCA1 CpG methylation was associated with the increased
recruitment of DNMT1 and the loss of PolII at the BRCA1
gene. These observations are in accordance with those of a previous
study reporting promoter hypermethylation and silencing of other
DNA repair (MLH1 and MSH2) genes in arsenic-exposed
populations (51). The
reprogramming of DNA methylation elicited by NaAsIII has
been previously linked to increased growth rate (52). In keeping with these earlier
reports, in this study, we noted that MCF7 treated for 6 weeks with
NaAsIII displayed increased proliferative capacity and
were refractory to TAM.
The injection of NaAsIII-preconditioned
MCF7 cells into the mammary fat pad of nude mice provided in
vivo evidence that the prior exposure to NaAsIII may
alter the behavior of ERα-positive breast cancer cells. Xenografted
MCF7NaAsIII cells produced a larger tumor volume
compared to control MCF7 cells and were more refractory to
treatment with TAM. We attributed this resilience of
MCF7NaAsIII to TAM, at least in part, to the reduced
expression of ERα associated with the CpG hypermethylation of
ESR1. The reduced expression of ERα in
MCF7NaAsIII tumors was paralleled by the lower
expression and hypermethylation of BRCA1, further supporting
the hypothesis that exposure to NaAsIII may contribute
to breast tumorigenesis by hampering DNA repair capacity controlled
by BRCA1 and altering the crosstalk between BRCA1 and ERα.
In agreement with previous findings (45), we noted that the expression of
MTHFR in MCF7 cells treated in vitro with
NaAsIII was markedly downregulated. Thus, exposure to
inorganic arsenic may deplete the pool of methyl groups and
interfere with folate metabolism with consequences on DNA synthesis
and repair. The reduced expression of MTHFR has been previously
associated with breast cancer development (53). Conversely, in this study, we noted
in MCF7 cells in culture that exposure to NaAsIII had a
biphasic effect on the expression of FOLR1, a membrane-bound
protein involved in transport of folate into cells. Short-term
exposure to NaAsIII reduced FOLR1 expression, whereas a
stimulatory effect on FOLR1 levels was observed after long-term
exposure. The upregulation of FOLR1 was confirmed in mammary tumors
that developed from xenografted MCF7NaAsIII cells. The
upregulation of FOLR1 has been interpreted as an adaptive response
triggered by cellular depletion of methyl groups by metabolism of
NaAsIII (45).
Moreover, recent studies reported that the increased expression of
FOLR1 was associated with a higher risk of recurrence in patients
with TNBC (54), which were
significantly enriched in FOLR1 compared to ERα- and human
epidermal growth factor receptor 2-positive breast tumors (46). Whereas it remains unknown whether
NaAsIII affects expression of MTHFR and FOLR1 through
epigenetic mechanisms, a possible translational implication of our
data is that breast cancer patients exposed to NaAsIII
and undergoing treatment with TAM may benefit from combination
therapy with anti-FOLR1 agents (54).
Taken together, the data of the present study
provide novel in vitro and mammary tumor xenograft evidence
that exposure to inorganic trivalent arsenic, such as
NaAsIII may increase resistance to endocrine therapy
based on TAM through reduction in BRCA1 and ERα expression. Future
studies with ERα-positive breast cancer patients residing in
geographical regions at high risk of exposure to AsIII
are warranted to investigate whether the dysregulation of CpG
hypermethylation of BRCA1 and ESR1 causes persistent
genomic instability (55), and
variations in efficacy of therapies based on antagonists of the
ERα. As DNA methylation changes are potentially reversible, they
may offer a novel target for combination therapies of ER-positive
breast tumors with epigenetic drugs.
Funding
This study was supported by a Pilot Project grant
from the National Cancer Institute of the National Institutes of
Health under the award for the Partnership of Native American
Cancer Prevention U54CA143924 (UACC); Cancer Biology Training Grant
T32CA009213; and Cancer Center Support Grant P30CA023074.
Availability of data and materials
All data generated during this study are included
in this published article.
Authors' contributions
OIS and DFR conceived the study and drafted the
manuscript. MGD contributed to laboratory experiments, data
analysis, and writing of the manuscript. OIS conducted cellular and
molecular measurements with cell lines and tumor xenografts. BS and
GDPM contributed to designing and performing the xenograft
experiments and review of data. All authors have read and approved
the final manuscript.
Ethics approval and consent to
participate
All mouse xenograft experiments were performed
under the #07–029 protocol approved by the University of Arizona
Institutional Animal Care and Use Committee approved on 02/22/2016.
All procedures were performed in compliance with the standard
operating procedures and relevant guidelines of the University of
Arizona Animal Care.
Patient consent for publication
Not applicable
Competing interests
The authors declare they have no competing
interests.
Acknowledgments
The authors wish to acknowledge the support of The
Experimental Mouse Shared Resource Core of the University of
Arizona Cancer Center.
Abbreviations:
|
AsIII
|
trivalent arsenite
|
|
AsV
|
pentavalent arsenate
|
|
BRCA1
|
breast cancer 1
|
|
CCND1
|
cyclin D1
|
|
ChIP
|
chromatin immunoprecipitation
|
|
DMEM/F12
|
Dulbecco's modified Eagle's/F12
medium
|
|
DNMT1
|
DNA methyltransferase 1
|
|
E2
|
17β-estradiol
|
|
ERα
|
estrogen receptor-α
|
|
FCS
|
fetal calf serum; FOLR1, folate
receptor 1
|
|
GAPDH
|
glyceraldehyde 3-phosphate
dehydrogenase
|
|
GEN
|
genistein
|
|
M
|
methylated-specific primers
|
|
MTHFR
|
methylenetetrahydrofolate
reductase
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
NaAsIII
|
sodium arsenite
|
|
OVX
|
ovariecromized
|
|
PBS
|
phosphate-buffered saline
|
|
PolII
|
RNA polymerase II
|
|
PR
|
progesterone receptor
|
|
qPCR
|
quantitative polymerase chain
reaction
|
|
TAM
|
tamoxifen
|
|
TNBC
|
triple-negative breast cancers
|
|
U
|
unmethylated-specific primers
|
References
|
1
|
Ayotte JD, Belaval M, Olson SA, Burow KR,
Flanagan SM, Hinkle SR and Lindsey BD: Factors affecting temporal
variability of arsenic in groundwater used for drinking water
supply in the United States. Sci Total Environ. 505:1370–1379.
2015. View Article : Google Scholar
|
|
2
|
Sorg TJ, Chen AS and Wang L: Arsenic
species in drinking water wells in the USA with high arsenic
concentrations. Water Res. 48:156–169. 2014. View Article : Google Scholar
|
|
3
|
Agusa T, Trang PT, Lan VM, Anh DH, Tanabe
S, Viet PH and Berg M: Human exposure to arsenic from drinking
water in Vietnam. Sci Total Environ. 488–489:562–569. 2014.
View Article : Google Scholar
|
|
4
|
Dummer TJ, Yu ZM, Nauta L, Murimboh JD and
Parker L: Geostatistical modelling of arsenic in drinking water
wells and related toenail arsenic concentrations across Nova
Scotia, Canada. Sci Total Environ. 505:1248–1258. 2015. View Article : Google Scholar
|
|
5
|
George CM, Sima L, Arias MH, Mihalic J,
Cabrera LZ, Danz D, Checkley W and Gilman RH: Arsenic exposure in
drinking water: An unrecognized health threat in Peru. Bull World
Health Organ. 92:565–572. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
García-Esquinas E, Pollán M, Umans JG,
Francesconi KA, Goessler W, Guallar E, Howard B, Farley J, Best LG
and Navas-Acien A: Arsenic exposure and cancer mortality in a
US-based prospective cohort: The strong heart study. Cancer
Epidemiol Biomarkers Prev. 22:1944–1953. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Saint-Jacques N, Parker L, Brown P and
Dummer TJ: Arsenic in drinking water and urinary tract cancers: a
systematic review of 30 years of epidemiological evidence. Environ
Health. 13:442014. View Article : Google Scholar
|
|
8
|
Gentry PR, Clewell HJ III, Greene TB,
Franzen AC and Yager JW: The impact of recent advances in research
on arsenic cancer risk assessment. Regul Toxicol Pharmacol.
69:91–104. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Stoica A, Pentecost E and Martin MB:
Effects of arsenite on estrogen receptor-alpha expression and
activity in MCF-7 breast cancer cells. Endocrinology.
141:3595–3602. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ren X, McHale CM, Skibola CF, Smith AH,
Smith MT and Zhang L: An emerging role for epigenetic dysregulation
in arsenic toxicity and carcinogenesis. Environ Health Perspect.
119:11–19. 2011. View Article : Google Scholar :
|
|
11
|
Gentry PR, McDonald TB, Sullivan DE, Shipp
AM, Yager JW and Clewell HJ III: Analysis of genomic dose-response
information on arsenic to inform key events in a mode of action for
carcinogenicity. Environ Mol Mutagen. 51:1–14. 2010.
|
|
12
|
Kijima I, Itoh T and Chen S: Growth
inhibition of estrogen receptor-positive and aromatase-positive
human breast cancer cells in monolayer and spheroid cultures by
letrozole, anastrozole, and tamoxifen. J Steroid Biochem Mol Biol.
97:360–368. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Khanjani N, Jafarnejad AB and Tavakkoli L:
Arsenic and breast cancer: A systematic review of epidemiologic
studies. Rev Environ Health. 32:267–277. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Musgrove EA and Sutherland RL: Biological
determinants of endocrine resistance in breast cancer. Nat Rev
Cancer. 9:631–643. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schiff R, Massarweh S, Shou J and Osborne
CK: Breast cancer endocrine resistance: how growth factor signaling
and estrogen receptor coregulators modulate response. Clin Cancer
Res. 9(1 Pt 2): 447S–454S. 2003.PubMed/NCBI
|
|
16
|
Johnston SR, Saccani-Jotti G, Smith IE,
Salter J, Newby J, Coppen M, Ebbs SR and Dowsett M: Changes in
estrogen receptor, progesterone receptor, and pS2 expression in
tamoxifen-resistant human breast cancer. Cancer Res. 55:3331–3338.
1995.PubMed/NCBI
|
|
17
|
Kuukasjärvi T, Kononen J, Helin H, Holli K
and Isola J: Loss of estrogen receptor in recurrent breast cancer
is associated with poor response to endocrine therapy. J Clin
Oncol. 14:2584–2589. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Osborne CK and Schiff R: Mechanisms of
endocrine resistance in breast cancer. Annu Rev Med. 62:233–247.
2011. View Article : Google Scholar
|
|
19
|
Muñoz A, Chervona Y, Hall M, Kluz T,
Gamble MV and Costa M: Sex-specific patterns and deregulation of
endocrine pathways in the gene expression profiles of Bangladeshi
adults exposed to arsenic contaminated drinking water. Toxicol Appl
Pharmacol. 284:330–338. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Peremartí J, Ramos F, Marcos R and
Hernández A: Arsenic exposure disrupts the normal function of the
FA/BRCA repair pathway. Toxicol Sci. 142:93–104. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Parodi DA, Greenfield M, Evans C, Chichura
A, Alpaugh A, Williams J and Martin MB: Alteration of mammary gland
development and gene expression by in utero exposure to arsenic.
Reprod Toxicol. 54:66–75. 2015. View Article : Google Scholar :
|
|
22
|
Tokar EJ, Diwan BA, Ward JM, Delker DA and
Waalkes MP: Carcinogenic effects of 'whole-life' exposure to
inorganic arsenic in CD1 mice. Toxicol Sci. 119:73–83. 2011.
View Article : Google Scholar
|
|
23
|
Waalkes MP, Liu J, Ward JM and Diwan BA:
Animal models for arsenic carcinogenesis: Inorganic arsenic is a
transplacental carcinogen in mice. Toxicol Appl Pharmacol.
198:377–384. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schrauzer GN, White DA, McGinness JE,
Schneider CJ and Bell LJ: Arsenic and cancer: Effects of joint
administration of arsenite and selenite on the genesis of mammary
adenocarcinoma in inbred female C3H/St mice. Bioinorg Chem.
9:245–253. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu Y, Hock JM, Sullivan C, Fang G, Cox
AJ, Davis KT, Davis BH and Li X: Activation of the p38
MAPK/Akt/ERK1/2 signal pathways is required for the protein
stabilization of CDC6 and cyclin D1 in low-dose arsenite-induced
cell proliferation. J Cell Biochem. 111:1546–1555. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu Y, Tokar EJ and Waalkes MP:
Arsenic-induced cancer cell phenotype in human breast epithelia is
estrogen receptor-independent but involves aromatase activation.
Arch Toxicol. 88:263–274. 2014. View Article : Google Scholar :
|
|
27
|
Tong D, Ortega J, Kim C, Huang J, Gu L and
Li GM: Arsenic Inhibits DNA Mismatch Repair by Promoting EGFR
Expression and PCNA Phosphorylation. J Biol Chem. 290:14536–14541.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bhattacharjee P, Banerjee M and Giri AK:
Role of genomic instability in arsenic-induced carcinogenicity. A
review. Environ Int. 53:29–40. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Benbrahim-Tallaa L, Webber MM and Waalkes
MP: Acquisition of androgen independence by human prostate
epithelial cells during arsenic-induced malignant transformation.
Environ Health Perspect. 113:1134–1139. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Baylin SB and Jones PA: A decade of
exploring the cancer epigenome - biological and translational
implications. Nat Rev Cancer. 11:726–734. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cardenas A, Houseman EA, Baccarelli AA,
Quamruzzaman Q, Rahman M, Mostofa G, Wright RO, Christiani DC and
Kile ML: In utero arsenic exposure and epigenome-wide associations
in placenta, umbilical artery, and human umbilical vein endothelial
cells. Epigenetics. 10:1054–1063. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chou WC, Chung YT, Chen HY, Wang CJ, Ying
TH, Chuang CY, Tseng YC and Wang SL: Maternal arsenic exposure and
DNA damage biomarkers, and the associations with birth outcomes in
a general population from Taiwan. PLoS One. 9:e863982014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cui X, Wakai T, Shirai Y, Hatakeyama K and
Hirano S: Chronic oral exposure to inorganic arsenate interferes
with methylation status of p16INK4a and RASSF1A and induces lung
cancer in A/J mice. Toxicol Sci. 91:372–381. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ramirez T, Brocher J, Stopper H and Hock
R: Sodium arsenite modulates histone acetylation, histone
deacetylase activity and HMGN protein dynamics in human cells.
Chromosoma. 117:147–157. 2008. View Article : Google Scholar
|
|
35
|
Lu G, Xu H, Chang D, Wu Z, Yao X, Zhang S,
Li Z, Bai J, Cai Q and Zhang W: Arsenic exposure is associated with
DNA hyper-methylation of the tumor suppressor gene p16. J Occup Med
Toxicol. 9:422014. View Article : Google Scholar
|
|
36
|
Zhang TC, Schmitt MT and Mumford JL:
Effects of arsenic on telomerase and telomeres in relation to cell
proliferation and apoptosis in human keratinocytes and leukemia
cells in vitro. Carcinogenesis. 24:1811–1817. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jiang R, Li Y, Zhang A, Wang B, Xu Y, Xu
W, Zhao Y, Luo F and Liu Q: The acquisition of cancer stem
cell-like properties and neoplastic transformation of human
keratinocytes induced by arsenite involves epigenetic silencing of
let-7c via Ras/NF-κB. Toxicol Lett. 227:91–98. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Romagnolo DF, Donovan MG, Papoutsis AJ,
Doetschman TC and Selmin OI: Genistein prevents BRCA1 CpG
methylation and proliferation in human breast cancer cells with
activated aromatic hydrocarbon receptor. Curr Dev Nutr.
1:e0005622017. View Article : Google Scholar
|
|
39
|
Papoutsis AJ, Borg JL, Selmin OI and
Romagnolo DF: BRCA-1 promoter hypermethylation and silencing
induced by the aromatic hydrocarbon receptor-ligand TCDD are
prevented by resveratrol in MCF-7 cells. J Nutr Biochem.
23:1324–1332. 2012. View Article : Google Scholar
|
|
40
|
Jeffy BD, Hockings JK, Kemp MQ, Morgan SS,
Hager JA, Beliakoff J, Whitesell LJ, Bowden GT and Romagnolo DF: An
estrogen receptor-alpha/p300 complex activates the BRCA-1 promoter
at an AP-1 site that binds Jun/Fos transcription factors:
Repressive effects of p53 on BRCA-1 transcription. Neoplasia.
7:873–882. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lothrop N, Wilkinson ST, Verhougstraete M,
Sugeng A, Loh MM, Klimecki W and Beamer PI: Home Water Treatment
Habits and Effectiveness in a Rural Arizona Community. Water.
7:1217–1231. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hough RL, Fletcher T, Leonardi GS,
Goessler W, Gnagnarella P, Clemens F, Gurzau E, Koppova K, Rudnai
P, Kumar R, et al: Lifetime exposure to arsenic in residential
drinking water in Central Europe. Int Arch Occup Environ Health.
83:471–481. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kumar A, Adak P, Gurian PL and Lockwood
JR: Arsenic exposure in US public and domestic drinking water
supplies: A comparative risk assessment. J Expo Sci Environ
Epidemiol. 20:245–254. 2010. View Article : Google Scholar
|
|
44
|
Raessler M: The Arsenic Contamination of
Drinking and Groundwaters in Bangladesh: Featuring Biogeochemical
Aspects and Implications on Public Health. Arch Environ Contam
Toxicol. 75:1–7. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ruiz-Ramos R, López- Carrillo L, Albores
A, Hernández-Ramírez RU and Cebrian ME: Sodium arsenite alters cell
cycle and MTHFR, MT1/2, and c-Myc protein levels in MCF-7 cells.
Toxicol Appl Pharmacol. 241:269–274. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Necela BM, Crozier JA, Andorfer CA,
Lewis-Tuffin L, Kachergus JM, Geiger XJ, Kalari KR, Serie DJ, Sun
Z, Moreno-Aspitia A, et al: Correction: Folate receptor-α (FOLR1)
expression and function in triple negative tumors. PLoS One.
10:e01271332015. View Article : Google Scholar
|
|
47
|
Hosey AM, Gorski JJ, Murray MM, Quinn JE,
Chung WY, Stewart GE, James CR, Farragher SM, Mulligan JM, Scott
AN, et al: Molecular basis for estrogen receptor alpha deficiency
in BRCA1-linked breast cancer. J Natl Cancer Inst. 99:1683–1694.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lips EH, Mulder L, Oonk A, van der Kolk
LE, Hogervorst FB, Imholz AL, Wesseling J, Rodenhuis S and Nederlof
PM: Triple-negative breast cancer: BRCAness and concordance of
clinical features with BRCA1-mutation carriers. Br J Cancer.
108:2172–2177. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Nakareangrit W, Thiantanawat A,
Visitnonthachai D, Watcharasit P and Satayavivad J: Sodium arsenite
inhibited genomic estrogen signaling but induced pERα (Ser118) via
MAPK pathway in breast cancer cells. Environ Toxicol. 31:1133–1146.
2016. View Article : Google Scholar
|
|
50
|
Birgisdottir V, Stefansson OA,
Bodvarsdottir SK, Hilmarsdottir H, Jonasson JG and Eyfjord JE:
Epigenetic silencing and deletion of the BRCA1 gene in sporadic
breast cancer. Breast Cancer Res. 8:R382006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bhattacharjee P, Sanyal T, Bhattacharjee S
and Bhattacharjee P: Epigenetic alteration of mismatch repair genes
in the population chronically exposed to arsenic in West Bengal,
India. Environ Res. 163:289–296. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Riedmann C, Ma Y, Melikishvili M, Godfrey
SG, Zhang Z, Chen KC, Rouchka EC and Fondufe-Mittendorf YN:
Inorganic Arsenic-induced cellular transformation is coupled with
genome wide changes in chromatin structure, transcriptome and
splicing patterns. BMC Genomics. 16:2122015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zara-Lopes T, Gimenez-Martins AP,
Nascimento-Filho CH, Castanhole-Nunes MM, Galbiatti-Dias AL,
Padovani-Júnior JA, Maniglia JV, Francisco JL, Pavarino EC and
Goloni-Bertollo EM: Role of MTHFR C677T and MTR A2756G
polymorphisms in thyroid and breast cancer development. Genet Mol
Res. 15:gmr82222016. View Article : Google Scholar
|
|
54
|
Ginter PS, McIntire PJ, Cui X, Irshaid L,
Liu Y, Chen Z and Shin SJ: Folate receptor alpha expression is
associated with increased risk of recurrence in triple-negative
breast cancer. Clin Breast Cancer. 17:544–549. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Mauro M, Caradonna F and Klein CB:
Dysregulation of DNA methylation induced by past arsenic treatment
causes persistent genomic instability in mammalian cells. Environ
Mol Mutagen. 57:137–150. 2016. View Article : Google Scholar :
|