1. Introduction
Calcium (Ca2+) signaling regulates
various physiological processes, including muscle contraction,
neuron excitability, cellular secretion and cell migration
(1,2). At the cellular level, fine-tuned
regulation of intracellular Ca2+ levels is essential for
maintaining cell survival, cell function and mitochondrial
dynamics, which are the focus of the first part of this review.
Total cellular Ca2+ levels are maintained
through the sum of Ca2+ influx that occurs through the
plasma membrane using cation channels and the release of
Ca2+ from intracellular Ca2+ stores. The
endoplasmic reticulum (ER) and mitochondria are the main
Ca2+ stores. The role of mitochondria in calcium
homeostasis is important because of its function as the main
cellular chamber involved in ATP production, and because there is a
link between mitochondrial levels of calcium and mitochondrial
dynamics, function and metabolism. Mitochondria are also involved
in cellular microdomains that regulate vital cellular processes
(3,4).
Cancer remodels the systems that maintain cellular
calcium homeostasis, including Ca2+ transporters and
their regulators, Ca2+-dependent enzyme activity and
accessory molecules that will support tumor development. These
processes are discussed in the second part of the present
review.
2. Mitochondrial calcium homeostasis
Mitochondrial calcium homeostasis is regulated
through a fine-tuned process that defines the capacity of the
mitochondria for handling calcium. This process is critical for the
regulation of aerobic metabolism and cell survival (5). In this manner, mitochondria an
important role in modulating the amplitude and timing of
intracellular Ca2+ signals through two mechanisms
(6): Mitochondrial calcium
buffering capacity and mitochondrial functional interactions with
other channels or organelles.
Mitochondria possess a calcium buffering capacity
that can handle levels of intracellular calcium between 50 and 500
nM in numerous types of normal cells (7-9).
Through this buffering activity, mitochondria may dynamically
manage transient alterations in calcium concentrations and may
buffer severe Ca2+ overloads, which are often associated
with pathological conditions (9).
Notably, mitochondria can functionally interact with
calcium-channel gating in the cell membrane of T lymphocytes. A
prolonged elevation of cytosolic Ca2+ is required for
T-cell activation, and functional mitochondria prevent a local
accumulation of Ca2+ near sites that govern channel
activation and maintain an opened Ca2+ release-activated
Ca2+ (CRAC)-channel (10).
The close contact between mitochondria and cellular
Ca2+ gates present in the ER and the cell membrane
creates microdomains that allow the increase of mitochondrial
calcium concentration ([Ca2+]m) in parallel
to cytosolic Ca2+ signals, despite the low affinity of
[Ca2+]m uptake systems (11). Numerous close contacts have been
observed between mitochondria and the ER, where, upon opening of
the inositol 1,4,5-triphosphate (IP3)-gated channels in
the ER, the surface of the mitochondria is exposed to higher
concentrations of Ca2+ than the bulk cytosol, which
increases [Ca2+]m (Fig. 1) (12,13).
In addition, microdomains between the mitochondria and cell
membrane allow the entrance of extracellular calcium through cell
membrane channels to the mitochondria, providing a link between
cellular activation and mitochondrial function (Fig. 1) (9). The rapid diffusion of intracellular
Ca2+ avoids organelle overload (12).
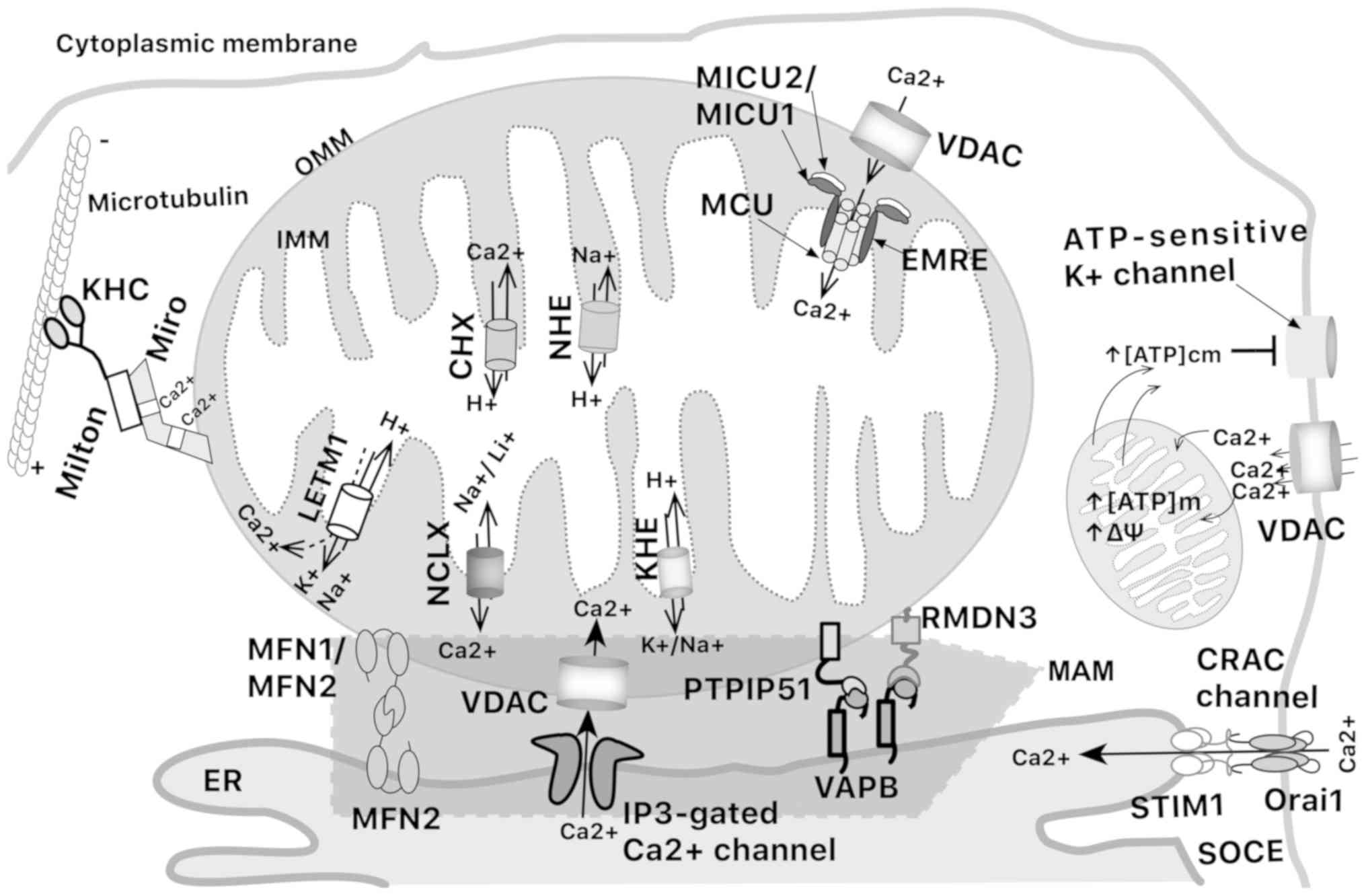 | Figure 1Schematic representation of
mitochondrial calcium interplay. Calcium can be directly
transported from the cytoplasmic membrane into mitochondria or ER
using VDAC and CRAC channels. VDAC/MCU and STIM1 introduce calcium
into mitochondria and ER, respectively. IP3Rs export
calcium from ER to mitochondria at MAMs where various pairs of
molecules support the union between mitochondria and ER. A
fine-tuning interplay exists among different cations that
participate in mitochondrial calcium extrusion. Dotted arrow
represents pH-dependent transport of Ca2+. CHX,
Ca2+/H+ exchanger; CRAC, calcium
release-activated channels; EMRE, essential MCU regulator; ER,
endoplasmic reticulum; IP3R, inositol
1,4,5-trisphosphate receptor; KHC, kinesin heavy chain; KHE,
mitochondrial K+/H+ exchanger; LETM1, leucine
zipper-EF-hand containing transmembrane protein 1; MAM,
mitochondria-associated membrane; MCU, mitochondrial calcium
uniporter; MICU1/2, mitochondrial calcium uptake 1 and 2; NCLX,
Na+/Ca2+ exchange; NHE,
Na+/H+ exchanger; PTPIP51, protein tyrosine
phosphatase interacting protein 51; RMDN3, regulator of microtubule
dynamics 3; SOCE, store-operated Ca2+ entry channels;
STIM1, stromal interaction molecule 1; VAPB, vesicle-associated
membrane protein associated protein B; VDAC, voltage-dependent
anion channels. |
3. Mitochondrial calcium transport
Calcium can be transported into the mitochondria
using voltage-dependent anion channels (VDACs) that exist on the
outer mitochondrial membrane (OMM) in excitable cells, including
neurons, muscle cells and fibroblasts. VDACs can form a large
conductance channel in lipid bilayers (14). Furthermore, VDACs exist in the cell
membrane of neurons and retinal amacrine cell (15,16).
VDACs can also transport other small molecules and ions;
Ca2+ can regulate VDAC activity, thus controlling
permeability to ions and small molecules through the OMM.
Elevations of [Ca2+] to the micromolar range induce
higher conductance and sustain VDAC opening. Notably,
[Ca2+]m uptake is often driven by spikes and
oscillations in [Ca2+], which are characterized by
short-lasting events; under physiological ionic conditions VDACs
often present closed conformations (Fig. 1) (14).
Calcium is transported by the mitochondrial calcium
uniporter (MCU), a ruthenium-red-sensitive uniporter that utilizes
the negative potential across the inner mitochondrial membrane
(IMM) (17). MCU pentamerizes in
the IMM as part of a larger molecular weight complex; the other
subunits are mitochondrial calcium uptake 1 and 2 (MICU1 and
MICU2), and the essential MCU regulator (EMRE). MICU1 and MICU2
serve as calcium sensors that allow Ca2+-dependent
opening of the MCU pore. EMRE is a single-pass membrane protein
required to allow Ca2+ access (Fig. 1). When Ca2+
concentration around the MCU is >1 mM, the channel opens and
allows Ca2+ to enter into the matrix that is driven by
the large negative membrane potential of the IMM (18). In endothelial cells exposed to
various glucose concentrations (2-30 mM), MCU and MICU1 are
upregulated in a dose- and time-dependent manner. Therefore,
endothelial cells under hyperglycemic stress exhibit cell
dysfunction associated with [Ca2+]m overload,
which results in an increase in reactive oxygen species (ROS) that
promotes apoptosis (19).
The primary mechanism underlying calcium extrusion
in excitable cells is the
Na+/Ca2+/Li+ exchanger (NCLX);
this transporter exchanges mitochondrial matrix calcium for
external sodium or lithium, hence its name (20,21).
Therefore, to allow activity of the Na+/Ca2+
antiporter, matrix Na+ must be exchanged for external
H+ by a Na+/H+ exchanger (22). In addition, Ca2+ efflux
might occur via a Ca2+/H+ exchanger, which is
expressed in acidic organelles, including mitochondria (22,23).
The mitochondrial K+/H+ exchanger (KHE) also
participates in mitochondrial calcium dynamics; however, KHE
discriminates poorly between K+ and other monovalent
cations, extruding Na+ by H+. Another IMM
protein that has K+/H+ exchange activity is
the leucine zipper-EF-hand containing transmembrane protein 1
(LETM1). Lowering LETM1 expression via short hairpin RNA hampers
mitochondrial K+ or Na+/H+
exchange. Furthermore, LETM1 downregulation decreases
Na+/Ca2+ exchange mediated by the NCLX
(22). Furthermore, LETM1 is able
to transport Ca2+ in a pH-dependent manner. When the pH
shifts from acidic to alkaline, LETM1 opens a central cavity
allowing Ca2+/H+ antiporter (24). Therefore, a fine-tuning interplay
exists among [Ca2+], [Na+], [Li+],
[K+] and [H+] at the mitochondrial matrix and
in the intermembrane space where calcium is a key player (Fig. 1).
4. Calcium interplay between ER and
mitochondria shapes mitochondrial function and dynamics
Mitochondria associated membrane (MAM) is
a key microdomain in calcium homeostasis
At the cellular level, calcium has a role in how
mitochondria interact with other organelles, including the ER,
which can also affect mitochondrial function and dynamics. The
spatially restricted domain created between the ER and the
mitochondria is known as the MAM. Notably, ~20% of the mitochondria
surface is in close contact with the ER, where the ER protein
vesicle-associated membrane protein associated protein B (VAPB) is
the scaffold that tethers the ER to the mitochondrial protein,
protein tyrosine phosphatase interacting protein 51 (Fig. 1) (13). Another molecule that participates
in tethering the ER and mitochondria together is mitofusin 2
(MFN2), which also has GTPase activity (25), wherein B-cell lymphoma-2 (Bcl-2)
related protein ovarian killer increases MFN2 to cause tethering of
the ER and mitochondria to facilitate mitochondrial fission
(26). Additionally, MAM functions
as a critical membrane contact site for lipid synthesis and
exchange (26). The importance of
MAM communication is underlined in various human diseases,
including cancer (13,26-29).
Notably, the ER supplies Ca2+ directly to
mitochondria via the MAM through the use of IP3
receptors (IP3Rs) (13). IP3Rs are tetrameric
ER-resident Ca2+ channels that release Ca2+
from the ER into the cytosol in response to IP3
(Fig. 1) (30). Three isoforms comprise the
IP3R family and the majority of cell types express two
or even all three isoforms. For example, secretory granules of
human astrocytes can contain all three IP3R isoforms
(31). Although, IP3R-1
is the most widely expressed and appears to be ubiquitous in animal
tissues, it can be highly expressed in cells from the mouse central
nervous system (32).
IP3R-2 and -3 are highly expressed in human
hematopoietic and lymphatic cells (33). In this regard, IP3Rs
seem to be essential for constitutive Ca2+ release from
the ER to mitochondria, in order to maintain sufficient
mitochondrial NADH production to support mitochondrial
bioenergetics. In DT40-KO cells, which is a chicken B cell line in
which the three IP3R isoforms are genetically deleted,
the absence of Ca2+ transfer inhibits pyruvate
dehydrogenase and activates 5′ AMP-activated protein kinase (AMPK),
which activates pro-survival autophagy via a mammalian target of
rapamycin (mTOR)-independent mechanism. AMPK phosphorylates
substrates to limit anabolic pathways that consume ATP and to
initiate catabolic pathways that support oxidative phosphorylation
(OXPHOS) (34).
Role of Ca2+ in mitochondrial
function
Increasing evidence has revealed how calcium
regulates mitochondrial function. Notably, high glucose levels
promote a sustained increase in [Ca2+]m,
cytoplasmic calcium concentration ([Ca2+]c)
and insulin secretion; these effects can be abolished by inhibitors
of voltage-gated Ca2+ channels in INS-1 pancreatic
β-cells. The increase in calcium levels promotes the synthesis of
mitochondrial ATP, accompanied by increased levels of NADH and
NADPH cofactors (35).
Calcium-associated activation of mitochondrial dehydrogenases,
including pyruvate and 2-oxoglutarate dehydrogenase complexes,
NAD+-isocitrate dehydrogenase and inorganic
pyrophosphatase, may sustain the reduction of NAD+ and
NADP+ cofactors. All these enzymes are highly sensitive
to alterations in calcium concentration inside the mitochondrial
matrix (9,11,35,36).
For example, [Ca2+]m <1 mM is required to
induce the full activation of pyruvate dehydrogenase
(PDH)-phosphate phosphatase, which removes the phosphate group from
PDH-phosphate, and increases the proportion of active and
dephosphorylated PDH. Two other examples include 2-oxoglutarate
dehydrogenase that catalyzes the irreversible reaction of the
oxidative decarboxylation of 2-oxoglutarate and
NAD+-isocitrate dehydrogenase that responds to changes
in [Ca2+]m varying between 1 and 20 mM
(37).
Notably, in single islet β-cells, extracellular
glucose concentrations (3-30 mM) increase [ATP]m in the
mitochondria located close to the cytoplasmic membrane (cm). This
phenomenon leads to increased [ATP]cm, which is
essential for sustained closure of the ATP-sensitive K+
channels that additionally causes Ca2+ influx into the
cell. The mitochondria located close to the cytoplasmic membrane
also have access to a privileged elevation of
[Ca2+]cm. This calcium enters mitochondria,
progressively activating Ca2+-dependent mitochondrial
enzymes to increase [ATP]m that establishes a
feed-forward process (Fig. 1)
(36).
Mitochondrial matrix calcium can also activate ATP
synthesis and adenylate transport (38). The increase in [ATP] depends on the
amplitude of Ca2+ elevations in the mitochondrial matrix
and the availability of mitochondrial substrates. Notably, a
Ca2+ increase induces a long-lasting effect that
persists even after the elevation has finished, which denotes a
cellular memory that allows prolonged metabolic activation in
stimulated cells (11).
Activation of ATP synthesis by
[Ca2+]m is concomitantly accompanied by a
rise in mitochondrial membrane potential, which is the driving
force for Ca2+ accumulation (36,37).
This has been shown in cerebellar granule neurons, where in
vitro neuronal stimulation induced by mitochondrial
depolarization with high concentrations of KCl results in the entry
of cytosolic Ca2+ into the mitochondria (3). Conversely, a decrease in
mitochondrial membrane potential inhibits
[Ca2+]m uptake (3).
In HL-1 cardiomyocytes, sustained elevation of
intracellular [Ca2+] can cause hypertrophy. During such
Ca2+ elevations, mitochondria accumulate calcium
rapidly, which induces the opening of permeability transition pores
(PTP), matrix swelling, OMM rupture and cell death (39). High concentrations of calcium (300
mM) decrease ATP production >2-fold in cardiomyocytic
mitochondria; however, incubation of these mitochondria with
Ca2+ and muscle fructose 1,6-bisphosphatase (FBP) allows
recovery of ATP levels, thus suggesting that FBP blocks the
inhibitory effects of high [Ca2+] (39). Muscle FBP has two predominant
oligomeric states that determine its subcellular localization. Only
dimeric FBP interacts with mitochondria and protects them against
stress stimuli (40). Therefore,
as a survival mechanism associated with the elevation of calcium,
cardiomyocytes promote the binding of FBP to mitochondria. FBP, in
addition to being a regulatory enzyme of glyconeogenesis, can also
join to mitochondria preventing their dysfunction by calcium
stress. FBP interacts with mitochondrial proteins involved in
regulating membrane permeability by reducing
Ca2+-induced PTP opening; FBP also interacts with
mitochondrial proteins involved in energy homeostasis, which causes
stimulation of ATP synthesis (39).
Role of Ca2+ in mitochondrial
dynamics
Ca2+ influx into mitochondria has
important consequences on the organelles themselves. For example,
Ca2+ influx through VDAC and MCU induces rapid
mitochondrial fission by phosphorylation at serine 600 of the
dynamin-related protein 1 (DRP1) in neurons (41). DRP1 also has GTPase activity and
serves a critical role in mitochondrial fission (42). In addition, the increase in
[Ca2+]c promotes the translocation of DRP1 to
mitochondria in cardiomyocytes and in cadmium-treated human and rat
liver cells (43,44). Mitochondrial fission events have
been located at the MAM compartment (26), where Ca2+ is a critical
regulator of mitochondrial morphology since mitochondrial
Ca2+ overload leads to mitochondrial fragmentation,
whereas mitochondrial Ca2+ depletion results in
mitochondrial hyperfusion (45).
Mitochondria depend on microtubules for their
intracellular motility via a Ca2+-regulated mechanism.
This movement capacity of mitochondria allows appropriate
distribution of these organelles for local ATP supply and
Ca2+ buffering. Using this movement, mitochondria is
settled in perinuclear clusters or in clusters near the cytoplasmic
membrane. In this regard, the KHC/Milton/Miro complex regulates
Ca2+-dependent mitochondrial motility. The Miro protein
joins to the OMM. Miro contains a pair of EF-hand
Ca2+-binding motifs that will permit interaction with
the motor domain of kinesin-1 through the Milton adaptor protein
(Fig. 1) (46). Calcium-dependent arrest of
mitochondrial motility is required to position mitochondria where
the cell needs Ca2+-buffering or an increased ATP
supply, as has been observed in neurons and other cells (46,47).
For example, motility of astrocytic mitochondria is inversely
related to intracellular calcium concentrations
([Ca2+]i); elevated
[Ca2+]i immobilizes astrocytic mitochondria,
whereas low [Ca2+]i under resting conditions
causes mitochondria to be highly mobile (47). The mitochondrial fission-fusion
machinery is also associated with the mitochondrial motility
machinery.
Ca2+ participation in mitochondrial
dynamics also includes mitophagy, which is a highly specialized
process that removes dysfunctional mitochondria. This has been
observed in neurodegenerative diseases, and calcium dysregulation
and ROS production are potential mitophagy inducers (48,49).
Mitophagy protects cells from damage that could occur from a
disordered mitochondrial metabolism. Phosphatase and tensin homolog
deleted on chromosome 10-induced kinase 1 (PINK1) is a
mitochondrial protein that preserves mitochondrial integrity, and
also participates in mitophagy activation. Upon mitochondrial
depolarization, PINK1 and beclin 1 (BECN1) selectively relocalize
to the surface of the damaged mitochondria, particularly at the MAM
contact. This anchoring of PINK1 and BECN1 recruits proteins, such
as ubiquitin and PARK2/Parkin, giving rise to autophagosome
formation, thus resulting in mitophagy of damaged mitochondria
(50). PINK1 localized at MAM also
modulates mitochondrial calcium levels (48,50).
5. Calcium signals control cell migration
and apoptosis
In migrating eukaryotic cells, calcium signals
control cell migration through the regulation of forward movement
and cell adhesion (2). For
example, Ca2+-influx channels regulate trailing edge
contraction in the migration of growth factor-induced fibroblast
cells and spontaneously polarized macrophages (1,51).
In particular, inhibition of the extracellular Ca2+
influx in RAW macrophages induces the loss of phosphoinositide
3-kinase (PI3K) activity at the leading edge, disassembly of
filamentous actin (F-actin) and cessation of ruffling.
Ca2+-sensitive enzyme protein kinase Ca (PKCa), which is
an actin regulatory protein, is enriched at the leading edge of
macrophages. This PKCa enrichment is affected by blockade of
Ca2+ influx, inhibition of PI3K activity or F-actin
depolymerization (51). PKCa can
synergistically regulate migration by phosphorylating myosin, by
regulating the cytoskeleton or by turning over integrin complexes
(2).
Calcium signals, in addition to regulating forward
movement, can also control cell adhesion. Pulsatile front
retraction and cell adhesion of human umbilical vein endothelial
cells are locally supported by Ca2+ pulses near the
leading edge caused by ER Ca2+ depletion and stromal
interaction molecule 1 (STIM1) activation. STIM1 likely acts
locally on adhesion by enhancing Ca2+ influx and
reloading ER Ca2+ stores in the front of the cell to
permit local Ca2+ pulses (Fig. 1) (2). Cancer cell migration will be
described below.
Ca2+ transfer between the ER and
mitochondria represents a critical signal in the induction of
apoptosis (28). One of the
best-characterized mechanisms is via the p53 pathway. The p53
protein is a master tumor-suppressor protein that promotes
apoptosis through two mechanisms: As a transcription factor, p53
controls cell-death programs within the nucleus (52), or through a
Ca2+-dependent mechanism modeling MAM cross talk. The
p53 protein interacts with sarco/ER Ca2+-ATPase (SERCA)
pumps causing an alteration in their oxidative state. This leads to
an increased Ca2+ load that is transferred directly to
the mitochondria that causes changes in mitochondrial morphology
and induction of apoptosis (28).
Depolarization of the mitochondria triggered by high
Ca2+ levels is followed by a decrease in ATP synthesis,
leading to cell death (39).
Morphological alterations in mitochondria caused by Ca2+
overload result in the opening of PTPs, mitochondrial fragmentation
and cytochrome c release, which are all factors that lead to
apoptotic cell death (28).
6. Calcium interplay is remodeled by cancer:
Alterations in MAM and mitochondrial dynamics
Cancer alters the architecture of MAM to
support survival and aggressiveness
The present review discussed the mechanisms where
the role of Ca2+ is essential for sustaining cellular
homeostasis. However, in cancer, increasing evidence has revealed a
link between alterations in mitochondrial Ca2+
homeostasis and malignant phenotypes, including cell survival,
invasiveness, migration, apoptosis evasion and drug resistance
(53,54).
Notably, MAM domains are involved in various
important processes in tumor cells that allow their survival,
including adhesion, motility, invasion, metastasis, autophagy and
apoptosis (28,55,56).
Cancer cells remodel MAM architecture by modifying the expression
levels of ER and mitochondrial Ca2+ channel proteins,
their associated binding partners, or their regulators (53). For example, in HeLa cells, a
detailed microscopy study demonstrated that a fraction of the
mitochondrial pool is in close contact with the ER cisternae in
these highly localized MAM domains (12). These contacts are also increased
between mitochondria and ER in prostate cancer cells that have
acquired cancer stem-like properties (57).
Numerous tumor suppressor proteins have been
demonstrated to be localized to the ER and MAMs where they regulate
cell death (Table I). Some of
these suppressor proteins directly affect calcium flux; for
example, silencing of ER oxidoreductin-1α significantly inhibits
calcium release from the ER, inducing cell death (58). A reduction in the
Bcl-2/Bcl-2-associated X, apoptosis regulator ratio also leads to
ER Ca2+ depletion, which induces cytosolic
Ca2+ overload and causes the loss of mitochondrial
membrane potential (59).
 | Table ITumor suppressor proteins and
oncoproteins that localize to the mitochondria-associated
membrane. |
Table I
Tumor suppressor proteins and
oncoproteins that localize to the mitochondria-associated
membrane.
| A, Tumor suppressor
proteins | | |
|---|
| First author,
year | Name | Reference |
|---|
| Raturi et
al, 2016 | Thioredoxin-related
transmembrane protein | (101) |
| Nutt et al,
2002; Scorrano et al, 2003 | Bcl-2-associated X,
apoptosis regulator | (102,103) |
| Nutt et al,
2002; Scorrano et al, 2003 | Bcl-2
antagonist/killer 1 | (102,103) |
| Echeverry et
al, 2013 | Bcl-2 related
protein ovarian killer | (104) |
| Giorgi et
al, 2010 | Promyelocytic
leukemia protein | (105) |
| Bononi et
al, 2013 | Phosphatase and
tensin homolog deleted on chromosome 10 | (106) |
| Verfaillie et
al, 2012 | RNA-dependent
protein kinase-like ER kinase | (107) |
| Li et al,
2008 | ER-associated
apoptosis-involved protein | (108) |
| Seervi et
al, 2013 | ER
oxidoreductin-1α | (58) |
| Iwasawa et
al, 2011 | Fission,
mitochondrial 1 | (109) |
| Iwasawa et
al, 2011 | B cell
receptor-associated protein 31 | (109) |
| Namba et al,
2013 | Cell death inducing
p53 target 1 | (110) |
|
| B, Anti-apoptotic
molecules | | |
|
| Name | Reference |
|
| Betz et al,
2013 | Mammalian target of
rapamycin complex 2 | (55) |
| Monaco et
al, 2015 | Bcl-2 | (62) |
IP3R-3 is an oncoprotein differentially
expressed in colorectal carcinoma that is absent in normal
colorectal mucosa (Fig. 2).
Increased expression of IP3R-3 is strongly correlated
with metastasis and decreased 5-year survival (60). In addition, IP3R-3, but
not the type 1 or 2 isoforms, is overexpressed in gastric cancer
cell lines established from malignant ascites (61). Conversely, IP3R-3 is
involved in mediating pro-apoptotic Ca2+ signals from
the ER to mitochondria in HeLa cells (62). Although IP3R-3
colocalizes more closely with mitochondria more efficiently
transmitting Ca2+ signals, in colorectal cancer,
IP3R-3 expression inhibits rather than promotes
apoptosis, possibly by participating in a localized signaling
domain mechanism that remains to be clarified (60).
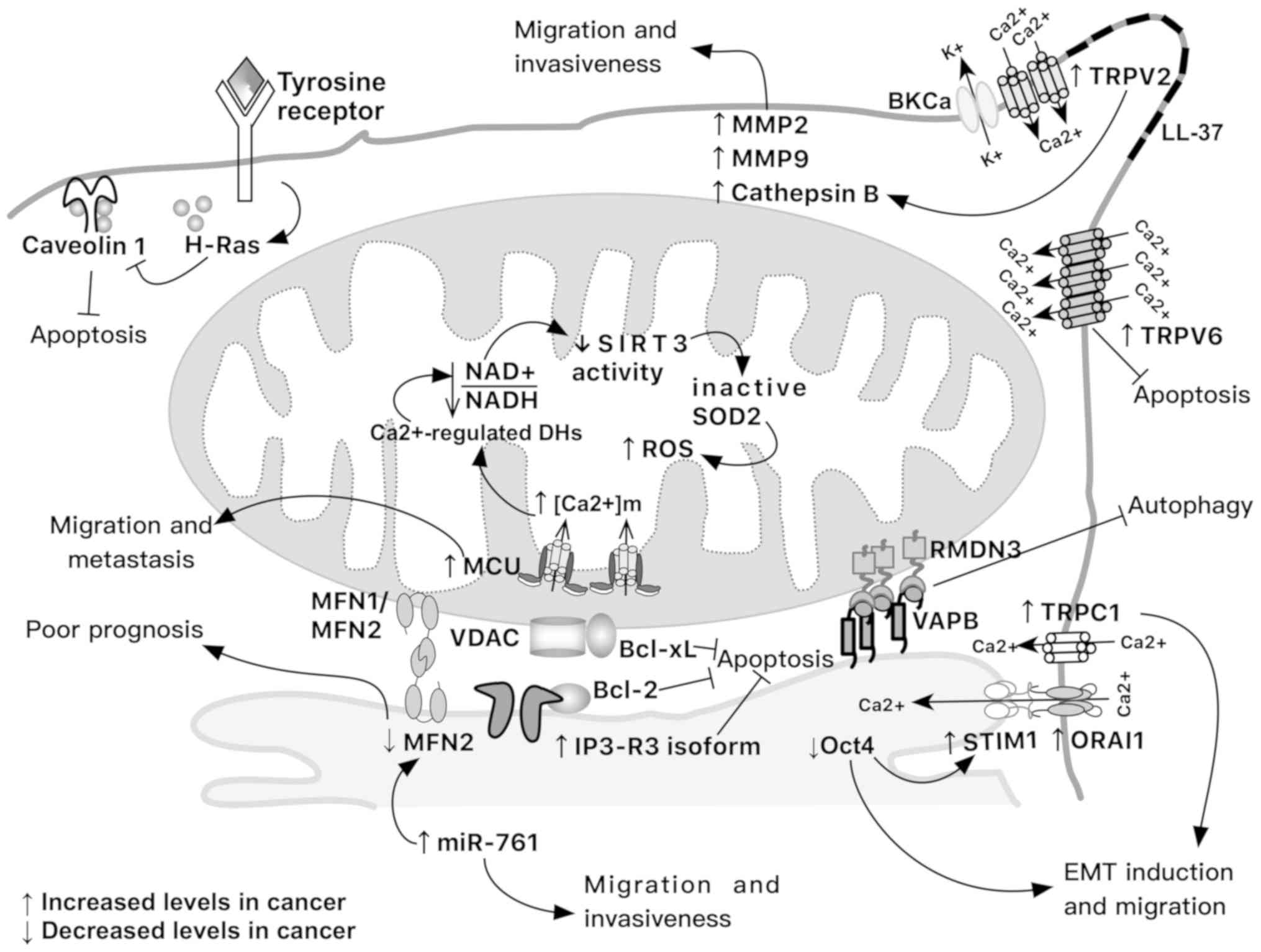 | Figure 2Tumor modifications in mitochondrial
calcium interplay that support invasiveness, migration, apoptosis
evasion and induction of epithelial-to-mesen-chymal transition.
Note that not all these modifications occur at the same time and in
the same tumor. MMP, matrix metalloproteinase; BKCa,
large-conductance Ca2+-activated K+ channel;
DH, dehydrogenase; EMT, epithelial-to-mesenchymal transition; Oct4,
octamer-binding transcription factor 4; SIRT3, sirtuin 3; SOD2,
superoxide dismutase 2; TRPC1, transient receptor potential cation
channel C1; TRPV, transient receptor potential cation channel
subfamily vanilloid. |
Some anti-apoptotic molecules are also localized to
the MAM and modulate cell death (Table
I). mTOR complex 2 (mTORC2) controls MAM integrity and
mitochondrial function via protein kinase B (AKT)-mediated
phosphorylation of IP3R and hexokinase 2 (55). In non-small cell lung carcinoma
(NSCLC) cells resistant to erlotinib, a tyrosine kinase inhibitor
(TKI), epidermal growth factor receptor (EGFR)-TKI resistance is
reduced in response to rituximab, an anti-cluster of
differentiation 20 monoclonal antibody. Briefly, NSCLC cells that
carry the EGFR T790M-mutation are resistant to erlotinib treatment;
however, the addition of rituximab significantly inhibits
proliferation and cell survival. The proposed mechanism involves a
reduction in the phosphorylation of EGFR, mTOR and mTORC2, which is
associated with inhibition of mTORC2 localization to MAM;
consequently, cell growth is inhibited (63).
Bcl-2 and Bcl-extra large (Bcl-XL) proteins exert
part of their anti-apoptotic functions by directly binding to
ER-localized IP3Rs and to OMM-localized VDAC1,
respectively. The joining of Bcl-2 and Bcl-XL to these
Ca2+ transport systems will negatively affect
Ca2+ transfer and apoptosis (Fig. 2) (62).
Calcium levels affect mitochondrial
dynamics
To satisfy cellular demands, mitochondria
continually change their shape and function; therefore, the
combined actions of fusion and fission determine the balance
between oxidative and glycolytic metabolisms. In cancer, the
ability of tumor cells to survive has been associated with
unbalanced mitochondrial dynamics, as it has been demonstrated in
lung, pancreatic, breast and oncocytic thyroid cancers, among
others (64-67). Notably, feedback regulation has
been established between calcium signaling and mitochondrial
dynamics to coordinate regulation of numerous tumor cellular
processes (68). For example, in
melanoma cells treated with three compounds, a mitochondrial
Na+/Ca2+ exchanger inhibitor (CGP-37157) and
two OXPHOS inhibitors (antimycin A and FCCP), a fast and persistent
rise in [Ca2+]m is detected. These compounds
increase tumor necrosis factor-related apoptosis-inducing ligand
(TRAIL) sensitivity, switching from apoptosis to nonapoptotic cell
death. The [Ca2+]m overload increases
mitochondrial fragmentation, whereas [Ca2+]m
removal induces mitochondrial hyperfusion. Notably, both actions
affecting mitochondrial dynamics exacerbate TRAIL-induced
mitochondrial rearrangements and cytotoxicity. This study indicated
that an appropriate level of mitochondrial calcium is essential for
maintaining mitochondrial dynamics associated with tumor cell
survival (45).
In liver cancer cells, increased
[Ca2+]c promotes mitochondrial fission by
upregulating DRP1 and fission, mitochondrial 1 genes via nuclear
factor of activated T cells 2 (NFATC2) and c-Myc activation,
respectively. NFATC2 is a calcium responsive transcription factor
that it is overactivated in several types of cancers (68).
MFN2 is a protein that promotes mitochondrial
fusion. This molecule is downregulated in patients with liver
cancer and low levels of MFN2 are associated with a poorer
prognosis. Notably, MFN2 expression is lower in tumor tissues than
in adjacent noncancer tissues. In vitro upregulation of MFN2
in the liver cancer cell line HepG2 triggers Ca2+ influx
from the ER into mitochondria, increasing
[Ca2+]m, ROS production and reducing
mitochondrial membrane potential; all these phenomena lead to tumor
cell apoptosis. These findings highlight the role of MFN2 as a
tumor suppressor protein (69).
Furthermore, it has been described that microRNA (miR)-761 binds
the 3′-untranslated region of MFN2. The upregulation of miR-761 is
associated with MFN2 downregulation; therefore, it may support cell
proliferation, cell migration and invasiveness of liver tumors.
Inhibition of miR-761 affects mitochondrial function and inhibits
cell proliferation, migration and invasion of liver cancer in
vivo and in vitro (Fig.
2) (70).
7. Mitochondrial calcium and mitoflashes in
epithelial-to-mesenchymal transition
Ca2+ signals regulate proliferation in a
spatial- and temporal-dependent manner. Whereas nuclear
Ca2+ signals may regulate cell proliferation, the
increase of [Ca2+]m may regulate apoptosis in
epithelial cells (62,71). Nevertheless, calcium signaling may
support loss of the balance between cell proliferation and
apoptosis, favoring cellular proliferation and inhibiting apoptosis
in cancer. Enhanced cytosolic Ca2+ signaling has been
suggested to contribute to the transition from normal colonic
epithelial cells to adenoma, which finally transforms to carcinoma
(60). The
epithelial-to-mesenchymal transition (EMT) is a critical process
during progression of tumor metastasis (72). The loss of cell-cell adhesion is
promoted by the transformation of polarized epithelial cells into
highly motile mesenchymal cells (73). In MCF-7 breast cancer cells,
inhibition of the Oct4 transcription factor or treatment with
transforming growth factor β1 (TGFβ1) promotes migration and
invasion. This EMT induction is accompanied by upregulation of
STIM1 and ORAI calcium release-activated calcium modulator 1
(Orai1), two major components of store-operated Ca2+
entry channels (SOCE). STIM1 serves as a Ca2+-sensor,
and Orai1 is an essential pore-forming component of SOCE channel
(Fig. 2) (74). Blockage of Ca2+ influx
by silencing STIM1 partially rescues the EMT initiated by Oct4
downregulation, indicating that other calcium channels possibly
take part in increased calcium influx (72). The SOCE pathway is activated by the
release of Ca2+ from the ER that causes the
oligomerization of STIM1. STIM1 binds the plasma membrane
Ca2+ channel Orai, which causes it to open (75). Nevertheless, this mechanism
requires functional mitochondria and the inhibition of MCU slowly
deactivates SOCE (6).
In addition to TGFβ, EGF is associated with EMT
induction in the breast cancer cell line MDA-MB-468, through
upregulating the mRNA expression levels of the ATP-binding cassette
C3 (ABCC3). The EGF-induced transcriptional upregulation of ABCC3
is dependent on calcium signals. This calcium-dependent regulation
of ABCC3 occurs via the calcium permeable ion channel transient
receptor potential cation channel C1 (TRPC1) (76). Furthermore, a previous study
suggested that the TRPC1 channel contributes to Orai1-mediated
Ca2+ influx in the EMT of MDA-MB-468 cells (Fig. 2) (77). Additionally, TRPC1-mediated calcium
influx has been linked to EGF-stimulated EGFR activation in
non-small cell lung cancer cells by promoting cellular
proliferation (78).
Mitochondrial calcium levels also regulate certain
discrete and stochastic mitochondrial events called mitoflashes,
which comprise a burst of superoxide production accompanied by
transient depolarization and alkalization in the mitochondrial
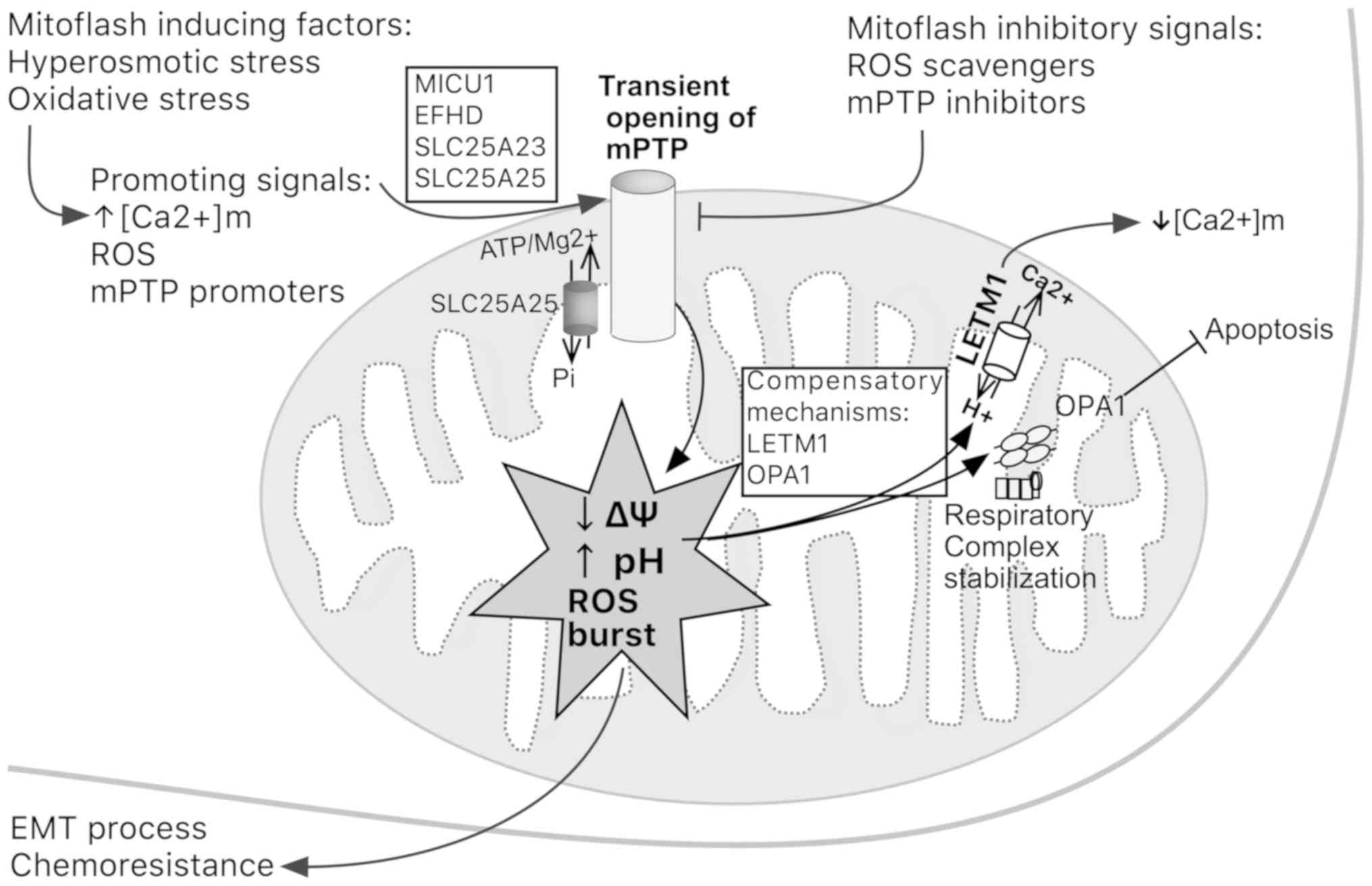
matrix (79). Transient opening of
the mitochondrial PTP (mPTP) seems to precede the mitoflash.
Mitoflash biogenesis can be activated by
[Ca2+]m increase, ROS and mPTP promoters, but
suppressed by ROS scavengers and mPTP inhibitors (80). Hyperosmotic or oxidative stress and
ROS-dependent apoptosis are associated with an increased frequency
of mitoflashes, which arises as a bioindicator of cellular
dysfunction (81). The frequency
of mitoflashes has been reported to transiently increase during the
early stages of somatic cell reprogramming, establishing a link
between the role of mitoflashes and the acquisition of pluripotency
(82). Other factors that may
modulate mitoflash occurrence rate are aging and cancer (80).
Proteins such as EF-hand domain family member D
(EFHD), solute carrier family 25 member 25 (SLC25A25), solute
carrier family 25 member 23 and MICU1 are considered mitoflash
activators. Notably, all of them contain Ca2+-binding
motifs. In addition, LETM1 is considered a mitoflash inhibitor that
responds to hyperosmotic stress (Fig.
3). Recently, optic atrophy 1 protein (OPA1) has emerged as a
regulator of the occurrence of mitoflashes, which is an independent
process from fusion; OPA1 stabilizes respiratory chain
supercomplexes in a conformation that enables mitochondria to
compensate a fall in mitochondrial membrane potential by an
explosive-pH flash (Fig. 3)
(81).
EFHD1 has been involved in the alteration of
mitochondrial function to control cell survival or neural
differentiation. EFHD1 is also known as mitocalcin and has
Ca2+ binding characteristics; this molecule acts as a
Ca2+ sensor enhancing mitoflash responses (79). Another example is SLC25A25, which
functions as a Ca2+-regulated shuttle of
ATP-Mg2+ and pyrophosphate across the IMM. SLC25A25 has
been reported to be upregulated by 3.2-fold in the RC77T/E human
prostate tumor cell line (83). In
this sense, the expression of the long noncoding RNA relating to
SLC25A25 (SLC25A25-AS1), which inhibits SLC25A25 translation, is
significantly decreased in tumor tissues and serum of patients with
colorectal cancer. In vitro overexpression of SLC25A25-AS1
significantly inhibits proliferation and colony formation in
colorectal cancer cell lines; in addition, downregulation of
SLC25A25-AS1 enhances chemoresistance and promotes the EMT process
(84).
8. Calcium: A critical regulator of tumor
migration, invasiveness and metastasis
Ca2+ signals regulate polarization,
speed and the direction of movement of migrating cells. Cancer
cells migrate by applying rearward forces against extracellular
media. Freely migrating cancer cells move by inducing the formation
of lamellipodia that are the extension of leading edge protrusions
(85). Contractility-driven bleb
formation has recently emerged as another mechanism of motility in
mesenchymal precursors (86). Two
sublines of WC256 carcinosarcoma cells (LC and BC) were transformed
to observe mesenchymal migration and ameboid migration, and to find
differences in electrotactic motility. LC and BC WC256 cells can
efficiently drive electrotactic migration by forming lamellipodia
and membrane blebs, respectively (87). Lamellipodia attach to the
substratum, and contraction of the trailing rear edge moves the
cell toward the lamellipodia. Blebs, on the other hand, are
cellular protrusions expanded by hydrostatic pressure generated by
a contractile actomyosin cortex. Bleb location appears to be
directly controlled by the Rho pathway and by alterations in
intracellular Ca2+ concentrations (87). Membrane rigidity is increased at
the leading edge of pseudopodia and retracting tail in breast
cancer cell lines by locally binding LL-37 to cytoplasmic membrane.
LL-37 is a peptide released from the C-terminus of the human
cathelicidin antimicrobial protein hCAPT18. LL-37 membrane binding
activates PI3K/AKT signaling. Subsequently, AKT induces the
recruitment of the transient receptor potential cation channel
subfamily vanilloid member 2 (TRPV2) transporter from intracellular
vesicles to plasma membrane of pseudopodia (Fig. 2). TRPV2 increases calcium that is
accompanied by K+ efflux through the BKCa channel
(88). These local Ca2+
signals govern forward movement by regulating lamellipodia
retraction and strengthen local adhesion to the extracellular
matrix. Each Ca2+ pulse triggers contraction of actin
filaments that activates myosin light-chain kinase and myosin II
behind the leading edge (85).
Activation of Ca2+-dependent kinases can also indirectly
influence actin dynamics (51).
MCU upregulation significantly increases migration
in vitro in MCF-7 breast cancer cells. Consistently, MCU
protein levels are increased in biopsy samples from breast cancer
patients who present with metastasis (Fig. 2) (89). Blocking Ca2+ influx
using Ni2+ inhibits migration of mouse 4T1 and human
MDA-MB-231 breast tumor cells, highlighting a link between
Ca2+ influx and migration (1).
Metastasis is a multistep process where cancer
cells from a primary tumor site spread to distant organs and form
new tumors, and calcium is a critical regulator of this process
(74,90). Using matrix metalloproteinases
(MMPs) and cathepsins, invasive cells degrade surrounding tissue,
and Ca2+ influx can affect the activity of these
enzymes. In human prostate cancer, the development and progression
of cancer to an aggressive castration-resistant phenotype are
characterized by de novo expression of the TRPV2 channel.
The role of TRPV2 is to maintain elevated cytosolic Ca2+
and to induce expression of MMP2, MMP9 and cathepsin B, enhancing a
cell migration-invasive phenotype (Fig. 2) (91).
The metastatic capacity of tumor cells is also
enhanced by upregulation of MCU expression. MCU increases
mitochondrial Ca2+ uptake, which promotes ROS
production, favoring a metabolic shift from oxidative to glycolytic
metabolism. For example, MCU overexpression promotes MMP2
expression and cell motility of liver cancer metastasis in a nude
mouse model, where intrahepatic and distal lung metastasis are
promoted through a ROS-activated c-Jun N-terminal kinase pathway
(54). Briefly, mitochondrial
Ca2+ elevation increases the activity of
Ca2+-sensitive dehydrogenases, which diminishes the
NAD+/NADH ratio. The NAD+-dependent
deacetylase activity of sirtuin 3 (SIRT3) deacetylates superoxide
dismutase 2 (SOD2), and the deacetylated and active form of SOD2
decreases ROS levels. However, low levels of NAD+,
caused by Ca2+-sensitive dehydrogenases, inhibit SIRT3
activity, and the inactive form of SOD2 allows accumulation of ROS,
which activates the c-Jun N-terminal kinase pathway (Fig. 2) (54). Furthermore,
[Ca2+]m levels are also increased in HepG2
cells that are exposed to high glucose levels (33 mM).
Nevertheless, when MCU is inhibited, high glucose levels induce
decreased generation of ROS and protection against inflammatory
effects (92). Silencing MCU in
cultured HeLa cells severely abrogates mitochondrial
Ca2+ uptake, whereas mitochondrial respiration and
membrane potential remain fully intact (17). Under these conditions, an oxidative
metabolism might be favored and cell motility could be
diminished.
The suppression of Ca2+ influx inhibits
the AKT pathway in breast cancer cells (4T1, TUBO-P2J and HCC70
cell lines), thus resulting in the prevention of migration
(90). Higher MCU expression has
also been detected in MDA-MB-231 cells compared with in MCF-7 cells
(89). MDA-MB-231 cells, which are
more invasive than other breast cancer cells (4), have an elevated
[Ca2+]m uptake that is essential for the
metabolic shift from oxidative to glycolytic metabolism that occurs
during cancer metastasis (89).
miR-340 can target and inhibit MCU, which consequently decreases
[Ca2+]m accumulation, metastatic cell
motility and the matrix invasiveness of breast cancer cells
(89). Additionally, STIM1 and
Orai1 inhibition reduces serum-induced in vitro cell
migration and in vivo metastasis formation of MDA-MB-231
breast cancer cells (74).
Molecular components of the SOCE also regulate breast cancer
metastasis by replenishing depleted ER Ca2+ reserves.
Inhibition of SERCA with cyclopiazonic acid or by agonist
stimulation with ATP (77), or
signaling mechanisms such as activated G-protein-coupled or
tyrosine kinase receptors (4), may
deplete ER Ca2+ stores. As further proof, inhibition of
MCU by ruthenium red or specific small interfering (si)RNAs induces
mitochondrial depolarization in MDA-MB-231 cells, which inhibits
SOCE through CRAC channels and abrogates cancer cell migration
(4).
9. Mitochondrial calcium in tumor apoptotic
failure and tumor autophagy
Tumor cells have developed mechanisms where
alterations in mitochondrial metabolism inhibit the intrinsic
apoptotic pathway. In this context, the oncoprotein Ras promotes
the failure of mitochondrial-dependent apoptosis, which induces
neoplasia via an interaction with caveolin-1 (Cav-1). This tumor
suppressor directly regulates Ca2+ influx machineries
and the Ca2+-dependent apoptotic pathway. Cav-1 is a
plasma membrane protein that also acts as a scaffolding protein for
signaling molecules, including Ca2+-signaling members
and Ras. Ras proteins are GTPases activated by receptor tyrosine
kinases that transduce extracellular signals to the nucleus. In
3T3NIH fibroblasts transformed with a retrovirus expressing the
oncogenic H-Ras (H-Ras12V), it was revealed that
H-Ras12V accumulates at the MAM and plasma
membrane-associated membranes (PAM), causing altered intracellular
Ca2+ homeostasis. This compartmentalization of H-Ras
compromises the cooperation between MAM and PAM, promoting the loss
of Cav-1 (Fig. 2) (56).
Another example includes BRAF mutations that can
reprogram melanoma metabolism. Briefly, BRAF inhibitors (BRAFi)
hamper glucose uptake prior to the complete elimination of melanoma
tumor cells. BRAFi-treated melanomas rapidly became
OXPHOS-dependent to survive. BRAFi induces increased mitochondrial
activity and biogenesis, and an increased capacity for
mitochondrial Ca2+ buffering associated with
mitochondrial network remodeling. A close interaction between the
ER and mitochondria in BRAFi-exposed melanoma facilitates
mitochondrial Ca2+ uptake after its release from the ER
that prevents ER-mediated cell death (93). Furthermore, the inhibition of
Ca2+ transfer from the ER to mitochondria may preserve
mitochondrial integrity and protect against
Ca2+-mediated apoptosis in human malignant pleural
mesothelioma. A severe dysregulation of Ca2+ signaling
is associated with resistance to apoptotic stimuli in mesothelioma
cell lines and short-term cell cultures obtained from patients with
malignant pleural mesothelioma (94).
The upregulation of some calcium transporters or
their regulators has also been associated with resistance to
apoptosis. Upregulation of MCU regulator 1 (MCUR1) has been
associated with increased survival of liver cancer cells. MCUR1
overexpression indirectly inhibits mitochondrial-dependent
intrinsic apoptosis due to AKT/MDM2 proto-oncogene-mediated p53
degradation by the elevated production of mitochondrial ROS
(95). Another example includes
TRPV6, which is a highly selective Ca2+ channel involved
in prostate carcinogenesis. Increased expression of TRPV6 is
strongly correlated with cancer progression and with the resistance
to apoptosis observed in hormone-sensitive prostate cancer cells
(Fig. 2) (96). These aforementioned studies refer
to the failure of apoptotic machinery that may favor phenotypes
that resist traditional chemotherapy treatments.
Autophagy is a conserved process for the delivery
of cellular material to lysosomes for degradation. This process
recycles molecules for the synthesis of proteins, nucleic acids,
lipids and carbohydrates, or prevents accumulation of protein
aggregates and damaged organelles, maintaining cellular homeostasis
(30). Intracellular levels of
calcium have also been identified as a potential regulator of
autophagy. This process can be induced by rapamycin and Torin 1;
both molecules selectively inhibit mTOR. Alternatively, starvation
can induce autophagy; this process is more complex but it also
involves mTOR and various upstream nutrient-sensing molecules,
including AMPK and calcium/calmodulin-dependent protein kinase
kinase β (CAMKK-β) (13). In HeLa
cells, Ca2+ mobilizing agents (ionomycin, ATP and
thapsigargin) stimulate autophagy via CAMKK, which directly
activates AMPK to inhibit mTOR (97). In addition, Ca2+
signaling promotes increased mitochondrial fission that leads to
autophagy in liver cancer cells (68).
Previous studies have reported that disruption of
IP3R-mediated Ca2+ delivery stimulates
autophagy in various types of cancer (98,99).
In this context, vesicle-associated membrane protein-associated
protein B (VAPB), an integral ER protein, binds to the regulator of
microtubule dynamics 3 (RMDN3), an OMM protein, to form one set of
tethers. Under non-stressing conditions, VAPB and RMDN3
overexpression favors the tether between the ER and mitochondria,
thus inhibiting autophagy (Fig.
1); however, blocking the ER-mitochondria Ca2+
exchange using MCU-targeted antagonist siRNAs abrogates the effect
of VAPB and RMDN3 overexpression, favoring autophagy. This
highlights the role of VAPB-RMDN3 in ER-mitochondria
Ca2+ delivery (13).
Paradoxically, starvation-induced autophagy requires
Ca2+ release from sensitized IP3Rs (30), thus suggesting that the autophagy
process depends on spatial and temporal parameters of
Ca2+ signaling, in addition to nutrient status and the
availability of growth factors (99). Autophagy favors cancer development
due to the energetic supply provided by organelle degradation
typically driven by this process, which allows tumor cells to
become resistant to stressing conditions, such as anticancer
therapies. Therefore, autophagy inhibition has emerged as a
potential cancer treatment strategy (100).
10. Conclusions
Through several mechanisms, mitochondria serve an
essential role in maintaining cellular calcium levels. These
processes are meticulously regulated because calcium acts as a
trigger of regulatory cascades that affect the mitochondria
themselves and, in consequence, metabolism, migration and cell
survival.
Tumor cells may remodel the systems that maintain
cellular calcium homeostasis, in order to promote their survival
and metastasis. Understanding the integral metabolic importance of
Ca2+ signaling in tumor cells, which includes the
participation of various organelles, will allow the development of
numerous therapies that may benefit patients with cancer.
Funding
No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
SRG conceived, designed and wrote the manuscript.
HPG was involved in writing and critically reviewing the
manuscript, and figure design.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Authors' information
Dr Susana Romero-Garcia, Department of
Chronic-Degenerative Diseases, National Institute of Respiratory
Diseases ‘Ismael Cosío Villegas’, Tlalpan 4502, Col. Sección XVI,
CP 14080 Mexico City, Mexico. Phone: (+52-55) 5487-1703; Fax:
(+52-55) 5665-4623. ORCID: 0000-0003-4539-0578. Dr Heriberto
Prado-Garcia, Department of Chronic-Degenerative Diseases, National
Institute of Respiratory Diseases ‘Ismael Cosío Villegas’, CP 14080
Mexico City, Mexico. ORCID: 0000-0002-4932-3244
Acknowledgments
Not applicable.
References
|
1
|
Yang S and Huang XY: Ca2+
influx through L-type Ca2+ channels controls the
trailing tail contraction in growth factor-induced fibroblast cell
migration. J Biol Chem. 280:27130–27137. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tsai FC, Seki A, Yang HW, Hayer A,
Carrasco S, Malmersjö S and Meyer T: A polarized Ca2+,
diacylglycerol and STIM1 signalling system regulates directed cell
migration. Nat Cell Biol. 16:133–144. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xiong J, Camello PJ, Verkhratsky A and
Toescu EC: Mitochondrial polarisation status and
[Ca2+]i signalling in rat cerebellar granule
neurones aged in vitro. Neurobiol Aging. 25:349–359. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tang S, Wang X, Shen Q, Yang X, Yu C, Cai
C, Cai G, Meng X and Zou F: Mitochondrial Ca2+ uniporter
is critical for store-operated Ca2+ entry-dependent
breast cancer cell migration. Biochem Biophys Res Commun.
458:186–193. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen L, Sun Q, Zhou D, Song W, Yang Q, Ju
B, Zhang L, Xie H, Zhou L, Hu Z, et al: HINT2 triggers
mitochondrial Ca2+ influx by regulating the
mitochondrial Ca2+ uniporter (MCU) complex and enhances
gemcitabine apoptotic effect in pancreatic cancer. Cancer Lett.
411:106–116. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Deak AT, Blass S, Khan MJ, Groschner LN,
Waldeck- Weiermair M, Hallström S, Graier WF and Malli R:
IP3-mediated STIM1 oligomerization requires intact mitochondrial
Ca2+ uptake. J Cell Sci. 127:2944–2955. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Imbert N, Cognard C, Duport G, Guillou C
and Raymond G: Abnormal calcium homeostasis in Duchenne muscular
dystrophy myotubes contracting in vitro. Cell Calcium. 18:177–186.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Budd SL and Nicholls DG: A reevaluation of
the role of mitochondria in neuronal Ca2+ homeostasis. J
Neurochem. 66:403–411. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hartmann J and Verkhratsky A: Relations
between intracellular Ca2+ stores and store-operated
Ca2+ entry in primary cultured human glioblastoma cells.
J Physiol. 513:411–424. 1998. View Article : Google Scholar
|
|
10
|
Hoth M, Button DC and Lewis RS:
Mitochondrial control of calcium-channel gating: A mechanism for
sustained signaling and transcriptional activation in T
lymphocytes. Proc Natl Acad Sci USA. 97:10607–10612. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jouaville LS, Pinton P, Bastianutto C,
Rutter GA and Rizzuto R: Regulation of mitochondrial ATP synthesis
by calcium: Evidence for a long-term metabolic priming. Proc Natl
Acad Sci USA. 96:13807–13812. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rizzuto R, Pinton P, Carrington W, Fay FS,
Fogarty KE, Lifshitz LM, Tuft RA and Pozzan T: Close contacts with
the endoplasmic reticulum as determinants of mitochondrial
Ca2+ responses. Science. 280:1763–1766. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gomez-Suaga P, Paillusson S, Stoica R,
Noble W, Hanger DP and Miller CC: The ER-mitochondria tethering
complex VAPB-PTPIP51 regulates autophagy. Curr Biol. 27:371–385.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Báthori G, Csordás G, Garcia-Perez C,
Davies E and Hajnóczky G: Ca2+-dependent control of the
permeability properties of the mitochondrial outer membrane and
voltage-dependent anion- selective channel (VDAC). J Biol Chem.
281:17347–17358. 2006. View Article : Google Scholar
|
|
15
|
Tekmen M and Gleason E: Multiple
Ca2+-dependent mechanisms regulate L-type
Ca2+ current in retinal amacrine cells. J Neurophysiol.
104:1849–1866. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hiester BG, Bourke AM, Sinnen BL, Cook SG,
Gibson ES, Smith KR and Kennedy MJ: L-type voltage-gated
Ca2+ channels regulate synaptic-activity-triggered
recycling endosome fusion in neuronal dendrites. Cell Rep.
21:2134–2146. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baughman JM, Perocchi F, Girgis HS,
Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L,
Goldberger O, Bogorad RL, et al: Integrative genomics identifies
MCU as an essential component of the mitochondrial calcium
uniporter. Nature. 476:341–345. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tsai CW, Wu Y, Pao PC, Phillips CB,
Williams C, Miller C, Ranaghan M and Tsai MF: Proteolytic control
of the mitochondrial calcium uniporter complex. Proc Natl Acad Sci
USA. 114:4388–4393. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen W, Yang J, Chen S, Xiang H, Liu H,
Lin D, Zhao S, Peng H, Chen P, Chen AF, et al: Importance of
mitochondrial calcium uniporter in high glucose-induced endothelial
cell dysfunction. Diab Vasc Dis Res. 14:494–501. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Luongo TS, Lambert JP, Gross P, Nwokedi M,
Lombardi AA, Shanmughapriya S, Carpenter AC, Kolmetzky D, Gao E,
van Berlo JH, et al: The mitochondrial
Na+/Ca2+ exchanger is essential for
Ca2+ homeostasis and viability. Nature. 545:93–97. 2017.
View Article : Google Scholar
|
|
21
|
Roy S, Dey K, Hershfinkel M, Ohana E and
Sekler I: Identification of residues that control Li+
versus Na+ dependent Ca2+ exchange at the
transport site of the mitochondrial NCLX. Biochim Biophys Acta Mol
Cell Res. 1864:997–1008. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Austin S, Tavakoli M, Pfeiffer C, Seifert
J, Mattarei A, De Stefani D, Zoratti M and Nowikovsky K:
LETM1-mediated K+ and Na+ homeostasis
regulates mitochondrial Ca2+ efflux. Front Physiol.
8:8392017. View Article : Google Scholar
|
|
23
|
Melchionda M, Pittman JK, Mayor R and
Patel S: Ca2+/H+ exchange by acidic
organelles regulates cell migration in vivo. J Cell Biol.
212:803–813. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shao J, Fu Z, Ji Y, Guan X, Guo S, Ding Z,
Yang X, Cong Y and Shen Y: Leucine zipper-EF-hand containing
transmembrane protein 1 (LETM1) forms a
Ca2+/H+ antiporter. Sci Rep. 6:341742016.
View Article : Google Scholar
|
|
25
|
Koshiba T, Detmer SA, Kaiser JT, Chen H,
McCaffery JM and Chan DC: Structural basis of mitochondrial
tethering by mitofusin complexes. Science. 305:858–862. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ausman J, Abbade J, Ermini L, Farrell A,
Tagliaferro A, Post M and Caniggia I: Ceramide-induced BOK promotes
mitochondrial fission in preeclampsia. Cell Death Dis. 9:2982018.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gutiérrez T, Parra V, Troncoso R, Pennanen
C, Contreras- Ferrat A, Vasquez-Trincado C, Morales PE,
Lopez-Crisosto C, Sotomayor-Flores C, Chiong M, et al: Alteration
in mitochondrial Ca(2+) uptake disrupts insulin signaling in
hypertrophic cardio-myocytes. Cell Commun Signal. 12:682014.
|
|
28
|
Giorgi C, Bonora M, Sorrentino G,
Missiroli S, Poletti F, Suski JM, Galindo Ramirez F, Rizzuto R, Di
Virgilio F, Zito E, et al: p53 at the endoplasmic reticulum
regulates apoptosis in a Ca2+-dependent manner. Proc
Natl Acad Sci USA. 112:1779–1784. 2015. View Article : Google Scholar
|
|
29
|
Park SJ, Lee SB, Suh Y, Kim SJ, Lee N,
Hong JH, Park C, Woo Y, Ishizuka K, Kim JH, et al: DISC1 modulates
neuronal stress responses by gate-keeping ER-mitochondria
Ca2+ transfer through the MAM. Cell Rep. 21:2748–2759.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Decuypere JP, Welkenhuyzen K, Luyten T,
Ponsaerts R, Dewaele M, Molgó J, Agostinis P, Missiaen L, De Smedt
H, Parys JB, et al: Ins(1,4,5)P3 receptor-mediated Ca2+
signaling and autophagy induction are interrelated. Autophagy.
7:1472–1489. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hur YS, Kim KD, Paek SH and Yoo SH:
Evidence for the existence of secretory granule (dense-core
vesicle)-based inositol 1,4,5-trisphosphate-dependent
Ca2+ signaling system in astrocytes. PLoS One.
5:e119732010. View Article : Google Scholar
|
|
32
|
Furuichi T, Simon-Chazottes D, Fujino I,
Yamada N, Hasegawa M, Miyawaki A, Yoshikawa S, Guénet JL and
Mikoshiba K: Widespread expression of inositol 1,4,5-trisphosphate
receptor type 1 gene (Insp3r1) in the mouse central nervous system.
Receptors Channels. 1:11–24. 1993.PubMed/NCBI
|
|
33
|
Sugiyama T, Yamamoto-Hino M, Miyawaki A,
Furuichi T, Mikoshiba K and Hasegawa M: Subtypes of inositol
1,4,5-trisphosphate receptor in human hematopoietic cell lines:
Dynamic aspects of their cell-type specific expression. FEBS Lett.
349:191–196. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cárdenas C, Miller RA, Smith I, Bui T,
Molgó J, Müller M, Vais H, Cheung KH, Yang J, Parker I, et al:
Essential regulation of cell bioenergetics by constitutive InsP3
receptor Ca2+ transfer to mitochondria. Cell.
142:270–283. 2010. View Article : Google Scholar
|
|
35
|
Kennedy ED, Rizzuto R, Theler JM, Pralong
WF, Bastianutto C, Pozzan T and Wollheim CB: Glucose-stimulated
insulin secretion correlates with changes in mitochondrial and
cytosolic Ca2+ in aequorin-expressing INS-1 cells. J
Clin Invest. 98:2524–2538. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kennedy HJ, Pouli AE, Ainscow EK,
Jouaville LS, Rizzuto R and Rutter GA: Glucose generates sub-plasma
membrane ATP microdomains in single islet beta-cells. Potential
role for strategically located mitochondria. J Biol Chem.
274:13281–13291. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rutter GA, Burnett P, Rizzuto R, Brini M,
Murgia M, Pozzan T, Tavaré JM and Denton RM: Subcellular imaging of
intramitochondrial Ca2+ with recombinant targeted
aequorin: Significance for the regulation of pyruvate dehydrogenase
activity. Proc Natl Acad Sci USA. 93:5489–5494. 1996. View Article : Google Scholar
|
|
38
|
Territo PR, Mootha VK, French SA and
Balaban RS: Ca(2+) activation of heart mitochondrial oxidative
phosphorylation: Role of the F(0)/F(1)-ATPase. Am J Physiol Cell
Physiol. 278:C423–C435. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gizak A, Pirog M and Rakus D: Muscle
FBPase binds to cardiomyocyte mitochondria under glycogen synthase
kinase-3 inhibition or elevation of cellular Ca2+ level.
FEBS Lett. 586:13–19. 2012. View Article : Google Scholar
|
|
40
|
Wiśniewski J, Piróg M, Hołubowicz R,
Dobryszycki P, McCubrey JA, Rakus D and Gizak A: Dimeric and
tetrameric forms of muscle fructose-1,6-bisphosphatase play
different roles in the cell. Oncotarget. 8:115420–115433. 2017.
View Article : Google Scholar
|
|
41
|
Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y,
Tomizawa K, Nairn AC, Takei K, Matsui H and Matsushita M: CaM
kinase I alpha-induced phosphorylation of Drp1 regulates
mitochondrial morphology. J Cell Biol. 182:573–585. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ji WK, Hatch AL, Merrill RA, Strack S and
Higgs HN: Actin filaments target the oligomeric maturation of the
dynamin GTPase Drp1 to mitochondrial fission sites. eLife.
4:e115532015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xu S, Pi H, Chen Y, Zhang N, Guo P, Lu Y,
He M, Xie J, Zhong M, Zhang Y, et al: Cadmium induced
Drp1-dependent mitochondrial fragmentation by disturbing calcium
homeostasis in its hepatotoxicity. Cell Death Dis. 4:e5402013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pennanen C, Parra V, López-Crisosto C,
Morales PE, Del Campo A, Gutierrez T, Rivera-Mejías P, Kuzmicic J,
Chiong M, Zorzano A, et al: Mitochondrial fission is required for
cardiomyocyte hypertrophy mediated by a Ca2+-calcineurin
signaling pathway. J Cell Sci. 127:2659–2671. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ohshima Y, Takata N, Suzuki-Karasaki M,
Yoshida Y, Tokuhashi Y and Suzuki-Karasaki Y: Disrupting
mitochondrial Ca2+ homeostasis causes tumor-selective
TRAIL sensitization through mitochondrial network abnormalities.
Int J Oncol. 51:1146–1158. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang X and Schwarz TL: The mechanism of
Ca2+ -dependent regulation of kinesin-mediated
mitochondrial motility. Cell. 136:163–174. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kremneva E, Kislin M, Kang X and Khiroug
L: Motility of astrocytic mitochondria is arrested by
Ca2+-dependent interaction between mitochondria and
actin filaments. Cell Calcium. 53:85–93. 2013. View Article : Google Scholar
|
|
48
|
Gandhi S, Wood-Kaczmar A, Yao Z,
Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi
SJ, Wood NW, et al: PINK1-associated Parkinson's disease is caused
by neuronal vulnerability to calcium-induced cell death. Mol Cell.
33:627–638. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Dagda RK, Cherra SJ III, Kulich SM, Tandon
A, Park D and Chu CT: Loss of PINK1 function promotes mitophagy
through effects on oxidative stress and mitochondrial fission. J
Biol Chem. 284:13843–13855. 2009. View Article : Google Scholar
|
|
50
|
Gelmetti V, De Rosa P, Torosantucci L,
Marini ES, Romagnoli A, Di Rienzo M, Arena G, Vignone D, Fimia GM
and Valente EM: PINK1 and BECN1 relocalize at
mitochondria-associated membranes during mitophagy and promote
ER-mitochondria tethering and autophagosome formation. Autophagy.
13:654–669. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Evans JH and Falke JJ: Ca2+
influx is an essential component of the positive-feedback loop that
maintains leading-edge structure and activity in macrophages. Proc
Natl Acad Sci USA. 104:16176–16181. 2007. View Article : Google Scholar
|
|
52
|
Gottlieb TM, Leal JF, Seger R, Taya Y and
Oren M: Cross-talk between Akt, p53 and Mdm2: Possible implications
for the regulation of apoptosis. Oncogene. 21:1299–1303. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Missiroli S, Danese A, Iannitti T,
Patergnani S, Perrone M, Previati M, Giorgi C and Pinton P:
Endoplasmic reticulum- mitochondria Ca2+ crosstalk in
the control of the tumor cell fate. Biochim Biophys Acta Mol Cell
Res. 1864:858–864. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ren T, Zhang H, Wang J, Zhu J, Jin M, Wu
Y, Guo X, Ji L, Huang Q, Zhang H, et al: MCU-dependent
mitochondrial Ca2+ inhibits NAD+/SIRT3/SOD2
pathway to promote ROS production and metastasis of HCC cells.
Oncogene. 36:5897–5909. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Betz C, Stracka D, Prescianotto-Baschong
C, Frieden M, Demaurex N and Hall MN: Feature Article: mTOR complex
2-Akt signaling at mitochondria-associated endoplasmic reticulum
membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad
Sci USA. 110:12526–12534. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Rimessi A, Marchi S, Patergnani S and
Pinton P: H-Ras-driven tumoral maintenance is sustained through
caveolin-1-dependent alterations in calcium signaling. Oncogene.
33:2329–2340. 2014. View Article : Google Scholar
|
|
57
|
Matsumoto T, Uchiumi T, Monji K, Yagi M,
Setoyama D, Amamoto R, Matsushima Y, Shiota M, Eto M and Kang D:
Doxycycline induces apoptosis via ER stress selectively to cells
with a cancer stem cell-like properties: Importance of stem cell
plasticity. Oncogenesis. 6:3972017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Seervi M, Sobhan PK, Joseph J, Ann Mathew
K and Santhoshkumar TR: ERO1α-dependent endoplasmic
reticulum-mitochondrial calcium flux contributes to ER stress and
mitochondrial permeabilization by procaspase-activating compound-1
(PAC-1). Cell Death Dis. 4:e9682013. View Article : Google Scholar
|
|
59
|
Wu LF, Guo YT, Zhang QH, Xiang MQ, Deng W,
Ye YQ, Pu ZJ, Feng JL and Huang GY: Enhanced antitumor effects of
adenoviral-mediated siRNA against GRP78 gene on adenosine- induced
apoptosis in human hepatoma HepG2 cells. Int J Mol Sci. 15:525–544.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Shibao K, Fiedler MJ, Nagata J, Minagawa
N, Hirata K, Nakayama Y, Iwakiri Y, Nathanson MH and Yamaguchi K:
The type III inositol 1,4,5-trisphosphate receptor is associated
with aggressiveness of colorectal carcinoma. Cell Calcium.
48:315–323. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sakakura C, Hagiwara A, Fukuda K,
Shimomura K, Takagi T, Kin S, Nakase Y, Fujiyama J, Mikoshiba K,
Okazaki Y, et al: Possible involvement of inositol
1,4,5-trisphosphate receptor type 3 (IP3R3) in the peritoneal
dissemination of gastric cancers. Anticancer Res. 23:3691–3697.
2003.PubMed/NCBI
|
|
62
|
Monaco G, Decrock E, Arbel N, van Vliet
AR, La Rovere RM, De Smedt H, Parys JB, Agostinis P, Leybaert L,
Shoshan- Barmatz V, et al: The BH4 domain of anti-apoptotic Bcl-XL,
but not that of the related Bcl-2, limits the voltage-dependent
anion channel 1 (VDAC1)-mediated transfer of pro-apoptotic
Ca2+ signals to mitochondria. J Biol Chem.
290:9150–9161. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Xu ZH, Liu CH, Hang JB, Gao BL and Hu JA:
Rituximab effectively reverses tyrosine kinase inhibitors (TKIs)
resistance through inhibiting the accumulation of rictor on
mitochondria- associated ER-membrane (MAM). Cancer Biomark.
20:581–588. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Rehman J, Zhang HJ, Toth PT, Zhang Y,
Marsboom G, Hong Z, Salgia R, Husain AN, Wietholt C and Archer SL:
Inhibition of mitochondrial fission prevents cell cycle progression
in lung cancer. FASEB J. 26:2175–2186. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zhao J, Zhang J, Yu M, Xie Y, Huang Y,
Wolff DW, Abel PW and Tu Y: Mitochondrial dynamics regulates
migration and invasion of breast cancer cells. Oncogene.
32:4814–4824. 2013. View Article : Google Scholar
|
|
66
|
Ferreira-da-Silva A, Valacca C, Rios E,
Pópulo H, Soares P, Sobrinho-Simões M, Scorrano L, Máximo V and
Campello S: Mitochondrial dynamics protein Drp1 is overexpressed in
oncocytic thyroid tumors and regulates cancer cell migration. PLoS
One. 10:e01223082015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Pan L, Zhou L, Yin W, Bai J and Liu R:
miR-125a induces apoptosis, metabolism disorder and
migrationimpairment in pancreatic cancer cells by targeting
Mfn2-related mitochondrial fission. Int J Oncol. 53:124–136.
2018.PubMed/NCBI
|
|
68
|
Huang Q, Cao H, Zhan L, Sun X, Wang G, Li
J, Guo X, Ren T, Wang Z, Lyu Y, et al: Mitochondrial fission forms
a positive feedback loop with cytosolic calcium signaling pathway
to promote autophagy in hepatocellular carcinoma cells. Cancer
Lett. 403:108–118. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wang W, Xie Q, Zhou X, Yao J, Zhu X, Huang
P, Zhang L, Wei J, Xie H, Zhou L, et al: Mitofusin-2 triggers
mitochondria Ca2+ influx from the endoplasmic reticulum
to induce apoptosis in hepatocellular carcinoma cells. Cancer Lett.
358:47–58. 2015. View Article : Google Scholar
|
|
70
|
Zhou X, Zhang L, Zheng B, Yan Y, Zhang Y,
Xie H, Zhou L, Zheng S and Wang W: MicroRNA-761 is upregulated in
hepatocellular carcinoma and regulates tumorigenesis by targeting
Mitofusin-2. Cancer Sci. 107:424–432. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Rodrigues MA, Gomes DA, Leite MF, Grant W,
Zhang L, Lam W, Cheng YC, Bennett AM and Nathanson MH:
Nucleoplasmic calcium is required for cell proliferation. J Biol
Chem. 282:17061–17068. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Hu J, Qin K, Zhang Y, Gong J, Li N, Lv D,
Xiang R and Tan X: Downregulation of transcription factor Oct4
induces an epithelial-to-mesenchymal transition via enhancement of
Ca2+ influx in breast cancer cells. Biochem Biophys Res
Commun. 411:786–791. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Cho KB, Cho MK, Lee WY and Kang KW:
Overexpression of c-myc induces epithelial mesenchymal transition
in mammary epithelial cells. Cancer Lett. 293:230–239. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Yang S, Zhang JJ and Huang XY: Orai1 and
STIM1 are critical for breast tumor cell migration and metastasis.
Cancer Cell. 15:124–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Prakriya M, Feske S, Gwack Y, Srikanth S,
Rao A and Hogan PG: Orai1 is an essential pore subunit of the CRAC
channel. Nature. 443:230–233. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Stewart TA, Azimi I, Thompson EW,
Roberts-Thomson SJ and Monteith GR: A role for calcium in the
regulation of ATP-binding cassette, sub-family C, member 3 (ABCC3)
gene expression in a model of epidermal growth factor-mediated
breast cancer epithelial-mesenchymal transition. Biochem Biophys
Res Commun. 458:509–514. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Davis FM, Peters AA, Grice DM, Cabot PJ,
Parat MO, Roberts-Thomson SJ and Monteith GR: Non-stimulated,
agonist- stimulated and store-operated Ca2+ influx in
MDA-MB-468 breast cancer cells and the effect of EGF-induced EMT on
calcium entry. PLoS One. 7:e369232012. View Article : Google Scholar
|
|
78
|
Tajeddine N and Gailly P: TRPC1 protein
channel is major regulator of epidermal growth factor receptor
signaling. J Biol Chem. 287:16146–16157. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Hou T, Jian C, Xu J, Huang AY, Xi J, Hu K,
Wei L, Cheng H and Wang X: Identification of EFHD1 as a novel
Ca(2+) sensor for mitoflash activation. Cell Calcium. 59:262–270.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Li W, Sun T, Liu B, Wu D, Qi W, Wang X, Ma
Q and Cheng H: Regulation of mitoflash biogenesis and signaling by
mitochondrial dynamics. Sci Rep. 6:329332016. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Rosselin M, Santo-Domingo J, Bermont F,
Giacomello M and Demaurex N: L-OPA1 regulates mitoflash biogenesis
independently from membrane fusion. EMBO Rep. 18:451–463. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ying Z, Chen K, Zheng L, Wu Y, Li L, Wang
R, Long Q, Yang L, Guo J, Yao D, et al: Transient activation of
mitoflashes modulates nanog at the early phase of somatic cell
reprogramming. Cell Metab. 23:220–226. 2016. View Article : Google Scholar
|
|
83
|
Burch TC, Rhim JS and Nyalwidhe JO:
Mitochondria biogenesis and bioenergetics gene profiles in isogenic
prostate cells with different malignant phenotypes. BioMed Res Int.
2016:17852012016. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Li Y, Huang S, Li Y, Zhang W, He K, Zhao
M, Lin H, Li D, Zhang H, Zheng Z, et al: Decreased expression of
LncRNA SLC25A25-AS1 promotes proliferation, chemoresistance, and
EMT in colorectal cancer cells. Tumour Biol. 37:14205–14215. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Tsai FC and Meyer T: Ca2+
pulses control local cycles of lamel-lipodia retraction and
adhesion along the front of migrating cells. Curr Biol. 22:837–842.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
de Lucas B, Bernal A, Pérez LM, San Martín
N and Gálvez BG: Membrane blebbing is required for mesenchymal
precursor migration. PLoS One. 11:e01500042016. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Sroka J, Krecioch I, Zimolag E, Lasota S,
Rak M, Kedracka-Krok S, Borowicz P, Gajek M and Madeja Z:
Lamellipodia and membrane blebs drive efficient electrotactic
migration of rat walker carcinosarcoma cells WC 256. PLoS One.
11:e01491332016. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Gambade A, Zreika S, Guéguinou M, Chourpa
I, Fromont G, Bouchet AM, Burlaud-Gaillard J, Potier-Cartereau M,
Roger S, Aucagne V, et al: Activation of TRPV2 and BKCa channels by
the LL-37 enantiomers stimulates calcium entry and migration of
cancer cells. Oncotarget. 7:23785–23800. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Yu C, Wang Y, Peng J, Shen Q, Chen M, Tang
W, Li X, Cai C, Wang B, Cai S, et al: Mitochondrial calcium
uniporter as a target of microRNA-340 and promoter of metastasis
via enhancing the Warburg effect. Oncotarget. 8:83831–83844.
2017.PubMed/NCBI
|
|
90
|
Park JH, Kim HK, Jung H, Kim KH, Kang MS,
Hong JH, Yu BC, Park S, Seo SK, Choi IW, et al: NecroX-5 prevents
breast cancer metastasis by AKT inhibition via reducing
intracellular calcium levels. Int J Oncol. 50:185–192. 2017.
View Article : Google Scholar
|
|
91
|
Monet M, Lehen'kyi V, Gackiere F, Firlej
V, Vandenberghe M, Roudbaraki M, Gkika D, Pourtier A, Bidaux G,
Slomianny C, et al: Role of cationic channel TRPV2 in promoting
prostate cancer migration and progression to androgen resistance.
Cancer Res. 70:1225–1235. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Panahi G, Pasalar P, Zare M, Rizzuto R and
Meshkani R: MCU-knockdown attenuates high glucose-induced
inflammation through regulating MAPKs/NF-κB pathways and ROS
production in HepG2 cells. PLoS One. 13:e01965802018. View Article : Google Scholar
|
|
93
|
Corazao-Rozas P, Guerreschi P, André F,
Gabert PE, Lancel S, Dekiouk S, Fontaine D, Tardivel M, Savina A,
Quesnel B, et al: Mitochondrial oxidative phosphorylation controls
cancer cell's life and death decisions upon exposure to MAPK
inhibitors. Oncotarget. 7:39473–39485. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Patergnani S, Giorgi C, Maniero S,
Missiroli S, Maniscalco P, Bononi I, Martini F, Cavallesco G,
Tognon M and Pinton P: The endoplasmic reticulum mitochondrial
calcium cross talk is downregulated in malignant pleural
mesothelioma cells and plays a critical role in apoptosis
inhibition. Oncotarget. 6:23427–23444. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Ren T, Wang J, Zhang H, Yuan P, Zhu J, Wu
Y, Huang Q, Guo X, Zhang J, Ji L, et al: MCUR1-Mediated
Mitochondrial Calcium Signaling Facilitates Cell Survival of
Hepatocellular Carcinoma via Reactive Oxygen Species-Dependent P53
Degradation. Antioxid Redox Signal. 28:1120–1136. 2018. View Article : Google Scholar
|
|
96
|
Lehen'kyi V, Flourakis M, Skryma R and
Prevarskaya N: TRPV6 channel controls prostate cancer cell
proliferation via Ca(2+)/NFAT-dependent pathways. Oncogene.
26:7380–7385. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Høyer-Hansen M, Bastholm L, Szyniarowski
P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N,
Elling F, Rizzuto R, et al: Control of macroautophagy by calcium,
calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell.
25:193–205. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Luyten T, Welkenhuyzen K, Roest G, Kania
E, Wang L, Bittremieux M, Yule DI, Parys JB and Bultynck G:
Resveratrol- induced autophagy is dependent on IP3Rs and on
cytosolic Ca2. Biochim Biophys Acta Mol Cell Res.
1864:947–956. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Cárdenas C, Müller M, McNeal A, Lovy A,
Jaňa F, Bustos G, Urra F, Smith N, Molgó J, Diehl JA, et al:
Selective vulnerability of cancer cells by inhibition of Ca(2+)
transfer from endoplasmic reticulum to mitochondria. Cell Rep.
14:2313–2324. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Martin KR, Celano SL, Solitro AR, Gunaydin
H, Scott M, O'Hagan RC, Shumway SD, Fuller P and MacKeigan JP: A
potent and selective ULK1 inhibitor suppresses autophagy and
sensitizes cancer cells to nutrient stress. iScience. 8:74–84.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Raturi A, Gutiérrez T, Ortiz-Sandoval C,
Ruangkittisakul A, Herrera-Cruz MS, Rockley JP, Gesson K, Ourdev D,
Lou PH, Lucchinetti E, et al: TMX1 determines cancer cell
metabolism as a thiol-based modulator of ER-mitochondria
Ca2+ flux. J Cell Biol. 214:433–444. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Nutt LK, Pataer A, Pahler J, Fang B, Roth
J, McConkey DJ and Swisher SG: Bax and Bak promote apoptosis by
modulating endoplasmic reticular and mitochondrial Ca2+
stores. J Biol Chem. 277:9219–9225. 2002. View Article : Google Scholar
|
|
103
|
Scorrano L, Oakes SA, Opferman JT, Cheng
EH, Sorcinelli MD, Pozzan T and Korsmeyer SJ: BAX and BAK
regulation of endoplasmic reticulum Ca2+: A control
point for apoptosis. Science. 300:135–139. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Echeverry N, Bachmann D, Ke F, Strasser A,
Simon HU and Kaufmann T: Intracellular localization of the BCL-2
family member BOK and functional implications. Cell Death Differ.
20:785–799. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Giorgi C, Ito K, Lin HK, Santangelo C,
Wieckowski MR, Lebiedzinska M, Bononi A, Bonora M, Duszynski J,
Bernardi R, et al: PML regulates apoptosis at endoplasmic reticulum
by modulating calcium release. Science. 330:1247–1251. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Bononi A, Bonora M, Marchi S, Missiroli S,
Poletti F, Giorgi C, Pandolfi PP and Pinton P: Identification of
PTEN at the ER and MAMs and its regulation of Ca(2+) signaling and
apoptosis in a protein phosphatase-dependent manner. Cell Death
Differ. 20:1631–1643. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Verfaillie T, Rubio N, Garg AD, Bultynck
G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A,
et al: PERK is required at the ER-mitochondrial contact sites to
convey apoptosis after ROS-based ER stress. Cell Death Differ.
19:1880–1891. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Li C, Liu Q, Li N, Chen W, Wang L, Wang Y,
Yu Y and Cao X: EAPF/Phafin-2, a novel endoplasmic
reticulum-associated protein, facilitates TNF-alpha-triggered
cellular apoptosis through endoplasmic reticulum-mitochondrial
apoptotic pathway. J Mol Med (Berl). 86:471–484. 2008. View Article : Google Scholar
|
|
109
|
Iwasawa R, Mahul-Mellier AL, Datler C,
Pazarentzos E and Grimm S: Fis1 and Bap31 bridge the
mitochondria-ER interface to establish a platform for apoptosis
induction. EMBO J. 30:556–568. 2011. View Article : Google Scholar
|
|
110
|
Namba T, Tian F, Chu K, Hwang SY, Yoon KW,
Byun S, Hiraki M, Mandinova A and Lee SW: CDIP1-BAP31 complex
transduces apoptotic signals from endoplasmic reticulum to
mitochondria under endoplasmic reticulum stress. Cell Rep.
5:331–339. 2013. View Article : Google Scholar : PubMed/NCBI
|