Introduction
Helicobacter pylori (HP) is
etiologically active in the occurrence of gastric cancer, stomach
inflammation and peptic ulcer disease (1), infecting ~50% of the whole global
population in 2015 (2). In cases
of chronic or acute HP infection, inflammation is a major
cause of peptic ulcer disease and gastric malignancy (3). The pathogenesis of gastric cancer is
multi-factorial, with HP infection being the probable
leading cause (4). The
International Agency for Research on Cancer, World Health
Organization in 1994 defined HP as a class I carcinogen and
a cause of human cancer (5).
Studies on HP infection in rodents have independently
provided evidence of the role of the bacterial pathogen in gastric
cancer development (6-8). A previous study reported HP as
the single leading risk factor of gastric cancer development and is
estimated to cause ~75% of all cases of gastric cancer globally in
2015 (9).
Long-term inflammation has been demonstrated to
intensify gastric barrier penetrability, further injure lamina
propria (10) and potentially
contribute to numerous extra-gastric dysfunctions (11-13).
HP utilizes virulence factors in gastric epithelial cells to
target the signaling pathways that modulate cell cycle, apoptosis
and other special survival processes (14). Among the several reported HP
factors involved in gastric lining disturbance, the most studied
virulence factors include cag pathogenicity island (cagPAI) and
vacuolating cytotoxin gene (vacA) (15). CagPAI encodes a type IV secretion
system, which is activated in human cancer through the deliverance
of the oncoprotein cytotoxin-associated gene A (CagA) into host
cells. Chronic inflammation due to the virulence factor CagA is
considered to be the mechanism of HP-associated gastric
cancer occurrence (16). However,
the mechanisms of tissue damage caused by the aforementioned
factors remain unknown.
The present study focused on experiments on the
carcinogenesis of HP infection in normal gastric epithelial
cells and investigated the specific molecular mechanisms of
carcinogenesis in the HP-infected GES-1 cells.
Materials and methods
GES-1 cell culture
Normal epithelial GES-1 cells of human gastric
mucosa (Cell Resource Center of Shanghai Academy of Sciences,
Chinese Academy of Sciences, Shanghai, China) were cultured in
wells or flasks at 37°C in an atmosphere containing 5%
CO2 in Dulbecco’s modified Eagle’s medium (DMEM; GE
Healthcare Life Sciences, Little Chalfont, UK). The medium
contained 10% (v/v) fetal bovine serum (FBS; GE Healthcare Life
Sciences), 0.25 µg/ml amphotericin B, 0.1 mg/ml streptomycin
and 100 U/ml penicillin.
HP culture
The wild-type HP strain 26695 was obtained
from the Chinese Center for Disease Control and Prevention
(Beijing, China). HP was cultured for 72 h at 37°C in an
atmosphere containing 5% CO2 on commercial HP
culture plates.
GES-1 and HP co-culture
Following washing once with PBS, GES-1 cells were
placed in 6-well plastic plates at a density of 3×105
cells/well in 1 ml FBS-free DMEM. HP was restored by a swab
from the agar plates and suspended in DMEM at an optical density of
0.6 at 600 nm, which corresponded to 3×106
colony-forming U/ml. The HP was added to cells in DMEM
without FBS and antibiotics at a multiplicity of infection (MOI) of
100:1, and co-cultured at 37°C and 5% CO2 for 24 or 48
h. The morphology of HP-infected GES-1 cells was observed by
Olympus CKX53 Inverted Fluorescence Microscope (magnification, ×40;
Olympus Corporation, Tokyo, Japan).
Apoptosis assay
Apoptosis was measured using an Annexin V-FITC
Apoptosis Detection kit (BD Biosciences; Becton, Dickinson and
Company, Franklin Lakes, NJ, USA), according to the manufacturer’s
protocols and analyzed by FlowJo 7.6 software (Tree Star, Inc.,
Ashland, OR, USA). Specifically, following washing twice with PBS,
cells were re-suspended in 100 µl 1X binding buffer (BD
Biosciences; Becton, Dickinson and Company), and then incubated for
15 min with fluorescein isothiocyanate Annexin V and propidium
iodide (each 5 µl; BD Biosciences; Becton, Dickinson and
Company) at 37°C in the dark. Subsequently, 400 µl 1X
binding buffer was added to each tube, followed by detection on a
FACS Canto II flow cytometer (BD Biosciences; Becton, Dickinson and
Company).
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
The primers of β-actin, cyclooxygenase-2 (COX-2),
inducible nitric oxide synthase (iNOS), p16, c-Myc, p53 and p21
designed by Primer Premier 5.0 (Premier Biosoft, Palo Alto, CA,
USA) and primers sequences were purchased from Invitrogen (Thermo
Fisher Scientific, Inc., Waltham, MA, USA).
Total RNA was isolated from HP-infected GES-1 cells
using a TRIzol® reagent (Thermo Fisher Scientific, Inc.)
and detected spectrophotometrically. The cDNA was prepared using
M-MLV reverse transcriptase (Promega Corporation, Madison, WI,
USA). A mixture consisting of RNA (500 ng/µl; 2 µl),
5X M-MLV RT buffer (12 µl), dNTP mixes (2.5 mM; 6
µl), RNase inhibitor (30 U/µl; 1 µl), M-MLV RT
(5 U/µl; 4 µl), Oligo dT(18) primer (500 ng/µl; 3
µl) and diethyl pyrocarbonate water (17 µl) was
cultured at 37°C for 1 h. RT-qPCR was performed on an Eppendorf PCR
system (Eppendorf, Hamburg, Germany). cDNA (1 µl), forward
and reverse primers (each 0.5 µl), SYBR® Premix
ExTaq (8 µl; Takara Biotechnology Co., Ltd., Dalian, China)
and ddH2O (9 µl) were added to the mix, followed
by 32 cycles of amplification as follows: Denaturation at 94°C for
2 min; 32 cycles of 94°C for 20 sec, 55°C for 35 sec, 72°C for 25
sec; and extension at 72°C for 5 min. The following primers were
used: iNOS, forward, 5′-GAG-CTTCTACCTCAAGCTATC-3′, and reverse,
5′-CCTGATGTTGCCATTGTTGGT-3′; COX-2, forward,
5′-AGATCATCTCTGCCTGAGTATCTT-3′, and reverse,
5′-TTCAAATGAGATTGTGGGAAAATTGCT-3′; p21, forward,
5′-CCATGATGTTGATGCCCTAC-3′, and reverse, 5′-TTGCCTGCCTTCCTTTCT-3′;
p53, forward, 5′-CAGTCTACCTCCCGCCATAA-3′, and reverse,
5′-CTCCCAAACATCCCTCACAG-3′; c-Myc, forward,
5′-GGGCTTTATCTAACTCGCTGTA-3′, and reverse,
5′-GCTATGGGCAAAGTTTCGTG-3′; p16, forward,
5′-GAAGAAAGAGGAGGGGCTGG-3′, and reverse,
5′-CTGCAGACCCTCTACCCACC-3′; and β-actin, forward, 5′- CC TG G
CACCCAG CACA AT-3′, a nd reverse, 5′-GCTGATCCACATCTGCTGGAA-3′.
Subsequently, fluorescence was detected. Specific amplification was
guaranteed by dissociation curves. Each sample was tested in
triplicate, and the mean Cq value was calculated (17).
Western blotting
The GES-1 cells from each group were collected,
washed with PBS twice on ice and incubated with a
Radioimmunoprecipitation Assay protein lysate (Beyotime Institute
of Biotechnology, Beijing, China; cat. no. P0013B) containing 1%
benzene sulfonyl fluoride for 30 min at 4°C. The protein
concentration in the supernatant was then measured with a BCA
protein concentration kit (Beyotime Institute of Biotechnology;
cat. no. P0010). Proteins (30 µg) were treated by 10%
SDS-PAGE electrophoresis and transferred onto polyvinylidene
difluoride membranes. The membranes were then immunoblotted with
primary antibodies against β-actin (dilution, 1:2,000; cat. no.
MAB8929), Cox-2 (dilution, 1:1,000; cat. no. AF4198), iNOS
(dilution, 1:1,200; cat. no. MAB9502), p16 (dilution, 1:800; cat.
no. AF5779), c-Myc (dilution, 1:1,000; cat. no. MAB3696), p53
(dilution, 1:1,600; cat. no. AF1355) and p21 (dilution, 1:1,000;
cat. no. AF1047; R&D Systems, Inc., Minneapolis, MN, USA)
overnight at 4°C, followed by cultivation with goat IgG horseradish
peroxidase-conjugated antibody (R&D Systems, Inc.; dilution,
1:1,000; cat. no. HAF019) at 37°C for 60 min and Enhanced
Chemiluminescence Western Blotting Detection reagents (cat. no.
RPN2209; GE Healthcare Life Sciences). The bands were visualized
using a LAS-4000 system (GE Healthcare Life Sciences) and were
analyzed quantitatively using ImageJ v1.8.0 software (National
Institutes of Health, Bethesda, MD, USA).
Cytokine quantification by ELISA
GES-1 cells cultured in 6-well plates were processed
in lipopolysaccharide (LPS; 50 ng/ml; Beijing Solarbio Science
& Technology Co., Ltd., Beijing, China; cat. no. L8880) or
HP (MOI of 100) medium for 24 or 48 h. The concentration of
interleukin-8 (IL-8) and IL-23 in supernatants of stimulated cells
was detected by IL-8 ELISA kit and IL-23 ELISA kits (Wuhan Boster
Biological Technology, Ltd., Wuhan, China; cat. nos. EK0413 and
EK1612, respectively), according to the manufacturer’s protocols.
Cytokine concentrations were determined by a standard curve.
Wound-healing and Matrigel assays in
vitro
Following 24 or 48 h of culture at 37°C, the GES-1
cells were transferred using trypsinization, counted and seeded at
1×105 cells/ml into 6-well plates, followed by overnight
culture at 37°C to form confluent single layers. Wounds were then
induced using a 100 µl pipette tip and imaged at 0, 24 or 48
h under Olympus CKX53 Inverted Fluorescence Microscope at
magnification, ×100. The migration distance of the monolayer from
the wounded area during this time period was detected and expressed
as a migration index (the migrating distance following a stimulus
relative to that of control cells). Each assay was conducted in
triplicate and repeated at least five times.
Invasion assays were conducted in vitro,
using Transwell plates (Costar; Corning, Inc., Corning, NY, USA)
with 8-µm pores. After 2×104 GES-1 cells were
processed in LPS (50 ng/ml) or HP (MOI of 100) medium for 48
h, they were added to the upper chamber of Transwell plates with
Matrigel (Costar; Corning, Inc.) and DMEM in the lower chamber of
Transwell plate contained with 5% FBS. Following 24 or 48 h
incubation, cells on the upper surface were removed using cotton
wool and cells attached to the bottom were fixed with 75% methanol
at 37°C for 15 min and stained with 0.5% crystal violet at 37°C for
30 min. Images were captured and the cells were counted using
Olympus CKX53 Inverted Fluorescence Microscope at ×200
magnification.
Flow cytometry
GES-1 cultivated in 6-well plates were infected with
HP for 24 or 48 h, and then harvested and rinsed twice with
PBS containing 0.2% bovine serum albumin (cat. no. B7542;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Following being
stained with phycoerythrin-labeled monoclonal cluster of
differentiation 44 (CD44; dilution, 1:200; cat. no. 562818; BD
Biosciences; Becton, Dickinson and Company) or
allophycocyanin-labeled CD54 (dilution, 1:200; cat. no. 559771; BD
Biosciences; Becton, Dickinson and Company) antibodies or the
isotype controls (dilution, 1:200; cat. no. 560787; BD Biosciences;
Becton, Dickinson and Company) at 37°C for 30 min, the cells were
washed twice with PBS and fixed in 10% (v/v) formaldehyde-PBS at
37°C. Finally, cells were sorted and analyzed using a BD
FACSCalibur with four-color fluorescence detection system (BD
Biosciences; Becton, Dickinson and Company). Statistical data were
analyzed by FlowJo 7.6 software. Mean fluorescence intensity and
the percentage of positive cells were estimated after the values
for the isotype controls were subtracted.
Immunohistochemical (IHC) analysis
Tissues from 6 normal male patients, 6 male patients
with HP+ gastritis and 6 male patients with
HP+ gastric cancer (stomach adenocarcinoma) aged 32-65
years (mean ± SD, 44.3±8.9 years) were collected from Chongqing
Cancer Institute (Chongqing, China) or Kunshan First People’s
Hospital Affiliated to Jiangsu University (Suzhou, China) from
January 2013 and January 2017. The HP infection status of normal
patients, patients with gastritis and patients with gastric cancer
was confirmed by a Carbon 13 breathing experiment because this
experiment could indicate that individuals were suffering from HP
infection. All protocols were approved by the Ethics Committee of
the Chinese Hospital Association (Beijing, China) and written
informed consent was provided by all participants in all
experiments.
CD44 and CD54 in the membrane of gastric tissue
cells from different participants were analyzed with an IHC assay.
Briefly, all tissues were fixed with 4% formaldehyde (Beyotime
Institute of Biotechnology; cat. no. P0099) at 37°C for 30 min, and
then were embedded in paraffin and cut into slices with a
histotome. Following xylene-deparaffinizing and rehydration through
AR grade absolute ethanol to distilled water at 37°C, the
5-mm-thick microarray sections were stained with Hydrogen Peroxide
Block (cat. no. ab64218; Abcam, Cambridge, UK). Following cultured
with rabbit anti-CD44 (dilution, 1:100; cat. no. 550392; BD
Biosciences; Becton, Dickinson and Company) and anti-CD54
antibodies (dilution, 1:100; cat. no. BBA17; R&D Systems, Inc.)
at 37°C for 40 min, the sections were washed with PBS three times
and cultivated with Biotin goat anti-rabbit IgG (dilution, 1:100;
cat. no. 550338; BD Biosciences; Becton, Dickinson and Company) for
30 min at 37°C. Subsequently, following colorization with
3,3′-diaminobenzidine, the nuclei were counterstained lightly with
hematoxylin at 37°C. Images were captured and the cells were
counted using Olympus CKX53 Inverted Fluorescence microscope, at
magnification, ×200. The immunostained sections were evaluated by
two independent pathologists.
Statistical analysis
Results from three independent experiments are
presented as the mean ± standard error of the mean (SEM). Data were
processed on SPSS 16.0 for Windows (SPSS,Inc., Chicago, IL, USA)
and tested using one-way analysis of variance (ANOVA). The one-way
ANOVA and post-hoc Tukey’s test were conducted on GraphPad Prism 6
(GraphPad Software, Inc., La Jolla, CA, USA) based on an assumption
of normal distribution. P<0.05 was considered to indicate a
statistically significant difference.
Results
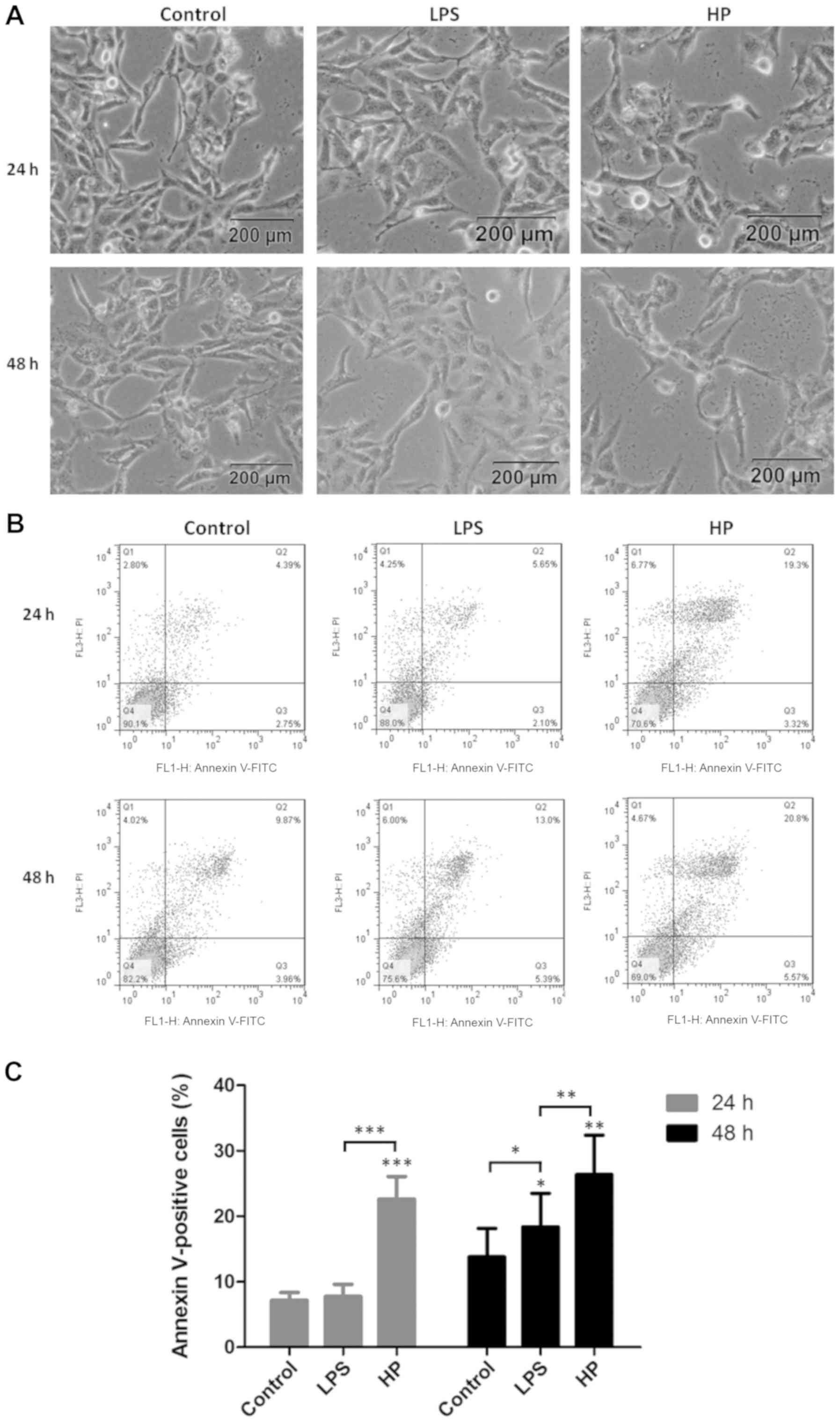
Apoptosis of HP-infected GES-1 cells
To investigate the effects of HP on cell
survival and physiology, the morphology and viability of
HP-infected GES-1 cells was analyzed by microscopy and flow
cytometry, respectively (Fig. 1).
In vitro, the majority of GES-1 cells were in good
condition, and only a small portion underwent HP
infection-induced apoptosis, indicating that the virulence factor
toxicity of HP could induce extensive apoptosis.
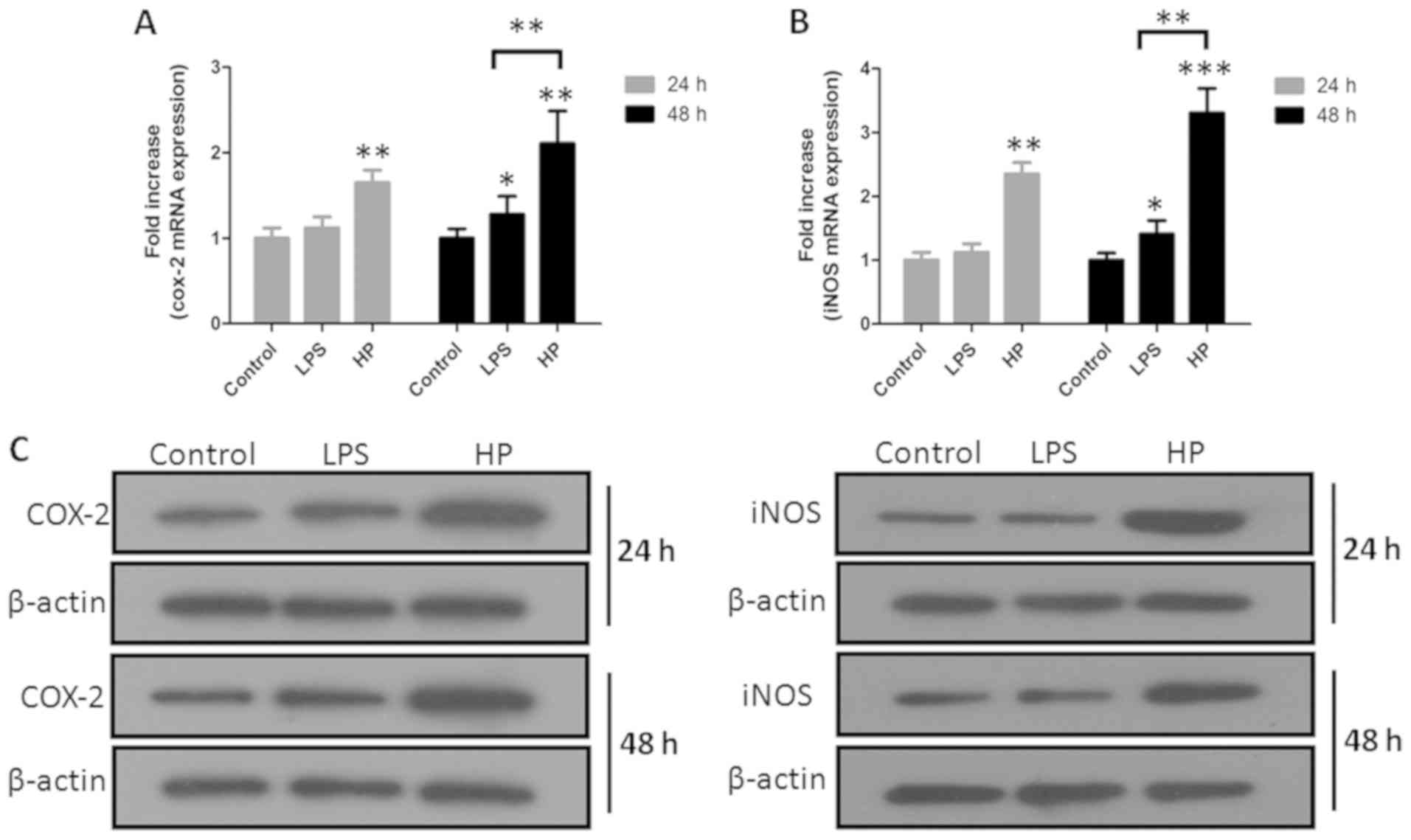
Inflammation-associated genes and
proteins iNOS and COX-2 in GES-1 cells influenced by HP
The host cells employ reactive oxygen species and
nitric oxide to eliminate an invading pathogen, but this reaction
mechanism could also regulate the inflammatory response of host
cells (18). A total of 2 major
enzymes (iNOS and COX-2) involved in the metabolism of
HP-infected GES-1 cells (Fig.
2) were therefore examined using RT-qPCR and western blotting.
The gray of bands was scanned on ImageJ, with β-actin as the
standard. The gene and protein expression of iNOS and COX-2 were
determined to be upregulated following infection.
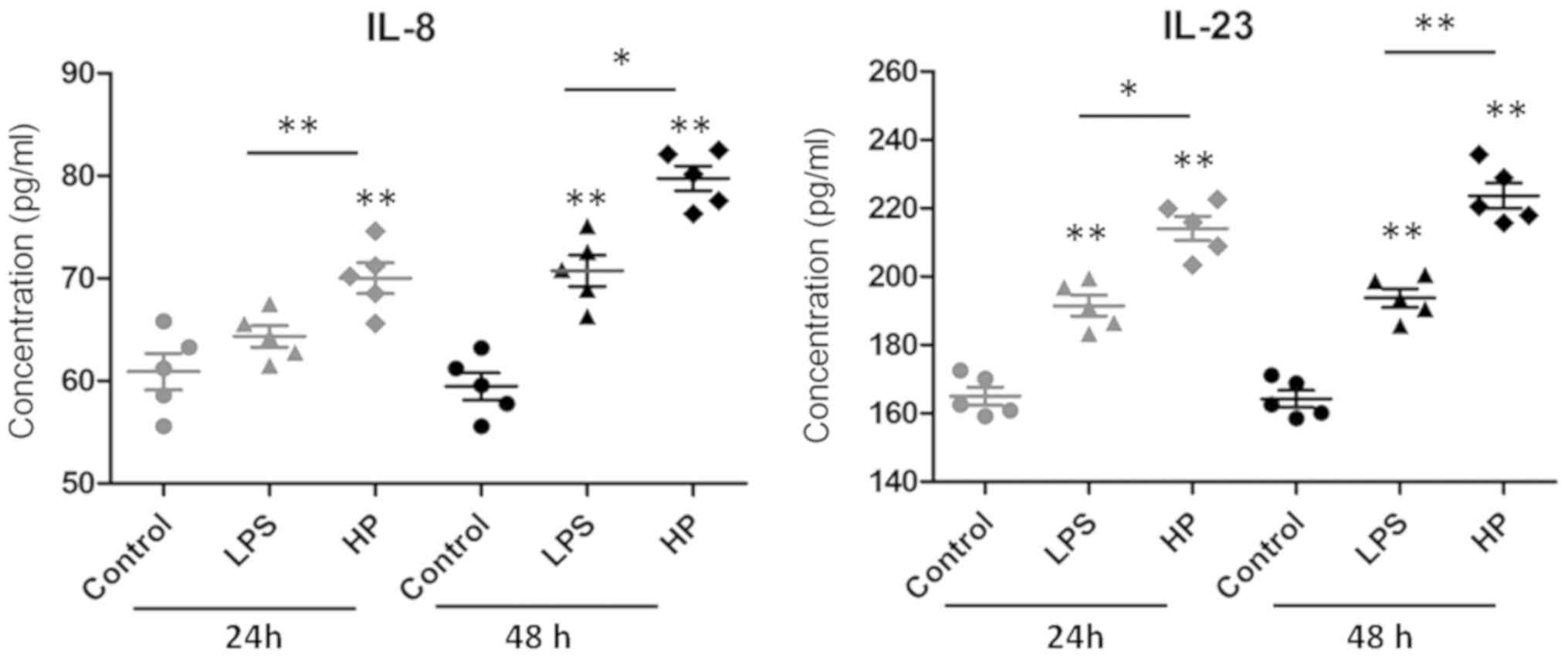
Inflammatory factors IL-8 and IL-23
regulated by HP
Since immune cells are the main secretor of
cytokines, the inflammatory response capacity of GES-1 cells
following HP infection was detected by measuring
inflammatory factors (IL-8 and IL-23) in the supernatant using
ELISA (Fig. 3). IL-8 and IL-23
levels were significantly increased in infected cells, compared
with the control or LPS-treated groups. These results indicated
that normal gastric mucosal cells can also secrete a number of
cytokines to inflammatorily respond to and resist the bacterial
invasion.
Expression of cell carcinoma-associated
genes and proteins induced by HP
Since long-term repeated HP infection could
induce chronic gastritis, gastric ulcer or gastric cancer, the
expression of cancer-associated genes and proteins, including p16,
c-Myc, p53 and p21, was analyzed in HP-infected GES-1 cells
using RT-qPCR and western blotting (Fig. 4). It was determined that the gene
and protein expression levels of p16, c-Myc, p53 and p21 were all
enhanced following HP infection, compared with the control
or LPS-treated groups, indicating that HP may be a risk
factor of gastric cancer.
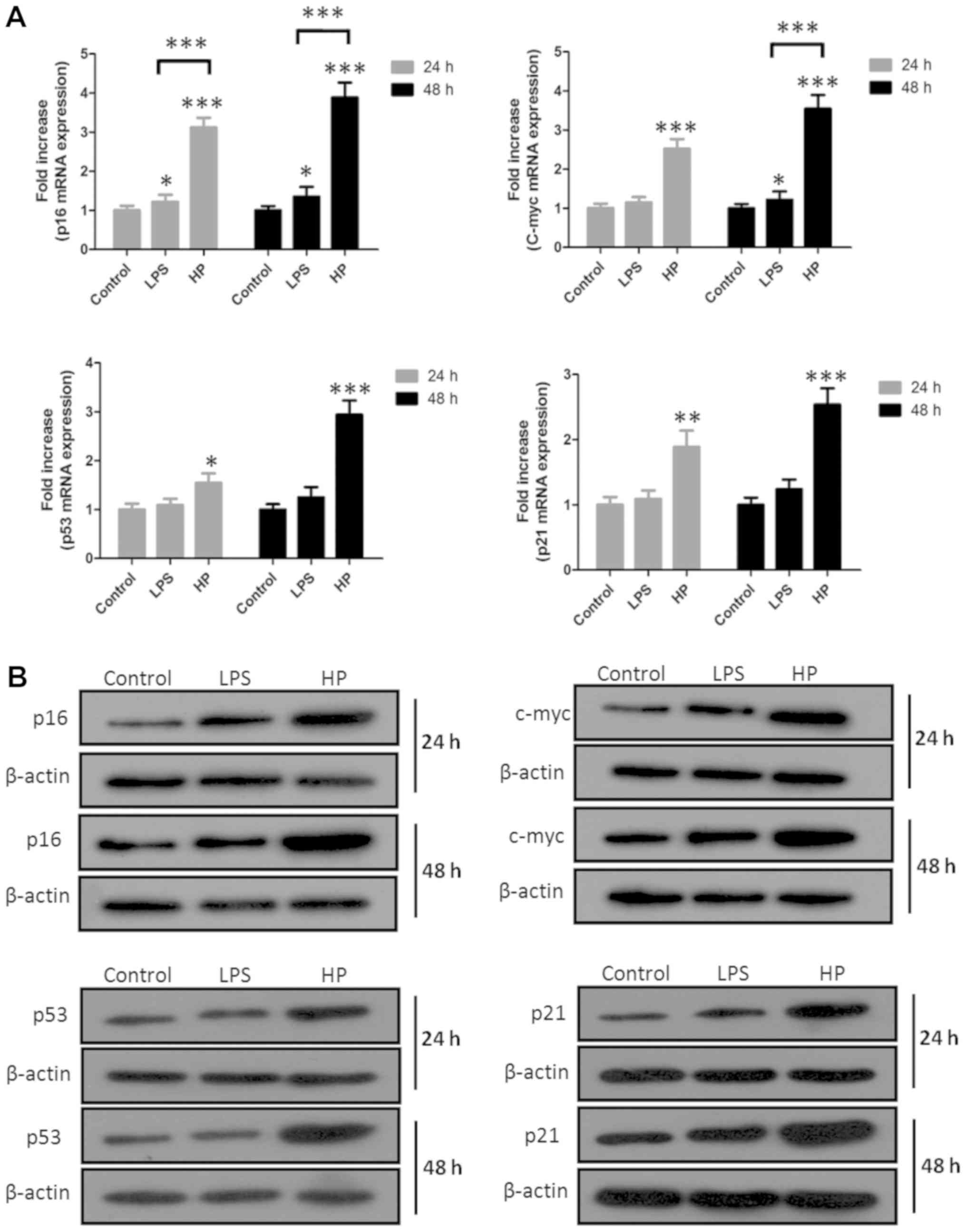 | Figure 4Cancer-associated genes and proteins
p16, c-Myc, p53 and p21 in HP-infected GES-1 cells were
analyzed by RT-qPCR and western blotting. (A) RT-qPCR results of
p16, c-Myc, p53 and p21 genes. (B) Western blotting results for
p16, c-Myc, p53 and p21 proteins in GES-1 cells. Quantification of
p16, c-Myc, p53 and p21 expression was conducted and data are
presented as the mean ± standard error of the mean of three
independent experiments (*P<0.05 vs. control,
**P<0.01 vs. control and ***P<0.001 vs.
control or LPS). RT-qPCR, reverse transcription-quantitative
polymerase chain reaction; LPS, lipopolysac-charides; HP,
Helicobacter pylori. |
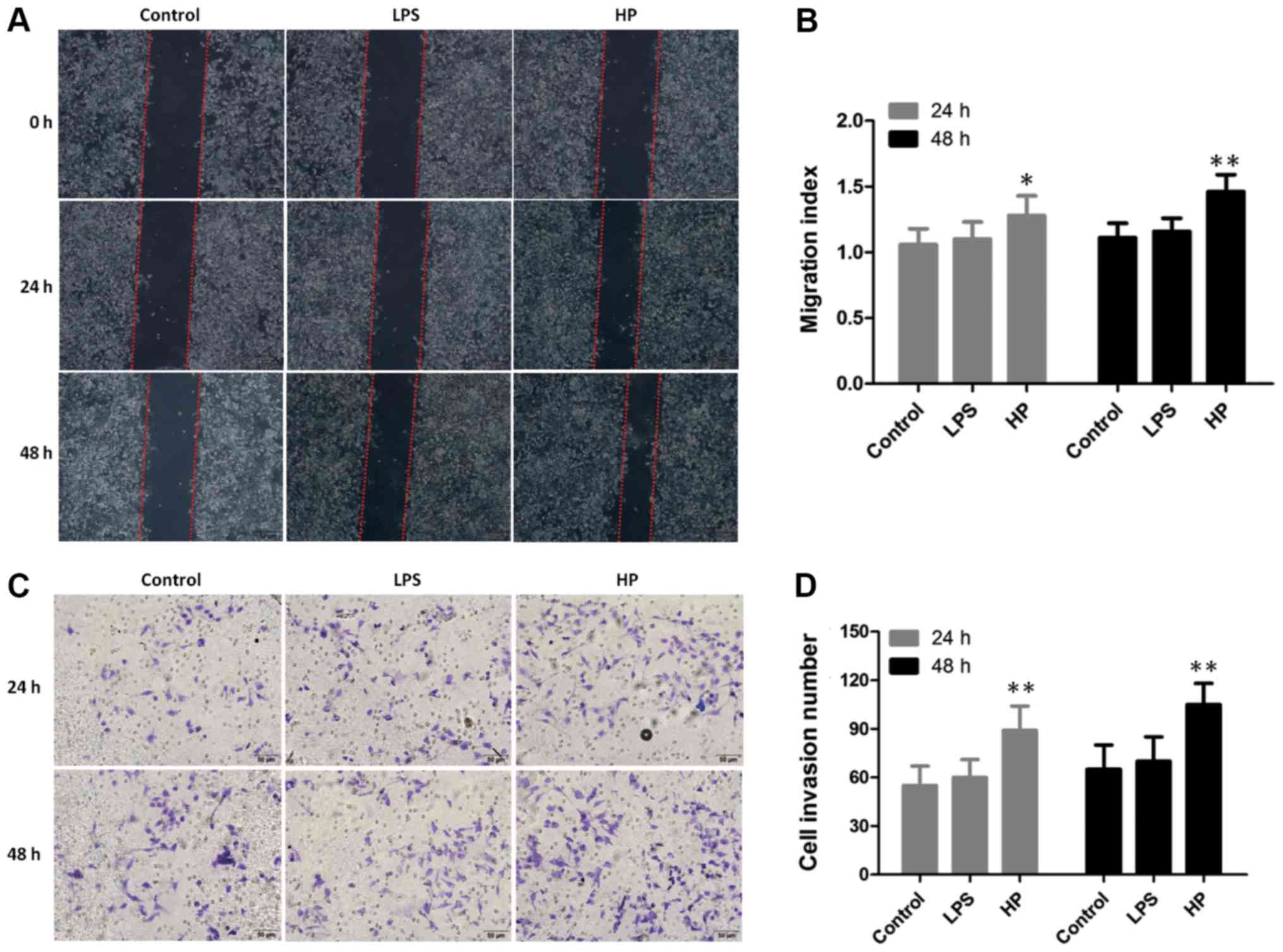
Cell migration and invasion capabilities
of HP-infected GES-1
Since HP infection could change the
expression of cancer-associated genes, causing the host cells to
become cancerous, the cell migration and invasion abilities of
HP-infected GES-1 cells were investigated by wound-healing
experiments (Fig. 5A and B) and
Matrigel assay (Fig. 5C and D). As
expected, the migration and invasion abilities of
HP-infected GES-1 cells were significantly increased,
compared with the control or LPS-treated groups.
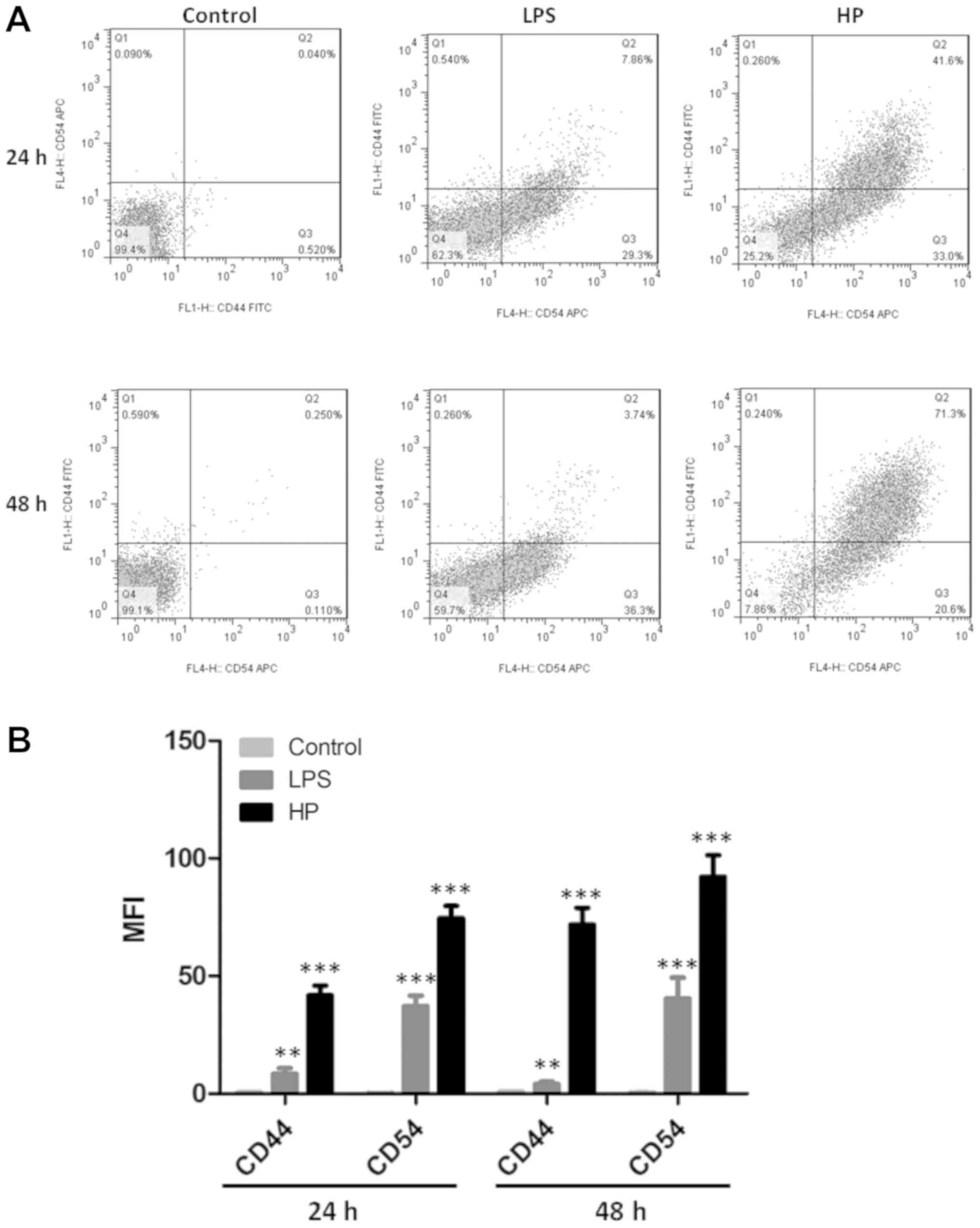
Expression of CD44 and CD54 in
HP-infected GES-1 cells by flow cytometry
CD44 and CD54 are associated with tumor invasion and
metastasis, and are highly expressed in various malignant tumor
types, including gastric cancer (19). The CD44/54 protein expression
levels in HP-infected GES-1 cells were quantified by flow
cytometry (Fig. 6). Results
demonstrated that CD44 and CD54 were upregulated in
HP-infected cells, and that CD54 protein levels were all
overexpressed.
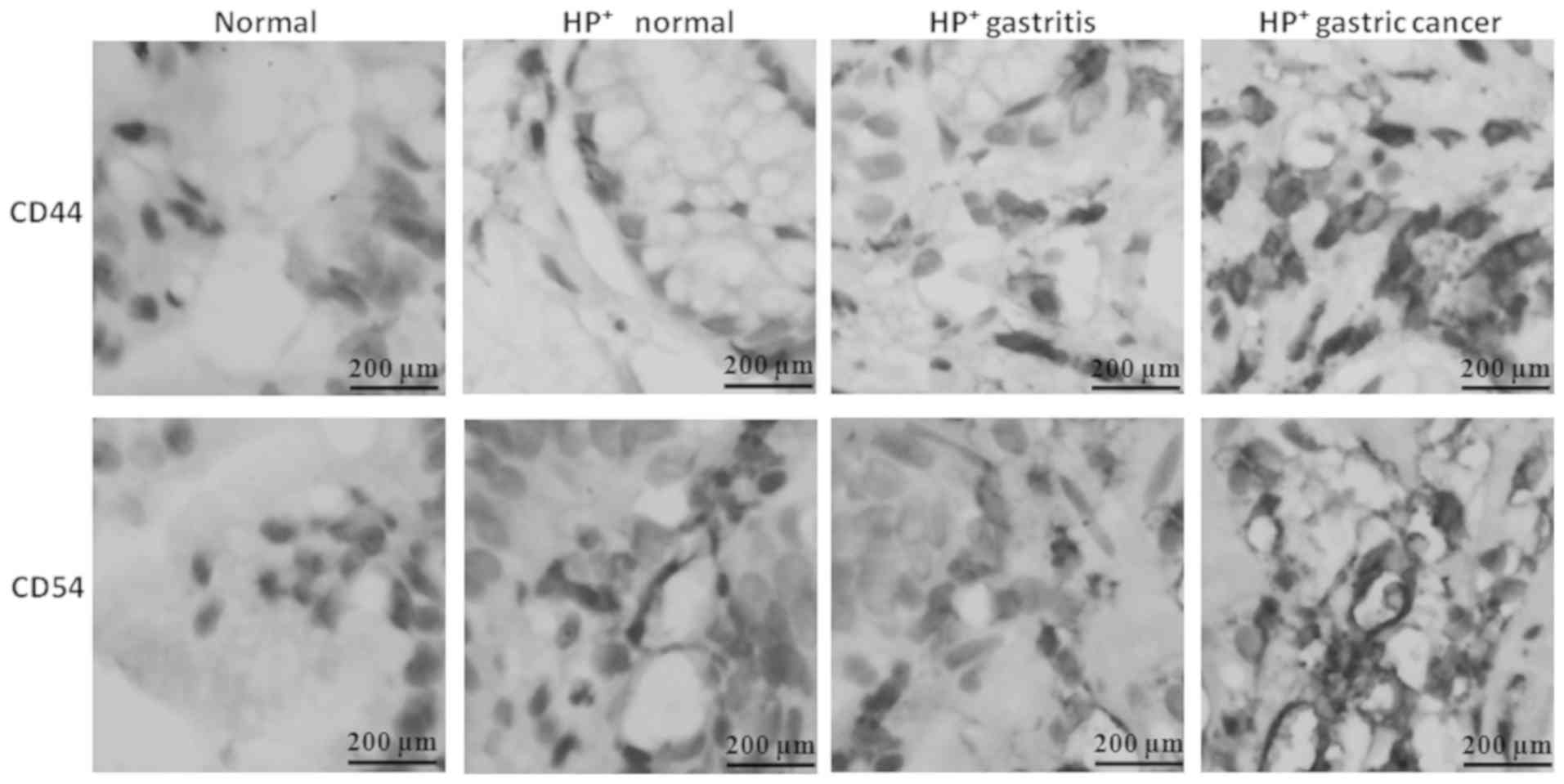
Expression of CD44 and CD54 in
HP+ gastric mucosa tissues
The expression of CD44 and CD54 proteins in the
tissues of different participants was quantified by IHC (Fig. 7) and it was determined that they
were increased in HP-infected tissues. The increased CD44
and CD54 were positively associated with the severity of the
disease, indicating that HP infection may critically affect
the expression of CD44/54.
Discussion
HP, a well-known gastric pathogen, can induce
the generation of inflammatory factors, including IL-1β, IL-6,
IL-8, IL-23 and tumor necrosis factor-α (20-23),
resulting in gastroduodenal inflammation, peptic ulcer disease or
gastric cancer (24,25). IL-8 in the gastric mucosa following
HP infection has been associated with the intensity of
gastritis and risk of gastric cancer (26,27).
IL-23 generated in the gastric mucosa could imply the occurrence of
chronic gastritis, and in the existence of HP, may be
outputted from the inflammatory gastric mucosa following the
kinetics of IL-1β (28).
VacA-activated p38 signaling pathway could induce (COX-2 expression
and thereby increase the formation of prostaglandin E2 (29). It was determined that IL-8 and
IL-23 were markedly upregulated in HP-infected GES-1 cells,
causing an inflammatory response, which in turn induced the
apoptosis of GES-1 cells. HP infection can increase the
expression of iNOS and COX-2, which regulate cellular inflammatory
responses, thereby contributing to the inflammatory response
(30).
HP possesses various virulence factors,
including VacA and CagA, that allow it to chronically survive in
the gastric mucosa (31). VacA and
CagA, two major virulence factors, have been reported to induce
inflammation in the gastric mucosa and be associated with gastric
cancer (32). Accumulative
evidence supports CagA as a pro-oncogenic factor, and these
observations are from mouse transgenic experiments, in which CagA
overexpression results in uniform hypertrophy, low frequency and
late onset focal tumorigenesis of the gastric epithelium, notably
without significant induction of gastritis or atrophy (33,34).
VacA has a variety of biological activities, including the
induction of apoptosis and gastric inflammation, and contribution
to gastric carcinogenesis (35-37).
HP upregulates the expression of p16-INK4 via
its promoter in SGC-7901 cells, and activates its promoter with the
involvement of specificity protein 1 (38). Telomerase mobility and c-Myc levels
in gastric diseases may be affected by HP infection,
particularly chronic atrophic gastritis (39). Similarly, in vivo p53
expression is significantly increased in the HP-positive
gastritis group, compared with the non-gastritis group, and the
expression levels of Ki-67 and p21 were increased significantly in
the HP-positive gastritis group(40). HP increases the
proliferation and increases the expression of p53, but not p21, in
gastric mucosa, indicating that the action of p53 may be
independent of p21 activity (41).
In a previous study, Mongolian gerbils were infected with
HP for a number of hours, and HP induced an acute
accumulation of p53 in the gastric mucosa, followed by a relatively
low plateau for a number of weeks, and then a second peak (42). This dynamic change may depend on
the balance between the p53 degradation induced by HP and
the intracellular self-defense mechanism. The aberrant activation
of oncogenes, DNA damage and high level of inflammation induced by
HP may trigger the intrinsic cellular protection mechanisms,
which upregulate the p53 protein (43). Notably, salt and stress synergize
HP-induced gastric lesions, cell proliferation, and p21
expression without p53 increases in Mongolian gerbils (44). HP infection could inhibit
the proliferation and induce the apoptosis of endothelial cells
through increased phosphorylated p53, p21 and B-cell lymphoma
2-associated X expression, which may contribute to gastric mucosal
injury and to delay healing of gastric lesions (45).
Notably, cell carcinogenesis-associated genes and
proteins, including p16, c-Myc, p53 and p21, were determined to be
upregulated in HP-infected gastric epithelial cells, and it
was considered that HP induced the inflammatory response of
GES-1 cells and induced the expression of p16, c-Myc, p53 and p21
in GES-1 cells at the initial stage of infection. However, as the
duration of infection continues, HP carcinogenic factor
proteins CagA and VacA enter into host cells to activate various
signaling pathways, including nuclear factor (NF-κB) NF-κB and
Janus kinase-signal transducer and activator of transcription, in
cells to induce cell carcinogenesis through changing the expression
of tumor suppressor genes, including p53 and p21 (46-50).
Additionally, HP infection significantly promoted the
migration and invasion abilities of gastric epithelial cells. These
changes may eventually result in inflammation-associated
oncogenesis. However, further experiments are required to
investigate the process of inflammation-induced carcinogenesis and
its specific mechanisms.
The inflammatory events caused by HP
infection can contribute to gastric cancer occurrence. The
circulating immune cells move to the infected mucosa via the
interaction between their ligands and receptors in the endothelial
zone (51). CD44, a surface marker
associated with cancer stem cells, is upregulated in severe gastric
lesions (52). Following
co-culturing with cytotoxin-associated gene pathogen
island-positive HP, the steady-state mRNA levels and surface
CD54 in epithelial cells were upregulated. Additionally, HP
stimulated the CD54 promoter via the NF-κB binding site and induced
CD54 expression in gastric epithelial cells in an NF-κB-dependent
manner that may advocate leukocyte linking upon inflammation
(53). CD44 and CD54 were
upregulated in epithelial cells following HP infection and
abnormally expressed in the mucosa tissues of patients with
HP-infected gastritis or gastric cancer.
Experiments demonstrated that the infection of
HP subjected normal gastric epithelial cells to significant
inflammatory responses and primary cancerous reactions, indicating
that HP is a risk factor for gastric cancer and the
inflammatory responses induced by HP-infection were
associated with cell carcinogenesis. Nevertheless, the mechanism of
carcinogenesis of gastric epithelial cells associated with the
inflammation response induced by HP should be further
investigated.
Funding
The present study was supported by grants from the
science and technology development of Kunshan project (grant no.
KS1717) and the Innovation Team of Kunshan First People’s Hospital
(grant no. KYC007).
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors’ contributions
JW and LT conceived and designed the experiments. YY
and SL performed the experiments. QZ and YY analyzed the data. JW
and LT drafted and revised the manuscript. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
This research was approved by the Ethics Committee
of Kunshan First People’s Hospital, Affiliated to Jiangsu
University.
Patient consent for publication
All patients whose tissues were used for
immunohistochemical analysis in the present study consented to
publication.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Blaser MJ and Berg DE: Helicobacter pylori
genetic diversity and risk of human disease. J Clin Invest.
107:767–773. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Safavi M, Sabourian R and Foroumadi A:
Treatment of Helicobacter pylori infection: Current and future
insights. World J Clin Cases. 4:5–19. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Robinson K, Argent RH and Atherton JC: The
inflammatory and immune response to Helicobacter pyloriinfection.
Best Pract Res Clin Gastroenterol. 21:237–259. 2007. View Article : Google Scholar
|
|
4
|
Conteduca V, Sansonno D, Lauletta G, Russi
S, Ingravallo G and Dammacco F: H. pylori infection and gastric
cancer: State of the art (review). Int J Oncol. 42:5–18. 2013.
View Article : Google Scholar
|
|
5
|
International Agency for Research on
Cancer: IARC Monographs on the Evaluation of Carcinogenic Risk to
Humans, Vol 61: Schistosomotes, Liver Flukes and Helicobacter
pylori. IARC; Lyon: 1994
|
|
6
|
Watanabe T, Tada M, Nagai H, Sasaki S and
Nakao M: Helicobacter pylori infection induces gastric cancer in
mongolian gerbils. Gastroenterology. 115:642–648. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Honda S, Fujioka T, Tokieda M, Satoh R,
Nishizono A and Nasu M: Development of Helicobacter pylori-induced
gastric carcinoma in Mongolian gerbils. Cancer Res. 58:4255–4259.
1998.PubMed/NCBI
|
|
8
|
Franco AT, Israel DA, Washington MK,
Krishna U, Fox JG, Rogers AB, Neish AS, Collier-Hyams L,
Perez-Perez GI, Hatakeyama M, et al: Activation of beta-catenin by
carcinogenic Helicobacter pylori. Proc Natl Acad Sci USA.
102:10646–10651. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Montano V, Didelot X, Foll M, Linz B,
Reinhardt R, Suerbaum S, Moodley Y and Jensen JD: Worldwide
population structure, long-term demography, and local adaptation of
Helicobacter pylori. Genetics. 200:947–963. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Miszczyk E, Rudnicka K, Moran AP, Fol M,
Kowalewicz-Kulbat M, Druszczyńska M, Matusiak A, Walencka M,
Rudnicka W and Chmiela M: Interaction of Helicobacter pylori with
C-type lectin dendritic cell-specific ICAM grabbing nonintegrin. J
Biomed Biotechnol. 2012:2064632012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pacifico L, Osborn JF, Tromba V,
Romaggioli S, Bascetta S and Chiesa C: Helicobacter pylori
infection and extragastric disorders in children: A critical
update. World J Gastroenterol. 20:1379–1401. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chmiela M, Gajewski A and Rudnicka K:
Helicobacter pylori vs coronary heart disease - searching for
connections. World J Cardiol. 7:187–203. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Buzás GM: Metabolic consequences of
Helicobacter pylori infection and eradication. World J
Gastroenterol. 20:5226–5234. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hu Y, He C, Liu JP, Li NS, Peng C, Yang-Ou
YB, Yang XY, Lu NH and Zhu Y: Analysis of key genes and signaling
pathways involved in Helicobacter pylori-associated gastric cancer
based on The Cancer Genome Atlas database and RNA sequencing data.
Helicobacter. 23:e125302018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen S, Duan G, Zhang R and Fan Q:
Helicobacter pylori cytotoxin-associated gene A protein upregulates
α-enolase expression via Src/MEK/ERK pathway: Implication for
progression of gastric cancer. Int J Oncol. 45:764–770. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Khatoon J, Prasad KN, Prakash Rai R,
Ghoshal UC and Krishnani N: Association of heterogenicity of
Helicobacter pylori cag pathogenicity island with peptic ulcer
diseases and gastric cancer. Br J Biomed Sci. 74:121–126. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Δ Δ C(T)) Method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
18
|
Lin SL, Yeh JL, Tsai PC, Chang TH, Huang
WC, Lee ST, Wassler M, Geng YJ and Sulistyowati E: Inhibition of
neointima hyperplasia, inflammation, and reactive oxygen species in
balloon-injured arteries by HVJ envelope vector-mediated delivery
of superoxide dismutase gene. Transl Stroke Res. Sep 6–2018, (Epub
ahead of print) http://doi.org/10.1007/s12975-018-0660-9urisimpledoi.org/10.1007/s12975-018-0660-9.
View Article : Google Scholar
|
|
19
|
Jia Q, Feng M, Wang Y and Xue S: Gastric
cancer cells in collagen gel matrix: Three-dimensional growth and
differential expression of adhesion molecules (CD44s, CD54,
E-cadherin). J Biomed Mater Res A. 84:917–925. 2008. View Article : Google Scholar
|
|
20
|
Tafreshi M, Guan J, Gorrell RJ, Chew N,
Xin Y, Deswaerte V, Rohde M, Daly RJ, Peek RM Jr, Jenkins BJ, et
al: Helicobacter pylori type IV secretion system and its adhesin
subunit, CagL, mediate potent inflammatory responses in primary
human endothelial cells. Front Cell Infect Microbiol. 8:222018.
View Article : Google Scholar :
|
|
21
|
Sierra JC, Asim M, Verriere TG, Piazuelo
MB, Suarez G, Romero-Gallo J, Delgado AG, Wroblewski LE, Barry DP,
Peek RM Jr, et al: Epidermal growth factor receptor inhibition
downregulates Helicobacter pylori-induced epithelial inflammatory
responses, DNA damage and gastric carcinogenesis. Gut.
67:1247–1260. 2018. View Article : Google Scholar
|
|
22
|
Müller A and Solnick JV: Inflammation,
immunity, and vaccine development for Helicobacter pylori.
Helicobacter. 16(Suppl 1): 26–32. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nguyen TT, Kim SJ, Park JM, Hahm KB and
Lee HJ: Repressed TGF-β signaling through CagA-Smad3 interaction as
pathogenic mechanisms of Helicobacter pylori-associated gastritis.
J Clin Biochem Nutr. 57:113–120. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Blaser MJ and Atherton JC: Helicobacter
pylori persistence: Biology and disease. J Clin Invest.
113:321–333. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang J, Wu J, Cheng Y, Jiang Y and Li G:
Over-expression of microRNA-223 inhibited the proinflammatory
responses in Helicobacter pylori-infection macrophages by
down-regulating IRAK-1. Am J Transl Res. 8:615–622. 2016.PubMed/NCBI
|
|
26
|
Panpetch W, Spinler JK, Versalovic J and
Tumwasorn S: Characterization of Lactobacillus salivarius strains
B37 and B60 capable of inhibiting IL-8 production in Helicobacter
pylori-stimulated gastric epithelial cells. BMC Microbiol.
16:2422016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chongruksut W, Limpakan Yamada S,
Chakrabandhu B, Ruengorn C and Nanta S: Correlation of Helicobacter
pylori and interleukin-8 mRNA expression in high risk gastric
cancer population prediction. World J Gastrointest Oncol.
8:215–221. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Koussoulas V, Vassiliou S,
Giamarellos-Bourboulis EJ, Tassias G, Kotsaki A, Barbatzas C and
Tzivras M: Implications for a role of interleukin-23 in the
pathogenesis of chronic gastritis and of peptic ulcer disease. Clin
Exp Immunol. 156:97–101. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ohnishi N, Yuasa H, Tanaka S, Sawa H,
Miura M, Matsui A, Higashi H, Musashi M, Iwabuchi K, Suzuki M, et
al: Transgenic expression of Helicobacter pylori CagA induces
gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl
Acad Sci USA. 105:1003–1008. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Park HS, Wijerathne CUB, Jeong HY, Seo CS,
Ha H and Kwun HJ: Gastroprotective effects of Hwanglyeonhaedok-tang
against Helicobacter pylori-induced gastric cell injury. J
Ethnopharmacol. 216:239–250. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Akeel M, Shehata A, Elhafey A, Elmakki E,
Aboshouk T, Ageely H and Mahfouz M: Helicobacter pylorivacA, cagA
and iceA genotypes in dyspeptic patients from southwestern region,
Saudi Arabia: distribution and association with clinical outcomes
and histopathological changes. BMC Gastroenterol. 19:162019.
View Article : Google Scholar
|
|
32
|
Lee KS, Kalantzis A, Jackson CB, O’Connor
L, Murata-Kamiya N, Hatakeyama M, Judd LM, Giraud AS and Menheniott
TR: Helicobacter pylori CagA Triggers expression of the
bactericidal lectin REG3c via gastric STAT3 activation. PLoS One.
7:e307862012. View Article : Google Scholar
|
|
33
|
Palframan SL, Kwok T and Gabriel K:
Vacuolating cytotoxin A (VacA), a key toxin for Helicobacter pylori
pathogenesis. Front Cell Infect Microbiol. 2:922012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Raju D, Hussey S, Ang M, Terebiznik MR,
Sibony M, Galindo-Mata E, Gupta V, Blanke SR, Delgado A,
Romero-Gallo J, et al: Vacuolating cytotoxin and variants in
Atg16L1 that disrupt autophagy promote Helicobacter pylori
infection in humans. Gastroenterology. 142:1160–1171. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rassow J and Meinecke M: Helicobacter
pylori VacA: A new perspective on an invasive chloride channel.
Microbes Infect. 14:1026–1033. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hisatsune J, Yamasaki E, Nakayama M,
Shirasaka D, Kurazono H, Katagata Y, Inoue H, Han J, Sap J, Yahiro
K, et al: Helicobacter pylori VacA enhances prostaglandin E2
production through induction of cyclooxygenase 2 expression via a
p38 mitogen-activated protein kinase/activating transcription
factor 2 cascade in AZ-521 cells. Infect Immun. 75:4472–4481. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Amieva MR and El-Omar EM: Host-bacterial
interactions in Helicobacter pylori infection. Gastroenterology.
134:306–323. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang P, Mei J, Zhang N, Tao J, Tian H and
Fu GH: Helicobacter pylori upregulates the expression of p16(INK4)
in gastric cancer cells. Hepatogastroenterology. 58:846–853.
2011.PubMed/NCBI
|
|
39
|
Zhang GX, Gu YH, Zhao ZQ, Xu SF, Zhang HJ,
Wang HD and Hao B: Coordinate increase of telomerase activity and
c-Myc expression in Helicobacter pylori-associated gastric
diseases. World J Gastroenterol. 10:1759–1762. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Saf C, Gulcan EM, Ozkan F, Cobanoglu Saf
SP and Vitrinel A: Assessment of p21, p53 expression, and Ki-67
proliferative activities in the gastric mucosa of children with
Helicobacter pylori gastritis. Eur J Gastroenterol Hepatol.
27:155–161. 2015. View Article : Google Scholar
|
|
41
|
Petersson F, Franzén LE and Borch K:
Characterization of the gastric cardia in volunteers from the
general population Type of mucosa, Helicobacter pylori infection,
inflammation, mucosal proliferative activity, p53 and p21
expression, and relations to gastritis. Dig Dis Sci. 55:46–53.
2010. View Article : Google Scholar
|
|
42
|
Wei J, Nagy TA, Vilgelm A, Zaika E, Ogden
SR, Romero-Gallo J, Piazuelo MB, Correa P, Washington MK, El-Rifai
W, et al: Regulation of p53 tumor suppressor by Helicobacter pylori
in gastric epithelial cells. Gastroenterology. 139:1333–1343. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xu H, Chaturvedi R, Cheng Y, Bussiere FI,
Asim M, Yao MD, Potosky D, Meltzer SJ, Rhee JG, Kim SS, et al:
Spermine oxidation induced by Helicobacter pylori results in
apoptosis and DNA damage: Implications for gastric carcinogenesis.
Cancer Res. 64:8521–8525. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gamboa-Dominguez A, Ubbelohde T,
Saqui-Salces M, Romano-Mazzoti L, Cervantes M, Domínguez-Fonseca C,
de la Luz Estreber M and Ruíz-Palacios GM: Salt and stress
synergize H. pylori-induced gastric lesions, cell proliferation,
and p21 expression in Mongolian gerbils. Dig Dis Sci. 52:1517–1526.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kurosawa A, Miwa H, Hirose M, Tsune I,
Nagahara A and Sato N: Inhibition of cell proliferation and
induction of apoptosis by Helicobacter pylori through increased
phosphorylated p53, p21 and Bax expression in endothelial cells. J
Med Microbiol. 51:385–391. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Suzuki M, Mimuro H, Kiga K, Fukumatsu M,
Ishijima N, Morikawa H, Nagai S, Koyasu S, Gilman RH, Kersulyte D,
et al: Helicobacter pylori CagA phosphorylation-independent
function in epithelial proliferation and inflammation. Cell Host
Microbe. 5:23–34. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Murata-Kamiya N, Kurashima Y, Teishikata
Y, Yamahashi Y, Saito Y, Higashi H, Aburatani H, Akiyama T, Peek RM
Jr, Azuma T, et al: Helicobacter pylori CagA interacts with
E-cadherin and deregulates the beta-catenin signal that promotes
intestinal transdifferentiation in gastric epithelial cells.
Oncogene. 26:4617–4626. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gagnaire A, Nadel B, Raoult D, Neefjes J
and Gorvel JP: Collateral damage: Insights into bacterial
mechanisms that predispose host cells to cancer. Nat Rev Microbiol.
15:109–128. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chauhan N, Tay ACY, Marshall BJ and Jain
U: Helicobacter pylori VacA, a distinct toxin exerts diverse
functionalities in numerous cells: An overview. Helicobacter.
24:e125442019. View Article : Google Scholar
|
|
50
|
Kumar S and Dhiman M: Inflammasome
activation and regulation during Helicobacter pylori pathogenesis.
Microb Pathog. 125:468–474. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lundgren A, Trollmo C, Edebo A,
Svennerholm AM and Lundin BS: Helicobacter pylori-specific
CD4+ T cells home to and accumulate in the human
Helicobacter pylori-infected gastric mucosa. Infect Immun.
73:5612–5619. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Garay J, Piazuelo MB, Majumdar S, Li L,
Trillo-Tinoco J, Del Valle L, Schneider BG, Delgado AG, Wilson KT,
Correa P, et al: The homing receptor CD44 is involved in the
progression of precancerous gastric lesions in patients infected
with Helicobacter pylori and in development of mucous metaplasia in
mice. Cancer Lett. 371:90–98. 2016. View Article : Google Scholar :
|
|
53
|
Maeda S, Akanuma M, Mitsuno Y, Hirata Y,
Ogura K, Yoshida H, Shiratori Y and Omata M: Distinct mechanism of
Helicobacter pylori-mediated NF-kappa B activation between gastric
cancer cells and monocytic cells. J Biol Chem. 276:44856–44864.
2001. View Article : Google Scholar : PubMed/NCBI
|