Prostate cancer (PC) is a major global health
burden, with a worldwide incidence of 1.1 million in 2012 (1). While typical active treatments may
involve surgery, chemotherapy, brachytherapy and/or
androgen-deprivation therapy, these often fail to be curative, with
many patients developing metastatic disease or therapeutic
resistance. Aggressive prostate disease, such as metastatic
castration-resistant PC (mCRPC), is usually lethal.
The DNA damage response (DDR) is an essential
pathway that ensures the survival of both normal and malignant
prostate cells and includes many important genes, such as breast
cancer susceptibility gene (BRCA)1/2, ataxia telangiectasia
mutated (ATM) and partner and localizer of BRCA2
(PALB2) (2). Efficient and
specific repair of DNA damage maintains the genomic integrity of
the cell and ensures its ability to persist and proliferate.
Mutations in DDR genes contribute to destabilizing PC cells, often
making them more susceptible to cell death (3). Poly (ADP)-ribose polymerase (PARP)
inhibitors represent an emerging therapeutic approach to target the
DDR pathway in malignant cells (3). In cancer cells that already harbor
multiple other genetic mutations (e.g. BRCA1/2 mutations),
PARP inhibition can render the cell unable to repair DNA damage,
leading to cell death (3).
In PC, mutations in genes involved in the DDR
pathway are relatively common, particularly in the advanced stages
of the disease (4). Several
ongoing international PC clinical trials are exploring PARP
inhibitors that target the DDR pathway, and the contribution of DDR
gene mutations to improving therapeutic outcomes in the context of
PARP inhibitor therapy is becoming more evident (Swift et
al, unpublished data).
Treatment guidelines and consensus statements from
international clinical organizations have recently been published
to reflect the importance of DDR mutation screening for the
management of PC, although the precise genes or gene panels are not
always in accordance. The National Comprehensive Cancer Network
(NCCN) 2018 guidelines for PC state that a strong family history
consists of: Brother, father or multiple family members diagnosed
with prostate cancer by at least 60 years of age; and known
germline DNA repair gene abnormalities, especially BRCA2
mutation. These guidelines advise clinicians as follows: 'Consider
testing for mutations in these genes (germline and somatic):
BRCA1, BRCA2, ATM, PALB2, FANCA;
refer to genetic counseling if positive. At present, this
information may be useful for genetic counseling, early use of
platinum chemotherapy, or eligibility for clinical trials (e.g.
PARP inhibitors)' (5). The
Philadelphia Prostate Cancer Consensus Conference 2017 recommended
that all patients with familial and metastatic PC (mPC) consider
genetic testing [encompassing ATM, BRCA1,
BRCA2, nibrin (NBN) and DNA mismatch repair genes]
(6): 'There was strong consensus
to factor BRCA2 mutations into [prostate cancer] screening
discussions. BRCA2 achieved moderate consensus for factoring
into early stage management discussion, with stronger consensus in
high-risk/advanced and metastatic setting[s]' (6). The St Gallen Advanced Prostate Cancer
Consensus Conference 2017 reported 'that BRCA1, BRCA2
and ATM mutations should be reported [for mCRPC] because
that knowledge will likely influence management decisions'
(7). It is important that
national/regional healthcare providers and decision makers be kept
informed of the burden of DDR mutations in PC.
The authors of the present study undertook a
systematic review of data to identify and summarize the prevalence
of DDR mutations in the unselected (general) population of patients
with PC (including mPC, mCRPC and CRPC). Selected subgroup data for
familial PC were also presented, since this population is currently
considered a key focus for genetic testing guidelines. Studies that
reported on other selected subgroups (including young-onset PC,
lethal PC, ductal PC, patients receiving pre-specified treatment
regimens, and Ashkenazi Jewish and African-American populations)
were identified but were not the focus of the review.
Methodological factors and limitations of the included studies that
may have led to variation in the prevalence rates reported are
noted and discussed.
This systematic review was carried out in accordance
with the methodologies recommended by the Cochrane Collaboration
(8) and the Centre for Reviews and
Dissemination (9). The review
adhered to a pre-defined protocol, which stipulated the methodology
provided below. Studies that reported the prevalence of mutated DDR
genes in men with PC, castration-resistant PC (CRPC), mPC or mCRPC
were included. Detailed inclusion criteria (including DDR
definition) are provided in Data
S1, Appendix S1.
In order to identify relevant studies, a range of
electronic databases (n=11) were searched from their inception to
December 2017, including Medline (Ovid), Embase (Ovid), CINAHL
(EBSCO) and the Cochrane Database of Systematic Reviews (Wiley).
Searches used a combination of text and database thesaurus terms.
No restrictions on language or publication status were applied.
Searches of conference abstracts and reference lists of articles
were conducted. Full details of the search methods employed, the
databases searched, and the Embase search strategy are provided in
Data S1, Appendix S2. Titles and abstracts were
independently screened by two reviewers. Full paper copies were
independently examined in detail by two reviewers to determine
whether or not they met the inclusion criteria. Data were extracted
from the included studies using a specifically designed and piloted
data extraction sheet developed using Microsoft Excel 2016
(Microsoft Corporation). Details of the study methods, population
characteristics, risk of bias, and outcome data were extracted from
each study by one reviewer and checked for accuracy against the
original publication by a second reviewer. Criteria used to assess
the risk of bias were taken from the Joanna Briggs Institute
Critical Appraisal Checklist for studies reporting prevalence data
(10). Any disagreements or
discrepancies in study selection or data extraction were resolved
by consensus or through consultation with a third reviewer.
A narrative summary of all included studies was
produced. The results prioritized the following countries:
Australia, Canada, France, Germany, Israel, Italy, Japan, Korea,
Russia, Spain, the UK and the USA. Multinational studies were
summarized separately. Analysis of prevalence was primarily
conducted for the unselected PC population and familial PC
subgroups only; data for additional subgroups identified during
screening (including young-onset PC, ductal PC, lethal PC,
African-American with PC, Ashkenazi Jewish with PC or patients
receiving pre-specified treatment regimens) were also extracted.
Studies were excluded from the analysis of prevalence if they
involved: i) Specific mutation(s) in a given gene rather than all
mutations for a given gene (unless the study was familial); ii)
<50 participants (unless the study was familial), to focus on
well powered studies (11,12); or iii) did not clearly report
whether a mutation was germline or somatic. Such studies are
summarized in Data S1, Appendix S3, together with reported
prevalence values. The median prevalence and range were reported
for each gene of interest or combination of genes (DDR). If
multiple definitions were available for a given gene mutation, then
the broadest definition was included where possible. The median
prevalence rates were compared between germline and somatic
mutations or between prostate subgroups if the dataset was greater
than 500 combined participants, to focus on data with a large
sample size.
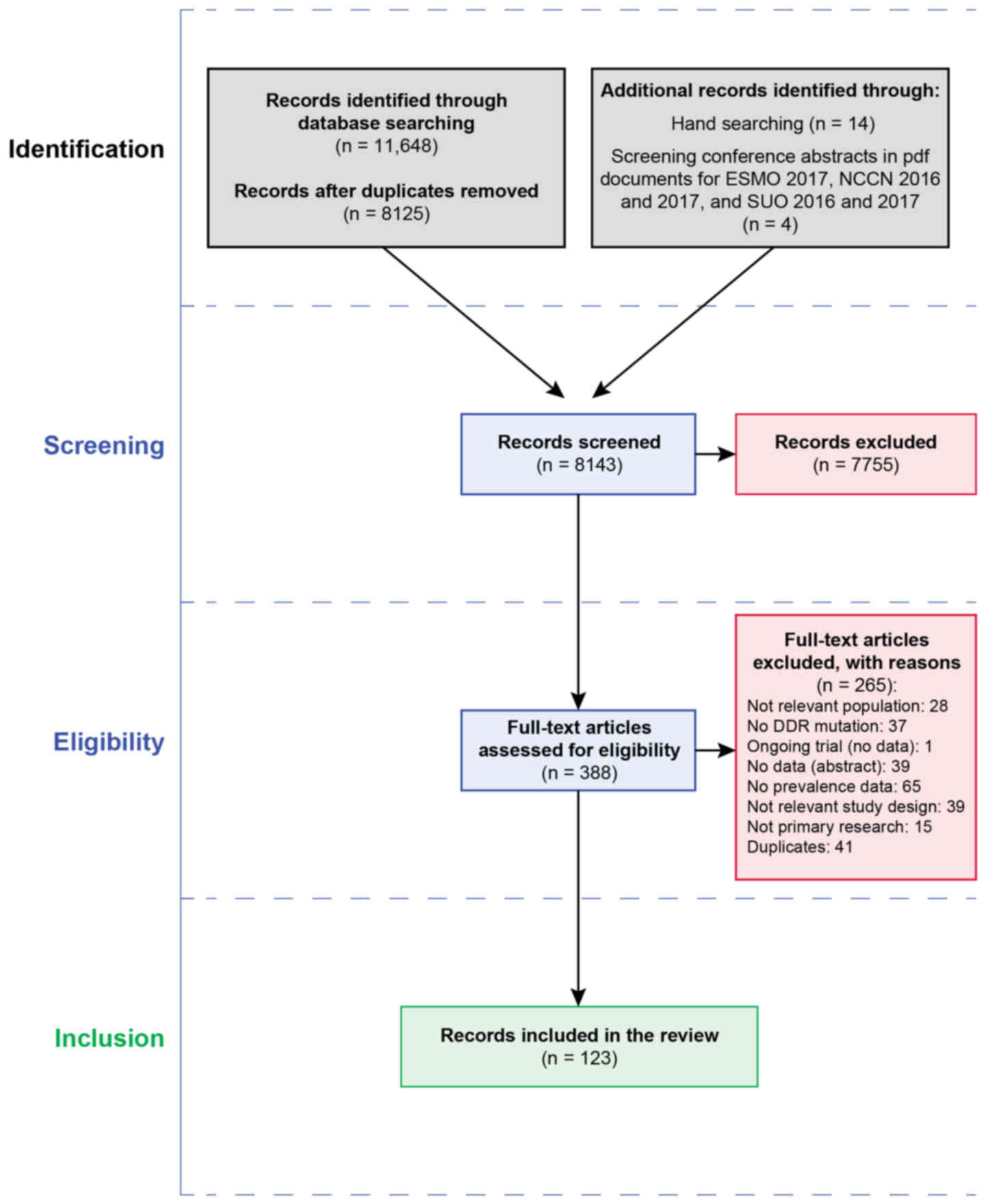
In total, 11,648 titles were retrieved from the
database searches. A total of 14 articles were identified from hand
and citation searching. Four articles were identified from
conference abstracts. Fig. 1
summarizes the flow of studies through the search and screening
process. A total of 265 articles were excluded after the full
records were read (Fig. 1 and
Data S1, Appendix S3). There were 123 records
identified for inclusion in the review, though 20 records were not
from countries of interest and were not considered further
(Data S1, Appendix S3). For the prioritized
countries, 103 records from 80 studies (some of which had multiple
records or publications) were identified for inclusion in the
present review. A total of 59 records focused on unselected patient
populations, and 13 records focused on familial PC patients.
Several records reporting on additional selected
subgroups were identified, including 11 for Ashkenazi Jewish
patients, 10 for patients receiving pre-specified treatment
regimens, five for young-onset (≤55 or 65 years) PC, five for
lethal PC, two for ductal PC and one for African-American patients
with PC. The results for these subgroups are presented in Data S1, Appendices S3 and S4, together with reported prevalence
values.
A total of 47 studies (59 records) were identified
for the unselected population. Out of the 47 studies, the majority
(22 studies) were conducted exclusively in the USA (Table I). A total of five studies were
conducted in the UK, three in Canada and two in Japan. Denmark,
Germany, Israel and Spain each provided a single study. A total of
nine studies recruited patients from multiple locations; the
country of recruitment was not reported for two studies (both
abstracts).
In total, 37 of the 47 primary studies reported
results for patients with unselected PC, five of which focused on
primary or localized PC (the authors' descriptions are reported
where the definitions of disease terms were not made more
explicit). Four studies focused on mPC and six studies focused on
mCRPC.
The numbers of patients in the studies varied
considerably, ranging from 8 to 39,014. A total of 10 studies
included <50 patients in their cohort, 23 studies included
between 50 and 500 patients, and 14 studies included >500
patients. The majority of studies failed to report recruitment or
enrollment dates (32 studies). Of the remaining studies, 11
commenced recruitment between 1990 and 1998, and only four
recruited patients after the year 2000.
A total of 11 primary studies (and two related
publications) focused on patients with familial PC (Table II). As with the studies targeting
unselected populations, the majority included patients from the USA
(six studies, including one multinational study) and the UK (two
studies). Two studies were identified for Germany (including the
single multinational study), and one study each included patients
from Australia and Japan. All 11 studies exclusively reported
results for patients with unselected PC. For the selected familial
PC subgroup, specific DDR mutations are described in Data S1, Appendix S4.
The source of DNA and the methods of genetic
analysis varied considerably among studies (Data S1, Appendix S6). The major sources of DNA
were blood (39 studies, 48.1%) and tumor tissue (28 studies,
34.6%). Six samples were sourced from cell-free DNA (7.4%), five
from paraffin-embedded tumor tissue (6.2%), and four from buccal
swabs/saliva (4.9%). One study sourced DNA from a single-cell
suspension of a primary tumor (1.2%). Nine studies did not report
the source of the DNA analyzed (11.1%).
The two most frequently used methods of mutational
analysis were PCR (29 studies, 35.8%) and next-generation
sequencing (19 studies, 23.5%). Seven studies performed whole-exome
sequencing (8.6%), three used Sanger sequencing (3.7%), three used
capture sequencing (3.7%) and two used fluorescence in situ
hybridization (FISH; 2.5%). A total of 10 studies used methods
other than those specified within this report (12.3%), whereas six
studies (7.4%) did not report which methods were used.
DDR genes could be separated into three general
categories: Those that focused on specific mutations in a specific
gene (21 studies, 25.9%); those that focused on all, or undefined,
mutations in a specific gene (22 studies, 27.1%); and those that
focused on undefined mutations in multiple DDR genes (38 studies,
46.9%). Definitions for individual gene mutations or DDR
combinations varied considerably from study to study.
None of the studies were free from the risk of bias
based on the Joanna Briggs Institute Critical Appraisal Checklist;
88% (of the 80 included studies) had at least one domain judged to
be high risk (Data S1, Appendix S7 and Fig. S1). The majority of studies had a
sample size of >50 participants (79% of studies), an analysis
with sufficient coverage/clearly described mutations (84%), and
appropriate statistical analysis with a clear prevalence
calculation (74%). Overall, however, the quality of reporting was
poor and did not allow for judgment of how well participants were
recruited or how data were acquired; consequently, 76% of studies
were judged to have an unclear risk of bias. Likewise, the criteria
for the diagnosis of PC (e.g. the Gleason score) were unreported in
the majority of studies (74%), and it was unclear in most studies
(91%) whether a pathologist had performed the diagnosis.
Confounding factors were not taken into consideration for
prevalence reporting in the majority of studies (71%). In addition,
the majority of studies (73%) did not describe one or more of the
participant baseline characteristics for this review.
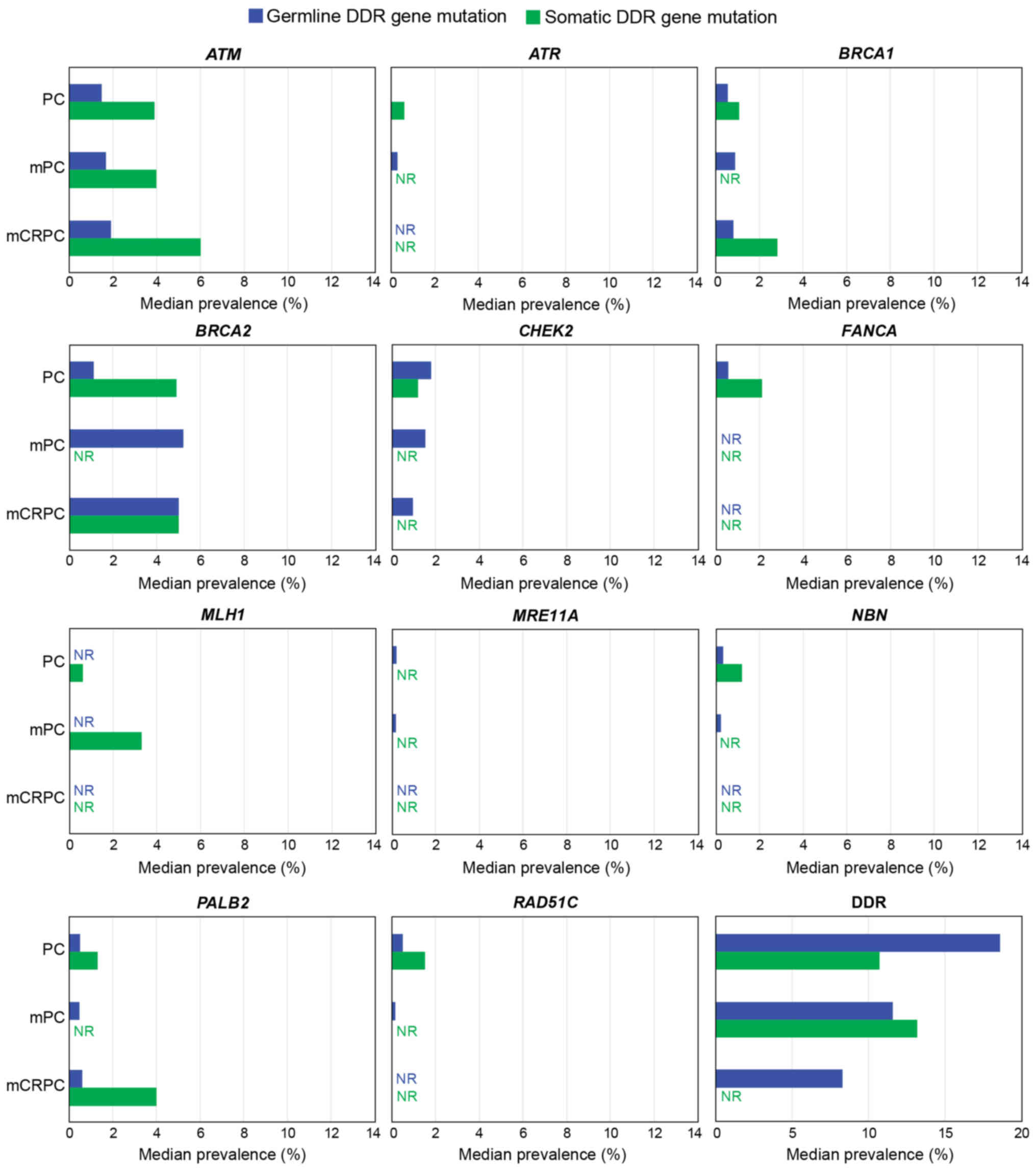
In unselected PC populations and datasets including
>500 patients, the frequency of somatic mutations was higher
than that of germline mutations in ATM (3.9% vs. 1.5%,
respectively), ATR (0.6% vs. 0%), BRCA1 (1.1% vs.
0.6%), BRCA2 (4.9% vs. 1.1%), MLH1 (0.6% vs. 0%),
NBN (1.2% vs. 0.3%), PALB2 (1.3% vs. 0.5%) and
RAD51C (1.5% vs. 0.5%). By contrast, germline mutations were
more common than somatic mutations in CHEK2 (1.8% vs. 1.2%,
respectively) and MRE11A (0.2% vs. 0%). In mCRPC, somatic
mutations were more common than germline mutations for BRCA1
(2.8% vs. 0.8%, respectively), whereas somatic and germline
mutations were equally common for BRCA2 (5% for each group).
The prevalence rates for other prostate subgroups were based on
<500 patients and were not summarized.
Germline mutations were more common in patients with
mPC (based on datasets that included >500 patients) than in
patients with general PC for ATM (1.7% vs. 1.5%,
respectively), ATR (0.3% vs. 0%), BRCA1 (0.9% vs.
0.6%) and BRCA2 (5.2% vs. 1.1%), but not for CHEK2
(1.5% vs. 1.8%), NBN (0.2% vs. 0.3%), PALB2 (0.5% for
both groups) or RAD51c (0.2% vs. 0.5%). Germline mutations
were more common in mCRPC than in general PC for BRCA1 (0.8%
vs. 0.6%) and BRCA2 (5.0% vs. 1.1%). Fewer than 500 patients
were available for other prostate subgroups, and prevalence in
those groups was therefore not summarized.
Somatic mutations were more common in the mCRPC
population (based on datasets including >500 patients) than in
the unselected PC population for BRCA1 (2.8% vs. 1.1%),
whereas similar rates of somatic mutations were seen in the two
populations for BRCA2 (5.0% vs. 4.9%).
The prevalence for DDR genes as a combined term was
much higher than for the individual genes. This was expected, as
the definition was based on the presence of multiple gene mutations
(Table III and Data S1, Appendix S8). In general PC, the
prevalence of somatic DDR gene mutations was in the range of
4.9-22%, while germline DDR mutation rates ranged between 17.2 and
19%. In mPC, the prevalence of somatic DDR mutations ranged between
10 and 16.4%, while germline mutation rates ranged between 11.4 and
11.8%. In mCRPC, the prevalence of germline DDR mutations was in
the range of 7.5-9.1%; no somatic mutations were reported. In
general PC, based on datasets including >500 patients, germline
mutations had a higher prevalence than somatic mutations (18.6% vs.
10.7%, respectively); other somatic subgroups included <500
patients, and so prevalence rates were not analyzed. Germline DDR
mutation rates were higher in patients with general PC (18.6%) than
in those with mPC (11.6%) or mCRPC (8.3%).
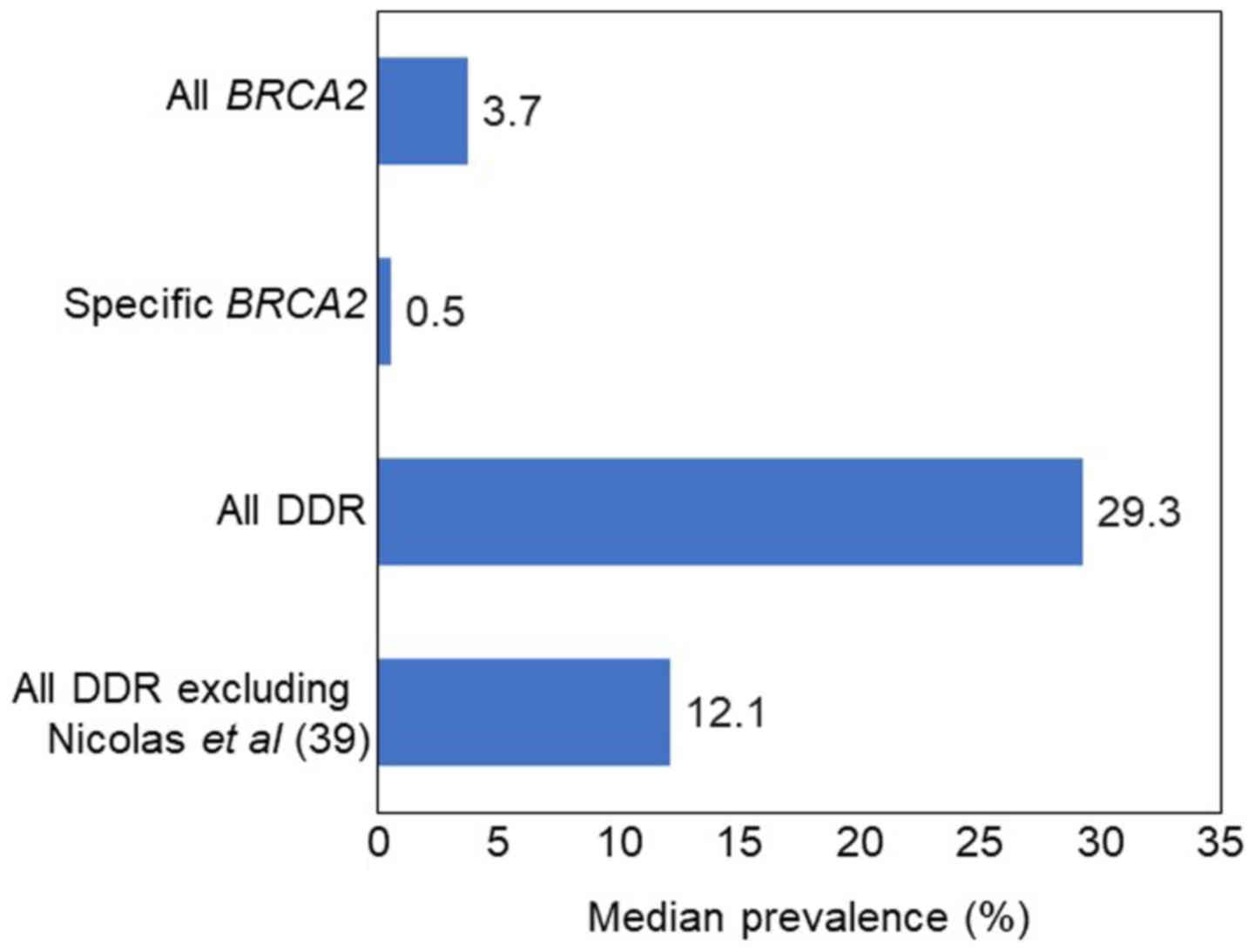
The median prevalence of germline DDR mutations in
familial PC was 29.3% (range, 7.3-91.67%), based on three studies
(n=327). It is noteworthy that one other study (39) provided two definitions for 'DNA
damage response or androgen-signaling gene variants' (Data S1, Appendix S6); one definition referred to
any affected gene (91.7%), whereas the other referred to two or
more affected genes (41.7%). The inclusion of 'androgen-signaling
gene variants' in the definition of DDR was inconsistent with the
definitions of DDR in the other studies, and therefore this study
was excluded from a sensitivity analysis, leading to a median
prevalence of 12.1% (range, 7.3-16.9%).
To the best of our knowledge, this is the first
review that has used rigorous systematic review methods (8,9) to
report a comprehensive summary of recently published data on the
prevalence of DDR genes in PC (including in mPC, mCRPC and selected
subgroups).
The most common mutations (measured by median
prevalence) in all unselected populations were ATM,
BRCA2 and PALB2. The highest median reported rates
for germline mutations in BRCA2 were found in mPC and mCRPC.
The highest median reported rates for somatic mutations in mCRPC
were found for ATM and BRCA2.
Overall, the median prevalence for DDR germline
mutations was 18.6% in general PC (range, 17.2-19%; n=1,712), 11.6%
in mPC (range, 11.4-11.8%; n=1,261), and 8.3% in mCRPC (range,
7.5-9.1%; n=738). The median prevalence for DDR somatic mutations
was 10.7% in general PC (range, 4.9-22%; n=1,537) and 13.2% in mPC
(range, 10-16.4%; n=105).
These results should be interpreted with caution
given the limited reporting of baseline characteristics and the
heterogeneity of sequencing methods and mutational definitions. In
particular, the patients included in the 'unselected PC' population
varied widely. For example, unselected PC cases in Abida et
al (17) represented a mixed
population of cancer types (local and metastatic) dominated by
metastatic samples (77%), whereas patients from the Cancer Genome
Atlas (18) were described as
primary since they were derived from prostatectomies, but in fact
included many high-grade cancers that were likely to be
metastatic.
No other systematic reviews on the prevalence of DNA
repair gene mutations and PC were identified. Two recent papers by
Isaacsson Velho et al (43)
and Quigley et al (44)
identified similar mutational rates to those reported in this
review. Isaacsson Velho et al (43) found a germline mutational rate of
BRCA2 in mPC of 6%, compared with the median prevalence of
5.2% reported here. Quigley et al (44) found germline and/or somatic
BRCA2 rates in mCRPC of 10%, compared with the rate of 12.7%
(4) reported here. It is likely
that the differences in rates may be explained by differences in
sequencing methodology, precise definitions of mutations, and/or
differences in populations. Another recent paper (45) used The Cancer Genome Atlas
(18) tissues and those reported
by Armenia et al (40) to
investigate the influence of Gleason score and tumor stage. The
overall prevalence of somatic DDR gene mutations in localized
tumors was 8%, which is within the range reported here (4.9-22%).
Marshall et al (45)
identified an increase in DDR mutation prevalence in patients with
Gleason grade ≥3 and clinical stage ≥cT3 disease.
The findings of the present review provide evidence
in support of the testing of DDR germline mutations in advanced
disease, in line with NCCN and Philadelphia Prostate Cancer
Consensus recommendations, and the St Gallen Advanced Prostate
Cancer Consensus Conference 2017 (5-7).
Some germline DDR mutations may present an increased healthcare
burden, since family members may also be at risk of PC. Depending
on which DDR gene mutations are present, family members may also
have an increased risk of breast, ovarian, pancreatic and
colorectal cancer, melanoma and other cancer types (5). Given that somatic mutations were
found to have a higher or similar prevalence compared with germline
mutations in patients with mCRPC, somatic mutations may provide
useful genetic information to guide participation in clinical
trials or additional mutation testing. These findings support the
testing of all patients with metastatic disease and not just those
with familial disease. BRCA2 was identified as having the
highest mutation rate of any individual DDR gene, supporting the
use BRCA2 screening as recommended across all consensus
conferences and NCCN (5-7).
Of note, some of the included studies provided
limited baseline details. Where reported, there was evident
heterogeneity in terms of the period of data collection, data
sources, previous treatments, sequencing/screening methods and the
risk of bias. Prevalence data based on low patient/study numbers
combined with limited information about and/or variation in study
characteristics prevented a thorough interrogation of the results,
and the reasons for variation in prevalence could not always be
determined. The repository data in many of the studies included in
this review were unaccompanied by baseline patient details
(13), and the source of the
samples was often mentioned only incidentally within the results.
This hampered data extraction and increased the risk of double
counting (using the same data source twice). For example, among the
three sets of tissues analyzed in the 2016 report of Decker et
al (30), one was the same
tumors used in the 2015 study of Robinson et al (4). Despite this apparent overlap, the
data presented in the two papers do not double count for any result
presented. To avoid double counting, only original data were
extracted from each paper. Study sizes were limited, and only eight
studies included >500 participants (20,21,25,31,32,34,36,46).
Definitions for individual gene mutations or DDR
combinations varied considerably from study to study. Sources of
tissue varied (being either fresh or paraffin-embedded), as did
methods of mutational analysis. Different DNA sequencing/screening
methods used for the detection of DDR mutation may explain the
variations observed in the prevalences reported; this was
demonstrated by the two linked publications of Williams et
al (47) and Gao et al
(48). Both publications used the
same 23 patients with PC, but reported using different techniques
to identify BRCA1 mutations. The first (48) detected loss of heterozygosity,
using PCR, at the BRCA1 locus in 22% of samples, whereas the
second paper (47), using FISH,
found that the loss was not in the BRCA1 gene but was
distal, and therefore reported a 0% prevalence. Small gene panels
and circulating DNA analysis may have lower coverage of large tumor
suppressor genes such as ATM, BRCA1 and BRCA2
compared to whole genome or whole exome sequencing.
Differences in the definitions of what is
considered a mutation can also arise, such as the use of
monoallelic versus biallelic approaches to categorizing DDR
mutations compared with non-mutations. One study (49) reported a comparison between DDR
mutations (defined as biallelic mutations in DDR genes) compared
with non-mutations (defined as wild-type or monoallelic mutations
in DDR genes, which the authors considered non-deleterious). By
contrast, all other studies (with one partial exception) reported
any mutation in a DDR gene (whether it affected one or both
alleles) as a bona fide DDR mutation. The exception involved
a study (13) that provided two
definitions for DDR, alternately based on one or both alleles being
defective.
The use of different reference genome builds as
comparator sequences introduces uncertainty. It is standard
practice to use such builds as normal comparator sequences in order
to identify mutations, but these reference sequences may change
over time (with improved methodologies), which may influence what
is defined as a genuine mutation. The use of different sequencing
depths introduces uncertainty for between-study comparisons. For
example, studies that perform next-generation sequencing to an
extended depth [e.g. x265 (38)]
may achieve more accurate sequencing (lower error rates) than
studies that sequence to a relatively minimal depth [e.g. x100
(27)]. The use of different
frequency cut-off thresholds for the inclusion of mutations
introduces uncertainty for between-study comparisons. For example,
studies that apply a relatively high frequency threshold when
analyzing exonic mutations (e.g. excluding mutations with
frequencies >2% in the observed population) (37) will include a larger pool of
mutations than studies that apply a more restrictive frequency
cutoff (e.g. excluding mutations with a population allele frequency
≥0.5%) (50).
There is a risk of sampling bias with somatic
mutations, given that PC is a very heterogeneous disease (51). This heterogeneity means that within
a patient tumor there are multiple sub-tumors and normal tissues
with different genotypes (different mutational profiles) or clones
(52,53). For example, PC can have areas of
moderately differentiated tumor and undifferentiated tumor, and
each clone will have a different prognosis. Tumor heterogeneity
impacts biomarker studies, since the sample taken for analysis may
not be the same as the sample given to the pathologist, and false
associations between biomarker and histological stage can arise.
Any method other than micro-dissected tissue confirmed by a
pathologist for tumor grade is at risk of sampling error.
In the present study, no prevalence data were
available for CRPC populations with >50 participants. There was
insufficient evidence to judge whether patterns of prevalence were
influenced by the country of recruitment. While there was some
evidence for all the DDR genes on which the study focused, evidence
was particularly limited for ATR, FANCA, MLH1
and MRE11A, as mutations in these genes were reported in
five or fewer studies in total.
Future research should focus on other subgroups
(including African-Americans, and young-onset, ductal and lethal
prostate cancer) that may have DDR mutation prevalences that are
different from that of the wider PC population. In addition, future
work should consider the contribution of founder mutations in
unselected populations and a consideration of mutations that are
prevalent in other countries. A more focused review would be
required to examine in detail whether mutations were pathogenic or
were variants of uncertain significance.
In future research, authors should better clarify
patient baseline characteristics, the diagnostic methods employed,
and whether the somatic samples used for analysis were diagnosed
directly or inferred from other pathology samples. Assessment of
data according to cancer stage and grade rather than broad terms
such as 'PC' may also be more useful. Definitions of what types of
genetic changes constitute 'mutations' (e.g. single-copy deletion
compared with homozygous deletion) need to be standardized, and
pathogenic classification should be routinely incorporated into the
reporting of mutations. Finally, reviews of biomarker studies need
to consider the aforementioned scientific limitations of these
studies, as well as study design limitations. Future
guidelines/consensus are required for greater standardization of
sequencing methods used for genetic association studies.
Given the generally poor quality and poor reporting
of studies showing prevalence data, there is a need for future
epidemiological studies that use recognized methodologies and
reporting tools. Authors should be encouraged to adhere to the
following reporting guidelines for prevalence studies: The Simon
et al (54) guidelines for
the use of archived specimens in the evaluation of prognostic and
predictive biomarkers; Biospecimen Reporting for Improved Study
Quality (55); Strengthening the
Reporting of Genetic Association Studies, an extension of the
STROBE statement (56); and the
Joanna Briggs Institute Critical Appraisal Checklist for Studies
Reporting Prevalence Data (to reduce the risk of bias) (10).
The present review highlighted variations in
definitions and methodology among studies investigating biomarkers,
including differences in tumor sampling, tumor heterogeneity,
sequencing depth and mutational frequency thresholds, and
comparability of mutational definitions (genetic terminology).
Further large, well-reported prevalence studies in PC are needed to
provide international prevalence data on the burden of DDR
dysfunction in patients with PC.
The present study was sponsored by Pfizer, Inc.
All data analyzed during this study are included in
this published article.
SHL, SLS, JK, RGWQ contributed to the conception
and design, acquisition of data, analysis and interpretation of
data. HW contributed to the acquisition of data, analysis and
interpretation of data. KM designed and performed the search
strategies. All authors contributed to the writing of the
manuscript and its final approval.
Not applicable.
Not applicable.
SHL, SLS, HW, KM and JK are employees of Kleijnen
Systematic Reviews Ltd., who were paid consultants to Pfizer, Inc.
in connection with the development of this manuscript; they have no
other competing interests. RGWQ is an employee of Pfizer, Inc..
Editorial and medical writing support was provided
by Dr Ying Jean, Dr Gautam Bijur, Ms. Mary Kacillas and Mr. Joshua
Safran, and was funded by Pfizer, Inc.
|
1
|
Ferlay J, Soerjomataram I, Ervik M,
Dikshit R, Eser S, Mathers C, Rebelo M, Parkin D, Forman D and Bray
F: GLOBOCAN 2012 v10, Cancer Incidence and Mortality Worldwide IARC
CancerBase No. 11. International Agency for Research on Cancer;
Lyon: 2013, http://globocan.iarc.fr.
Accessed September 3 , 2018.
|
|
2
|
Castro E, Mateo J, Olmos D and de Bono JS:
Targeting DNA repair: The role of PARP inhibition in the treatment
of castration-resistant prostate cancer. Cancer J. 22:353–356.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lord CJ and Ashworth A: PARP inhibitors:
Synthetic lethality in the clinic. Science. 355:1152–1158. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Robinson D, Van Allen EM, Wu YM, Schultz
N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC,
Attard G, et al: Integrative clinical genomics of advanced prostate
cancer. Cell. 161:1215–1228. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
National Comprehensive Cancer Network
Inc.: NCCN Clinical Practice Guidelines in Oncology (NCCN
Guidelines): Prostate cancer (Version 4. 2018 8/15/18). NCCN; Fort
Washington, PA: 2018, https://www.nccn.org/professionals/physician_gls/pdf/prostate.pdf.
Accessed September 3 , 2018.
|
|
6
|
Giri VN, Knudsen KE, Kelly WK, Abida W,
Andriole GL, Bangma CH, Bekelman JE, Benson MC, Blanco A, Burnett
A, et al: Role of genetic testing for inherited prostate cancer
risk: Philadelphia Prostate Cancer Consensus Conference 2017. J
Clin Oncol. 36:414–424. 2018. View Article : Google Scholar :
|
|
7
|
Gillessen S, Attard G, Beer TM, Beltran H,
Bossi A, Bristow R, Carver B, Castellano D, Chung BH, Clarke N, et
al: Management of patients with advanced prostate cancer: The
report of the Advanced Prostate Cancer Consensus Conference APCCC
2017. Eur Urol. 73:178–211. 2018. View Article : Google Scholar
|
|
8
|
Higgins JPT and Green S: Cochrane handbook
for systematic reviews of interventions [Internet] The Cochrane
Collaboration, Version 5.1.0. updated March 2011. 2011, http://handbook.cochrane.org/.
Accessed November 22 , 2017.
|
|
9
|
Centre for Reviews and Dissemination
Systematic Reviews: CRD's guidance for undertaking reviews in
health care. University of York; York: 2017, http://www.york.ac.uk/inst/crd/SysRev/!SSL!/WebHelp/SysRev3.htm.
Accessed November 22 , 2017.
|
|
10
|
Munn Z, Moola S, Lisy K, Riitano D and
Tufanaru C: Methodological guidance for systematic reviews of
observational epidemiological studies reporting prevalence and
cumulative incidence data. Int J Evid-Based Healthc. 13:147–153.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ioannidis JPA: Why most published research
findings are false. PLoS Med. 2:e1242005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Button KS, Ioannidis JP, Mokrysz C, Nosek
BA, Flint J, Robinson ES and Munafò MR: Power failure: Why small
sample size undermines the reliability of neuroscience. Nat Rev
Neurosci. 14:365–376. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Timms KM, Cuzick J, Neff C, Reid J,
Solimeno C, Sangale Z, Pruss D, Gutin A, Lanchbury JS and Stone S:
The molecular landscape of genome instability in prostate cancer.
Proceedings of the European Society for Medical Oncology Congress;
Copenhagen. 2016, http://discovery.northernlight.com/document.php?datasource=PHE&docid=PE20161109050002300&context=WK%40northernlight.com"xrefwindow" href="https://doi.org/10.1093/annonc/mdw363.63" id="d7e1504" name="d7e1504" shape="rect">https://doi.org/10.1093/annonc/mdw363.63.
Accessed December 7 , 2017.
|
|
14
|
Jefferies M, Cox A, Clarke A and Kynaston
H: Targeted next-generation sequencing analysis of primary prostate
cancer identifies potential therapeutic targets. J Urol. 197(Suppl.
4): e594–e595. 2017. View Article : Google Scholar
|
|
15
|
Gourdin TS and Lilly MB: Genomic profiling
of metastatic prostate cancer through analysis of circulating tumor
DNA (ctDNA). J Clin Oncol. 34(Suppl 2): 1742016. View Article : Google Scholar
|
|
16
|
Pritchard CC, Morrissey C, Kumar A, Zhang
X, Smith C, Coleman I, Salipante SJ, Milbank J, Yu M, Grady WM, et
al: Complex MSH2 and MSH6 mutations in hypermutated microsatellite
unstable advanced prostate cancer. Nat Commun. 5:49882014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Abida W, Armenia J, Gopalan A, Brennan R,
Walsh M, Barron D, Danila D, Rathkopf D, Morris M, Slovin S, et al:
Prospective genomic profiling of prostate cancer across disease
states reveals germline and somatic alterations that may affect
clinical decision making. JCO Precis Oncol. 2017:1–16. 2017.
|
|
18
|
Abeshouse A, Ahn J, Akbani R, Ally A, Amin
S, Andry CD, Annala M, Aprikian A, Armenia J, Arora A, et al:
Cancer Genome Atlas Research Network: The molecular taxonomy of
primary prostate cancer. Cell. 163:1011–1025. 2015. View Article : Google Scholar
|
|
19
|
Nicolosi PLW, Michalski ST, Freschi B,
O'Leary E, Quintana R, Wilson I, Powers MP and Sartor O: Need for
re-evaluation of current guidelines based on results from germline
genetic testing in prostate cancer. J Clin Oncol. 35(Suppl 15):
50092017. View Article : Google Scholar
|
|
20
|
Nelson P, Mateo J, Beltran H, De Sarkar N,
Elemento O, Rubin MA, Vinson JN, Filipenko J, Robinson DR,
Chinnaiyan AM, et al: Inherited mutations in DNA repair genes in
men with metastatic castration-resistant prostate cancer. J Clin
Oncol. 34(Suppl 15): 50092016. View Article : Google Scholar
|
|
21
|
Pritchard CC, Mateo J, Walsh MF, De Sarkar
N, Abida W, Beltran H, Garofalo A, Gulati R, Carreira S, Eeles R,
et al: Inherited DNA-repair gene mutations in men with metastatic
prostate cancer. N Engl J Med. 375:443–453. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Romero Laorden N, Lozano Mejorada R,
Piulats Rodriguez J, Vallespin E, Montesa A, Lorente Estelles D,
Villaguzman J, Grau G, Rodriquez-Vida A, Ibanez K, et al: 826P -
Prevalence and baseline clinico-pathological associations of
germline deleterious mutations in DNA repair genes (gmDDR) in a
metastatic castration resistant prostate cancer (mCRPC) prospective
spanish cohort (PROREPAIR-B study). Presented at European Society
for Medical Oncology (ESMO) 2017 Congress; 1-12 Sep 2017; Madrid:
Spain. https://cslide.ctimeetingtech.com/library/esmo/browse/search/2yZL#2Bb5p0SK
Accessed February 28 , 2018.
|
|
23
|
Struss WJ, Annala M, Warner EW, Beja K,
Vandekerkhove G, Wong A, Gleave M, Chi KN and Wyatt AW: Germline
DNA repair mutations in metastatic castration-resistant prostate
cancer: Therapy response and applicability of circulating tumor
DNA. J Clin Oncol. 35(Suppl 6): 1402017. View Article : Google Scholar
|
|
24
|
Lu C, Xie M, Wendl MC, Wang J, McLellan
MD, Leiserson MDM Huang KL, Wyczalkowski MA, Jayasinghe R, Banerjee
T, et al: Patterns and functional implications of rare germline
variants across 12 cancer types. Nat Commun. 6:100862015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Na R, Zheng SL, Han M, Yu H, Jiang D, Shah
S, Ewing CM, Zhang L, Novakovic K, Petkewicz J, et al: Germline
mutations in ATM and BRCA1/2 distinguish risk for lethal and
indolent prostate cancer and are associated with early age at
death. Eur Urol. 71:740–747. 2017. View Article : Google Scholar :
|
|
26
|
Grasso CS, Wu YM, Robinson DR, Cao X,
Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC,
et al: The mutational landscape of lethal castration-resistant
prostate cancer. Nature. 487:239–243. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Manson-Bahr D, Ball R, Gundem G, Sethia K,
Mills R, Rochester M, Goody V, Anderson E, O'Meara S, Flather M, et
al: Mutation detection in formalin-fixed prostate cancer biopsies
taken at the time of diagnosis using next-generation DNA
sequencing. J Clin Pathol. 68:212–217. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Evans JR, Zhao SG, Chang SL, Tomlins SA,
Erho N, Sboner A, Schiewer MJ, Spratt DE, Kothari V, Klein EA, et
al: Patient-level DNA damage and repair pathway profiles and
prognosis after prosta-tectomy for high-risk prostate cancer. JAMA
Oncol. 2:471–480. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Patel VL, Busch E, D'Amico AV and Rebbeck
TR: BRCA2 mutations in prostate cancer assort into cluster regions.
Int J Radiat Oncol. 96:E569–E570. 2016. View Article : Google Scholar
|
|
30
|
Decker B, Karyadi DM, Davis BW, Karlins E,
Tillmans LS, Stanford JL, Thibodeau SN and Ostrander EA: Biallelic
BRCA2 mutations shape the somatic mutational landscape of
aggressive prostate tumors. Am J Hum Genet. 98:818–829. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Castro E, Goh CL, Olmos D, Leongamornlert
D, Saunders E, Tymrakiewicz M, Mahmud N, Dadaev T, Govindasami K,
Guy M, et al: Correlation of germ-line BRCA2 mutations with
aggressive prostate cancer and outcome. J Clin Oncol. 29(Suppl 15):
15172011. View Article : Google Scholar
|
|
32
|
Leongamornlert D, Mahmud N, Tymrakiewicz
M, Saunders E, Dadaev T, Castro E, Goh C, Govindasami K, Guy M,
O'Brien L, et al: UKGPCS Collaborators: Germline BRCA1 mutations
increase prostate cancer risk. Br J Cancer. 106:1697–1701. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sonpavde G, Nagy RJ, Sartor AO, Pond GR,
Gourdin TS, Nandagopal L, Ledet EM, Agarwal N, Carroll E, Naik G,
et al: Circulating tumor (ct)-DNA alterations in metastatic
castration-resistant prostate cancer (mCRPC): Association with
outcomes and evolution with therapy. J Clin Oncol. 35(Suppl 6):
1492017. View Article : Google Scholar
|
|
34
|
Lara P, McPherson J, Heyer W, Hartmaier R,
DeVere White R, Ching J, Ali S and Dall'Era M: 827P - Comprehensive
char-acterization of BRCA1 and BRCA2 alterations in circulating
tumor DNA and tumor tissue in men with prostate cancer:
Implications for clinical care. Presented at European Society for
Medical Oncology (ESMO) 2017 Congress; 1-12 Sep 2017; Madrid:
Spain. 2017, https://cslide.ctimeetingtech.com/library/esmo/browse/search/2ape#2Bb5p0Vn.
Accessed February 28 , 2018.
|
|
35
|
Myers CE, Feldman R, Abbott BL, Reddy SK
and Castro M: Frequency of BRCA mutations and co-occurring
alterations in prostate cancer. J Clin Oncol. 34(Suppl 2): 2892016.
View Article : Google Scholar
|
|
36
|
Akbari MR, Wallis CJD, Toi A, Trachtenberg
J, Sun P, Narod SA and Nam RK: The impact of a BRCA2 mutation on
mortality from screen-detected prostate cancer. Br J Cancer.
111:1238–1240. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Maier C, Herkommer K, Luedeke M, Rinckleb
A, Schrader M and Vogel W: Subgroups of familial and aggressive
prostate cancer with considerable frequencies of BRCA2 mutations.
Prostate. 74:1444–1451. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fontugne J, Ramazanoglu S, Knudsen K, De
Bono J, Feng F, Sboner A and Rubin M: DNA damage response genes in
prostate cancer: Development of a novel targeted sequencing
platform. Lab Invest. 95:220A–221A. 2015.
|
|
39
|
Nicolas E, Arora S, Zhou Y, Serebriiskii
IG, Andrake MD, Handorf ED, Bodian DL, Vockley JG, Dunbrack RL,
Ross EA, et al: Systematic evaluation of underlying defects in DNA
repair as an approach to case-only assessment of familial prostate
cancer. Oncotarget. 6:39614–39633. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Armenia J, Wankowicz SAM, Liu D, Gao J,
Kundra R, Reznik E, Chatila WK, Chakravarty D, Han GC, Coleman I,
et al: PCF/SU2C International Prostate Cancer Dream Team: The long
tail of oncogenic drivers in prostate cancer. Nat Genet.
50:645–651. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Leongamornlert D, Saunders E, Dadaev T,
Tymrakiewicz M, Goh C, Jugurnauth-Little S, Kozarewa I, Fenwick K,
Assiotis I, Barrowdale D, et al: UKGPCS Collaborators: Frequent
germline deleterious mutations in DNA repair genes in familial
prostate cancer cases are associated with advanced disease. Br J
Cancer. 110:1663–1672. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hayano T, Matsui H, Nakaoka H, Ohtake N,
Hosomichi K, Suzuki K and Inoue I: Germline variants of prostate
cancer in Japanese families. PLoS One. 11:e01642332016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Isaacsson Velho P, Silberstein JL,
Markowski MC, Luo J, Lotan TL, Isaacs WB and Antonarakis ES:
Intraductal/ductal histology and lymphovascular invasion are
associated with germline DNA-repair gene mutations in prostate
cancer. Prostate. 78:401–407. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Quigley DA, Dang HX, Zhao SG, Lloyd P,
Aggarwal R, Alumkal JJ, Foye A, Kothari V, Perry MD, Bailey AM, et
al: Genomic hallmarks and structural variation in metastatic
prostate cancer. Cell. 174:758–769.e9. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Marshall CH, Fu W, Wang H, Baras AS, Lotan
TL and Antonarakis ES: Prevalence of DNA repair gene mutations in
localized prostate cancer according to clinical and pathologic
features: Association of Gleason score and tumor stage. Prostate
Cancer Prostatic Dis. 22:59–65. 2019. View Article : Google Scholar :
|
|
46
|
Petrovics G, Ravindranath L, Chen Y, Ying
K, Ali A, Young D, McLeod D, Sesterhenn I, Rosner I, Dahut W, et
al: Higher frequency of germline BRCA1 and BRCA2 mutations in
African American prostate cancer. J Urol. 195(Suppl. 4): e5482016.
View Article : Google Scholar
|
|
47
|
Williams BJ, Jones E, Zhu XL, Steele MR,
Stephenson RA, Rohr LR and Brothman AR: Evidence for a tumor
suppressor gene distal to BRCA1 in prostate cancer. J Urol.
155:720–725. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gao X, Zacharek A, Salkowski A, Grignon
DJ, Sakr W, Porter AT and Honn KV: Loss of heterozygosity of the
BRCA1 and other loci on chromosome 17q in human prostate cancer.
Cancer Res. 55:1002–1005. 1995.PubMed/NCBI
|
|
49
|
Hussain M, Daignault-Newton S, Twardowski
PW, Albany C, Stein MN, Kunju LP, Siddiqui J, Wu YM, Robinson D,
Lonigro RJ, et al: Targeting androgen receptor and DNA repair in
metastatic castration-resistant prostate cancer: Results from NCI
9012. J Clin Oncol. 36:991–999. 2018. View Article : Google Scholar :
|
|
50
|
Annala M, Struss WJ, Warner EW, Beja K,
Vandekerkhove G, Wong A, Khalaf D, Seppälä IL, So A, Lo G, et al:
Treatment outcomes and tumor loss of heterozygosity in germline DNA
repair-deficient prostate cancer. Eur Urol. 72:34–42. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cyll K, Ersvær E, Vlatkovic L, Pradhan M,
Kildal W, Avranden Kjær M, Kleppe A, Hveem TS, Carlsen B, Gill S,
et al: Tumour heterogeneity poses a significant challenge to cancer
biomarker research. Br J Cancer. 117:367–375. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bedard PL, Hansen AR, Ratain MJ and Siu
LL: Tumour heterogeneity in the clinic. Nature. 501:355–364. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Stanta G and Bonin S: A practical approach
to tumor heterogeneity in clinical research and diagnostics.
Pathobiology. 85:7–17. 2018. View Article : Google Scholar
|
|
54
|
Simon RM, Paik S and Hayes DF: Use of
archived specimens in evaluation of prognostic and predictive
biomarkers. J Natl Cancer Inst. 101:1446–1452. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Moore HM, Kelly AB, Jewell SD, McShane LM,
Clark DP, Greenspan R, Hayes DF, Hainaut P, Kim P, Mansfield E, et
al: Biospecimen reporting for improved study quality (BRISQ). J
Proteome Res. 10:3429–3438. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Little J, Higgins JP, Ioannidis JP, Moher
D, Gagnon F, von Elm E, Khoury MJ, Cohen B, Davey-Smith G, Grimshaw
J, et al: STrengthening the REporting of Genetic Association
Studies: STrengthening the REporting of Genetic Association Studies
(STREGA): An extension of the STROBE statement. PLoS Med.
6:e222009. View Article : Google Scholar
|
|
57
|
Nam RK, Zhang WW, Jewett MA, Trachtenberg
J, Klotz LH, Emami M, Sugar L, Sweet J, Toi A and Narod SA: The use
of genetic markers to determine risk for prostate cancer at
prostate biopsy. Clin Cancer Res. 11:8391–8397. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Näslund-Koch C, Nordestgaard BG and
Bojesen SE: Increased risk for other cancers in addition to breast
cancer for CHEK2*1100delC heterozygotes estimated from
the Copenhagen General Population Study. J Clin Oncol.
34:1208–1216. 2016. View Article : Google Scholar
|
|
59
|
Maier C, Wiest I, Luedeke M, Surowy H,
Rinckleb A, Herkommer K, Kuefer R and Vogel W: BRCA2 mutation
analysis in familial and early onset prostate cancer. Med Genetik.
22:97–98. 2010.
|
|
60
|
Vazina A, Baniel J, Yaacobi Y, Shtriker A,
Engelstein D, Leibovitz I, Zehavi M, Sidi AA, Ramon Y, Tischler T,
et al: The rate of the founder Jewish mutations in BRCA1 and BRCA2
in prostate cancer patients in Israel. Br J Cancer. 83:463–466.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Tanaka Y, Zaman MS, Majid S, Liu J,
Kawakami K, Shiina H, Tokizane T, Dahiya AV, Sen S and Nakajima K:
Polymorphisms of MLH1 in benign prostatic hyperplasia and sporadic
prostate cancer. Biochem Biophys Res Commun. 383:440–444. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Uchida T, Wang C, Sato T, Gao J, Takashima
R, Irie A, Ohori M and Koshiba K: BRCA1 gene mutation and loss of
heterozygosity on chromosome 17q21 in primary prostate cancer. Int
J Cancer. 84:19–23. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Cendón Flórez Y, Nombela Blanco P, Medina
A, Romero Laorden N, Puente J, López Casas P, Gutierrez Pecharromán
A, Sanchez-Escribano R, Magraner L, Gallardo Diaz E, et al: 1660P -
ATM role in prostate cancer (PrCa) progression and survival.
Presented at European Society for Medical Oncology (ESMO) 2017
Congress; 1-12 Sep 2017; Madrid: Spain. https://cslide.ctimeetingtech.com/library/esmo/browse/search/2yZz.
Accessed February 28 , 2018.
|
|
64
|
Centro Nacional de Investigaciones
Oncologicas CARLOS III: Prospective multicentre cohort study
PROREPAIR-B (mCRPC) NCT03075735. WHO International Clinical Trials
Registry Platform (ICTRP) [Internet] Geneva: World Health
Organization (WHO); 2017, https://clinicaltrials.gov/show/NCT03075735.
Accessed December 12 , 2017.
|
|
65
|
Angèle S, Falconer A, Edwards SM, Dörk T,
Bremer M, Moullan N, Chapot B, Muir K, Houlston R, Norman AR, et
al: ATM polymorphisms as risk factors for prostate cancer
development. Br J Cancer. 91:783–787. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Dawson NA, Heath EI, Feldman R, Reddy SK,
Spetzler D, Poste GH and Raghavan D: Use of panomic assessment to
reveal DNA repair alterations and to predict potential therapeutic
response to taxaneplatinum combination therapy in prostate cancer.
J Clin Oncol. 34(Suppl 15): pp. 50402016, View Article : Google Scholar
|
|
67
|
Xia S, Kohli M, Du M, Dittmar RL, Lee A,
Nandy D, Yuan T, Guo Y, Wang Y, Tschannen MR, et al: Plasma genetic
and genomic abnormalities predict treatment response and clinical
outcome in advanced prostate cancer. Oncotarget. 6:16411–16421.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Abida W, Curtis KR, Taylor BS, Arcila ME,
Brennan R, Danila DC, Rathkopf DE, Morris MJ, Slovin SF, Solit DB,
et al: Genomic characterization of primary and metastatic prostate
cancer (PC) using a targeted next-generation sequencing assay. J
Clin Oncol. 33(Suppl 15): 50622015. View Article : Google Scholar
|
|
69
|
Abida W, Walsh MF, Armenia J, Vijai J,
Gopalan A, Brennan R, Curtis K, Arcila M, Danila D, Arnold A, et
al: Next generation sequencing of prostate cancer reveals germline
and somatic alterations detected at diagnosis and at metastasis
that may impact clinical decision making. Cancer Res.
76:LB-0702016.
|
|
70
|
Abida W, Brennan R, Armenia J, Curtis KR,
Gopalan A, Arcila ME, Danila DC, Rathkopf DE, Morris MJ, Slovin SF,
et al: Genomic characterization of primary and metastatic prostate
cancer (PC) using a targeted next-generation sequencing assay. J
Clin Oncol. 34(Suppl 2): 2542016. View Article : Google Scholar
|
|
71
|
Cheng ML, Abida W, Rathkopf DE, Arcila ME,
Barron D, Autio KA, Zehir A, Danila DC, Morris MJ, Gopalan A, et
al: Next-generation sequencing (NGS) of tissue and cell free DNA
(cfDNA) to identify somatic and germline alterations in advanced
prostate cancer. J Clin Oncol. 35(Suppl 15): 15102017. View Article : Google Scholar
|
|
72
|
Feldman RA, Dan Basu G, Xiu J, Arguello D,
Millis SZ, Bender R, Gatalica Z, Paul L and Myers CE: Molecular
profiling of advanced refractory prostate cancer. J Clin Oncol.
32(Suppl 4): 1072014. View Article : Google Scholar
|
|
73
|
Palapattu GS, Cani AK, Huang J, Hovelson
DH, Lu D, Margolis D, Natarajan S, Mehra R, Montgomery JS, Morgan
TM, et al: Progression of low-to high-grade prostate cancer:
Molecular profiling of tissue obtained by serial targeted biopsy. J
Clin Oncol. 33(Suppl 15): 50172015. View Article : Google Scholar
|
|
74
|
Browning RE IV, Li H, Shinohara ET, Cai Q,
Chen H, Courtney R, Cao C, Zheng W and Lu B: ATM polymorphism
IVS62+60G>A is not associated with disease aggressiveness in
prostate cancer. Urology. 67:1320–1323. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Patel V, Busch E, D'Amico A and Rebbeck T:
BRCA2 mutations in prostate cancer assort into cluster regions. J
Radiat Oncol. 6:229–230. 2017.
|
|
76
|
Wu X, Dong X, Liu W and Chen J:
Characterization of CHEK2 mutations in prostate cancer. Hum Mutat.
27:742–747. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Dall'Era M, Glass A, Lara P, Hartmaier R,
DeVere White R and McPherson J: Frequency of DNA repair gene
mutations in localized and metastatic prostate cancer. J Clin
Oncol. 35(Suppl 6): 102017. View Article : Google Scholar
|
|
78
|
Glass A, Lara P, Hartmaier R, DeVere White
R, McPherson J and Dall'Era M: Frequency of DNA repair gene
mutations in localized and metastatic prostate cancer. J Urol.
197(Suppl 4): e56–e57. 2017. View Article : Google Scholar
|
|
79
|
Beltran H, Eng K, Mosquera JM, Sigaras A,
Romanel A, Rennert H, Kossai M, Pauli C, Faltas B, Fontugne J, et
al: Whole-exome sequencing of metastatic cancer and biomarkers of
treatment response. JAMA Oncol. 1:466–474. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Beltran H, Sboner A, Mosquera JM, Rickman
D, Eng K, Prandi D, Kossai M, Faltas B, Pauli C, Fontugne J, et al:
Precision medicine program for whole-exome sequencing (WES)
provides new insight on platinum sensitivity in advanced prostate
cancer (PCa). J Clin Oncol. 33(Suppl 7): 1582015. View Article : Google Scholar
|
|
81
|
Beltran H, Yelensky R, Frampton GM, Park
K, Downing SR, MacDonald TY, Jarosz M, Lipson D, Tagawa ST, Nanus
DM, et al: Targeted next-generation sequencing of advanced prostate
cancer identifies potential therapeutic targets and disease
heterogeneity. Eur Urol. 63:920–926. 2013. View Article : Google Scholar :
|
|
82
|
Gambhira R, Ledet EM, Dotiwala A, Mandal D
and Sartor AO: Copy number variations in AR-associated and DNA
repair genes from plasma cell-free DNA of metastatic CRPC patients.
J Clin Oncol. 34(Suppl 2): 2812016. View Article : Google Scholar
|
|
83
|
Daniel S, Gornstein E, Frampton GM, Sun J,
Morley S, Heilmann A, Reddy P, Chung J, Suh J, Ramkissoon S, et al:
BRCA1/2 reversion mutations in prostate cancer identified from
clinical tissue and liquid biopsy samples. J Clin Oncol. 35(Suppl
15): 50242017. View Article : Google Scholar
|
|
84
|
Robbins CM, Tembe WA, Baker A, Sinari S,
Moses TY, Beckstrom-Sternberg S, Beckstrom-Sternberg J, Barrett M,
Long J, Chinnaiyan A, et al: Copy number and targeted mutational
analysis reveals novel somatic events in metastatic prostate
tumors. Genome Res. 21:47–55. 2011. View Article : Google Scholar :
|
|
85
|
Hebbring SJ, Fredriksson H, White KA,
Maier C, Ewing C, McDonnell SK, Jacobsen SJ, Cerhan J, Schaid DJ,
Ikonen T, et al: Role of the Nijmegen breakage syndrome 1 gene in
familial and sporadic prostate cancer. Cancer Epidemiol Biomarkers
Prev. 15:935–938. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Baca SC, Prandi D, Lawrence MS, Mosquera
JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi
M, et al: Punctuated evolution of prostate cancer genomes. Cell.
153:666–677. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Liu Z, Zhou M, Lepor H, Zoino R, Rajoria G
and Klimek S: Mutational analysis of prostate cancer using next
generation cancer hotspot panel. Lab Invest. 96:246A–247A.
2016.
|
|
88
|
Cheng Y, Thorne H, Bolton D, Clouston D,
Willems A, Li J, Niedermeyer E, Fox S and Mitchell G: Altered
significance of D'Amico risk assessment in BRCA2 positive vs
negative patients from high risk breast cancer families. J Urol.
185(Suppl. 4): e622011. View Article : Google Scholar
|
|
89
|
Gayther SA, de Foy KA, Harrington P,
Pharoah P, Dunsmuir WD, Edwards SM, Gillett C, Ardern-Jones A,
Dearnaley DP, Easton DF, et al: The Cancer Research
Campaign/British Prostate Group United Kingdom Familial Prostate
Cancer Study Collaborators: The frequency of germ-line mutations in
the breast cancer predisposition genes BRCA1 and BRCA2 in familial
prostate cancer. Cancer Res. 60:4513–4518. 2000.PubMed/NCBI
|
|
90
|
Zuhlke KA, Johnson AM, Okoth LA, Stoffel
EM, Robbins CM, Tembe WA, Salinas CA, Zheng SL, Xu J, Carpten JD,
et al: Identification of a novel NBN truncating mutation in a
family with hereditary prostate cancer. Fam Cancer. 11:595–600.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
LaDuca H, Espenschied C, Dolinsky JS,
Smith LP, Fulk K, Pronold M, Horton C, Couch FJ and Davis BT:
Hereditary cancer panel results identify gaps in knowledge of
cancer risks and limitations in current guidelines. In: Presented
at the American Society of Human Genetics Annual Meeting; 17 - 21
Oct , 2017; Orlando: United States. 2017
|
|
92
|
Ledet EM, Ernst EM, Schiff J, Lin S, Lewis
BE and Sartor AO: Germline variants and family history in caucasian
and African-American prostate cancer. J Clin Oncol. 35(Suppl 15):
pp. e165482017, View Article : Google Scholar
|
|
93
|
Lin S, Ledet EM, Schiff J, Ernst EM,
Garvey CE, Lewis BE and Sartor O: Inherited pathologic mutations
and family history in patients with prostate cancer. J Clin Oncol.
35(Suppl 6): pp. 1852017, View Article : Google Scholar
|
|
94
|
Marshall M, Tully D, Susswein L, Theobald
K, Murphy P and Klein R: Panel testing in men with prostate cancer
meeting NCCN genetic testing criteria (AB2017-59). J Natl Compr
Canc Netw. 15:e15–e16. 2017.
|
|
95
|
Tischkowitz MD, Yilmaz A, Chen LQ, Karyadi
DM, Novak D, Kirchhoff T, Hamel N, Tavtigian SV, Kolb S, Bismar TA,
et al: Identification and characterization of novel SNPs in CHEK2
in Ashkenazi Jewish men with prostate cancer. Cancer Lett.
270:173–180. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Hamel N, Kotar K and Foulkes WD: Founder
mutations in BRCA1/2 are not frequent in Canadian Ashkenazi Jewish
men with prostate cancer. BMC Med Genet. 4:72003. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Damaraju S, Murray D, Dufour J, Carandang
D, Myrehaug S, Fallone G, Field C, Greiner R, Hanson J, Cass CE, et
al: Association of DNA repair and steroid metabolism gene
polymorphisms with clinical late toxicity in patients treated with
conformal radiotherapy for prostate cancer. Clin Cancer Res.
12:2545–2554. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Meyer A, Wilhelm B, Dörk T, Bremer M,
Baumann R, Karstens JH and Machtens S: ATM missense variant P1054R
predisposes to prostate cancer. Radiother Oncol. 83:283–288. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Nientiedt C, Heller M, Endris V, Volckmar
AL, Zschäbitz S, Tapia-Laliena MA, Duensing A, Jäger D, Schirmacher
P, Sültmann H, et al: Mutations in BRCA2 and taxane resistance in
prostate cancer. Sci Rep. 7:45742017. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Hubert A, Peretz T, Manor O, Kaduri L,
Wienberg N, Lerer I, Sagi M and Abeliovich D: The Jewish Ashkenazi
founder mutations in the BRCA1/BRCA2 genes are not found at an
increased frequency in Ashkenazi patients with prostate cancer. Am
J Hum Genet. 65:921–924. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
101
|
Giusti RM, Rutter JL, Duray PH, Freedman
LS, Konichezky M, Fisher-Fischbein J, Greene MH, Maslansky B,
Fischbein A, Gruber SB, et al: A twofold increase in BRCA mutation
related prostate cancer among Ashkenazi Israelis is not associated
with distinctive histopathology. J Med Genet. 40:787–792. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Kote-Jarai Z, Leongamornlert D, Saunders
E, Tymrakiewicz M, Castro E, Mahmud N, Guy M, Edwards S, O'Brien L,
Sawyer E, et al: UKGPCS Collaborators: BRCA2 is a moderate
penetrance gene contributing to young-onset prostate cancer:
Implications for genetic testing in prostate cancer patients. Br J
Cancer. 105:1230–1234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Edwards SM, Kote-Jarai Z, Meitz J, Hamoudi
R, Hope Q, Osin P, Jackson R, Southgate C, Singh R, Falconer A, et
al: Cancer Research UK/Bristish Prostate Group UK Familial Prostate
Cancer Study Collaborators; British Association of Urological
Surgeons Section of Oncology: Two percent of men with early-onset
prostate cancer harbor germline mutations in the BRCA2 gene. Am J
Hum Genet. 72:1–12. 2003. View
Article : Google Scholar
|
|
104
|
Agalliu I, Karlins E, Kwon EM, Iwasaki LM,
Diamond A, Ostrander EA and Stanford JL: Rare germline mutations in
the BRCA2 gene are associated with early-onset prostate cancer. Br
J Cancer. 97:826–831. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Gallagher DJ, Cronin AM, Milowsky MI,
Morris MJ, Bhatia J, Scardino PT, Eastham JA, Offit K and Robson
ME: Germline BRCA mutation does not prevent response to
taxane-based therapy for the treatment of castration-resistant
prostate cancer. BJU Int. 109:713–719. 2012. View Article : Google Scholar
|
|
106
|
Agalliu I, Gern R, Leanza S and Burk RD:
Associations of high-grade prostate cancer with BRCA1 and BRCA2
founder mutations. Clin Cancer Res. 15:1112–1120. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Kirchhoff T, Kauff ND, Mitra N, Nafa K,
Huang H, Palmer C, Gulati T, Wadsworth E, Donat S, Robson ME, et
al: BRCA mutations and risk of prostate cancer in Ashkenazi Jews.
Clin Cancer Res. 10:2918–2921. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Lehrer S, Fodor F, Stock RG, Stone NN, Eng
C, Song HK and McGovern M: Absence of 185delAG mutation of the
BRCA1 gene and 6174delT mutation of the BRCA2 gene in Ashkenazi
Jewish men with prostate cancer. Br J Cancer. 78:771–773. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Cesaretti JA, Stock RG, Atencio DP, Peters
SA, Peters CA, Burri RJ, Stone NN and Rosenstein BS: A genetically
determined dose-volume histogram predicts for rectal bleeding among
patients treated with prostate brachytherapy. Int J Radiat Oncol
Biol Phys. 68:1410–1416. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Zhu Y, Marshall D, Garrett-Mayer E, Zhou D
and Jenrette J: Association between urinary morbidity/erectile
dysfunction induced by radiotherapy and SNPs of
radiosensitivity-relevant genes in prostate cancer patients. Clin
Chem. 56:A902010.
|
|
111
|
Pomerantz MM, Spisák S, Jia L, Cronin AM,
Csabai I, Ledet E, Sartor AO, Rainville I, O'Connor EP, Herbert ZT,
et al: The association between germline BRCA2 variants and
sensitivity to platinum-based chemotherapy among men with
metastatic prostate cancer. Cancer. 123:3532–3539. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Hart SN, Ellingson MS, Schahl K, Vedell
PT, Carlson RE, Sinnwell JP, Barman P, Sicotte H, Eckel-Passow JE,
Wang L, et al: Determining the frequency of pathogenic germline
variants from exome sequencing in patients with castrate-resistant
prostate cancer. BMJ Open. 6:e0103322016. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Antonarakis ES, Lu C, Luber B, Liang C,
Wang H, Chen Y, Silberstein JL, Piana D, Lai Z, Chen Y, et al:
Germline DNA-repair gene mutations and outcomes in men with
metastatic castration-resistant prostate cancer receiving
first-line abiraterone and enzalutamide. Eur Urol. 74:218–225.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Nastiuk KL, Mansukhani M, Terry MB,
Kularatne P, Rubin MA, Melamed J, Gammon MD, Ittmann M and
Krolewski JJ: Common mutations in BRCA1 and BRCA2 do not contribute
to early prostate cancer in Jewish men. Prostate. 40:172–177. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Schweizer MT, Cheng HH, Tretiakova MS,
Vakar-Lopez F, Klemfuss N, Konnick EQ, Mostaghel EA, Nelson PS, Yu
EY, Montgomery B, et al: Mismatch repair deficiency may be common
in ductal adenocarcinoma of the prostate. Oncotarget.
7:82504–82510. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Stephens PJ, Gay LM, Ali SM,
Hoffman-Censits JH, Elvin JA, Vergilio JA, Suh J, Mian B, Fisher
HAG, Nazeer T, et al: Comprehensive genomic profiling of
neuroendocrine carcinoma of the prostate. J Clin Oncol. 34(Suppl
15): 1872016. View Article : Google Scholar
|