Introduction
Cervical cancer is one of the most prevalent cancer
worldwide with an incidence of 500,000 new cases and mortality of
270,000 women recorded in 2018 (1). Patients with high levels of estrogen
and the estrogen receptor alpha (ERα) are at increased risk of
developing this type of tumor (2).
Recently, our group described the tumor-promoting effect of
17β-estradiol (E2) through the induction of changes in cervical
cancer cells metabolism (3). It
remains to define how E2 produces these modifications.
Metabolism is one of the most complex processes in
biological systems. The effect of the metabolism in cancer cells
was first described by Otto Warburg in 1924 (4). Several hypotheses have been proposed
to explain how tumor cells activate glycolysis under aerobic
conditions. Asgari et al (5) described seven subsystems that form
the basis of the Warburg effect, including glutamine metabolism,
nucleotides, glycolysis, oxidative phosphorylation system (OXPHOS),
pentose phosphate pathway, tricarboxylic acid (TCA) cycle and
pyruvate metabolism. Even though Warburg postulated impaired
mitochondrial respiration in tumor cells (4), the role of mitochondria in the
malignant transformation is still not well understood.
Mitochondria are organelles with vital roles in
cellular energy production. Functionally, they are best known for
their ability to generate the majority of ATP and free radicals
from NADH and FADH using molecular oxygen (O2) via
electron transfer coupled with the OXPHOS (5). Also, they are involved in the
modulation of various cellular processes, such as the intracellular
calcium homeostasis, fatty acid oxidation, urea cycle, biosynthesis
of amino acids, lipids, hemes and purines, and other central
metabolic pathways. A close link between attenuated mitochondrial
bioenergetics and enhanced glycolysis dependency is present in
human tumor cells (6).
Estrogen has been shown to influence mitochondrial
structure, biogenesis and activity (7). The majority of the biological effects
of E2 are mediated via two ERs, namely ERα and ERβ. Both ERs are
localized in the mitochondria and they play an important role in
the regulation of the organelle structure and function (7,8).
Under various stress conditions, the major function
of E2 is to maintain OXPHOS in mitochondria, explaining the
neuroprotective and cardioprotective effects of estrogens (9,10).
The stressed mitochondria produce excessive amounts of reactive
oxygen species (ROS), which can damage in the lipid bilayers,
mutate DNA and alter the activity of specific enzymes critical for
the maintenance of oxidative function (11). Estrogens act as free-radical
scavengers, and modify the cytosolic and mitochondrial influx of
calcium ions (Ca2+) potentially providing protection to
the cell from toxic Ca2+ influx (12). Collectively, these effects indicate
that the hormone protects from mitochondrial membrane potential
collapse, avoids the loss of the inner mitochondrial membrane
integrity and inhibits the release of pro-apoptotic factors.
In tumor cells, there is a change in the metabolic
profile to support proliferation and the increased biosynthetic
demands. These cells have an alteration of the mitochondrial
activity and a high rate of glycolysis, followed by increased
lactic acid production under aerobic conditions (13). In this work, we tested the
association between the activity of E2 and its ability to moderate
the Warburg effect and the mitochondrial function in cell lines
derived from cervical cancer with the aim to define the role of
this hormone in the process of cervical carcinogenesis.
Materials and methods
Reagents
E2, phorbol myristate acetate (PMA; cat. no. PI585-1
mg), ionomycin calcium salt (cat. no. IO634), cisplatin (cat. no.
479306), sulfuric acid (cat. no. 7664-93-9), phosphotungstic acid
hydrate (cat. no. 12501-23-4), 2-thio-barbituric acid (TBA; cat.
no. 504-17-6) and metformin were purchased from Calbiochem (cat.
no. D150959; Merck KGaA). 1-[4-(6-bromobenzo[1,3](8)dioxol-5yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl]-ethanone
(G-1; cat. no. 10008933) was obtained from Cayman Chemical Company.
DMEM, charcoal stripped fetal bovine serum (FBS) and antibiotics
were purchased from Gibco (Thermo Fisher Scientific, Inc.).
mitoCapture™ (cat. no. K250) was acquired through BioVision, Inc.,
and 1-butanol and Baker Analyzed™ A.C.S. were from Avantor,
Inc.
Cell lines
Cervical carcinoma cell lines were obtained from the
American Type Culture Collection, including HeLa [positive to human
papillomavirus (HPV) 18], SiHa (positive to HPV 16) and C33A
(negative to HPV). Non-tumorigenic HaCaT was cultured as mock
control; cells maintained normal properties of differentiation and
were kindly provided by Dr. Petra Boukamp (German Cancer Research
Center; DKFZ). Cell lines were tested and authenticated by the
Multiplexion's Multiplex Cell Line Authentication tests in
September 2018 (www.multiplexion.de).
Cell culture
Cells were cultured in DMEM containing 1% antibiotic
[penicillin G (10,000 U/ml) and streptomycin (10,000
µg/ml)], amphotericin B (250 µg/ml) and 10% of
charcoal-stripped FBS at 37°C in a humidified atmosphere with 5%
CO2. Cells were grown until reaching 70-80% confluence.
Three to four passages were achieved before the experiments were
performed.
Mitochondrial permeability
Cell lines were cultured on 8-well slides (cat. no.
177402; Sigma-Aldrich; Merck KGaA) at 5×103/well for
HeLa and SiHa or at 10×103/well for C33A and HaCaT.
Cells were stimulated with E2 (10 nM), as previously determined
(3) and incubated for 24 h.
Staining was performed using the MitoCapture™ staining according to
the manufacturer's instructions and examined under a fluorescence
microscope coupled to a digital camera (magnification, ×10 and
×40). In healthy cells, dye aggregates and stains the mitochondria
red; in abnormal cells, the mitochondrial membrane potential
collapses and the dye remains in the cytoplasm in its green
fluorescent monomeric form. As in recent years evidence suggested
that low concentrations of chemotherapeutic agents, like cisplatin,
facilitate malignancy of carcinoma cells (14,15),
cisplatin (0.5 µg/ml) was used as control of cellular and
mitochondrial damages and were compared with a basal control (no
treatment). The evaluation of the mean optical density was
determined using Image-pro Plus 6.0 (Media Cybernetics, Inc.) based
on the red staining; data were normalized to the fluorescence of
vehicle-treated cells. Each sample was tested by triplicate in
three independent experiments.
Calcium pathway
The effect of E2 (10 nM) on intracellular
Ca2+ concentration was measured using the Fluo-4 Calcium
kit (cat. no. F36206; Molecular Probes; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. The supernatant
of the cells was obtained 48 h after applying E2. Ionomycin (750
ng/ml) and PMA (10 ng/ml) stimuli were applied for 5 min at 37°C
before the analysis of the samples in the spectrophotometer.
Excitation and emission wavelengths were 494 and 516 nm,
respectively. Readings were recorded every minute over 16 min. G1
(1 µM) was used as a positive control alone or in
combination with PMA (10 ng/ml) and ionomycin (750 ng/ml) and were
compared with a basal control.
Nitric oxide (NO) concentration
Cells (5×103) were seeded in 96-well
plates with DMEM containing 10% charcoal-stripped FBS. Cells were
stimulated with E2 (10 nM) and/or metformin (10 mM) and were
compared with a basal control. After 48 h, nitrite concentrations
were measured using the Measure-iT™ High-Sensitivity Nitrite assay
(Invitrogen; Thermo Fisher Scientific, Inc.). The amount of nitrite
is extrapolated to NO concentrations following the manufacturer's
instructions. Reagent (Measure-iT™ nitrite quantitation reagent;
2.0 ml of 100X in 0.62 M HCl; 1:100; 100 µl) was added to
the wells and samples (10 µl) were added. After 10 min at
room temperature, 5 µl developer solution was added and the
fluorescence was measured using a spectrophotometer (excitation and
emission wavelength, 360 and 460 nm, respectively).
Lipid peroxidation
The levels of lipid peroxides were calculated via
determining the malondialdehyde (MDA) concentrations. Cells were
stimulated with E2 (10 nM) and/or metformin (10 mM) and were
compared with a basal control. After 48 h, 300 µl cell
supernatant was mixed with 2 ml sulfuric acid (N/12), 0.3 ml 10%
phosphotungstic acid and 1 ml 0.6% TBA. The mixture was placed in a
boiling water bath at 95°C for 1 h, followed by cooling at room
temperature. Then, 1.3 ml of n-butane was added, mixed vigorously
and centrifuged at 1,400 × g for 15 min. The absorption of the
organic phase was recorded at 534 nm.
Concentration of iron and ferritin
Cell supernatants were used to determine iron
concentrations and to evaluated ferritin levels. Cells were
stimulated with E2 (10 nM) and incubated for 48 h and were compared
with a basal control. The VITROS Fe slide method was performed
using the VITROS Fe slides and the VITROS Calibrator 4 kit (Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. The difference in the density of the reflectance
after 1-5 min is proportional to the concentration of iron in the
samples. The determination of the ferritin levels was determined
using the same samples with the IMMULITE® kit (cat. no.
L2KFE2; Siemens Healthineers) following the manufacturer's
instructions.
Concentration of glucose
Cells were stimulated with E2 (10 nM) and/or
metformin (10 mM) and were compared with a basal control. The
supernatant was obtained after 48 h. Glucose concentrations were
measured in the BS-120 Clinical Chemistry analyzer (Shenzhen
Mindray Bio-Medical Electronics Co., Ltd.) using the glucose
oxidase method (COD11803; BioSystems S.A.) following the
manufacturer's instructions. Glucose in the samples generated a
colored complex that was quantified spectrophotometrically at 500
nm.
Concentration of lactic acid
Cells were stimulated with E2 (10 nM) and/or
metformin (10 mM) and were compared with a basal control. The
supernatant was obtained after 48 h. The VITROS LAC Slide method
was performed using the VITROS LAC slides and the VITROS Calibrator
kit 1 (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Lactate in samples is oxidized to
pyruvate and hydrogen peroxide (H2O2). The
generated H2O2 oxidizes 4-aminoantipy-rine,
which forms a complex with 1,7-dihydroxynaphthalene. Absorbance at
540 nm was determined spectrophotometrically.
RNA extraction
Cells were stimulated with E2 (10 nM) and incubated
for 48 h and were compared with a basal control. Total RNA was
extracted from SiHa and HeLa using the RNeasy Plus Mini kit (cat.
no. 74136; Qiagen, Inc.), according to the manufacturer's
instructions. RNA was quantified measuring the absorbance at
260/280 nm.
RNA sequencing
Transcriptome sequencing was performed using RNAseq
technology. The samples (SiHa and HeLa cells) stimulated with E2
were sent to the Genome Institute (Novogene Corporation, Inc.),
where the Illumina HiSeq 4000 system was used; the sequencing
strategy included a 250-300 bp insert cDNA library, HiSeq platform,
paired-end 150 and 50 bp single-end. The RNA quality was measured
and selection criteria were: Amount ≥1 µg; volume ≥20
µl; concentration ≥50 ng/µl; RNA integrity number
value ≥7; 28S/18S >1; no degradation or pollution. Subsequently,
fragmentation of mRNA and synthesis of cDNA of the first and second
chain was performed. For the mRNA isolation and fragmentation, 200
ng total RNA was purified using oligo-dT beads and poly
(A)-containing mRNA were fragmented into small pieces with Fragment
Buffer. For the cDNA synthesis, first-strand cDNA was generated
using the First Strand Master Mix and Super Script II (Invitrogen;
Thermo Fisher Scientific, Inc.) for 25°C for 10 min, 42°C for 50
min and 70°C for 15 min. Then Second Strand Master Mix was added to
synthesize the second-strand cDNA (16°C for 1 h).
cDNA fragments were purified, resolved for the final
repair and the adenine single nucleotide addition was performed.
cDNA fragments were ligated using adapters and fragments with a
suitable size were selected for PCR amplification. The library
construct was prepared for the quantification and qualification of
libraries. For the end repair, the purified fragmented cDNA was
combined with End Repair Mix and incubated at 30°C for 30 min. The
end-repaired DNA was purified with Ampure XP beads (Thermo Fisher
Scientific, Inc.) and incubated with A-Tailing Mix at 37°C for 30
min. For the adapter ligation, the adenylated 3'-DNAs were combined
and RNA Index Adapter and Ligation Mix was added at 30°C for 10
min. The end-repaired DNA was purified with Ampure XP beads.
Several rounds of PCR amplification with the PCR Primer Cocktail
and PCR Master Mix were performed to enrich the cDNA fragments and
products were purified with the Ampure XP beads.
For the validation of the library, quantitation was
performed in two ways: (i) Determination of the mean molecule
length using the Agilent 2100 bioanalyzer (Agilent Technologies,
Inc.); (ii) reverse transcription-quantitative PCR (TaqMan Probe;
Illumina, Inc.). Library sequencing was performed as follows: (i)
Amplification within the flow cell on the Bot instrument for
cluster generation (HiSeq® 4000 PE Cluster Kit;
Illumina, Inc.); and (ii) loading of the clustered flow cell onto
the HiSeq 4000 Sequencer for paired-end sequencing (Illumina; Inc.)
with recommended read lengths of 100 bp.
Downstream analysis was performed using a
combination of programs, including FLEXBAR version 2.5 for
filtration (16). Alignments of
readings with index hg38 were parsed employing Kallisto version
0.44.0 (17). Differential
expression analysis between groups (two biological replicates per
group) was performed using the DESeq2 R package version 1.24.0
(18). The resulting P-values were
adjusted applying the Benjamini-Hochberg approach to control the
false discovery rate (FDR). Genes with an adjusted P<0.05 were
considered as differentially expressed. An absolute fold change of
1 was considered in the significant differential expression. Gene
Ontology (19) and KEGG enrichment
(20,21) were implemented by the Cluster
Profiler R package (https://www.R-project.org). A corrected P<0.05 was
considered significantly enriched by differential expressed genes
(DEGs).
Cell proliferation assay
Cell proliferation of HeLa, SiHa, C33A and HaCaT
cells was assessed using the xCELLigence platform (Roche Applied
Science). Cells were cultured at 0.25×104/well in
96-well E-plates (cat. no. 058416; ACEA Biosciences, Inc.) and were
introduced into the xCELLigence reading station. After 4 h, cells
were stimulated with 10 nM E2, 10 mM metformin, 1 µg/ml
cisplatin alone or in combination (E2+metformin, E2+cisplatin and
E2+metformin+cisplatin), a vehicle-stimulated (DMEM+ethanol) cells
and were compared with basal control. The proliferation rate was
determined in real time every 30 min over 72 h. Cisplatin
stimulation was used as a cell death control. Three independent
experiments were performed with three replicates in each case.
Statistical analysis
Statistical analyses were made using SPSS 20 (IBM
Corp.) and GraphPad Prism 6 (GraphPad Software, Inc.). Experiments
were performed in triplicate and tested in three independent
trials. One-way ANOVA with Bonferroni correction was used to
determine significance following Levene's test for homogeneity of
variances. Data are presented as the mean ± standard error of the
mean. P<0.05 was considered to indicate a statistically
significant difference.
Results
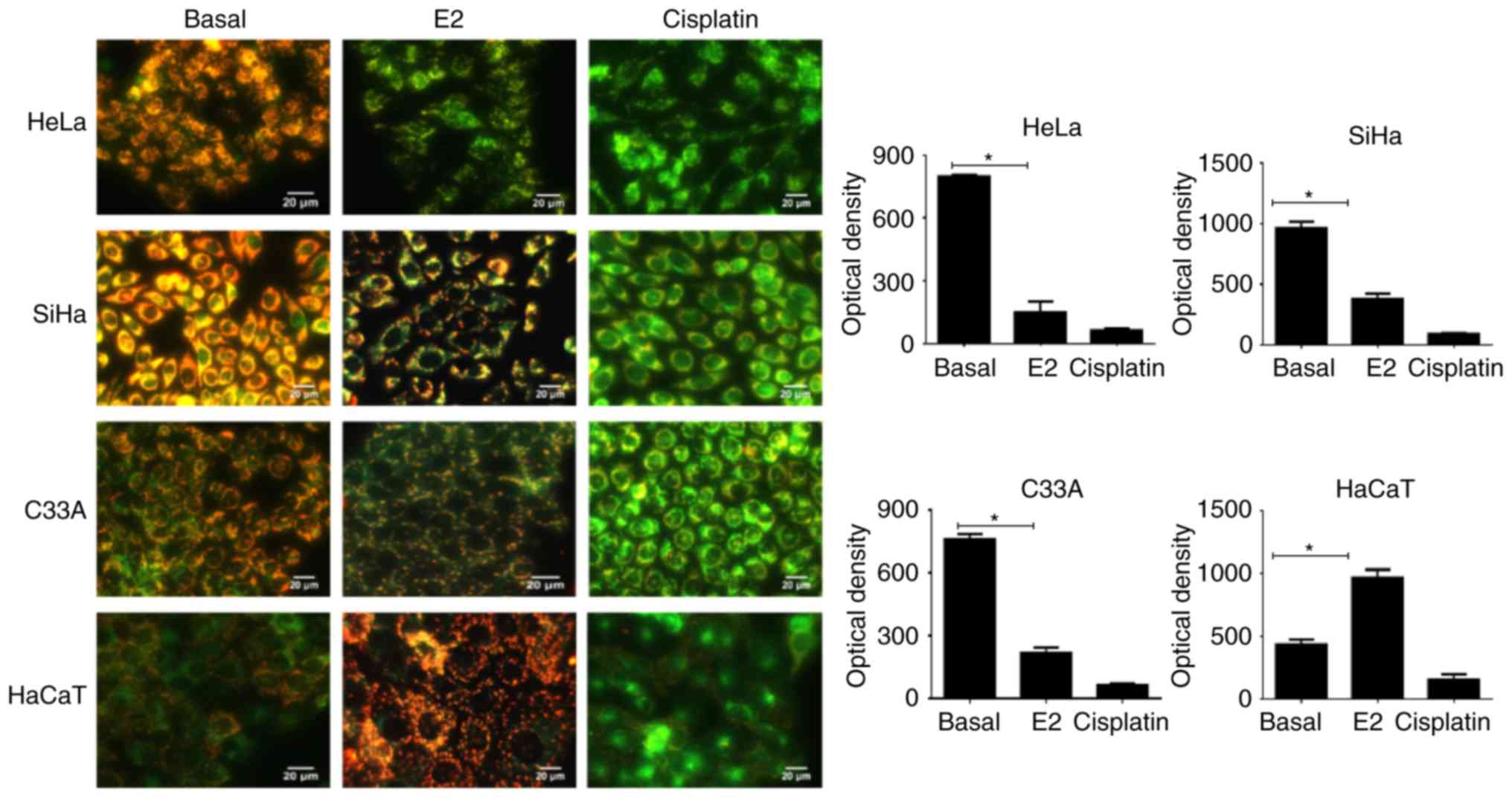
E2 decreases the mitochondrial membrane
permeability in cervical cancer cell lines
The effect of E2 on the mitochondrial permeability
of cervical cancer and HaCaT cell lines was evaluated using a
selective dye. In HeLa, SiHa and C33A, E2 significantly decreased
the mitochondrial permeability compared with the basal control
(P<0.0001; Fig. 1). The green
color indicated that the dye accumulated in the cytoplasm. In HaCaT
cells, the opposite effect was observed with increased
mitochondrial permeability following E2 treatment as suggested by
the red color (P<0.0001).
E2 alters the calcium pathways in SiHa
and HeLa
E2 had no significant effect on the calcium
concentrations in cervical cancer cells or HaCaT compared with the
basal control (P>0.05; Fig. 2).
The use of ionomycin plus PMA significantly increased the ion
concentration compared with the basal control (HeLa, P=0.003; SiHa,
P=0.001; C33A, P=0.001). Exposure to E2, ionomycin and PMA
significantly decreased the calcium concentration in SiHa and HeLa
compared with the associated control (P<0.05), suggesting that
E2 altered the calcium signaling pathway. In C33A and HaCaT, E2 did
not significantly modify the cellular response to stimulation with
ionomycin plus PMA (P>0.05). G1, a selective agonist of the ER
in the membrane (GPR30) associated with non-genomic effects, such
as calcium mobilization, was used as positive control. E2 and G1
exhibited similar behaviors in all cell lines.
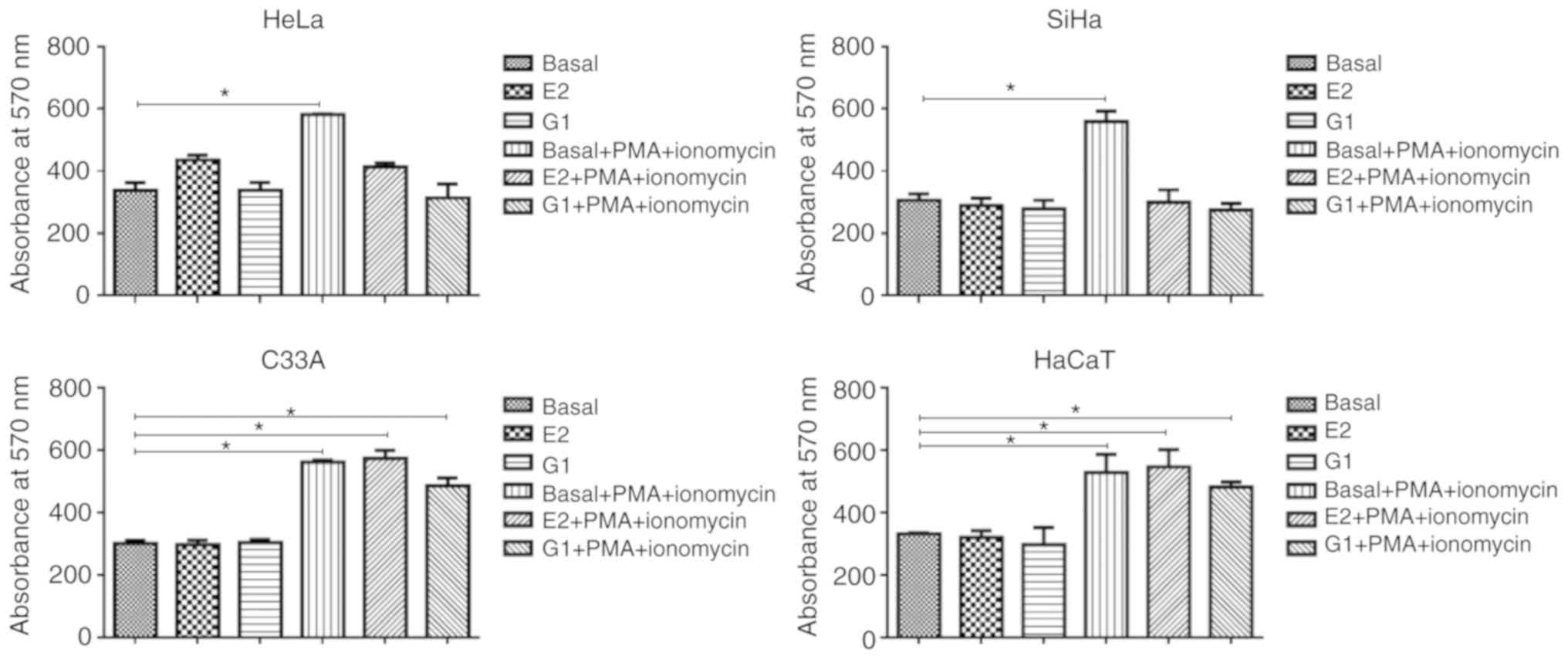 | Figure 2E2 affects the calcium regulation in
cervical cancer cells. Ca2+ levels were determined in
HeLa, SiHa and C33A cervical cancer cells and HaCaT following
various treatments. Experiments were performed in triplicate and
with three repeats. *P<0.05. E2, 17β-estradiol; PMA,
1,1,3,3-tetramethoxypropane; G1,
1-[4-(6-bromobenzo[1,3]dioxol-5yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl]-ethanone. |
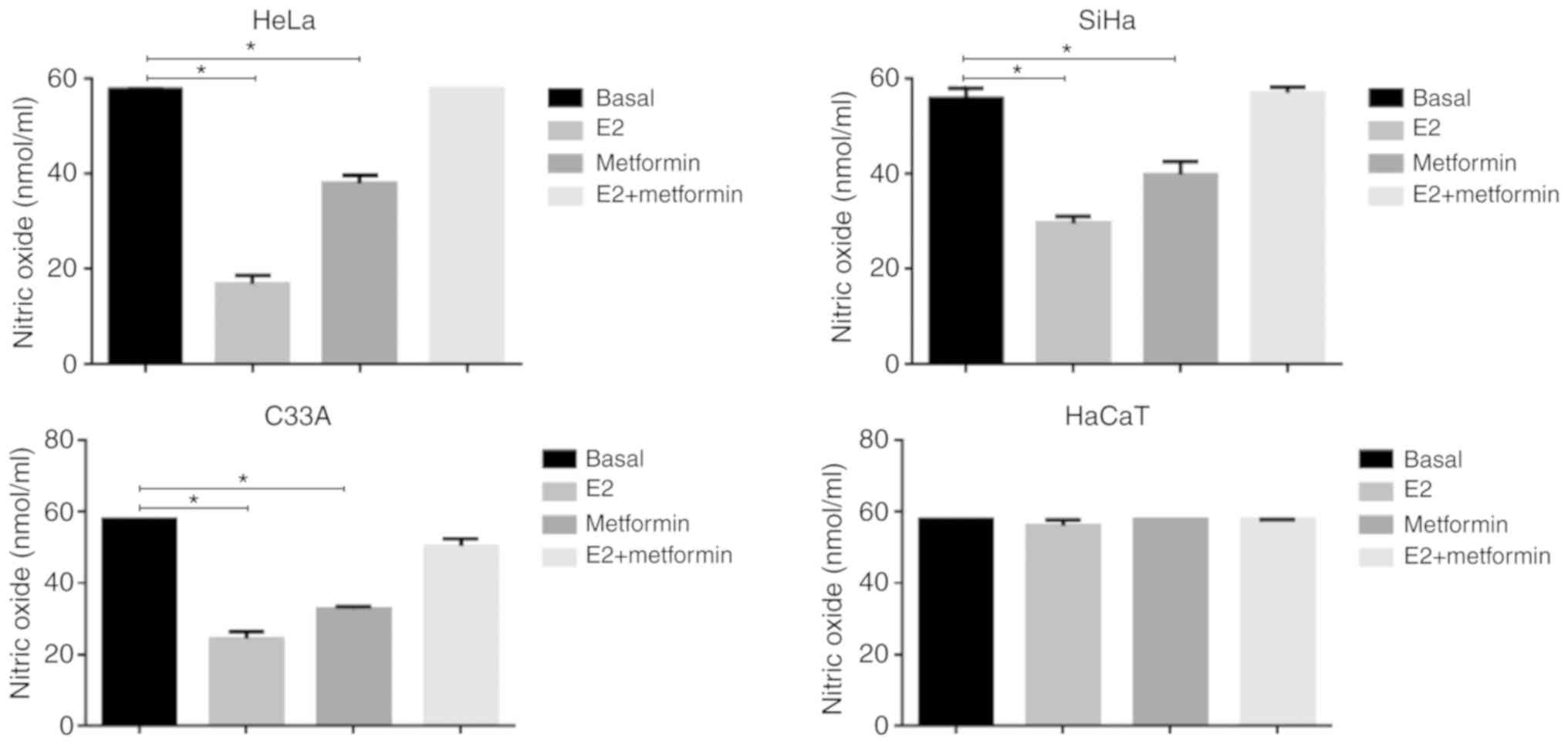
E2 has an antioxidant effect in cervical
cancer cell lines
To evaluate the participation of E2 in other
mechanisms involved in the mitochondrial activity, we analyzed
whether E2 modulated oxidative stress. First, we determined the
effect of E2 on the concentration of NO in cervical cancer cells,
as well as in HaCaT. E2 significantly decreased NO levels in HeLa,
SiHa and C33A compared with the basal control (P=0.0002, P=0.0014
and P=0.0003; respectively; Fig.
3). This effect was significantly reversed when treating cells
with metformin and E2 (P<0.05). In HaCaT cells, no differences
were observed following treatment with E2, metformin or a
combination of these compared with the control.
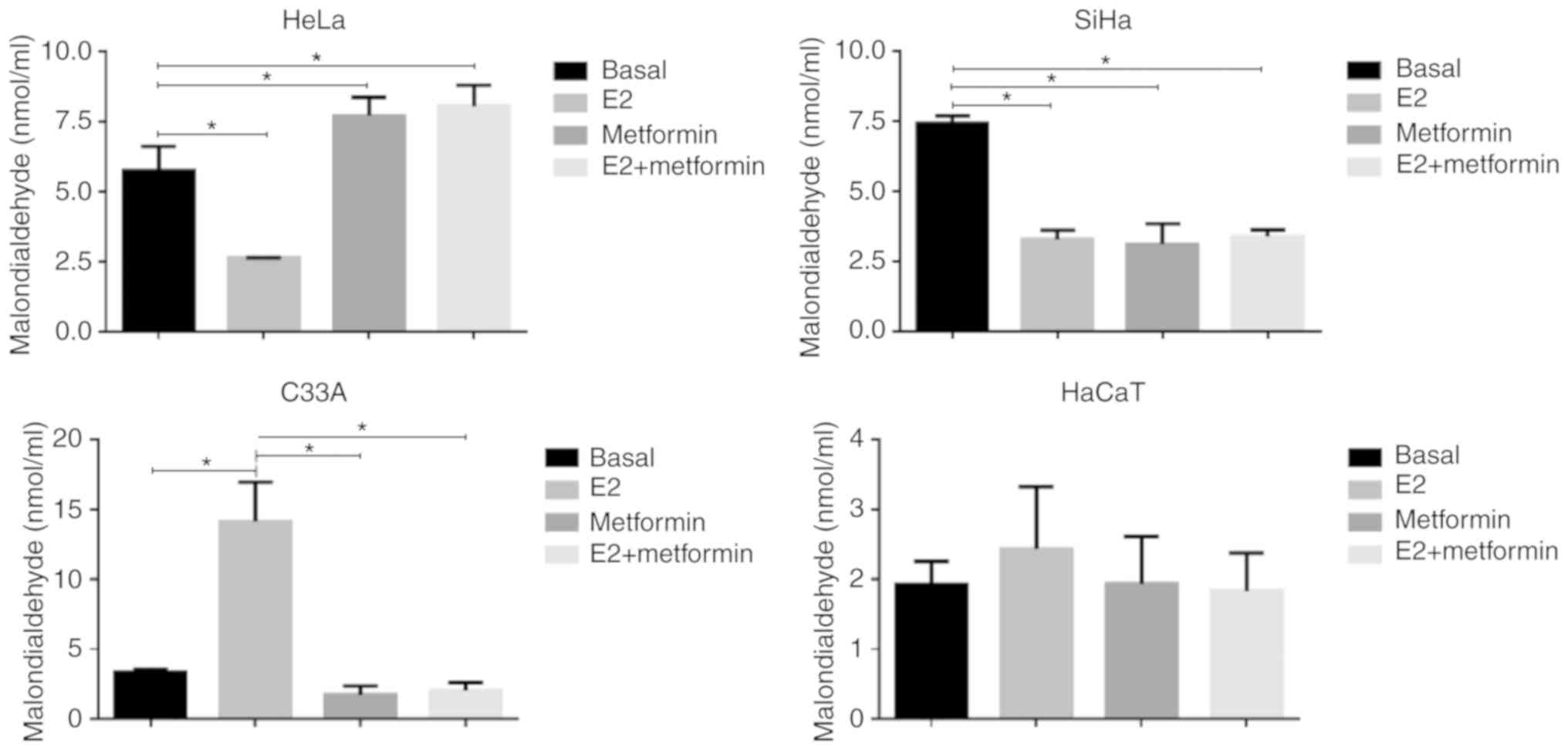
Furthermore, we analyzed the concentration of MDA,
which is a final product of the lipid peroxidation and is used as a
direct indicator of cell damage (22). E2 induced a significant decrease in
lipid peroxidation in HeLa (P=0.0004) and SiHa (P=0.0005; Fig. 4) compared with the basal control.
In C33A, the effect was the opposite and lipid peroxidation was
significantly increased following E2 treatment (P=0.0002).
Treatment with metformin significantly alleviated E2 effects in
HeLa and C33A cells (P<0.05), but no significant effects were
observed in SiHa (P>0.05). In HaCaT cells, no significant
differences were observed following treatment with E2, metformin or
a combination of these compared with the control.
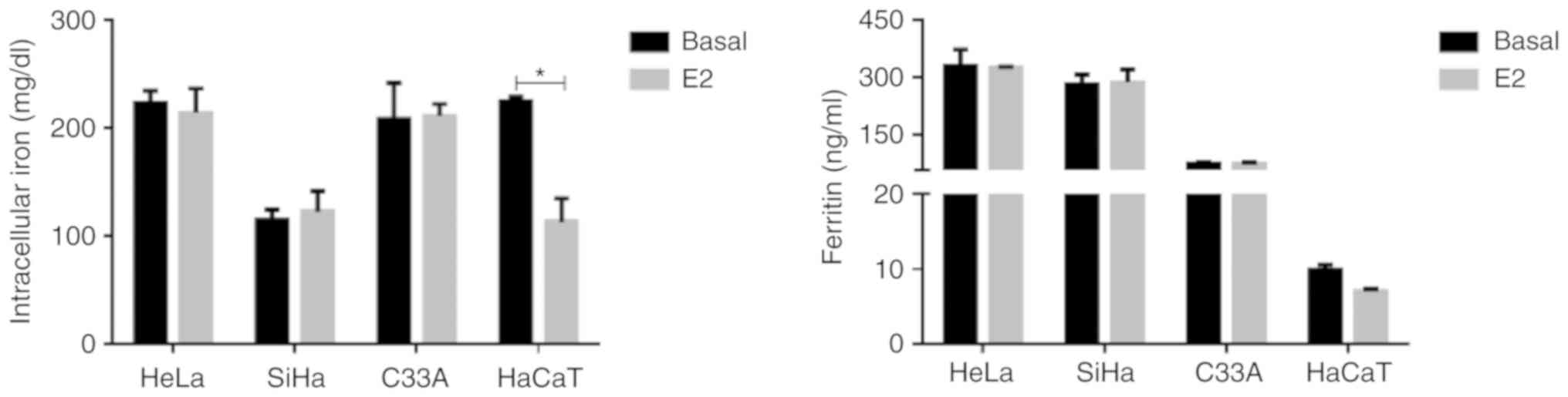
E2 does not modify iron metabolism in
cervical cancer cells
The role of iron in lipid peroxidation has been
associated with a form of cell death other than apoptosis or
necrosis. Ferritin binds to iron and this complex is vital in
controlling ROS generation (22).
E2 stimulus did not significantly modify intracellular iron or
ferritin levels in cervical cancer cells compared with the basal
control (P>0.05; Fig. 5). In
HaCaT, intracellular iron was significantly decreased following E2
treatment compared with the basal control (P=0.007).
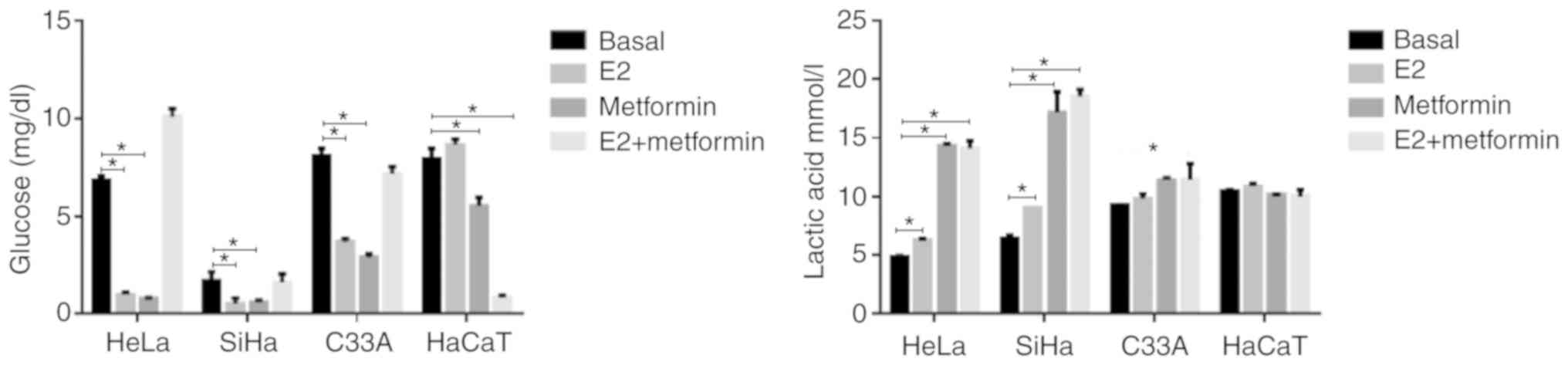
E2 favors the Warburg effect
The mitochondrial bioenergetics dysfunction favors
the activation of the glycolytic pathway in different types of
tumors (13). In HeLa, SiHa and
C33A supernatants, there was a significant decrease in glucose
levels following E2 stimulus (P=0.0001, P=0.002 and P=0.0003;
respectively; Fig. 6), suggesting
that the cells increased the uptake of glucose. A similar
significant decrease was observed using metformin treatment
(P<0.01). The combination of both treatments showed glucose
levels similar to those observed in the basal control (P>0.05).
It was hypothesized that the hypoglycemic effect of metformin was
associated with E2. Furthermore, E2 significantly increased lactic
acid levels in HeLa and SiHa compared with the basal control
(P=0.0001 and P=0.004; respectively). The effects were more
pronounced with metformin or a combination treatment with E2
(P<0.001).
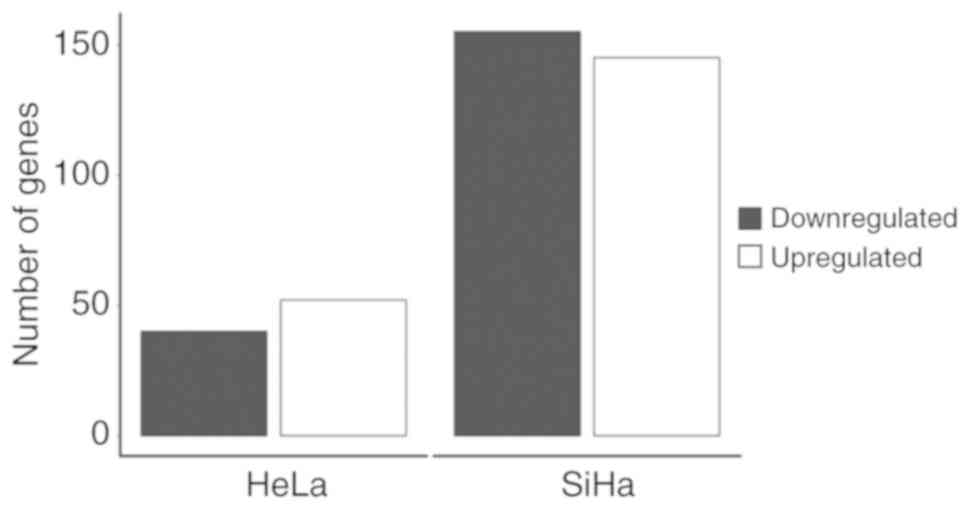
E2 modulates DEGs in SiHa and HeLa
Considering that E2 modified the metabolism of the
cervical cancer cells, we analyzed the effects of E2 on the
expression of molecules associated with metabolic pathways using
the RNA sequencing technology. The total number of DEGs in SiHa
following E2 treatment was ~3-fold higher than that in HeLa cells
(Fig. 7). E2 stimulus had an
impact on various metabolic pathways, including the upregulation of
genes involved in the synthesis of proteins associated with the
mitochondrial electron transport chain, which may affect OXPHOS and
the energy metabolism (Table I).
Additionally, upregulation of genes associated with glycolysis was
observed (Table I and II), supporting the theory of the role
played by E2 in the regulation of the Warburg effect associated
with cervical cancer (Fig. 8).
Defects in the metabolism of lipids, amino acids, proteoglycans,
nucleotides, inositol phosphate, and choline have been reported in
different types of cancer (23-25).
E2 stimulation modulated various metabolic pathways (Table I). Table II summarized the DEGs shared by
HeLa and SiHa.
 | Table IDifferential expression of genes
associated with the metabolism in HeLa and SiHa cells following
17β-estradiol stimulation. |
Table I
Differential expression of genes
associated with the metabolism in HeLa and SiHa cells following
17β-estradiol stimulation.
A, HeLa
|
|---|
| Pathway | N | Downregulated
genes | Upregulated
genes |
|---|
| Oxidative
phosphorylation | 24 | NDUFV1, COX6B2,
TCIRG1 | COX6C, NDUFAB1,
NDUFA4, NDUFS5, COX5B, COX7C, COX7B, ATP5F1A, ATP5F1B, ATP5F1E,
ATP5MC2, APT5MC3, ATP5MG, ATP5PB, ATP5PO, COX6A1, COX6B1, COX8A,
UQCRB, UQCRH, UQCRQ |
|
Glycolysis/gluconeogenesis | 6 | PFKP | GAPDH, LDHA,
LDHB, PGK1, TPI1 |
| Glycan biosynthesis
and metabolism | 18 | NDST1, SGSH,
LSS, MGAT5B, PIGQ, FUK, MGAT1, GPAA1, GAA, MAN2A2, MOGS, PIGQ,
B4GALT2 | EXT1, PIGK,
GALNT1, RPN2, TUSC3 |
| Lipid
metabolism | 14 | AGPS, FASN,
ACADVL, LSS, ACSL4, DGKD, SPHK1, MGAT1 | PTDSS1, PTGES3,
CDS2, HMGCS1, PLA2G4A, PPT1, CPS1 |
| Metabolism of
cofactors and vitamins | 8 | RDH10, NADK,
NAPRT | MTHFD2, NAMPT,
EPRS, RFK, PANK3 |
| Amino acid
metabolism | 12 | DNMT1, ANPEP,
OPLAH | MDH1, CPS1,
LDHA, HMGCS1, ODC1, TPI1, LDHB, GAPDH, CPS1 |
| Nucleotide
metabolism | 10 | POLD1, POLE,
CAD, AMPD2, IMPDH1 | PAICS, DUT,
GMPS, HPRT1, PAPSS2 |
| Choline
metabolism | 4 | DGKD,
PIP5K1C | PLA2G4A,
PPT1 |
| Inositol phosphate
metabolism | 4 | PLCD3, PIP5K1C,
INPPL1 | TPI1 |
|
B, SiHa
|
| Pathway | N | Downregulated
genes | Upregulated
genes |
|
| Oxidative
phosphorylation | 45 | ATP5F1D,
ATP6AP1, ATP6V0C, ATP6V0E1, ATP6V0E2, CYC1, NDUFA1, NDUFA11,
NDUFS6, NDUFS7 | ATP6V1E1,
ATP5MC3, NDUFA4, NDUFA5, NDUFA8, NDUFAB1, NDUFB3, NDUFB4, NDUFS5,
NDUFV2, ATP6V1B2, ATP6V1C1, COX15, ATP5F1C, ATP5F1E, ATP5MC2,
ATP5ME, ATP5MG, ATP5PB, ATP5PD, ATP5PF, ATP5PO, COX5A, COX5B,
COX6A1, COX6C, COX7B, COX7C, ATP5F1A, ATP5F1B, SDHB, SDHC, UQCRB,
UQCRC2, UQCRH |
|
Glycolysis/gluconeogenesis | 13 | ALDH1A3,
ALDH3B1, ALDOA, PFKL, PFKP | DLAT, ALDH7A1,
PGK1, TPI1, PKM, LDHA, LDHB, ADH5 |
| Glycan biosynthesis
and metabolism | 40 | ALG12, ALG14,
B3GALT6, B3GAT3, B4GALNT1, B4GALT7, CHPF, GAA, GALNT2, GPAA1,
HYAL2, IDUA, LSS, MAN1B1, MAN2A2, MGAT4B, MGAT5B, MOGS, NAGLU,
NDST1, SGSH, ST3GAL1, ST3GAL2, ST6GALNAC4, ST6GALNAC6, XYLT1,
XYLT2 | B4GALT1, FKTN,
C1GALT1C1, ALG8, EXT1, EXTL2, CSGALNACT2, PIGF, PIGK, PIGN, DPM1,
STT3A, TUSC3 |
| Lipid
metabolism | 59 | LSS, ACADVL,
ADO, AGPAT2, AGPS, B4GALNT1, CAD, CBR1, CDIPT, CKB, DGKQ, DGKZ,
DHCR7, FASN, GPAT4, HSD17B1, MCAT, MGAT1, MGLL, PAFAH1B3 PGP, PGS1,
PISD, PLPP1, PLPP2, PNPLA2, PTDSS2, PTGES2, SGMS2, SMPD4, SPHK1,
SPHK2, ST3GAL1, ST3GAL2, TKFC, ST6GALNAC4, ST6GALNAC6 | LTA4H, ACSL3,
ACADSB, PTGES3, PAFAH1B1, ELOVL5, HACD3, HADH, HADHB, CRLS1, ETNK1,
KDSR, SPTLC2, CYP51A1, CPS1, B4GALT1, ALDH7A1, ACAT1, HADHA, PPT1,
SMS, PLA2G4A |
| Metabolism of
cofactors and vitamins | 21 | ALPL, ALPP,
COASY, FPGS, NADK, NAPRT, PANK4, PDXK, PDXP, PNPO, SHMT1,
SHMT2 | ACP1, DHFR, NNT,
MTHFD1, PANK1, EPRS, PNP, PSAT1, COX15 |
| Amino acid
metabolism | 56 | ALDH6A1, ALDOA,
AMD1, AMDHD1, ASS1, BCAT2, SRM, BCKDHA, CAD, CBS, CHDH, DNMT3A,
FAHD1, GAMT, GLUL, GPT2, MPST, MRI1, NAT8L, OAT, OGDH, OPLAH, PGP,
PHGDH, PYCR1, PYCR3, SHMT1, SHMT2 | GOT2, PSAT1,
ASNS, AHCYL2, RIMKLB, P4HA1, SAT1, ODC1, MAT2B, GCLM, MAT2A, KYNU,
HIBADH, MCCC2, ACAA2, ATIC, IDH1, PGK1, TPI1, PKM, LDHA, LDHB,
GCSH, DBT, HIBCH, HADHA, ACADSB, CPS1 |
| Nucleotide
metabolism | 39 | IMPDH1, ADK,
AMPD2, PFAS, GUK1, POLD1, POLD2 TK1, POLR1A, POLR2E, POLR2F,
POLR2L, POLR3H, NME3, NME4, POLD4, DCTPP1, TYMP | GMPS, HPRT1,
ADSL, ADSS, PAICS, HPRT1, IMPDH2, GART, POLA1, POLD3, POLE3,
POLR2B, POLR2G, PRIM2, ME1, NME7, NT5C3A, CMPK1, UMPS, PKM,
NME1 |
| Choline
metabolism | 8 | CKB, DGKQ, DGKZ,
PLPP1, PLPP2, PIP5K1C | PLA2G4A,
PPPI |
| Inositol phosphate
metabolism | 4 | PLCD3, PIP5K1C,
INPPL1 | TPI1 |
| Table IIDifferentially expressed genes shared
by HeLa and SiHa associated with the metabolism following
stimulation with 17β-estradiol. |
Table II
Differentially expressed genes shared
by HeLa and SiHa associated with the metabolism following
stimulation with 17β-estradiol.
| Pathway | N | Downregulated
genes | Upregulated
genes |
|---|
| Oxidative
phosphorylation | 19 | TCIRG1 | COX6C, NDUFAB1,
NDUFA4, NDUFS5, COX5B, COX7C, COX7B, ATP5F1A, ATP5F1B, ATP5F1E,
ATP5MC2, ATP5MG, ATP5PB, ATP5PO, COX6A1, UQCRB, UQCRH,
MDH1 |
|
Glycolysis/gluconeogenesis | 5 | PFKP | LDHA, LDHB,
PGK1, TPI1 |
| Glycan biosynthesis
and metabolism | 10 | GAA, NDST1,
SGSH, PIGQ, MGAT5B, GPAA1, MGAT1 | EXT1, PIGK,
TUSC3 |
| Lipid
metabolism | 10 | MGAT1, AGPS,
FASN, ACADVL, CAD, SPHK1 | CPS1, PTGES3,
PLA2G4A, PPT1 |
| Metabolism of
cofactors and vitamins | 3 | NADK,
NAPRT | EPRS |
| Amino acid
metabolism | 9 | ANPEP, OPLAH,
CAD | MDH1, LDHA,
LDHB, ODC1, PGK1, TPI1, CPS1 |
| Nucleotide
metabolism | 9 | FUK, POLD1,
AMPD2, IMPDH1, PFAS | PAICS, GMPS,
HPRT1, NME1 |
| Choline
metabolism | 3 | PIP5K1C | PLA2G4A,
PPT1 |
| Inositol phosphate
metabolism | 4 | PLCD3, PIP5K1C,
INPPL1 | TPI1 |
E2 regulation of the differential expression of
transcripts with functional roles in the processes of energy
production was demonstrated by evaluating the impact on OXPHOS,
glycolysis/gluconeogenesis, lipid processing, vitamins and
cofactors, as well as on the metabolism of amino acids,
nucleotides, phosphatidylinositol and choline (Table I). E2 regulated the expression of
genes involved in mitochondrial complexes as follows: (i) Complex
I, NDUFAB1, NDUFA4, and NDUFS5 upregulated; (ii)
complex III, UQCRB and UQCRH upregulated; (iii)
complex IV, COX6C, COX5B, COX7C, COX7B and COX6A1
upregulated; (iv) complex V, ATP5F1A, ATP5F1E, ATP5F1B, ATP5MC2,
ATP5MG, ATP5PB and ATP5PO upregulated, and TCIRG1
downregulated.
Absolute changes in DEGs ranged between −0.4985 and
0.2496. A total of 5 genes associated with glycolysis were modified
in both HeLa and SiHa by E2 stimulus, including LDHA, LDHB,
PGK1, TPI1 and PFKP (absolute changes, −0.1893 to
0.2598). Other gene alterations modulated by E2 include: (i)
Metabolism and synthesis of glycans (n=10), with GAA, NDST1,
SGSH, PIGQ, MGAT5B, GPAA1 and MGAT1 downregulated and
EXT1, PIGK and TUSC3 upregulated; (ii) lipid
metabolism (n=10), with MGAT1, AGPS, FASN, ACADVL, CAD and
SPHK1 upregulated and CPS1, PTGES3, PLA2G4A and
PPT1 downregulated; (iii) metabolism of amino acids and
nucleotides (n=9); (iv) metabolism of inositol phosphate (n=4); and
(v) metabolism of choline, vitamins and cofactors (n=3; Table II and Fig. 8).
A total of 12 signaling pathways were affected by
E2, including hypoxia-inducible factor (HIF)-1, Ras,
mitogen-activated protein kinase (MAPK), vascular endothelial
growth factor (VEGF), phospholipase D, 5'-AMP-activated protein
kinase (AMPK), cGMP-dependent protein kinase (PKG), calcium,
appeline, phosphatidylinositol, PI3K-Akt and pathways mediated by
sphingolipid (Table III). It
should be noted that of these, genes that affected HIF-1, Ras,
MAPK, VEGF and phospholipase D pathways that were upregulated
included PGK1, LDHA and PLA2G4A (absolute
changes, 0.1606, 0.2167 and 0.2494, respectively). Decreased genes
following E2 stimulus included PFKP (−0.1893), FASN
(−0.5459), CAD (−0.3228), SPHK1 (−0.3164),
PLCD3 (−0.5829), INPPL1 (−0.7263) and LAMA5
(−0.7294), corresponding to the AMPK, cGMP-PKG, calcium,
sphingolipid-mediated, appeline, phosphatidylinositol and PI3K-Akt
signaling pathways (Table
III).
| Table IIIDifferentially expressed genes
affecting signaling pathways associated with metabolism shared by
HeLa and SiHa after stimulation with 17β-estradiol. |
Table III
Differentially expressed genes
affecting signaling pathways associated with metabolism shared by
HeLa and SiHa after stimulation with 17β-estradiol.
A, Downregulated
genes
|
|---|
| Signaling
pathway | Gene | P-value |
|---|
| VEGF | SPHK1 |
1.6706x10-2 |
| Phospholipase
D | SPHK1 |
1.6706x10-2 |
| PIP5K1C |
7.1061x10-3 |
| AMPK | PFKP |
1.0293x10-2 |
| FASN |
1.2382x10-34 |
| cGMP-PKG | CAD |
1.5215x10-4 |
| Calcium | SPHK1 |
1.6706x10-2 |
| PLCD3 |
8.8258x10-7 |
| Sphingolipid | SPHK1 |
1.6706x10-2 |
| Apelin | SPHK1 |
1.6706x10-2 |
|
Phosphatidylinositol | PLCD3 |
8.8258x10-7 |
| PIP5K1C |
7.1062x10-3 |
| INPPL1 |
1.7603x10-11 |
| PI3K-Akt | LAMA5 |
1.1881x10-85 |
|
B, Upregulated
genes
|
| Signaling
pathway | Gene | P-value |
|
| HIF-1 | PGK1, |
4.7152x10-4 |
| LDHA |
6.4214x10-7 |
| Ras | PLA2G4A |
9.7518x10-3 |
| MAPK | PLA2G4A |
9.7518x10-7 |
| VEGF | PLA2G4A |
9.7518x10-7 |
| Phospholipase
D | PLA2G4A |
9.7518x10-7 |
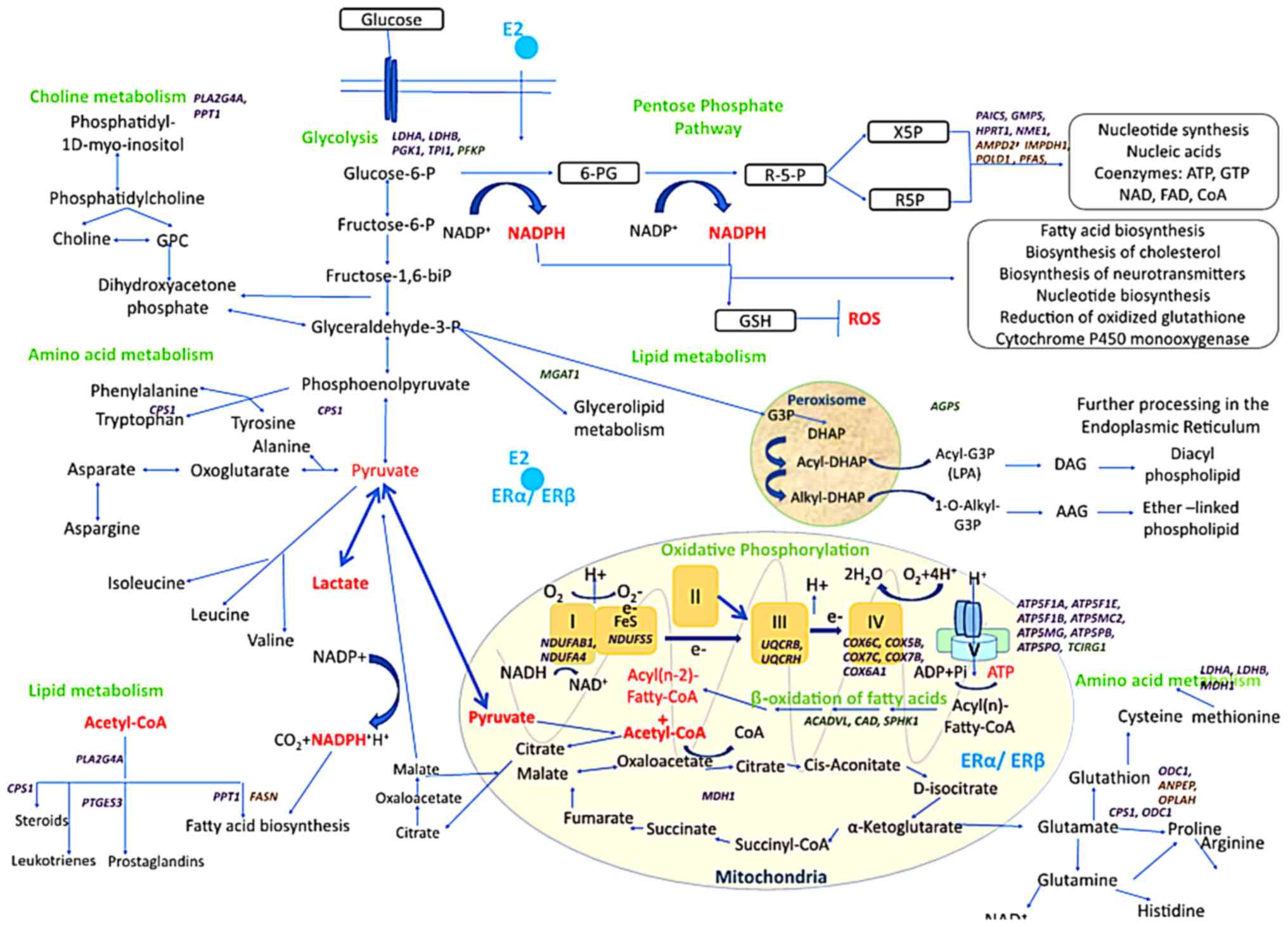
Fig. 8 outlines
important DEGs regulated by E2 and those directly involved in
mitochondrial function, metabolism and associated signaling
pathways. The impact of E2 in the pathophysiology of cervical
cancer was suggested to be associated with the dysregulation of the
respiratory mitochondrial electron chain, increasing OXPHOS, and
activating glycolysis and the pentose phosphate pathway (Warburg
effect). E2 was also suggested to inhibit the degradation of amino
acids and provide metabolites for the synthesis of nucleotides,
nucleic acids and coenzymes. Furthermore, it was hypothesized that
E2 stimulates the synthesis of fatty acids associated with
steroids, leukotrienes and prostaglandins metabolites, while it
reduces the β-oxidation of lipids. It was suggested that E2 reduces
oxidized glutathione, which regulates the oxidative state of tumor
cells.
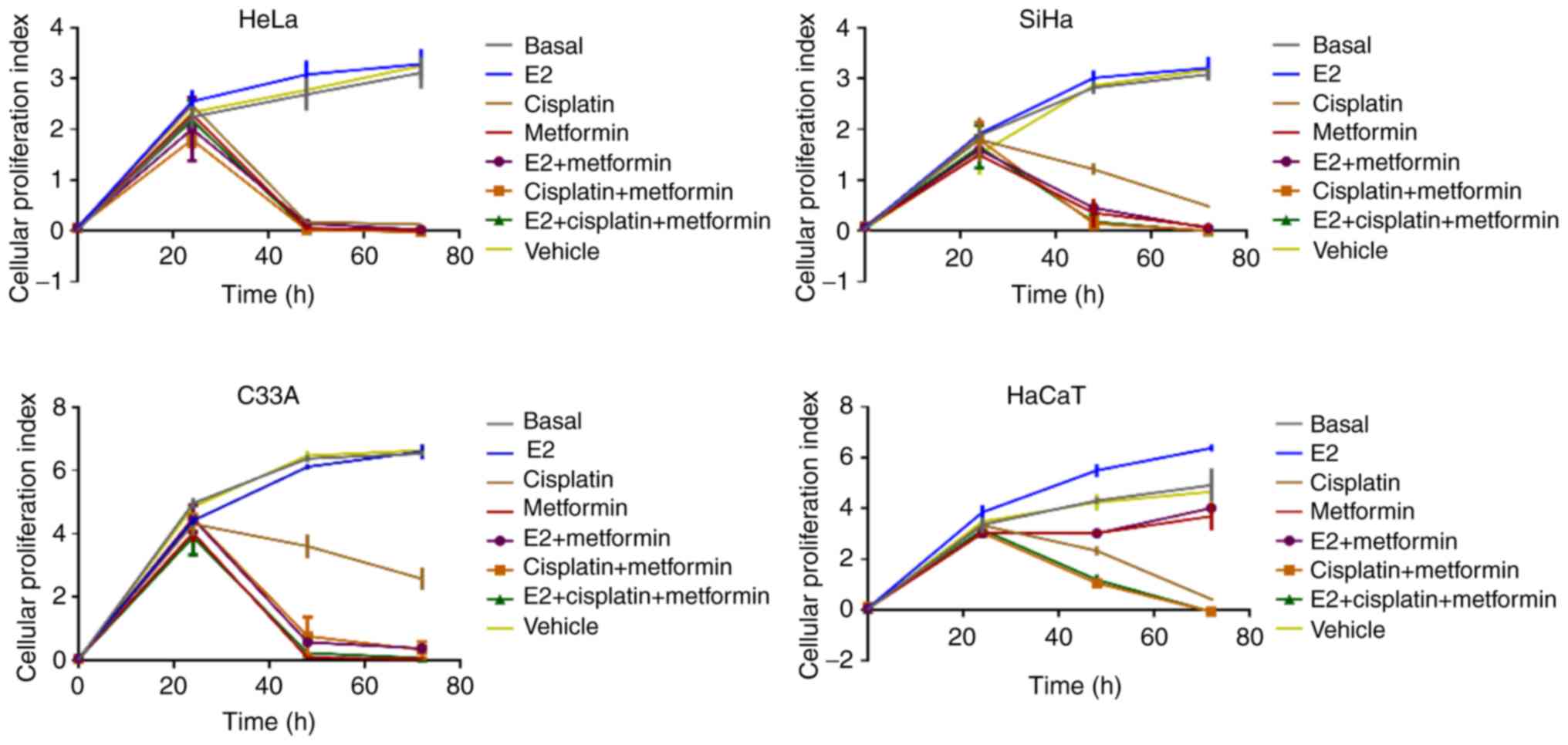
E2 does not modify metformin
antiproliferative effect
Metformin is proposed as an antitumor drug (26). The most relevant mechanism of its
activity is the regulation of cellular metabolism (27). The potential role of E2 in
metformin-induced cell death in cervical cancer was assessed in
this study. In cervical cancer cells treated with metformin a
significant decrease in proliferation was observed compared with
the basal control at incubation times >20 h (P=0.0001; Fig. 9). In HeLa, the effect of metformin
was similar to that induced by cisplatin, thus metformin was
suggested to be a potential anti-carcinogen. E2 treatment did not
markedly alleviate effects exerted by metformin.
Discussion
Chronic and excessive exposure to estrogen is
associated with an increased cancer risk incidence and the
classical mechanism to explain this association is that estrogen
affects the cell proliferation (28). Previously, effects of E2 on the
mitochondrial metabolism were shown by increased mitochondrial
activity based on MTT assays, which are known to alter the complex
IV levels and therefore inferred a modification in mitochondrial
metabolism (3). However, in this
study we approached other aspects associated with this alteration
in cervical cancer, such as the influence of the hormone in
mitochondrial permeability, calcium mobilization, NO levels, the
Warburg effect and modifications of pathways important in
tumorigenesis.
The presence of E2 decreased the mitochondrial
membrane permeability, which was in response to several factors,
such as calcium concentration, oxidative stress and ATP depletion.
It has been shown that estrogen affects the mitochondrial function
through the modulation of Ca2+ levels. Excessive
Ca2+ loading coupled with oxidative stress triggers a
collapse of the mitochondrial membrane potential (Δψm) accompanied
by edema of the matrix and initiation of apoptosis due to the
release of cytochrome c (10). E2
protects mitochondria by preventing Δψm collapse and this may
explain the described ability of estrogen to prevent the release of
apoptotic factors (8).
Intracellular calcium signals are critical for
mitochondrial-nuclear crosstalk and the maintenance of cellular
homeostasis (12). In the central
nervous system, E2 exhibits neuroprotective characteristics
preventing the influx of cytosolic and mitochondrial
Ca2+ during excitotoxic stimulation (9,10).
Additionally, the repression of Ca2+ mobilization by E2
may serve to protect cells from death during oxidative stress. In
previous studies, it was observed that the use of E2 inhibits the
effect of Ca2+ levels on pro-oxidant substances, such as
3-nitropropionic acid and H2O2 (9,11).
In the present study, E2 inhibited the response induced by
ionomycin and PMA with regards to the mobilization of calcium and
it was hypothesized that E2 caused alterations in the calcium
signaling pathway. Furthermore, the results suggested that E2 may
protect cervical cancer cells.
NO is a well-known intracellular messenger in a
range of physiological processes and a regulator of cell death and
survival. It modulates apoptotic pathways and long-term exposure to
µmolar NO can induce cell death by apoptosis (13,28).
When NO is combined with superoxide radicals, it generates
compounds, such as peroxynitrite, that lead to irreversible protein
nitration, enzymatic inactivation, DNA damage, disruption of the
mitochondrial integrity and cell apoptosis either by direct contact
or by inducing lipid peroxidation (29). It has been reported that E2 has an
antioxidant mechanism by increasing a blockage of the sensitivity
of mitochondria to calcium, enhancing antioxidant systems and
decreasing the expression of NO synthetase (10–12,30–32);
this may explain why no lipid peroxidation was observed in HeLa and
SiHa. However other mechanisms may further be involved. The
protective effect of E2 on the survival of the cervical cancer
cells was demonstrated in our study by reporting a decrease in NO
levels following E2 stimulus.
Excessive iron availability in mitochondria
exacerbates oxidative stress and promotes membrane lipid
peroxidation, affecting the integrity of the inner mitochondrial
membrane and the mitochondrial function (33). Several studies showed the
participation of estrogen in iron metabolism. In general, the
hormone appears to protect the cells from a distinct nonapoptotic
cell death (ferroptosis) because it lowers the levels of iron and
ferritin, and it regulates the expression of other proteins that
participate in iron metabolism (22,34–36).
This effect was not observed in the present study, as we did not
find any significant impact of E2 on iron and ferritin levels in
cervical cancer cells. It is known that for rapid proliferation,
cells enhance metabolic activity allowing for increased DNA
biosynthesis; this may describe why HaCaT cells to have high iron
levels and require less ferritin. However, in SiHa, oxidative
stress may be controlled through maintaining lower free iron
levels, as intracellular iron may be harmful due to its ability to
produce ROS (37–40).
The metabolic networks that give rise to metabolic
phenotypes in most tumors are unknown. In this study, an essential
role of E2 favoring the Warburg effect was demonstrated. It
increased the glucose consumption and lactic acid levels in
cervical cancer cell lines. Glucose utilization provides tumor
cells with a constant energy supply, as well as precursors for
de novo macromolecular biosynthesis that are essential for
cell growth and proliferation (13). Estrogen substantially increased the
glycolytic activities and glucose utilization in the uterus of
ovariectomized rats in concert with the uterine growth (41). The hormone increases the glucose
uptake in muscle and adipose tissues through the regulation of the
expression of the glucose transporter 4 and decreases
gluconeogenesis in the liver (36,42).
Although the Warburg effect predominantly focuses on
glycolysis and suggesting attenuation in the mitochondrial
metabolism, more recent works suggested that certain TCA cycle
intermediates are elevated and important in the context of cancer
(43–45). Thus, while glycolysis is
drastically upregulated in most cancer cells, mitochondrial
respiration continues to operate normally at rates proportional to
oxygen supply.
To gain a better understanding of estrogen effects
in the metabolism of cervical cancer, we performed a detailed
analysis of the metabolic gene expression modulated by E2 in SiHa
and HeLa cells, which are representative of ~99% of cervical tumors
(46). E2 upregulated various
genes associated with the protein components of the electron
respiratory chain complexes. Consistent with these observations,
previous work suggested that female liver mitochondria have greater
capabilities of OXPHOS than the mitochondria from males (47). Mitochondrial (mt) DNA contains
sequences similar to the estrogen response element (ERE) and
glucocorticoid responsive elements (48). Chen et al (49) observed that recombinant human ERα
and ERβ bind to mtEREs and this binding is enhanced by E2. Studies
in which MTT is used, reported an increase in the conversion of MTT
to formazan in the presence of E2 (50,51).
Furthermore, nuclear genes that participate in the transcription of
proteins of the respiratory chain of electrons are suggested to be
modulated by estrogen. The expression of the genes involved in
mitochondrial ATP synthase subunit β and F complex is upregulated
by E2 in prostatic dysplasia (52), and the subunit c isoform of the F0
complex and mitochondrial ribosomal protein 3 are other nuclear
estrogen-regulated genes identified in ERα-positive breast cancer
cells (53).
In neuroblastoma, E2 has a protective effect against
ATP depletion, Δψm decline and the generation of ROS induced by an
inhibitor of the succinate dehydrogenase complex (54). There is potential that E2 mediated
these effects via stimulating the synthesis of protein components
of the electron respiratory chain complexes, and contributed to the
preservation and regulation of the mitochondrial structure and
functions. The presence of ERs has been associated with the
preservation of ATP levels via enhancing OXPHOS and reducing ATPase
activity, and thus, increasing mitochondrial respiration efficiency
in neurons (55).
In association with these effects, E2 enhances the
antioxidant systems in the brain by inducing the expression of
glutathione, thioredoxin and superoxide dismutase proteins
(30,32). With these observations and the
findings reported in our bioenergetics analysis at the gene level,
we hypothesized that in cervical cancer, E2 protects tumor cells
from stress-induced death during malignant transformation partially
by modulating mitochondrial respiratory chain structure and
function.
Mitochondrial alterations as part of the adaptation
of tumor cells to their microenvironment favor the Warburg effect
(43). In this study, E2 modulated
numerous genes associated with an increase in the expression of
glycolytic proteins. The only gene downregulated by the hormone
shared by HeLa and SiHa in this metabolic pathway was PFKP.
The conversion of fructose-6-phosphate to fructose-1,6-bisphosphate
catalyzed by phosphofructokinase-1 is the most important control
step in mammalian glycolysis (56,57).
Downregulation of PFKP potentiates cancer cell survival
under metabolic stress through the maintenance of pentose phosphate
pathway flow (57). Dynamic
regulation of glucose flux between aerobic glycolysis and the
pentose phosphate pathway provides the tumor cell with the
metabolites needed to proliferate. NADPH production is an important
strategy against oxidative stress and oxidative stress, not ATP
depletion, is the primary trigger of cell death during metabolic
stress (57). Downregulation of
PFKP by E2 constituted a transcendent mechanism of evasion
of cell death in difficult situations.
Our bioinformatics analysis suggested other
metabolic pathways modulated by E2. Abnormal choline metabolism has
emerged as a hallmarks of cancer, and increases in choline kinase-α
expression and activity, a higher rate of choline transport and
increased phosphatidylcholine-specific phospholipase C and D
activities have been well documented within the pathophysiology of
several cancers (57). The
aberrant choline phospholipid metabolism of breast cancer cells and
its correlation with the malignant progression has been reported
(58). In our study, E2 was
suggest to upregulate the expression of enzymes associated with the
metabolism of choline and other lipids, which can affect the
cholesterol metabolism and the synthesis of steroids,
prostaglandins and leukotrienes. Physiologically, it has been
reported that estrogen, in general, inhibits de novo
lipogenesis in the liver (59),
but increase cholesterol biosynthesis for cell proliferation
(60).
In one study, a metabolomics approach for evaluating
the estrogenic effects in uterus established that the TCA cycle,
lipid metabolism, choline and amino acid metabolisms are disturbed
(61). The reported alterations
dependent on the ERs include: (i) Phenylalanine, tyrosine and
tryptophan biosynthesis; (ii) alanine, aspartate, glutamate,
glycerophospholipids and pyruvate metabolism; (iii) the citrate
cycle; and (iv) glycolysis or gluconeogenesis. These observations
are in accordance with our results and led to the hypothesis that
E2 may exert physiological functions in an erroneous context within
the pathophysiology of cervical cancer.
Another remarkable finding of our work was that E2
modulated signaling pathways that are essential for metabolism and
tumorigenesis. The role of HIF-1 (62) and Ras-MAPK in the metabolism of
cancer has been well documented (63,64).
AMPK is a pivotal intracellular energy sensor in normal and tumor
cells. Studies have been reported that high glucose levels reduce
AMPK phosphorylation and activate the MAPK signaling pathway in
cancer cells (64,65). Inactivation of AMPK accelerates
tumorigenesis (66). There are no
conclusive results about the effects of estrogen on AMPK expression
in different tumors; some evidence suggests AMPK activation by ERα
(67), while other evidence
considers that the hormone inhibits AMPK by repressing the
expression of its activating proteins (68). Interestingly, our study showed that
E2 was suggested to decrease the expression of genes that
indirectly repress AMPK.
Further observation of this work included that E2
modulated the differential expression of several genes associated
with metabolic pathways. The number of DEGs in SiHa was 3 times
greater than in HeLa; this may be explained due to these cells
being derived from different epithelial cells (squamous cell
carcinoma and adenocarcinoma) and that they are infected by two
different virus genotypes (16 and 18, respectively) that may modify
DEGs involved in signaling pathways, and therefore, affected
pathogenicity mechanisms and treatment responses. Furthermore, our
results indicated that, although from the same anatomical site,
SiHa and HeLa displayed marked differences following treatment with
E2. Regional differences in HPV genotypes may affect DEGs and the
phenotype of the cervical cells. The difference in HPV genotype
leads to different treatment responses with different clinical
characteristics, pathogenicity mechanisms and histopathological
subtypes (69). These results are
essential for the design of new therapeutic models against cervical
cancer.
Metformin has gained considerable attention as a
potential anticancer agent. One of its main mechanisms of action is
the suppression of the mitochondrial respiration by inhibiting the
respiratory complex I (70). In
human breast adipose stromal cells, it activates the liver kinase
B1-AMPK signaling pathway and inhibits aromatase expression
(71). Furthermore, the negative
modulation of metformin on ERs, in particular ERα, has been
demonstrated in numerous studies (72–74).
In this work, the effect of metformin on E2 metabolic actions was
evaluated. There are specific molecular features that control the
uptake of biguanides into mitochondria (70), so we suggested that modifications
in the mitochondrial permeability by E2 may be another factor that
conditioned the entry of metformin in the organelle. In addition,
although there are no specific transporters for biguanides, uptake
is tissue-specific due to varying levels of distinct transporter
expression (70). This evidence
supported the different effects of metformin in the metabolic
regulation of various cell lines, even when derived from the same
type of tumor.
Measuring the metabolic responses of cervical cancer
cell lines to E2 stimulus, allowed us to identify a distinct
bioenergetics phenotype in cervical cancer that was the result of
alterations in OXPHOS and the glycolytic energy metabolism. We
concluded that E2 induced changes in the energy metabolism that
allowed cell survival in high-stress conditions. The antioxidant
and cytoprotective power of E2 in the context of cancer was
demonstrated in this work.
Funding
This work was supported by CB 2017-2018 CONACYT
(grant no. A1-S-51207) and the Health Research Fund from IMSS
(grant no. FIS/IMSS/PROT/PRIO/15/046).
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
ARL and PCOL performed the experimental work,
searched for scientific literature, contributed to figures and the
writing of the manuscript. LFJS, ARdA and JFMV participated in
contributed to the data analysis and reviewed the manuscript. AAL,
YMOG, CABG, RSM and SLdA performed experiments. APS conceived and
designed the study and wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Chen Q, Cao HZ and Zheng PS: LGR5 promotes
the proliferation and tumor formation of cervical cancer cells
through the Wnt/β-catenin signaling pathway. Oncotarget.
5:9092–9105. 2014.PubMed/NCBI
|
|
2
|
Chung SH, Wiedmeyer K, Shai A, Korach KS
and Lambert PF: Requirement for estrogen receptor alpha in a mouse
model for human papillomavirus-associated cervical cancer. Cancer
Res. 68:9928–9934. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Riera-Leal A, Ramirez De Arellano A,
Ramirez-Lopez IG, Lopez-Pulido EI, Davila Rodriguez JR,
Macias-Barragan JG, Ortiz-Lazareno PC, Jave-Suárez LF,
Artaza-Irigaray C, Del Toro Arreola S, et al: Effects of 60 kDa
prolactin and estradiol on metabolism and cell survival in cervical
cancer: Coexpression of their hormonal receptors during cancer
progression. Oncol Rep. 40:3781–3793. 2018.PubMed/NCBI
|
|
4
|
Otto AM: Warburg effect(s)-a biographical
sketch of otto warburg and his impacts on tumor metabolism. Cancer
Metab. 4:52016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Asgari Y, Zabihinpour Z, Salehzadeh-Yazdi
A, Schreiber F and Masoudi-Nejad A: Alterations in cancer cell
metabolism: The warburg effect and metabolic adaptation. Genomics.
105:275–281. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu M, Neilson A, Swift AL, Moran R,
Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J,
et al: Multiparameter metabolic analysis reveals a close link
between attenuated mitochondrial bioenergetic function and enhanced
glycolysis dependency in human tumor cells. Am J Physiol Cell
Physiol. 292:C125–C136. 2007. View Article : Google Scholar
|
|
7
|
Chen JQ, Yager JD and Russo J: Regulation
of mitochondrial respiratory chain structure and function by
estrogens/estrogen receptors and potential
physiological/pathophysiological implications. Biochim Biophys
Acta. 1746:1–17. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Paterni I, Granchi C, Katzenellenbogen JA
and Minutolo F: Estrogen receptors alpha (ERα) and beta (ERβ):
Subtype-selective ligands and clinical potential. Steroids.
90:13–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen JQ, Cammarata PR, Baines CP and Yager
JD: Regulation of mitochondrial respiratory chain biogenesis by
estrogens/estrogen receptors and physiological, pathological and
pharmacological implications. Biochim Biophys Acta. 1793:1540–1570.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Simpkins JW, Yang SH, Sarkar SN and Pearce
V: Estrogen actions on mitochondria-physiological and pathological
implications. Mol Cell Endocrinol. 290:51–59. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ribas V, Drew BG, Zhou Z, Phun J, Kalajian
NY, Soleymani T, Daraei P, Widjaja K, Wanagat J, de Aguiar Vallim
TQ, et al: Skeletal muscle action of estrogen receptor alpha is
critical for the maintenance of mitochondrial function and
metabolic homeostasis in females. Sci Transl Med. 8:334ra542016.
View Article : Google Scholar
|
|
12
|
Lobaton CD, Vay L, Hernandez-Sanmiguel E,
Santodomingo J, Moreno A, Montero M and Alvarez J: Modulation of
mitochondrial Ca(2+) uptake by estrogen receptor agonists and
antagonists. Br J Pharmacol. 145:862–871. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Palsson-McDermott EM and O'Neill LA: The
Warburg effect then and now: From cancer to inflammatory diseases.
BioEssays. 35:965–973. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu YQ, Zhang GA, Zhang BC, Wang Y, Liu Z,
Jiao YL, Liu N and Zhao YR: Short low concentration cisplatin
treatment leads to an epithelial mesenchymal transition-like
response in DU145 prostate cancer cells. Asian Pac J Cancer Prev.
16:1025–1028. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen Y, Wang S, Bu S, Xu M and Lai D:
Low-dose cisplatin-induced CXCR4 expression promotes proliferation
of ovarian cancer stem-like cells. Acta Biochim Biophys Sin
(Shanghai). 48:282–289. 2016. View Article : Google Scholar
|
|
16
|
Dodt M, Roehr JT, Ahmed R and Dieterich C:
FLEXBAR-flexible barcode and adapter processing for next-generation
sequencing platforms. Biology (Basel). 1:895–905. 2012.
|
|
17
|
Bray NL, Pimentel H, Melsted P and Pachter
L: Near-optimal probabilistic RNA-seq quantification. Nat
Biotechnol. 34:525–527. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15:5502014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mi H, Muruganujan A, Ebert D, Huang X and
Thomas PD: PANTHER version 14: More genomes, a new PANTHER GO-slim
and improvements in enrichment analysis tools. Nucleic Acids Res.
47:D419–D426. 2019. View Article : Google Scholar :
|
|
20
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar
|
|
21
|
Kanehisa M, Sato Y, Furumichi M, Morishima
K and Tanabe M: New approach for understanding genome variations in
KEGG. Nucleic Acids Res. 47:D590–D595. 2019. View Article : Google Scholar :
|
|
22
|
Latunde-Dada GO: Ferroptosis: Role of
lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta
Gen Subj. 1861:1893–1900. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen K, Liu H, Liu Z, Bloomer W, Amos CI,
Lee JE, Li X, Nan H and Wei Q: Genetic variants in glutamine
metabolic pathway genes predict cutaneous melanoma-specific
survival. Mol Carcinog. 2019.Epub ahead of print. View Article : Google Scholar
|
|
24
|
Cheng M, Bhujwalla ZM and Glunde K:
Targeting phospholipid metabolism in cancer. Front Oncol.
6:2662016. View Article : Google Scholar
|
|
25
|
Zou RC, Xiao SF, Shi ZT, Ke Y, Tang HR, Wu
TG, Guo ZT, Ni F, Li WX and Wang L: Identification of
metabolism-associated pathways and genes involved in male and
female liver cancer patients. J Theor Biol. 480:218–228. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ikhlas S and Ahmad M: Metformin: Insights
into its anticancer potential with special reference to AMPK
dependent and independent pathways. Life Sci. 185:53–62. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tyszka-Czochara M, Bukowska-Strakova K and
Majka M: Metformin and caffeic acid regulate metabolic
reprogramming in human cervical carcinoma SiHa/HTB-35 cells and
augment anticancer activity of Cisplatin via cell cycle regulation.
Food Chem Toxicol. 106:260–272. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang T, Liu B, Guan Y, Gong M, Zhang W,
Pan J, Liu Y, Liang R, Yuan Y and Ye L: Melatonin inhibits the
proliferation of breast cancer cells induced by bisphenol A via
targeting estrogen receptor-related pathways. Thorac Cancer.
9:368–375. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vallance P and Chan N: Endothelial
function and nitric oxide: Clinical relevance. Heart. 85:342–350.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chiueh C, Lee S, Andoh T and Murphy D:
Induction of anti-oxidative and antiapoptotic thioredoxin supports
neuroprotective hypothesis of estrogen. Endocrine. 21:27–31. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ronchetti SA, Machiavelli LI, Quinteros
FA, Duvilanski BH and Cabilla JP: Nitric oxide plays a key role in
ovariectomy-induced apoptosis in anterior pituitary: Interplay
between nitric oxide pathway and estrogen. PLoS One.
11:e01624552016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schmidt AJ, Krieg J and Vedder H:
Differential effects of gluco-corticoids and gonadal steroids on
glutathione levels in neuronal and glial cell systems. J Neurosci
Res. 67:544–550. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schapira A and Lodi R: Assessment of in
vitro and in vivo mitochondrial function in Friedreich's ataxia and
Huntington's disease. Methods Mol Biol. 277:293–307.
2004.PubMed/NCBI
|
|
34
|
Yang X, Xu MM, Wang J and Xie JX: Effect
of estrogen on iron metabolism in mammals. Sheng Li Xue Bao.
68:637–643. 2016.PubMed/NCBI
|
|
35
|
Chen B, Li GF, Shen Y, Huang XI and Xu YJ:
Reducing iron accumulation: A potential approach for the prevention
and treatment of postmenopausal osteoporosis. Exp Ther Med.
10:7–11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xie Q, Xi G, Keep RF and Hua Y: Effects of
gender and estrogen receptors on iron-induced brain edema
formation. Acta Neurochir Suppl. 121:341–345. 2016. View Article : Google Scholar
|
|
37
|
Asano M, Yamasaki K, Yamauchi T, Terui T
and Aiba S: Epidermal iron metabolism for iron salvage. J Dermatol
Sci. 87:101–109. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
El-Rifaie AA, Sabry D, Doss RW, Kamal MA
and Abd El Hassib DM: Heme oxygenase and iron status in exosomes of
psoriasis patients. Arch Dermatol Res. 310:651–656. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Miniaci MC, Irace C, Capuozzo A, Piccolo
M, Di Pascale A, Russo A, Lippiello P, Lepre F, Russo G and
Santamaria R: Cysteine prevents the reduction in keratin synthesis
induced by iron deficiency in human keratinocytes. J Cell Biochem.
117:402–412. 2016. View Article : Google Scholar
|
|
40
|
Pelle E, Jian J, Zhang Q, Muizzuddin N,
Yang Q, Dai J, Maes D, Pernodet N, Yarosh DB, Frenkel K and Huang
X: Menopause increases the iron storage protein ferritin in skin. J
Cosmet Sci. 64:175–179. 2013.PubMed/NCBI
|
|
41
|
Roberts and Szego CM: The influence of
steroids on uterine respiration and glycolysis. J Biol Chem.
201:21–30. 1953.PubMed/NCBI
|
|
42
|
Faulds MH, Zhao C, Dahlman-Wright K and
Gustafsson JÅ: The diversity of sex steroid action: Regulation of
metabolism by estrogen signaling. J Endocrinol. 212:3–12. 2012.
View Article : Google Scholar
|
|
43
|
Chen X, Qian Y and Wu S: The Warburg
effect: Evolving interpretations of an established concept. Free
Radic Biol Med. 79:253–263. 2015. View Article : Google Scholar
|
|
44
|
Liberti MV and Locasale JW: The warburg
effect: How does it benefit cancer cells? Trends Biochem Sci.
41:211–218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Potter M; Newport E; Morten KJ: The
warburg effect: 80 years on. Biochem Soc Trans. 44:1499–1505. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Jhingran A, Russell AH, Seiden MV, Duska
LR, Goodman A, Lee SL, et al: 84-Cancers of the Cervix, Vulva, and
Vagina. Niederhuber JE, Armitage JO, Kastan MB, Doroshow JH and
Tepper JE: Abeloff's Clinical Oncology (Sixth Edition)
Philadelphia: Content Repository Only; 2020, pp. 1468–1507.e8
|
|
47
|
Valle A, Català-Niell A, Colom B,
García-Palmer FJ, Oliver J and Roca P: Sex-related differences in
energy balance in response to caloric restriction. Am J Physiol
Endocrinol Metab. 289:E15–E22. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Demonacos CV, Karayanni N, Hatzoglou E,
Tsiriyiotis C, Spandidos DA and Sekeris CE: Mitochondrial genes as
sites of primary action of steroid hormones. Steroids. 61:226–232.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chen JQ, Eshete M, Alworth WL and Yager
JD: Binding of MCF-7 cell mitochondrial proteins and recombinant
human estrogen receptors alpha and beta to human mitochondrial DNA
estrogen response elements. J Cell Biochem. 93:358–373. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhai P, Eurell TE, Cooke PS, Lubahn DB and
Gross DR: Myocardial ischemia-reperfusion injury in estrogen
receptor-alpha knockout and wild-type mice. Am J Physiol Heart Circ
Physiol. 278:H1640–H1647. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhai P, Eurell TE, Cotthaus R, Jeffery EH,
Bahr JM and Gross DR: Effect of estrogen on global myocardial
ischemia-reperfusion injury in female rats. Am J Physiol Heart Circ
Physiol. 279:H2766–H2775. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Thompson CJ, Tam NN, Joyce JM, Leav I and
Ho SM: Gene expression profiling of testosterone and estradiol-17
beta-induced prostatic dysplasia in Noble rats and response to the
antiestrogen ICI 182-780. Endocrinology. 143:2093–2105. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Weisz A, Basile W, Scafoglio C, Altucci L,
Bresciani F, Facchiano A, Sismondi P, Cicatiello L and De Bortoli
M: Molecular identification of ERalpha-positive breast cancer cells
by the expression profile of an intrinsic set of estrogen regulated
genes. J Cell Physiol. 200:440–450. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wang J, Green PS and Simpkins JW:
Estradiol protects against ATP depletion, mitochondrial membrane
potential decline and the generation of reactive oxygen species
induced by 3-nitroproprionic acid in SK-N-SH human neuroblastoma
cells. J Neurochem. 77:804–811. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Nilsen J and Brinton RD: Mitochondria as
therapeutic targets of estrogen action in the central nervous
system. Curr Drug Targets CNS Neurol Disord. 3:297–313. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Berg JM, Tymoczko JL, Gatto GJ and Stryer
L: Biochemistry. 2015.
|
|
57
|
Kim NH, Cha YH, Lee J, Lee SH, Yang JH,
Yun JS, Cho ES, Zhang X, Nam M, Kim N, et al: Snail reprograms
glucose metabolism by repressing phosphofructokinase PFKP allowing
cancer cell survival under metabolic stress. Nat Commun.
8:143742017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Glunde K, Jie C and Bhujwalla ZM:
Molecular causes of the aberrant choline phospholipid metabolism in
breast cancer. Cancer Res. 64:4270–4276. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Palmisano BT, Zhu L and Stafford JM: Role
of estrogens in the regulation of liver lipid metabolism. Adv Exp
Med Biol. 1043:227–256. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Di Croce L, Vicent GP, Pecci A, Bruscalupi
G, Trentalance A and Beato M: The promoter of the rat
3-hydroxy-3-methylgl-utaryl coenzyme A reductase gene contains a
tissue-specific estrogen-responsive region. Mol Endocrinol.
13:1225–1236. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wang D, Zhu W, Wang Y, Yan J, Teng M, Miao
J and Zhou Z: Metabolomics approach to investigate estrogen
receptor-dependent and independent effects of o, p'-DDT in the
uterus and brain of immature mice. J Agric Food Chem. 65:3609–3616.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Marín-Hernández A, Gallardo-Pérez JC,
Ralph SJ, Rodríguez-Enríquez S and Moreno-Sánchez R: HIF-1alpha
modulates energy metabolism in cancer cells by inducing
over-expression of specific glycolytic isoforms. Mini Rev Med Chem.
9:1084–1101. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhang X, Ma L, Qi J, Shan H, Yu W and Gu
Y: MAPK/ERK signaling pathway-induced hyper-O-GlcNAcylation
enhances cancer malignancy. Mol Cell Biochem. 410:101–110. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Han J, Zhang L, Guo H, Wysham WZ, Roque
DR, Willson AK, Sheng X, Zhou C and Bae-Jump VL: Glucose promotes
cell proliferation, glucose uptake and invasion in endometrial
cancer cells via AMPK/mTOR/S6 and MAPK signaling. Gynecol Oncol.
138:668–675. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cao M, Jiang J, Du Y and Yan P:
Mitochondria-targeted antioxidant attenuates high glucose-induced
P38 MAPK pathway activation in human neuroblastoma cells. Mol Med
Rep. 5:929–934. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Faubert B, Boily G, Izreig S, Griss T,
Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, et
al: AMPK is a negative regulator of the Warburg effect and
suppresses tumor growth in vivo. Cell Metab. 17:113–124. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lipovka Y, Chen H, Vagner J, Price TJ,
Tsao TS and Konhilas JP: Oestrogen receptors interact with the
α-catalytic subunit of AMP-activated protein kinase. Biosci Rep.
35:e002642015. View Article : Google Scholar
|
|
68
|
Linher-Melville K, Zantinge S and Singh G:
Liver kinase B1 expression (LKB1) is repressed by estrogen receptor
alpha (ERα) in MCF-7 human breast cancer cells. Biochem Biophys Res
Commun. 417:1063–1068. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Artaza-Irigaray C, Molina-Pineda A,
Aguilar-Lemarroy A, Ortiz-Lazareno P, Limón-Toledo LP,
Pereira-Suárez AL, Rojo-Contreras W and Jave-Suárez LF: E6/E7 and
E6* from HPV16 and HPV18 upregulate IL-6 expression independently
of p53 in keratinocytes. Front Immunol. 10:16762019. View Article : Google Scholar
|
|
70
|
Bridges HR, Sirviö VA, Agip AN and Hirst
J: Molecular features of biguanides required for targeting of
mitochondrial respiratory complex I and activation of AMP-kinase.
BMC Biol. 14:652016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Brown KA, Hunger NI, Docanto M and Simpson
ER: Metformin inhibits aromatase expression in human breast adipose
stromal cells via stimulation of AMP-activated protein kinase.
Breast Cancer Res Treat. 123:591–596. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zhang J, Zhang B, Yin Z, Chen F, Liu T, Xu
H, Liu Y and Zhou X: Effects of metformin on the estrogen-induced
proliferation and the expression of ER in human endometrial cancer
cells. Zhonghua Fu Chan Ke Za Zhi. 49:932–937. 2014.In Chinese.
|
|
73
|
Kim J, Lee J, Jang SY, Kim C, Choi Y and
Kim A: Anticancer effect of metformin on estrogen receptor-positive
and tamoxifen-resistant breast cancer cell lines. Oncol Rep.
35:2553–2560. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Scherbakov AM, Sorokin DV, Tatarskiy VV
Jr, Prokhorov NS, Semina SE, Berstein LM and Krasil'nikov MA: The
phenomenon of acquired resistance to metformin in breast cancer
cells: The interaction of growth pathways and estrogen receptor
signaling. IUBMB Life. 68:281–292. 2016. View Article : Google Scholar : PubMed/NCBI
|