Introduction
Lung carcinoma, which is caused by both genetic and
environmental factors, is the leading cause (18%) of cancer-related
mortality worldwide (1,2). Non-small cell lung cancer (NSCLC),
such as large cell carcinoma, squamous cell carcinoma and
adenocarcinoma, constitutes >80% of all lung cancer cases
(3). The 5-year overall survival
rate of NSCLC is poor despite great advances being made in
diagnosis and treatment (4).
Although cigarette smoking is the major risk factor for lung
cancer, numerous genes have been reported to participate in lung
carcinogenesis (5–7).
Semaphorin family proteins, which contain a
conserved N-terminal Sema domain of 400–500 amino acids (8), comprise 8 classes with
membrane-anchored and cleaved extracellular domain forms (9). The cleaved extracellular domain, such
as SEMA3E (10), is modified from
the membrane-anchored forms by proteolytic effects (11,12).
This proteolytic process is executed by the A disintegrin and
metalloprotease (ADAM) family. For example, SEMA3C can be cleaved
by ADAMTS1 (13) and SEMA5B can be
cleaved by ADAM17 (14).
Initially, the semaphorin family proteins were
discovered to regulate axon growth and neuronal migration (15,16).
Recently, several studies have found that semaphorins are involved
in cardiac/skeletal development (17), the immune response (18), the regulation of angiogenesis
(19) and tumor growth, as well as
metastasis (20). Class 3
semaphorins, such as SEMA3B and SEMA3F have been reported to be
regulated by DNA methylation (21,22)
and are related to various carcinogenic processes in the lungs, for
example angiogenesis and metastasis (15,23-25)
and tumor suppressor activities (26-28).
However, whether other classes of the semaphorin family are subject
to methylation regulation or are involved in carcinogenic processes
in the lungs remains unclear.
Previously, a comprehensive analysis of the gene
expression signature performed by the authors in non-smoking women
with lung adenocarcinoma revealed that the downregulation of
SEMA5A was associated with a poor overall survival (29). SEMA5A belonging to class V of the
semaphorin family, is an integral membrane protein containing the
Sema domain, 7 thrombospondin type-1 repeats and a short
cytoplasmic domain (23,30). SEMA5A has been reported to have
both a membrane-bound (8,31) and cleaved extracellular domain
(32). Sheddases, which are the
members of the ADAM protein families, are known to be majorly
involved in ectodomain shedding by cleaving the extracellular
portions of transmembrane proteins (33). However, the role of sheddase
responsible for releasing the ectodomain from membrane-bound SEMA5A
has yet to be identified.
In addition, as regards the function of
SEMA5A, it was implicated as a susceptible gene related to
Cri-du-chat syndrome (34) and
autism (35). SEMA5A has also been
reported to promote angiogenesis by increasing endothelial cell
proliferation and decreasing apoptosis (36), and to have high tumorigenic and
metastatic potential in pancreatic and gastric tumors (37-39).
On the other hand, some studies have demonstrated that SEMA5A also
plays the role of a tumor suppressor. For instance, SEMA5A
maintains the epithelial phenotype of malignant pancreatic cancer
cells (40), and inhibits glioma
cell motility through the RhoGDIα-mediated inactivation of RAC1
GTPase (41) and the actin
cytoskeleton (42). However,
little is known about the mechanisms and functional role of the
downregulation of SEMA5A in lung adenocarcinoma cells.
Therefore, this study aimed to elucidate the mechanisms associated
with low endogenous expression levels of SEMA5A in lung
adenocarcinoma and to determine functional roles in lung
carcinogenesis.
Materials and methods
Cells and cell culture
Cancerous lung cell lines (CL1-0, CL1-5, A549, and
H1299) (gifts from Dr Pan-Chyr Yang) and normal cells (BEAS-2B) (a
gift from Dr Pan-Chyr Yang) were cultured in RPMI-1640 medium
(Gibco; Thermo Fisher Scientific) with 1% streptomycin/puromycin
(Biological Industries) and 10% fetal bovine serum (FBS; Biological
Industries). Brain metastatic lung adenocarcinoma Bm7 cells
(generated in the authors′ laboratory) were cultured in DME/F12
plus 10% FBS (43). The cultured
plates were maintained at 37°C in a humidified atmosphere with 5%
CO2. Another normal lung cell line (MRC-5) and human
bronchial epidermal cells (16HBE) (a gift from Dr. Kuo-Ting Chang)
were grown in Eagle's Minimum Essential Medium (Gibco; Thermo
Fisher Scientific) under the same conditions.
Cell line authentication
Cell experiments were performed on cells that were
passaged <20 times, and were routinely tested for mycoplasma
using the PCR Mycoplasma Detection kit (ABM Inc., Vancouver,
Canada). The identity of the cell lines was authenticated by
short-tandem repeat (STR) analysis (Mission Biotech Inc., Taipei,
Taiwan) in February, 2018.
Endogenous expression of SEMA5A
To quantify the transcriptional expression of
SEMA5A in different cellular models, total RNA was isolated
using TRIzol reagent (Ambion) and purified by precipitation with
isopropanol (Sigma-Aldrich). A NanoDrop™ 2000 spectrophotometer
(Thermo Fisher Scientific) was used to assess the purity and
quantity of the RNA. A high Capacity cDNA Reverse Transcription kit
(Thermo Fisher Scientific) was used to synthesize the cDNA from 1
µg of total RNA of each cell line. The final cDNA products
were used as the templates for expression analysis using
quantitative PCR (qPCR).
qPCR
The quality and quantity of the RNA were measured
using a NanoDrop™ 2000 spectrophotometer (Thermo Fisher
Scientific). A total of 1 µg of total RNA from each cell
line was reverse transcribed using the High Capacity cDNA Reverse
Transcription kit (Thermo Fisher Scientific). The reaction was
carried using thermal cycling conditions, including annealing at
25°C for 5 min, an extension temperature at 42°C for 1 h and
inactivation temperature at 70°C for 15 min. The final cDNA
products were used as the templates for subsequent qPCR with the
following thermal cycling conditions. Denaturation temperature at
95°C for 15 sec, anneal/extend temperature: 60°C for 1 min for 40
cycles. RT-qPCR was performed using SYBR-Green (Roche) on an ABI
7900 system (Life Technologies; Thermo Fisher Scientific) according
to standard protocols. All individual experiments were carried out
in triplicate. Relative quantification, ΔΔCq (cycle threshold)
(44), was applied using
glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as an
internal control and empty group as a reference control.
Impact of methylation on gene
expression
As a number of tumor suppressor genes are
inactivated via hypermethylation within the promoter region
(45,46), this study examined the role of
methylation in regulating the expression of SEMA5A. The
A549, H1299 and BEAS-2B cells were seeded on a 6-well plate, and
after 24 h, the cells were treated with 5 or 10 µM of
5-aza-2′-deoxycytidine (5-aza; Sigma-Aldrich). The quantification
of the expression levels was carried after 3 days of treatment.
To quantify the methylation levels of multiple CpG
sites in the 5′ untranslated region of SEMA5A, bisulfite
treatment was first performed with an EpiTect Fast bisulfite
conversion kit (Qiagen) according to the manufacturer's
instructions, and a pre-designed PyroMark CpG assay
(Hs_SEMA5A_03_PM PyroMark CpG assay, PM00021203, Qiagen) was
used. Specific CpG sites, including Chr5: 9545395, 9545400,
9545407, 9545409 and 9545417 were examined.
Overexpression of SEMA5A in lung
adenocarcinoma cells
Full-length SEMA5A cDNA (3,225 bp) tagged
with a Flag epitope was inserted into a pZeoSV2+ viral
expression vector with the NotI and AsiSI restriction
enzymes (Addgene). The pZeoSV2+-SEMA5A-Flag
plasmid was transiently transfected into the A549 and H1299 cell
lines using TransIT-2020 transfection reagent (MirusBio) according
to the manufacturer's instructions. The empty pZeoSV2+
viral expression vector was used as a control. All sequences were
verified by Sanger sequencing (First Core Laboratory, College of
Medicine, National Taiwan University). mRNA levels were quantified
by RT-qPCR using SEMA5A-specific primers (forward,
5′-GTCTATACTTACTGCCAGCG-3′ and reverse,
5′-GTTAAATGCCTTGATGGCCTC-3′) and GAPDH-specific primers
(forward, 5′-TGCACCACCAACTGCTTAG-3′ and reverse,
5′-GATGCAGGGATGATGTTC-3′). Protein levels were examined by western
blot analysis.
Cleavage of SEMA5A by ADAM17
To examine the proteolytic effects of ADAM17 on
SEMA5A, the H1299 cells were transfected with SEMA5A
overexpression plasmid and treated with active recombinant ADAM17
(BioVision). The amount of administering ADAM17 was 0.66 µg
for 6-cm dish. Cell lysates and media were collected at 2 days
following transfection. Bovine serum albumin (BSA) was added
externally to the culture media as the spike-in control, and all
proteins in the medium were concentrated using a 10K Acrodisc
syringe filter (Pall Life Sciences).
Western blot analysis
Total cell lysates of A549 and H1299 cells
transfected with pZeoSV2+-SEMA5A-Flag plasmid or
empty vector were prepared. Proteins (30 µg) were separated
by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE), and then electrotransferred onto polyvinylidene
difluoride (PVDF) membranes (Bio-Rad Laboratories). After blocking
with 5% milk, the membranes were incubated with anti-SEMA5A
(1:1,000, Cat. no. PA5-26066, Thermo Fisher Scientific), anti-GAPDH
antibody (1:20,000, Cat. no. GTX100118, GeneTex), or anti-BSA
antibody (1:5,000, Cat. no. GTX79812, GeneTex). Following
incubation overnight at 4°C, the membranes were then incubated with
horseradish peroxidase-conjugated anti-IgG (1:5,000, Cat. no.
GTX213110, GeneTex) at room temperature for 1 h, and the blots were
developed with the chemiluminescent western blotting reagent
(Millipore). Western blot images were further analyzed using
Gel-Pro Analyzer v6.3 software (Meyer Instruments) to obtain the
optical density values of SEMA5A and GAPDH antibodies.
Isolation and amplification of total RNA
for gene expression profiling
The mRNA detection and analysis were performed as
previously described (47).
Briefly, the cDNA was synthesized from total RNA primed with T7
Oligo(dT) and amplified using an Illumina TotalPre RNA
Amplification kit (Ambion). The second strand of cDNA was
synthesized by employing DNA polymerase and RNAase H to
simultaneously degrade the RNA and synthesize the second strand of
cDNA. Following cleanup, in vitro transcription was
conducted to synthesize biotinylated complementary RNA (cRNA).
Following amplification, the cRNA was hybridized to Illumina Human
HT-12 v4 BeadChips (Illumina) for 16 h. Following hybridization,
the BeadChip was washed and stained with streptavidin-Cy3 dye. The
intensity of the beads′ fluorescence was detected by HiScan SQ
(Illumina), and the results were analyzed using BeadStudio v2011.1
software.
After scanning, the intensity data of Illumina
BeadChips were analyzed using Partek v7.0 software (Partek).
Background-adjusted signals were normalized by a quantile
normalization algorithm. Student′s t-tests and Bonferroni P-value
adjustment were utilized to identify differentially expressed
genes. Principle component analysis (PCA) was utilized to evaluate
the similarity of the gene expression profiles. Hierarchical
clustering analysis and the Genesis program were used to generate
visual representation of expression profiles. All data have been
deposited at the Gene Expression Omnibus (GEO, GSE114578).
Furthermore, ingenuity pathway analysis (Ingenuity Systems Inc.)
was applied to comprehend the biological functions and signaling
pathways of differentially expressed genes.
Analysis of cell proliferation
A total of 3,000 lung adenocarcinoma cells (A549
& H1299) were seeded in 96-well plates in triplicate and
incubated for 12 h at 37°C in a CO2 incubator. The day
after seeding, the cells were transfected with
pZeoSV2+-SEMA5A-Flag plasmid or empty vectors.
Following transfection, the cells proliferative activity was
measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT) (EMD Biosciences) assay at 24, 48 and 72 h,
respectively. The absorbance of the A549 and H1299 cells was then
measured using a microtiter plate reader (BioTek) at 570 nm.
Immunohistochemistry
The A549 (3,000 cells) or H1299 (3,000 cells) cells
were seeded on silane coated micro slides (Muto pure chemicals Co.)
with the same cell density and transfection concentration as MTT
assay. The cells were fixed with 4% paraformaldehyde
(Sigma-Aldrich) at 48 h following transfection. The cell membrane
was damaged by 0.1% Triton X-100 (Mallinckrodt Specialty Chemicals
Co) and blocked with 2% bovine serum albumin (Sigma-Aldrich). Ki-67
primary antibody (1:400, #9129, Cell Signaling Technology) and
anti-rabbit IgG secondary antibody (1:800, #8889, Cell Signaling
Technology) were used for Ki-67 detection at a wavelength of
550/580 nm. The incubation conditions for the primary antibody were
4°C overnight, and for the secondary antibody they were 2 h at room
temperature. Cell nuclei were stained with Hoechst 33342 (#B2261, 1
µg/ml, Sigma-Aldrich) and detected at wavelength of 360/460
nm. The incubation conditions for Hoechst 33342 were 30 min at room
temperature. Images were acquired using the Zeiss AxioImager A1
system (Carl Zeiss).
Clonogenic assay
The A549 (300 cells), H1299 (300 cells) and Bm7 (300
cells) cells were first seeded in 6-well plates for 24 h, and
transfected with pZeoSV2+-SEMA5A-Flag plasmid or
empty vectors. After 2 weeks, the cells were fixed using 3:1
methanol-acetic acid and stained using 0.1% crystal violet
(Sigma-Aldrich) for 10 min at room temperature. Finally, the
stained plates were dried and used for image acquisition using
microscopic (×100 magnification) evaluation. Colonies containing
>50 cells under a stereomicroscope were counted.
Gap closure assay
Following 24 h of transfection, 2×104
cells of A549 and H1299 were seeded into 24-well plates with a
sterile culture insert and grown to ~90% confluence in RPMI-1640
medium containing 10% FBS and changed to 2% FBS when removing the
cassettes. The culture inserts were removed and an image of the gap
at 0 h was captured. The cells were further incubated at 37°C in a
CO2 incubator for 36 h, and images were captured at 24
and 36 h to measure the progress of gap closure.
Cell migration
Migration assays were carried out using a 24-well
Transwell unit (Corning, Inc.). The upper chamber of the Transwell
unit was loaded with 4×104 cells/well in 0.2 ml
serum-free RPMI-1640 medium, and the lower chambers were loaded
with 0.6 ml RPMI-1640 containing 10% FBS as a chemoattractant. The
A549 and H1299 cells were then incubated for 24 h at 37°C. A
methanol-acetic acid (3:1) mixture was then added to the lower
chamber to fix the cells for 20 min at room temperature followed by
staining with 0.1% crystal violet for a further 20 min at room
temperature. Cells on the upper side of the membrane surface were
removed by scraping with a cotton swab, and the cells that passed
through the filter were de-stained using 10% acetic acid. The
absorbance was measured at 570 nm with an enzyme-linked
immunosorbent assay (ELISA) reader (BioTek Instruments). Cells
transfected with empty vector were used as the controls.
Time-lapse migration assay
The experiment was performed as previously described
(43). Briefly, cells were
cultured on dishes coated with collagen (10 µg/ml) overnight
and then cultured in serum-free conditioned medium. Cell movement
was detected under A-Plan objectives (X5; 0.55 NA) using an
inverted microscope (Axio Observer Z1, Zeiss) in 37°C chambers.
Images were collected from CCD video cameras (AxioCam MRm, Zeiss)
at 20-min intervals for a total of 16 h using MetaMorph software
(Molecular Devices Corp.). The accumulated distance was measured by
tracking each cell nuclei for 30 individual cancer cells in each
group using the Track Point function of NIH ImageJ v1.43
software.
Lung cancer animal model
The SEMA5A-Flag plasmid was transiently
transfected into Bm7 lung cancer cells with stable luciferase
expression. The following day, the Bm7 cells (2×106
cells in 50 µl of PBS) were mixed with 50 µl of
Matrigel and then implanted into 6–8 week-old male SCID mice
(weighing 20–25 g; BioLASCO) by the subcutaneous route at two
separate sites (right and left side) of the back (each group had 4
mice; 2 groups). Mice were housed in specific pathogen-free rooms
with one group in one cage at room temperature (20–23°C), 40–60%
relative humidity and under a 12-h light-dark cycle, with free
access to food and water. No cachexia and ascites in mice were
observed in this study. As Bm7 lung tumors had luciferase, the
tumor volume was detected using an IVIS Spectrum Imaging system
(Xenogen). Prior to image acquisition, the mice were subsequently
administered D-luciferin via intraperitoneal injection and photons
emitted from the mice were detected using an imaging system. Tumor
size and distribution in vivo were quantified as
photons/second. The maximum length and width exhibited by a single
subcutaneous tumor in this study did not exceed 1.5 cm. The maximum
tumor volume calculated by signal was 500 mm3 using the
formula, (length × width2)/2. Anesthesia was induced and
maintained with 2.5% isoflurane (Panion & BF Biotech Inc.) in
100% oxygen in an anesthetic chamber when mice were monitored by an
IVIS Spectrum Imaging system. The mice were sacrificed by using
carbon dioxide (CO2) at a displacement rate from 10 to
30% of the chamber volume per minute for mouse euthanasia and death
was verified by physical methods following euthanasia on day 21,
including no heartbeat, no pupillary response to light and no
respiratory pattern.
This animal model followed protocols approved by the
Institutional Animal Care and Use Committee of China Medical
University and Hospital (animal protocol no. 2016-102). Suitable
humane endpoints were included in the approved animal experiments.
In this study, the mouse experiment was terminated by euthanasia
(using carbon dioxide) on day 21 or animals that reached humane
endpoints when the tumor size was near 1,000 mm3 or
unexpected circumstances, such as illnesses (infection, difficulty
breathing, etc.) and injuries (necrosis or bleeding in tumors). The
mice were sacrificed by using carbon dioxide (CO2) at a
displacement rate from 10 to 30% of the chamber volume per minute
for mouse euthanasia and death was verified by methods, including
no heartbeat, no pupillary response to light, and no respiratory
pattern for at least 5 min.
Kaplan-Meier survival analysis
Kaplan-Meier survival analyses were conducted using
Kaplan-Meier Plotter for lung cancer (48,49),
an online platform (http://www.kmplot.com/lung), using SEMA5A probe
(Affy ID: 229427_at) and adenocarcinoma in histology restriction.
In total, Kaplan-Meier Plotter analyzed 1,715 patients for survival
analysis, which included 1,120 patients in 7 GEO datasets (GSE4573,
GSE14814, GSE8894, GSE19188, GSE3141, GSE31210, GSE29013 and
GSE37745), 462 patients in the caArray, and 133 patients in The
Cancer Genome Atlas (TCGA). The log rank test was used to determine
differences in the survival rate between the high and low
expression groups.
Statistical analysis
Data are expressed as the means ± standard deviation
(SD) of at least 3 independent experiments. Student's t-tests and
Bonferroni P-value adjustment were utilized to identify
differentially expressed genes. P-values <0.05 were considered
to indicate statistically significant differences.
Results
Association of SEMA5A expression in lung
cancer tissues with a poor overall survival
In previous genomic studies, it was found that the
downregulation of SEMA5A in non-smoking women with lung
adenocarcinoma was associated with a poor overall survival
(29,50,51).
To validate this phenomenon in a Caucasian population, another
publicly available dataset was examined (52). As shown in Fig. 1A, SEMA5A was also
significantly (P<0.01) downregulated in cancer tissues as
compared to adjacent normal control tissues. Furthermore, the
survival of lung adenocarcinoma patients in the new dataset for
>15 years was examined using Kaplan-Meier plotter (www.kmplot.com/lung) (49). In total, 1,715 patients were used
for survival analysis, which included 1,120 patients in 7 GEO
datasets (GSE4573, GSE14814, GSE8894, GSE19188, GSE3141, GSE31210,
GSE29013 and GSE37745), 462 patients in the caArray, and 133
patients in The Cancer Genome Atlas (TCGA). Patients were divided
into the high and low expression groups based on the median
expression value of SEMA5A. The results of Kaplan-Meier
survival analysis revealed that the lung adenocarcinoma patients
with higher expression levels of SEMA5A had a lower risk of
death (hazard ratio, 0.65), i.e., a higher overall survival
probability (Fig. 1B), indicating
the potential utility of SEMA5A as a prognostic marker for
patients with lung adenocarcinoma.
Mechanisms responsible for the lower
SEMA5A expression in lung adenocarcinoma cells
Since SEMA5A was downregulated in non-smoking
female lung adenocarcinoma patients (29), the endogenous expression levels of
SEMA5A were then examined in 4 lung adenocarcinoma cell
lines (CL1-5, CL1-0, A549 and H1299), 2 normal lung cell lines
(MRC-5 and BEAS-2B) and human bronchial epithelial cells (16HBE).
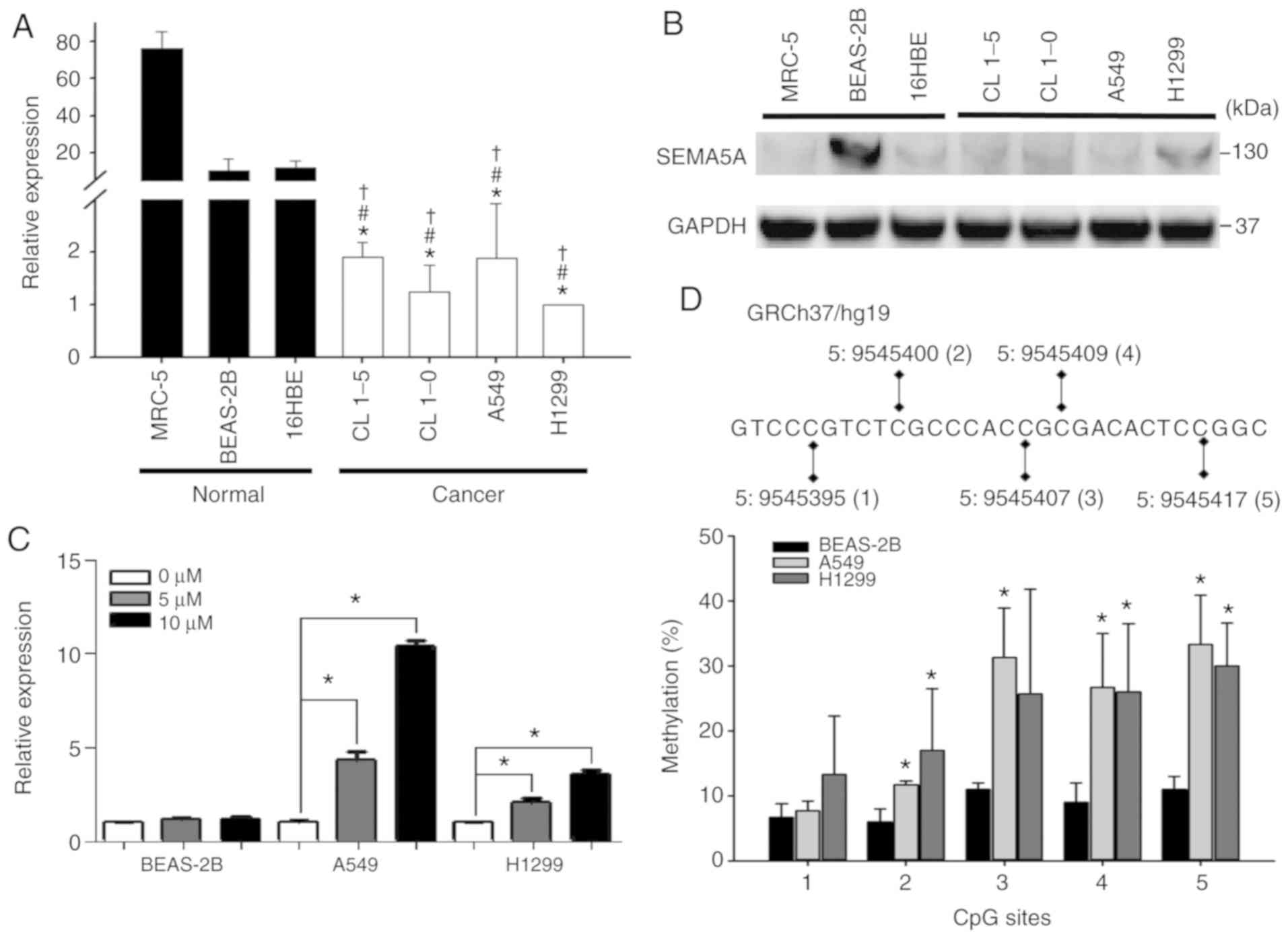
The results of RT-qPCR revealed that SEMA5A was
significantly (P<0.01) downregulated in all lung adenocarcinoma
cell lines (Fig. 2A). The results
of western blot showed that the endogenous SEMA5A proteins in lung
adenocarcinoma cell lines were lower than that of BEAS-2B (Fig. 2B); however, it was not
downregulated the 2 other normal control cell lines (MRC-5 and
16HBE). Therefore, the A549, H1299 and BEAS-2B cells were
arbitrarily selected for use in the following experiments.
Subsequently, to investigate the mechanisms leading
to a lower SEMA5A expression in lung adenocarcinoma cells,
we first examined the role of methylation in regulating endogenous
expression by treating the A549, H1299 and BEAS-2B cell lines with
5-aza. The expression levels of SEMA5A were significantly
(P<0.01) increased in a dose-dependent manner in the A549 and
H1299 lung adenocarcinoma cell lines, whereas they were not altered
in the BEAS-2B normal cells (Fig.
2C). Moreover, pyrosequencing analysis was performed to
identify the specific CpG sites of methylation in SEMA5A. In
total, 5 CpG sites in the 5′untranslated region of SEMA5A
were examined (Fig. 2D top panel).
The 4 CpG sites in the A549 cells and 3 CpG sites in the H1299
cells revealed a significantly higher methylation percentage as
compared to the normal BEAS-2B cells (Fig. 2D bottom panel), indicating that
methylation plays a role in regulating the expression of
SEMA5A in lung adenocarcinoma cells.
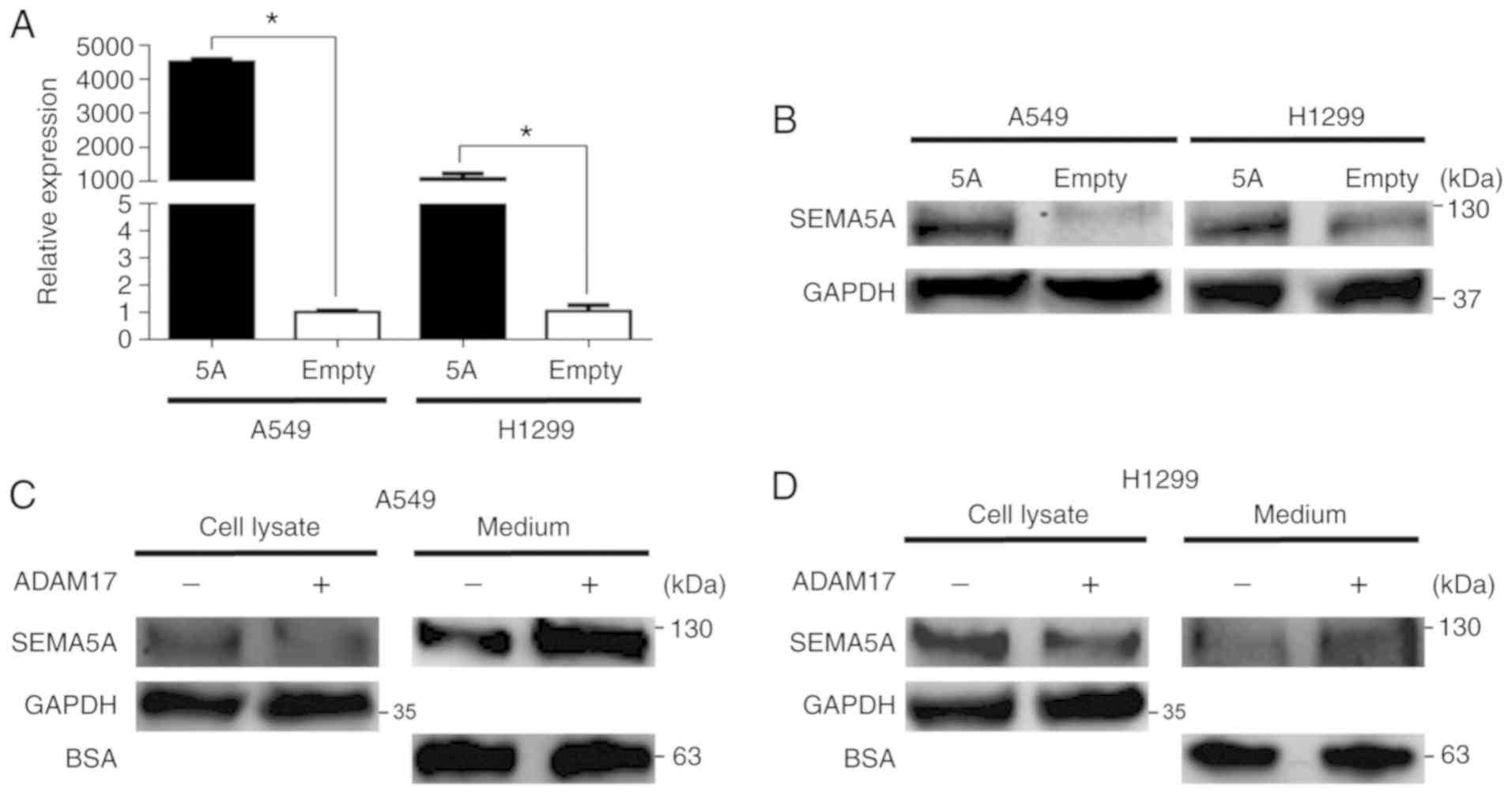
Subsequently, SEMA5A was transiently
overexpressed in the A549 and H1299 cells. As shown in Fig. 3A, the mRNA levels of SEMA5A
in the A549 and H1299 cells following transfection were
significantly (P<0.01) increased. Western blot analysis also
validated that the SEMA5A protein levels increased in both the A549
and H1299 cells upon transfection with SEMA5A expression
plasmid (Fig. 3B). Since a
previous study demonstrated that the mature form of SEMA5B was
proteolytically processed by ADAM17 (14), this study also examined whether
SEMA5A can be cleaved by ADAM17. H1299 cells overexpressing
SEMA5A were treated with active recombinant ADAM17. As shown
in Fig. 3C and D, the amount of
SEMA5A in the cell lysate from the A549 (Fig. 3C) and H1299 (Fig. 3D) cells was lower in the presence
of ADAM17. By contrast, the amount of SEMA5A in the medium
increased (Fig. 3C and D),
suggesting that membrane-bound SEMA5A was released into the medium
in the presence of ADAM17.
Identification and function of
SEMA5A-regulated genes using microarray
In order to investigate the functional role of
SEMA5A, the functions of SEMA5A-regulated genes were
first investigated. Total RNA was extracted 24 h following the
transfection of SEMA5A plasmid into the A549 cells.
Differentially expressed genes regulated by SEMA5A were
identified by Illumina Human HT-12 v4 Bead Chips. The criteria for
the selection of differentially expressed genes consisted of a fold
change ≥3 and significant differences (P<0.05). In total, 350
differentially expressed probes were identified. The volcano plot
illustrated in Fig. 4A represents
350 differentially expressed probes (a.k.a. genes), with 296 probes
upregulated (red points) and 54 ones downregulated (green points)
(Fig. 4A and Table SI). Genes that did not meet the
criteria of differentially expressed genes were shown in gray
(Fig. 4A). Principal component
analysis, a visualization tool to used illustrate that the
expressional profiling of the 350 differentially expressed probes
between the SEMA5A group and control group, indicated that
the distribution between samples overexpressing SEMA5A (cyan
spots) and the controls (black spots) was separated, indicating
different expressional profiling of the selected genes between the
SEMA5A group and the control group (Fig. 4B).
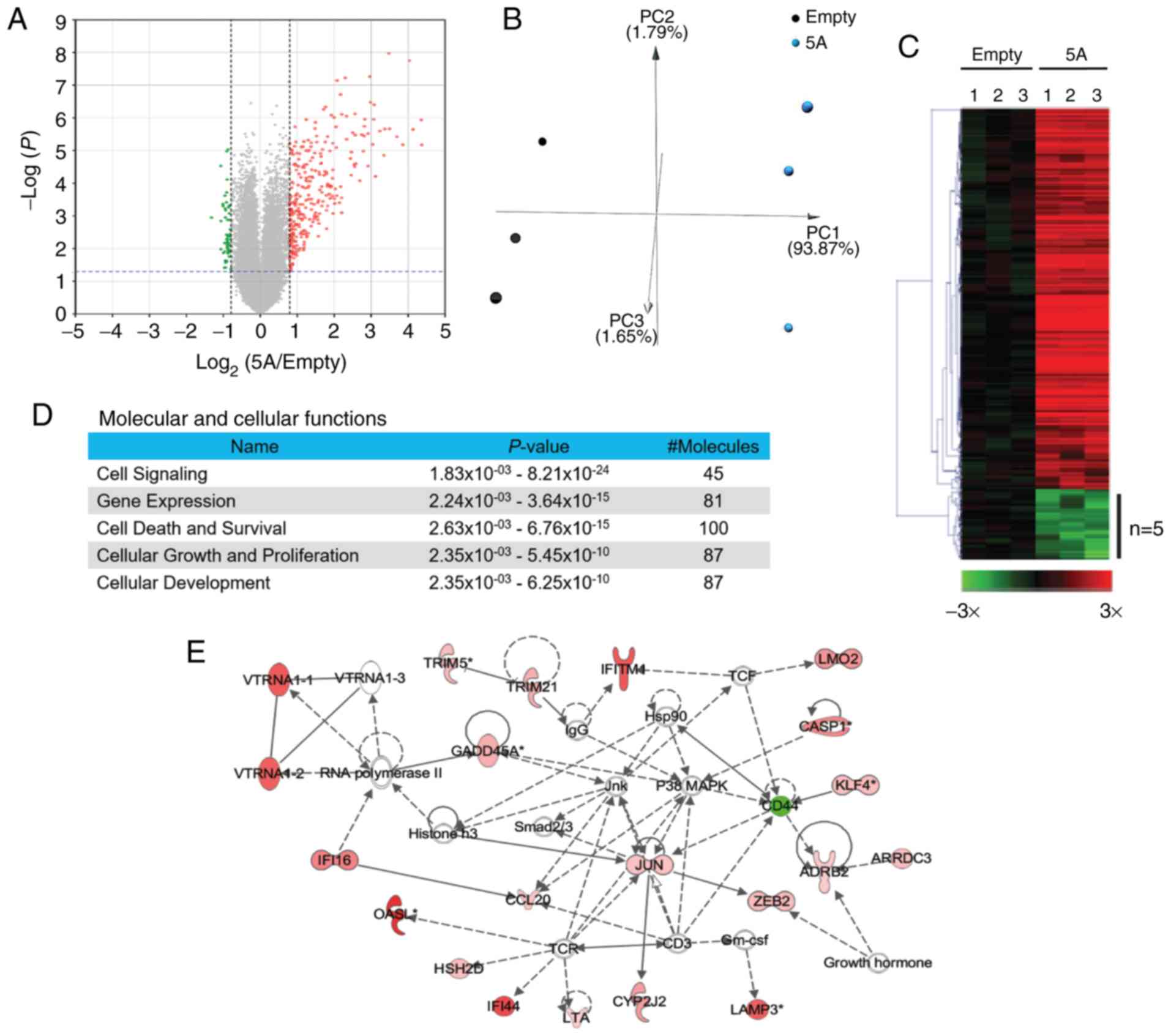 | Figure 4Functions of SEMA5A-regulated
genes are involved in cellular growth and proliferation in lung
adenocarcinoma cells. (A) Volcano plots of differentially expressed
genes. Total RNA was extracted 24 h following transfection and
genomic profiling was examined by Illumina Human HT-12 v4 Bead
Chips. The criteria for the selection of SEMA5A-regulated
genes were a fold change ≥3 and P<0.05. Red points, upregulated
genes in cells overexpressing SEMA5A; green points,
downregulated genes; gray points, genes without significant
changes. (B) Principal component analysis (PCA) of A549 cells
overexpressing SEMA5A. PCA was plotted using the expression
of differentially expressed probes following quantile
normalization. Each dot represents each sample. (C) Heatmap and
hierarchical cluster analysis of SEMA5A-regulated genes. The
rows represent genes, and the columns represent samples. Red,
upregulated genes in cells overexpressing SEMA5A; green,
downregulated genes; black, unaltered expression as compared to the
empty control. (D) Molecular and cellular functions of
SEMA5A-regulated genes using ingenuity pathway analysis. (E)
Network of SEMA5A-regulated genes involved in cellular
movement, cellular growth and proliferation. The solid lines
indicate direct evidence of an interaction between the 2 genes
according to published literature reports, whereas the dashed lines
indicate indirect interactions between molecules as supported by
information in the Ingenuity knowledge base. Red areas denote genes
that were upregulated in cells overexpressing SEMA5A and
green areas indicate downregulated genes. SEMA5A, semaphorin
5A. |
A heatmap with hierarchical clustering, a common
method of visualizing the relative intensity of gene expression by
arranging genes together based on the similarity of their
expressional levels, is presented in Fig. 4C. The relative intensities of the
296 upregulated probes are shown in red, and those of the 54
downregulated ones are shown in green.
Furthermore, network analyses were performed by
ingenuity pathway analysis. The dashed lines indicate indirect
interactions between molecules as supported by information in the
Ingenuity knowledge base. The results of pathway analysis revealed
that these genes were involved in cell signaling, gene expression,
cell death and survival, cellular growth and proliferation
(Fig. 4D and Table SII). Network analysis also
revealed the interactions among some of the differentially
expressed genes. In one of the representative networks, although
SEMA5A was not enriched in this network, contained genes
which were involved in cellular movement, cellular growth and
proliferation, as shown in Fig.
4E, which was later validated by further functional
analysis.
Functional roles of SEMA5A in lung
adenocarcinoma
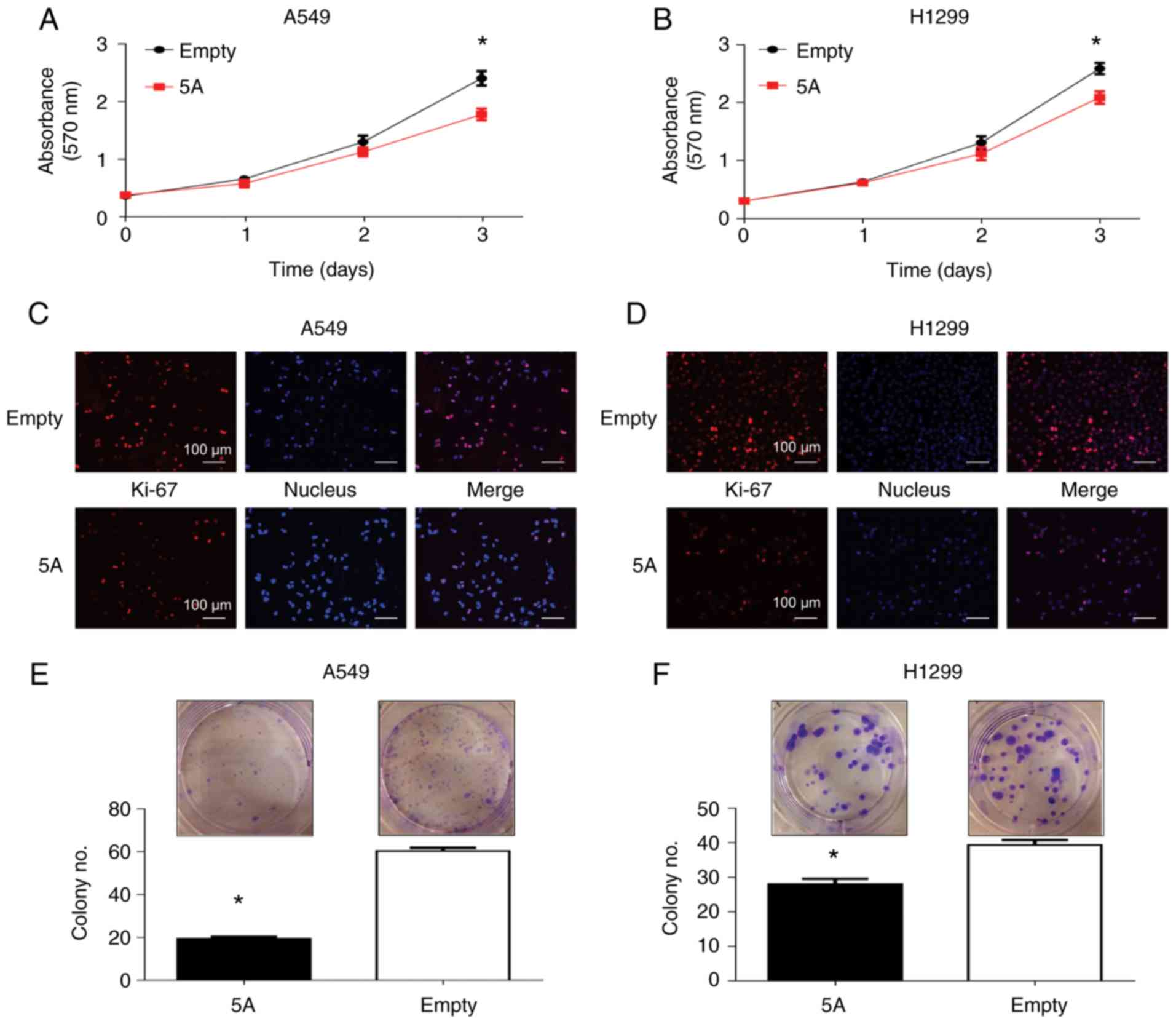
Based on the function of SEMA5A-regulated
genes, the effects of SEMA5A on tumor growth by MTT assays were
hence examined. The results revealed a significant decrement in the
proliferation of both the A549 and H1299 cells overexpressing
SEMA5A (Fig. 5A and B). In
agreement with these results, immunohistochemistry assay of Ki-67,
a cellular marker for proliferation, also revealed a markedly
decreased amount of Ki-67 and cell numbers following transfection
with the SEMA5A expression plasmid (Fig. 5C and D). Furthermore, SEMA5A
overexpression reduced colony formation in both the A549 (Fig. 5E) and H1299 (Fig. 5F) cells. These results demonstrated
the suppressive effects of SEMA5A on the proliferation and colony
formation of lung adenocarcinoma cells.
Subsequently, the effects of SEMA5A on the mobility
of lung adenocarcinoma cells were investigated by gap closure assay
and Transwell migration assays. Both assays revealed that the
overexpression of SEMA5A significantly suppressed the
migratory abilities of both the A549 (Fig. 5G and I) and H1299 (Fig. 5H and J) cells. Previously, SEMA5A
has been shown to impede the motility of human gliomas via the
indirect inactivation of RAC1 and FSCN1 (41,42).
However, the transcriptional levels of RAC1 and FSCN1
were not altered significantly in this study (Fig. S1).
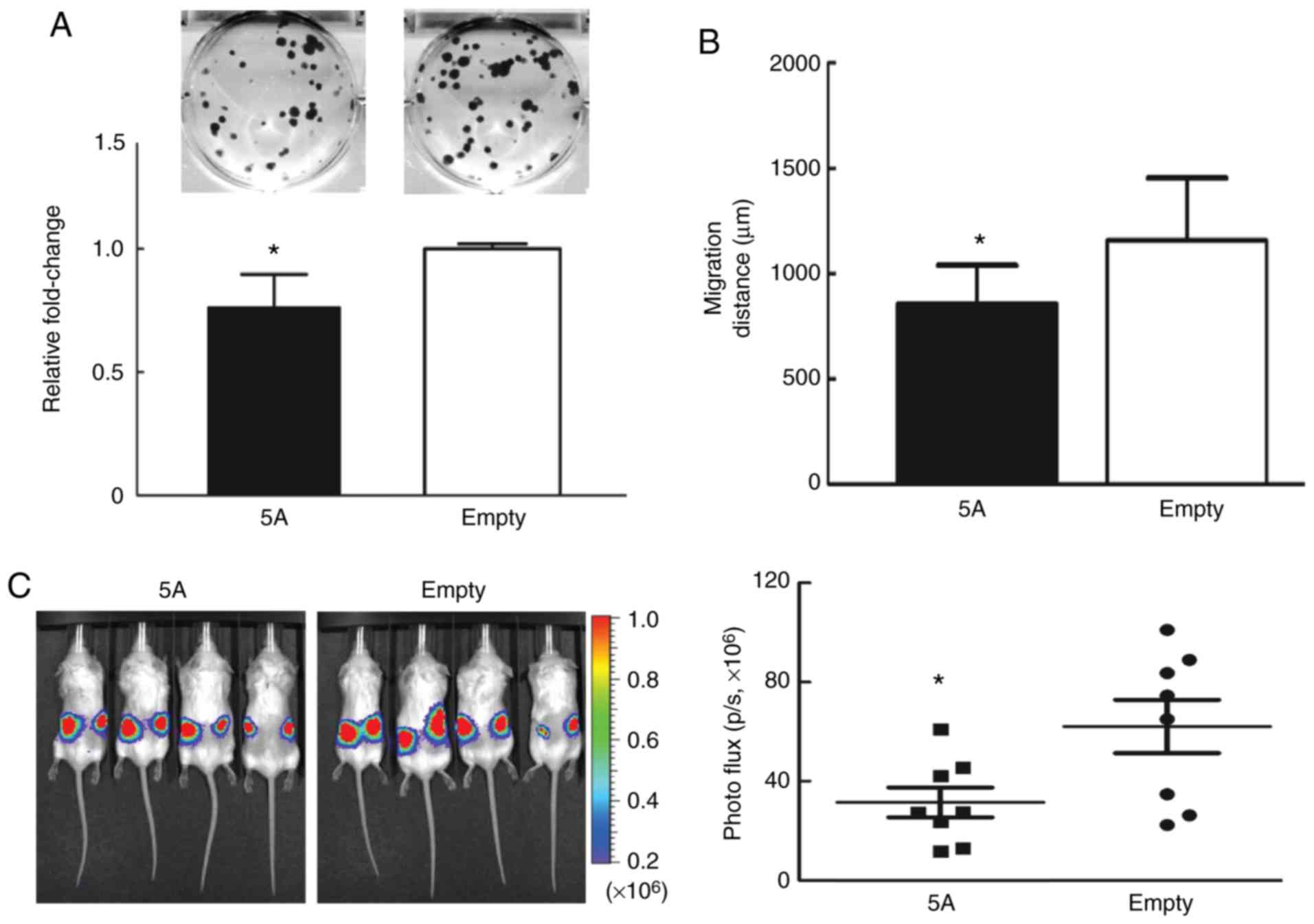
Lastly, the role of SEMA5A in severe combined
immunodeficiency (SCID) mice was examined. Since the growth of
tumors in SCID mice using either A549 or H1299 lung cancer cells
was not successful in pilot studies by the authors (data not
shown), brain metastatic Bm7 lung cancer cells (43) were used to demonstrate the role of
SEMA5A in in vivo experiments. Similar results were observed
in the in vitro experiments (Fig. 6). SEMA5A overexpression
reduced colony formation (Fig. 6A)
and cell migration examined by time-lapse video microscopy
(Fig. 6B). In the tumor xenograft
assays, Bm7 cells overexpressing SEMA5A were injected
subcutaneously into the backs of SCID mice to develop subcutaneous
tumors. After 13 days, the tumor size was found to be significantly
smaller in the SEMA5A group (Fig. 6C). No lymph nodes or distant organ
metastases were detected during the experimental period.
Discussion
In a previous genomic study by the authors,
SEMA5A was identified as a prognostic biomarker in
non-smoking Taiwanese women with lung adenocarcinoma (29). In this study, it was confirmed that
the downregulation of SEMA5A in lung adenocarcinoma tissues
was associated with a poor overall survival in different ethnic
groups. In addition, lower levels of SEMA5A in lung adenocarcinoma
cells were the result of both hypermethylation in the
5′untranslated region at the genetic level and cleavage to the
secretory form. In addition, microarray analyses revealed that
SEMA5A-regulated genes were involved in growth and
proliferation. Finally, in vitro and in vivo analyses
revealed the suppressive effects of SEMA5A overexpression on
lung adenocarcinoma cell lines in terms of proliferation, colony
formation and migration.
Semaphorins represent a large family of proteins,
many of which are promising targets for interfering with cancer
progression due to their roles in tumor angiogenesis, tumor growth
and metastasis (27,53,54).
In particular, previous studies have reported the roles of SEMA5A
in the development of several types of cancer, such as pancreatic
cancer, gastric cancer, ovarian cancer and gliomas (37,41,42,55,56).
SEMA5A has been reported to enhance the invasion and metastasis of
gastric cancer and pancreatic cancer cells (39,57)
and has been shown to be associated with a poor survival in ovarian
cancer (56). In contrast to this
finding, a previous investigation by the authors revealed that the
incidence of lung adenocarcinoma was associated with the
downregulation of SEMA5A expression in non-smoking female
lung adenocarcinoma patients, and that this down-regulation was
associated with a poor overall survival (29). To investigate these seemingly
opposing roles of SEMA5A in different types of cancer, this study
first examined the endogenous expression levels of SEMA5A in
lung adenocarcinoma cell lines. Consistent with the expression
pattern in clinical tissues, the expression of SEMA5A was
downregulated in lung adenocarcinoma cells as compared to their
normal counterparts (Fig. 2A).
Furthermore, the inactivation of several tumor
suppressor genes has been shown to be partly due to
hypermethylation within the promoter region (45,46).
Epigenetically disrupted gene expression can further alter various
cancer-related processes, such as cell proliferation, apoptosis and
angiogenesis (58,59). The abnormal DNA methylation of
genes may be associated with clinical outcomes in lung cancer
patients (60). This study found
that hypermethylation in the upstream genetic loci was partly
responsible for the downregulation of SEMA5A in lung
adenocarcinoma cells, as has been previously reported for other
tumor suppressor genes (45,46).
In this study, when the cells were treated with the methylation
inhibitor, 5-aza, the upregulation of SEMA5A was observed
only in the cancer cell lines. Pyrosequencing results further
identified the methylated CpG sites modulating the expression of
SEMA5A in these cell lines, suggesting that aberrant
methylation changes result in the inactivation of SEMA5A in
lung adenocarcinoma cells (Fig. 2B and
C). However, additional studies using larger numbers of tissue
samples that contain clinical features are required to validate
whether methylation changes of SEMA5A are involved in
tumorigenesis.
Furthermore, this study demonstrated that SEMA5A
can be possibly cleaved by ADAM17, which belongs to the protein
family of disintegrins and metalloproteases (Fig. 3C and D). ADAM17 is involved in the
release of a soluble ectodomain from membrane-bound pro-proteins.
It has also been reported to be upregulated in non-small cell lung
cancer (61), and this
upregulation of ADAM17 can be caused due to ionizing radiation
(62). Moreover, the silencing of
ADAM17 has been shown to suppress the migration and invasion
of A549 cells in vitro, and tumor growth in vivo
(63). Since previous studies have
demonstrated that the mature form of SEMA5B is proteolytically
processed by ADAM17 (14), and
that the cleaved extracellular domain of SEMA5A decreases following
the silencing ADAM17 (32),
this study assessed the proteolytic effect of recombinant ADAM17 on
SEMA5A. The results suggested that the membrane-bound SEMA5A was
exported to the medium following cleavage by ADAM17. However,
further clarifications are required to conclude whether the
endogenous ADAM17 in lung adenocarcinoma cells has enough
proteolytic activity to shed membrane-bound SEMA5A.
The extracellular domain of SEMA5A is involved in
angiogenesis (36). There is
evidence to indicate an increase in proliferation and the
upregulation of anti-apoptotic genes (e.g., BCL-2 and
BIRC5) following treatment of endothelial cells with the
extracellular domain of recombinant SEMA5A (36). In addition, pancreatic cells
transfected with the extracellular domain of SEMA5A exhibit a
greater metastatic potential and an enhanced endothelial cell
proliferative ability (64). These
results suggest that the extracellular domain of SEMA5A plays a
potential role in carcinogenesis.
However, this study found that the total amount of
membrane-bound SEMA5A in lung adenocarcinoma cells was
downregulated compared to normal lung cells. The transient
overexpression of SEMA5A in lung adenocarcinoma cells had
tumor-suppressive effects, such as decreasing cell proliferation,
colony formation and migration (Figs.
5 and 6), although not
increasing apoptosis (data not shown). Furthermore, lower
expression levels of SEMA5A were found to be associated with
a worse prognosis (Fig. 1B), which
suggested the tumor suppressive role of SEMA5A in lung
adenocarcinoma. Consistent with the findings of this study, lower
endogenous SEMA5A levels have been found to be associated with
increased invasiveness in glioma. Furthermore, SEMA5A has been
shown to impede the motility of human gliomas upon its interaction
with Plexin-B3 via the indirect inactivation of Rac1 through
RhoGDIα, and the inactivation of protein kinase C (PKC) to
phosphorylate fascin-1 (41,42).
However, the transcriptional levels of RAC1 and FSCN1
did not alter significantly in this study (Fig. S1), and whether the levels and
activity of these proteins are altered remains to be determined in
lung adenocarcinoma cells.
On the contrary, a high expression of SEMA5A
protein has been shown to be associated with poor overall survival
outcomes in metastatic ovarian cancer (56) and to be associated with progression
and metastasis in gastric cancer and pancreatic cancer (39,57).
In explaining the discrepancy of the functions of SEMA5A, it was
thus speculated that the cleaved extracellular domain and
full-length of SEMA5A may carry out opposite functions in different
cancer types. The alternative explanation is that receptor-ligand
interactions generate simultaneous bidirectional signals (i.e.,
forward signaling and reverse signaling) with opposite functions
(65-67). That is, the full-length of SEMA5A
on the membrane may function as both a receptor and ligand to
generate simultaneous forward and reverse signals, whereas the
cleaved extracellular domain may only initiate reverse signaling by
serving as ligand for receptors on other cells. Therefore, it was
hypothesized that these phenomena may mainly be due to different
experimental settings and tumor types. Yet, further studies are
warranted to explore the function of SEMA5A in different cancer
types.
Finally, the functions of SEMA5A in lung
adenocarcinoma were investigated by identifying the downregulated
related genes using microarrays. Both pathway analysis and network
analysis revealed that one function of SEMA5A-regulated
genes was cell growth and proliferation (Fig. 4). Among these genes involved in
growth and proliferation, a number of genes have been reported with
similar functions in other types of cancer. For example,
ARRDC3 and CASP1 were found to be upregulated in this
study. ARRDC3 has been reported to suppress breast carcinoma
invasion (68). The downregulation
of the expression of CASP1 has been shown to result in the
proliferation and invasion of breast cancer cells (69). In this study, the functions of
SEMA5A were further validated in in vitro (Fig. 5) and in vivo (Fig. 6) experiments, demonstrating that
SEMA5A truly plays a tumor-suppressive role in the proliferation
and migration of lung adenocarcinoma cells. On the whole, the
findings of this study may thus contribute to the development of
novel therapeutic regimens for lung adenocarcinoma in the
future.
Supplementary Data
Funding
This study was supported by grants from the
Ministry of Science and Technology (NSC 98-2320-B-002-044-MY3 and
MOST 106-2320-B-002-016-MY3) and China Medical University
(CMU107-TU-09).
Availability of data and materials
All data have been deposited in the Gene Expression
Omnibus (GEO, GSE114578). The other data and materials are
available upon request from the corresponding authors.
Authors' contributions
PHK, GL, YPS and LCL conceived and designed the
experiments. PHK, GL and YPS performed the experiments. PHK, GL,
YAC, MHT and EYC analyzed the data. MHT, EYC and LCL contributed
reagents, materials and/or analysis tools. PHK, GL, YPS and LCL
wrote the manuscript. All authors have reviewed and approved the
final manuscript.
Ethics approval and consent to
participate
This animal model followed protocols approved by
the Institutional Animal Care and Use Committee of China Medical
University and Hospital (animal protocol no. 2016-102).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Dr Melissa Stauffer
for providing editorial assistance.
References
|
1
|
Zhang N, Wei X and Xu L: miR-150 promotes
the proliferation of lung cancer cells by targeting P53. FEBS Lett.
587:2346–2351. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Molina JR, Yang P, Cassivi SD, Schild SE
and Adjei AA: Non-small cell lung cancer: Epidemiology, risk
factors, treatment, and survivorship. Mayo Clin Proc. 83:584–594.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: The impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 61:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Boyero L, Sánchez-Palencia A, Miranda-León
MT, Hernández-Escobar F, Gómez-Capilla JA and Fárez-Vidal ME:
Survival, classifications, and desmosomal plaque genes in non-small
cell lung cancer. Int J Med Sci. 10:1166–1173. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shaoyan X, Juanjuan Y, Yalan T, Ping H,
Jianzhong L and Qinian W: Downregulation of EIF4A2 in
non-small-cell lung cancer associates with poor prognosis. Clin
Lung Cancer. 14:658–665. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vendetti FP and Rudin CM: Epigenetic
therapy in non-small-cell lung cancer: Targeting DNA
methyltransferases and histone deacetylases. Expert Opin Biol Ther.
13:1273–1285. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Adams RH, Betz H and Püschel AW: A novel
class of murine semaphorins with homology to thrombospondin is
differentially expressed during early embryogenesis. Mech Dev.
57:33–45. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Capparuccia L and Tamagnone L: Semaphorin
signaling in cancer cells and in cells of the tumor
microenvironment-two sides of a coin. J Cell Sci. 122:1723–1736.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Christensen C, Ambartsumian N, Gilestro G,
Thomsen B, Comoglio P, Tamagnone L, Guldberg P and Lukanidin E:
Proteolytic processing converts the repelling signal Sema3E into an
inducer of invasive growth and lung metastasis. Cancer Res.
65:6167–6177. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Adams RH, Lohrum M, Klostermann A, Betz H
and Püschel AW: The chemorepulsive activity of secreted semaphorins
is regulated by furin-dependent proteolytic processing. EMBO J.
16:6077–6086. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hattori M, Osterfield M and Flanagan JG:
Regulated cleavage of a contact-mediated axon repellent. Science.
289:1360–1365. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Esselens C, Malapeira J, Colome N, Casa l
C, Rodríguez-Manzaneque JC, Canals F and Arribas J: The cleavage of
semaphorin 3C induced by ADAMTS1 promotes cell migration. J Biol
Chem. 285:2463–2473. 2010. View Article : Google Scholar :
|
|
14
|
Browne K, Wang W, Liu RQ, Piva M and
O′Connor TP: Transmembrane semaphorin5B is proteolytically
processed into a repulsive neural guidance cue. J Neurochem.
123:135–146. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Potiron VA, Roche J and Drabkin HA:
Semaphorins and their receptors in lung cancer. Cancer Lett.
273:1–14. 2009. View Article : Google Scholar :
|
|
16
|
Chedotal A, Kerjan G and Moreau-Fauvarque
C: The brain within the tumor: New roles for axon guidance
molecules in cancers. Cell Death Differ. 12:1044–1056. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Behar O, Golden JA, Mashimo H, Schoen FJ
and Fishman MC: Semaphorin III is needed for normal patterning and
growth of nerves, bones and heart. Nature. 383:525–528. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hall KT, Boumsell L, Schultze JL,
Boussiotis VA, Dorfman DM, Cardoso AA, Bensussan A, Nadler LM and
Freeman GJ: Human CD100, a novel leukocyte semaphorin that promotes
B-cell aggregation and differentiation. Proc Natl Acad Sci USA.
93:11780–11785. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Miao HQ, Soker S, Feiner L, Alonso JL,
Raper JA and Klagsbrun M: Neuropilin-1 mediates
collapsin-1/semaphorin III inhibition of endothelial cell motility:
Functional competition of collapsin-1 and vascular endothelial
growth factor-165. J Cell Biol. 146:233–242. 1999.PubMed/NCBI
|
|
20
|
Christensen CR, Klingelhofer J, Tarabykina
S, Hulgaard EF, Kramerov D and Lukanidin E: Transcription of a
novel mouse semaphorin gene, M-semaH, correlates with the
metastatic ability of mouse tumor cell lines. Cancer Res.
58:1238–1244. 1998.PubMed/NCBI
|
|
21
|
Chen R, Zhuge X, Huang Z, Lu D, Ye X, Chen
C, Yu J and Lu G: Analysis of SEMA3B methylation and expression
patterns in gastric cancer tissue and cell lines. Oncol Rep.
31:1211–1218. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gao X, Tang C, Shi W, Feng S, Qin W, Jiang
T and Sun Y: Semaphorin-3F functions as a tumor suppressor in
colorectal cancer due to regulation by DNA methylation. Int J Clin
Exp Pathol. 8:12766–12774. 2015.
|
|
23
|
Kruger RP, Aurandt J and Guan KL:
Semaphorins command cells to move. Nat Rev Mol Cell Biol.
6:789–800. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nagai H, Sugito N, Matsubara H, Tatematsu
Y, Hida T, Sekido Y, Nagino M, Nimura Y, Takahashi T and Osada H:
CLCP1 interacts with semaphorin 4B and regulates motility of lung
cancer cells. Oncogene. 26:4025–4031. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nasarre P, Kusy S, Constantin B,
Castellani V, Drabkin HA, Bagnard D and Roche J: Semaphorin SEMA3F
has a repulsing activity on breast cancer cells and inhibits
E-cadherin-mediated cell adhesion. Neoplasia. 7:180–189. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Castro-Rivera E, Ran S, Thorpe P and Minna
JD: Semaphorin 3B (SEMA3B) induces apoptosis in lung and breast
cancer, whereas VEGF165 antagonizes this effect. Proc Natl Acad Sci
USA. 101:11432–11437. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tomizawa Y, Sekido Y, Kondo M, Gao B,
Yokota J, Roche J, Drabkin H, Lerman MI, Gazdar AF and Minna JD:
Inhibition of lung cancer cell growth and induction of apoptosis
after reexpression of 3p21.3 candidate tumor suppressor gene
SEMA3B. Proc Natl Acad Sci USA. 98:13954–13959. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Brambilla E, Constantin B, Drabkin H and
Roche J: Semaphorin SEMA3F localization in malignant human lung and
cell lines: A suggested role in cell adhesion and cell migration.
Am J Pathol. 156:939–950. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lu TP, Tsai MH, Lee JM, Hsu CP, Chen PC,
Lin CW, Shih JY, Yang PC, Hsiao CK, Lai LC and Chuang EY:
Identification of a novel biomarker, SEMA5A, for non-small cell
lung carcinoma in nonsmoking women. Cancer Epidemiol Biomarkers
Prev. 19:2590–2597. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kolodkin AL, Matthes DJ and Goodman CS:
The semaphorin genes encode a family of transmembrane and secreted
growth cone guidance molecules. Cell. 75:1389–1399. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bahri SM, Chia W and Yang X:
Characterization and mutant analysis of the Drosophila sema 5c
gene. Dev Dyn. 221:322–330. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gras C, Eiz-Vesper B, Jaimes Y, Immenschuh
S, Jacobs R, Witte T, Blasczyk R and Figueiredo C: Secreted
semaphorin 5A activates immune effector cells and is a biomarker
for rheumatoid arthritis. Arthritis Rheumatol. 66:1461–1471. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Arribas J and Borroto A: Protein
ectodomain shedding. Chem Rev. 102:4627–4638. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Simmons AD, Overhauser J and Lovett M:
Isolation of cDNAs from the Cri-du-chat critical region by direct
screening of a chromosome 5-specific cDNA library. Genome Res.
7:118–127. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mosca-Boidron AL, Gueneau L, Huguet G,
Goldenberg A, Henry C, Gigot N, Pallesi-Pocachard E, Falace A,
Duplomb L, Thevenon J, et al: A de novo microdeletion of SEMA5A in
a boy with autism spectrum disorder and intellectual disability.
Eur J Hum Genet. 24:838–843. 2016. View Article : Google Scholar :
|
|
36
|
Sadanandam A, Rosenbaugh EG, Singh S,
Varney M and Singh RK: Semaphorin 5A promotes angiogenesis by
increasing endothelial cell proliferation, migration, and
decreasing apoptosis. Microvasc Res. 79:1–9. 2010. View Article : Google Scholar
|
|
37
|
Sadanandam A, Varney ML, Singh S, Ashour
AE, Moniaux N, Deb S, Lele SM, Batra SK and Singh RK: High gene
expression of semaphorin 5A in pancreatic cancer is associated with
tumor growth, invasion and metastasis. Int J Cancer. 127:1373–1383.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sadanandam A, Varney ML, Kinarsky L, Ali
H, Mosley RL and Singh RK: Identification of functional cell
adhesion molecules with a potential role in metastasis by a
combination of in vivo phage display and in silico analysis. OMICS.
11:41–57. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pan G, Zhang X, Ren J, Lu J, Li W, Fu H,
Zhang S and Li J: Semaphorin 5A, an axon guidance molecule,
enhances the invasion and metastasis of human gastric cancer
through activation of MMP9. Pathol Oncol Res. 19:11–18. 2013.
View Article : Google Scholar
|
|
40
|
Saxena S, Purohit A, Varney ML, Hayashi Y
and Singh RK: Semaphorin-5A maintains epithelial phenotype of
malignant pancreatic cancer cells. BMC Cancer. 18:12832018.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li X and Lee AY: Semaphorin 5A and
plexin-B3 inhibit human glioma cell motility through
RhoGDIα-mediated inactivation of Rac1 GTPase. J Biol Chem.
285:32436–32445. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li X, Law JW and Lee AY: Semaphorin 5A and
plexin-B3 regulate human glioma cell motility and morphology
through Rac1 and the actin cytoskeleton. Oncogene. 31:595–610.
2012. View Article : Google Scholar
|
|
43
|
Lin CY, Chen HJ, Huang CC, Lai LC, Lu TP,
Tseng GC, Kuo TT, Kuok QY, Hsu JL, Sung SY, et al: ADAM9 promotes
lung cancer metastases to brain by a plasminogen activator-based
pathway. Cancer Res. 74:5229–5243. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
45
|
Feinberg AP and Tycko B: The history of
cancer epigenetics. Nat Rev Cancer. 4:143–153. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002. View
Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lin HC, Yeh CC, Chao LY, Tsai MH, Chen HH,
Chuang EY and Lai LC: The hypoxia-responsive lncRNA NDRG-OT1
promotes NDRG1 degradation via ubiquitin-mediated proteolysis in
breast cancer cells. Oncotarget. 9:10470–10482. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
The PONES Correction: Online survival
analysis software to assess the prognostic value of biomarkers
using transcriptomic data in non-small-cell lung cancer. PLoS One.
9:e1118422014. View Article : Google Scholar
|
|
49
|
Győrffy B, Surowiak P, Budczies J and
Lánczky A: Online survival analysis software to assess the
prognostic value of biomarkers using transcriptomic data in
non-small-cell lung cancer. PLoS One. 8:e822412013. View Article : Google Scholar
|
|
50
|
Bild AH, Yao G, Chang JT, Wang Q, Potti A,
Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, et al:
Oncogenic pathway signatures in human cancers as a guide to
targeted therapies. Nature. 439:353–357. 2006. View Article : Google Scholar
|
|
51
|
Director's Challenge Consortium for the
Molecular Classification of Lung Adenocarcinoma. Shedden K, Taylor
JM, Enkemann SA, Tsao MS, Yeatman TJ, Gerald WL, Eschrich S,
Jurisica I, Giordano TJ, et al: Gene expression-based survival
prediction in lung adenocarcinoma: A multi-site, blinded validation
study. Nat Med. 14:822–827. 2008.PubMed/NCBI
|
|
52
|
Selamat SA, Chung BS, Girard L, Zhang W,
Zhang Y, Campan M, Siegmund KD, Koss MN, Hagen JA, Lam WL, et al:
Genome-scale analysis of DNA methylation in lung adenocarcinoma and
integration with mRNA expression. Genome Res. 22:1197–1211. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Roche J, Boldog F, Robinson M, Robinson L,
Varella-Garcia M, Swanton M, Waggoner B, Fishel R, Franklin W,
Gemmill R and Drabkin H: Distinct 3p213 deletions in lung cancer
and identification of a new human semaphorin. Oncogene.
12:1289–1297. 1996.PubMed/NCBI
|
|
54
|
Xiang RH, Hensel CH, Garcia DK, Carlson
HC, Kok K, Daly MC, Kerbacher K, van den Berg A, Veldhuis P, Buys
CH and Naylor SL: Isolation of the human semaphorin III/F gene
(SEMA3F) at chromosome 3p21, a region deleted in lung cancer.
Genomics. 32:39–48. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Pan G, Lv H, Ren H, Wang Y, Liu Y, Jiang H
and Wen J: Elevated expression of semaphorin 5A in human gastric
cancer and its implication in carcinogenesis. Life Sci. 86:139–144.
2010. View Article : Google Scholar
|
|
56
|
Wang DG, Yousif NG, Sadiq AM, Schilling MM
and Danielson X: Critical role of SEMA5A expression in invasion and
metastasis of ovarian cancer cell. Am J Biomed. 2:292–305.
2014.
|
|
57
|
Saxena S, Hayashi Y, Wu L, Awaji M, Atri
P, Varney ML, Purohit A, Rachagani S, Batra SK and Singh RK:
Pathological and functional significance of Semaphorin-5A in
pancreatic cancer progression and metastasis. Oncotarget.
9:5931–5943. 2017.
|
|
58
|
Baylin SB, Esteller M, Rountree MR,
Bachman KE, Schuebel K and Herman JG: Aberrant patterns of DNA
methylation, chromatin formation and gene expression in cancer. Hum
Mol Genet. 10:687–692. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lenka G, Tsai MH, Hsiao JH, Lai LC and
Chuang EY: Overexpression of methylation-driven DCC suppresses
proliferation of lung cancer cells. Translational Cancer Res.
5:169–175. 2016. View Article : Google Scholar
|
|
60
|
Lenka G, Tsai MH, Lin HC, Hsiao JH, Lee
YC, Lu TP, Lee JM, Hsu CP, Lai LC and Chuang EY: Identification of
methylation-driven, differentially expressed STXBP6 as a novel
biomarker in lung adenocarcinoma. Sci Rep. 7:425732017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Baumgart A, Seidl S, Vlachou P, Michel L,
Mitova N, Schatz N, Specht K, Koch I, Schuster T, Grundler R, et
al: ADAM17 regulates epidermal growth factor receptor expression
through the activation of Notch1 in non-small cell lung cancer.
Cancer Res. 70:5368–5378. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sharma A, Bender S, Zimmermann M,
Riesterer O, Broggini-Tenzer A and Pruschy MN: Secretome signature
identifies ADAM17 as novel target for radiosensitization of
non-small cell lung cancer. Clin Cancer Res. 22:4428–4439. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lv X, Li Y, Qian M, Ma C, Jing H, Wen Z
and Qian D: ADAM17 silencing suppresses the migration and invasion
of non-small cell lung cancer. Mol Med Rep. 9:1935–1940. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sadanandam A, Sidhu SS, Wullschleger S,
Singh S, Varney ML, Yang CS, Ashour AE, Batra SK and Singh RK:
Secreted semaphorin 5A suppressed pancreatic tumour burden but
increased metastasis and endothelial cell proliferation. Br J
Cancer. 107:501–507. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Hamada K, Oike Y, Ito Y, Maekawa H, Miyata
K, Shimomura T and Suda T: Distinct roles of ephrin-B2 forward and
EphB4 reverse signaling in endothelial cells. Arterioscler Thromb
Vasc Biol. 23:190–197. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhang H, Yan D, Shi X, Liang H, Pang Y,
Qin N, Chen H, Wang J, Yin B, Jiang X, et al: Transmembrane
TNF-alpha mediates 'forward' and 'reverse' signaling, inducing cell
death or survival via the NF-kappaB pathway in Raji Burkitt
lymphoma cells. J Leukoc Biol. 84:789–797. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Godenschwege TA, Hu H, Shan-Crofts X,
Goodman CS and Murphey RK: Bi-directional signaling by Semaphorin
1a during central synapse formation in Drosophila. Nat Neurosci.
5:1294–1301. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
68
|
Arakaki AKS, Pan WA, Lin H and Trejo J:
The alpha-arrestin ARRDC3 suppresses breast carcinoma invasion by
regulating G protein-coupled receptor lysosomal sorting and
signaling. J Biol Chem. 293:3350–3362. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Sun Y and Guo Y: Expression of Caspase-1
in breast cancer tissues and its effects on cell proliferation,
apoptosis and invasion. Oncol Lett. 15:6431–6435. 2018.PubMed/NCBI
|