Introduction
Lung cancer is the leading cause of
cancer-associated mortality worldwide (1-3).
More than 80% of patients with lung cancer suffer from non-small
cell lung cancer (NSCLC), and the majority have advanced disease at
diagnosis with a poor prognosis (4).
Adenocarcinoma is the most common subtype of NSCLC.
In addition to epidermal growth factor receptor (EGFR)
mutations that occur in approximately 50% of patients, the oncogene
KRAS, responsible for tumor formation, and the tumor
suppressor gene TP53, implicated in cell cycle regulation,
cell proliferation and apoptosis, are also frequently mutated in
this type of tumor, with EGFR and KRAS mutations
generally mutually exclusive (1).
The role of such mutations in the selection of the anticancer
treatment is still under debate, even though it appears that they
may be associated with differential sensitivity patterns to
currently available therapies (5,6).
Specific targeted therapies are available for
patients with advanced disease harboring EGFR mutations or
anaplastic lymphoma kinase (ALK) translocation, and
immunotherapy has also been increasingly adopted on the basis of
the programmed death protein 1 (PD-1) level. It is still not clear
how the remaining mutations could drive the selection of
anti-cancer treatment. The American Society of Clinical Oncology
and National Comprehensive Cancer Network (NCCN) recommends a
combination of two cytotoxic drugs for the first-line therapy of
patients who are not eligible for biological treatment (7). The antifolate drug, pemetrexed, is
frequently used in combination with cisplatin, since an improved
overall survival has been observed in patients treated with this
combination compared to those undergoing the cisplatin-gemcitabine
regimen (8). However, acquired
drug resistance mechanisms are common and compromise the efficacy
of chemotherapy for lung cancer (8-11).
The mechanism of action of cisplatin relies on the induction of DNA
damage (12), resulting in cell
cycle arrest and subsequent apoptosis. Neoplastic cells activate
several genes involved in DNA damage repair to withstand
platinum-based chemotherapy. Among these, cyclin-dependent kinase
inhibitor 1A (CDKN1A) plays a central role in cell cycle
progression, regulating both G1/S and G2/M checkpoints. Upon DNA
damage, CDKN1A is transiently recruited to target sites to
facilitate their repair. However, CDKN1A activity is dependent on
the p53 status. In fact, upon p53 loss-of-function
mutation, CDKN1A overexpression drives cells to acquire a more
aggressive phenotype that is capable of escaping cell block,
senescence and apoptosis (13).
The aim of the present study was to identify novel
potential biomarkers involved in the onset of resistance to the
cisplatin-pemetrexed combination in an EGFR-wild type (wt)
NSCLC cell line (RAL) harboring KRAS and TP53
mutations.
Materials and methods
Cells and cell culture
The NSCLC cell line, RAL, is derived from a
metastatic lesion of lung adenocarcinoma of a 52-year-old female
previously treated with cisplatin (14). The identity of the patient was
irreversibly anonymized prior to specimen processing. The cell line
is characterized by the following: EGFR-wt, KRAS
mutation at exon 1 (p.G12C, missense, not functional, deleterious),
TP53 mutation at exon 7 (p.G244C, missense, not functional,
deleterious) and no ALK rearrangement. The cells were grown
in Dulbecco's modified Eagle's medium/HAM F12 (1:1) supplemented
with 10% fetal bovine serum, 2 mM of L-glutamine (EuroClone) and 10
µg/ml of insulin (Sigma-Aldrich; Merck KGaA) in a humidified
atmosphere with 5% CO2 at 37°C. The population doubling
time was 65 h, as previously described (14).
Chemosensitivity assay
Cisplatin (Ebewe Pharma-Sandoz) and pemetrexed (Eli
Lilly Italia Spa) were tested at plasma peak concentrations (10 and
320.5 µM, respectively). Cell viability was assessed by MTS
assay according to the manufacturer's protocol (CellTiter
96® AQueous One Solution Cell Proliferation assay,
Promega Corp.). Experiments were run in octuplicate and each
experiment was repeated 3 times. To ensure that the exposure time
was compatible with the half-life of the drugs administered as in a
clinical setting, the cells were treated with pemetrexed or
cisplatin alone and washed out with drug-free medium after 3 and 6
h of exposure, respectively, followed by up to 96 h of recovery, as
previously described (15). For
the combination treatment, the cells were exposed to cisplatin and
pemetrexed for 6 h and washed out with drug-free medium, followed
by 96 h of recovery (96 h-post wo).
Flow cytometry
Untreated cells, and those at 96-h post wo and 21
days post-treatment washout (21 days-post wo) were collected and
fixed with ice-cold 70% ethanol. The cells at 21 days-post wo were
allowed to recover by replacement of the fresh medium 3 times/week.
No split was carried out, in order to permit the RAL cells to form
resistant clones and repopulate the flask. Flow cytometric analysis
was performed using a FACSCanto flow cytometer (BD Biosciences).
Data acquisition and analysis were performed using FACSDiva
software (Version 6.1.3, BD Biosciences). Samples were run in
triplicate (10,000 events for each replica). Data report the mean
value of 3 experiments with standard deviations <5%.
TUNEL assay
Cells were fixed in 1% paraformaldehyde in
phosphate-buffered saline (PBS) on ice for 15 min, suspended in
cold ethanol (70%) and stored overnight at −20°C. The cells were
then washed twice in PBS, resuspended in PBS containing 0.1% Triton
X-100 for 5 min at 4°C, incubated in 50 µl of solution
containing TdT and FITC-conjugated dUTP deoxynucleotides 1:1 (Roche
Diagnostics GmbH) in a humidified atmosphere for 90 min at 37°C in
the dark, washed in PBS, counterstained with propidium iodide (PI,
200 µg/ml, MP Biomedicals) and RNase (10,000 U/ml,
Sigma-Aldrich; Merck KGaA) for 30 min at 4°C in the dark, and
analyzed by flow cytometry.
Cell cycle distribution analysis
The cells were fixed in ethanol (70%) and stained in
a solution containing 100 µg/ml of PI (Sigma-Aldrich; Merck
KGaA), 10,000 U/ml of RNase (Sigma-Aldrich; Merck KGaA) and 0.01%
NP40 (Sigma-Aldrich; Merck KGaA). After 24 h, the samples were
analyzed by flow cytometry. Data were elaborated using ModFit (DNA
Modelling System) software (Version 4.1.7, Verity Software House
Inc.), and expressed as fractions of cells in the different cell
cycle phases.
Annexin V assay
The cells were washed once in PBS, incubated with
250 µl of Annexin V-FITC in binding buffer (eBioscience) for
15 min at 37°C in a humidified atmosphere in the dark, and then
washed in PBS and suspended in binding buffer. Immediately before
flow cytometric analysis, PI was added to a final concentration of
5 µg/ml to distinguish between total apoptotic cells
(Annexin V+ and PI− or PI+) and
necrotic cells (Annexin V− and PI+).
siRNA transfection
CDKN1A Silencer Select Validated siRNA (Thermo
Fisher Scientific, Inc.) and a validated Negative Universal
Control™ (Invitrogen; Thermo Fisher Scientific, Inc.) were used.
The cells were seeded in 25 cm2-flasks at a density of
2.5×105 cells. The transfections were carried out using
the TransIT-X2 Dynamic Delivery System (Mirus Bio LCC) and Opti-MEM
GlutaMax medium (Invitrogen; Thermo Fisher Scientific, Inc.)
without antibiotics. siRNA/TransIT-X2 was incubated with the cells
for 25 min. The total incubation time before the combinational
treatment was 24 h.
Cellular fractionation, protein
extraction and western blot analysis
Cells (7×106) that had either been
treated with cisplatin and pemetrexed or under the control
condition were washed in hypotonic buffer (10 mM Tris, 5 mM
MgCl2). Triton X-100 (10% stock) was added to a 0.3%
final concentration. The suspension was passed twice through a
22-gauge needle with a syringe and centrifuged at 800 × g for 10
min at 4°C to remove debris. The supernatant was collected.
Protease/phosphatase inhibitor cocktail (Sigma-Aldrich; Merck KGaA)
was added and protein lysate was saved as the cytosolic fraction.
Nuclei were isolated following 2 subsequent centrifugations at 800
× g for 10 min at 4°C and lysed in RIPA buffer containing 1
µg/ml of DNAse (Voden Medical Instruments). Western blot
analysis was performed as previously described (16). The list of the specific antibodies
and conditions used is available upon request.
RNA extraction, purification and reverse
transcription
Total RNA was extracted using TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and purified
with the RNeasy MinElute Cleanup kit (Qiagen GmbH) according to the
manufacturer's instructions. DNase enzyme digestion (Qiagen GmbH),
was performed to exclude genomic DNA contamination. Purified RNA
was eluted with 14 µl of RNase-free water (Qiagen GmbH). A
total of 500 ng of purified RNA were reverse transcribed into cDNA
using the RT2 First Strand kit (SABiosciences Corp.) in
a volume of 20 µl reaction and diluted with 91 µl of
nuclease-free water, following the manufacturer's instructions.
RT2 Profiler™ PCR array and
quantitative (real-time) PCR (qPCR)
The Human Oncogenes and Tumor Suppressor genes
RT2 Profiler™ PCR Array (complete list of genes is
presented in Table SI) was used
to profile the expression of 84 different key genes that promote
oncogenesis. A mix containing 102 µl of cDNA, 1,248
µl of water and 1,350 µl of 2X RT2 SYBR
Green Master Mix (SABiosciences Corp.) was prepared and 25
µl were added to each well. qPCR was performed using the
Real-Time Cycler 7500 (Applied Biosystems) with the following
thermal profile: 95°C for 10 min; 40 cycles: 95°C for 15 sec and
60°C for 1 min; dissociation curve at 95°C for 1 min, 55°C 30 sec
and 95°C for 30 sec. Raw data from qPCR were exported and analyzed
using online software at the GeneGlobe Data Analysis Center website
(Qiagen GmbH). Fold changes in gene expression were calculated
using the 2−ΔΔCq method (17).
Differentially expressed genes (with >2-fold
changes in expression) were validated with 3 biological independent
repli-cates by RT-qPCR (Table
SIIA), using 1 µl of cDNA, 0.4 µM of specific
primers, 12.5 µl of RT2 SYBR-Green Master Mix and
water up to a final volume of 25 µl. The relative gene
expression was calculated as previously mentioned.
A protein-protein interaction (PPI) network and
functional module were established using the Search Tool for the
Retrieval of Interacting Genes (STRING) (18). A confidence score of ≥0.4 and a
maximum number of interactors of (=) 0 were set as the cut-off
criteria.
Bisulfite sequencing
CDKN1A and RASSF1 gene promoter
methylation was analyzed by bisulfite sequencing (BS). A total of 1
µg of genomic DNA was bisulfite-treated using the EZ DNA
Methylation Kit (Zymo Research), which induces the chemical
conversion of unmethylated cytosine to uracil, and eluted in 20
µl of elution buffer. A total of 3 µl of
bisulfite-treated DNA were amplified by PCR in a total volume of 20
µl, with 4 mM of MgCl2, 0.5 mM of each dNTP, 0.2
µM of each primer, and 0.5 unit of Taq Hot Start
Thermostable DNA polymerase (EuroClone). The primers were designed
using the Methyl Primer Express® Software v1.0 (Applied
Biosystems) and their sequences are presented in Table SIIB. Amplifications were performed
using the following thermal profile: 95°C for 2 min followed by 38
cycles at 95°C for 30 sec, 59°C for 30 sec and 72°C for 30 sec. The
PCR products were purified from agarose gel using the EuroGold Gel
Extraction kit (EuroClone) and cloned using the pGEM-T Easy Vector
System (Promega Corp.) following the manufacturer's instructions.
In total, 10 white colonies for each time point were resuspended in
50 µl of water and tested by colony PCR using the same PCR
conditions as before, adding an extra step at 95°C for 10 min to
induce cell lysis. PCR products were then purified by QIAquick PCR
Purification kit (Qiagen GmbH). Sequencing of PCR products was
performed using the Big Dye Terminator Cycle Sequencing kit
(Applied Biosystems) on the Applied Biosystems 3130 Avant Genetic
Sequencer.
Chromatin immunoprecipitation (ChIP)
ChIP assay was performed as previously described
(19). Washes of
immuno-precipitated (IP) samples were carried out for 1 min using
the HulaMixer® Sample Mixer (Thermo Fisher Scientific,
Inc.), setting the rotation as follows: Orbital (rpm) 58-1,
Reciprocal (deg) 13°-01, Vibro (pause) 5° off. IP purified DNA and
inputs were amplified using genomic primer sets specific for
RASSF1 and CDKN1A gene promoters designed to overlap
the regions investigated by BS (Table
SIIB). RT-qPCR was performed in a total volume of 20 µl
including 5 µl of IP-DNA, 0.1 µM of each primer and
14 µl of SsoAdvanced™ Universal SYBR®-Green
Supermix (Bio-Rad Laboratories, Inc.). Amplification was carried
out in the CFX Connect™ Real-Time PCR Detection System (Bio-Rad
Laboratories, Inc.) using a two-step protocol consisting of 95°C
for 3 min, 95°C for 10 sec and 64°C for 30 sec, 40 cycles, followed
by a melting curve step to confirm the specificity of the PCR
products. ChIP data were analyzed using the Percent Input Method:
input Ct values corresponding to 1% of initial chromatin were
adjusted to 100% and used to normalized Ct values of IP-DNA
samples.
Immunocytochemistry (ICC)
Cell blocks were prepared by fixing untreated cells,
and cells at 96-h post wo and 21 days- post wo in 10%
neutral-buffered formalin for 24 h and then embedding them in
paraffin using Bio Agar gel (Bio Optica). Sections
4-µm-thick were cut and placed on positive-charged slides
(Bio Optica). Immunostaining was performed with the Ventana
Benchmark XT system (Ventana Medical Systems, Tucson, AZ) and the
Optiview DAB Detection kit (Ventana Medical Systems), an indirect,
biotin-free system for mouse IgG, mouse IgM and rabbit primary
antibodies detection. The kit aims to identify targets by
immunohistochemistry (IHC) and ICC in formalin-fixed,
paraffin-embedded sections that are stained on the Ventana
automated slide stainers and visualized by light microscopy.
Incubation time and temperature are set by the stainer
automatically. Antibodies against CDKN1A (DCS-60.2, Cell Marque,
Ventana Medical Systems), thymidylate synthase (TS; clone 4H4B1,
Life Technologies; Thermo Fisher Scientific, Inc.) and the DNA
excision repair cross-complementation group 1 (ERCC1; clone 8F1,
Neomarkers) were used. The CDKN1A antibody was pre-diluted by the
supplier and incubated at room temperature for 16 min. For TS and
ERCC1 detection, antibodies diluted at 1:300 and 1:100 were used,
respectively, and incubated at room temperature for 32 min.
Sections were automatically counterstained at room temperature for
16 min with hematoxylin II (Ventana Medical Systems). Hematoxylin
and eosin staining, at room temperature for 16 and 1 min
respectively, was also performed for each sample. Biomarker
expression was semi-quantitatively analyzed as the percentage of
immunopositive tumor cells out of the total number of tumor cells.
All samples were evaluated by a trained pathologist and analyzed
using an upright light microscope in bright field (Axioscope,
Zeiss).
Statistical analysis
Three biological replicates for each time-point
(untreated cells, and cells at 96 h-post wo and 21 days-post wo)
were analyzed for each experiment and the results were reported as
the mean values and standard deviation (SD) or standard error
(SEM). Time points were compared using the non-parametric
Friedman's test followed by the Bonferroni post-hoc test. Reported
P-values were two-sided and P<0.05 was used as the threshold for
significance. Correction for multiple testing was not performed due
to the explorative aim of this analysis. Statistical analyses were
carried out using STATA/MP 15.0 for Windows (Stata Corp. LP).
Results
Cytotoxic effect analysis
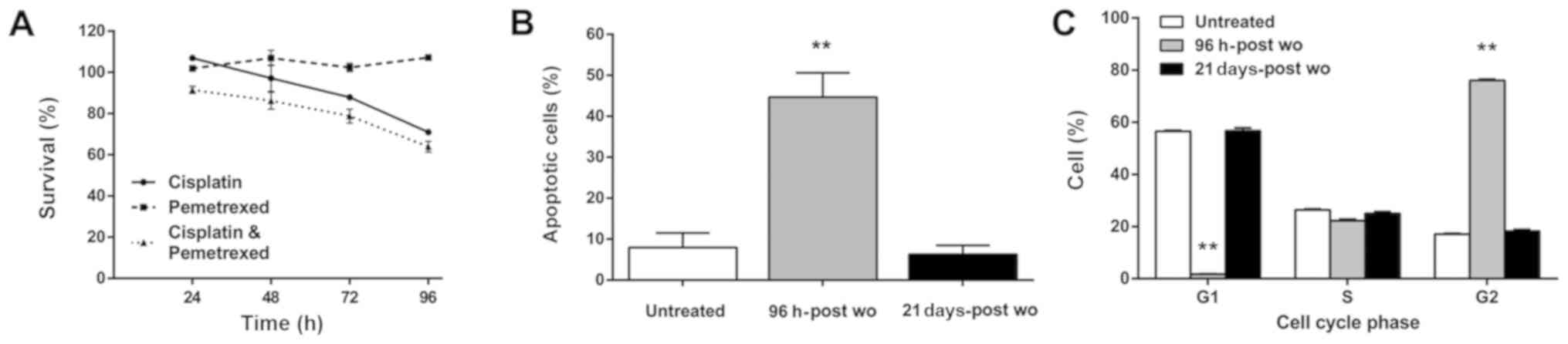
The anti-proliferative effects of cisplatin and
pemetrexed used as single agents or in combination were evaluated
using the RAL cells by MTS assay at 24, 48, 72 and 96 h after drug
washout. The RAL cells exhibited no or only slight sensitivity to
pemetrexed and cisplatin, with 100 and 71% of cell survival at 96
h, respectively. Although a higher cytotoxic effect was observed at
the end of the exposure time to both drugs, the half-maximal
inhibitory concentration (IC50) value was never reached
(64% of cell survival at 96 h-post wo) (Fig. 1A).
TUNEL assay (Fig.
1B) revealed a percentage of apoptotic cells consistent with
that of non-surviving cells detected by MTS assay when the two
drugs were used in combination. In particular, a significant
increase in the number of apoptotic cells was observed at 96 h-post
wo (44.6%, P=0.002). Notably, the cells had fully recovered 21 days
after the end of treatment and began to grow and divide normally.
Cell cycle distribution analysis (Fig.
1C) revealed a significant decrease in the number of cells in
the G0G1 phase (1.7%) and a marked increase
in the number of cells in the G2M phase at 96 h-post wo
(76%), whereas no differences with respect to the untreated samples
were detected in the cells in the S phase. These cell cycle
perturbations had fully recovered at 21 days-post wo when the cell
percentages in the G0G1 or G2M
phase were similar to those of the untreated cells (56.7% vs.
56.5%, and 18.2% vs. 17.1%, respectively).
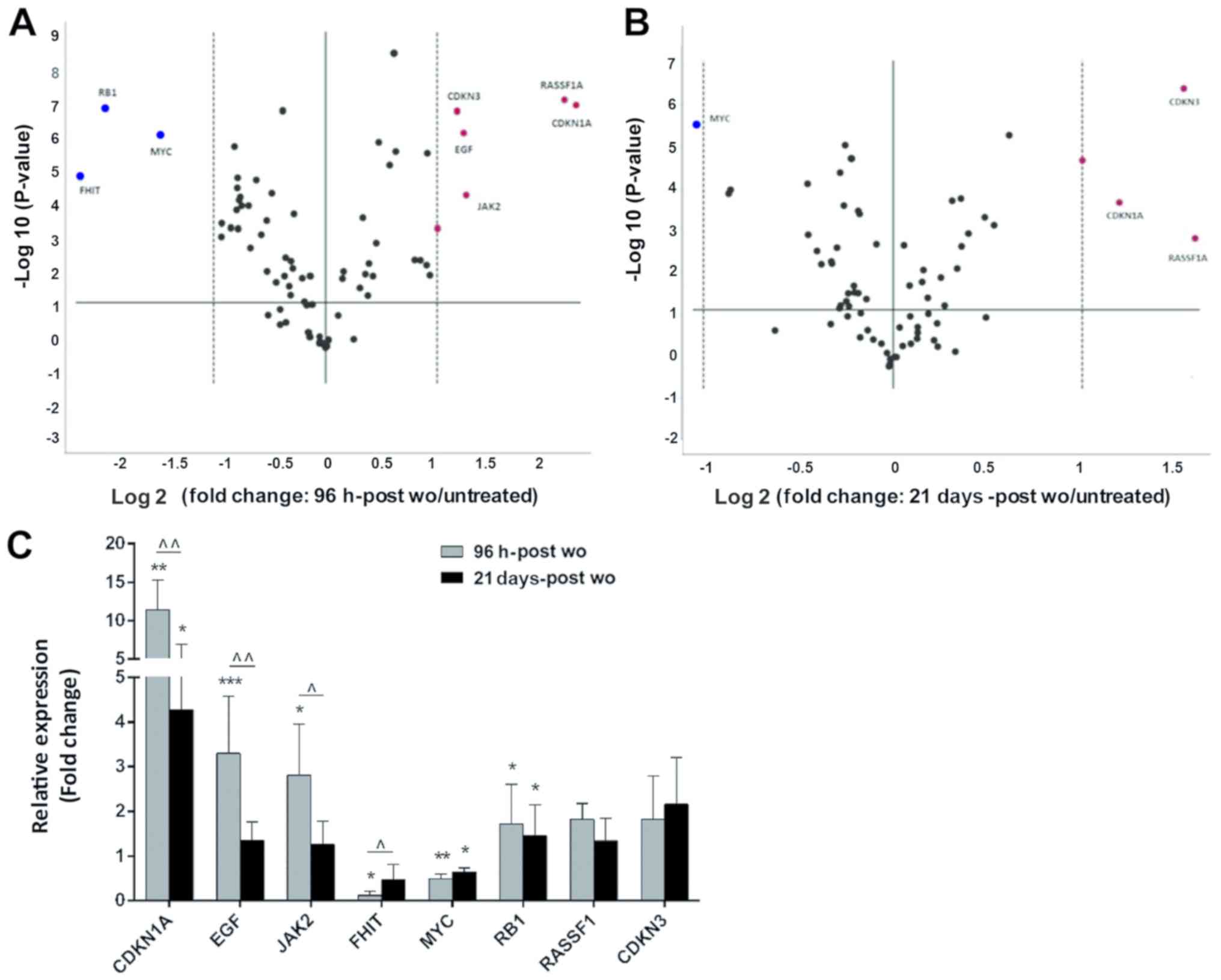
Gene expression analysis
The expression of 84 oncogenes and tumor suppressor
genes (TSGs) was analyzed in the untreated cells, and in the cells
at 96 h-post wo and 21 days-post wo. In total, 11/84 genes were not
expressed (ESR1, FOXD3, HGF, KIT, MOS, MYCN, ROS1, RUNX3, TNF,
TP73 and WT1) and 2/84 were unevaluable (BCL2 and
WWOX). Among the genes included in the panel, 8 (CDKN1A,
CDKN3, EGF, FHIT, JAK2, MYC, RASSF1 and RB1) and 3
(CDKN1A, CDKN3 and RASSF1) were differentially
expressed by >2-fold in the cells at 96 h-post wo and 21
days-post wo, respectively (Fig. 2A
and B). In particular, RT-qPCR analysis of the cells at 96
h-post wo confirmed that CDKN1A (P= 0.002),
EGF (P= 0.001) and JAK2 (P=0.009) were significantly
upregulated with a >2 fold change, whereas FHIT was
downregulated (P=0.008). A significant increase in CDKN1A
mRNA expression was also maintained and confirmed in the cells at
21 days-post wo (P=0.011) (Fig.
2C). The STRING database used to visualize protein-protein
interaction (PPI) revealed a network with high degree of
connectivity between the differentially expressed genes,
KRAS and AKT, with a PPI enrichment P-value equal to
0.00106 (Fig. S1).
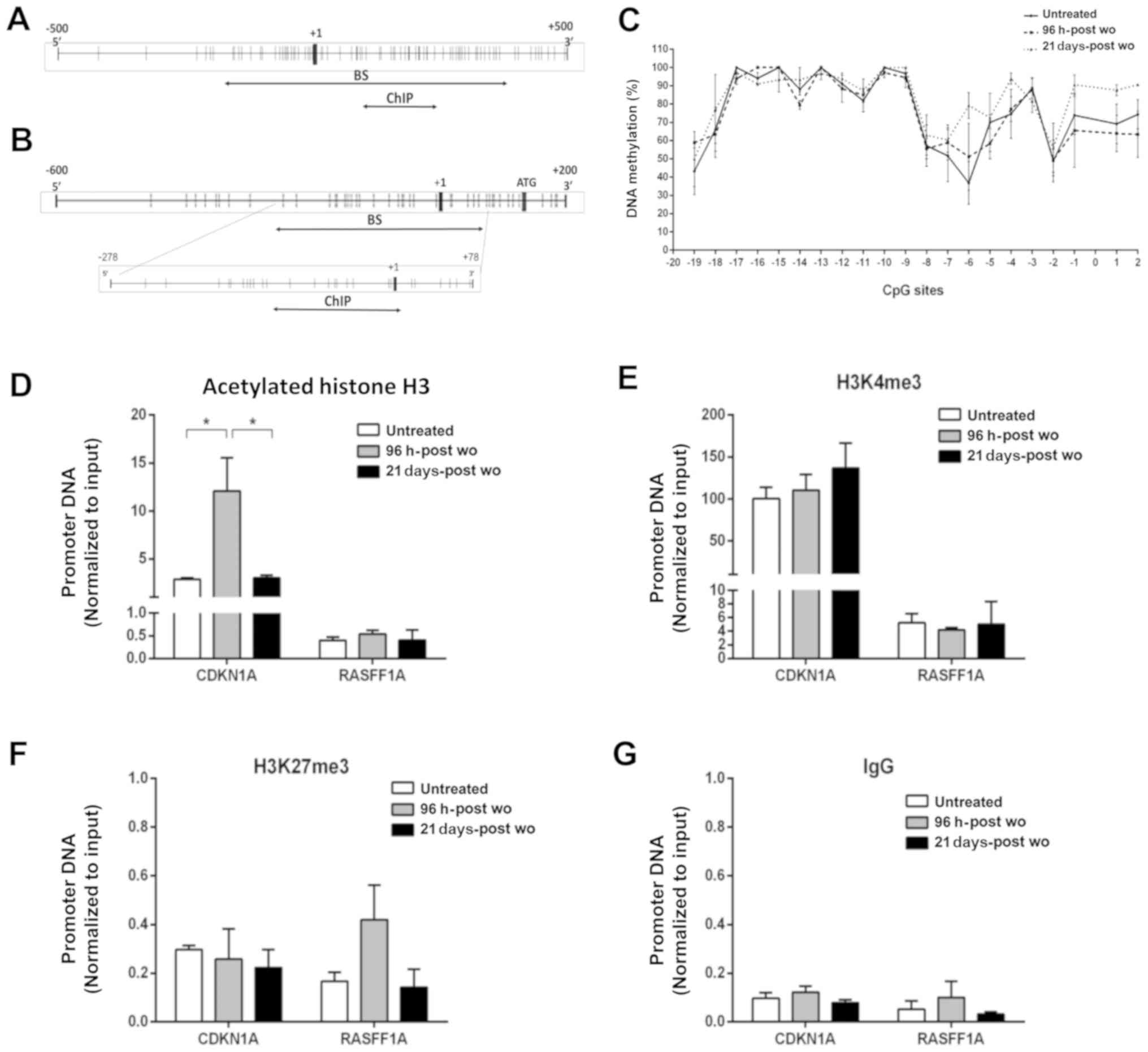
Epigenetic modifications associated with
the CDKN1A promoter region
Subsequently, it was investigated whether epigenetic
modifications (DNA methylation and histone marks) are associated
with gene expression, by performing BS and ChIP on the RAL cells at
96 h and 21 days following the combined treatment. The results for
the CDKN1A gene were compared with those obtained for
RASSF1, a tumor suppressor gene often silenced and
hypermethylated in RAL cells (15). The promoter regions of
CDKN1A and RASSF1 are shown in Fig. 3A and B, respectively.
DNA methylation analysis was performed by BS in 10
clones corresponding to the untreated cells, and cells at 96 h-post
wo and 21 days-post wo. The methylation percentage of each cytosine
was calculated by the average of the methylation status of the 10
clones. The promoter region of the CDKN1A gene was
completely unmethylated (data not shown), whereas the RASSF1
gene promoter exhibited a hypermethylated (>40%) CpG island
(Fig. 3C). No significant
differences were detected among the treated and untreated
cells.
Three post-transcriptional histone modifications
were investigated by ChIP assay: Two of these were associated with
transcriptional active chromatin, i.e., the acetylated form of the
H3 histone tail (acH3) and trimethyl-Lysine 4 of H3 histone tail
(H3K4me3), and one enriched in transcriptional silenced chromatin
domains, trimethyl-Lysine 27 of H3 histone tail (H3K27me3). The
chromatin histone marks (acH3 and H3K4me3) corresponding to a
transcriptionally open chromatin region were ≥7-fold enriched in
the promoter region of the CDKN1A gene compared to
RASSF1 (Fig. 3D and E).
Conversely, the repressive histone mark H3K27me3 showed comparable
results in the two genes (Fig.
3F). Isotype rabbit IgG was used as the background control
(Fig. 3G). No substantial
differences between the untreated cells and cells surviving the
combination treatment were detected in histone modifications
associated with the promoters of both genes, with the exception of
a significant 4-fold increase in acH3 enrichment in the
CDKN1A promoter detected in the cells at 96 h-post wo with
respect to the untreated cells, and cells at 21 days-post wo
(Fig. 3D).
Effects of cisplatin and pemetrexed on
CDKN1A, TS and ERCC1 protein expression and subcellular
localization
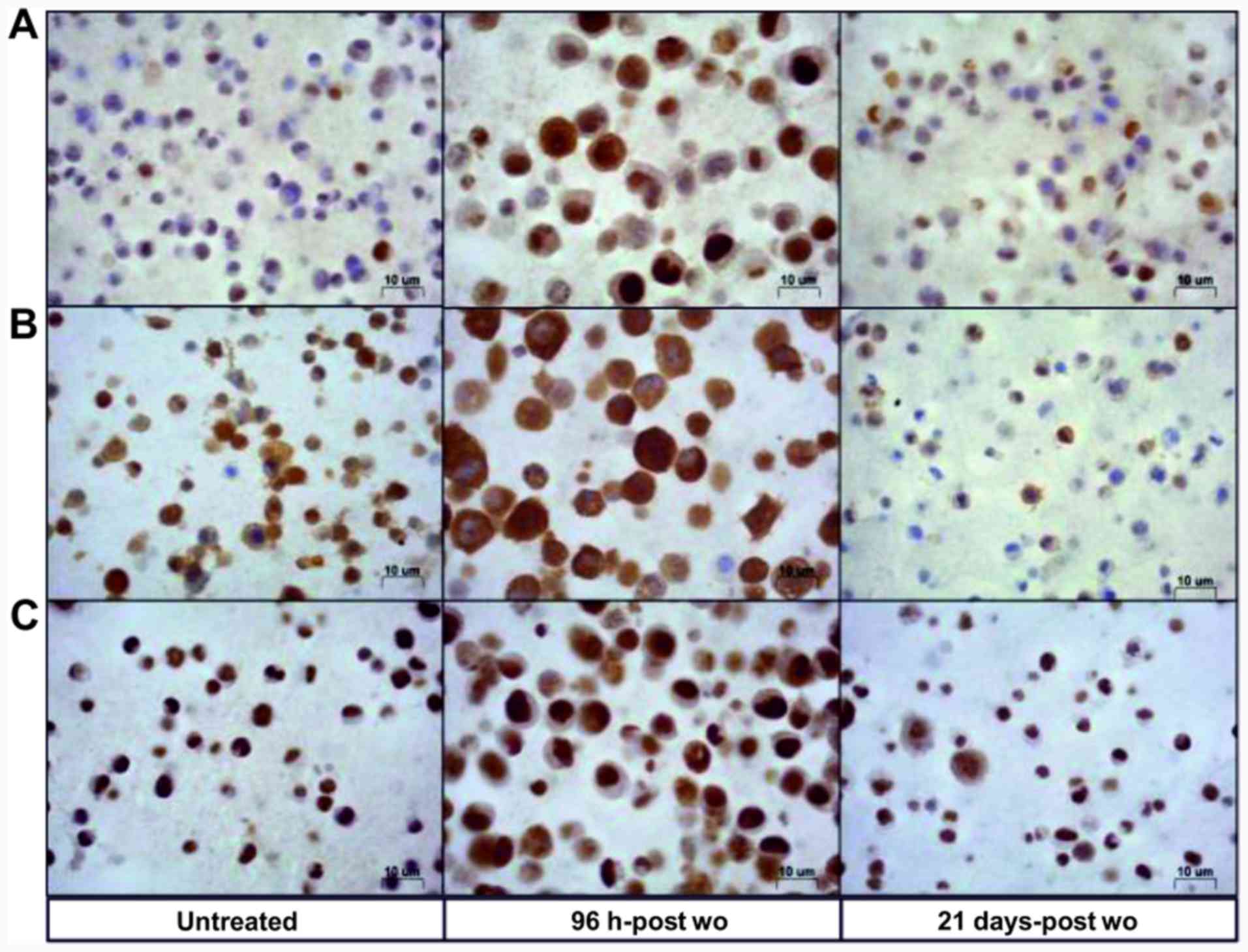
An upregulation of CDKN1A protein expression was
found in the cells at 96 h-post wo with respect to the controls (85
and 10% immunopositive cells, respectively), whereas CDKN1A
expression in the cells at 21 days-post wo was equal to baseline
(5% immunopositive cells) (Fig.
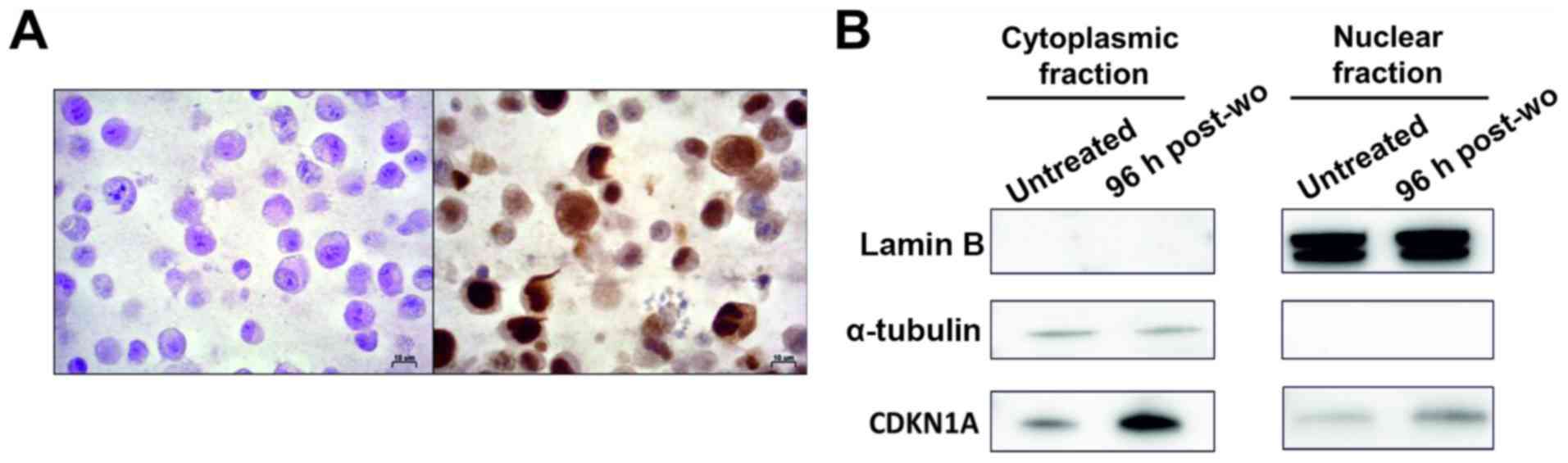
4A). In particular, it was observed that 10% of cells at 96
h-post wo exhibited a strong cytoplasmic expression (Fig. 5A) that was virtually absent in the
untreated cells, and in cells at 21 days-post wo.
The results of western blot analysis of whole
lysates (Fig. S2) were in line
with the ICC results, confirming that the cells at 96 h-post wo
exhibited a marked upregulation of CDKN1A protein expression, while
in the cells at 21 d-post wo, the protein level decreased with
respect to the cells at 96 h-post wo.
Moreover, western blot analysis of protein lysates
from subcellular compartments revealed that CDKN1A overexpression
observed following treatment was mainly related to a protein
increment in the cytoplasmic fraction. Again, the increased CDKN1A
expression was more evident in the cytoplasm of the cells at 96
h-post wo (Fig. 5B).
We also characterized the RAL cells for the
expression of TS and ERCC1, known to be potential markers of
response to pemetrexed and cisplatin, respectively. The RAL cells
exhibited a high basal nuclear and cytoplasmic expression of TS,
80% of which were strongly immunopositive. Strong TS stain
intensity was also observed in the cytoplasm (Fig. 4B). An additional upregulation of
cytoplasmic TS was observed in the cells at 96 h-post wo, whereas
only 40% of the cells were immunopositive at 21d-post wo. As
regards ERCC1 protein expression, almost 100% of the untreated
cells and cells at 96 h-post wo were strongly positive, whereas the
cells at 21 days-post wo exhibited a somewhat lower positivity
(75%) (Fig. 4C). The TS and ERCC1
protein expression levels were also confirmed by western blot
analysis (Fig. S2). In all the
immunocyto-chemical evaluations, the cells at 96 h-post wo appeared
larger than their untreated counterparts, according to the
well-known acute effects of cisplatin treatment (20,21).
Promotion of cell death by CDKN1A
knockdown in the cells treated with the cisplatin-pemetrexed
treatment combination
The role of CDKN1A gene in the cellular
response to cisplatin and pemetrexed was investigated by transient
silencing. The CDKN1A mRNA levels decreased significantly
with respect to those observed in cells transfected with an
appropriate negative control (scrambled oligonucleotide),
confirming CDKN1A knockdown by siRNA. Moreover, its protein
level was undetectable in cells transfected with CDKN1A
siRNA (Fig. S3).
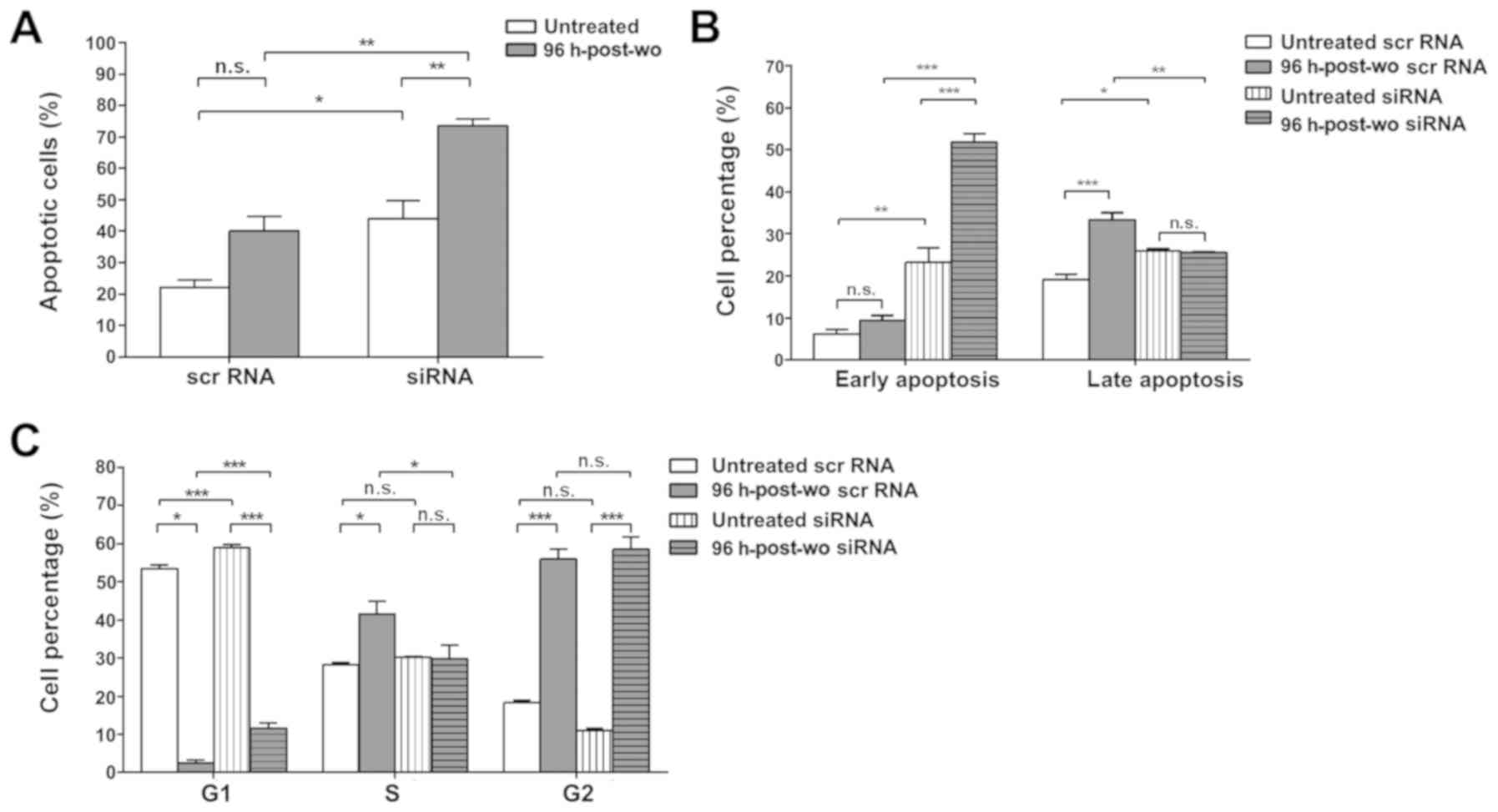
The RAL cells in which CDKN1A was silenced
and which were exposed to the cisplatin-pemetrexed combination and
evaluated 96 h after washout exhibited a consistently significant
increase in cell death compared to the scramble control-transfected
cells at 96 h-post wo. TUNEL assay revealed that there were 73.6%
of apoptotic cells when CDKN1A was knocked down compared
with 44% of apoptotic cells in the untreated cells transfected with
CDKN1A siRNA (Figs. 6A and
S4). As was expected, when
scramble RNA was used instead of the CDKN1A siRNA probes,
the cisplatin-pemetrexed combination induced a considerable
increase in cell death compared to the untreated control conditions
(40.1% vs. 22.1%, respectively) (Fig.
6A). The increase in cell death detected by TUNEL assay in the
cells at 96 h-post wo with CDKN1A silencing was also
confirmed by Annexin V assay, which identified both early and late
apoptosis. In the 96 h-post wo condition, CDKN1A silencing
was associated with 51.9% of cells in early apoptosis compared with
23.2% cells in the untreated siRNA counter-parts and 9.4% in the
cells at 96 h-post wo transfected with the scramble control
(Figs. 6B and S4).
Finally, a cell cycle distribution analysis revealed
that the cisplatin-pemetrexed combination led to a significant G2
phase block (Figs. 6C and S4) that was more evident in the cells at
96 h-post wo than in the untreated cells transfected with either
CDKN1A siRNA (58.6% in 96 h-post wo vs. 10.9% in untreated
cells) or scramble RNA (56% in 96 h-post wo vs. 18.2% in untreated
cells). Furthermore, as CDKN1A inhibits the activity of
cyclin-CDK2, -CDK1 and -CDK4/6 complexes, functioning as a
regulator of cell cycle progression during G1 and S phases, a
significant increase in the cells in the G1-phase was also
detectable in the cells transfected with siRNA at 96 h-post wo with
respect to their scramble control counter-parts (11.7 and 2.4%,
respectively). No significant differences in percentages of S phase
cells were detected in the untreated cells. Treatment with the
cisplatin-pemetrexed combination caused an increase in the number
of cells in the S phase (41.6% vs. 28.3% in the cells at 96 h-post
wo and untreated cells, respectively). When CDKN1A was
knocked down, the percentage of cells in the S phase decreased to
the level of the untreated cells (29.7%).
Discussion
Platinum-based regimens used in combination with
other chemotherapeutic compounds, such as pemetrexed, are still the
optimal treatment selection for the adjuvant treatment of NSCLC and
as the first-line therapy of advanced disease in patients whose
tumor molecular signature limits the usefulness of biological
therapies (22). However, the
majority of patients are susceptible to disease progression due to
acquired resistance.
The aim of the present study was to identify novel
biomarkers of sensitivity/resistance to the cisplatin-pemetrexed
treatment combination using the RAL cell line, an in vitro
model representative of a subset of NSCLC patients who cannot
benefit from biological therapies. The data of the present study
revealed a significant increase in antitumor activity when both
drugs were used in combination, rather than as single agents. For
this reason, the present study aimed to perform a more in depth
investigation of the combination of the two drugs, with no further
investigations on single drugs. The observed antitumor activity was
transient and 21 days after the treatment washout, the RAL cells
had fully resumed their proliferation capacity.
Firstly, TS and ERCC1 protein expression was
analyzed, since it is known that these markers may play a role in
the response to pemetrexed and cisplatin respectively, even though
contradictory results have been observed (23-27).
In the present study, both markers were highly expressed either in
the untreated cells and in the cells at 96 h-post wo, thus
confirming their limited utility as response markers in this cell
line.
Subsequently, the expression of genes involved in
different biological pathways was analyzed to identify the genes
implicated in the resistance of RAL cells to the
cisplatin-pemetrexed treatment.
To this purpose, the gene profiling of 84 TSGs and
oncogenes was performed followed by the validation of altered gene
expression by qPCR. Among the genes confirmed as differentially
expressed in the cells at 96 h-post wo, FHIT was the only
one downregulated, perhaps contributing to platinum drug resistance
via the serine threonine kinase AKT, particularly in
TP53-mutated patients (28-30).
The CDKN1A, EGF and JAK2 mRNA expression
levels were >2-fold higher in the cells at 96 h-post wo compared
to the untreated cells. Both EGF and JAK2 are
upstream regulators of STAT3 activity. STAT3 acts as a
transcription factor, inducing the expression of genes involved in
cancer progression, apoptosis resistance, cell proliferation and
sustained angiogenesis (31). The
significant overexpression of EGF and JAK2 genes
suggests that the activation of the JAK2/STAT3 signaling pathway
may play a role in the resistance to chemotherapy observed in the
model in the present study, as reported elsewhere (32).
Since it was the most upregulated gene in the cells
at 96 h-post wo and the only gene still upregulated at 21 d-post
wo, the role of CDKN1A was further investigated. The CDKN1A
gene plays a crucial role in DNA repair, cell differentiation and
apoptosis through the regulation of the cell cycle. It blocks DNA
replication by binding to proliferating cell nuclear antigen and
inhibits the activity of cyclin-CDK4 or -CDK2 complexes. As a
result, the cell cycle is arrested in the G1/S or G2/M transitions
(33,34). The role of CDKN1A in
apoptosis is multifaceted and remains unclear (35-37).
Some studies have suggested that it may mediate cell cycle arrest
following DNA damage in either a p53-dependent or -independent
manner, leading to an increase in apoptosis (38,39).
Others have found that the inhibition of cell cycle progression,
associated with elevated levels of CDKN1A gene expression,
prevents apoptosis (34,40). Such a positive or a negative
influence on cell growth and survival may result from its
subcellular localization (34,41).
Its cytoplasmic localization may favor cell proliferation as it
acts as a chaperone for cyclin E, particularly when p53 is damaged
or absent (42). Conversely,
CDKN1A nuclear localization is associated with its better known
function of modulator of cell cycle arrest and negative regulator
of DNA repair pathways, resulting in unresolved DNA damage and
growth inhibition (36).
Shoji et al reported that CDKN1A
expression, in a large fraction of patients with completely
resected pathologic stage I-IIIA NSCLC, was associated with a
favorable prognosis (43).
Moreover, a number of studies have demonstrated that CDKN1A
modulation is involved in the response to different therapies in
many cancer types (44-51). For example, cytoplasmic CDKN1A
localization in testicular embryonal carcinoma leads to cisplatin
resistance. In particular, high cytoplasmic CDKN1A levels exert a
protective effect against cisplatin in embryonal carcinoma cell
lines (52). The present study
demonstrated that only 10% of the untreated RAL cells expressed
CDKN1A, in line with the weak positive expression reported by
Rosetti et al (46). The
cytoplasmic expression revealed by western blot analysis (Fig. 5B) in untreated cells could be
linked to its oncogenic activity in such an aggressive cell line,
which is metastasis-derived.
In the present study, a significant CDKN1A
upregulation was detected in the cells at 96 h-post wo, both at the
mRNA and protein level, having a 10-fold higher mRNA expression and
a marked cytosolic CDKN1A upregulation with respect to the
untreated cells. In the cells at 21 d-post wo, CDKN1A remained
upregulated, even though at a lower extent (Figs. 2C and S2).
Chromatin accessibility to transcriptional
machinery is a crucial regulator of gene expression known to be
involved in the clinical course of lung cancer (53). The analysis of epigenetic
modifications associated with the CDKN1A gene promoter
revealed that its gene expression regulation was not mediated by
DNA methylation, but rather by chromatin histone marks H3K4me3 and
acH3, representative of a transcriptionally open and active
chromatin structure (Fig. 3).
Although the increased level of association of acH3 in the promoter
region of CDKN1A in 96 h-post wo cells compared to untreated
cells correlated with increased gene expression, it was not
sufficient to confirm epigenetics as the main molecular mechanism
involved in the modulation of CDKN1A gene expression.
Taking into account the subcellular localization of
CDKN1A in RAL cells surviving the combined chemotherapeutic action
of cisplatin and pemetrexed, the high cytoplasmic expression levels
of the protein appear consistent with the observed reduction in
drug efficacy. It has been demonstrated that the phosphorylation of
CDKN1A and its translocation to the cytosol, with the consequent
progression of cell cycle through G1/S phase (54) and activation of anti-apoptotic
pathways, may be AKT-mediated (55) and interfere with the cytotoxic
effect of chemotherapeutic drugs (54-59).
Preclinical and clinical data have also highlighted that
PI3K/AKT/mTOR pathway inhibitors may be used in combination with
other agents for the treatment of NSCLC (60).
In the present study, it was demonstrated that in
the NSCLC cell line harboring KRAS G12C and TP53
G244C mutations, CDKN1A silencing increased apoptosis and
G1/S cell cycle arrest, thus increasing the efficacy of the
cisplatin-pemetrexed combination. These findings support the
hypothesis that CDKN1A functions as an oncogene, promoting
cancer cell proliferation by inhibiting apoptosis, and highlight
its potential role as a therapeutic response indicator of the
pemetrexed-cisplatin combination in NSCLC.
A retrospective study to evaluate CDKN1A
gene expression and localization in association with patient
response to standard chemotherapy could also confirm CDKN1A
role as a predictive biomarker of response to therapy in
KRAS- and TP53-mutated NSCLC.
Supplementary Data
Acknowledgements
The authors would like to thank Mrs. Gráinne
Tierney and Mr. Cristiano Verna of Istituto Scientifico Romagnolo
per lo Studio e la Cura dei Tumori, for providing editorial
assistance.
Funding
The present study was partially supported by
Istituto Oncologico Romagnolo.
Availability of data and materials
All data generated or analyzed during this study
are included in this published article or are available from the
corresponding author on reasonable request.
Authors' contributions
AZ, AP, EG, DC, AT, PU, AD and CM conceived the
idea for and designed the study. AZ, AP, CM, FP, SR and FFa were
responsible for data acquisition and interpretation. FFo performed
the statistical analysis. AZ, CM and AP drafted the manuscript. EG,
GM, DC, AT, PU and AD critically revised the manuscript for
important intellectual content. All authors read and approved the
present version of the manuscript for submission and the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors confirm that they have no competing
interests.
References
|
1
|
Herbst RS, Heymach JV and Lippman SM: Lung
cancer. N Engl J Med. 359:1367–1380. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
3
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Novello S, Barlesi F, Califano R, Cufer T,
Ekman S, Levra MG, Kerr K, Popat S, Reck M, Senan S, et al:
Metastatic non-small-cell lung cancer: ESMO clinical practice
guidelines for diagnosis, treatment and follow-up. Ann Oncol.
27(Suppl 5): Sv1–Sv27. 2016. View Article : Google Scholar
|
|
5
|
Ricciuti B, Leonardi GC, Metro G, Grignani
F, Paglialunga L, Bellezza G, Baglivo S, Mencaroni C, Baldi A,
Zicari D and Crinò L: Targeting the KRAS variant for treatment of
non-small cell lung cancer: Potential therapeutic applications.
Expert Rev Respir Med. 10:53–68. 2016. View Article : Google Scholar
|
|
6
|
Shajani-Yi Z, de Abreu FB, Peterson JD and
Tsongalis GJ: Frequency of somatic TP53 mutations in combination
with known pathogenic mutations in colon adenocarcinoma, non-small
cell lung carcinoma, and gliomas as identified by next-generation
sequencing. Neoplasia. 20:256–262. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Azzoli CG, Baker S Jr, Temin S, Pao W,
Aliff T, Brahmer J, Johnson DH, Laskin JL, Masters G, Milton D, et
al: American society of clinical oncology clinical practice
guideline update on chemotherapy for stage IV non-small-cell lung
cancer. J Clin Oncol. 27:6251–6266. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Scagliotti GV, Parikh P, von Pawel J,
Biesma B, Vansteenkiste J, Manegold C, Serwatowski P, Gatzemeier U,
Digumarti R, Zukin M, et al: Phase III study comparing cisplatin
plus gemcitabine with cisplatin plus pemetrexed in
chemotherapy-naive patients with advanced-stage non-small-cell lung
cancer. J Clin Oncol. 26:3543–3551. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hsu WH, Zhao X, Zhu J, Kim IK, Rao G,
McCutcheon J, Hsu ST, Teicher B, Kallakury B, Dowlati A, et al:
Checkpoint kinase 1 inhibition enhances cisplatin cytotoxicity and
overcomes cisplatin resistance in SCLC by promoting mitotic cell
death. J Thorac Oncol. 14:1032–1045. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sarin N, Engel F, Kalayda GV, Mannewitz M,
Cinatl J Jr, Rothweiler F, Michaelis M, Saafan H, Ritter CA, Jaehde
U and Frötschl R: Cisplatin resistance in non-small cell lung
cancer cells is associated with an abrogation of cisplatin-induced
G2/M cell cycle arrest. PLoS One. 12:e01810812017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cai Y, Yan X, Zhang G, Zhao W and Jiao S:
The predictive value of ERCC1 and p53 for the effect of
panobinostat and cisplatin combination treatment in NSCLC.
Oncotarget. 6:18997–19005. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shtivelman E, Hensing T, Simon GR, Dennis
PA, Otterson GA, Bueno R and Salgia R: Molecular pathways and
therapeutic targets in lung cancer. Oncotarget. 5:1392–1433.
2014.PubMed/NCBI
|
|
13
|
Georgakilas AG, Martin OA and Bonner WM:
P21: A two-faced genome guardian. Trends Mol Med. 23:310–319. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gasperi-Campani A, Roncuzzi L, Ricotti L,
Lenzi L, Gruppioni R, Sensi A, Zini N, Zoli W and Amadori D:
Molecular and biological features of two new human squamous and
adenocarcinoma of the lung cell lines. Cancer Genet Cytogenet.
107:11–20. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pasini A, Paganelli G, Tesei A, Zoli W,
Giordano E and Calistri D: Specific biomarkers are associated with
docetax-eland gemcitabine-resistant NSCLC cell lines. Transl Oncol.
5:461–468. 2012. View Article : Google Scholar
|
|
16
|
Pignatta S, Arienti C, Zoli W, Di Donato
M, Castoria G, Gabucci E, Casadio V, Falconi M, De Giorgi U,
Silvestrini R and Tesei A: Prolonged exposure to (R)-bicalutamide
generates a LNCaP subclone with alteration of mitochondrial genome.
Mol Cell Endocrinol. 382:314–324. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-delta delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
18
|
Szklarczyk D, Morris JH, Cook H, Kuhn M,
Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al:
The STRING database in 2017: Quality-controlled protein-protein
association networks, made broadly accessible. Nucleic Acids Res.
45:D362–D368. 2017. View Article : Google Scholar
|
|
19
|
Pasini A, Bonafe F, Govoni M, Guarnieri C,
Morselli PG, Sharma HS, Caldarera CM, Muscari C and Giordano E:
Epigenetic signature of early cardiac regulatory genes in native
human adipose-derived stem cells. Cell Biochem Biophys. 67:255–262.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fang K, Chiu CC, Li CH, Chang YT and Hwang
HT: Cisplatin-induced senescence and growth inhibition in human
non-small cell lung cancer cells with ectopic transfer of p16INK4a.
Oncol Res. 16:479–488. 2007. View Article : Google Scholar
|
|
21
|
Kung ML, Hsieh CW, Tai MH, Weng CH, Wu DC,
Wu WJ, Yeh BW, Hsieh SL, Kuo CH, Hung HS and Hsieh S: Nanoscale
characterization illustrates the cisplatin-mediated biomechanical
changes of B16-F10 melanoma cells. Phys Chem Chem Phys.
18:7124–7131. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Masters GA, Temin S, Azzoli CG, Giaccone
G, Baker S Jr, Brahmer JR, Ellis PM, Gajra A, Rackear N, Schiller
JH, et al: Systemic therapy for stage IV non-small-cell lung
cancer: American society of clinical oncology clinical practice
guideline update. J Clin Oncol. 33:3488–3515. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kasai D, Ozasa H, Oguri T, Miyazaki M,
Uemura T, Takakuwa O, Kunii E, Ohkubo H, Maeno K and Niimi A:
Thymidylate synthase gene copy number as a predictive marker for
response to pemetrexed treatment of lung adenocarcinoma. Anticancer
Res. 33:1935–1940. 2013.PubMed/NCBI
|
|
24
|
Yoon JY, Park CK, Choi YD, Oh IJ and Kim
YC: Predictive factors for long-term responders of pemetrexed
maintenance treatment in non-small cell lung cancer. Thorac Cancer.
10:942–949. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Friboulet L, Olaussen KA, Pignon JP,
Shepherd FA, Tsao MS, Graziano S, Kratzke R, Douillard JY, Seymour
L, Pirker R, et al: ERCC1 isoform expression and DNA repair in
non-small-cell lung cancer. N Engl J Med. 368:1101–1110. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lord RV, Brabender J, Gandara D, Alberola
V, Camps C, Domine M, Cardenal F, Sánchez JM, Gumerlock PH, Tarón
M, et al: Low ERCC1 expression correlates with prolonged survival
after cisplatin plus gemcitabine chemo-therapy in non-small cell
lung cancer. Clin Cancer Res. 8:2286–2291. 2002.PubMed/NCBI
|
|
27
|
Vilmar A and Sorensen JB: Excision repair
cross-complementation group 1 (ERCC1) in platinum-based treatment
of non-small cell lung cancer with special emphasis on carboplatin:
A review of current literature. Lung Cancer. 64:131–139. 2009.
View Article : Google Scholar
|
|
28
|
Andriani F, Perego P, Carenini N, Sozzi G
and Roz L: Increased sensitivity to cisplatin in non-small cell
lung cancer cell lines after FHIT gene transfer. Neoplasia. 8:9–17.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cortinovis DL, Andriani F, Livio A, Fabbri
A, Perrone F, Marcomini B, Pilotti S, Mariani L, Bidoli P, Bajetta
E, et al: FHIT and p53 status and response to platinum-based
treatment in advanced non-small cell lung cancer. Curr Cancer Drug
Targets. 8:342–348. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu DW, Lee MC, Hsu NY, Wu TC, Wu JY, Wang
YC, Cheng YW, Chen CY and Lee H: FHIT loss confers cisplatin
resistance in lung cancer via the AKT/NF-KB/Slug-mediated PUMA
reduction. Oncogene. 34:3882–3883. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Harada D, Takigawa N and Kiura K: The role
of STAT3 in non-small cell lung cancer. Cancers (Basel). 6:708–722.
2014. View Article : Google Scholar
|
|
32
|
Hu Y, Hong Y, Xu Y, Liu P, Guo DH and Chen
Y: Inhibition of the JAK/STAT pathway with ruxolitinib overcomes
cisplatin resistance in non-small-cell lung cancer NSCLC.
Apoptosis. 19:1627–1636. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bunz F, Dutriaux A, Lengauer C, Waldman T,
Zhou S, Brown JP, Sedivy JM, Kinzler KW and Vogelstein B:
Requirement for p53 and p21 to sustain G2 arrest after DNA damage.
Science. 282:1497–1501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Abbas T and Dutta A: P21 in cancer:
Intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu S, Bishop WR and Liu M: Differential
effects of cell cycle regulatory protein p21(WAF1/Cip1) on
apoptosis and sensitivity to cancer chemotherapy. Drug Resist
Updat. 6:183–195. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Karimian A, Ahmadi Y and Yousefi B:
Multiple functions of p21 in cell cycle, apoptosis and
transcriptional regulation after DNA damage. DNA Repair (Amst).
42:63–71. 2016. View Article : Google Scholar
|
|
37
|
Kreis NN, Louwen F and Yuan J: The
multifaceted p21 (Cip1/Waf1/CDKN1A) in cell differentiation,
migration and cancer therapy. Cancers (Basel). 11. pp. E12202019,
View Article : Google Scholar
|
|
38
|
Wu L and Levine AJ: Differential
regulation of the p21/WAF-1 and mdm2 genes after high-dose UV
irradiation: P53-dependent and p53-independent regulation of the
mdm2 gene. Mol Med. 3:441–451. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang K, Zheng XY, Qin J, Wang YB, Bai Y,
Mao QQ, Wan Q, Wu ZM and Xie LP: Up-regulation of p21WAF1/Cip1 by
saRNA induces G1-phase arrest and apoptosis in T24 human bladder
cancer cells. Cancer Lett. 265:206–214. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cazzalini O, Scovassi AI, Savio M, Stivala
LA and Prosperi E: Multiple roles of the cell cycle inhibitor
p21(CDKN1A) in the DNA damage response. Mutat Res. 704:12–20. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Coqueret O: New roles for p21 and p27
cell-cycle inhibitors: A function for each cell compartment? Trends
Cell Biol. 13:65–70. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Abella N, Brun S, Calvo M, Tapia O, Weber
JD, Berciano MT, Lafarga M, Bachs O and Agell N: Nucleolar
disruption ensures nuclear accumulation of p21 upon DNA damage.
Traffic. 11:743–755. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shoji T, Tanaka F, Takata T, Yanagihara K,
Otake Y, Hanaoka N, Miyahara R, Nakagawa T, Kawano Y, Ishikawa S,
et al: Clinical significance of p21 expression in non-small-cell
lung cancer. J Clin Oncol. 20:3865–3871. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lincet H, Poulain L, Remy JS, Deslandes E,
Duigou F, Gauduchon P and Staedel C: The p21(cip1/waf1)
cyclin-dependent kinase inhibitor enhances the cytotoxic effect of
cisplatin in human ovarian carcinoma cells. Cancer Lett. 161:17–26.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mitsuuchi Y, Johnson SW, Selvakumaran M,
Williams SJ, Hamilton TC and Testa JR: The phosphatidylinositol
3-kinase/AKT signal transduction pathway plays a critical role in
the expression of p21WAF1/CIP1/SDI1 induced by cisplatin and
paclitaxel. Cancer Res. 60:5390–5394. 2000.PubMed/NCBI
|
|
46
|
Rosetti M, Zoli W, Tesei A, Ulivi P,
Fabbri F, Vannini I, Brigliadori G, Granato AM, Amadori D and
Silvestrini R: Iressa strengthens the cytotoxic effect of docetaxel
in NSCLC models that harbor specific molecular characteristics. J
Cell Physiol. 212:710–716. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dabrowska M, Mosieniak G, Skierski J,
Sikora E and Rode W: Methotrexate-induced senescence in human
adenocarcinoma cells is accompanied by induction of p21(waf1/cip1)
expression and lack of polyploidy. Cancer Lett. 284:95–101. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wu MF, Hsiao YM, Huang CF, Huang YH, Yang
WJ, Chan HW, Chang JT and Ko JL: Genetic determinants of pemetrexed
responsiveness and nonresponsiveness in non-small cell lung cancer
cells. J Thorac Oncol. 5:1143–1151. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Huang WY, Yang PM, Chang YF, Marquez VE
and Chen CC: Methotrexate induces apoptosis through
p53/p21-dependent pathway and increases E-cadherin expression
through downregulation of HDAC/EZH2. Biochem Pharmacol. 81:510–517.
2011. View Article : Google Scholar
|
|
50
|
Liu Z, Sun M, Lu K, Liu J, Zhang M, Wu W,
De W, Wang Z and Wang R: The long noncoding RNA HOTAIR contributes
to cisplatin resistance of human lung adenocarcinoma cells via
downregualtion of p21(WAF1/CIP1) expression. PLoS One.
8:e772932013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang H, Zhu LJ, Yang YC, Wang ZX and Wang
R: MiR-224 promotes the chemoresistance of human lung
adenocarcinoma cells to cisplatin via regulating G1/S
transition and apoptosis by targeting p21(WAF1/CIP1). Br J Cancer.
111:339–354. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Koster R, di Pietro A, Timmer-Bosscha H,
Gibcus JH, van den Berg A, Suurmeijer AJ, Bischoff R, Gietema JA
and de Jong S: Cytoplasmic p21 expression levels determine
cisplatin resistance in human testicular cancer. J Clin Invest.
120:3594–3605. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Pasini A, Delmonte A, Tesei A, Calistri D
and Giordano E: Targeting chromatin-mediated transcriptional
control of gene expression in non-small cell lung cancer therapy:
Preclinical rationale and clinical results. Drugs. 75:1757–1771.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhou BP, Liao Y, Xia W, Spohn B, Lee MH
and Hung MC: Cytoplasmic localization of p21Cip1/WAF1 by
akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat
Cell Biol. 3:245–252. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Li Y, Dowbenko D and Lasky LA: AKT/PKB
phosphorylation of p21Cip/WAF1 enhances protein stability of
p21Cip/WAF1 and promotes cell survival. J Biol Chem.
277:11352–11361. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Heliez C, Baricault L, Barboule N and
Valette A: Paclitaxel increases p21 synthesis and accumulation of
its AKT- phosphorylated form in the cytoplasm of cancer cells.
Oncogene. 22:3260–3268. 2003. View Article : Google Scholar
|
|
57
|
Perez-Tenorio G, Berglund F, Esguerra
Merca A, Nordenskjöld B, Rutqvist LE, Skoog L and Stål O:
Cytoplasmic p21WAF1/CIP1 correlates with akt activation and poor
response to tamoxifen in breast cancer. Int J Oncol. 28:1031–1042.
2006.PubMed/NCBI
|
|
58
|
Vincent AJ, Ren S, Harris LG, Devine DJ,
Samant RS, Fodstad O and Shevde LA: Cytoplasmic translocation of
p21 mediates NUPR1-induced chemoresistance: NUPR1 and p21 in
chemoresistance. FEBS Lett. 586:3429–3434. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Xia X, Ma Q, Li X, Ji T, Chen P, Xu H, Li
K, Fang Y, Weng D, Weng Y, et al: Cytoplasmic p21 is a potential
predictor for cisplatin sensitivity in ovarian cancer. BMC Cancer.
11:3992011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Fumarola C, Bonelli MA, Petronini PG and
Alfieri RR: Targeting PI3K/AKT/mTOR pathway in non small cell lung
cancer. Biochem Pharmacol. 90:197–207. 2014. View Article : Google Scholar : PubMed/NCBI
|