Introduction
Lung cancer is the most common malignant tumor, and
its incidence and mortality rates are ~11.6 and ~18.4% worldwide,
which are continuously rising (1).
Lung cancer is insidious and difficult to detect in the early
stage, and this disease seriously threatens patient health. The
treatment strategy for advanced non-small-cell lung cancer (NSCLC)
is mainly based on platinum-based chemotherapy (2,3).
However, the curative effect of chemotherapy is limited, and the
effective rate has reached a plateau that has been difficult to
overcome (4). Chemotherapy drugs
are highly toxic and can cause serious side effects, and thus are
intolerable to patients (4). In
the past decade, epidermal growth factor receptor-tyrosine kinase
inhibitors (EGFR-TKIs) have influenced the therapeutic strategies
for advanced NSCLC, providing notable survival benefits to patients
with EGFR-sensitive mutations, including exon 19 deletion and exon
21 L858R mutation (5,6). However, numerous patients cannot
benefit or continue to benefit from EGFR-TKI monotherapy due to
primary and secondary drug resistance (7).
Currently, there is no standard strategy available
for treating patients who are EGFR-TKI resistant. The common
therapeutic strategies usually include: Platinum-based
chemotherapy, high-dose EGFR-TKIs, platinum-based chemotherapy
combined with EGFR-TKIs, new targeted drugs monotherapy (7), such as AZD929 (EGFR-TKI inhibitor)
(8), afatinib (dual EGFR and human
EGFR 2 inhibitor) (9), vandetanib
(dual EGFR and vascular EGFR inhibitor) (10) and crizotinib (for patients with
anaplastic lymphoma kinase mutations) (11). Furthermore, common therapeutic
strategies involve new agents combined with EGFR-TKI, such as
apatinib (12), AXL receptor
tyrosine kinase degradation (13)
and Crizotinib (14). However,
most of these agents are still in clinical trials, and the
combination therapy of EGFR-TKIs and platinum-based chemotherapy is
the preferred choice for most patients. Previous studies have
reported that such combination therapy could improve the overall
survival and progression-free survival of patients (15-18).
The synergistic mechanism between EGFR-TKIs and platinum remains
unknown, and no biomarkers are currently available to predict the
benefit of the combination therapy. If the phenotype of sensitive
combinations could be determined, patients may benefit from the
combination strategy. However, it is not possible to accurately
determine the selection of appropriate patients without
understanding the mechanisms of interactions between EGFR-TKIs and
chemotherapy agents. Thus, biomarkers could only be identified when
the synergistic mechanisms between EGFR-TKIs and platinum-based
chemotherapy are understood.
The present study investigated the interaction
between gefitinib (an EGFR-TKI) and cisplatin (a main
platinum-based drug) to identify the mechanisms of their
synergistic effects in NSCLC. Gefitinib was developed to target
EGFR (19,20), and cisplatin is a cytotoxic drug
that directly targets DNA and promotes the DNA double helix to form
hinges that limit DNA unwinding, inhibit DNA replication and induce
tumor necrosis (21,22). Previous studies have revealed that
some of the agents affecting cisplatin sensitivity are mainly DNA
repair proteins (23,24), such as DNA-dependent protein kinase
(DNA-PK) (25), ERCC excision
repair 1, endonuclease non-catalytic subunit (ERCC1) (26) and BRCA1 DNA repair associated
(BRCA1) (27). DNA-PK is the
pivotal kinase protein involved in non-homologous end joining
pathways of DNA double-strand-break repair (25). Previous studies have also reported
that intracellular EGFR could interact with DNA-PK (28,29).
Moreover, EGFR in the cytoplasm can enter nucleus and regulate the
transcription of DNA repair-related genes (30,31),
assisting in the repair of DNA breaks (29,32).
The aim of the present study was to determine the synergistic
mechanism of gefitinib and cisplatin.
Materials and methods
Chemicals and antibodies
The selective EGFR-TKI gefitinib (GD760; Iressa),
kindly provided by AstraZeneca PLC, was dissolved in pure DMSO to a
working concentration of 20 mM. Cisplatin, MTT, Nucleopriein
Extraction kit and AnnexinV-FITC Apoptosis Detection kit were
obtained from Sigma-Aldrich (Merck KGaA). The caspase-3 Activity
Assay kit was purchased from Beyotime Institute of Biotechnology.
The DNA-PK inhibitor NU7441 (KU-57788) was purchased from Selleck
Chemicals. Antibodies against Akt (cat. no. 4691), phosphorylated
(p)-Akt (Thr308; cat. no. 13038), ERK1/2 (cat. no. 9102), p-ERK1/2
(Thr202/Tyr204; cat. no. 4370), PTEN (cat. no. 9188), BRCA1 (cat.
no. 9010), p-BRCA1 (Ser1524; cat. no. 9009), DNA-PK (cat. no.
38168), p-DNA-PK (Ser2056; cat. no. 68716), EGFR (cat. no. 4267),
p-EGFR (Tyr1068; cat. no. 3777), ERCC1 (cat. no. 12345), caspase-3
(cat. no. 9662), Cleaved caspase-3 (cat. no. 9661), β-actin (cat.
no. 4970) and Lamin B1 (cat. no. 13435), purchased from Cell
Signaling Technology, Inc., which were diluted to 1:1,000 for
western blot analysis and Co-immunoprecipitation (Co-IP)
analysis.
Cell lines
The human NSCLC cell lines were used: A549, H1299
and H1975 (Cell Bank of Chinese Academy of Sciences) with different
EGFR and Kirsten rat sarcoma 2 viral oncogene homolog
(K-Ras) gene statuses [exons 18-21 of the EGFR gene
and exons 2-3 of the K-Ras gene were sequenced, the A549
cell line harbors the pathogenic mutation of K-Ras (exon 2
G12S), the H1975 cell line carries EGFR L858 and T790M and
the EGFR and K-Ras of the H1299 cell line are all
wild-type]. Cell lines were maintained in RPMI-1640 media (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% fetal calf
serum (Gibco; Thermo Fisher Scientific, Inc.) and were cultured at
37°C in a 5% CO2 atmosphere in a humidified
incubator.
Sequencing of EGFR and K-ras genes
The exons encoding the intracellular domain of
EGFR and K-ras were amplified by PCR [the source DNA
was genomic DNA of cell lines, the DNA polymerase was pfu DNA
polymerase (Promega Corproation); thermocycling conditions were as
follows: Initial denaturation at 95°C for 1.5 min, followed by 30
cycles at 95°C for 40 sec, 52-60°C for 30 sec and 72°C for 2 min,
with a final extension step at 72°C for 5 min, and help at 4°C] and
sequenced by bidirectional sequence. The EGFR exon 18-21 and K-ras
exon 2-3 were amplified from H1299, A549 and H1975 cDNA, and the
PCR products were isolated and sequenced. The primer sequences are
listed in Table I. Tests were
performed at least twice for every sample.
 | Table IPrimers for EGFR (exon 18, 19,
20 and 21) and K-Ras (exon 2 and 3). |
Table I
Primers for EGFR (exon 18, 19,
20 and 21) and K-Ras (exon 2 and 3).
| Gene | Exon | Forward
(5′→3′) | Reverse
(5′→3′) |
|---|
| EGFR | 18 |
CAAATGAGCTGGCAAGTGCCGTGT |
GAGTTTCCCAAACACTCAGTGAAA |
| 19 |
GCAATATCAGCCTTAGGTGCGGCTC |
CATAGAAAGTGAACATTTAGGATGTG |
| 20 |
ACTTCACAGCCCTGCGTAAAC |
ATGGGACAGGCACTGATTTGT |
| 21 |
CTAACGTTCGCCAGCCATAAGTCC |
GCTGCGAGCTCACCCAGAATGTCTGG |
| K-Ras | 2 |
CTTAAGCGTCGATGGAGGAG |
CCCTGACATACTCCCAAGGA |
| 3 |
TGGGTATGTGGTAGCATCTCA |
AATCCCAGCACCACCACTAC |
Cell apoptosis analysis
H1299, A549 and H1975 cells were seeded in 6-well
plates at 2×105 cells per well and incubated at 37°C
overnight. After treatment with 10 µM gefitinib or 15
µM cisplatin at 37°C for 48 h, the cells were collected and
assessed using an Annexin V-FITC Apoptosis Detection kit
(Sigma-Aldrich; Merck KGaA), according to the manufacturer's
instructions. Then, cells were detected by a BD FACSCalibur Flow
Cytometer (BD Biosciences) and analyzed with FlowJo software
(Ver.10 for windows; BD Biosciences). The activation of caspase-3
was detected using the caspase-3 Activity Assay kit (Beyotime
Institute of Biotechnology) according to the manufacturer's
instructions. Cells were washed twice in PBS and lysed in Lysis
Buffer (containing 1 mM DTT, 1 mM PMSF and protease inhibitor) on
ice for 20-60 min. Lysates were centrifuged at 15,000 × g for 1 min
at 4°C. Appropriate protein extracts (100-200 µg) of cell
lysates was treated with Reaction Buffer and Caspase-3 Substrate
37°C for 4 h. Absorbance was measured at 400 nm wavelength using a
multiwall spectrophotometer (Bio-Rad Laboratories, Inc.). Caspase-3
Activity was expressed as the percentage of agents-treated cells
vs. control cells, whose activity was considered 0%. The
experiments were independently performed three times.
MTT assay
Cells grown to 70-80% confluence were harvested,
seeded in 96-well microtiter plates at 3,000 cells per well and
incubated at 37°C overnight. Cells were then treated with various
concentrations (the specific concentration is listed in the
corresponding figures) of gefitinib or cisplatin at 37°C for 72 h.
When measuring cell viability, 5 mg/ml MTT was added into the media
and cells were cultured at 37°C for 4 h. Plate centrifugation was
performed in the speed of 1,000 rpm for 15 min under normal
temperature. Then, the supernatant was removed by pipette and the
formazan crystals were dissolved in 200 ml DMSO for 15 min.
Absorbance was measured at 570 nm wavelength using a multiwall
spectrophotometer (Bio-Rad Laboratories, Inc.). Cell viability was
expressed as the percentage of surviving agents-treated cells vs.
control cells whose viability was considered 100%. The combination
index (CI) values of the two drugs were calculated using the
Chou-Talalay median method as previously described (33). To assess the interaction between
cisplatin and gefitinib on cell lines, cells were pretreated with
15 µM cisplatin at 37°C for 72 h before gefitinib was
administered or pretreated with 10 µM gefitinib before
cisplatin was administered. To examine whether gefitinib could
inhibit DNA-PK activation, cells were treated with 20 nM NU7441
(DNA-PK specific inhibitor), 10 µM gefitinib and a
combination of cisplatin (the specific concentration is listed in
the corresponding figures) at 37°C for 72 h. These experiments were
independently performed in triplicate.
Western blot analysis
Whole cell protein and nuclear protein were obtained
by respectively treating cells with RIPA buffer (Beyotime Institute
of Biotechnology) and Nucleopriein Extraction kit (Sigma-Aldrich;
Merck KGaA), containing protease inhibitor cocktail and phosphatase
inhibitor (Bimake. com) for 30 min on ice. Proteins were then
determined using a bicinchoninic acid assay kit (Thermo Fisher
Scientific, Inc.). Appropriate protein extracts (40-60 µg)
of cell lysates were loaded per lane and fractionated through
SDS-PAGE (5% blocking gel at 4°C for 30 min; 8-12% separating gel)
and were electro-transferred to PVDF membranes (EMD Millipore). The
membranes were immersed in the blocking reagent (5% non-fat milk)
and shaken slowly on a shaker at room temperature for 1 h.
Membranes were then probed with various primary antibodies (the
aforementioned antibodies) and horseradish peroxidase-conjugated
secondary antibodies (cat. no. FD-GAR007; 1:5,000; Fude Biological
Technology Co., Ltd.), and were visualized using ECL detection
reagents (EMD Millipore). The molecular weights of the
immunoreactive proteins were estimated based on the PageRuler
Prestained Protein ladder (Fermentas; Thermo Fisher Scientific,
Inc.). Experiments were repeated ≥3 times.
Tumorsphere formation and growth
assays
After adding 200 µl Matrigel (BD Bioscience)
per well in 24-well plate (Corning, Inc.) at 37°C in an incubator
for 30 min, cells (100 µl per well) were seeded onto the
24-well plate at 1,000 cells/ml and cultured in the tumorsphere
medium: RPMI-1640 medium supplemented with Matrigel at 10% volume
ratio. NSCLC cells were treated with low-concentration 6 µM
gefitinib and high-concentration 20 µM cisplatin at 37°C in
an incubator according to the following treatment regimens: i)
Control group (Control), RPMI-1640 media without drug for 18 days;
ii) Cisplatin monotherapy group (cisplatin), RPMI-1640 media
without drug for 12 days and 20 µM cisplatin for 6 days;
iii) Gefitinib combined with cisplatin group (gefitinib +
cisplatin), RPMI-1640 media without drug-treated tumorspheres for
12 days and 20 µM cisplatin combined with 6 µM
gefitinib for 6 days; and iv) Gefitinib pretreatment group
(gefitinib → cisplatin), RPMI-1640 media without drugs for 6 days,
6 µM gefitinib for 6 days and 20 µM cisplatin for 6
days. The volumes of tumorspheres were measured using a light
microscope rule (magnification, ×20) once every 6 days and were
calculated [Volume (mm3)=π × length ×
width2/6]. The experiments were independently performed
in triplicate.
Co-IP assays
Cells were washed twice in PBS and lysed in a IP
buffer (containing 1 mM DTT, 100 mmol/l NaCl and 1 mM
MgCl2) and protease inhibitor cocktails (Bimake.com).
Lysates were centrifuged at 12,000 × g for 10 min at 4°C. The total
cell lysates were used for IP with primary antibodies (anti-EGFR or
anti-DNA-PKCS) on protein A + G mix beads (Thermo Fisher
Scientific, Inc.) at 4°C overnight The immunoprecipitates were
collected and prepared for western blot analysis, which was
performed as aforementioned. In total, 100 µl cell lysates
were used as the input control for western blot analysis.
Orthotopic xenograft assay
A total of 16 female athymic mice
(BALB/cnu/nu; age, 5 weeks; weight, 15-18 g) were
ordered from the Experimental Animal Center of Zhejiang Chinese
Medical University. Mice were housed in cages with wood chip
beddings in a temperature-controlled room (68-72 uF; 24°C) with a
12 h light-dark cycle and 45-55% relative humidity and were
permitted free access to food and drinking water. All of the animal
experiments described in this study were approved by the
Institutional Animal Care and USE Committee (IACUC) at Zhejiang
University. All animals were maintained in accordance with the
IACUC guideline.
These mice were randomized into four groups as
follows: i) Control group (Control), intraperitoneal injection of
normal saline 0.01 ml/g with DMSO; ii) Gefitinib alone group
(gefitinib alone), intraperitoneal injection of 35 mg/kg gefitinib;
iii) Cisplatin alone group (cisplatin alone), intraperitoneal
injection of 5 mg/kg cisplatin; and iv) Combination of gefitinib
and cisplatin group (gefitinib + cisplatin): Intraperitoneal
injection of 35 mg/kg gefitinib and 5 mg/kg cisplatin. Mice were
injected with 2×106 H1299, A549 and H1975 cells
suspension in 100 ml RPMI-1640 medium containing 50% Matrigel (BD
Bioscience) subcutaneously into the right flank. The mice were
randomly assigned to experimental and control groups (4 mice/group)
when the tumors reached the size of 50 mm3. The mice
were fasted overnight and then administered 35 mg/kg gefitinib
and/or 5 mg/kg cisplatin or 0.9% physiological saline (vehicle) by
intraperitoneal injection once every 3 days for 15 day. The
treatment was started on day 7 and stopped on day 22. Tumors were
measured using calipers once every 3 days, and the tumor volumes
were calculated [Volume (mm3)=π x length x
width2/6]. According to the Animal Ethics of IACUC at
Zhejiang University, the experiment ended when the largest tumor
volume approached 2,000 mm3. The mice were sacrificed by
cervical dislocation under deep inhalation anesthesia with 2%
isoflurane at the end of the experiment. At the end of study, tumor
tissues were harvested, fixed in 10% formalin at room temperature
for 24 h and then embedded in paraffin. Tumor sections (thickness,
2-3 µm) were subjected to standard hematoxylin for 10 min
and eosin for 10 sec at room temperature (H&E) staining using a
light microscope (magnification, ×100). The experiment was
conducted in triplicate.
Immunofluorescence
After treatment with 10 µM gefitinib or 15
µM cisplatin at 37°C for 48 h, cells were fixed in 4%
paraformaldehyde at room temperature for 15 min, incubated in 0.1%
Triton X-100 at room temperature for 10 min and washed with PBS.
Cells were blocked with goat serum (Gibco; Thermo Fisher
Scientific, Inc.) at room temperature for 60 min, stained with
primary EGFR antibody or DNA-PK antibody (afore-mentioned
antibodies; 1:250) at 4°C overnight and secondary antibodies (cat.
no. 102-095-003; Jackson ImmunoResearch Laboratories; 1:50) at 37°C
for 60 min. Then, cells were counterstained by DAPI (Sigma-Aldrich;
Merck KGaA) at room temperature for 15 min. The images were
acquired by confocal laser-scanning microscopy (magnification,
×200; Zeiss LSM710; Zeiss AG).
Statistical analysis
Data are presented as the mean ± standard error of
mean obtained from ≥3 experiments. The two-tailed Student's t-tests
were used for analyzing statistical differences between two groups
and one-way ANOVA was used with a post hoc Tukey for multiple
comparisons when comparing >2 groups using SPSS software (Ver.22
for Mac; SPSS, Inc.). Graphs were generated using GraphPad Prism
software (Ver.6 for Mac; GraphPad Software, Inc.). P<0.05 was
considered to indicate a statistically significant difference.
Results
Evaluation of the antiproliferative
effect of gefitinib and cisplatin on NSCLC cell lines with
different EGFR and K-ras mutations
EGFR and K-ras genes were sequenced to identify that
H1299 cells are wild-type, that A549 cells carry K-ras G12S
mutation and that H1975 cells carry EGFR L858R and T790M mutations
(Table II).
| Table IICytotoxicity of gefitinib or
cisplatin and the mutation status of EGFR and K-Ras
gene in human non-small-cell lung cancer cell lines. |
Table II
Cytotoxicity of gefitinib or
cisplatin and the mutation status of EGFR and K-Ras
gene in human non-small-cell lung cancer cell lines.
| Cell line | Cell type | EGFR
gene | K-Ras
gene | Gefitinib
IC50 (µmol/l) | Cisplatin
IC50 (µmol/l) |
|---|
| A549 | Adenocarcinoma | WT | G12S exon 2 | 13.2±2.69 | 11.9±2.77 |
| H1299 | Large cell | WT | WT | 17.5±4.54 | 11.6±1.88 |
| H1975 | Adenocarcinoma | L858R, T790M | WT | 16.6±2.38 | 14.6±6.45 |
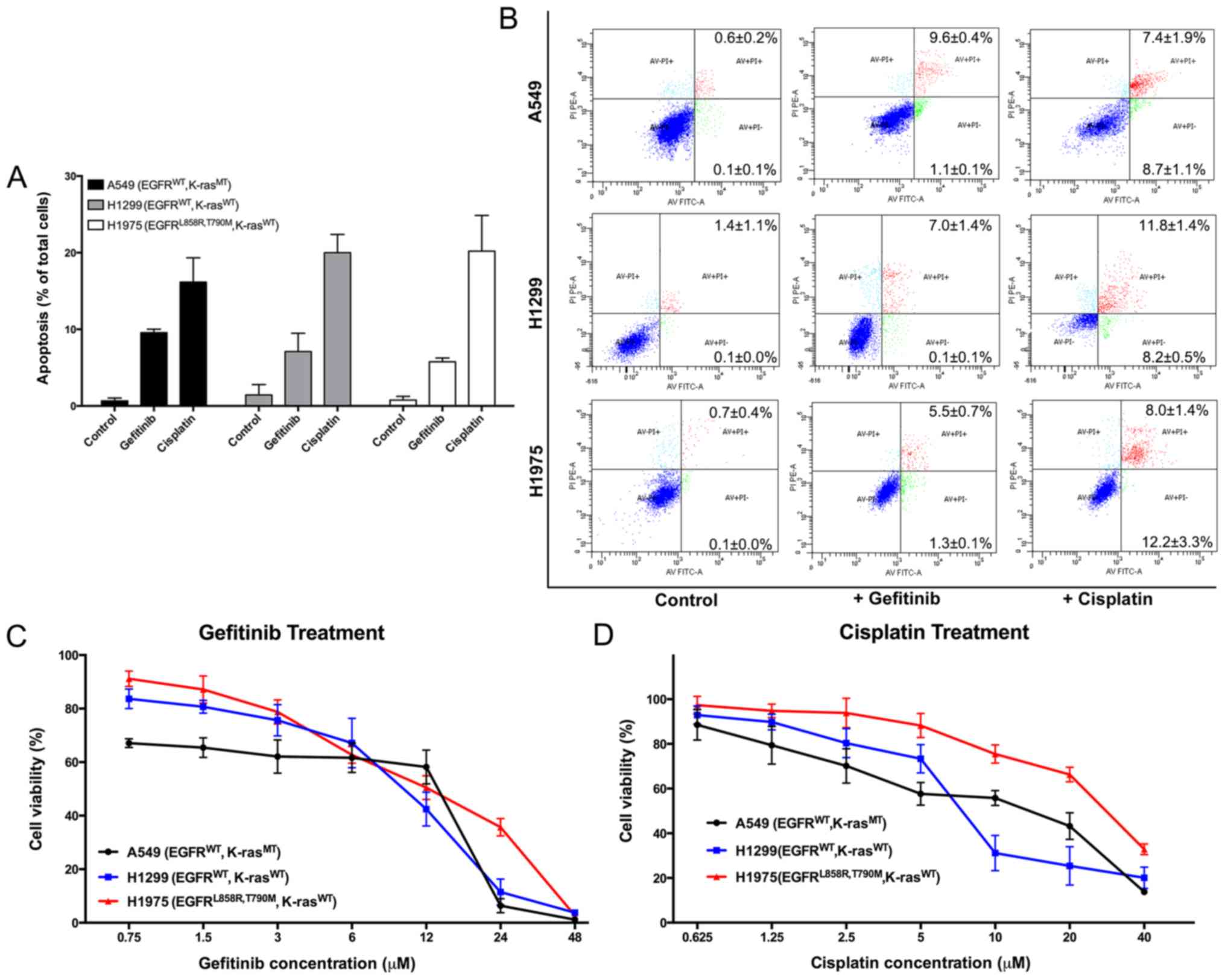
A549, H1299 and H1975 cells were treated with 10
µM gefitinib and 15 µM cisplatin for 48 h to
investigate the sensitivity of the three types of NSCLC cells with
different EGFR and K-ras mutants. Apoptosis was assessed using
Annexin V-FITC/propidium iodide (PI) dual staining by FACSCalibur
Flow Cytometer. Apoptosis in the treatment groups was markedly
higher compared with the control group of each cell line, and no
significant difference in sensitivity of gefitinib and cisplatin
was found between the three cell lines (Fig. 1A and B). A MTT assay was performed,
and the results suggested that the three cell lines were similarly
resistant to gefitinib (The IC50 values of gefitinib in
Fig. 1C are presented in Table II), and the cisplatin
IC50 values were also similar among the cell lines (The
IC50 values of cisplatin in Fig. 1D are presented in Table II).
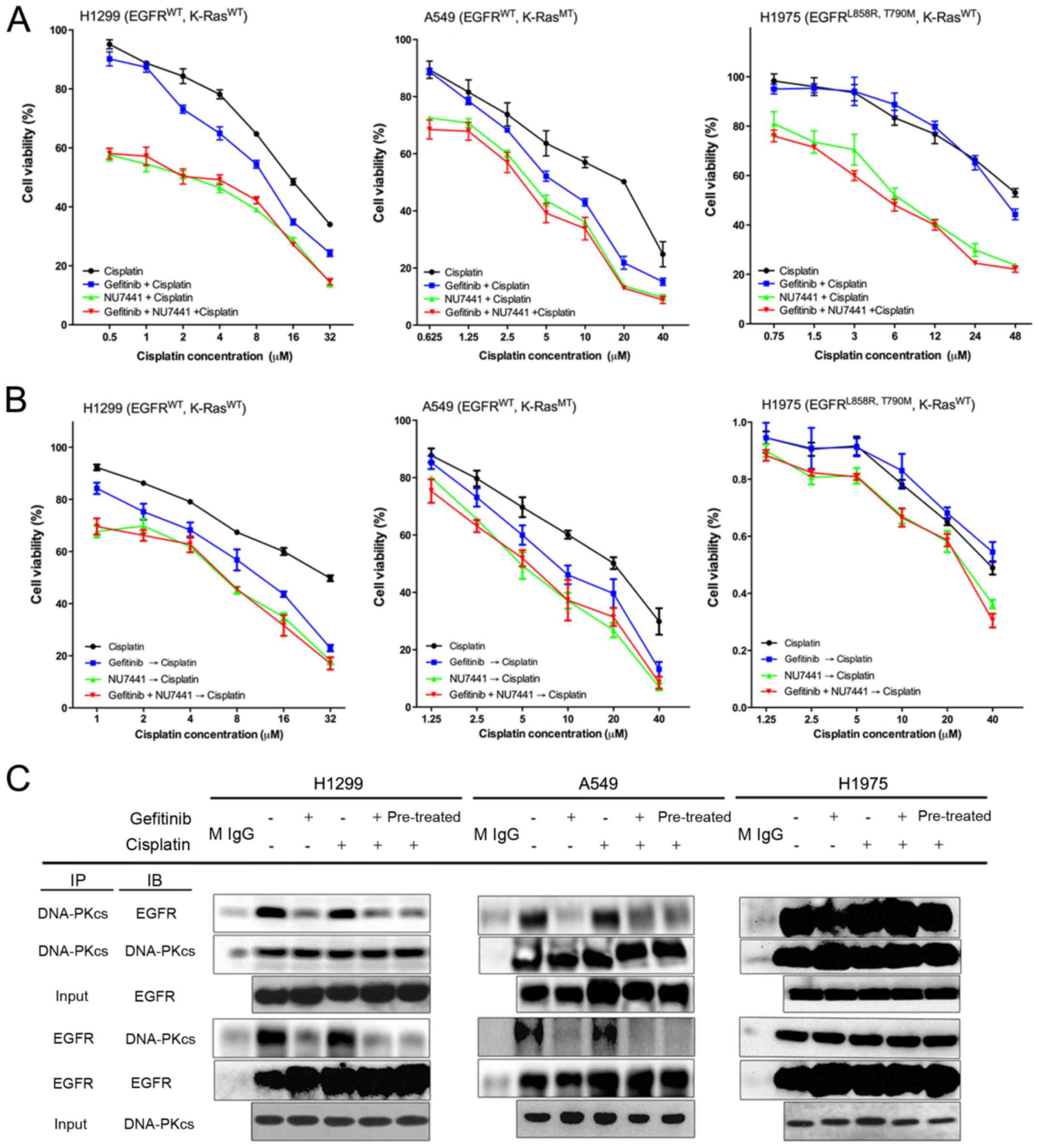
Synergistic and antagonistic
antiproliferative effects of gefitinib and cisplatin
combination
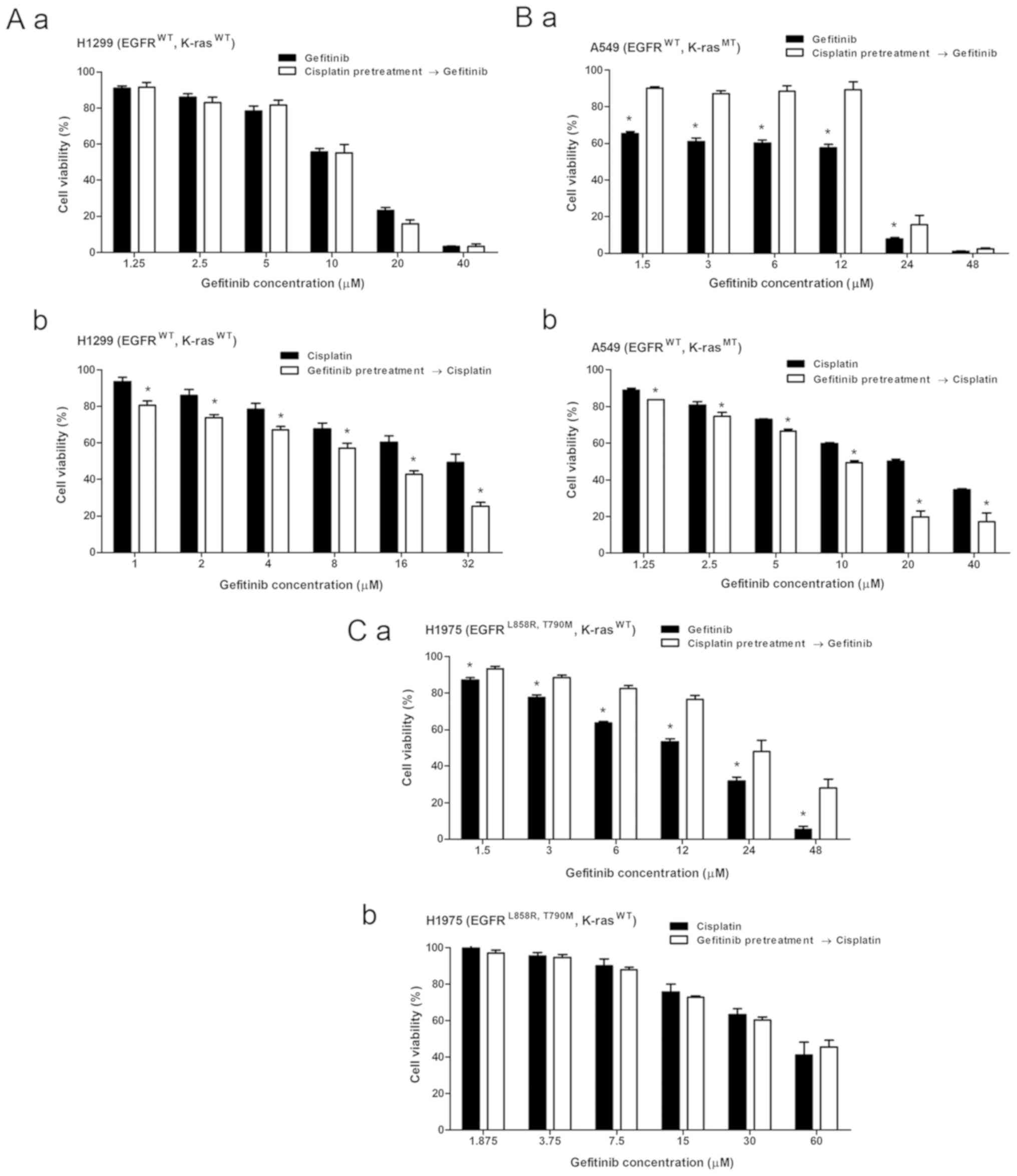
Each cell line was treated with the combination of
cisplatin and gefitinib for 72 h with relatively suitable
concentration (Fig. 2A-a, B-a and
C-a) based on the IC50 ratio of the two agents to
investigate the combined effects of cisplatin and gefitinib in the
three types of NSCLC with different EGFR and K-ras genotypes. Cell
viability was measured by using the MTT method, and the combination
index (CI) values of the two drugs were calculated using the
Chou-Talalay median method as previously described (33). A marked decrease in cell viability
was observed after cisplatin was combined with gefitinib in H1299
cells (EGFR wild-type; K-ras wild-type; Fig. 2A-a). The CI values of cisplatin and
gefitinib at any inhibition rate were <1, and the CI value
decreased as the inhibition rate increased (Fig. 2A-b). This finding indicates that
the combined effect of cisplatin and gefitinib in H1299 cells was
synergistic, and the synergistic effect was stronger as the
combination concentration of the two agents was increased.
A decrease in cell viability was observed after
combination treatment in A549 cells (EGFR wild-type; K-ras exon-2
G12S mutation; Fig. 2B-a).
However, the CI value in A549 cells spanned over one (Fig. 2B-b), suggesting that the combined
effect in A549 cells was mixed. For instance, the effect was
antagonistic when the drugs were combined at low concentrations but
was synergistic as the concentrations increased.
The combined effect in H1975 cells (EGFR exon-21
L858R; exon-20 T790M mutation; K-ras wild-type) was antagonistic.
No significant decrease in cell viability was found in the
combination treatment group in H1975 cells (Fig. 2C-a). The CI value was >1 at most
inhibition rate, indicating that the antagonist effect was stronger
as the combination concentration of the two drugs increased
(Fig. 2C-b). These three different
NSCLC cells demonstrated different cisplatin and gefitinib combined
effects (Table III).
| Table IIICombination and sequential effect of
gefitinib and cisplatin on human non-small-cell lung cancer cell
lines. |
Table III
Combination and sequential effect of
gefitinib and cisplatin on human non-small-cell lung cancer cell
lines.
| Cell line | EGFR
gene | K-Ras
gene | G + C | G→C | C→G |
|---|
| A549 | WT | G12S exon 2 |
Synergistic/antagonistic | Sensitive | Inhibitive |
| H1299 | WT | WT | Synergistic | Sensitive | No effect |
| H1975 | L858R, T790M | WT | Antagonistic | No effect | Inhibitive |
Different sequential effects of gefitinib
and cisplatin on different NSCLC cells
NSCLC cells were pretreated with 15 µM
cisplatin for 72 h before gefitinib was administered or pretreated
with 10 µM gefitinib before cisplatin was administered to
further assess the interaction between cisplatin and gefitinib on
H1299, A549 and H1975 cells. No significant difference in gefitinib
sensitivity on H1299 cells was observed with or without cisplatin
pretreatment (P>0.05; Fig.
3A-a). However, after 10 µM gefitinib pretreatment, the
sensitivity of H1299 cells to cisplatin was significantly higher
compared with the cells without gefitinib pretreatment (P<0.05;
Fig. 3A-b). Thus, in H1299 cells,
cisplatin pretreatment did not affect the antiproliferative effect
of gefitinib, while gefitinib could enhance the inhibition of
cisplatin. The sensitivity of gefitinib on cisplatin-pretreated
A549 cells was significantly reduced (Fig. 3B-a), while cisplatin sensitivity
was increased after gefitinib pretreatment (Fig. 3B-b). In H1975 cells, cisplatin
reduced the sensitivity of gefitinib (Fig. 3C-a), and gefitinib had no
significant effect on cisplatin (Fig.
3C-b). Moreover, the three types of NSCLC cells demonstrated
different sequential effects of the two drugs (Table III).
Gefitinib selectively promotes
cisplatin-induced apoptosis on H1299 and A549 cells
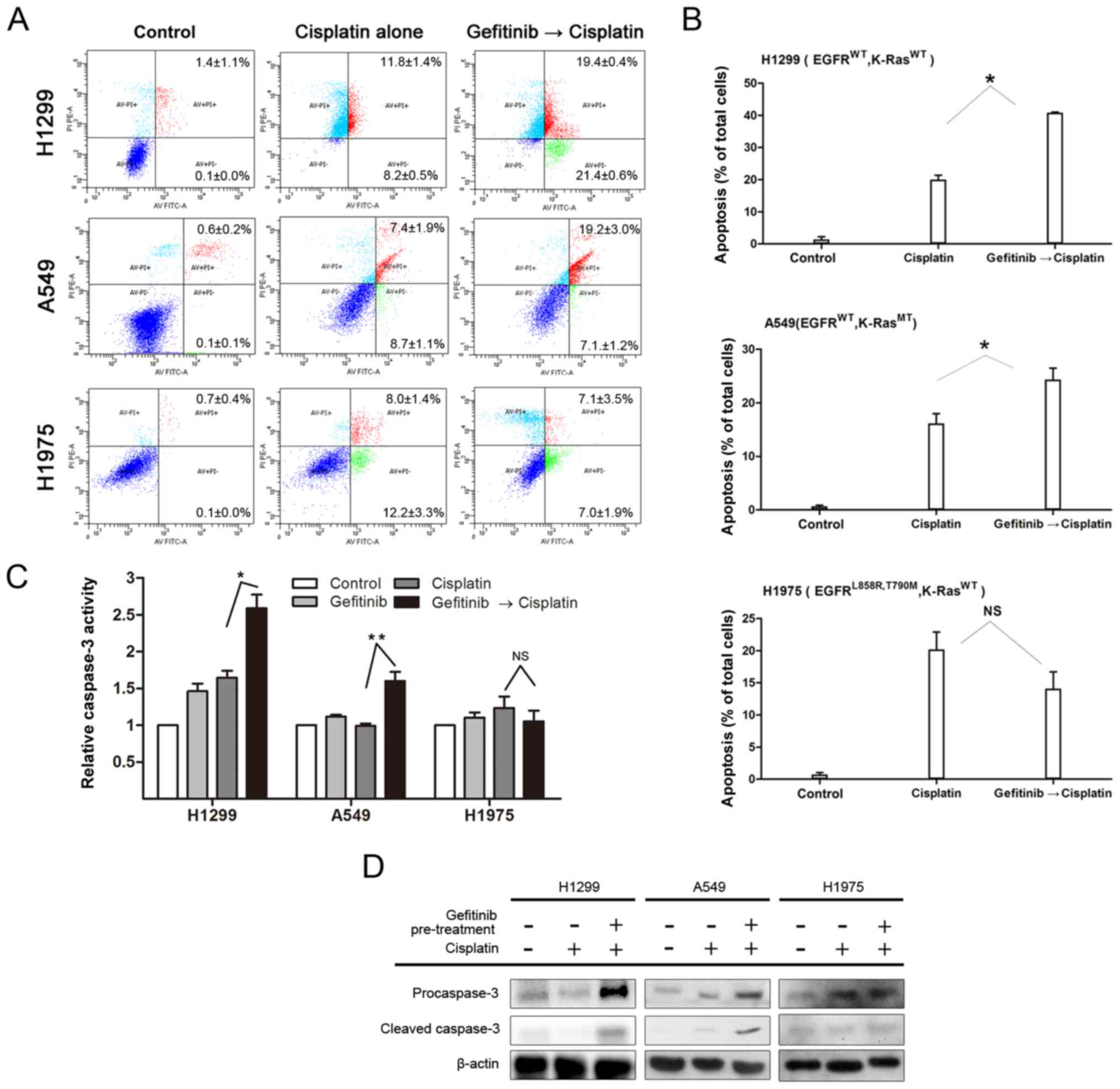
Cells were pretreated with 10 µM gefitinib
for 48 h and then treated with 15 µM cisplatin for 48 h to
investigate whether gefitinib could promote cisplatin-induced
apoptosis on H1299, A549 and H1975 cells. Apoptotic cells were
detected via the AnnexinV-FITC/PI flow cytometry assay. For H1299
and A549 cells, the apoptotic rate of gefitinib pretreatment group
was significantly higher compared with the cisplatin monotherapy
group (P<0.05). However, no significant difference in H1975
cells was observed between the two groups (P>0.05; Fig. 4A and B). Therefore, the results
suggested that gefitinib enhanced the apoptotic ability of
cisplatin on H1299 and A549 cells.
The activation of caspase-3 was detected via the
caspase-3 Activity Assay kit according to the manufacturer's
instructions, and the expression levels of caspase-3 precursor
protein and cleaved caspase-3 were detected by western blot
analysis to determine whether caspase-3 activity after cisplatin
administration could be enhanced by gefitinib pretreatment. The
results demonstrated that caspase-3 activity was more active and
the expression levels of procaspase-3/cleaved caspase-3 proteins
were increased after cisplatin treatment in the gefitinib
pretreatment group compared with the cisplatin monotherapy group
(Fig. 4C and D; P<0.05).
However, no enhancement was observed in H1975 cells.
Gefitinib sensitizes the antitumor effect
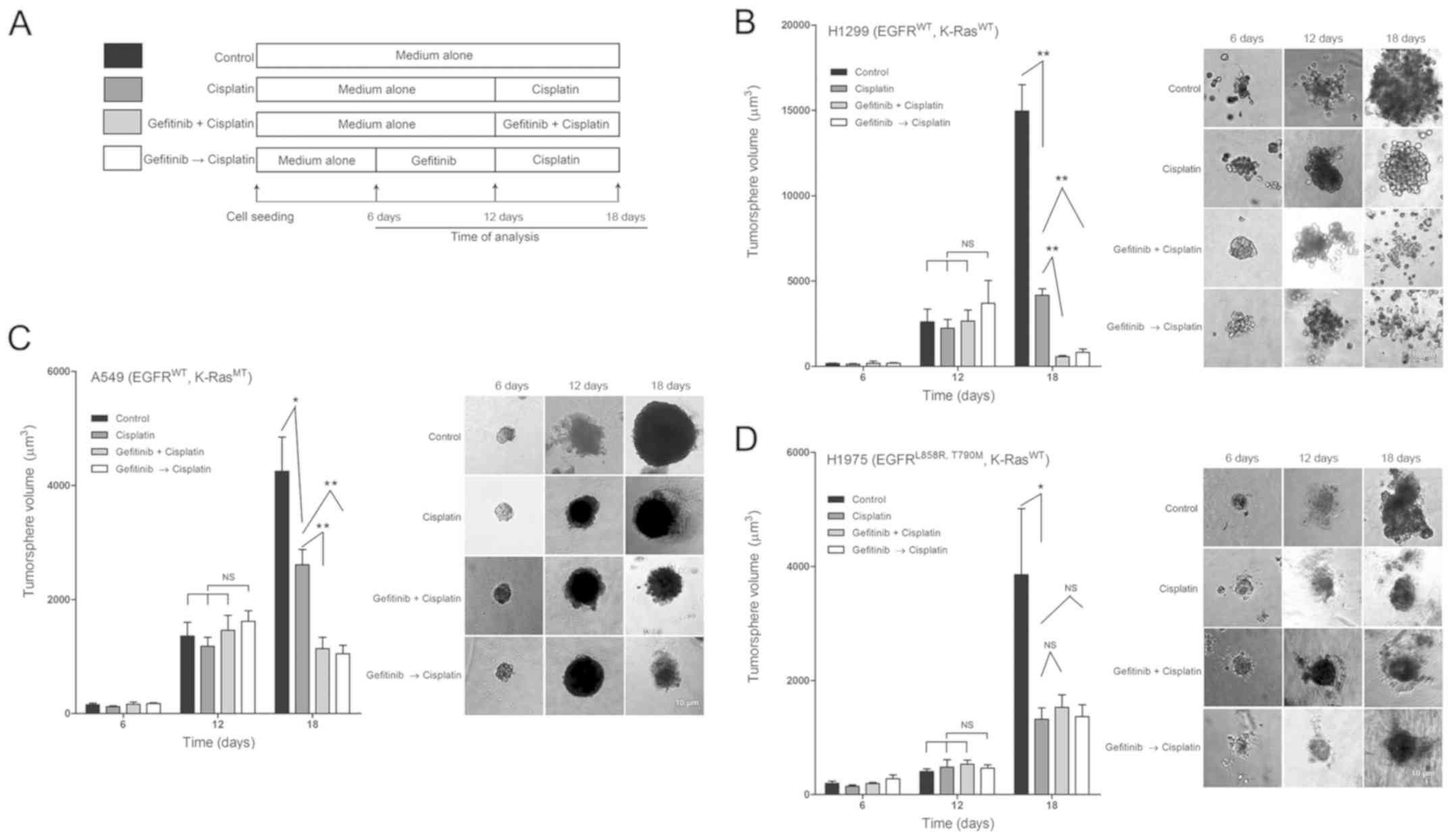
of cisplatin on H1299 and A549 tumorspheres
The 3D tumorspheres were cultured to simulate the
cell growth environment in vitro, and the combination effect
of cisplatin and gefitinib was monitored to assess the promotion of
gefitinib on cisplatin-induced tumor inhibition.
Based on previous studies (34), RPMI-1640 medium was replaced with
Matrigel at 10% volume ratio every 2 days to ensure that cells
could survive >20 days. NSCLC cells were treated with
low-concentration 6 µM gefitinib and high-concentration 20
µM cisplatin, according to the treatment regimens (Fig. 5A). According to the growth trend of
tumors in control group, the tumor growth was in a logarithmic rise
phase on the 18th day (Fig. 5B-D).
The tumor lengths were measured on 6, 12 and 18 days, and the
tumorsphere volumes were also calculated. No significant difference
in the tumor-sphere volumes among the four groups were identified
on the 6 and 12th day (Fig. 5B-D).
However, on the 18th day, the tumorsphere volumes of H1299 in the
four groups were as follows: 14,966.4±3,442.1, 4,072.3±561.5,
570.0±203.2 and 871.5±472.6 µm3, respectively.
The tumor volumes of cisplatin monotherapy group were significantly
reduced compared with the control group (Fig. 5B; P<0.01). Furthermore, the
volumes of G + C and the G → C groups were significantly reduced
compared with the cisplatin monotherapy group (Fig. 5B; both P<0.01). The volumes of
A549 cells in the four groups were as follows: 4,252.7±2,383.4,
2,610.2±997.4, 1,138.6±592.3 and 1,051.4±562.2
µm3. The tumor volumes of G + C with those of the
G → C pretreatment groups were significantly reduced compared with
the cisplatin monotherapy group (Fig.
5C; P<0.01). The volumes of H1975 tumors were as follows:
3,856.7±3,661.2, 1,321.6±915.8, 1,530.5±977.3 and 1,368.6±963.7
µm3. No significant difference in tumor volumes
between G + C or G → C pretreatment groups and the cisplatin
monotherapy groups were observed (Fig.
5D; P>0.05). Collectively, the results indicated that
gefitinib could selectively enhance the antitumor effect of
cisplatin, which was present in H1299 and A549 cells but not H1975
cells.
Synergistic antiproliferative effect of
gefitinib and cisplatin combination on H1299 and A549
xenografts
A xenograft mouse model was used to study the effect
on xenograft tumor growth in nude mice in vivo. H1299, A549
and H1975 cell suspensions were injected into the right flank of
each mouse.
The mean ± SEM of largest diameters of H1299 tumors
in the control, gefitinib alone, cisplatin alone and gefitinib +
cisplatin groups was: 16.31±3.58, 15.02±1.49, 12.9±1.73 and
9.04±2.64 mm respectively. The mean ± SEM in A549 tumors were:
8.14±1.42, 8.62±0.65, 6.99±1.55 and 5.78±1.86 mm. Furthermore,
those in H1975 tumors were: 18.98±0.76, 18.33±1.77, 13.76±1.78 and
13.77±1.62 mm. The volumes and qualities of H1299 and A549 tumors
were significantly reduced in the gefitinib + cisplatin group
combination compared with gefitinib alone and cisplatin alone
groups (Fig. 6A-a and b and B-a and
b). However, in H1975 tumors, the combination of gefitinib +
cisplatin did not significantly inhibit tumor growth compared with
cisplatin alone group (Fig. 6A-c and
B-c).
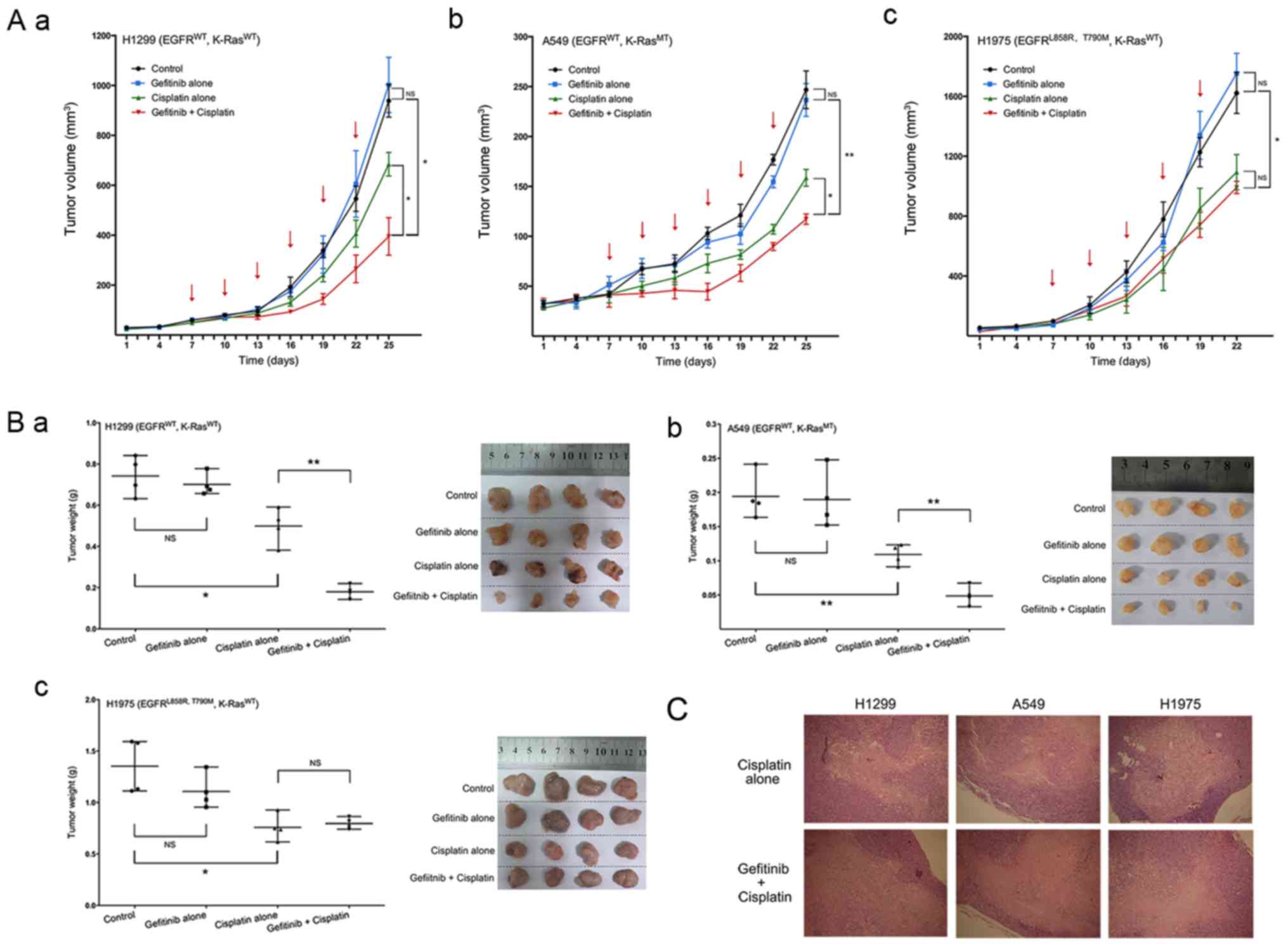 | Figure 6Effect of gefitinib and cisplatin
combination on NSCLC xenografts. Tumor volume curves of each
treatment group on three NSCLC xenografts, with in (A-a) H1299,
(A-b) A549 and (A-c) H1975 cells. Tumor weights and images of three
NSCLC xenografts with in (B-a) H1299, (B-b) A549 and (B-c) H1975
cells. (C) Paraffin-embedded tissue sections of the tumors were
subjected to hematoxylin and eosin staining. Magnification, ×40.
The red arrows indicate drug administration time. Data are
presented as the mean ± SEM. *P<0.05,
**P<0.01. NS, not significant; MT, mutation type; WT,
wild-type; K-ras, Kirsten rat sarcoma 2 viral oncogene homolog;
NSCLC, non-small-cell lung cancer. |
Subsequently, the results of tumor H&E staining
indicated that in H1299 and A549 tumors, the tissue necrosis was
more serious after a combination of gefitinib + cisplatin treatment
compared with cisplatin monotherapy. However, no notable difference
was observed in necrotic areas for H1975 tumors (Fig. 6C).
Gefitinib selectively inhibits DNA-PK
activity on H1299 and A549 cells
The NSCLC cells were treated with 15 µM
cisplatin and 10 µM gefitinib for 72 h to examine the
mechanism of interaction between cisplatin and gefitinib. In
addition, the activation and expression levels of the key proteins
of the EGFR pathway and platinum-sensitive associated proteins,
such as Akt, Erk1/2, PTEN, BRCA1 and ERCC1, were detected via
western blot analysis. The results identified that both gefitinib
alone and in combination with cisplatin could decrease the
expression levels of p-Akt and p-Erk1/2 in H1299, A549 and H1975
cells in comparison with the control group, while cisplatin alone
had no effect on the expression levels of p-Akt and p-Erk1/2.
Cisplatin monotherapy, gefitinib monotherapy and combination
therapy of the two drugs had no effect on the expression of PTEN
and ERCC1 proteins in the three cells. Moreover, cisplatin
monotherapy and cisplatin + gefitinib could increase the expression
of p-BRCA1 in the three cells, but gefitinib monotherapy did not
have the same effect. The changes in the activation and expression
levels of the above proteins were all consistent in H1299, A549 and
H1975 cells (Fig. 7A).
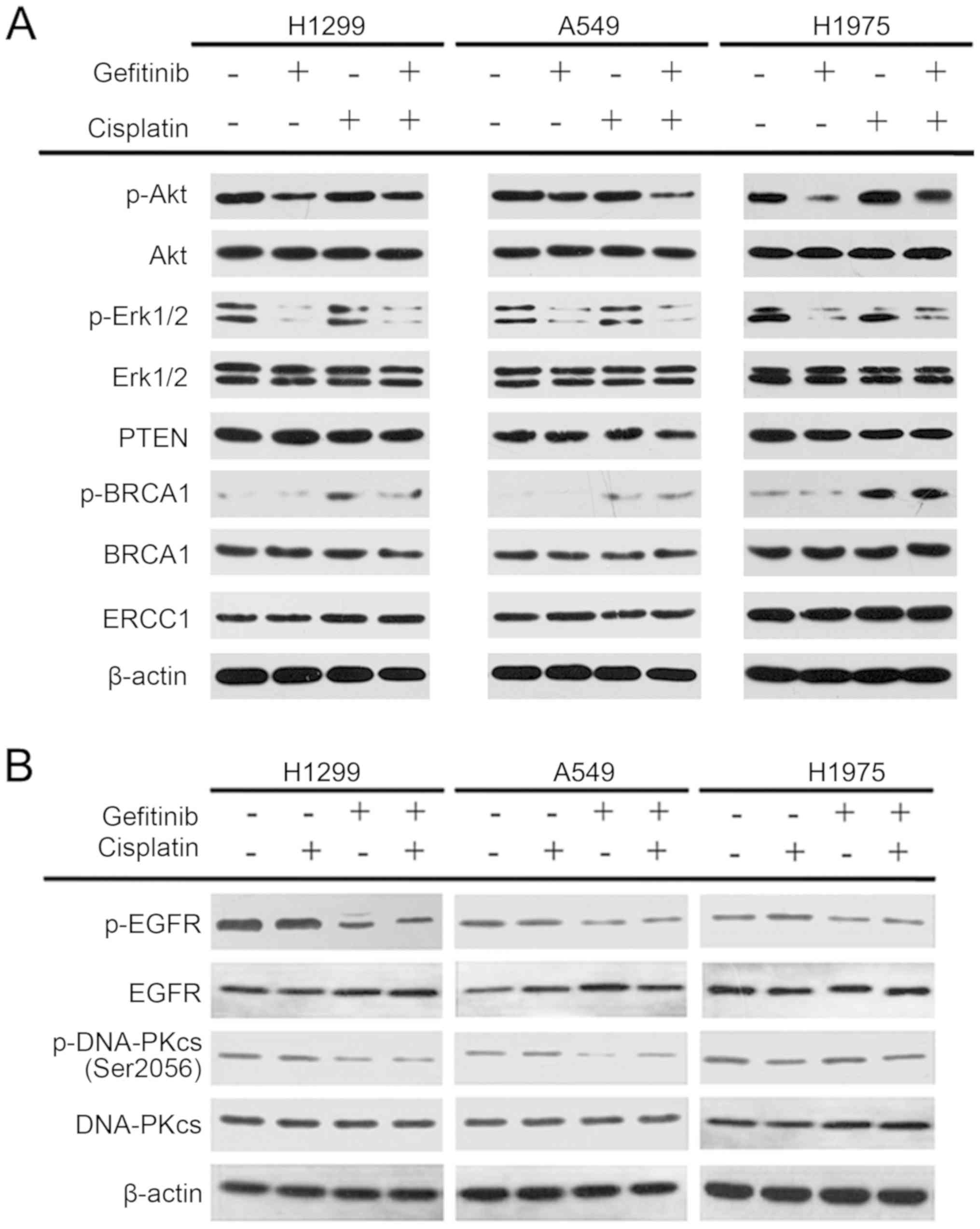 | Figure 7Activation and expression levels of
Akt, Erk1/2, BRCA1, PTEN, ERCC1, EGFR and DNA-PKcs on the three
non-small-cell lung cancer cells after different treatments. (A)
Effects of Akt, Erk1/2, BRCA1, PTEN, and ERCC1 on cisplatin
combined with gefitinib. (B) Expression levels and activation of
EGFR and DNA-PKcs in the three NSCLC cells after the combination of
gefitinib and cisplatin. p-, phosphorylated; BRCA1, BRCA1 DNA
repair associated; ERCC1, ERCC excision repair 1, endonuclease
non-catalytic subunit; DNA-PK, DNA-dependent protein kinase; EGFR,
epidermal growth factor receptor. |
Subsequently, the expression and activation status
of EGFR and DNA-PK in the three cell lines were examined. After 15
µM cisplatin treatment alone for 72 h, the expression levels
and phosphorylation of EGFR and DNA-PK did not change in these
three NSCLC cells. Although the EGFR and DNA-PK expression levels
of the three cells did not change significantly after 10 µM
gefitinib monotherapy for 72 h, the expression levels of p-EGFR and
p-DNA-PK decreased in H1299 and A549 cells. p-EGFR in H1975 cells
was also inhibited, but no notable change was observed in DNA-PK
phosphorylation. After the combination of 15 µM cisplatin
and 10 µM gefitinib, the expression levels of EGFR and
DNA-PK in the three NSCLC cells remain unchanged, and the
phosphorylation levels of EGFR and DNA-PK of the H1299 and A549
cells were reduced. Furthermore, p-EGFR in H1975 cells was also
inhibited, but p-DNA-PK was unchanged (Fig. 7B). Therefore, it was speculated
that gefitinib selectively inhibited the pathway of DNA-PK
phosphorylation in H1299 and A549 cells, and this inhibition was
not associated with EGFR phosphorylation.
Gefitinib selectively inhibits EGFR
binding to DNA-PK in H1299 and A549 cells
To further examine whether gefitinib could inhibit
DNA-PK activation, NSCLC cells were treated with 20 nM NU7441
(DNA-PK specific inhibitor), 10 µM gefitinib and a
combination of different cisplatin concentrations at 37°C in an
incubator for 72 h. In H1299 and A549 cells, the effect of 20 nM
NU7441 combined with cisplatin on cell viability had a synergistic
effect similar to that of the combination of gefitinib + cisplatin.
Moreover, the synergistic effect of the combination of NU7441,
gefitinib and cisplatin was not stronger than that of the combined
NU7441 and cisplatin. However, no synergy in the combination of
gefitinib and cisplatin group was observed in the H1975 cells
(Fig. 8A). In addition, H1299 and
A549 cells pretreated with 20 nM NU7441 were highly sensitive to
cisplatin, and this effect was similar to that of the gefitinib
pretreatment (10 µM gefitinib at 37°C for 72 h). However,
the effect could not be enhanced by treatment with NU7441 and
gefitinib simultaneously (Fig.
8B). Therefore, the data indicated that the synergistic effect
of gefitinib combined with cisplatin was achieved via the gefitinib
inhibition of the DNA-PK activity (Fig. 8B).
The interaction of EGFR and DNA-PK was further
detected via IP after treatment with 15 µM cisplatin and 10
µM gefitinib for 72 h. The results suggested that the IP of
EGFR and DNA-PK was positive in all three NSCLC cells without any
drug treatment (Fig. 8C). After
treatment with cisplatin alone, the IP results of EGFR and DNA-PK
of these three cells were not notably different from those of the
untreated cells. However, the IP of EGFR and DNA-PK in H1299 and
A549 cells was markedly attenuated after gefitinib treatment, but
no notable change in H1975 cells was observed (Fig. 8C). Thus, an interaction may occur
between EGFR and DNA-PK in the NSCLC cells, and gefitinib could
selectively inhibit the inter-action between EGFR and DNA-PK in
H1299 and A549 cells.
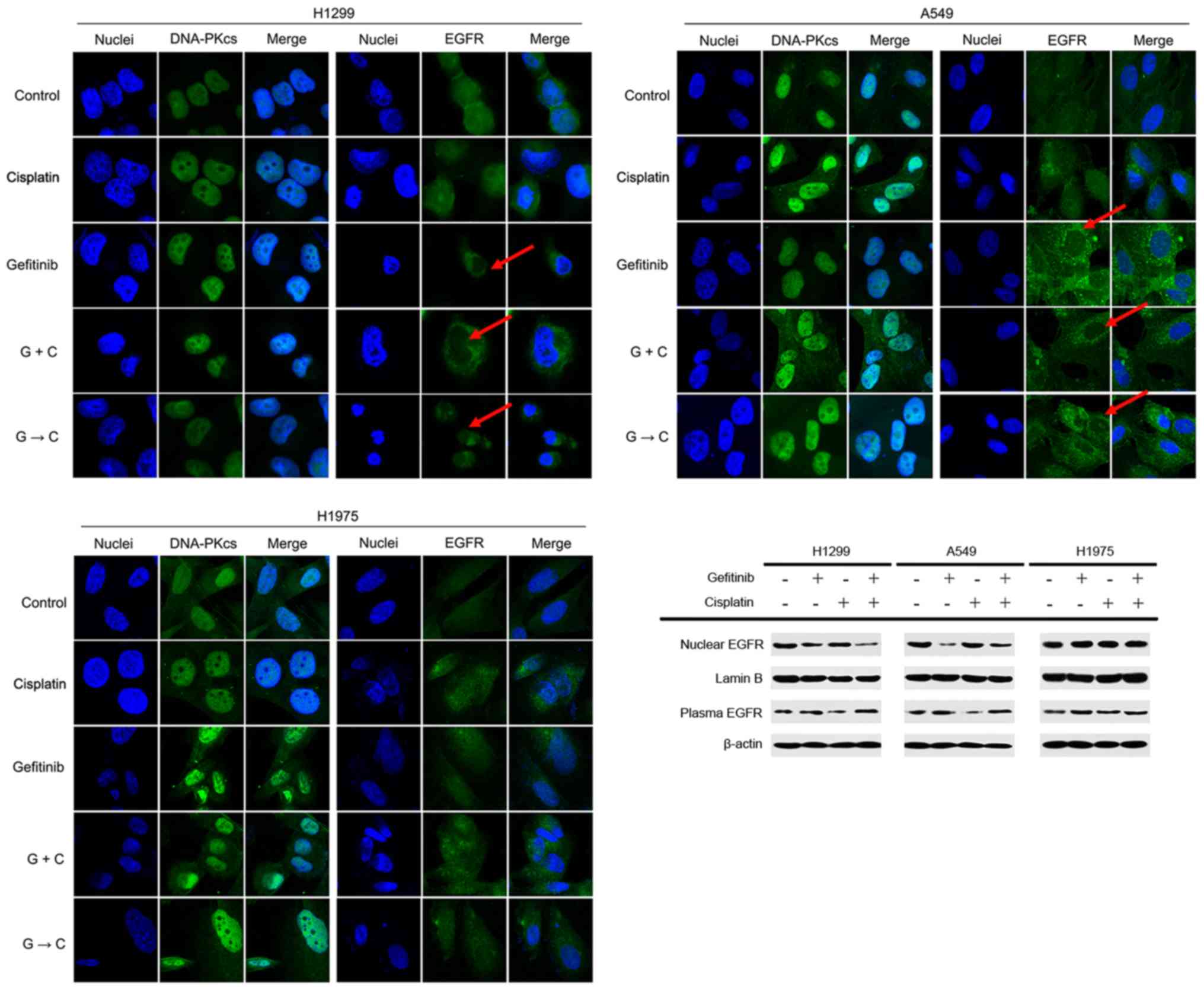
Gefitinib selectively blocks EGFR
translocation into the nucleus of H1299 and A549 cells
It was found that DNA-PK was mainly distributed in
the nucleus, and cisplatin + gefitinib could not interfere with the
DNA-PK distribution. Furthermore, EGFR was evenly distributed in
the cytoplasm and nucleus of these cell lines, and EGFR in the
nucleus of H1299 and A549 cells was relatively decreased after
treatment containing gefitinib (Fig.
9A and B). However, gefitinib did not interfere with EGFR in
the nucleus of H1975 cells (Fig.
9C). Western blot-ting results demonstrated that
gefitinib-containing treatment reduced the nuclear EGFR expression
of H1299 and A549 cells, and the corresponding cytosolic EGFR
expression levels were increased. However, gefitinib had no effect
on EGFR in H1975 cells (Fig. 9D).
The results indicated that DNA-PK was mainly distributed in the
nucleus of NSCLC cells, while EGFR was almost evenly distributed in
the cytoplasm and nucleus. Thus, gefitinib could selectively
prevent the nuclear enrichment of EGFR in H1299 and A549 cells, but
could not prevent trans-location of EGFR into the nucleus of the
H1975 cells.
Discussion
The sensitivity of NSCLC cells to gefitinib depends
on whether EGFR genes harbor sensitive mutations, such as EGFR 19
exon deletion and exon 21 L858R mutation. Furthermore, patients
with these mutations are likely to benefit from EGFR-TKIs drugs
(5,6). However, Westover et al
(35) reported that wild-type
EGFR, K-ras mutation and secondary EGFR mutation (exon 20 T790M)
were associated with resistance to EGFR-TKIs. Zhang et al
(36) also revealed that the
genotypes of EGFR and K-ras in the NSCLC cell line A549 were EGFR
wild type and K-ras mutation (exon 2 G12S), which could explain the
resistance of A549 to EGFR-TKI (37,38).
The genotypes of EGFR-TKI-resistant H1299 cell line have been shown
to be wild-type EGFR and wild-type K-ras (39). In addition, the H1975 cell line
with secondary EGFR mutation (exon 20 T790M) and exon 21 L858R
mutation (36) developed a
secondary resistance to EGFR-TKIs (40). All three cell lines have been
analyzed and verified with the gefitinib-resistant NSCLC models,
and it has been reported that the EGFR and K-ras genotypes of A549,
H1299 and H1975 cell lines are different. For example, A549 and
H1299 cells with wild-type EGFR are primarily resistant to
gefitinib (39,41,42),
while H1975 harbors EGFR L858R-sensitive mutation in exon 21
(43) and secondary resistant
mutation T790M. The mutation sites of A549, H1299 and H1975 cells
in the present study were consistent with the literature (44). It has been reported that the cells
resistance to EGFR-TKIs if IC50 >2 µM
(38,45), but patients with wild-type EGFR may
still benefit from EGFR-TKIs in the clinical practice (46). The present results suggested that
these three types of NSCLC cell models were all EGFR-TKI resistant,
and their IC50 values were ~10 µM.
The combined effects of cisplatin and gefitinib
were shown to be inconclusive in previous studies (40,47).
For instance, Tsai et al (47) reported that these two drugs had no
synergetic effects on NSCLC cells, but Laurila and Koivunen
(48) suggested otherwise. In the
present study, it was found that cisplatin + gefitinib combination
produced different effects among the three NSCLC cell lines. It was
demonstrated that gefitinib monotherapy significantly inhibited the
viability of three NSCLC cells in vitro. Moreover, cisplatin
improved the antiproliferative effect of gefitinib in H1299 and
A549 cells in vivo and in vitro, but such effect was
not observed in H1975 cells.
Zhang et al (49) reported that some patients with
advanced NSCLC with wild-type EGFR could benefit from the
combination therapy of EGFR-TKI and platinum-based drugs. The
present results suggested that the combination effects of gefitinib
and cisplatin in H1299 and A549 varied, despite both cells
containing wild-type EGFR. For example, H1299 cells had a
consistent synergistic effect in vitro, while A549 cells had
a complex mixture effect with regards to synergy. Thus, it was
speculated that wild-type EGFR could not be used as a single
biomarker to predict the combined effect of cisplatin and
gefitinib. Hierarchical analyses of most previous clinical trials
are based on EGFR genotype, which may explain the contradictory
conclusions from the literature (9,14,15,49).
Moreover, the biological difference of tumors may cause this
inconsistency. The most obvious differences between the H1299 and
A549 cells is that H1299 contains wild-type K-ras, but A549 has the
most common mutant of exon 2 G12S. Previous studies have revealed
that the K-ras mutation was a mechanism for primary resistance to
EGFR-TKI (50), and this could be
an indicator for excluding the combination of erlotinib and
platinum-based chemo-therapy (51). In the present study, gefitinib and
cisplatin had extensive synergistic effects in A549 cells with
mutant K-ras (exon 2 G12S), but their effects were completely
different in H1975 and H1299 cells with wild-type K-ras. Thus, both
the present study and previous reports (47,51,52)
suggested that the EGFR and K-ras gene status are not reliable
indictors to assess the sensitivity of combination treatments.
The Iressa Mutation-Positive Multicenter Treatment
Beyond ProgRESsion Study (IMPRESS) trial reported that
gefitinib-platinum combination therapy was ineffective in patients
with secondary resistance and who were carriers of the EGFR T790M
mutation, but the treatment was effective in patients with
secondary resistance and who were non-carriers of the T790M
mutation (53). Moreover,
biomarker analyses suggested that this effect may be driven by the
T790M-positive status (53). In
the present study, an antagonistic effect was observed in the H1975
cells with the EGFR T790M mutation and this observation was
consistent with IMPRESS results, which that may be explained via
the synergistic mechanisms between gefitinib and cisplatin that
were investigated in the current study. However, no biomarker is
available to predict the benefit of the combination therapy.
In the current study, it was indicated that the
synergistic effect of cisplatin and gefitinib depended on the
increased sensitivity of cisplatin induced by gefitinib. The two
drugs demonstrated antagonistic effect in A549 cells when combined
at low concentrations, but the effect became synergistic at high
concentrations. Therefore, sensitization of gefitinib on cispl-atin
was dominant at high concentrations. The mechanisms of
sensitization could provide novel ideas for investigating new
biomarkers for combination therapy.
The present results demonstrated that gefitinib
enhanced cisplatin-induced apoptosis in H1299 and A549 cells and
activation of caspase-3, but this phenomenon was not observed in
H1975 cells. Previous studies have shown that gefitinib could
enhance the antitumor ability of cisplatin in clinical practice
(54), and some patients with
NSCLC with failed platinum-based chemotherapy could resume
chemotherapy after treatment of gefitinib for a period of time
(55). In the present study,
cisplatin inhibited the sensitivity of gefitinib, and this result
was different from previous findings (40,56).
It was speculated that this difference may be associated with the
biological background of NSCLC models. Therefore, the inhibition
mechanism of cisplatin on gefitinib requires further examination in
future studies.
EGFR has two main downstream signal path-ways: The
Ras/Raf/Mitogen-activated protein kinase
kinase/ERK/Mitogen-activated protein kinase pathway (57) and the PI3K/Akt/mTOR pathway
(58). The core agents of these
two pathways are ERK1/2 and Akt proteins, respectively, and
EGFR-TKI can inhibit the activation of EGFR downstream pathways via
EGFR activation inhibition (45).
PTEN is a tumor suppressor protein that blocks the activation of
Akt and its related proteins (59), and the deletion or inhibition of
PTEN is associated with EGFR-TKI resistance (60-62).
Previous studies have shown that the mechanism of the synergistic
effects between cisplatin and gefitinib was associated with
cisplatin-induced activation of EGFR pathway (40). However, in the current study, this
pathway was not observed in all three NSCLC cells.
The present results indicated that DNA-PK
phosphorylation could be inhibited by gefitinib in H1299 and A549
cells but not in H1975 cells. Previous studies also revealed that
gefitinib inhibited DNA-PK activity in breast cancer cells and
decreased DNA-PK phosphorylation (63,64).
However, gefitinib had no effect on BRCA1 activation and ERCC1
expression. Moreover, BRCA1 was activated by cisplatin, which may
be the stress response of cisplatin. Thus, it was speculated that
the synergistic effect between gefitinib and cisplatin in NSCLC
cells may be achieved via the inhibition of the DNA-PK pathway.
An interaction between EGFR and DNA-PK was
identified in the NSCLC cell nucleus, and the interaction could be
selectively inhibited by gefitinib. In addition, the present
results suggested that the amount of EGFR in the nucleus could be
reduced by gefitinib. Therefore, gefitinib may prevent EGFR from
entering the nucleus, thus activating DNA-PK and sensitizing
cisplatin. The synergistic effect between gefitinib and cisplatin
may due to the enhanced cytotoxicity of cisplatin by gefitinib.
Similar mechanisms have also been reported in previous studies,
which showed that EGFR could move into the nucleus and bind to
DNA-PK to assist in DNA double-strand repair, enhancing tumor cell
repair (28,29,63,65).
Cetuximab, an EGFR antibody, has also been showed to prevent EGFR
from entering the nucleus and inhibiting DNA-PK activity, which
increases the sensitivity of chemotherapy (65,66).
Nuclear EGFR can regulate the translation of cell
signaling proteins (67).
Furthermore, not all types of EGFR can enter the nucleus. It has
been revealed that cisplatin and radiation facilitate the wild-type
EGFR and EGFR vIII entering the nucleus, but not EGFR with the
L858R multination (32). EGFR vIII
is an EGFR mutation and it lacks the extracellular ligand-binding
region factor (32). The inability
of ligand binding inhibits the intracellular activation of EGFR and
causes the resistance to EGFR-targeted drugs (68). As H1299 and A549 cells contain
wild-type EGFR, and H1975 harbors L858R mutation, it was speculated
that wild-type EGFR may be an essential agent for the sensitization
of cisplatin by gefitinib.
In conclusion, the present results suggested that
gefitinib can selectively inhibit DNA-PK activity and sensitize the
cytotoxicity of cisplatin in NSCLC cells by inhibiting nuclear EGFR
to active DNA-PK, which is the synergistic mechanism between
platinum-based chemotherapy and EGFR-TKI. In addition, wild-type
EGFR may be a potential biomarkers for the selection of combination
therapy of cisplatin-based chemo-therapy and gefitinib; however,
further research is required to identify other biomarkers.
Acknowledgments
The authors would like to thank Dr Qinghua Lv, Dr
Qi Dong and Dr Jiaping Peng (Cancer Institute, Zhejiang University)
for technical support in the laboratory. The authors would also
like to thank Dr Xiangyin Wei (Food and Drug Administration) for
language modification of the manuscript.
Funding
This work was supported by research grants from the
Zhejiang Provincial Natural Science Foundation of China (grant nos.
LQ17H160012, LQ19H160041 and LY17H160013), the China Natural
Science Foundation projects (grant nos. 81602516, 81402365,
81502598 and 81602716), the Foundation of Education Department of
Zhejiang Province (grant no. Y201636729), the Key Program of the
Natural Science Foundation of Zhejiang Province (grant no.
LZ16H160002) and the Zhejiang Provincial Program for the
Cultivation of High-level Innovative Health Talents to YDC,
Training Program of the Major Research Plan of the National Science
Foundation of China (grant no. 91229104).
Availability of data and materials
The datasets used or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JJH and SZZ conceived the concept and designed the
study. CP, HJD, YNW, CPZ, CHY, YD, DML, CG, DQW, YYW, XHF, JX, ML,
YDC, TP and WT performed the experiments. CP and HJD wrote the
paper. YDC, TP and WHX reviewed and edited the manuscript. All
authors read and approved the manuscript.
Ethics approval and consent to
participate
All of the animal experiments described in this
study were approved by the Institutional Animal Care and USE
Committee (IACUC) at Zhejiang University. All animals were
maintained in accordance with the IACUC guideline.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferlay J, Colombet M, Soerjomataram I,
Mathers C, Parkin DM, Piñeros M, Znaor A and Bray F: Estimating the
global cancer incidence and mortality in 2018: GLOBOCAN sources and
methods. Int J Cancer. 144:1941–1953. 2019. View Article : Google Scholar
|
|
2
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 coun-tries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Burdett S, Stewart L and Pignon JP:
Chemotherapy in non-small cell lung cancer: An update of an
individual patient data-based meta-analysis. J Thorac Cardiovasc
Surg. 129:1205–1206. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
NSCLC Meta-Analyses Collaborative Group:
Chemotherapy in addition to supportive care improves survival in
advanced non-small-cell lung cancer: A systematic review and
meta-analysis of individual patient data from 16 randomized
controlled trials. J Clin Oncol. 26:4617–4625. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baselga J and Averbuch SD: ZD1839
('Iressa') as an anticancer agent. Drugs. 60(Suppl 1): S33–S42.
2000. View Article : Google Scholar
|
|
6
|
Singh M and Jadhav HR: Targeting non-small
cell lung cancer with small-molecule EGFR tyrosine kinase
inhibitors. Drug Discov Today. 23:745–753. 2018. View Article : Google Scholar
|
|
7
|
Wu SG and Shih JY: Management of acquired
resistance to EGFR TKI-targeted therapy in advanced non-small cell
lung cancer. Mol Cancer. 17:382018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jänne PA, Yang JC, Kim DW, Planchard D,
Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, et al: AZD9291
in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J
Med. 372:1689–1699. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang JC, Hirsh V, Schuler M, Yamamoto N,
O'Byrne KJ, Mok TS, Zazulina V, Shahidi M, Lungershausen J, Massey
D, et al: Symptom control and quality of life in LUX-Lung 3: A
phase III study of afatinib or cisplatin/pemetrexed in patients
with advanced lung adenocarcinoma with EGFR mutations. J Clin
Oncol. 31:3342–3350. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Takeda H, Takigawa N, Ohashi K, Minami D,
Kataoka I, Ichihara E, Ochi N, Tanimoto M and Kiura K: Vandetanib
is effective in EGFR-mutant lung cancer cells with PTEN deficiency.
Exp Cell Res. 319:417–423. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Costa DB, Shaw AT, Ou SH, Solomon BJ,
Riely GJ, Ahn MJ, Zhou C, Shreeve SM, Selaru P, Polli A, et al:
Clinical experience with crizotinib in patients with advanced
ALK-rearranged non-small-cell lung cancer and brain metastases. J
Clin Oncol. 33:1881–1888. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang Z, Luo F, Zhang Y, Ma Y, Hong S,
Yang Y, Fang W, Huang Y, Zhang L and Zhao H: The ACTIVE study
protocol: Apatinib or placebo plus gefitinib as first-line
treatment for patients with EGFR-mutant advanced non-small cell
lung cancer (CTONG1706). Cancer Commun (Lond). 39:692019.
View Article : Google Scholar
|
|
13
|
Kim D, Bach DH, Fan YH, Luu TT, Hong JY,
Park HJ and Lee SK: AXL degradation in combination with EGFR-TKI
can delay and overcome acquired resistance in human non-small cell
lung cancer cells. Cell Death Dis. 10:3612019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang W, Wang H, Lu P, Yu Z, Xu C, Zhuang W
and Song Z: Crizotinib with or without an EGFR-TKI in treating
EGFR-mutant NSCLC patients with acquired MET amplification after
failure of EGFR-TKI therapy: A multicenter retrospective study. J
Transl Med. 17:522019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou H, Zeng C, Wang LY, Xie H, Zhou J,
Diao P, Yao WX, Zhao X and Wei Y: Chemotherapy with or without
gefitinib in patients with advanced non-small-cell lung cancer: A
meta-anal-ysis of 6,844 patients. Chin Med J (Engl). 126:3348–3355.
2013.
|
|
16
|
Fang H, Lin RY, Sun MX, Wang Q, Zhao YL,
Yu JL, Tian Y and Wang XY: Efficacy and survival-associated factors
with gefitinib combined with cisplatin and gemcitabine for advanced
non-small cell lung cancer. Asian Pac J Cancer Prev.
15:10967–10970. 2014. View Article : Google Scholar
|
|
17
|
Lee S, Joo J, Kwak M, Sohn K and Chon S:
Role of chemo-therapy with epidermal growth factor
receptor-tyrosine kinase inhibitor (EGFR-TKI) rechallenge in small
cell transformation after EGFR-TKI failure: A case report. Onco
Targets Ther. 11:3943–3947. 2018. View Article : Google Scholar :
|
|
18
|
Ding T, Zhou F, Chen X, Zhang S, Liu Y,
Sun H, Ren S, Li X, Zhao C, Wang H and Zhou C: Continuation of
gefitinib plus chemo-therapy prolongs progression-free survival in
advanced non-small cell lung cancer patients who get acquired
resistance to gefitinib without T790M mutations. J Thorac Dis.
9:2923–2934. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Anderson NG, Ahmad T, Chan K, Dobson R and
Bundred NJ: ZD1839 (Iressa), a novel epidermal growth factor
receptor (EGFR) tyrosine kinase inhibitor, potently inhibits the
growth of EGFR-positive cancer cell lines with or without erbB2
overexpression. Int J Cancer. 94:774–782. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Anido J, Matar P, Albanell J, Guzmán M,
Rojo F, Arribas J, Averbuch S and Baselga J: ZD1839, a specific
epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor,
induces the formation of inactive EGFR/HER2 and EGFR/HER3
heterodimers and prevents heregulin signaling in
HER2-overexpressing breast cancer cells. Clin Cancer Res.
9:1274–1283. 2003.PubMed/NCBI
|
|
21
|
Roberts JJ and Friedlos F: Quantitative
estimation of cisplatin-induced DNA interstrand cross-links and
their repair in mammalian cells: Relationship to toxicity.
Pharmacol Ther. 34:215–246. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dijt FJ, Fichtinger-Schepman AM, Berends F
and Reedijk J: Formation and repair of cisplatin-induced adducts to
DNA in cultured normal and repair-deficient human fibroblasts.
Cancer Res. 48:6058–6062. 1988.PubMed/NCBI
|
|
23
|
Pera MF Jr, Rawlings CJ and Roberts JJ:
The role of DNA repair in the recovery of human cells from
cisplatin toxicity. Chem Biol Interact. 37:245–261. 1981.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chu G and Berg P: DNA cross-linked by
cisplatin: A new probe for the DNA repair defect in xeroderma
pigmentosum. Mol Biol Med. 4:277–290. 1987.PubMed/NCBI
|
|
25
|
Shao CJ, Fu J, Shi HL, Mu YG and Chen ZP:
Activities of DNA-PK and Ku86, but not Ku70, may predict
sensitivity to cisplatin in human gliomas. J Neurooncol. 89:27–35.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ryu JS, Memon A and Lee SK: ERCC1 and
personalized medicine in lung cancer. Ann Transl Med.
2:322014.PubMed/NCBI
|
|
27
|
Wang LR, He LJ, Wang Y, Li YY, Lou Y,
Zhang GB, Li Y and Chen J: Correlation between BRCA1 and TopBP1
protein expression and clinical outcome of non-small cell lung
cancer treated with platinum-based chemotherapy. Cancer Chemother
Pharmacol. 76:163–170. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang SC and Hung MC: Nuclear translocation
of the epidermal growth factor receptor family membrane tyrosine
kinase receptors. Clin Cancer Res. 15:6484–6489. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rodemann HP, Dittmann K and Toulany M:
Radiation-induced EGFR-signaling and control of DNA-damage repair.
Int J Radiat Biol. 83:781–791. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang SC, Nakajima Y, Yu YL, Xia W, Chen
CT, Yang CC, McIntush EW, Li LY, Hawke DH, Kobayashi R and Hung MC:
Tyrosine phosphorylation controls PCNA function through protein
stability. Nat Cell Biol. 8:1359–1368. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Das AK, Chen BP, Story MD, Sato M, Minna
JD, Chen DJ and Nirodi CS: Somatic mutations in the tyrosine kinase
domain of epidermal growth factor receptor (EGFR) abrogate
EGFR-mediated radioprotection in non-small cell lung carcinoma.
Cancer Res. 67:5267–5274. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liccardi G, Hartley JA and Hochhauser D:
EGFR nuclear translocation modulates DNA repair following cisplatin
and ionizing radiation treatment. Cancer Res. 71:1103–1114. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lee GY, Kenny PA, Lee EH and Bissell MJ:
Three-dimensional culture models of normal and malignant breast
epithelial cells. Nat Methods. 4:359–365. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Westover D, Zugazagoitia J, Cho BC, Lovly
CM and Paz-Ares L: Mechanisms of acquired resistance to first- and
second-generation EGFR tyrosine kinase inhibitors. Ann Oncol.
29(Suppl 1): i10–i19. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang Q, Kanterewicz B, Shoemaker S, Hu Q,
Liu S, Atwood K and Hershberger P: Differential response to
1α,25-dihydroxyvitamin D3 (1α,25(OH)2D3) in non-small cell lung
cancer cells with distinct oncogene mutations. J Steroid Biochem
Mol Biol. 136:264–270. 2013. View Article : Google Scholar
|
|
37
|
Takezawa K, Okamoto I, Yonesaka K,
Hatashita E, Yamada Y, Fukuoka M and Nakagawa K: Sorafenib inhibits
non-small cell lung cancer cell growth by targeting B-RAF in KRAS
wild-type cells and C-RAF in KRAS mutant cells. Cancer Res.
69:6515–6521. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li T, Ling YH, Goldman ID and Perez-Soler
R: Schedule-dependent cytotoxic synergism of pemetrexed and
erlotinib in human non-small cell lung cancer cells. Clin Cancer
Res. 13:3413–3422. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang W, Peyton M, Xie Y, Soh J, Minna JD,
Gazdar AF and Frenkel EP: Histone deacetylase inhibitor romidepsin
enhances anti-tumor effect of erlotinib in non-small cell lung
cancer (NSCLC) cell lines. J Thorac Oncol. 4:161–166. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Van Schaeybroeck S, Kyula J, Kelly DM,
Karaiskou-McCaul A, Stokesberry SA, Van Cutsem E, Longley DB and
Johnston PG: Chemotherapy-induced epidermal growth factor receptor
activation determines response to combined gefitinib/chemotherapy
treatment in non-small cell lung cancer cells. Mol Cancer Ther.
5:1154–1165. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim GW, Song JS, Choi CM, Rho JK, Kim SY,
Jang SJ, Park YS, Chun SM, Kim WS, Lee JS, et al: Multiple
resistant factors in lung cancer with primary resistance to EGFR-TK
inhibitors confer poor survival. Lung Cancer. 88:139–146. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Karachaliou N, Rosell R, Molina MA and
Viteri S: Predicting resistance by selection of signaling pathways.
Transl Lung Cancer Res. 3:107–115. 2014.
|
|
43
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, et al: Activating mutations in the
epidermal growth factor receptor underlying responsiveness of
non-small-cell lung cancer to gefitinib. N Engl J Med.
350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Denis MG, Vallée A and Théoleyre S: EGFR
T790M resistance mutation in non small-cell lung carcinoma. Clin
Chim Acta. 444:81–85. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sordella R, Bell DW, Haber DA and
Settleman J: Gefitinib-sensitizing EGFR mutations in lung cancer
activate anti-apoptotic pathways. Science. 305:1163–1167. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Vitale MG, Riccardi F, Mocerino C, Barbato
C, Monaco R, Galloro P, Gagliardi N and Cartenì G:
Erlotinib-induced complete response in a patient with epidermal
growth factor receptor wild-type lung adenocarcinoma after
chemotherapy failure: A case report. J Med Case Rep. 8:1022014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tsai CM, Chen JT, Stewart DJ, Chiu CH, Lai
CL, Hsiao SY, Chen YM and Chang KT: Antagonism between gefitinib
and cisplatin in non-small cell lung cancer cells: Why randomized
trials failed? J Thorac Oncol. 6:559–568. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Laurila N and Koivunen JP: EGFR inhibitor
and chemotherapy combinations for acquired TKI resistance in
EGFR-mutant NSCLC models. Med Oncol. 32:2052015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhang Y, Yang H, Yang X, Deng Q, Zhao M,
Xu X and He J: Erlotinib with pemetrexed/cisplatin for patients
with EGFR wild-type lung adenocarcinoma with brain metastases. Mol
Clin Oncol. 2:449–453. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Pao W, Wang TY, Riely GJ, Miller VA, Pan
Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG and Varmus HE: KRAS
mutations and primary resistance of lung adenocarcinomas to
gefitinib or erlotinib. PLoS Med. 2:e172005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Eberhard DA, Johnson BE, Amler LC, Goddard
AD, Heldens SL, Herbst RS, Ince WL, Jänne PA, Januario T, Johnson
DH, et al: Mutations in the epidermal growth factor receptor and in
KRAS are predictive and prognostic indicators in patients with
non-small-cell lung cancer treated with chemotherapy alone and in
combination with erlotinib. J Clin Oncol. 23:5900–5909. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Rosetti M, Zoli W, Tesei A, Ulivi P,
Fabbri F, Vannini I, Brigliadori G, Granato AM, Amadori D and
Silvestrini R: Iressa strengthens the cytotoxic effect of docetaxel
in NSCLC models that harbor specific molecular characteristics. J
Cell Physiol. 212:710–716. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mok TSK, Kim SW, Wu YL, Nakagawa K, Yang
JJ, Ahn MJ, Wang J, Yang JC, Lu Y, Atagi S, et al: Gefitinib plus
chemotherapy versus chemotherapy in epidermal growth factor
receptor mutation-positive non-small-cell lung cancer resistant to
first-line gefitinib (IMPRESS): Overall survival and biomarker
analyses. J Clin Oncol. 35:4027–4034. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Melisi D, Troiani T, Damiano V, Tortora G
and Ciardiello F: Therapeutic integration of signal transduction
targeting agents and conventional anti-cancer treatments. Endocr
Relat Cancer. 11:51–68. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Fujiwara K, Kiura K, Gemba K, Ogata Y,
Hotta K, Kishino D, Tabata M, Ueoka H and Tanimoto M: Gefitinib
('Iressa', ZD1839) may restore chemosensitivity in NSCLC patients?
Anticancer Res. 25:547–549. 2005.PubMed/NCBI
|
|
56
|
Dai Q, Ling YH, Lia M, Zou YY, Kroog G,
Iwata KK and Perez-Soler R: Enhanced sensitivity to the
HER1/epidermal growth factor receptor tyrosine kinase inhibitor
erlotinib hydro-chloride in chemotherapy-resistant tumor cell
lines. Clin Cancer Res. 11:1572–1578. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Hilger RA, Scheulen ME and Strumberg D:
The Ras-Raf-MEK-ERK pathway in the treatment of cancer. Onkologie.
25:511–518. 2002.
|
|
58
|
Cheng GZ, Park S, Shu S, He L, Kong W,
Zhang W, Yuan Z, Wang LH and Cheng JQ: Advances of AKT pathway in
human oncogenesis and as a target for anti-cancer drug discovery.
Curr Cancer Drug Targets. 8:2–6. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Sharrard RM and Maitland NJ: Regulation of
protein kinase B activity by PTEN and SHIP2 in human
prostate-derived cell lines. Cell Signal. 19:129–138. 2007.
View Article : Google Scholar
|
|
60
|
Kokubo Y, Gemma A, Noro R, Seike M,
Kataoka K, Matsuda K, Okano T, Minegishi Y, Yoshimura A, Shibuya M
and Kudoh S: Reduction of PTEN protein and loss of epidermal growth
factor receptor gene mutation in lung cancer with natural
resistance to gefitinib (IRESSA). Br J Cancer. 92:1711–1719. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Albitar L, Carter MB, Davies S and Leslie
KK: Consequences of the loss of p53, RB1, and PTEN: Relationship to
gefitinib resistance in endometrial cancer. Gynecol Oncol.
106:94–104. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yamamoto C, Basaki Y, Kawahara A,
Nakashima K, Kage M, Izumi H, Kohno K, Uramoto H, Yasumoto K,
Kuwano M and Ono M: Loss of PTEN expression by blocking nuclear
translocation of EGR1 in gefitinib-resistant lung cancer cells
harboring epidermal growth factor receptor-activating mutations.
Cancer Res. 70:8715–8725. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Friedmann BJ, Caplin M, Savic B, Shah T,
Lord CJ, Ashworth A, Hartley JA and Hochhauser D: Interaction of
the epidermal growth factor receptor and the DNA-dependent protein
kinase pathway following gefitinib treatment. Mol Cancer Ther.
5:209–218. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Friedmann B, Caplin M, Hartley JA and
Hochhauser D: Modulation of DNA repair in vitro after treatment
with chemotherapeutic agents by the epidermal growth factor
receptor inhibitor gefitinib (ZD1839). Clin Cancer Res.
10:6476–6486. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Dittmann K, Mayer C, Fehrenbacher B,
Schaller M, Raju U, Milas L, Chen DJ, Kehlbach R and Rodemann HP:
Radiation-induced epidermal growth factor receptor nuclear import
is linked to activation of DNA-dependent protein kinase. J Biol
Chem. 280:31182–31189. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Dittmann K, Mayer C and Rodemann HP:
Inhibition of radiation-induced EGFR nuclear import by C225
(Cetuximab) suppresses DNA-PK activity. Radiother Oncol.
76:157–161. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Dittmann K, Mayer C, Czemmel S, Huber SM
and Rodemann HP: New roles for nuclear EGFR in regulating the
stability and translation of mRNAs associated with VEGF signaling.
PLoS One. 12:e01890872017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Sok JC, Coppelli FM, Thomas SM, Lango MN,
Xi S, Hunt JL, Freilino ML, Graner MW, Wikstrand CJ, Bigner DD, et
al: Mutant epidermal growth factor receptor (EGFRvIII) contributes
to head and neck cancer growth and resistance to EGFR targeting.
Clin Cancer Res. 12:5064–5073. 2006. View Article : Google Scholar : PubMed/NCBI
|