Globally, lung cancer is the most prevalent type of
cancer, affecting 2.09 million individuals and was responsible for
1.76 million associated deaths in 2018 (1). Smoking is the leading cause of lung
cancer, as 86% of individuals with the disease have a history of
smoking (2). Lung cancer can be
divided into 2 major types, which are small cell lung cancer (SCLC)
and non-small cell lung cancer (NSCLC). NSCLC accounts for >85%
of all lung cancer cases and can be classified into adenocarcinoma
(50%), lung squamous cell carcinoma (SCC) (30-40%) and large cell
carcinomas (10%) (3). NSCLC is
often diagnosed at a late stage, resulting in poor therapeutic
responses and high mortality rates (4,5). The
1-year survival rate for patients with lung cancer is 44%, and the
5-year survival rate is only 17% (6). The treatment options for lung cancer
are surgery, radiotherapy, platinum-based chemotherapy and
neoadjuvant chemotherapy. However, the efficacy of these treatments
for some patients remains unsatisfactory, as lung cancer is a
heterogeneous disease. Moreover, these treatments can have
deleterious side-effects, while patients with metastatic tumors are
vulnerable to developing a post-treatment resistance to such
medications, rendering the treatment of lung cancer difficult
(7). However, improvements in
effectiveness and survival rates have been observed when
histology-guided chemotherapy, maintenance therapy, or vascular
endothelial growth factor (VEGF)-targeted therapy is combined with
platinum doublet therapy, which is the standard therapy for
unresectable and metastatic lung cancer (8,9).
Nowadays, novel therapies targeting molecular aberrations or driver
mutations have emerged as a therapy of choice due to the excellent
effectiveness and lower side-effects, owing to the completely
sequenced human genome allowing the identification of novel
mutations that play a key role in lung carcinogenesis. An example
of such a therapy, which has been tested and has yielded promising
results, is tyrosine kinase therapy, that specifically targets the
epidermal growth factor receptor (EGFR), a type of protein commonly
altered in NSCLC (10). However,
this novel treatment spectrum only includes a minority of patients
that harbor the mutation (11).
Therefore, targeting oncogenic pathways that play a central role in
cancer is a sensible strategy, as it is likely to be effective. One
such pathway that has recently become of interest in cancer therapy
is the Rho-associated kinase signaling pathway (ROCK).
ROCK plays an essential role in carcinogenesis,
particularly in promoting cancer cell motility that causes
metastasis. ROCK is an effector of the small GTPase Rho and has
been studied in various malignancies, such as breast (12), skin (13), liver (14) and lung cancer (15). Studies on lung cancer usually use
NSCLC cell lines or tissue biopsies from patients with NSCLC to
assess changes in the proliferation, migration, and growth of
cancer following the inhibition of knockdown of ROCK (16-21).
These studies have found that ROCK is responsible for promoting
lung cancer growth if upregulated or overexpressed, and this has
led researchers to suggest that ROCK may be a novel target for the
treatment of lung cancer.
In the present review, the role of ROCK in lung
cancer is discussed and the published evidence from in vitro
and in vivo studies that were performed to decode the
function of ROCK in lung cancer is summarized.
A literature search on ROCK and studies on lung
cancer was conducted from January 1, 2020, to November 15, 2020
using Scopus, PubMed and Web of Science, with the following
keywords: 'ROCK OR Rho' OR 'Rho-associated kinase' AND 'Lung
cancer' OR 'NSCLC' OR 'Non-small cell lung cancer' OR 'SCLC' OR
'small-cell lung cancer'. Only original research articles on the
roles of ROCK and ROCK inhibition in studies on lung cancer written
in the English language were selected for reading. The full text of
the articles concerned was retrieved following the screening of the
titles and abstracts.
ROCK is a member of the protein kinase A/protein
kinase G/protein kinase C (AGC) serine/threonine kinases family
that plays an essential role in promoting cell motility by
facilitating cytoskeleton contractility (22,23).
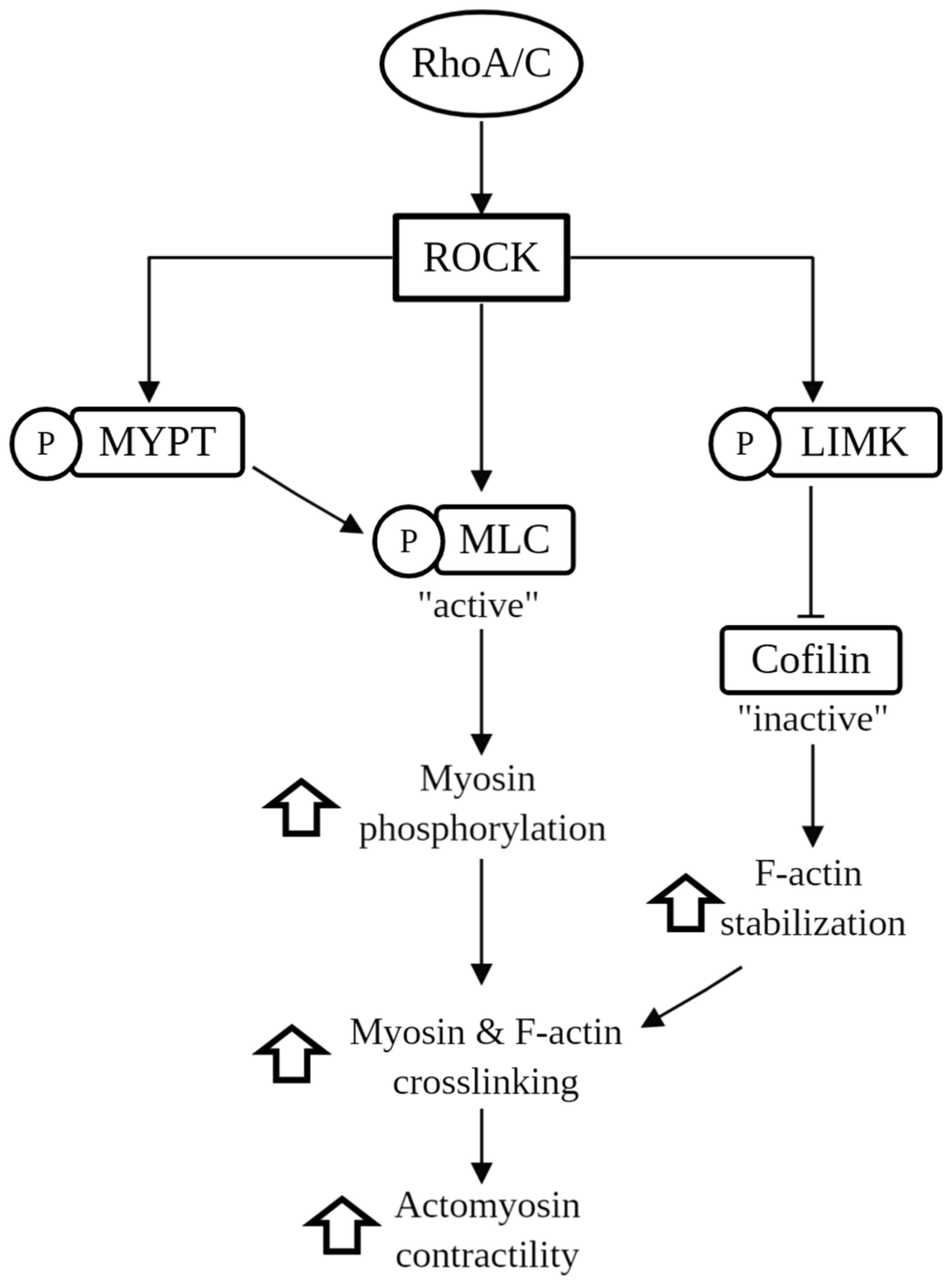
As shown in Fig. 1, ROCK is
activated by binding of the Rho GTPase (i.e., RhoA and RhoC) to its
Rho-binding domain (RBD), which leads to the activation by
phosphorylation of the myosin-binding subunit of the myosin light
chain phosphatase (MYPT) (24),
myosin light chain (MLC) (24) and
LIM kinases (LIMK) (25,26). The activation of the MLC substrate
leads to the activation of myosin II motor activity, leading to the
crosslinking of myosin to filamentous actin (F-actin), enhancing
actomyosin cytoskeleton contractility (27,28).
On the other hand, the activation of MYPT inhibits MLC
dephosphorylation, and the activation of LIMK phosphorylates
cofilin, which renders it inactive and unable to polymerize F-actin
(29). In brief, the
phosphorylation of MLC and MYPT leads to increased levels of
phospho-MLC, and thus promotes actomyosin contractility, which
alters the migratory behavior of cells. Enhanced contractility
resulting from ROCK activation also facilitates cancer cell
proliferation and regulates cell adhesion (28,30-32).
There are 2 ROCK homologs, namely ROCK1 and ROCK2.
Collectively they are referred to as ROCK. ROCK1 and ROCK2 consist
of 1,354 and 1,388 amino acids, respectively, and both contain an
N-terminally located kinase domain, a coiled-coil region followed
by a Rho-binding domain (33). The
homologs share ~65% similarity in their overall amino acid
sequences, and the sequence similarity increases to 92% if only
their kinase domains are compared (22). Although both ROCK proteins share
the same function, which is to regulate cytoskeleton contractility
and exert a redundant effect on MLC and MYPT phosphorylation
(34), they have also been found
to differ from each other in terms of tissue distribution and
subcellular localization. ROCK1 is ubiquitously expressed in
non-neuronal tissues such as the lungs, liver, thymus, stomach,
spleen, kidneys, testes, placenta and embryo (22,28).
ROCK1 has been found at the microtubule-organizing center,
cytoplasm, plasma membrane, and cell-cell adhesion sites (19,35,36).
Moreover, ROCK2 is abundant in the brain, muscle, placenta, lungs
and heart (22,28). It has been found to be localized at
the nuclei and pre-synapses, including active zones (31). Notably, different types of cancer
seem to have various needs for the ROCK proteins, specifically
expressing both ROCK1 and ROCK2, or either alone (37-40).
According to Kümper et al (34), both ROCK isoforms are independently
important to the cells since the cells that lack any of the ROCK
isoforms are still capable of proliferating. However, the cells
that lack both ROCK isoforms are unable to contract and become
flattened out. Eventually, cell division and growth are attenuated.
This evidence has demonstrated the essential role of ROCK in
maintaining cell survival, and the heterogeneity of ROCK expression
in different types of cancer has particularly suggested the need
for the development of a ROCK isoform-specific cancer therapy.
The increased stiffness of the extracellular matrix
(ECM) in a tumor is one of the factors recognized to activate Rho
GTPases, as demonstrated in a 3D culture system with varying levels
of tissue or substrate stiffness (12,52).
The aim of this ROCK activation is to counterbalance the external
force exerted on cells by increasing the contractility of the
internal cytoskeletal structure (53). The increased stiffness of lung
cancer is promoted by the increased production of cancer-associated
fibroblasts and various ECM proteins, such as fibrillary collagen,
fibronectin and tenascin C (54).
Collagen, the main component of the ECM, has been shown to increase
tensile strength in lung cancer and to interact with fibroblasts to
activate ROCK (55). ROCK is
activated by physical changes via the p-integrin receptor, a
transmembrane protein (56), which
phosphorylates focal adhesion kinase (FAK) (57). Activated FAK or p-FAK subsequently
activate the downstream substrates, RhoGTPase and ROCK (58). Apart from increased tissue
stiffness, tumor tissue also develops hypoxic regions with known
partial oxygen pressure in lung cancer of only 16.6 mmHg (59) and it has been demonstrated that
this condition can increase the expression of RhoA/ROCK in lung
cancer (60). Cancer cells in
hypoxic tumors are usually deprived of oxygen, which causes them to
migrate to less hypoxic microenvironments through hypoxia-inducible
factors (HIFs) and to later transactivate RhoA/ROCK. Since ROCK is
a key protein that promotes cell motility, its upregulation in the
hypoxic condition is crucial (61). During the process, cancer cells can
escape the basement barrier and gain access to circulation through
tumor vasculature by intravasation, which is the first step of
metastasis (62). Collectively,
this evidence indicates that both increased tissue stiffness and
the hypoxic environment of a tumor may lead to the ROCK-mediated
malignant transformation of lung cancer, indicating the important
role of these biophysical properties in the promotion of
carcinogenesis.
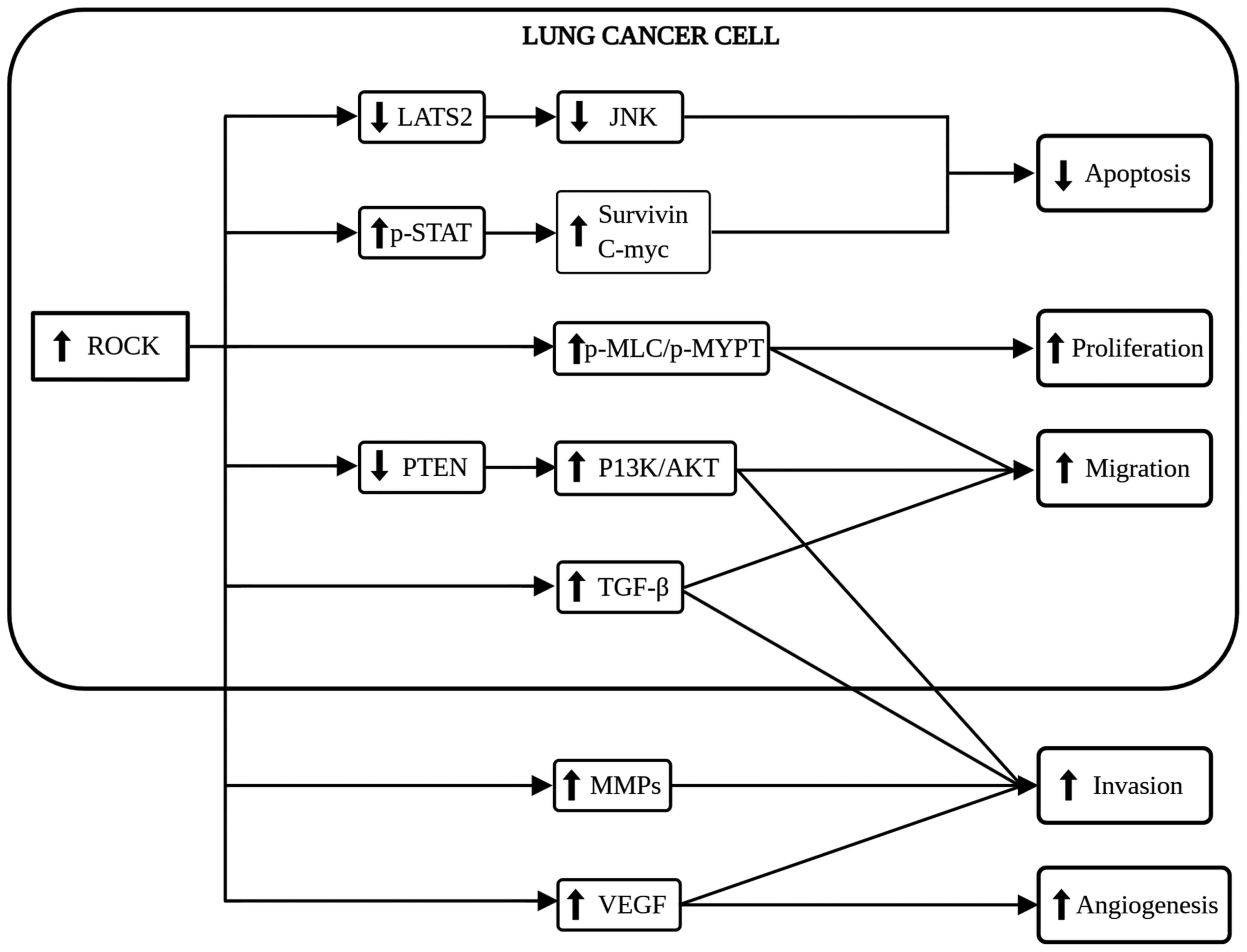
The ROCK signaling pathway plays multiple roles in
lung cancer carcinogenesis, including the suppression of apoptosis
and conferring immortality to cancer cells (30). ROCK activation inhibits caspase-3,
a crucial mediator of apoptosis, which eventually suppresses
MYC-induced apoptosis (63,64).
The reduction of caspase-3 has been suggested to inhibit the cell
cycle, thereby providing a path for NSCLC to bypass senescence
(65). The effect of ROCK on
apoptosis via caspase-3 regulation was confirmed by Yang et
al (66) who reported an
increased level of active caspase-3 in a SCLC cell line following
treatment with fasudil, a ROCK inhibitor. In addition, a previous
study by Xin et al (67)
reported that NSCLC cells treated with small interfering RNA
(siRNA) against ROCK1 or ROCK1 knockdown resulted in apoptosis
induced by the upregulation of the LATS2 and JNK signaling pathway,
suggesting the functional role of ROCK in the regulation of lung
cancer apoptosis. Another mechanism of the suppression of apoptosis
is through the increment of phospho-signal transducer and activator
of transcription 3 (p-STAT3) (64)
and nuclear factor-κB (NF-κB) (68), which both are initially activated
by RhoA (69). STAT3 upregulation
can upregulate its downstream target responsible for the
suppression pf apoptosis, such as c-MYC, cyclin D1 and survivin
(70). However, NF-κB plays a
critical role in desensitizing cells to apoptosis by suppressing
reactive oxygen species (ROS) and antagonizing p53 (68). According to Gu et al
(71), NF-κB expression was found
to be highly expressed and associated with poor survival outcomes
among patients with NSCLC, suggesting the essential role of NF-κB
in driving carcinogenesis. In conclusion, the ROCK signaling
pathway is capable of promoting the survival and growth of lung
cancer cells by suppressing apoptosis and bypassing senescence
through multiple mechanisms.
The regulation of cell proliferation is another
prerequisite for the development of lung cancer other than
apoptosis (72). RhoA/ROCK has
been shown to play an important role in promoting the proliferation
of NSCLC in in vitro (20,73-75)
and in vivo (34) studies.
The majority of these studies used MTT or MTS tetrazolium assays to
measure the level of cell proliferation. In ROCK-activated
fibroblasts, Ras/MAPK increased the expression of multiple
downstream signaling cascades, such as p27, cyclin D1 and cyclin A
(76), which are known to promote
cell cycle progression and cell proliferation (77). Notably, ROCK knockdown results in
the downregulation of cyclin D1 and cyclin E in NSCLC cells
(67), suggesting the functional
role of ROCK in promoting cell proliferation if overexpressed. In
addition, other studies have also attempted to elucidate the role
of ROCK in promoting cancer cell proliferation by using ROCK
inhibitors or knockdown of ROCK. The studies by Liu et al
(64) and Tang et al
(78) demonstrated that the
application of RhoA inhibitor and RhoE/rnd3 (RhoA competitor) was
capable of inhibiting ROCK activation, thus reducing the
proliferation of lung cancer cells. Of note, the study by Kümper
et al (34) using NSCLC
cells and animal models found that the depletion of both ROCKs led
to a cell proliferation defect by affecting MLC and MYPT
phosphorylation. However, the proliferation defect was not observed
in NSCLC cells lacking either ROCK1 or ROCK2, demonstrating the
functional role of both ROCK isoforms in cell proliferation. Thus,
these studies indicated that ROCK activated multiple proteins that
play a key role in promoting cell proliferation during lung cancer
development.
Since cell migration is a pivotal step in
metastasis, it is important to identify molecular pathways that
promote cancer cell motility or migration. Cancer cell migration is
a dynamic process involving several biochemical and morphological
changes. ROCK is the most well-known signaling pathway that
promotes cancer cell migration as it can regulate cytoskeletal
contractility (79). According to
Hu et al (37), ROCK1 has
been reported to enhance NSCLC cell migration by inhibiting
phosphatase and tensin homolog (PTEN) activation, which then
activates the phosphoinositide 3-kinase (PI3K)/AKT and FAK
signaling pathways. This hierarchy of events promotes cell
migration by increasing cytoskeletal contraction and by regulating
cell-cell adhesion. ROCK further promotes the motility of NSCLC
cells through the formation of lamellipodia at the edge of the cell
surface by increasing pFAK colocalization with actin (37) and by cofilin inactivation (80), resulting in plasma membrane
protrusion (81). Cancer cells
employ 2 migration phenotypes that ROCK is capable of performing,
the amoeboid and mesenchymal phenotype. ROCK is the most prominent
signaling pathway that regulates amoeboid migration (82), characterized by losing of cell-cell
and cell-ECM adhesion, eventually forming a bleb. The amoeboid type
of migration uses a bleb-driven mechanism to pass through the holes
in the surrounding 3D network of ECM filaments (82,83).
In comparison, the mesenchymal mode of migration induced by the EMT
process is characterized by the acquisition of mesenchymal
characteristics which causes the loss of intact cell-cell contact
and apical-basal polarity. EMT also mediates cytoskeleton
contractility, which results in a change in cell shape from a
cuboidal to a spindle-like shape that aids cell migration and has
been found to be caused by TGF-p1 released by RhoC and ROCK
activation in lung cancer (19,84-86).
Therefore, ROCK is suggested as a worthy target to decrease cell
migration since it plays an essential role in regulating the 2 main
types of cell migration that aid metastasis.
Concurrently, cancer cells begin to invade their
surroundings as they develop malignant phenotypes, particularly an
enhanced cell motility. Invasion processes include extracellular
matrix remodeling by matrix metalloproteinases (MMPs), which can
degrade the basement membrane and stromal ECM, thereby providing a
'path' for cancer cells to invade (87). In this regard, ROCK has been found
to play an essential role in the invasion of NSCLC cells by
increasing the MMP-2 and MMP-9 expression (17), which are also known to promote
angiogenesis and VEGF production (88). In addition, another family of MMPs,
including MMP-10 and MMP-13, has been found to play a key role in
the invasion of lung cancer by increasing the vascular permeability
that aids in cancer cell intravasation into blood vessels (89). Notably, a high expression of MMPs
has been reported in late-stage and metastatic lung cancer compared
to early-stage and non-metastatic NSCLC (90). Their expression is also directly
associated with the high potential of NSCLC invasion (91). This further supports to the role of
ROCK and MMPs in promoting invasion, which will inevitably lead to
metastasis.
RhoA/ROCK is responsible for the formation of a
vascular structure or angiogenesis necessary for the growth of the
tumor. The actin cytoskeleton regulated by ROCK plays a central
role in the angiogenesis process involving endothelial cells (EC)
proliferation, branching and sprouting (92). The study by Zhang et al
(81) demonstrated that lung
cancer cells that were conditioned with endothelial cells relied on
RhoA/ROCK signaling to invade and metastasizes by angiogenesis.
ROCK is also involved in the formation of a specific type of
vascular pattern in NSCLC known as vasculogenic mimicry (VM), which
preferably forms in the hypoxic environment of the tumor (93,94).
VM is a supplement to endothelial cell-dependent angiogenesis
induced by cancer cells (95).
ROCK-induced VM formation is associated with the expression of the
glycoprotein dimer known as Semaphorin 4D (Sema4D). Sema4D has been
shown to be highly expressed in various types of solid tumors,
including lung cancer (96). With
respect to the pro-angiogenic factor, ROCK has also been reported
to stimulate the secretion of VEGF, which is responsible for
vascular formation from cancer cells and infiltrating immune cells.
This regulation was reported by Zhu et al (17), who found a decreased VEGF
expression in NSCLC cells treated with fasudil. They also reported
decreased cell proliferation, migration, invasion and angiogenesis
in cells with a low VEGF expression, suggesting the essential role
of VEGF in lung cancer carcinogenesis. Similarly, another study by
Zahra et al (97) reported
decreased endothelial cell proliferation, migration and tube
formation that induced angiogenesis following VEGF inhibition by
RhoA knockdown. In summary, ROCK, Sema4D and VEGF are good targets
for anti-angiogenic therapy as they play an essential role in
angiogenesis. In particular, ROCK holds promise for use as a novel
anticancer therapy, as it is capable of regulating a number of
downstream signaling pathways and factors crucial in
carcinogenesis, as shown in Fig.
2.
Since the use of the first approved ROCK inhibitor
(fasudil) for the treatment of cerebral vasospasms (98), immense efforts have been made to
repurpose the therapeutic benefits of this agent for the treatment
of cancer (81,99-101). Apart from fasudil, a wide range
of compounds has also been tested against lung cancer, and as ROCK
overexpression or upregulation is associated with an enhanced
growth of lung cancer as discussed above, the current treatment
modality is to inhibit or reduce ROCK expression. Researchers have
been testing several inhibitors that may target ROCK upstream
effectors or downstream substrates in lung cancer, such as RhoA
inhibitor (64), RhoC inhibitor
(85,102), ROCK pan-inhibitor (Y27632)
(103,104) and LIMK inhibitor (BMS-5)
(105). As these targets are
intertwined in one signaling pathway, the inhibition of RhoA or
LIMK, for example, also yields a preferable result, as with the
inhibition of ROCK. Thus, the present review also includes studies
on ROCK-associated substrate inhibitors instead of only studies on
ROCK inhibition. Novel natural compounds have also been tested for
their inhibitory action against ROCK in lung cancer, with promising
results (46,80). Examples of the natural compounds
studied are zerumbone derived from ginger (80), glabridin from licorice (106), plumbagin from chitrak (46), deguelin from cork bush (107), p-escin from horse chestnut
(108), and XAP from Muruwa
(102). In recent years, natural
compounds have attracted the attention of researchers in developing
novel cancer treatment, since they are believed to exert a less
toxic effect on normal cells (109). p-escin (a RhoA/ROCK inhibitor)
for instance, has been evaluated to not cause significant body
weight loss or histologic cytotoxicity on normal mice after 34
weeks of treatment, which indicates that this natural compound is
unable to jeopardize general health (108). As reviewed by Surien et al
(110), a number of natural
compounds have been proven to be beneficial in the treatment of
lung cancer at pre-clinical studies. From the literature search,
the present review identified 6 different studies that evaluated
ROCK expression following treatment with natural compounds
(46,80,102,106-108). Notably, these natural compounds
have been documented to be capable of reducing RhoA, RhoC and ROCK
expression, all of which promote cell proliferation, migration,
invasion and angiogenesis.
The specificity of ROCK inhibition should also be
considered, as some ROCK inhibitors, such as Y-27632 have
additional off-target inhibitory activity against mitogen- and
stress-activated protein kinase 1 (MSK1) (111). Therefore, a more potent and
specific ROCK inhibitor should be developed to resolve these
issues. Some researchers have begun to investigate this possibility
by developing OXA-06, a potent ATP-competitive ROCK inhibitor that
is structurally distinct from Y-27632, a ROCK1/2 inhibitor
(15) and a Rho-kinase inhibitor
(RKI) (112), both of which have
been tested and proven to exhibit less in vitro off-target
protein kinase inhibitory activity. OXA-06 also has the ability to
block anchorage-independent cancer cell growth by causing cell
cycle arrest in G0/G1 (15). siRNA
and short hairpin RNA (shRNA) have also been used for the
inhibition of ROCK activation in lung cancer studies. These short
double-strand RNA molecules are a promising therapeutic approach
that can be programmed to silence a specific target. Their use,
either in vitro or in vivo, has allowed researchers
to compare the effects of ROCK inhibition with those of their novel
compounds and has enhanced our understanding of the underlying
mechanisms that regulate this pathway. For example, siRNA targeting
either ROCK1 or ROCK2 alone is capable of inhibiting ~90% of NSCLC
anchorage-independent cells. Moreover, non-specific siRNA that
targets both ROCKs effectively inhibits tumor growth as reflected
by near-complete colony formation suppression. These findings
demonstrate that siRNAs can specifically discriminate against ROCK
homologs and give a clear image of their inhibition effects on a
specific ROCK homolog (15).
Moreover, shRNA has been used to target RhoC (85) and ROCK1 (37) in lung cancer studies. These studies
reported that the inhibition of RhoC and ROCK1 by shRNA
significantly suppressed the EMT process and the migration of lung
cancer, as with other ROCK inhibitors.
The use of microRNAs (miRNAs or miRs) for the
treatment of lung cancer has also been documented. miRNAs are
small, non-protein-coding RNA molecules that have been shown to be
involved in carcinogenesis as either tumor suppressors or oncogenes
(113). miRNAs are regarded as
convenient biomarkers due to their better stability compared to
mRNAs. As reviewed by Iqbal et al (114), several miRNAs have been
identified to play a prominent role in lung cancer, and a few have
been identified to target RhoA/ROCK. Throughout the literature,
only 4 studies on miRNAs have been found that elucidate ROCK
expression. These miRNAs have been reported to be downregulated in
NSCLC tissue biopsies and their restoration in vitro has
been reported to reduce the proliferation, migration and invasion
of the A549, H1299 and SPC-A1 cell lines by decreasing ROCK
expression (19,20,115,116). Furthermore, a previous
meta-analysis and Kaplan-Meier data by Yang et al (117) and Wu et al (118) demonstrated that other miRNAs,
such as miRNA-21, miRNA-155, miRNA-19b and miRNA-146a, can be used
to predict recurrence, as a prognostic biomarker and to demonstrate
association with the survival rate of patients with lung
cancer.
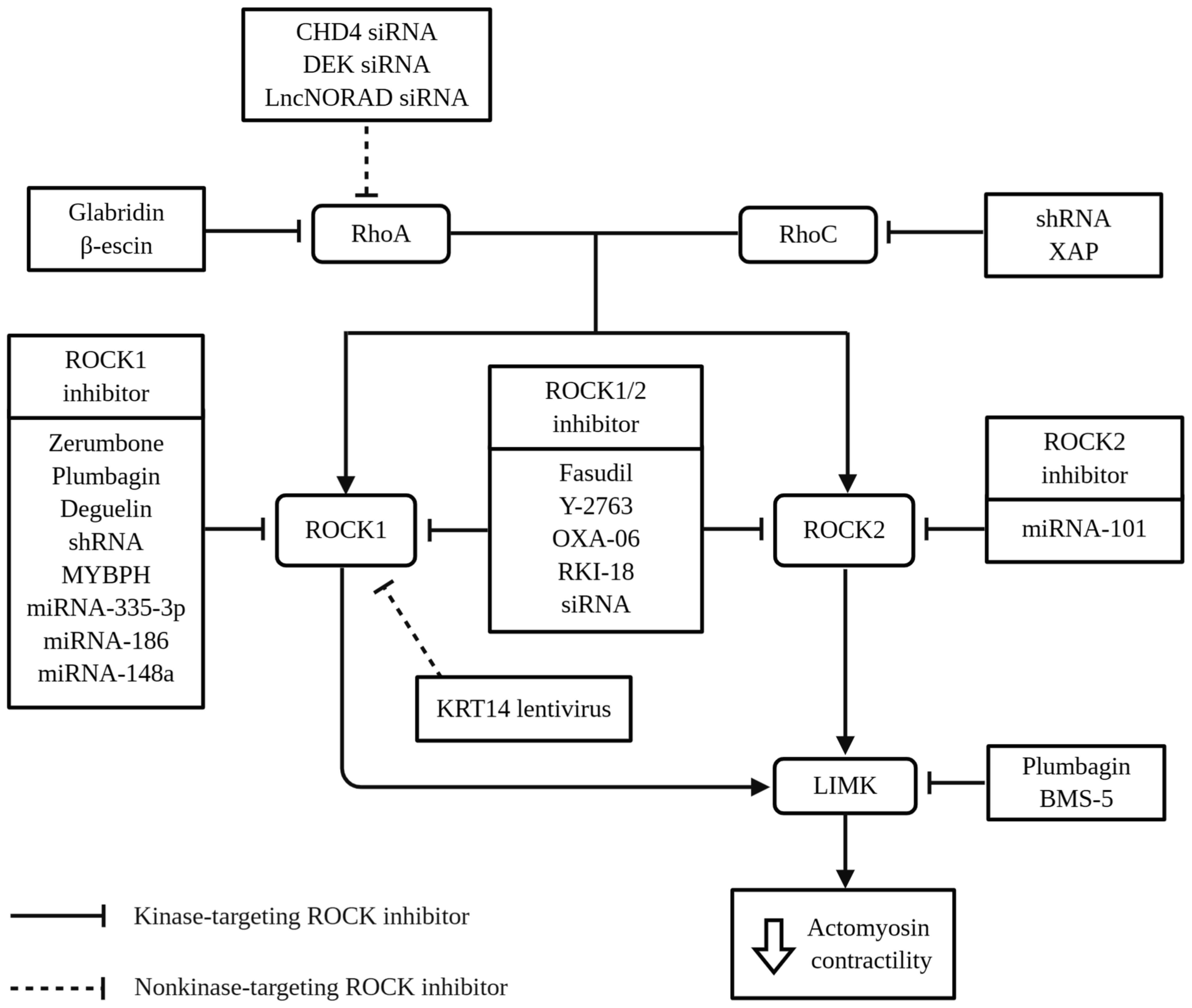
Generally, studies on ROCK and lung cancer in
literature can be divided into 2 major categories: Kinase-targeting
ROCK inhibition (Table I) and
non-kinase-targeting ROCK inhibition (Table II). Kinase-targeting ROCK
inhibitors target upstream effectors, downstream substrates, or
ROCK, whereas non-kinase-targeting ROCK inhibitors target signaling
pathways other than ROCK, such as proteins, oncogenes and
non-coding RNAs, but can still affect ROCK expression. The targets
of the 24 ROCK inhibitors identified in the literature are
presented in Fig. 3. Collectively,
these inhibitors can inhibit MLC/LIMK phosphorylation, which
activates cofilin and promotes F-actin depolymerization. As a
result, crosslinking between F-actin and myosin is reduced, leading
to a decrease in actomyosin contractility, which plays an essential
role in the transformation of malignant phenotype in cancer cells
(119). It was found that the
majority of the kinase-targeting ROCK inhibition studies were
conducted in vitro using NSCLC cell lines, while there is a
lack of studies conducted in vivo or by using tissue
biopsies of lung cancer patients. The most commonly used cell line
is human A459 lung adenocarcinoma cells, followed by H1299 and H460
cells, which are also categorized as NSCLC.
Targeting proteins, oncogenes, or non-coding RNAs
that coordinate Rho may also be a promising approach, as presented
in non-kinase-targeting ROCK inhibition studies (Table II). Generally, these studies have
reported that NSCLC tissue has a higher expression of DEK (73), keratin14 (KRT14) (120), chromodomain helicase DNA-binding
protein 4 (CHD4) (74) and long
non-coding RNA (lncRNA) NORAD (121); the depletion or disruption of
these has been observed to reduce RhoA, ROCK and phospho-myosin
expression in NSCLC cell lines. It has also been demonstrated that
the depletion of these proto-oncogenes is capable of inhibiting
NSCLC cell growth by promoting apoptosis and attenuating their
proliferation, migration and invasion (73,74,120,121). In addition, a high DEK and CHD4
expression has been shown to be associated with poor survival rates
among patients with NSCLC, as analyzed using Kaplan-Meier data, and
to be positively associated with TNM staging, differentiation and
nodal status (73,74). Therefore, it is suggested that DEK
and CHD4 expression may be critical factors and potential
biomarkers of NSCLC progression. Therefore, studies on DEK, CHD4,
KRT14 and lncNORAD expression in NSCLC tissues provide insight into
the possibility of these proteins to be translated into potential
therapeutic targets for treating lung cancer.
Some studies have found inconsistent effectiveness
in treating cancer either by targeting both ROCK isoforms, or only
one of them. Furthermore, the characterization of the role of
ROCK1/2 in lung cancer carcinogenesis is therefore essential.
However, ROCK1 tends to be more significant than ROCK2 in lung
cancer studies (34,37). ROCK1 is also the preferred isoform
in most of the kinase-targeting ROCK inhibition studies, as shown
in Fig. 3. It may be due to the
predominant role of ROCK1 over ROCK2 in regulating cytoskeletal
organization, which indicates isoform-specific regulation in lung
cancer. ROCK1 has been regarded as a positive regulator of cell
migration and invasion in several solid tumors, such as
osteosarcoma (38), breast
(12), pancreatic (122), gastric (123) and lung cancer (37,124,125). In addition, some ROCK inhibitors,
such as Y-27632, which can target both ROCK isoforms, have been
found to be more potent against ROCK1 than against ROCK2 (126). However, this should not be the
reason to discount the role of ROCK2 in lung cancer growth. Further
evidence is needed to demarcate the role of ROCK2 in lung cancer.
Overall, the aim of these ROCK inhibition studies, either
kinase-targeting or non-kinase-targeting ROCK inhibition, is to
determine the effects of ROCK inhibition that elucidates the ROCK
function in lung carcinogenesis. The majority of the studies
included cell migration assays to investigate cellular motility
changes following ROCK inhibition or knockdown, since it is a key
mediator that regulates cytoskeletal rearrangement that affects
cellular motility (37,104,116,125). Examples of the migration assays
employed are Transwell, collagen invasion, tube formation and
scratch wound healing assays. Their findings suggested that the
ROCK signaling pathway should be considered as a potential
therapeutic target for inhibiting lung cancer development, as ROCK
action is diffuse and its activation can promote multiple hallmarks
of cancer.
Identifying the genetic aberrations of lung cancer,
such as KRAS and EGFR mutations is important in tailoring the
appropriate therapy for the disease. NSCLC harboring KRAS mutations
has been found to be more vulnerable to RhoA inhibitor-induced
apoptosis compared to wild-type NSCLC (127). As KRAS mutations are common in
lung cancer, occurring in 30% of adenocarcinoma and 5% of SCC
cases, the inhibition if RhoA appears to be an excellent treatment
option for NSCLC (45,128). Furthermore, the KRAS-mutant NSCLC
cell line is also vulnerable to a combination of drug pairs that
inhibit polo-like kinase 1 (PLK1), a synthetic lethal partner of
Ras oncogene, and ROCK that exhibit marked apoptosis induction and
colony form inhibition (45).
Another common gene aberration that has been studied as a drug
target is EGFR mutations, which occur in 39% of adenocarcinoma and
58% of the SCC subtype of lung cancer (129). EGFR is a tyrosine kinase that
plays an essential role in SCC pathogenesis by dimerizing its
receptor (130). Therefore,
inhibiting EGFR dimerization can inhibit the pathogenesis, and
inhibiting RhoA by using lovastatin has been shown to yield the
same effect as an EGFR inhibitor, as RhoA is necessary for EGFR
localization and activation (131). Inhibiting EGFR activation can
also reduce programmed death-ligand 1 (PD-L1) expression associated
with the p-ERK1/2/p-c-Jun pathway (132). Reducing PD-1 and its interaction
with the ligand is important in restoring the proliferation of
T-cells and promoting the cytotoxic activity of immune cells to
cancer cells (133). Importantly,
an increased PD-L1 expression has been reported to be associated
with EGFR mutations (134,135)
and is currently the only approved biomarker for NSCLC immune
checkpoint inhibitors (136).
Targeting proteins that regulate the ROCK signaling
pathway has also been suggested to yield promising results.
Osteopontin (OPN), a major non-collagenous bone matrix protein, has
been found to be responsible for activating ROCK1, and to
subsequently increase LIMK and cofilin phosphorylation in NSCLC
cell lines, thereby promoting cancer cell migration and invasion
(103). It is suggested that OPN
is a worthy target as its inhibition can significantly reduce the
tumor weight and volume of NSCLC as studied by Cho et al
(137). In addition, OPN can
reduce lamellipodia formation and actin polymerization via the ROCK
signaling pathway following suppression with zerumbone and
plumbagin, respectively (46,80).
Since OPN plays an essential role in mediating tumor-stroma
interaction and contributes to tumor growth and metastasis
(138), targeting OPN may hold
promise for the prevention of lung cancer metastasis. In addition,
caveolin-1 (CAV1), an ECM-associated oncogenic membrane protein
that can activate the ROCK signaling pathway, may also be a
potential druggable target. High levels of stromal CAV1 have been
identified in various types of cancer, including lung cancer
(139,140) and its interactions with
Rho-GTPases have been demonstrated to promote metastasis through
Src, Ras and Erk activation (141). This interaction promotes cell
migration and invasion by regulating CAV1 tyrosine phosphorylation,
which can lead to the regulation of focal adhesion dynamics
(142).
The treatment of lung cancer has improved
substantially over the years, involving various strategies and
modalities, such as surgery, radiotherapy, chemotherapy,
immunotherapy and molecular-targeted therapy (143). The search for a suitable target
candidate to treat lung cancer is still ongoing. Herein, the ROCK
signaling pathway is suggested as one of the potential targets that
can be utilized for the treatment of lung cancer, since its
inhibition has resulted in promising outcomes to reduce cancer cell
proliferation, migration, and invasion in pre-clinical studies.
Repurposing the use of already licensed drugs, such as fasudil for
the treatment of lung cancer is a good start, as it provides a
rapid translation of pre-clinical data into effective therapies for
lung cancer patients. However, the use of fasudil is still
associated with certain drawbacks, such as the off-target effect.
On the other hand, other novel compounds may have insufficient
efficacy apart from the concerns of side-effects and the selective
binding of ROCK inhibitors, as the ROCK signaling pathway also
plays an essential role in normal cell homeostasis. Nevertheless,
the expression of ROCK in cancer is higher if compared to normal
cells or tissues; thus, the use of ROCK inhibitors in cancerous and
normal cells or tissues may yield different outcomes in term of
expression following treatment. Specific pathway inhibition,
limited to cancer cells, is preferable to prevent undesirable
side-effects caused by current chemotherapeutic drugs. Obtaining a
range of effective doses without causing adverse effects in
patients is also another challenge that requires a comprehensive
analysis of safety, efficacy and toxicity to be made. The majority
of the ROCK and lung cancer studies were found to be performed
in vitro only, while there is a lack of in vivo
studies or the use of tissue biopsies from patients with lung
cancer. Therefore, further research should also employ these more
complex settings to fully elucidate the mechanisms of the ROCK
signaling pathway in lung cancer.
To date, to the best of our knowledge, no single
inhibitor targeting the ROCK signaling pathway has been approved
for use in clinical trials against lung cancer. Nevertheless,
defactinib, a drug that can inhibit FAK, a mechanosensor that can
detect changes in the ECM and activate ROCK, has been tested in a
clinical trial for the treatment of KRAS mutant NSCLC. However,
defactinib has been shown to only yield modest and contrasting
results from pre-clinical studies (144). This may be due to the
insufficient efficacy and it being an unspecific target of FAK
inhibitor. Therefore, it is suggested that the targeting of ROCK
should be performed to yield a more profound impact in regulating
cellular phenotypes, as discussed herein, thus treating lung
cancer. A combination of agents in the treatment of lung cancer
should also be considered in future research, as supported by
better outcomes in pre-clinical studies targeting ROCK with EFGR or
PLK1 (45,104). Moreover, an agent that can affect
multiple oncogenic pathways and fine-tuning treatment strategies
based on molecular aberrations can provide more effective treatment
strategies, since cancer is highly adaptive and can acquire
resistance rapidly (145,146). Targeting ROCK can also help to
solve several issues, such as drug resistance seen in hypoxic
tumors. According to Murakami et al (147), gefitinib was found to be
ineffective in hypoxic EGFR mutation-positive NSCLC due to a
vascular inadequacy that dampens the bioavailability of the drug in
the target area. Therefore, targeting ROCK along with gefitinib can
help in restoring the role of the blood vessel to deliver oxygen
and increase the drug bioavailability. Taken together, the ROCK
signaling pathway plays a critical role in the carcinogenesis of
lung cancer, and is therefore suggested as a potential therapeutic
target in the treatment of lung cancer. Further in-depth research
is urgently required to enhance our understanding of this pathway,
and further attempts should be made to elucidate the biological
mechanisms between ROCK and lung cancer.
The present study was supported by the Ministry of
Higher Education (grant no. FRGS/1/2018/SKK06/UKM/03/1).
Not applicable.
MAZ and SFM contributed to the conceptualization,
drafting and writing of the manuscript. SFM, EWC, NFR and GTS
provided substantial contributions to the finalization, correction
and critical reviewing of the manuscript. All authors read and
approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
|
1
|
World Health Organization (WHO): Cancer.
2019.
|
|
2
|
Remen T, Pintos J, Abrahamowicz M and
Siemiatycki J: Risk of lung cancer in relation to various metrics
of smoking history: A case-control study in Montreal. BMC Cancer.
18:12752018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dela Cruz CS, Tanoue LT and Matthay RA:
Lung cancer: Epidemiology, etiology, and prevention. Clin Chest
Med. 32:605–644. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Perez-Moreno P, Brambilla E, Thomas R and
Soria JC: Squamous cell carcinoma of the lung: Molecular subtypes
and therapeutic opportunities. Clin Cancer Res. 18:2443–2451. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gandara DR, Hammerman PS, Sos ML, Lara PN
Jr and Hirsch FR: Squamous cell lung cancer: From tumor genomics to
cancer therapeutics. Clin Cancer Res. 21:2236–2243. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sosa Iglesias V, Giuranno L, Dubois LJ,
Theys J and Vooijs M: Drug resistance in non-small cell lung
cancer: A potential for NOTCH targeting? Front Oncol. 8:2672018.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gentzler RD and Johnson ML: Complex
decisions for first-line and maintenance treatment of advanced
wild-type non-small cell lung cancer. Oncologist. 20:299–306. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Baxevanos P and Mountzios G: Novel
chemotherapy regimens for advanced lung cancer: Have we reached a
plateau? Ann Transl Med. 6:1392018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thomas A, Rajan A and Giaccone G: Tyrosine
kinase inhibitors in lung cancer. Hematol Oncol Clin North Am.
26:589–605. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chung CH: EGFR tyrosine kinase inhibitor
therapy for lung cancer treatments and their clinical outcomes: A
cohort study in Taiwan. Oncol Lett. 18:6090–6100. 2019.PubMed/NCBI
|
|
12
|
Peng Y, Chen Z, Chen Y, Li S, Jiang Y,
Yang H, Wu C, You F, Zheng C, Zhu J, et al: ROCK isoforms
differentially modulate cancer cell motility by mechanosensing the
substrate stiffness. Acta Biomater. 88:86–101. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Masre SF, Rath N, Olson MF and Greenhalgh
DA: ROCK2/rasHa co-operation induces malignant
conversion via p53 loss, elevated NF-k B and tenascin C-associated
rigidity, but p21 inhibits ROCK2/NF-KB-mediated progression.
Oncogene. 36:2529–2542. 2017. View Article : Google Scholar
|
|
14
|
Zheng Y, Xiang L, Chen M and Xiang C:
MicroRNA-130a inhibits the proliferation, migration and invasive
ability of hepatocellular carcinoma cells by downregulating
Rho-kinase 2. Mol Med Rep. 18:3077–3084. 2018.PubMed/NCBI
|
|
15
|
Vigil D, Kim TY, Plachco A, Garton AJ,
Castaldo L, Pachter JA, Dong H, Chen X, Tokar B, Campbell SL and
Der CJ: ROCK1 and ROCK2 are required for non-small cell lung cancer
anchorage-independent growth and invasion. Cancer Res.
72:5338–5347. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang X, Zhang Y, Wang S and Shi W: Effect
of fasudil on growth, adhesion, invasion, and migration of 95D lung
carcinoma cells in vitro. Can J Physiol Pharmacol. 88:874–879.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhu F, Zhang Z, Wu G, Li Z, Zhang R, Ren J
and Nong L: Rho kinase inhibitor fasudil suppresses migration and
invasion though down-regulating the expression of VEGF in lung
cancer cell line A549. Med Oncol. 28:565–571. 2011. View Article : Google Scholar
|
|
18
|
Huo Z, Su Y, Dong Y, Zheng Y, Zhang Q,
Duan Y and Wang G: Rho-kinase inhibition by Fasudil promotes tumor
maturation and apoptosis in small-cell lung cancer. Am J Transl
Res. 12:4354–4370. 2020.PubMed/NCBI
|
|
19
|
Du W, Tang H, Lei Z, Zhu J, Zeng Y, Liu Z
and Huang JA: miR-335-5p inhibits TGF-|31-induced
epithelial-mesenchymal transition in non-small cell lung cancer via
ROCK1. Respir Res. 20:2252019. View Article : Google Scholar
|
|
20
|
Cui G, Cui M, Li Y, Liang Y, Li W, Guo H
and Zhao S: MiR-186 targets ROCK1 to suppress the growth and
metastasis of NSCLC cells. Tumor Biol. 35:8933–8937. 2014.
View Article : Google Scholar
|
|
21
|
Hosono Y, Yamaguchi T, Mizutani E,
Yanagisawa K, Arima C, Tomida S, Shimada Y, Hiraoka M, Kato S,
Yokoi K, et al: MYBPH, a transcriptional target of TTF-1, inhibits
ROCK1, and reduces cell motility and metastasis. EMBO J.
31:481–493. 2012. View Article : Google Scholar :
|
|
22
|
Nakagawa O, Fujisawa K, Ishizaki T, Saito
Y, Nakao K and Narumiya S: ROCK-I and ROCK-II, two isoforms of
Rho-associated coiled-coil forming protein serine/threonine kinase
in mice. FEBS Lett. 392:189–193. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Matsui T, Amano M, Yamamoto T, Chihara K,
Nakafuku M, Ito M, Nakano T, Okawa K, Iwamatasu A and Kaibuchi K:
Rho-associated kinase, a novel serine/threonine kinase, as a
putative target for small GTP binding protein Rho. EMBO J.
15:2208–2216. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Amano M, Ito M, Kimura K, Fukata Y,
Chihara K, Nakano T, Matsuura Y and Kaibuchi K: Phosphorylation and
activation of myosin by Rho-associated kinase (Rho-kinase). J Biol
Chem. 271:20246–20249. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Maekawa M, Ishizaki T, Boku S, Watanabe N,
Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K and Narumiya S:
Signaling from Rho to the actin cytoskeleton through protein
kinases ROCK and LIM-kinase. Science. 285:895–898. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ohashi K, Nagata K, Maekawa M, Ishizaki T,
Narumiya S and Mizuno K: Rho-associated kinase ROCK activates
LIM-kinase 1 by phosphorylation at threonine 508 within the
activation loop. J Biol Chem. 275:3577–3582. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hoon JL, Tan MH and Koh CG: The regulation
of cellular responses to mechanical cues by Rho GTPases. Cells.
5:172016. View Article : Google Scholar :
|
|
28
|
Julian L and Olson MF: Rho-associated
coiled-coil containing kinases (ROCK), structure, regulation, and
functions. Small GTPases. 5:e298462014. View Article : Google Scholar
|
|
29
|
Riento K and Ridley AJ: Rocks:
Multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol.
4:446–456. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rath N and Olson MF: Rho-associated
kinases in tumorigenesis: Re-considering ROCK inhibition for cancer
therapy. EMBO Rep. 13:900–908. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Samuel MS, Lopez JI, McGhee EJ, Croft DR,
Starchan D, Timpson P, Munro J, Schröder E, Zhou J, Brunton VG, et
al: Actomyosin-mediated cellular tension drives increased tissue
stiffness and ß-catenin activation to induce epidermal hyperplasia
and tumor growth. Cancer Cell. 19:776–791. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chow W, Amaya CN, Rains S, Chow M,
Dickerson EB and Bryan BA: Growth attenuation of cutaneous
angiosarcoma with propranolol-mediated ß-blockade. JAMA Dermatol.
151:1226–1229. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ishizaki T, Maekawa M, Fujisawa K, Okawa
K, Iwamatsu A, Fujita A, Watanabe N, Saito Y, Kakizuka A, Morii N
and Narumiya S: The small GTP-binding protein Rho binds to and
activates a 160 kDa Ser/Thr protein kinase homologous to myotonic
dystrophy kinase. EMBO J. 15:1885–1893. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kümper S, Mardakheh FK, McCarthy A, Yeo M,
Stamp GW, Paul A, Worboys J, Sadok A, Jprgensen C, Guichard S and
Marshall CJ: Rho-associated kinase (ROCK) function is essential for
cell cycle progression, senescence and tumorigenesis. Elife.
5:e129942016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chevrier V, Piel M, Collomb N, Saoudi Y,
Frank R, Paintrand M, Narumiya S, Bornens M and Job D: The
Rho-associated protein kinase p160ROCK is required for centrosome
positioning. J Cell Biol. 157:807–817. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Iizuka M, Kimura K, Wang S, Kato K, Amano
M, Kaibuchi K and Mizoguchi A: Distinct distribution and
localization of Rho-kinase in mouse epithelial, muscle and neural
tissues. Cell Struct Funct. 37:155–175. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hu C, Zhou H, Liu Y, Huang J, Liu W, Zhang
Q, Tang Q, Sheng F, Li G and Zhang R: ROCK1 promotes migration and
invasion of non-small-cell lung cancer cells through the
PTEN/PI3K/FAK pathway. Int J Oncol. 55:833–844. 2019.PubMed/NCBI
|
|
38
|
Liu X, Choy E, Hornicek FJ, Yang S, Yang
C, Harmon D, Mankin H and Duan Z: ROCK1 as a potential therapeutic
target in osteosarcoma. J Orthop Res. 29:1259–1266. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vieira GM, Roberto GM, Lira RC, Engel EE,
Tone LG and Brassesco MS: Prognostic value and functional role of
ROCK2 in pediatric Ewing sarcoma. Oncol Lett. 15:2296–2304.
2018.PubMed/NCBI
|
|
40
|
Zakaria MA, Rajab NF, Chua EW, Selvarajah
GT and Masre SF: The roles of tissue rigidity and its underlying
mechanisms in promoting tumor growth. Cancer Invest. 38:445–462.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Amaya CN, Mitchell DC and Bryan BA: Rho
kinase proteins display aberrant upregulation in vascular tumors
and contribute to vascular tumor growth. BMC Cancer. 17:4852017.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Masre SF, Rath N, Olson MF and Greenhalgh
DA: Epidermal ROCK2-induces AKT1/GSK3ß/ß-catenin, NFkB and dermal
tenascin-C; but enhanced differentiation and p53/p21 inhibit
papilloma. Carcinogenesis. 41:1409–1420. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li J, Bharadwaj SS, Guzman G, Vishnubhotla
R and Glover SC: ROCK I has more accurate prognostic value than met
in predicting patient survival in colorectal cancer. Anticancer
Res. 35:3267–3273. 2015.PubMed/NCBI
|
|
44
|
Kumar MS, Hancock DC, Molina-Arcas M,
Steckel M, East P, Diefenbacher M, Armenteros-Monterroso E,
Lassailly F, Matthews N, Nye E, et al: The GATA2 transcriptional
network is requisite for RAS oncogene-driven non-small cell lung
cancer. Cell. 149:642–655. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang J, Hu K, Guo J, Cheng F, Lv J, Jiang
W, Lu W, Liu J, Pang X and Liu M: Suppression of KRas-mutant cancer
through the combined inhibition of KRAS with PLK1 and ROCK. Nat
Commun. 7:113632016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kang CG, Im E, Lee HJ and Lee EO:
Plumbagin reduces osteopontin-induced invasion through inhibiting
the Rho-associated kinase signaling pathway in A549 cells and
suppresses osteopontin-induced lung metastasis in BalB/c mice.
Bioorg Med Chem Lett. 27:1914–1918. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yan SH, Gao HC, Meng HY, Cheng L, Zhe L,
Cao GS, Yan WQ and Xin H: Role of Rock 1 protein in non-small cell
lung cancer. Biomed Res. 28:2530–2534. 2017.
|
|
48
|
Zhang C, Qin S, Qin L, Liu L, Sun W, Li X,
Li N, Wu R and Wang X: Cigarette smoke extract-induced
p120-mediated NF-kB activation in human epithelial cells is
dependent on the RhoA/ROCK pathway. Sci Rep. 6:231312016.
View Article : Google Scholar
|
|
49
|
Duong-Quy S, Dao P, Hua-Huy T, Guilluy C,
Pacaud P and Dinh-Xuan AT: Increased Rho-kinase expression and
activity and pulmonary endothelial dysfunction in smokers with
normal lung function. Eur Respir J. 37:349–355. 2011. View Article : Google Scholar
|
|
50
|
Tan X and Chen M: MYLK and MYL9 expression
in non-small cell lung cancer identified by bioinformatics analysis
of public expression data. Tumor Biol. 35:12189–12200. 2014.
View Article : Google Scholar
|
|
51
|
Zhang D, Zhang JY, Dai SD, Liu SL, Liu Y,
Tang N and Wang EH: Co-expression of delta-catenin and RhoA is
significantly associated with a malignant lung cancer phenotype.
Int J Clin Exp Pathol. 7:3724–3732. 2014.PubMed/NCBI
|
|
52
|
Vishnubhotla R, Bharadwaj S, Sun S,
Metlushko V and Glover SC: Treatment with Y-27632, a ROCK
inhibitor, increases the proinvasive nature of SW620 cells on 3D
collagen type 1 matrix. Int J Cell Biol. 2012:2591422012.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Samuel MS, Rath N, Masre SF, Boyle ST,
Greenhalgh DA, Kochetkova M, Bryson S, Stevenson D and Olson MF:
Tissue-selective expression of a conditionally-active
ROCK2-estrogen receptor fusion protein. Genesis. 54:636–646. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Burgstaller G, Oehrle B, Gerckens M, White
ES, Schiller HB and Eickelberg O: The instructive extracellular
matrix of the lung: Basic composition and alterations in chronic
lung disease. Eur Respir J. 50:16018052017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Xu S, Xu H, Wang W, Li S, Li H, Li T,
Zhang W, Yu X and Liu L: The role of collagen in cancer: From bench
to bedside. J Transl Med. 17:3092019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Galbraith CG, Yamada KM and Sheetz MP: The
relationship between force and focal complex development. J Cell
Biol. 159:695–705. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wozniak MA, Desai R, Solski PA, Der CJ and
Keely PJ: ROCK-generated contractility regulates breast epithelial
cell differentiation in response to the physical properties of a
three-dimensional collagen matrix. J Cell Biol. 163:583–595. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Gkretsi V and Stylianopoulos T: Cell
adhesion and matrix stiffness : Coordinating cancer cell invasion
and metastasis. Front Oncol. 8:1452018. View Article : Google Scholar
|
|
59
|
Le QT, Chen E, Salim A, Cao H, Kong CS,
Whyte R, Donington J, Cannon W, Wakelee H, Tibshirani R, et al: An
evaluation of tumor oxygenation and gene expression in patients
with early stage non-small cell lung cancers. Clin Cancer Res.
12:1507–1514. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kataoka Y, Ohshio Y, Teramoto K, Igarashi
T, Asai T and Hanaoka J: Hypoxia-induced galectin-3 enhances RhoA
function to activate the motility of tumor cells in non-small cell
lung cancer. Oncol Rep. 41:853–862. 2019.
|
|
61
|
Gilkes DM, Xiang L, Lee SJ, Chaturvedi P,
Hubbi ME, Wirtz D and Semenza GL: Hypoxia-inducible factors mediate
coordinated RhoA-ROCK1 expression and signaling in breast cancer
cells. Proc Natl Acad Sci USA. 111:E384–E393. 2014. View Article : Google Scholar
|
|
62
|
Rofstad EK, Gaustad JV, Egeland TA,
Mathiesen B and Galappathi K: Tumors exposed to acute cyclic
hypoxic stress show enhanced angiogenesis, perfusion and metastatic
dissemination. Int J Cancer. 127:1535–1546. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Xue Y, Wu L, Liu Y, Ma Y, Zhang L, Ma X,
Yang Y and Chen J: ENTPD5 induces apoptosis in lung cancer cells
via regulating caspase 3 expression. PLoS One. 10:e01200462015.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Liu D, Mei X, Wang L and Yang X: RhoA
inhibits apoptosis and increases proliferation of cultured SPCA1
lung cancer cells. Mol Med Rep. 15:3963–3968. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Radziszewska A, Schroer SA, Choi D, Tajmir
P, Radulovich N, Ho JC, Wang L, Liadis N, Hakem R, Tsao MS, et al:
Absence of caspase-3 protects pancreatic p-cells from c-Myc-induced
apoptosis without leading to tumor formation. J Biol Chem.
284:10947–10956. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Yang X, Di J, Zhang Y, Zhang S, Lu J, Liu
J and Shi W: The Rho-kinase inhibitor inhibits proliferation and
metastasis of small cell lung cancer. Biomed Pharmacother.
66:221–227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Xin T, Lv W, Liu D, Jing Y and Hu F: ROCK1
knockdown inhibits non-small-cell lung cancer progression by
activating the LATS2-JNK signaling pathway. Aging (Albany NY).
12:12160–12174. 2020. View Article : Google Scholar
|
|
68
|
Chen W, Li Z, Bai L and Lin Y: NF-kappaB
in lung cancer, a carcinogenesis mediator and a prevention and
therapy target. Front Biosci (Landmark Ed). 16:1172–1185. 2011.
View Article : Google Scholar
|
|
69
|
Orgaz JL, Herraiz C and Sanz-Moreno V: Rho
GTPases modulate malignant transformation of tumor cells. Small
GTPases. 5:e290192014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kang JH, Jang YS, Lee HJ, Lee CY, Shin DY
and Oh SH: Inhibition of STAT3 signaling induces apoptosis and
suppresses growth of lung cancer: Good and bad. Lab Anim Res.
35:302019. View Article : Google Scholar
|
|
71
|
Gu L, Wang Z, Zuo J, Li H and Zha L:
Prognostic significance of NF-kB expression in non-small cell lung
cancer: A meta-analysis. PLoS One. 13:e01982232018. View Article : Google Scholar
|
|
72
|
Morgensztern D, Campo MJ, Dahlberg SE,
Doebele RC, Garon E, Gerber DE, Goldberg SB, Hammerman PS, Heist
RS, Hensing T, et al: Molecularly targeted therapies in
non-small-cell lung cancer annual update 2014. J Thorac Oncol. 10(1
Suppl 1): S1–S63. 2015. View Article : Google Scholar :
|
|
73
|
Wang J, Sun L, Yang M, Luo W, Gao Y, Liu
Z, Qiu X and Wang E: DEK depletion negatively regulates
Rho/ROCK/MLC pathway in non-small cell lung cancer. J Histochem
Cytochem. 61:510–521. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Xu N, Liu F, Wu S, Ye M, Ge H, Zhang M,
Song Y, Tong L, Zhou J and Bai C: CHD4 mediates proliferation and
migration of non-small cell lung cancer via the RhoA/ROCK pathway
by regulating PHF5A. BMC Cancer. 20:2622020. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Asnaghi L, Vass WC, Quadri R, Day PM, Qian
X, Braverman R, Papageorge AG and Lowry DR: E-cadherin negatively
regulates neoplastic growth in non-small cell lung cancer: Role of
Rho GTPases. Oncogene. 29:2760–2771. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Croft DR and Olson MF: The Rho GTPase
effector ROCK regulates cyclin A, cyclin D1, and p27Kip1 levels by
distinct mechanisms. Mol Cell Biol. 26:4612–4627. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Street CA and Bryan BA: Rho kinase
proteins-pleiotropic modulators of cell survival and apoptosis.
Anticancer Res. 31:3645–3657. 2011.PubMed/NCBI
|
|
78
|
Tang Y, Hu C, Yang H, Cao L, Li Y, Deng P
and Huang L: Rnd3 regulates lung cancer cell proliferation through
notch signaling. PLoS One. 9:e1118972014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Amano M, Nakayama M and Kaibuchi K:
Rho-Kinase/ROCK: A key regulator of the cytoskeleton and cell
polarity. Cytoskeleton (Hoboken). 67:545–554. 2010. View Article : Google Scholar
|
|
80
|
Kang CG, Lee HJ, Kim SH and Lee EO:
Zerumbone suppresses osteopontin-induced cell invasion through
inhibiting the FAK/AKT/ROCK pathway in human non-small cell lung
cancer a549 cells. J Nat Prod. 79:156–160. 2016. View Article : Google Scholar
|
|
81
|
Zhang Z, Ren JH, Li ZY, Nong L and Wu G:
Fasudil inhibits lung carcinoma-conditioned endothelial cell
viability and migration. Oncol Rep. 27:1561–1566. 2012.PubMed/NCBI
|
|
82
|
Gandalovicova A, Vomastek T, Rosel D and
Brabek J: Cell polarity signaling in the plasticity of cancer cell
invasiveness. Oncotarget. 7:25022–25049. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Charras G and Paluch E: Blebs lead the
way: How to migrate without lamellipodia. Nat Rev Mol cell Biol.
9:730–736. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Mittal V: Epithelial mesenchymal
transition in aggressive lung cancers. Adv Exp Med Biol. 890:37–56.
2016. View Article : Google Scholar
|
|
85
|
Lu X, Guo H, Chen X, Xiao J, Zou Y, Wang W
and Chen Q: Effect of RhoC on the epithelial-mesenchymal transition
process induced by TGF-ß1 in lung adenocarcinoma cells. Oncol Rep.
36:3105–3112. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Zhang Q, Li X, Li X, Li X and Chen Z:
LncRNA H19 promotes epithelial-mesenchymal transition (EMT) by
targeting miR-484 in human lung cancer cells. J Cell Biochem.
119:4447–4457. 2018. View Article : Google Scholar
|
|
87
|
Gialeli C, Theocharis AD and Karamanos NK:
Roles of matrix metalloproteinases in cancer progression and their
pharmacological targeting. FEBS J. 278:16–27. 2011. View Article : Google Scholar
|
|
88
|
Rundhaug JE: Matrix metalloproteinases and
angiogenesis. J Cell Mol Med. 9:267–285. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Huang Y, Song N, Ding Y, Yuan S, Li X, Cai
H, Shi H and Luo Y: Pulmonary vascular destabilization in the
premetastatic phase facilitates lung metastasis. Cancer Res.
69:7529–7537. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
El-Badrawy MK, Yousef AM, Shaalan D and
Elsamanoudy AZ: Matrix metalloproteinase-9 expression in lung
cancer patients and its relation to serum mmp-9 activity,
pathologic type, and prognosis. J Bronchology Interv Pulmonol.
21:327–334. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Laack E, Scheffler A, Burkholder I,
Boeters I, Andritzky B, Schuch G, Görn M, Vohwinkel G, Edler L,
Fiedler W and Hossfeld DK: Pretreatment vascular endothelial growth
factor (VEGF) and matrix metalloproteinase-9 (MMP-9) serum levels
in patients with metastatic non-small cell lung cancer (NSCLC).
Lung Cancer. 50:51–58. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Chang YW, Bean RR and Jakobi R: Targeting
RhoA/Rho kinase and p21-activated kinase signaling to prevent
cancer development and progression. Recent Pat Anticancer Drug
Discov. 4:110–124. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Paulis YW, Soetekouw PM, Verheul HM,
Tjan-Heijnen VC and Griffioen AW: Signalling pathways in
vasculogenic mimicry. Biochim Biophys Acta. 1806:18–28.
2010.PubMed/NCBI
|
|
94
|
Xia Y, Cai XY, Fan JQ, Zhang LL, Ren JH,
Li ZY, Zhang RG, Zhu F and Wu G: The role of sema4D in vasculogenic
mimicry formation in non-small cell lung cancer and the underlying
mechanisms. Int J Cancer. 144:2227–2238. 2019. View Article : Google Scholar
|
|
95
|
Maniotis AJ, Folberg R, Hess A, Seftor EA,
Gardner LM, Pe'er J, Trent JM, Meltzer PS and Hendrix MJ: Vascular
channel formation by human melanoma cells in vivo and in vitro:
Vasculogenic mimicry. Am J Pathol. 155:739–752. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Lontos K, Adamik J, Tsagianni A, Galson
DL, Chirgwin JM and Suvannasankha A: The role of semaphorin 4D in
bone remodeling and cancer metastasis. Front Endocrinol (Lausanne).
9:3222018. View Article : Google Scholar
|
|
97
|
Zahra FT, Sajib MS, Ichiyama Y, Akwii RG,
Tullar PE, Cobos C, Minchew SA, Doci CL, Zheng Y, Kubota Y, et al:
Endothelial RhoA GTPase is essential for in vitro endothelial
functions but dispensable for physiological in vivo angiogenesis.
Sci Rep. 9:116662019. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Shibuya M, Suzuki Y, Sugita K, Saito I,
Sasaki T, Takakura K, Nagata I, Kikuchi H, Takemae T, Hidaka H, et
al: Effect of AT877 on cerebral vasospasm after aneurysmal
subarachnoid hemorrhage: Results of a prospective
placebo-controlled double-blind trial. J Neurosurg. 76:571–577.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Yang X, Liu Y, Zong Z and Tian D: The Rho
kinase inhibitor fasudil inhibits the migratory behaviour of 95-D
lung carcinoma cells. Biomed Pharmacother. 64:58–62. 2010.
View Article : Google Scholar
|
|
100
|
Miyamoto C, Maehata Y, Motohashi K, Ozawa
S, Ikoma T, Hidaka K, Wada-Takahashi S, Takahashi SS, Yoshino F,
Yoshida A, et al: Fasudil, a Rho kinase inhibitor, suppresses tumor
growth by inducing CXCL14/BRAK in head and neck squamous cell
carcinoma. Biomed Res. 35:381–388. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Xia Y, Cai XY, Fan JQ, Zhang LL, Ren JH,
Chen J, Li ZY, Zhang RG, Zhu F and Wu G: Rho kinase inhibitor
fasudil suppresses the vasculogenic mimicry of B16 mouse melanoma
cells both in vitro and in vivo. Mol Cancer Ther. 14:1582–1590.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Lin SS, Li FF, Sun L, Fan W, Gu M, Zhang
LY, Qin S and Yuan ST: Marsdenia tenacissima extract suppresses
A549 cell migration through regulation of CCR5-CCL5 axis, Rho C,
and phosphorylated FAK. Chin J Nat Med. 14:203–209. 2016.PubMed/NCBI
|
|
103
|
Kang CG, Han HJ, Lee HJ, Kim SH and Lee
EO: Rho-associated kinase signaling is required for
osteopontin-induced cell invasion through inactivating cofilin in
human non-small cell lung cancer cell lines. Bioorganic Med Chem
Lett. 25:1956–1960. 2015. View Article : Google Scholar
|
|
104
|
Umelo IA, Wever OD, Kronenberger P, Noor
A, Teugels E, Chen G, Bracke M and Grève JD: Combined inhibition of
rho-associated protein kinase and EGFR suppresses the invasive
phenotype in EGFR-dependent lung cancer cells. Lung Cancer.
90:167–174. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Mardilovich K, Baugh M, Crighton D,
Kowalczyk D, Gabrielsen M, Munro J, Croft DR, Lourenco F, James D,
Kalna G, et al: LIM kinase inhibitors disrupt mitotic microtubule
organization and impair tumor cell proliferation. Oncotarget.
6:38469–38486. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Tsai YM, Yang CJ, Hsu YL, Wu LY, Tsai YC,
Hung JY, Lien CT, Huang MS and Kuo PL: Glabridin inhibits
migration, invasion, and angiogenesis of human non-small cell lung
cancer A549 cells by inhibiting the FAK/Rho signaling pathway.
Integr Cancer Ther. 10:341–349. 2011. View Article : Google Scholar
|
|
107
|
Zhao H, Jiao Y and Zhang Z: Deguelin
inhibits the migration and invasion of lung cancer A549 and H460
cells via regulating actin cytoskeleton rearrangement. Int J Clin
Exp Pathol. 8:15582–15590. 2015.
|
|
108
|
Patlolla JM, Qian L, Biddick L, Zhang Y,
Desai D, Amin S, Lightfoot S and Rao CV: ß-Escin inhibits
NNK-induced lung adenocarcinoma and ALDH1A1 and RhoA/Rock
expression in A/J mice and growth of H460 human lung cancer cells.
Cancer Prev Res (Phila). 6:1140–1149. 2013. View Article : Google Scholar
|
|
109
|
Aung TN, Qu Z, Kortschak RD and Adelson
DL: Understanding the effectiveness of natural compound mixtures in
cancer through their molecular mode of action. Int J Mol Sci.
18:6562017. View Article : Google Scholar :
|
|
110
|
Surien O, Ghazali AR and Masre SF: Lung
cancers and the roles of natural compounds as potential
chemotherapeutic and chemopreventive agents. Biomed Pharmacol J.
12:85–98. 2019. View Article : Google Scholar
|
|
111
|
Davies SP, Reddy H, Caivano M and Cohen P:
Specificity and mechanism of action of some commonly used protein
kinase inhibitors Stephen. Biochem J. 351:95–105. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Patel RA, Liu Y, Wang B, Li R and Sebti
SM: Identification of novel ROCK inhibitors with anti-migratory and
anti-invasive activities. Oncogene. 33:550–555. 2014. View Article : Google Scholar :
|
|
113
|
Zhang B, Pan X, Cobb GP and Anderson TA:
MicroRNAs as oncogenes and tumor suppressors. Dev Biol. 302:1–12.
2007. View Article : Google Scholar
|
|
114
|
Iqbal MA, Arora S, Prakasam G, Calin GA
and Syed MA: MicroRNA in lung cancer: Role, mechanisms, pathways
and therapeutic relevance. Mol Aspects Med. 70:3–20. 2019.
View Article : Google Scholar
|
|
115
|
Ye Z, Yin S, Su Z, Bai M, Zhang H, Hei Z
and Cai S: Downregulation of miR-101 contributes to
epithelial-mesenchymal transition in cisplatin resistance of NSCLC
cells by targeting ROCK2. Oncotarget. 7:37524–37535. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Li J, Song Y, Wang Y, Luo J and Yu W:
MicroRNA-148a suppresses epithelial-to-mesenchymal transition by
targeting ROCK1 in non-small cell lung cancer cells. Mol Cell
Biochem. 380:277–282. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Yang M, Shen H, Qiu C, Ni Y, Wang L, Dong
W, Liao Y and Du J: High expression of miR-21 and miR-155 predicts
recurrence and unfavourable survival in non-small cell lung cancer.
Eur J Cancer. 49:604–615. 2013. View Article : Google Scholar
|
|
118
|
Wu C, Cao Y, He Z, He J, Hu C, Duan H and
Jiang J: Serum levels of miR-19b and miR-146a as prognostic
biomarkers for non-small cell lung cancer. Tohoku J Exp Med.
232:85–95. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Rodriguez-Hernandez I, Cantelli G, Bruce F
and Sanz-Moreno V: Rho, ROCK and actomyosin contractility in
metastasis as drug targets. F1000Res. 5:F1000 Faculty Rev-783.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Wang X, Han L, Shan S, Sun Y and Mao Y:
KRT14 promoting invasion and migration of lung cancer cells through
ROCK-1 signaling pathway. Int J Clin Exp Pathol. 10:795–803.
2017.
|
|
121
|
Wu Y, Shen QW, Niu YX, Chen XY, Liu HW and
Shen XY: LncNORAD interference inhibits tumor growth and lung
cancer cell proliferation, invasion and migration by
down-regulating CXCR4 to suppress RhoA/ROCK signaling pathway. Eur
Rev Med Pharmacol Sci. 24:5446–5455. 2020.PubMed/NCBI
|
|
122
|
Whatcott CJ, Ng S, Barrett MT, Hostetter
G, Von Hoff DD and Han H: Inhibition of ROCK1 kinase modulates both
tumor cells and stromal fibroblasts in pancreatic cancer. PLoS One.
12:e01838712017. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Shin JY, Kim Y Il, Cho SJ, Lee MK, Kook
MC, Lee JH, Lee SS, Ashktorab H, Smoot DT, Ryu KW, et al: MicroRNA
135a suppresses lymph node metastasis through down-regulation of
ROCK1 in early gastric cancer. PLoS One. 9:e852052014. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Zhang R, Li G, Zhang Q, Tang Q, Huang J,
Hu C, Liu Y, Wang Q, Liu W, Gao N and Zhou S: Hirsutine induces
mPTP-dependent apoptosis through ROCK1/PTEN/PI3K/GSK3 ß pathway in
human lung cancer cells. Cell Death Dis. 9:5982018. View Article : Google Scholar
|
|
125
|
Lin L, Li M, Lin L, Xu X, Jiang G and Wu
L: FPPS mediates TGF-ß1-induced non-small cell lung cancer cell
invasion and the EMT process via the RhoA/Rock1 pathway. Biochem
Biophys Res Commun. 496:536–541. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Ishizaki T, Uehata M, Tamechika I, Keel J,
Nonomura K, Maekawa M and Narumiya S: Pharmacological properties of
Y-27632, a specific inhibitor of rho-associated kinases. Mol
Pharmacol. 57:976–983. 2000.PubMed/NCBI
|
|
127
|
Konstantinidou G, Ramadori G, Torti F,
Kangasniemi K, Ramirez RE, Cai Y, Behrens C, Dellinger MT, Brekken
RA, Wistuba II, et al: RHOA-FAK is a required signaling axis for
the maintenance of KRAS-driven lung adenocarcinomas. Cancer Discov.
3:444–457. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Tomasini P, Walia P, Labbe C, Jao K and
Leighl NB: Targeting the KRAS pathway in non-small cell lung
cancer. Oncologist. 21:1450–1460. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Korpanty GJ, Graham DM, Vincent MD and
Leighl NB: Biomarkers that currently effect clinical practice in
lung cancer: EGFR, ALK, MET, ROS-1 and KRAS. Front Oncol.
4:2042014. View Article : Google Scholar
|
|
130
|
Nicholson RI, Gee JM and Harper ME: EGFR
and cancer prognosis. Eur J Cancer. 37(Suppl 4): S9–S15. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Zhao TT, Le Francois BG, Goss G, Ding K,
Bradbury PA and Dimitroulakos J: Lovastatin inhibits EGFR
dimerization and AKT activation in squamous cell carcinoma cells:
Potential regulation by targeting rho proteins. Oncogene.
29:4682–4692. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Chen N, Fang W, Zhan J, Hong S, Tang Y,
Kang S, Zhang Y, He X, Zhou T, Qin T, et al: Upregulation of PD-L1
by EGFR activation mediates the immune escape in EGFR-driven NSCLC:
Implication for optional immune targeted therapy for NSCLC patients
with egfr mutation. J Thorac Oncol. 10:910–923. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Zitvogel L and Kroemer G: Targeting
PD-1/PD-L1 interactions for cancer immunotherapy. Oncoimmunology.
1:1223–1225. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Azuma K, Ota K, Kawahara A, Hattori S,
Iwama E, Harada T, Matsumoto K, Takayama K, Takamori S, Kage M, et
al: Association of PD-L1 overexpression with activating EGFR
mutations in surgically resected nonsmall-cell lung cancer. Ann
Oncol. 25:1935–1940. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Zhang M, Li G and Wang Y and Wang Y, Zhao
S, Haihong P, Zhao H and Wang Y: PD-L1 Expression in lung cancer
and its correlation with driver mutations: A meta-analysis. Sci
Rep. 7:102552017. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Kim H and Chung JH: PD-L1 testing in
non-small cell lung cancer: Past, present, and future. J Pathol
Transl Med. 53:199–206. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Cho WY, Hong SH, Singh B, Islam MA, Lee S,
Lee AY, Gankhuyag N, Kim JE, Yu KN, Kim KH, et al: Suppression of
tumor growth in lung cancer xenograft model mice by
poly(sorbitol-co-PEI)-mediated delivery of osteopontin siRNA. Eur J
Pharm Biopharm. 94:450–462. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Zhao H, Chen Q, Alam A, Cui J, Suen KC,
Soo AP, Eguchi S, Gu J and Ma D: The role of osteopontin in the
progression of solid organ tumour. Cell Death Dis. 9:3562018.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Nwosu ZC, Ebert MP, Dooley S and Meyer C:
Caveolin-1 in the regulation of cell metabolism: A cancer
perspective. Mol Cancer. 15:712016. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Joshi B, Strugnell SS, Goetz JG, Kojic LD,
Cox ME, Griffith OL, Chan SK, Jones SJ, Leung SP, Masoudi H, et al:
Phosphorylated caveolin-1 regulates Rho/ROCK-dependent focal
adhesion dynamics and tumor cell migration and invasion. Cancer
Res. 68:8210–8220. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Arpaia E, Blaser H, Quintela-Fandino M,
Duncan G, Leong HS, Ablack A, Nambiar SC, Lind EF, Silvester J,
Fleming CK, et al: The interaction between caveolin-1 and
Rho-GTPases promotes metastasis by controlling the expression of
alpha5-integrin and the activation of Src, Ras and Erk. Oncogene.
31:884–896. 2012. View Article : Google Scholar :
|
|
142
|
Urra H, Torres VA, Ortiz RJ, Lobos L, Diaz
MI, Diaz N, Härtel S, Leyton L and Quest AF: Caveolin-1-enhanced
motility and focal adhesion turnover require Tyrosine-14 but not
accumulation to the rear in metastatic cancer cells. PLoS One.
7:e330852012. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Reck M and Rabe KF: Precision diagnosis
and treatment for advanced non-small-cell lung cancer. N Engl J
Med. 377:849–861. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Gerber DE, Camidge DR, Morgensztern D,
Cetnar J, Kelly RJ, Ramalingam SS, Spigel DR, Jeong W, Scaglioni
PP, Zhang S, et al: Phase 2 study of the focal adhesion kinase
inhibitor defactinib (VS-6063) in previously treated advanced KRAS
mutant non-small cell lung cancer. Lung Cancer. 139:60–67. 2020.
View Article : Google Scholar :
|
|
145
|
Hanahan D: Rethinking the war on cancer.
Lancet. 383:558–563. 2014. View Article : Google Scholar
|
|
146
|
Malone ER, Oliva M, Sabatini PJB, Stockley
TL and Siu LL: Molecular profiling for precision cancer therapies.
Genome Med. 12:82020. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Murakami A, Takahashi F, Nurwidya F,
Kobayashi I, Minakata K, Hashimoto M, Nara T, Kato M, Tajima K,
Shimada N, et al: Hypoxia increases gefitinib-resistant lung cancer
stem cells through the activation of insulin-like growth factor 1
receptor. PLoS One. 9:e864592014. View Article : Google Scholar : PubMed/NCBI
|