Acute myeloid leukemia (AML) is the most common type
of adult leukemia, with a wide range of biological and clinical
characteristics (1). A total of
19,520 new cases of AML were reported in the US in 2018 (2), and 14,100 cases were reported in
China in 2015, according to survey data (3). Genetic and epigenetic abnormalities
have been identified to play key roles in the pathogenesis of AML
(4,5).
Epigenomics, which refers to the epigenetic changes
that modify the expression of a genotype into a particular
phenotype without any alteration of the genetic material, play key
roles in mammalian growth and maturation (6). Canonical epigenetics research had
previously focused on the modifications and variations of DNA in
chromatin, whereas epigenetic modifications of RNA, particularly
those involving non-coding RNAs, have been attracting increasing
attention recently. With the advancement of RNA deep sequencing
technologies and bioinformatics approaches, circular (circ)RNAs
have become increasingly significant among RNA species. Distinct
from linear RNAs, circRNAs have loop structures that are covalently
closed and lack 5′ caps and 3′ poly(A) tails due to back-splicing
(7). Due to their stability
(8), evolutionary conservatism
(9) and abundance (10), circRNAs act as microRNA
(miRNA/miR) sponges (4,11), RNA splicing factors (12) and parental gene expression
modulators (13). In addition,
circRNAs have been detected to serve as biomarkers for a wide range
of diseases, including gastric and hepatocellular cancers (14). Furthermore, studies have shown
that circRNAs are N6-methyladenosine (m6A)
methylated (15,16), and methyltransferase-like
(METTL)3/14 promotes their translation, whereas fat mass and
obesity-associated (FTO) gene inhibits their translation (15). Both circRNAs and m6A
participate in RNA processing, and both are associated with AML.
Therefore, the aim of the present review is to report the role of
canonical epigenetic effects in AML, summarize the progress of RNA
epigenetics and circRNAs, and propose a possible link between AML
and circRNA epigenetic modifications.
Epigenetic modifications are associated with
numerous important biological processes and serve key roles in the
development of an organism. Through epigenetic modifications, cells
that bear a similar genome can differentiate into various cell
types with different functions (17). The treatment of hematological
malignancies, including AML, is challenging. Hence, studies on the
association between AML and epigenetics may contribute to
elucidating the pathogenesis of this disease. The conventional
epigenetic processes include histone modification, chromatin
remodeling and DNA methylation. In this section, the role of these
epigenetic processes in AML pathogenesis is examined.
Another important group of epigenetic regulators
involved in hematopoietic development is the ten-eleven
translocation (TET) protein family. TET1 is commonly expressed in
embryonic stem cells, whereas TET2 and TET3 are found in most adult
tissues (38). TET2 is the most
commonly expressed of the three TET family members in the
hematopoietic lineage, and it is frequently mutated in
hematological malignancies. Tet2 knockout mice developed
splenomegaly, monocytosis and extramedullary hematopoiesis as a
result of bone marrow defects with enlargement of the HSC
compartment (39). HSCs with Tet2
deletion exhibited increased self-renewal capacity, allowing them
to outcompete wild-type counterparts and predominate in the
transplanted mice's peripheral blood (40). Furthermore, Tet2-/-HSCs showed a
transcriptional program similar to that of common myeloid
progenitors, but with enhanced expression of self-renewal
regulators Meis1 and Evi1, and decreased expression of
myeloid-specific factors Cebpa, Mpo and Csf1 (40). These findings suggested that TET2
is vital for HSC self-renewal and differentiation into the myeloid
lineage (39,40).
TET2 is commonly found to be aberrantly expressed in
AML, myelodysplastic syndromes/myeloproliferative neoplasms and
chronic myelomonocytic leukemia (41,42). Approximately 17% of patients with
AML have loss-of-function mutations of TET2 (43). TET2 mutations can predispose HSCs
to a pre-leukemic state, in which they retain the ability to
differentiate to a wide range of mature blood cells. However, after
acquiring additional genetic lesions, these pre-leukemic stem cells
may transform into leukemia-initiating cells (44,45). This suggests that while TET2
mutations can promote leukemic transformation, they are
insufficient for completing the process. TET2 mutations frequently
co-occur with other mutations in KRAS, CCAAT enhancer-binding
protein α, AML1, nucleophosmin 1, FMS-like tyrosine kinase 3 (FLT3)
and Janus kinase 2 in AML (46),
suggesting that TET2 inactivation works in tandem with these other
mutations to drive leukemogenesis. The findings that the
synergistic action of TET2 depletion and FLT3-internal tandem
duplication (ITD) mutation dysregulates DNA methylation and
interferes with normal hematopoietic cell differentiation, leading
to HSPC and granulocyte-monocyte progenitor accumulation (47), further substantiates this
hypothesis. Several hypermethylated regions of TET2 and FLT3-ITD
mutations are located at gene regulatory elements, triggering the
deregulation of self-renewal and differentiation genes (Gata1,
Gata2, inhibitor of differentiation 1, myeloproliferative leukemia
virus l and suppressor of cytokine signaling 2) (47). Furthermore, knocking out TET2 in
pre-leukemic cells with AML1-ETO yielded genome-wide DNA
hypermethylation, affecting ~25% of enhancer elements (48). As several hypermethylated
enhancers are linked to tumor suppressor genes, this suggests that
TET2 mutations play a role in leukemia development through an
epigenetic mechanism.
The structural unit of chromatin is a nucleosome
consisting of one H1, two H2A and H2B dimers, and one H3/H4
tetramer (49). Histone
modification, which is a set of covalent post-translational
modifications of histone proteins and modifications that commonly
involve acetylation, methylation, phosphorylation, sumoylation,
ubiquitination and ADP-ribosylation (49), has been shown to play a role in
stem cell differentiation (50).
For example, class I and II histone deacetylases (HDACs) that
contain the two catalytic domains, function as the mammalian
regulators of histone acetylation (50,51).
DNA methylation and histone modification are
significant epigenetic mechanisms for gene expression. DNA
hypermethylation in the promoter CGIs of tumor suppressor genes
that trigger transcriptional silencing is considered to be
essential in carcinogenesis (52-54). Histone proteins are assembled into
nucleosomes that act as both transcriptional regulators and DNA
packaging units. The histone amino-terminal tails protrude from the
nucleosome and are subject to chemical modifications, such as
acetylation, phosphorylation and methylation (55). Modifications to the
post-translational histone tail, added or removed by
histone-modifying proteins (HMPs), serve to control access to the
underlying DNA and alter gene expression by affecting the structure
of chromatin. It has been shown that altered HMP activity
contributes to leukemogenesis in AML via gene transcription
regulation and, since modifications of post-translational histones
are reversible, they may be considered as possible therapeutic
targets (56). In addition,
removal of the H3K4 methyl group via lysine-specific histone
demethylase 1A resulted in decreased expression of the tumor
suppressor gene. Similarly, the aberrant recruitment of HDACs to
promoters of hematopoietic genes was found in AML (56).
Chromatin remodeling is the chromatin architectural
modification that controls transcription through nucleosome
displacement and rearrangement. The chromatin remodeling mechanism
is powered by ATP (57), and
chromatin remodeling complexes comprise four main classes as
follows: Imitation SWI, switch/sucrose non-fermentable, INO80
complex ATPase subunit and chromodomain-helicase-DNA-binding
protein Mi-2 homolog (Mi2/CHDD) (58,59). Chromatin remodeling is fundamental
to transcription. Redner et al (60) outlined models of the normal
control of chromatin remodeling during gene-specific transcription,
and concluded that disruption of these mechanisms may lead to
transcriptional disorders and leukemic transformation. They further
suggested that chromatin therapy may emerge as a potential
antileukemic strategy in the future. In addition, chromatin
remodeler inhibition was reported to reduce the development of AML
and sensitize AML cells to genotoxic drugs through increased DNA
accessibility and impaired double-strand break repair (61).
The chromodomain-helicase-DNA-binding protein 4
(CHD4), an ATP-dependent chromatin remodeling factor, is part of
the nucleosome remodeling and histone deacetylation nucleosome
remodeling deacetylase complex and plays an important role in the
regulation of epigenetic transcriptional genes (62). CHD4 has been associated with
oncogenic processes, including cell cycle progression regulation
(63-65), cancer metastasis,
epithelial-to-mesenchymal transition, and epigenetic repression of
tumor suppressor genes (66).
Heshmati et al (67)
indicated that CHD4 is important for the proliferation of different
types of leukemic cells and AML development in vivo, but not
for normal primary hematopoietic cell proliferation and survival.
It was also confirmed that CHD4 was previously shown to be
important for the proliferation of a broad range of cancer cells
(67), as well as the capacity of
AML cells to form colonies (61),
suggesting that CHD4 may represent a cancer-specific dependency in
a wider tumor repertoire. In another study, the activity of
chromodomain-helicase DNA-binding protein-7 (CHD7), an
ATP-dependent chromatin remodeling factor, was found to interact
with the AML1/CBFβ-SMMHC complex and altering the expression of its
target genes. Chd7 deficiency in Chd7f/fMx1-CreCbfb+/56M mice
expressing the Cbfb-MYH11 fusion gene delayed Cbfb-MYH11-induced
leukemia in both primary and transplanted mice (68).
One mechanism via which miRNA dysregulation causes
AML is epigenetic alterations by altered expression of
transcription factors or oncogenic fusion proteins. Of note, the
expression of AML1-ETO causes heterochromatic silencing of genomic
regions that produce miR-223 by recruiting chromatin remodeling
enzymes at the (Runt-related transcription factor 1) RUNX1-binding
site of the pre-miR-223 gene (69). Furthermore, AML1-ETO induces
heterochromatic silencing at the RUNX1-binding sites of miR-193a by
recruiting chromatin remodeling enzymes and expanding the oncogenic
function of the fusion protein (70). Taken together, these data
demonstrated that chromatin remodeling may be crucial for
leukemogenesis, including AML, and may influence its pathogenesis
to a certain extent.
Epigenomics involves stable and inheritable gene
expression variations without changes to the sequence of DNA
(71). However, epigenetic
changes occur in DNA as well as in RNA, termed the
epitranscriptome; >100 forms of RNA modifications are involved
in the epitranscriptome (72),
and previous studies have identified RNA modifications mostly in
transfer (t)RNAs, ribosomal (r)RNAs and small nuclear (sn)RNAs,
whereas they are relatively infrequent in mRNAs (72,73). However, technological advancements
have been made in the last few years, increasing our ability to
recognize alterations to the mRNA, and recent cellular
transcriptome studies have focused attention on epitranscription
(74). Numerous studies indicate
that these modifications significantly enhance the role of RNA in
promoting genetic diversity (71-73), and the common RNA modifications
consist of N1-methyladenosine, pseudouridine,
5-methylcytosine (m5C), 7-methylguanosine,
m6A and 2′-O-ribosemethylation (72,75). The most common types of RNA
epigenetic modifications are summarized in this review.
During the development of an organism,
N6-methyladenosine plays a critical role, and changes in
m6A levels affect several life processes, including
tissue development, self-renewal (96,108) and differentiation of stem cells
(99). m6A can also
regulate the heat shock response (91), circadian clocks (98), as well as processes related to the
fate and function of RNAs, such as RNA stability, splicing,
transport, localization and translation (89,90,96,102,107,108), primary processing of miRNAs
(109,110) and RNA-protein interactions
(80,81,111). A substantial body of research
however, suggests a link between m6A and certain
diseases, including AML. m6A has been associated with
obesity, diabetes and cancer (112). m6A modifications may
be used in combination with tumor therapy. A study analyzed The
Cancer Genome Atlas (TCGA) datasets and discovered that changes in
m6A regulatory genes were linked to TP53 mutations in
patients with AML. Moreover, alterations in the m6A
regulatory genes were found to lower the survival rates of patients
with AML. Therefore, m6A regulatory genes may serve as
potential new molecular targets for AML therapy (113). In addition, Su et al
(114) reported the antitumor
activity of R-2-hydroxyglutarate in patients with AML harboring an
isocitrate dehydrogenase (IDH) mutation by blocking FTO to induce
MYC degradation. In tissue cells with an IDH mutation, TCGA data
showed high MYC and low FTO levels. Numerous studies have recently
investigated the regulation of mRNA metabolism by m6A
modifications, revealing m6A modification
characteristics and associated regulatory mechanisms in AML
(Table I) (100,115,116).
circRNAs are an abundant class of RNA species
formed from the ligation of a downstream splice donor to an
upstream acceptor. They have a cyclically ordered structure, and
are involved in a variety of physiological and pathological
processes (4,131), have structural stability,
sequence conservation and tissue-specific expression. circRNAs have
more recently become one of the most frequently studied RNA
species. Due to the aforementioned unique characteristics, circRNAs
are known to act as miRNA sponges (4,11),
and they are capable of being translated into proteins through an
internal ribosome entry site (IRES)-driven process (132). Furthermore, several circRNAs
have been suggested to serve as potential biomarkers for several
diseases, including several types of cancer (14). Although numerous biological
functions of circRNAs remain unclear, there is a continuous
exploration of this research field. In 2017, circRNAs were
identified to be widely methylated by m6A, and this was
determined by m6A immunoprecipitation of RNase R
exoribonuclease-treated RNA samples, and they were effectively
translated as IRESs in human cells via short sequences consisting
of the m6A site (15).
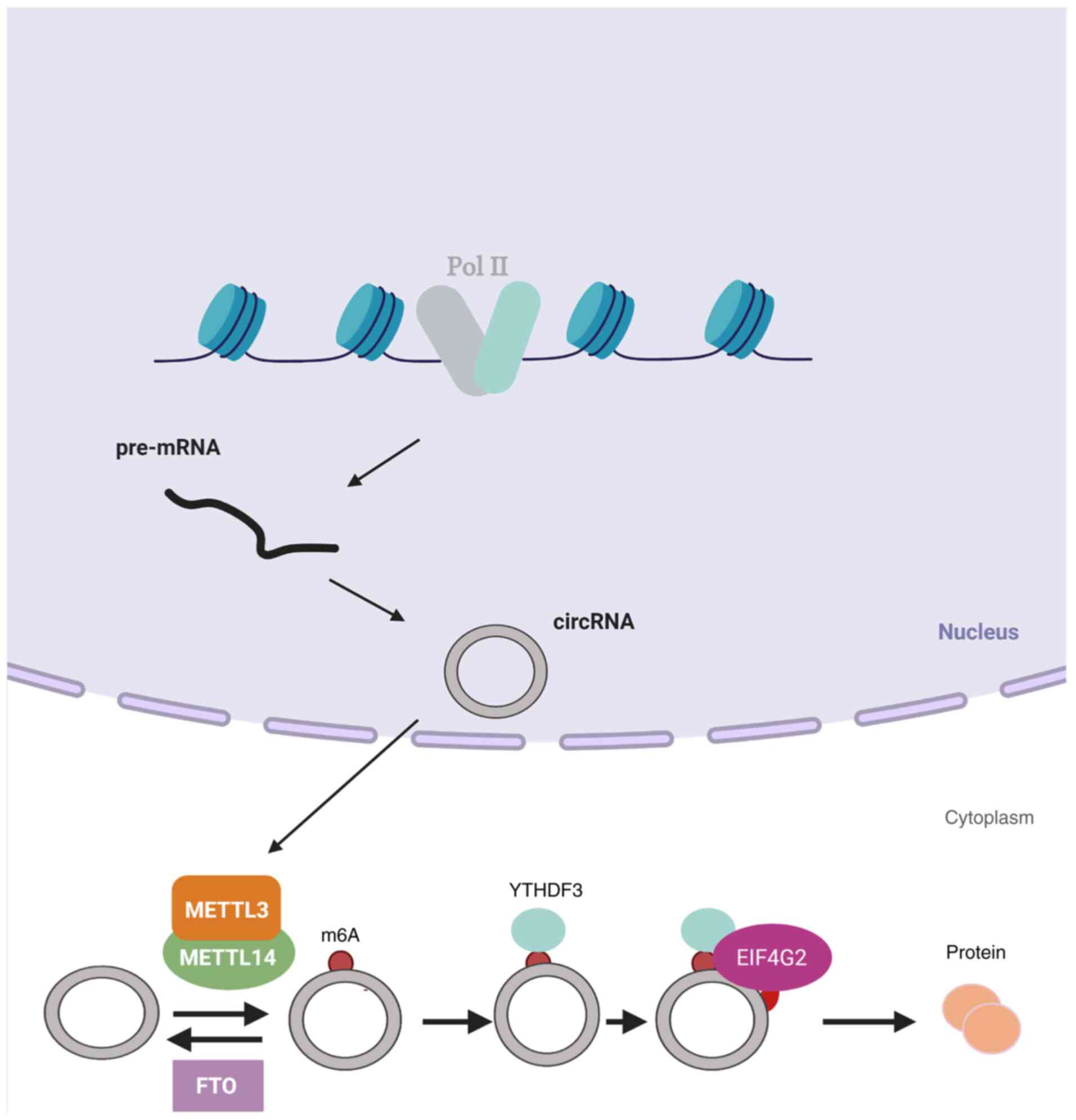
Initiation of this m6A-mediated translation involves the
eukaryotic translation initiation factor 4G2 and a YTH m6A
RNA-binding protein (YTHDF)3 reader, and their mechanism of
translation involves METTL3/14 and is inhibited by FTO (Fig. 1). In addition, that study detected
that when circRNAs were subjected to heat, their translational
function improved, suggesting that circRNA-encoded proteins may be
essential under conditions of stress (15). Other researchers also built an
AutoCirc computational pipeline to analyze RNA and m6A
immunoprecipitation results, and further confirmed that
m6A modifications are largely observed in circRNAs
(16). m6A circRNAs
were shown to possess highly cell-specific expression, and found
that circRNAs with m6A modifications also had long
single exons (16). Moreover,
m6A circRNAs and m6A mRNAs were compared by
the researchers, and it was validated that the methylated exons in
mRNAs were distinct from the exons that form m6A
circRNAs. In addition, they indicated that m6A circRNAs
were correlated with mRNA stability via the interaction with
YTHDF1/YTHDF2 (16).
Circular RNAs are naturally more stable than their
parental linear RNAs due to their closed circular structure, as
they are not the primary targets of foreign chemicals or
exonucleases. This was confirmed in several studies associated with
the characterization of circRNAs (138,139). In Actinomycin D and RNase R
treatment, circRNAs are rarely degraded before their corresponding
parental linear RNAs (140).
However, little is known about how circRNAs are degraded and what
factors contribute to circRNA degradation. One of the pathways by
which m6A-modified RNAs are degraded is the
endoribonucleolytic cleavage pathway. As emerging research in the
field of RNA research, m6A-modified circRNAs were also
discovered to be endoribonuclease-cleaved via a YTHDF2-HRSP12-RNase
P/MRP axis (141). HRSP12 is an
adaptor protein that connects YTHDF2 (m6A reader
protein) and RNase P/MRP (endoribonucleases) to form the
YTHDF2-HRSP12-RNase P/MRP complex, with YTHDF2 serving as the
guide. When an m6A-modified circRNA is recognized by
YTHDF2, regardless of whether it occupies an HRSP12-binding site,
RNase P/MRP always performs its endonuclease function. The only
difference is that the presence of the HRSP12 binding site improves
endoribonucleolytic cleavage efficiency significantly. The
m6A-modified circRNA is then selectively downregulated.
The biological function of circRNAs is altered as a result
(142). Thus, it can be deduced
that one of the means by which m6A modification
regulates circRNA biological function is by affecting their
degradation.
The accumulation of abnormal and immature
hematopoietic progenitor cells (HPCs) in the bone marrow and
peripheral blood is caused by a variety of genetic and epigenetic
abnormalities that arrest hematopoietic cell differentiation and
maturation. Lethal infection, organ infiltration and cytopenias are
frequently associated with these abnormalities (4,9).
The progression and pathogenesis of hematopoietic malignancies and
solid tumors including AML have been linked to aberrant circRNA
expression (Table II). This was
further validated in a recent study in which hundreds of circRNAs
were found to be differentially expressed in AML, and several of
these circRNAs were transcribed from genes implicated in leukemia
biology (144). miRNAs are short
stretches of RNA (~23 nt in length) that are linked to a variety of
biological processes (2), and
circRNAs have also been associated with tumorigenesis, metastasis
and drug resistance (145).
Interestingly, the most well-known mechanism of action of circRNAs
is their 'sponge' function, which involves binding to miRNAs
(15,146), proteins (139-141) or DNA (147,148). circRNAs modulate mRNA stability
and translation by sequestering the mRNA and protein transcripts,
and this is the most well-known role of circRNAs in AML (149,150). A brief review of several AML
studies suggests that circRNAs could become possible biomarkers in
AML (Table II) (150-156). Although the roles of circRNAs in
AML requires further exploration, it is evident that circRNA levels
are dynamically modulated in AML. Thus, these findings suggest that
circRNAs may play an important role in AML.
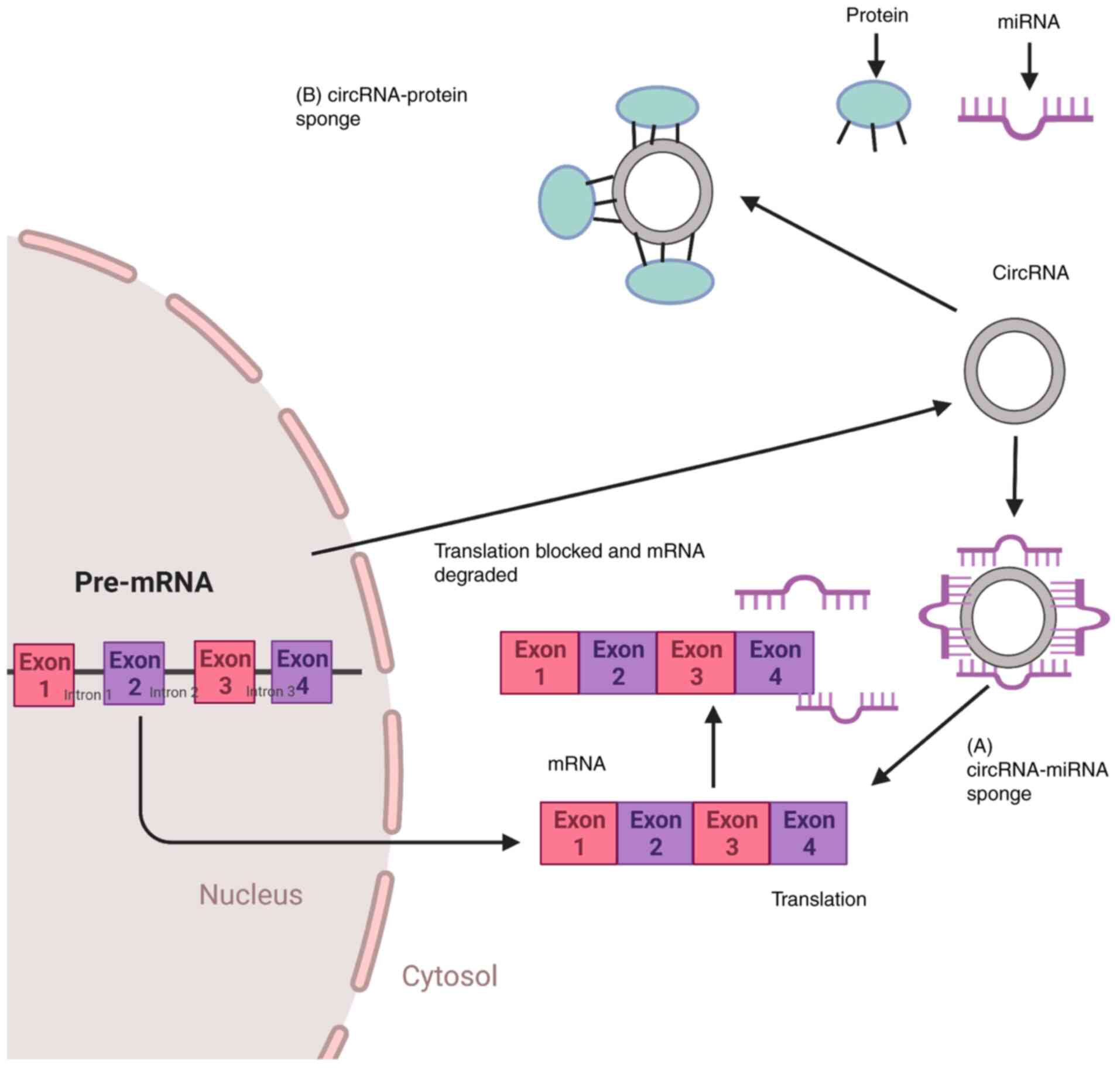
The first function of circRNAs, which was
discovered in 2013, was that of miRNA sponging, and the most
well-established function of circRNAs is to sponge miRNAs and
proteins. ciRS-7 has >70 conserved miR-7 binding sites, and it
can bind to the Argonaute (AGO) protein (11,147). The sequestration of miRNAs by
circRNAs supports the translational machinery to bind to the
specific mRNA, resulting in gene derepression in the case of
circRNA-miRNA sponge formation (Fig.
2A). Increased expression of genes that are involved in cell
proliferation, differentiation and migration may support the
development of leukemia (147).
Both cis-and trans-acting factors (157), the latter also termed as RBPs,
regulate circRNA biogenesis (147). Since RBPs are also involved in
cell cycle progression as well as the biogenesis of circRNAs,
circRNA-RBP interactions (Fig.
2B) or indirect circRNA-miRNA-RBP interactions by circRNAs may
also induce the development of leukemia (158).
The interaction between circRNAs and RBPs, as well
as the associated potential functional aspects, are becoming
increasingly clear (159). AGO
(4,11), RNA polymerase II (9), Muscleblind protein (12), Quaking I (147) and elongation initiation factor
4A3 (160) are some of the RBPs
that have been identified. These RBPs play a role in cellular
processes by regulating gene expression. Some upregulated
interacting RBPs serve key roles in RNA splicing and maintaining
the leukemic condition, according to CRISPR-Cas9-based RBP
screening in AML. When RBM39, the network's main regulator, is
knocked out, the splicing of essential mRNAs for AML is disrupted,
resulting in AML cell apoptosis (161). Furthermore, as comprehensively
reviewed previously (162),
mutational profiling of leukemic patients has revealed somatic
genetic mutations in RBPs that are linked to splicing. In addition,
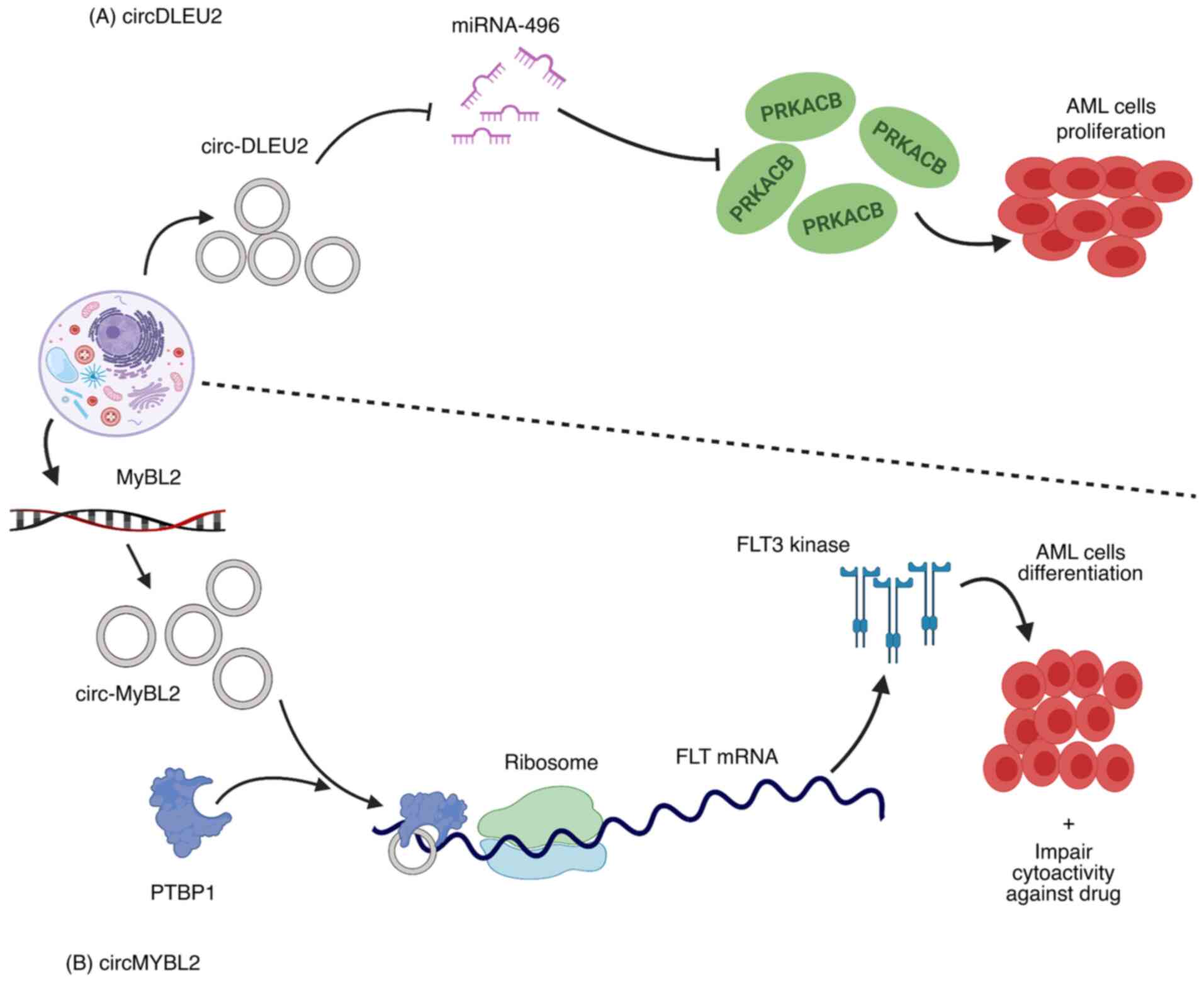
in patients with AML with ITD mutations in the FLT3 gene, high
expression of circMyb-related protein B (MYBL2), a product of the
MYBL2 gene, was reported. The circMYBL2 and FLT3-ITD mutant kinase
were found to have a positive regulatory relationship. circMYBL2
was identified to improve mutant FLT3 kinase protein expression, as
a result, FLT3-ITD-dependent signaling pathways were activated.
circMYBL2 enhanced FLT3 kinase translational efficiency by
promoting the binding of polypyrimidine tract-binding protein 1
(PTBP1) to mutant FLT3 kinase mRNA. In addition to inhibiting AML
cell proliferation and supporting differentiation in vitro
and in vivo, circMYBL2 knockdown compromised the
cytoactivity of cells with the FLT3-ITD mutation against
quizartinib (Fig. 3B) (163).
circRNAs are primarily located in the cytoplasm due
to their stable structure, nonetheless, some circular isoforms
(EIcircRNA) can also be found in the nucleus. These circular
isoforms bind to chromatin modifiers, causing the gene to be
repressed or activated (164,165). RNA polymerase II interacts with
certain EIcircRNAs, such as circEIF3J and circPAIP2, to recruit U1
small nuclear ribonucleoprotein to promote gene transcription
(13). Furthermore, some circRNAs
positively regulate the expression of their parent gene, as seen in
the case of circRNA, ci-ankrd52, which reduces the expression of
ankrd52 without affecting the expression of the surrounding genes
(9). By binding to its cognate
DNA, circRNA derived from the SEP3 gene controls expression of the
linear transcript. circRNA-SEP3 has a linear counterpart with the
same sequence that binds to DNA with a low affinity. Hence,
transcriptional repression together with the generation of a SEP3
linear transcript with exon skipping are likely outcomes of
circRNA-DNA formation (166).
Furthermore, promoter-associated RNA suppresses rRNA gene
expression by recruiting DNMT3b to the TTF-I (transcription factor)
target site via complementarity with the rDNA promoter. By binding
to genomic DNA and forming a DNA-RNA triplex, the circRNA, like
other RNA species, may affect DNA replication (167). These findings suggest that
circRNAs may bind to DNA to regulate gene expression and DNA
replication.
Even though circRNAs have an open reading frame,
they often lack essential translational components, such as a
poly(A) tail and a 7-methylguanosine cap (133). Nonetheless, mounting evidence
suggests that circRNAs are capable of translation (133). For example, the RNA modification
motif m6A, which is abundantly present in circRNAs, aids
circRNA translation in human cells (15). Other mechanisms exist for circRNA
translation. circRNAs containing an IRES which drives translation,
such as circ-ZNF609 and circMbl3, have been found to translate
proteins (132,133). Furthermore, Sun et al
(163) suggested that circMYBL2
regulated FLT3 translation by recruiting PTBP1 to enhance FLT3-ITD
AML progression. Generally, circRNA translational mechanisms in AML
are not well understood and require further investigation.
By acting as miRNA sponges, circRNAs are involved
in regulating RNA processing, such as alternative splicing, pre-RNA
splicing and RNA editing (11,168,169). Furthermore, aberrant circRNA
expression (mainly upregulation), has been identified as a
potential biomarker in AML (Table
II). The mechanisms by which circRNAs regulate AML remains
unclear. Previous findings suggest that circRNAs may regulate
tumorigenesis, at least partly via m6A modification
(139,140,143).
In conclusion, the role of circRNAs in
carcinogenesis, including AML, is currently a major focus of cancer
research. Although alterations in circRNA epigenetic modifications
may have an impact on hematopoiesis and AML development, further
studies are required to confirm this hypothesis. Therefore, it may
be necessary to identify alterations in circRNA epigenetic
modifications in AML, as well as the regulatory mechanisms behind
these modifications, which could further elucidate the specific
roles of circRNAs in this disease. These studies may provide new
insights into AML pathogenesis and therapy.
Not applicable.
MAI, DW and JS contributed to study
conceptualization. MAI was the primary contributor with support
from FZ, WZ, HF and HZ in writing the original draft, and it was
reviewed and edited by MAI, YL and RC. All the authors have read
and approved the final manuscript. Data authentication is not
applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
This study was supported in part by Grants-in-Aid from the
National Natural Science Foundation of China (grant nos. 81300428
and 81800167); Joints Funds for the Innovation of Science and
Technology, Fujian Province (grant nos. 2018Y9010 and 2018Y9205);
Qihang Foundation of Fujian Medical University (grant no.
2020QH2015); and the Construction project of Fujian Medical Center
of Hematology, Clinical Research Center for Hematological
Malignancies of Fujian Province (grant no. Min201704).
|
1
|
Chen LL and Yang L: Regulation of circRNA
biogenesis. RNA Biol. 12:381–388. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dong Y, He D, Peng Z, Peng W, Shi W, Wang
J, Li B, Zhang C and Duan C: Circular RNAs in cancer: An emerging
key player. J Hematol Oncol. 10:22017. View Article : Google Scholar
|
|
4
|
Memczak S, Jens M, Elefsinioti A, Torti F,
Krueger J, Rybak A, Maier L, Mackowiak S, Gregersen LH, Munschauer
M, et al: Circular RNAs are a large class of animal RNAs with
regulatory potency. Nature. 495:333–338. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang Y, Zhang XO, Chen T, Xiang JF, Yin
QF, Xing YH, Zhu S, Yang L and Chen LL: Circular intronic long
noncoding RNAs. Mol Cell. 51:792–806. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dupont C, Armant DR and Brenner CA:
Epigenetics: Definition, mechanisms and clinical perspective. Semin
Reprod Med. 27:351–357. 2009. View Article : Google Scholar :
|
|
7
|
Bolisetty MT and Graveley BR: Circuitous
route to transcription regulation. Mol Cell. 51:705–706. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Suzuki H and Tsukahara T: A view of
pre-mRNA splicing from RNase R resistant RNAs. Int J Mol Sci.
15:9331–9342. 2014. View Article : Google Scholar :
|
|
9
|
Jeck WR, Sorrentino JA, Wang K, Slevin MK,
Burd CE, Liu J, Marzluff WF and Sharpless NE: Circular RNAs are
abundant, conserved, and associated with ALU repeats. RNA.
19:141–157. 2013. View Article : Google Scholar
|
|
10
|
Gruner H, Cortés-López M, Cooper DA, Bauer
M and Miura P: CircRNA accumulation in the aging mouse brain. Sci
Rep. 6:389072016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hansen TB, Jensen TI, Clausen BH, Bramsen
JB, Finsen B, Damgaard CK and Kjems J: Natural RNA circles function
as efficient microRNA sponges. Nature. 495:384–388. 2013.
View Article : Google Scholar
|
|
12
|
Ashwal-Fluss R, Meyer M, Pamudurti NR,
Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N and
Kadener S: CircRNA biogenesis competes with pre-mRNA splicing. Mol
Cell. 56:55–66. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li Z, Huang C, Bao C, Chen L, Lin M, Wang
X, Zhong G, Yu B, Hu W, Dai L, et al: Exon-intron circular RNAs
regulate transcription in the nucleus. Nat Struct Mol Biol.
22:256–264. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Meng S, Zhou H, Feng Z, Xu Z, Tang Y, Li P
and Wu M: CircRNA: Functions and properties of a novel potential
biomarker for cancer. Mol Cancer. 16:942017. View Article : Google Scholar
|
|
15
|
Yang Y, Fan X, Mao M, Song X, Wu P, Zhang
Y, Jin Y, Yang Y, Chen LL, Wang Y, et al: Extensive translation of
circular RNAs driven by N6-methyladenosine. Cell Res. 27:626–641.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou C, Molinie B, Daneshvar K, Pondick
JV, Wang J, Van Wittenberghe N, Xing Y, Giallourakis CC and Mullen
AC: Genome-wide maps of m6A circRNAs identify widespread and
cell-type-specific methylation patterns that are distinct from
mRNAs. Cell Rep. 20:2262–2276. 2017. View Article : Google Scholar
|
|
17
|
Gapp K, Woldemichael BT, Bohacek J and
Mansuy IM: Epigenetic regulation in neurodevelopment and
neurodegenerative diseases. Neuroscience. 264:99–111. 2014.
View Article : Google Scholar
|
|
18
|
Trowbridge JJ, Snow JW, Kim J and Orkin
SH: DNA methyltransferase 1 is essential for and uniquely regulates
hematopoietic stem and progenitor cells. Cell Stem Cell. 5:442–449.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harman MF and Martín MG: Epigenetic
mechanisms related to cognitive decline during aging. J Neurosci
Res. 98:234–246. 2020. View Article : Google Scholar
|
|
20
|
Feinberg AP and Tycko B: The history of
cancer epigenetics. Nat Rev Cancer. 4:143–153. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jones PA: Functions of DNA methylation:
Islands, start sites, gene bodies and beyond. Nat Rev Genet.
13:484–492. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hájková H, Marková J, Haškovec C, Šárová
I, Fuchs O, Kostečka A, Cetkovský P, Michalová K and Schwarz J:
Decreased DNA methylation in acute myeloid leukemia patients with
DNMT3A mutations and prognostic implications of DNA methylation.
Leuk Res. 36:1128–1133. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bröske AM, Vockentanz L, Kharazi S, Huska
MR, Mancini E, Scheller M, Kuhl C, Enns A, Prinz M, Jaenisch R, et
al: DNA methylation protects hematopoietic stem cell multipotency
from myeloerythroid restriction. Nat Genet. 41:1207–1215. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bock C, Beerman I, Lien WH, Smith ZD, Gu
H, Boyle P, Gnirke A, Fuchs E, Rossi DJ and Meissner A: DNA
methylation dynamics during in vivo differentiation of blood and
skin stem cells. Mol Cell. 47:633–647. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hodges E, Molaro A, Dos Santos CO, Thekkat
P, Song Q, Uren PJ, Park J, Butler J, Rafii S, McCombie WR, et al:
Directional DNA methylation changes and complex intermediate states
accompany lineage specificity in the adult hematopoietic
compartment. Mol Cell. 44:17–28. 2011. View Article : Google Scholar
|
|
26
|
Hogart A, Lichtenberg J, Ajay SS, Anderson
S; NIH Intramural Sequencing Center; Margulies EH and Bodine DM:
Genome-wide DNA methylation profiles in hematopoietic stem and
progenitor cells reveal overrepresentation of ETS transcription
factor binding sites. Genome Res. 22:1407–1418. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tadokoro Y, Ema H, Okano M, Li E and
Nakauchi H: De novo DNA methyltransferase is essential for
self-renewal, but not for differentiation, in hematopoietic stem
cells. J Exp Med. 204:715–722. 2007. View Article : Google Scholar :
|
|
28
|
Jiang Y, Dunbar A, Gondek LP, Mohan S,
Rataul M, O'Keefe C, Sekeres M, Saunthararajah Y and Maciejewski
JP: Aberrant DNA methylation is a dominant mechanism in MDS
progression to AML. Blood. 113:1315–1325. 2009. View Article : Google Scholar :
|
|
29
|
Chen J, Odenike O and Rowley JD:
Leukaemogenesis: More than mutant genes. Nat Rev Cancer. 10:23–36.
2010. View Article : Google Scholar
|
|
30
|
Schoofs T, Berdel WE and Müller-Tidow C:
Origins of aberrant DNA methylation in acute myeloid leukemia.
Leukemia. 28:1–14. 2014. View Article : Google Scholar
|
|
31
|
Figueroa ME, Lugthart S, Li Y,
Erpelinck-Verschueren C, Deng X, Christos PJ, Schifano E, Booth J,
van Putten W, Skrabanek L, et al: DNA methylation signatures
identify biologically distinct subtypes in acute myeloid leukemia.
Cancer Cell. 17:13–27. 2010. View Article : Google Scholar :
|
|
32
|
Cole CB, Verdoni AM, Ketkar S, Leight ER,
Russler-Germain DA, Lamprecht TL, Demeter RT, Magrini V and Ley TJ:
PML-RARA requires DNA methyltransferase 3A to initiate acute
promyelocytic leukemia. J Clin Invest. 126:85–98. 2016. View Article : Google Scholar :
|
|
33
|
Ley TJ, Miller C, Ding L, Raphael BJ,
Mungall AJ, Robertson AG, Hoadley K, Triche TJ Jr, Laird PW, Batty
JD, et al: Genomic and epigenomic landscapes of adult de novo acute
myeloid leukemia. N Engl J Med. 368:2059–2074. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Thol F, Damm F, Lüdeking A, Winschel C,
Wagner K, Morgan M, Yun H, Göhring G, Schlegelberger B, Hoelzer D,
et al: Incidence and prognostic influence of DNMT3A mutations in
acute myeloid leukemia. J Clin Oncol. 29:2889–2896. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Marková J, Michková P, Burčková K,
Březinová J, Michalová K, Dohnalová A, Maaloufová JS, Soukup P,
Vítek A, Cetkovský P and Schwarz J: Prognostic impact of DNMT3A
mutations in patients with intermediate cytogenetic risk profile
acute myeloid leukemia. Eur J Haematol. 88:128–135. 2012.
View Article : Google Scholar
|
|
36
|
Alvarez S, Suela J, Valencia A, Fernández
A, Wunderlich M, Agirre X, Prósper F, Martín-Subero JI, Maiques A,
Acquadro F, et al: DNA methylation profiles and their relationship
with cytogenetic status in adult acute myeloid leukemia. PLoS One.
5:e121972010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Akalin A, Garrett-Bakelman FE, Kormaksson
M, Busuttil J, Zhang L, Khrebtukova I, Milne TA, Huang Y, Biswas D,
Hess JL, et al: Base-pair resolution DNA methylation sequencing
reveals profoundly divergent epigenetic landscapes in acute myeloid
leukemia. PLoS Genet. 8:e10027812012. View Article : Google Scholar :
|
|
38
|
Cimmino L, Dawlaty MM, Ndiaye-Lobry D, Yap
YS, Bakogianni S, Yu Y, Bhattacharyya S, Shaknovich R, Geng H,
Lobry C, et al: Erratum: TET1 is a tumor suppressor of
hematopoietic malignancy. Nat Immunol. 16:8892015. View Article : Google Scholar
|
|
39
|
Moran-Crusio K, Reavie L, Shih A,
Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar
A, Patel J, Zhao X, et al: Tet2 loss leads to increased
hematopoietic stem cell self-renewal and myeloid transformation.
Cancer Cell. 20:11–24. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li Z, Cai X, Cai CL, Wang J, Zhang W,
Petersen BE, Yang FC and Xu M: Deletion of Tet2 in mice leads to
dysregulated hematopoietic stem cells and subsequent development of
myeloid malignancies. Blood. 118:4509–4518. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Abdel-Wahab O, Mullally A, Hedvat C,
Garcia-Manero G, Patel J, Wadleigh M, Malinge S, Yao J, Kilpivaara
O, Bhat R, et al: Genetic characterization of TET1, TET2, and TET3
alterations in myeloid malignancies. Blood. 114:144–147. 2009.
View Article : Google Scholar :
|
|
42
|
Tefferi A, Lim KH, Abdel-Wahab O, Lasho
TL, Patel J, Patnaik MM, Hanson CA, Pardanani A, Gilliland DG and
Levine RL: Detection of mutant TET2 in myeloid malignancies other
than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML.
Leukemia. 23:1343–1345. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bacher U, Haferlach C, Schnittger S,
Kohlmann A, Kern W and Haferlach T: Mutations of the TET2 and CBL
genes: Novel molecular markers in myeloid malignancies. Ann
Hematol. 89:643–652. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sato H, Wheat JC, Steidl U and Ito K:
DNMT3A and TET2 in the pre-leukemic phase of hematopoietic
disorders. Front Oncol. 6:1872016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chan SM and Majeti R: Role of DNMT3A,
TET2, and IDH1/2 mutations in pre-leukemic stem cells in acute
myeloid leukemia. Int J Hematol. 98:648–657. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Weissmann S, Alpermann T, Grossmann V,
Kowarsch A, Nadarajah N, Eder C, Dicker F, Fasan A, Haferlach C,
Haferlach T, et al: Landscape of TET2 mutations in acute myeloid
leukemia. Leukemia. 26:934–942. 2012. View Article : Google Scholar
|
|
47
|
Shih AH, Jiang Y, Meydan C, Shank K,
Pandey S, Barreyro L, Antony-Debre I, Viale A, Socci N, Sun Y, et
al: Mutational cooperativity linked to combinatorial epigenetic
gain of function in acute myeloid leukemia. Cancer Cell.
27:502–515. 2015. View Article : Google Scholar
|
|
48
|
Rasmussen KD, Jia G, Johansen JV, Pedersen
MT, Rapin N, Bagger F, Porse BT, Bernard OA, Christensen J, Helin
K, et al: Loss of TET2 in hematopoietic cells leads to DNA
hypermethylation of active enhancers and induction of
leukemogenesis. Genes Dev. 29:910–922. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Berger SL: The complex language of
chromatin regulation during transcription. Nature. 447:407–412.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Podobinska M, Szablowska-Gadomska I,
Augustyniak J, Sandvig I, Sandvig A and Buzanska L: Epigenetic
modulation of stem cells in neurodevelopment: The role of
methylation and acetylation. Front Cell Neurosci. 11:232017.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang Y, Gilquin B, Khochbin S and
Matthias P: Two catalytic domains are required for protein
deacetylation. J Biol Chem. 281:2401–2404. 2006. View Article : Google Scholar
|
|
52
|
Uchida T, Kinoshita T, Nagai H, Nakahara
Y, Saito H, Hotta T and Murate T: Hypermethylation of the p15INK4B
gene in myelodysplastic syndromes. Blood. 90:1403–1409. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Melki JR, Vincent PC and Clark SJ:
Concurrent DNA hyper-methylation of multiple genes in acute myeloid
leukemia. Cancer Res. 59:3730–3740. 1999.PubMed/NCBI
|
|
54
|
Herman JG, Jen J, Merlo A and Baylin SB:
Hypermethylation-associated inactivation indicates a tumor
suppressor role for p15INK4B. Cancer Res. 56:722–727.
1996.PubMed/NCBI
|
|
55
|
Jenuwein T: Translating the histone code.
Science. 293:1074–1080. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
van Dijk AD, Hu CW, de Bont ESJM, Qiu Y,
Hoff FW, Yoo SY, Coombes KR, Qutub AA and Kornblau SM: Histone
modification patterns using RPPA-based profiling predict outcome in
acute myeloid leukemia patients. Proteomics. 18:17003792018.
View Article : Google Scholar
|
|
57
|
Zaghlool A, Halvardson J, Zhao JJ,
Etemadikhah M, Kalushkova A, Konska K, Jernberg-Wiklund H,
Thuresson AC and Feuk L: A role for the chromatin-remodeling factor
BAZ1A in neurodevelopment. Hum Mutat. 37:964–975. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Olave IA, Reck-Peterson SL and Crabtree
GR: Nuclear actin and actin-related proteins in chromatin
remodeling. Annu Rev Biochem. 71:755–781. 2002. View Article : Google Scholar
|
|
59
|
Choi KY, Yoo M and Han JH: Toward
understanding the role of the neuron-specific BAF chromatin
remodeling complex in memory formation. Exp Mol Med. 47:e1552015.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Redner RL, Wang J and Liu JM: Chromatin
remodeling and leukemia: New therapeutic paradigms. Blood.
94:417–428. 1999. View Article : Google Scholar
|
|
61
|
Sperlazza J, Rahmani M, Beckta J, Aust M,
Hawkins E, Wang SZ, Zu Zhu S, Podder S, Dumur C, Archer K, et al:
Depletion of the chromatin remodeler CHD4 sensitizes AML blasts to
genotoxic agents and reduces tumor formation. Blood. 126:1462–1472.
2015. View Article : Google Scholar
|
|
62
|
Denslow SA and Wade PA: The human
Mi-2/NuRD complex and gene regulation. Oncogene. 26:5433–5438.
2007. View Article : Google Scholar
|
|
63
|
D'Alesio C, Punzi S, Cicalese A, Fornasari
L, Furia L, Riva L, Carugo A, Curigliano G, Criscitiello C, Pruneri
G, et al: RNAi screens identify CHD4 as an essential gene in breast
cancer growth. Oncotarget. 7:80901–80915. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
O'Shaughnessy A and Hendrich B: CHD4 in
the DNA-damage response and cell cycle progression: Not so NuRDy
now. Biochem Soc Trans. 41:777–782. 2013. View Article : Google Scholar :
|
|
65
|
Polo SE, Kaidi A, Baskcomb L, Galanty Y
and Jackson SP: Regulation of DNA-damage responses and cell-cycle
progression by the chromatin remodelling factor CHD4. EMBO J.
29:3130–3139. 2010. View Article : Google Scholar :
|
|
66
|
Xia L, Huang W, Bellani M, Seidman MM, Wu
K, Fan D, Nie Y, Cai Y, Zhang YW, Yu LR, et al: CHD4 has oncogenic
functions in initiating and maintaining epigenetic suppression of
multiple tumor suppressor genes. Cancer Cell. 31:653–668.e7. 2017.
View Article : Google Scholar :
|
|
67
|
Heshmati Y, Türköz G, Harisankar A,
Kharazi S, Boström J, Dolatabadi EK, Krstic A, Chang D, Månsson R,
Altun M, et al: The chromatin-remodeling factor CHD4 is required
for maintenance of childhood acute myeloid leukemia. Haematologica.
103:1169–1181. 2018. View Article : Google Scholar :
|
|
68
|
Zhen T, Kwon EM, Zhao L, Hsu J, Hyde RK,
Lu Y, Alemu L, Speck NA and Liu PP: Chd7 deficiency delays
leukemogenesis in mice induced by Cbfb-MYH11. Blood. 130:2431–2442.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Fazi F, Racanicchi S, Zardo G, Starnes LM,
Mancini M, Travaglini L, Diverio D, Ammatuna E, Cimino G, Lo-Coco
F, et al: Epigenetic silencing of the myelopoiesis regulator
microRNA-223 by the AML1/ETO oncoprotein. Cancer Cell. 12:457–466.
2007. View Article : Google Scholar
|
|
70
|
Li Y, Gao L, Luo X, Wang L, Gao X, Wang W,
Sun J, Dou L, Li J, Xu C, et al: Epigenetic silencing of
microRNA-193a contributes to leukemogenesis in t(8;21) acute
myeloid leukemia by activating the PTEN/PI3K signal pathway. Blood.
121:499–509. 2013. View Article : Google Scholar
|
|
71
|
Berger SL, Kouzarides T, Shiekhattar R and
Shilatifard A: An operational definition of epigenetics. Genes Dev.
23:781–783. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Sun WJ, Li JH, Liu S, Wu J, Zhou H, Qu LH
and Yang JH: RMBase: A resource for decoding the landscape of RNA
modifications from high-throughput sequencing data. Nucleic Acids
Res. 44:D259–D265. 2016. View Article : Google Scholar :
|
|
73
|
Lee M, Kim B and Kim VN: Emerging roles of
RNA modification: m6A and U-tail. Cell. 158:980–987. 2014.
View Article : Google Scholar
|
|
74
|
Flamand MN and Meyer KD: The
epitranscriptome and synaptic plasticity. Curr Opin Neurobiol.
59:41–48. 2019. View Article : Google Scholar
|
|
75
|
Maden BE: The numerous modified
nucleotides in eukaryotic ribosomal RNA. Prog Nucleic Acid Res Mol
Biol. 39:241–303. 1990. View Article : Google Scholar
|
|
76
|
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han
D, Fu Y, Parisien M, Dai Q, Jia G, et al:
N6-methyladenosine-dependent regulation of messenger RNA stability.
Nature. 505:117–120. 2014. View Article : Google Scholar
|
|
77
|
Zhang X and Jia GF: RNA epigenetic
modification: N6-methyladenosine. Yi Chuan. 38:275–288. 2016.
|
|
78
|
Wei CM, Gershowitz A and Moss B:
Methylated nucleotides block 5′ terminus of HeLa cell messenger
RNA. Cell. 4:379–386. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Niu Y, Zhao X, Wu YS, Li MM, Wang XJ and
Yang YG: N6-methyl-adenosine (m6A) in RNA: An old modification with
a novel epigenetic function. Genomics Proteomics Bioinformatics.
11:8–17. 2013. View Article : Google Scholar
|
|
80
|
Meyer KD, Saletore Y, Zumbo P, Elemento O,
Mason CE and Jaffrey SR: Comprehensive analysis of mRNA methylation
reveals enrichment in 3′ UTRs and near stop codons. Cell.
149:1635–1646. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Dominissini D, Moshitch-Moshkovitz S,
Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K,
Jacob-Hirsch J, Amariglio N, Kupiec M, et al: Topology of the human
and mouse m6A RNA methylomes revealed by m6A-seq. Nature.
485:201–206. 2012. View Article : Google Scholar
|
|
82
|
Chen K, Lu Z, Wang X, Fu Y, Luo GZ, Liu N,
Han D, Dominissini D, Dai Q, Pan T and He C: High-resolution
N(6)-methyladenosine (m(6) A) map using photo-crosslinking-assisted
m(6) A sequencing. Angew Chemie Int Ed. 54:1587–1590. 2015.
View Article : Google Scholar
|
|
83
|
Linder B, Grozhik AV, Olarerin-George AO,
Meydan C, Mason CE and Jaffrey SR: Single-nucleotide-resolution
mapping of m6A and m6Am throughout the transcriptome. Nat Methods.
12:767–772. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Roundtree IA and He C: RNA
epigenetics-chemical messages for posttranscriptional gene
regulation. Curr Opin Chem Biol. 30:46–51. 2016. View Article : Google Scholar
|
|
85
|
Ping XL, Sun BF, Wang L, Xiao W, Yang X,
Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al: Mammalian WTAP is
a regulatory subunit of the RNA N6-methyladenosine
methyltransferase. Cell Res. 24:177–189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang
L, Jia G, Yu M, Lu Z, Deng X, et al: A METTL3-METTL14 complex
mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem
Biol. 10:93–95. 2014. View Article : Google Scholar :
|
|
87
|
Bokar JA, Rath-Shambaugh ME, Ludwiczak R,
Narayan P and Rottman F: Characterization and partial purification
of mRNA N6-adenosine methyltransferase from HeLa cell nuclei.
Internal mRNA methylation requires a multisubunit complex. J Biol
Chem. 269:17697–17704. 1994. View Article : Google Scholar
|
|
88
|
Schwartz S, Mumbach MR, Jovanovic M, Wang
T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N,
Cacchiarelli D, et al: Perturbation of m6A writers reveals two
distinct classes of mrna methylation at internal and 5′ sites. Cell
Rep. 8:284–296. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Ke S, Alemu EA, Mertens C, Gantman EC, Fak
JJ, Mele A, Haripal B, Zucker-Scharff I, Moore MJ, Park CY, et al:
A majority of m 6 A residues are in the last exons, allowing the
potential for 3′ UTR regulation. Genes Dev. 29:2037–2053. 2015.
View Article : Google Scholar
|
|
90
|
Meyer KD, Patil DP, Zhou J, Zinoviev A,
Skabkin MA, Elemento O, Pestova TV, Qian SB and Jaffrey SR: 5′ UTR
m6A promotes cap-independent translation. Cell. 163:999–1010. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR
and Qian SB: Dynamic m6A mRNA methylation directs translational
control of heat shock response. Nature. 526:591–594. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Pendleton KE, Chen B, Liu K, Hunter OV,
Xie Y, Tu BP and Conrad NK: The U6 snRNA m 6 A methyltransferase
METTL16 regulates SAM synthetase intron retention. Cell.
169:824–835.e14. 2017. View Article : Google Scholar
|
|
93
|
Dina C, Meyre D, Gallina S, Durand E,
Körner A, Jacobson P, Carlsson LMS, Kiess W, Vatin V, Lecoeur C, et
al: Variation in FTO contributes to childhood obesity and severe
adult obesity. Nat Genet. 39:724–726. 2007. View Article : Google Scholar
|
|
94
|
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang
Y, Yi C, Lindahl T, Pan T, Yang YG and He C: N6-Methyladenosine in
nuclear RNA is a major substrate of the obesity-associated FTO. Nat
Chem Biol. 7:885–887. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Zheng G, Dahl JA, Niu Y, Fedorcsak P,
Huang CM, Li CJ, Vågbø CB, Shi Y, Wang WL, Song SH, et al: ALKBH5
is a mammalian RNA demethylase that impacts RNA metabolism and
mouse fertility. Mol Cell. 49:18–29. 2013. View Article : Google Scholar :
|
|
96
|
Fu Y, Jia G, Pang X, Wang RN, Wang X, Li
CJ, Smemo S, Dai Q, Bailey KA, Nobrega MA, et al: FTO-mediated
formation of N6-hydroxymethyladenosine and N6-formyladenosine in
mammalian RNA. Nat Commun. 4:17982013. View Article : Google Scholar
|
|
97
|
Zhao X, Yang Y, Sun BF, Shi Y, Yang X,
Xiao W, Hao YJ, Ping XL, Chen YS, Wang WJ, et al: FTO-dependent
demethylation of N6-methyladenosine regulates mRNA splicing and is
required for adipogenesis. Cell Res. 24:1403–1419. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Hess ME, Hess S, Meyer KD, Verhagen LAW,
Koch L, Brönneke HS, Dietrich MO, Jordan SD, Saletore Y, Elemento
O, et al: The fat mass and obesity associated gene (Fto) regulates
activity of the dopaminergic midbrain circuitry. Nat Neurosci.
16:1042–1048. 2013. View Article : Google Scholar
|
|
99
|
Geula S, Moshitch-Moshkovitz S,
Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V,
Peer E, Mor N, Manor YS, et al: Stem cells. m6A mRNA methylation
facilitates resolution of naïve pluripotency toward
differentiation. Science. 347:1002–1006. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Li Z, Weng H, Su R, Weng X, Zuo Z, Li C,
Huang H, Nachtergaele S, Dong L, Hu C, et al: FTO plays an
oncogenic role in acute myeloid leukemia as a 6-methyladenosine RNA
demethylase. Cancer Cell. 31:127–141. 2017. View Article : Google Scholar
|
|
101
|
Jaffrey SR and Kharas MG: Emerging links
between m6A and misregulated mRNA methylation in cancer. Genome
Med. 9:22017. View Article : Google Scholar :
|
|
102
|
Zhang Z, Theler D, Kaminska KH, Hiller M,
de la Grange P, Pudimat R, Rafalska I, Heinrich B, Bujnicki JM,
Allain FHT and Stamm S: The YTH domain is a novel RNA binding
domain. J Biol Chem. 285:14701–14710. 2010. View Article : Google Scholar :
|
|
103
|
Xu C, Wang X, Liu K, Roundtree IA, Tempel
W, Li Y, Lu Z, He C and Min J: Structural basis for selective
binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol.
10:927–929. 2014. View Article : Google Scholar
|
|
104
|
Luo S and Tong L: Molecular basis for the
recognition of methylated adenines in RNA by the eukaryotic YTH
domain. Proc Natl Acad Sci USA. 111:13834–13839. 2014. View Article : Google Scholar
|
|
105
|
Zhu T, Roundtree IA, Wang P, Wang X, Wang
L, Sun C, Tian Y, Li J, He C and Xu Y: Crystal structure of the YTH
domain of YTHDF2 reveals mechanism for recognition of
N6-methyladenosine. Cell Res. 24:1493–1496. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Wang X, Zhao BS, Roundtree IA, Lu Z, Han
D, Ma H, Weng X, Chen K, Shi H and He C: N6-methyladenosine
modulates messenger RNA translation efficiency. Cell.
161:1388–1399. 2015. View Article : Google Scholar
|
|
107
|
Wang Y, Li Y, Toth JI, Petroski MD, Zhang
Z and Zhao JC: N6-methyladenosine modification destabilizes
developmental regulators in embryonic stem cells. Nat Cell Biol.
16:191–198. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Fustin JM, Doi M, Yamaguchi Y, Hida H,
Nishimura S, Yoshida M, Isagawa T, Morioka MS, Kakeya H, Manabe I
and Okamura H: RNA-methylation-dependent rna processing controls
the speed of the circadian clock. Cell. 155:793–806. 2013.
View Article : Google Scholar
|
|
109
|
Alarcón CR, Lee H, Goodarzi H, Halberg N
and Tavazoie SF: N6-methyladenosine marks primary microRNAs for
processing. Nature. 519:482–485. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Chen T, Hao YJ, Zhang Y, Li MM, Wang M,
Han W, Wu Y, Lv Y, Hao J, Wang L, et al: m6A RNA methylation is
regulated by MicroRNAs and promotes reprogramming to pluripotency.
Cell Stem Cell. 16:289–301. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Liu N, Dai Q, Zheng G, He C, Parisien M
and Pan T: N6-methyladenosine-dependent RNA structural switches
regulate RNA-protein interactions. Nature. 518:560–564. 2015.
View Article : Google Scholar
|
|
112
|
Klungland A and Dahl JA: Dynamic RNA
modifications in disease. Curr Opin Genet Dev. 26:47–52. 2014.
View Article : Google Scholar
|
|
113
|
Kwok CT, Marshall AD, Rasko JEJ and Wong
JJL: Erratum to: Genetic alterations of m6A regulators predict
poorer survival in acute myeloid leukemia. J Hematol Oncol.
10:492017. View Article : Google Scholar :
|
|
114
|
Su R, Dong L, Li C, Nachtergaele S,
Wunderlich M, Qing Y, Deng X, Wang Y, Weng X, Hu C, et al: R-2HG
exhibits anti-tumor activity by targeting FTO/m6A/MYC/CEBPA
signaling. Cell. 172:90–105.e23. 2018. View Article : Google Scholar
|
|
115
|
Vu LP, Pickering BF, Cheng Y, Zaccara S,
Nguyen D, Minuesa G, Chou T, Chow A, Saletore Y, MacKay M, et al:
The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid
differentiation of normal hematopoietic and leukemia cells. Nat
Med. 23:1369–1376. 2017. View Article : Google Scholar
|
|
116
|
Weng H, Huang H, Wu H, Qin X, Zhao BS,
Dong L, Shi H, Skibbe J, Shen C, Hu C, et al: METTL14 inhibits
hematopoietic stem/progenitor differentiation and promotes
leukemogenesis via mRNA m6A modification. Cell Stem Cell.
22:191–205.e9. 2018. View Article : Google Scholar
|
|
117
|
Chhabra R: miRNA and methylation: A
multifaceted liaison. Chembiochem. 16:195–203. 2015. View Article : Google Scholar
|
|
118
|
Hall RH: Isolation of 3-methyluridine and
3-methylcytidine from soluble ribonucleic acid. Biochem Biophys Res
Commun. 12:361–364. 1963. View Article : Google Scholar
|
|
119
|
Xu L, Liu X, Sheng N, Oo KS, Liang J,
Chionh YH, Xu J, Ye F, Gao YG, Dedon PC and Fu XY: Three distinct
3-methylcytidine (m3C) methyltransferases modify tRNA and mRNA in
mice and humans. J Biol Chem. 292:14695–14703. 2017. View Article : Google Scholar :
|
|
120
|
Glasner H, Riml C, Micura R and Breuker K:
Label-free, direct localization and relative quantitation of the
RNA nucleobase methylations m6A, m5C, m3U, and m5U by top-down mass
spectrometry. Nucleic Acids Res. 45:8014–8025. 2017. View Article : Google Scholar :
|
|
121
|
Li X, Zhu P, Ma S, Song J, Bai J, Sun F
and Yi C: Chemical pulldown reveals dynamic pseudouridylation of
the mammalian transcriptome. Nat Chem Biol. 11:592–597. 2015.
View Article : Google Scholar
|
|
122
|
Charette M and Gray MW: Pseudouridine in
RNA: What, where, how, and why. IUBMB Life. 49:341–351. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Ofengand J: Ribosomal RNA pseudouridines
and pseudouridine synthases. FEBS Lett. 514:17–25. 2002. View Article : Google Scholar
|
|
124
|
Jack K, Bellodi C, Landry DM, Niederer RO,
Meskauskas A, Musalgaonkar S, Kopmar N, Krasnykh O, Dean AM,
Thompson SR, et al: rRNA pseudouridylation defects affect ribosomal
ligand binding and translational fidelity from yeast to human
cells. Mol Cell. 44:660–666. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Kiss T, Fayet-Lebaron E and Jády BE: Box
H/ACA small ribonucleoproteins. Mol Cell. 37:597–606. 2010.
View Article : Google Scholar
|
|
126
|
Yu AT, Ge J and Yu YT: Pseudouridines in
spliceosomal snRNAs. Protein Cell. 2:712–725. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Karijolich J and Yu YT: Converting
nonsense codons into sense codons by targeted pseudouridylation.
Nature. 474:395–398. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Rosselló-Tortella M, Ferrer G and Esteller
M: Epitranscriptomics in hematopoiesis and hematologic
malignancies. Blood Cancer Discov. 1:26–31. 2020. View Article : Google Scholar
|
|
129
|
Alseth I, Dalhus B and Bjørås M: Inosine
in DNA and RNA. Curr Opin Genet Dev. 26:116–123. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Bass BL, Nishikura K, Keller W, Seeburg
PH, Emeson RB, O'Connell MA, Samuel CE and Herbert A: A
standardized nomenclature for adenosine deaminases that act on RNA.
RNA. 3:947–949. 1997.
|
|
131
|
Li X, Yang L and Chen LL: The biogenesis,
functions, and challenges of circular RNAs. Mol Cell. 71:428–442.
2018. View Article : Google Scholar
|
|
132
|
Legnini I, Di Timoteo G, Rossi F, Morlando
M, Briganti F, Sthandier O, Fatica A, Santini T, Andronache A, Wade
M, et al: Circ-ZNF609 is a circular rna that can be translated and
functions in myogenesis. Mol Cell. 66:22–37.e9. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Pamudurti NR, Bartok O, Jens M,
Ashwal-Fluss R, Stottmeister C, Ruhe L, Hanan M, Wyler E,
Perez-Hernandez D, Ramberger E, et al: Translation of CircRNAs. Mol
Cell. 66:9–21.e7. 2017. View Article : Google Scholar :
|
|
134
|
Haimov O, Sinvani H and Dikstein R:
Cap-dependent, scanning-free translation initiation mechanisms.
Biochim Biophys Acta. 1849:1313–1318. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Yang Y, Gao X, Zhang M, Yan S, Sun C, Xiao
F, Huang N, Yang X, Zhao K, Zhou H, et al: Novel role of FBXW7
Circular RNA in repressing glioma tumorigenesis. J Natl Cancer
Inst. 110:304–315. 2018. View Article : Google Scholar :
|
|
136
|
Zhang M, Huang N, Yang X, Luo J, Yan S,
Xiao F, Chen W, Gao X, Zhao K, Zhou H, et al: A novel protein
encoded by the circular form of the SHPRH gene suppresses glioma
tumorigenesis. Oncogene. 37:1805–1814. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Liang WC, Wong CW, Liang PP, Shi M, Cao Y,
Rao ST, Tsui SKW, Waye MMY, Zhang Q, Fu WM and Zhang JF:
Translation of the circular RNA circβ-catenin promotes liver cancer
cell growth through activation of the wnt pathway. Genome Biol.
20:842019. View Article : Google Scholar
|
|
138
|
Huang X, He M, Huang S, Lin R, Zhan M,
Yang D, Shen H, Xu S, Cheng W, Yu J, et al: Circular RNA circERBB2
promotes gallbladder cancer progression by regulating
PA2G4-dependent rDNA transcription. Mol Cancer. 18:1662019.
View Article : Google Scholar
|
|
139
|
Chen RX, Chen X, Xia LP, Zhang JX, Pan ZZ,
Ma XD, Han K, Chen JW, Judde JG, Deas O, et al: 6-methyladenosine
modification of circNSUN2 facilitates cytoplasmic export and
stabilizes HMGA2 to promote colorectal liver metastasis. Nat
Commun. 10:46952019. View Article : Google Scholar
|
|
140
|
Wu P, Fang X, Liu Y, Tang Y, Wang W, Li X
and Fan Y: N6-methyladenosine modification of circCUX1 confers
radio-resistance of hypopharyngeal squamous cell carcinoma through
caspase1 pathway. Cell Death Dis. 12:2982021. View Article : Google Scholar
|
|
141
|
Park OH, Ha H, Lee Y, Boo SH, Kwon DH,
Song HK and Kim YK: Endoribonucleolytic cleavage of m6A-containing
RNAs by RNase P/MRP complex. Mol Cell. 74:494–507.e8. 2019.
View Article : Google Scholar
|
|
142
|
Zhang L, Hou C, Chen C, Guo Y, Yuan W, Yin
D, Liu J and Sun Z: The role of N6-methyladenosine (m6A)
modification in the regulation of circRNAs. Mol Cancer. 19:1052020.
View Article : Google Scholar
|
|
143
|
Chen YG, Chen R, Ahmad S, Verma R, Kasturi
SP, Amaya L, Broughton JP, Kim J, Cadena C, Pulendran B, et al:
N6-methyladenosine modification controls circular RNA immunity. Mol
Cell. 76:96–109.e9. 2019. View Article : Google Scholar
|
|
144
|
Lux S, Blätte TJ, Gillissen B, Richter A,
Cocciardi S, Skambraks S, Schwarz K, Schrezenmeier H, Döhner H,
Döhner K, et al: Deregulated expression of circular RNAs in acute
myeloid leukemia. Blood Adv. 5:1490–1503. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Bell CC, Fennell KA, Chan YC, Rambow F,
Yeung MM, Vassiliadis D, Lara L, Yeh P, Martelotto LG, Rogiers A,
et al: Targeting enhancer switching overcomes non-genetic drug
resistance in acute myeloid leukaemia. Nat Commun. 10:27232019.
View Article : Google Scholar :
|
|
146
|
Arteaga CL and Engelman JA: ERBB
receptors: From oncogene discovery to basic science to
mechanism-based cancer therapeutics. Cancer Cell. 25:282–303. 2014.
View Article : Google Scholar :
|
|
147
|
Conn SJ, Pillman KA, Toubia J, Conn VM,
Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA and
Goodall GJ: The RNA binding protein quaking regulates formation of
circRNAs. Cell. 160:1125–1134. 2015. View Article : Google Scholar
|
|
148
|
L'Abbate A, Tolomeo D, Cifola I,
Severgnini M, Turchiano A, Augello B, Squeo G, D'Addabbo P,
Traversa D, Daniele G, et al: MYC-containing amplicons in acute
myeloid leukemia: Genomic structures, evolution, and
transcriptional consequences. Leukemia. 32:2152–2166. 2018.
View Article : Google Scholar
|
|
149
|
Guarnerio J, Bezzi M, Jeong JC, Paffenholz
SV, Berry K, Naldini MM, Lo-Coco F, Tay Y, Beck AH and Pandolfi PP:
Oncogenic role of fusion-circRNAs derived from cancer-associated
chromosomal translocations. Cell. 165:289–302. 2016. View Article : Google Scholar
|
|
150
|
Wu DM, Wen X, Han XR, Wang S, Wang YJ,
Shen M, Fan SH, Zhang ZF, Shan Q, Li MQ, et al: Role of circular
RNA DLEU2 in human acute myeloid leukemia. Mol Cell Biol.
38:e00259–e00218. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Ping L, Jian-Jun C, Chu-Shu L, Guang-Hua L
and Ming Z: Silencing of circ_0009910 inhibits acute myeloid
leukemia cell growth through increasing miR-20a-5p. Blood Cells Mol
Dis. 75:41–47. 2019. View Article : Google Scholar
|
|
152
|
Fan H, Li Y, Liu C, Liu Y, Bai J and Li W:
Circular RNA-100290 promotes cell proliferation and inhibits
apoptosis in acute myeloid leukemia cells via sponging miR-203.
Biochem Biophys Res Commun. 507:178–184. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Chen H, Liu T, Liu J, Feng Y, Wang B, Wang
J, Bai J, Zhao W, Shen Y, Wang X, et al: Circ-ANAPC7 is upregulated
in acute myeloid leukemia and appears to target the miR-181 family.
Cell Physiol Biochem. 47:1998–2007. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Li W, Zhong C, Jiao J, Li P, Cui B, Ji C
and Ma D: Characterization of hsa_circ_0004277 as a new biomarker
for acute myeloid leukemia via circular RNA profile and
bioinformatics analysis. Int J Mol Sci. 18:5972017. View Article : Google Scholar :
|
|
155
|
Shang J, Chen WM, Wang ZH, Wei TN, Chen ZZ
and Wu WB: CircPAN3 mediates drug resistance in acute myeloid
leukemia through the miR-153-5p/miR-183-5p-XIAP axis. Exp Hematol.
70:42–54.e3. 2019. View Article : Google Scholar
|
|
156
|
Hirsch S, Blätte TJ, Grasedieck S,
Cocciardi S, Rouhi A, Jongen-Lavrencic M, Paschka P, Krönke J,
Gaidzik VI, Döhner H, et al: Circular RNAs of the nucleophosmin
(NPM1) gene in acute myeloid leukemia. Haematologica.
102:2039–2047. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Chen LL: The biogenesis and emerging roles
of circular RNAs. Nat Rev Mol Cell Biol. 17:205–211. 2016.
View Article : Google Scholar
|
|
158
|
Okcanoğlu TB and Gündüz C: Circular RNAs
in leukemia (Review). Biomed Rep. 10:87–91. 2019.
|
|
159
|
Qu S, Yang X, Li X, Wang J, Gao Y, Shang
R, Sun W, Dou K and Li H: Circular RNA: A new star of noncoding
RNAs. Cancer Lett. 365:141–148. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Dudekula DB, Panda AC, Grammatikakis I, De
S, Abdelmohsen K and Gorospe M: CircInteractome: A web tool for
exploring circular RNAs and their interacting proteins and
microRNAs. RNA Biol. 13:34–42. 2016. View Article : Google Scholar
|
|
161
|
Wang E, Lu SX, Pastore A, Chen X, Imig J,
Lee SC, Hockemeyer K, Ghebrechristos YE, Yoshimi A, Inoue D, et al:
Targeting an RNA-binding protein network in acute myeloid leukemia.
Cancer Cell. 35:369–384.e7. 2019. View Article : Google Scholar
|
|
162
|
Yoshida K, Sanada M, Shiraishi Y, Nowak D,
Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et
al: Frequent pathway mutations of splicing machinery in
myelodysplasia. Nature. 478:64–69. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Sun YM, Wang WT, Zeng ZC, Chen TQ, Han C,
Pan Q, Huang W, Fang K, Sun LY, Zhou YF, et al: circMYBL2, a
circRNA from MYBL2, regulates FLT3 translation by recruiting PTBP1
to promote FLT3-ITD AML progression. Blood. 134:1533–1546. 2019.
View Article : Google Scholar
|
|
164
|
Guil S and Esteller M: Cis-acting
noncoding RNAs: Friends and foes. Nat Struct Mol Biol.
19:1068–1075. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Mercer TR and Mattick JS: Structure and
function of long noncoding RNAs in epigenetic regulation. Nat
Struct Mol Biol. 20:300–307. 2013. View Article : Google Scholar
|
|
166
|
Conn VM, Hugouvieux V, Nayak A, Conos SA,
Capovilla G, Cildir G, Jourdain A, Tergaonkar V, Schmid M, Zubieta
C and Conn SJ: A circRNA from SEPALLATA3 regulates splicing of its
cognate mRNA through R-loop formation. Nat Plants. 3:170532017.
View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Schmitz KM, Mayer C, Postepska A and
Grummt I: Interaction of noncoding RNA with the rDNA promoter
mediates recruitment of DNMT3b and silencing of rRNA genes. Genes
Dev. 24:2264–2269. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Starke S, Jost I, Rossbach O, Schneider T,
Schreiner S, Hung LH and Bindereif A: Exon circularization requires
canonical splice signals. Cell Rep. 10:103–111. 2015. View Article : Google Scholar
|
|
169
|
van Rossum D, Verheijen BM and Pasterkamp
RJ: Circular RNAs: Novel regulators of neuronal development. Front
Mol Neurosci. 9:742016. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Chen C, Yuan W, Zhou Q, Shao B, Guo Y,
Wang W, Yang S, Guo Y, Zhao L, Dang Q, et al:
N6-methyladenosine-induced circ1662 promotes metastasis of
colorectal cancer by accelerating YAP1 nuclear localization.
Theranostics. 11:4298–4315. 2021. View Article : Google Scholar
|
|
171
|
Dai F, Wu Y, Lu Y, An C, Zheng X, Dai L,
Guo Y, Zhang L, Li H, Xu W and Gao W: Crosstalk between RNA m6A
modification and non-coding RNA contributes to cancer growth and
progression. Mol Ther Nucleic Acids. 22:62–71. 2020. View Article : Google Scholar
|
|
172
|
Harding CV, Heuser JE and Stahl PD:
Exosomes: Looking back three decades and into the future. J Cell
Biol. 200:367–371. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Melo SA, Sugimoto H, O'Connell JT, Kato N,
Villanueva A, Vidal A, Qiu L, Vitkin E, Perelman LT, Melo CA, et
al: Cancer exosomes perform cell-independent MicroRNA biogenesis
and promote tumorigenesis. Cancer Cell. 26:707–721. 2014.
View Article : Google Scholar :
|
|
174
|
Boyiadzis M and Whiteside TL: Exosomes in
acute myeloid leukemia inhibit hematopoiesis. Curr Opin Hematol.
25:279–284. 2018. View Article : Google Scholar
|