Introduction
RNA modifications have emerged as a new layer of
epigenetic regulation and are anticipated to further deepen our
understanding of molecular complexity and to optimize therapeutic
inventions for patients. Currently, more than 170 RNA
modifications, including N6-methyladenosine
(m6A), N1-methyladenosine
(m1A), and 5-methylcytidine (m5C), have been
identified in nearly all classes of coding and non-coding RNAs
(1). Of these modifications,
m6A has been comprehensively characterized; it regulates
gene expression and various cellular processes, including RNA
folding, stability, splicing, transport, and translation.
Accumulating evidence suggests that RNA modifications mediate
various biological processes, and aberrant RNA modification has
been linked to numerous human diseases such as cancer and cognitive
dysfunctions (2-5).
Hepatocellular carcinoma (HCC) is the most common
type of primary liver cancer and the fourth leading cause of
cancer-related deaths worldwide (6). Similar to the evolution of other
cancers, hepatocarcinogenesis is a multistep process involving
genetic and epigenetic alterations (7-12).
In terms of RNA modulations in HCC, only a few prevalent mRNA
regulators have been examined. For example, m6A writer
methyltransferase-like 3 (METTL3) has been reported as an
unfavorable prognostic factor in HCC, and its overexpression
contributes to HCC progression by reducing cytokine signaling 2
(SOCS2) messenger RNA (mRNA) stability in an
m6A-dependent manner (13). Intriguingly, METTL3 has been
recruited to the transcriptional start sites, and the
promoter-bound METTL3 induces co-transcriptionally modified
m6A within the coding region of the transcript (14). Another m6A writer
member, METTL14, is associated with HCC metastasis by regulating
the processing of miR-126 (15).
Incidentally, miR-126 is downregulated in various cancers and is
known to play a role in carcinogenesis in various cancers,
including in HCC (16). The
m6A reader protein YTHDF2 has been shown to suppress
cancer cell proliferation by destabilizing epidermal growth factor
receptor (EGFR) mRNA in HCC (17).
To date, only a few studies have been conducted on
METTL6, a tRNA methylation enzyme, with limited knowledge about its
role and mechanism of action in cancer. One study showed that
METTL6 was upregulated in highly proliferative luminal breast
cancer (18) and another study
revealed that knockout of METTL6 altered cisplatin sensitivity in
lung cancer cells (19). Very
recently, Ignatova et al reported that loss of METTL6
impaired the pluripotency of mouse stem cells, and that this
molecular was a vital regulator of HCC cell proliferation (20). Another recently published paper
established a prognostic risk score based on five
metabolism-related genes, including that of METTL6, indicating
high-risk patients had a poor overall survival (OS) compared to
low-risk patients with HCC (21).
Although METTL6 has been identified in various human
cancers, the characterization of this tRNA modification and its
mechanisms of action in cancer remain unclear. Hence, the aim of
this study was to investigate the functional roles of METTL6 in
cancer development, and to study its potential mechanisms of
action. In the present study, we demonstrated that tumorigenicity
in HCC was due to the upregulation of METTL6. Furthermore, the
depletion of METTL6 mitigated HCC cell proliferation, migration,
invasion, and attachment ability, possibly through the regulation
of cell adhesion-related genes post-transcriptionally. In addition,
METTL6 was found to be localized in the cytosol. Our findings may
provide insights into the functional roles of tRNA and
post-transcriptional regulation in cancer, and may lead to the
development of a therapeutic strategy for HCC.
Materials and methods
Bioinformatics analysis
We retrieved multi-omics data from RNA-sequencing
(RNA-seq), DNA promoter methylation, DNA copy number, and clinical
information from The Cancer Genome Atlas (TCGA) using cBioPortal
(https://www.cbio-portal.org)
corresponding to a total of 372 HCC samples. The differential
expression analysis between HCC and non-HCC tissues and across four
different tumor stages, as well as OS and disease-free survival
(DFS) analyses were performed using the Gene Expression Profiling
Interactive Analysis (GEPIA) webserver (http://gepia.cancer-pku.cn/). In addition, GraphPad
Prism 8 software (GraphPad Software, Inc.) was used to analyze the
association between DNA copy number alterations (CNAs) and mRNA
expression. A one-way ANOVA test was performed to compare mRNA
expression levels between groups of different CNAs. Statistical
significance was set at P<0.05.
Plasmid DNA constructs
The lentiviral packaging plasmids pMD2.G and psPAX2
were obtained from Addgene (#12259 and #12260, Addgene, USA,
respectively). To generate inducible Cas9 nuclease-expressing cell
lines, we purchased the Edit-R inducible lentiviral plasmid
(#CAS11229, Dharmacon, UK). Five individual sgRNAs targeting the
METTL6 gene used in this study were as follows (5′→3′
orientation): sgRNA1: 5′-GGA GCT AAG ATC ATG TAG AG-3′, sgRNA2:
5′-ATA TGA TAC AGA AAG ATG CA-3′, sgRNA3: 5′-GTT TCA TAG GTA TTA
AAA CC-3′, sgRNA4: 5′-ATA ACA ACA TCC ACA GAC TC-3′ and sgRNA5:
5′-ACA GCA GAA ATT GGA ACA AG-3′. sgRNAs were designed and cloned
into the pLKO.1-puro U6 sgRNA BfuAI large stuffer plasmid (#52628,
Addgene). All plasmids were verified using Sanger sequencing.
Cell lines and culture conditions
The human HCC cell lines SNU-423 and SNU-475 were
purchased from the American Type Culture Collection (ATCC), and
tested and authenticated using DNA profiling for polymorphic short
tandem repeat (STR) markers analyzed by BEX Co. Ltd. using
GenePrint 10 systems (Promega Corp.) before starting the project
(Table SI). The human liver
cancer cell lines HepG2, Huh-7, and Li-7 were purchased from RIKEN
Bioresource Research Center (RIKEN BRC, Japan), and tested and
authenticated using DNA profiling for polymorphic short tandem
repeat (STR) markers analyzed by Applied Biosystems using
GeneMapper ID software before starting the project (Table SI). SNU-423, SNU-475, Huh-7, and
Li-7 cells were cultured as monolayers in RPMI-1640 supplemented
with 10% FBS and antibiotics, and HepG2 cells were cultured as
monolayers in MEM supplemented with 10% FBS, 0.1 mM NEAA and
antibiotics. All cells were maintained at 37°C in humid air with 5%
CO2. In the present study, SNU423 and SNU475 cells were
mainly used as HCC cell lines for various experiments. The reason
for this is as follows. i) HepG2 was originally considered a
hepatocellular carcinoma cell line, but it has been shown to be a
hepatoblastoma (22). In
addition, HepG2 cells were established from a younger boy (15 years
of age). Therefore, we decided not to use this line in further
studies. ii) As for Huh-7 cells, previous literature has indicated
that Huh-7 cells may produce adhesion-related factors and growth
factors autonomously (23).
Therefore, we considered Huh-7 cells as inappropriate because we
examined the role of adhesion-related genes with and without METTL6
expression. iii) Regarding Li-7 cells, a previous study showed that
of five hepatocellular carcinoma cells, only Li-7 cells showed a
population change after two months of culture (24). Then, we decided not to use this
line in further studies. iv) SNU-423 is an adult hepatocellular
carcinoma that was generated from a 40-year-old man. SNU-475 is
another adult hepatocellular carcinoma cell line generated from a
43-year-old man. In addition, we previously constructed a knockout
system for both SNU-423 and SNU475 cells using CRISPR/Cas9, and
confirmed that the target genes were properly knocked out (25). Hence, we decided to use these two
cell lines mainly in this study.
CRISPR/Cas9 gene editing system
A lentivirus transduction system was used to
generate CRISPR/Cas9 knockout cells. To produce lentiviruses, viral
vectors and packaging plasmids were co-transfected into 293T cells
using Lipofectamine 3000 (#L3000-008, Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. After 48 h, the
cell culture medium containing lentiviruses was collected and
filtered through a 0.45-µm filter. Target cell lines were
plated in 6-well plates and cultured with lentivirus-containing
medium for 3 days, which was carried out in the absence of
polybrene. Stable cell clones were selected using blasticidin S (10
µg/ml) (#029-18701, FUJIFILM Wako Pure Chemical Corporation,
Japan). For inducible CRISPR/Cas9 knockout experiments, conditional
Cas9 expression cells further underwent lentivirus transduction of
conditional sgRNA expression plasmid and selection in the presence
of both blasticidin S (10 µg/ml) and puromycin (2
µg/ml) (#ant-pr-1, InvivoGen). Knockout of METTL6 was
induced by adding doxycycline (Dox) (1 µg/ml) (#D9891,
Sigma-Aldrich; Merck KGaA) for 48 h.
Western blot analysis
SNU-423 and SNU-475 cells were lysed with CelLytic™
Reagent (#C2978, Sigma-Aldrich; Merck KGaA) using standard methods.
Protein lysates (15 µg) were separated on a 10% SDS-PAGE gel
and transferred to nitrocellulose membranes. The membranes were
blocked with 5% skimmed non-fat milk for 2 h at 25°C, and then the
membranes were incubated with rabbit polyclonal anti-METTL6 primary
antibody (#HPA035166, Sigma-Aldrich; Merck KGaA; dilution used in
WB: 1:2,000); rabbit polyclonal anti-METTL2 primary antibody
(#PA5-113304, Invitrogen/Thermo Fisher Scientific, Inc.; dilution
used in WB: 1:2,000); rabbit poly-clonal anti-METTL8 primary
antibody (#ab122273, Abcam; dilution used in WB: 1:2,000); rabbit
polyclonal anti-ITGA1 primary antibody (#22146-1-AP, Proteintech;
dilution used in WB: 1:2,000); chicken polyclonal anti-SPON1
primary antibody (#ab14271, Abcam; dilution used in WB: 1:1,000);
goat polyclonal anti-CLDN14 primary antibody (#ab19035, Abcam;
dilution used in WB: 1:1,000); and mouse monoclonal anti-α-tubulin
(DM1A, EMD Millipore; dilution used in WB: 1:1,000) antibodies at
4°C overnight. After primary antibody incubation, the membranes
were incubated with an HRP-linked donkey anti-rabbit secondary
antibody (#NA934V, GE Healthcare, USA; dilution used in WB:
1:5,000) and mouse anti-goat IgG-HRP (#sc-2354, Santa Cruz
Biotechnology, Inc.; dilution used in WB: 1:5,000) at 25°C for 1 h.
The signal was detected using the ECL system (#RPN2236, GE
Healthcare, USA), and NIH ImageJ software (version 1.53) (National
Institutes of Health) was used for quantification (26).
Colony formation assay
Cells were seeded in 6-well plates at a density of
1,000 cells/well and cultured at 37°C. The culture medium with or
without Dox was replaced every 3 days. After 14 days, cells were
fixed with 1% formaldehyde (#252549, Sigma-Aldrich; Merck KGaA) and
1% methanol (#137-01823, FUJIFILM Wako Pure Chemical Corporation),
stained with 0.05% crystal violet (#V5265, Sigma-Aldrich; Merck
KGaA) for 20 min, and then washed three times with Milli-Q water.
Colony numbers were quantified using the NIH ImageJ software
(26).
Cell proliferation assay
Cell viability was measured using the Cell Counting
Kit-8 (CCK-8) (#343-07623, Dojindo Laboratory, Japan). Cells were
seeded in 96-well plates at a density of 1.0×103
cells/well and cultured for 24, 48, 72 and 96 h. Then, 10 µl
of CCK-8 solution was added and the plates were incubated at 37°C
for 2 h. The absorbance of each well was measured at 450 nm using a
microplate reader (Multiskan FC, Thermo Fisher Scientific,
Inc.).
Cell scratch assay
Cells were seeded in 6-well plates at a density of
3×105 cells/well and incubated for 24 h. When the
cellular confluence reached 90%, a 200-µl pipette tip was
used to create wounds in the monolayer cells. The wells were then
rinsed with PBS three times to remove any detached cells, and fresh
serum-free medium was added. The wells were then placed in an
incubator, and visualized using a phase-contrast microscope
(Olympus CKX53, Japan) at 0 and 48 h. The percentage of wound area
was calculated using the ImageJ software.
Cell invasion assay
For the cell invasion assay, a 96-well Boyden
chamber coated with basement membrane extract (BME) was used
(#ab235697, Abcam). Prior to the assay, the cells were starved for
24 h in serum-free media. Then, cells were suspended in serum-free
medium and seeded into the upper chamber (8.0-µm pore size)
at a density of 50,000 cells per well. Further, 200 µl of
RPMI medium containing 10% serum was added to the lower chamber.
The cells were allowed to invade for 24 h at 37°C. A standard curve
was constructed for each cell type. After incubation, cells were
washed carefully, and those that did not invade the lower side of
the chamber were removed from the top side. Then, 100 µl of
the cell invasion dye and cell dissociation solution were added to
each well in the lower chamber. The chamber was incubated at 37°C
for 1 h, and the bottom wells were read at excitation and emission
wavelengths of 530/590 nm using a Synergy H1 Hybrid Multi-Mode
Reader (BioTrek).
Cell adhesion assay
Cells were seeded in 6-well plates at a density of
0.5×105 cells/well (SNU-423) and 1.0×105
cells/well (SNU-475). The cells were then incubated for 15, 30, 45,
and 60 min. Next, the wells were washed with PBS three times to
remove any detached cells, and images were obtained using a
phase-contrast microscope (Olympus CKX53, Japan) at 15, 30, 45, and
60 min. Cell numbers were calculated using ImageJ software.
RNA-seq and data analysis
Total RNA was extracted using the QIAzol lysis
reagent (#79306, Qiagen). Extracted RNA was treated with DNase I
(#043-31261, FUJIFILM Wako Pure Chemical Corporation) at 37°C for
30 min, followed by acid phenol:chloroform extraction (#AM9722,
Thermo Fisher Scientific, Inc.). Purified RNA was re-suspended in
Takara's buffer for mRNA amplification using 5′ template switching
PCR with Takara's SMART-Seq v4 Ultra Low Input RNA Kit (both from
Takara). The amplified cDNA was fragmented and appended with
dual-indexed barcodes using Illumina Nextera XT DNA Library Prep
Kits (Illumina, Inc.). Libraries were validated by electrophoresis,
pooled, and sequenced on an Illumina NovaSeq 6000 (150 base pairs,
paired ends) (Illumina, Inc.). RNA-seq data were mapped to the hg38
version of the human genome using the DRAGEN Bio-IT Platform
(v3.6.3) (Illumina, Inc.). Raw counts were converted into
transcripts per million reads. Genes with q≤0.05 and |log2_ratio|≥1
were identified as differentially expressed genes (DEGs) after the
knockout of METTL6 in SNU-423_KO2, SNU-423_KO5, SNU-475_KO3, and
SNU-475_KO4. Gene ontology (GO) and Kyoto Encyclopedia of Genes and
Genomes (KEGG) enrichment analyses of the DEGs were performed using
Metascape (27) with P<0.05,
indicating statistically significant enrichment. Reads from all
sequencing experiments were deposited into the DNA Data Bank of
Japan (DDBJ) with the accession number DRA012940.
Chromatin immunoprecipitation sequencing
and data analysis
Chromatin immunoprecipitation sequencing (ChIP-seq)
analyses were performed using the SimpleChIP Enzymatic Chromatin IP
Kit (#9003, Cell Signaling Technology, Inc.) with minor
modifications. Briefly, cells cultured in 10 cm2 dishes
were fixed in 1% formaldehyde for 10 min, and fixation was quenched
with the addition of glycine to 125 mM for an additional 5 min.
Cells were harvested by scraping from the plates and stored at
−80°C. During the solubilization of chromatin, fixed nuclei were
sonicated with an E220 focused ultrasonicator (Covaris, USA).
Immunoprecipitation antibodies were rabbit polyclonal anti-H3K27ac
(#ab4729, Abcam) and rabbit polyclonal anti-CTCF (#3418s, Cell
Signaling Technology, Inc.). Approximately 5 µg of chromatin
was incubated with the indicated antibody overnight at 4°C on a
rotator. Then, FG Beads HM Protein G (#TAB8848N3173, Tamagawa
Seiki, Japan) was added to the solution and washed with buffer, as
described previously (28).
Immunoprecipitated chromatin was eluted and reverse-crosslinked
according to the manufacturer's instructions (#9003, Cell Signaling
Technology, Inc.). Immunoprecipitated DNA was purified using a
QIAquick PCR Purification Kit (#28106, Qiagen). DNA libraries were
prepared using the QIAseq Ultralow Input Library Kit (#180492,
Qiagen) for Illumina. Library quality was checked using a
TapeStation 4200 instrument (Agilent Technologies, Inc.). DNA
libraries were sequenced using the NovaSeq 6000 system (Illumina).
Sequencing reads from ChIP-seq experiments were mapped to the hg38
version of the human genome using Bowtie (v2.2.9) and parameters
(local) (29). Duplicate reads
were removed using the Samtools (v1.3.1). The normalized ChIP-seq
signals were visualized using the Integrative Genomics Viewer (IGV;
v2.3.91) (30). Reads from all
sequencing experiments were deposited into the DNA Data Bank of
Japan (DDBJ) with the accession number DRA012940.
Immunofluorescence analysis
Cells were seeded at a density of 1×104
cells/well (SNU-423_KO2) with and without Dox treatment in 2-well
chamber culture slides (#154852, Lab-Tek™ II CC2™ Chamber Slide
System, Thermo Fisher Scientific, Inc.). After 4 days, the cells
were rinsed with ice-cold PBS and fixed with 4% paraformaldehyde
(#163-20145, Paraformaldehyde Phosphate Buffer Solution, FUJIFILM
Wako Pure Chemical Corporation) for 15 min at 25°C. Cells were then
blocked with 3% BSA (#A-9647, bovine serum albumin, Sigma-Aldrich;
Merck KGaA) and 0.3% Triton X-100 [#160-24751,
Polyoxyethylene(10) Octylphenyl
Ether, FUJIFILM Wako Pure Chemical Corporation] for 60 min. The
cells were subjected to immunofluorescence staining with rabbit
polyclonal METTL6 primary antibody (#HPA035166, Sigma-Aldrich;
Merck KGaA; dilution used: 1:200) overnight at 4°C. Next, the cells
were washed with cold PBS three times for 5 min each time, and
incubated with donkey anti-rabbit IgG (H+L) secondary antibody
Alexa Fluor 594 (#A21207, Thermo Fisher Scientific, Inc., dilution
used: 1:500) at 25°C for 1 h. The cells were washed with cold PBS
and mounted using mounting medium containing DAPI (#H-1200, Vector
Laboratories). The cells were examined using fluorescence
microscopy (Celldiscover 7, ZEISS) at 20× with 5×5 tiling.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism 8.0 (GraphPad Software, Inc.). The cut-off value for
METTL6 mRNA expression was determined at the median level.
Differences in METTL6 mRNA expression levels between HCC
samples and control tissues, as well as expression levels at
different stages, were calculated using one-way analysis of
variance (ANOVA). Survival curves were obtained using Kaplan-Meier
curves and log-rank tests. The association of METTL6 mRNA
expression levels with DNA CNAs was assessed using one-way ANOVA.
All quantitative results, including relative expression analysis by
western blot analysis, cell colony assay, cell proliferation assay,
cell scratch assay, and cell attachment assay, are presented as
means ± standard deviation (SD) of at least three separate
experiments. A two-tailed unpaired t-test was used to compare the
mean values between the two groups. Results were considered
significant when the P-value was *P<0.05,
**P<0.01, ***P<0.001, ****P<0.0001,
or NS=non-significant (as indicated with these symbols/initials in
the figures).
Results
METTL6 is elevated in HCC tissues and is
associated with poor prognosis
First, we established a list of 60 known RNA
regulators, including methyltransferases, demethylases, and
acetyltransferases. We then analyzed their mRNA expression levels
in HCC tumor samples and adjacent non-tumor tissues and examined
whether they might be correlated with HCC patient survival rates
based on the Cancer Genome Atlas (TCGA) and Genotype-Tissue
Expression (GTEx) (31) and
OncoLnc platforms based on TCGA independently (32). To assess the novelty of these
regulators, we conducted a literature survey of HCC-related studies
for each candidate (Table SII
and Fig. S1). Notably, these
results suggest that METTL6, which was recently identified as a
methylcytidine (m3C) methyltransferase on tRNA (33) might be a promising novel target
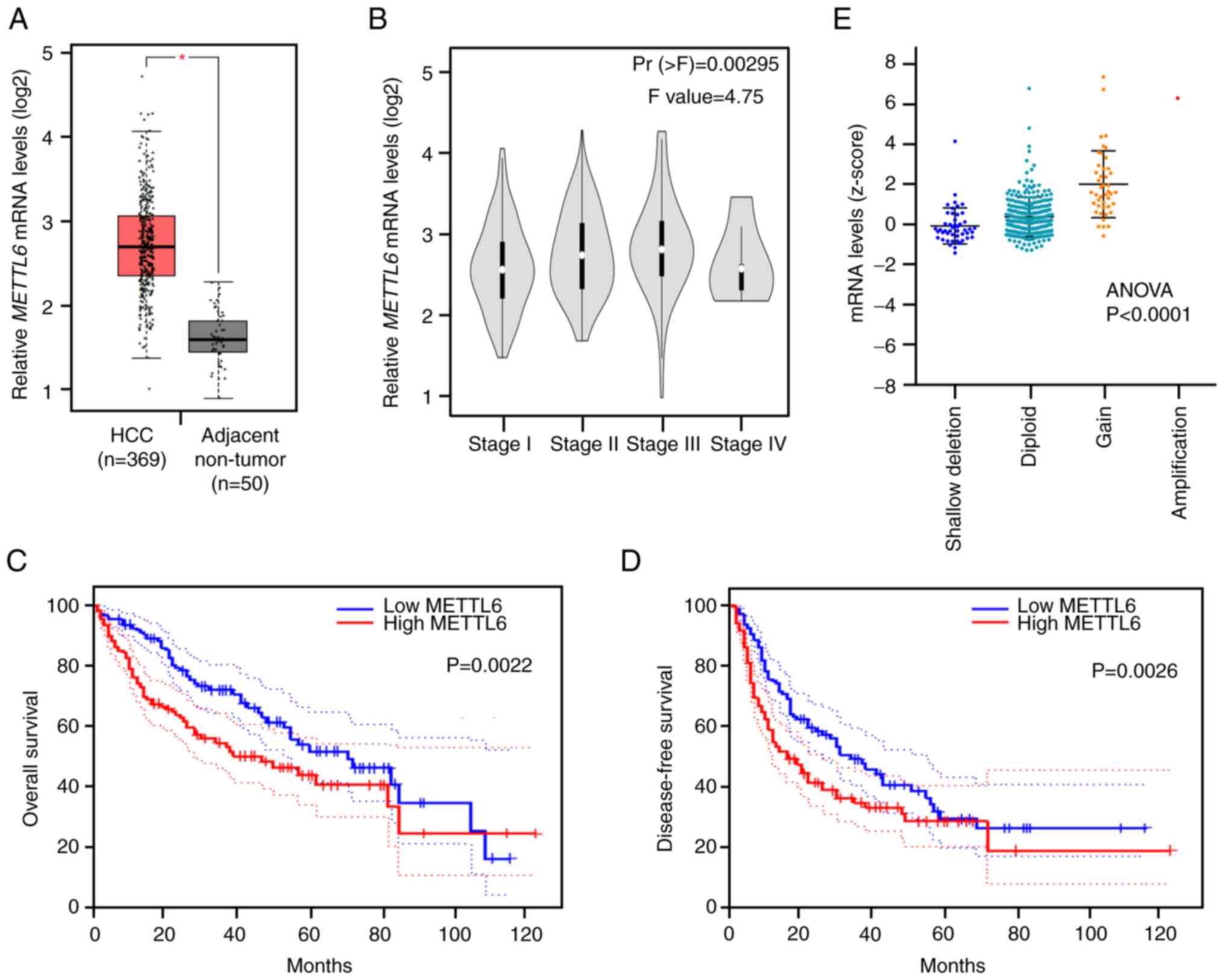
among all candidates. mRNA expression of METTL6 was
significantly upregulated in HCC tumor tissues compared to that in
adjacent non-tumor tissues (Fig.
1A). Meanwhile, a gradual increase in the mRNA level of
METTL6 was observed from tumor stages I to III, while a
slight decrease was observed in stage IV (Fig. 1B). This suggests that METTL6 is
highly expressed in the early stages of cancer and involved in the
development of cancer, and that there is no significant correlation
with the degree of cancer progression. Kaplan-Meier analysis
revealed that patients with higher METTL6 expression levels
had significantly poorer OS and DFS (Fig. 1C and D), suggesting that
METTL6 could be a potential prognostic indicator for
patients with HCC. In addition, we analyzed the TCGA_LIHC (liver
HCC) dataset, which contains CNAs and DNA methylation levels of
METTL6. The METTL6 copy number was positively
correlated with its mRNA levels (Fig.
1E), indicating that the CNAs of METTL6 might be
involved in HCC through alterations in METTL6 gene
expression levels.
Knockout of METTL6 inhibits HCC colony
formation, cell proliferation, and cell migration in vitro
To investigate the biological functions of METTL6 in
HCC and to assess the effect of METTL6 expression, we first
assessed its protein expression levels across five different HCC
cell lines (Fig. S2A). We
established stable METTL6 knockout models in SNU-423 and
SNU-475 cells using the CRISPR/Cas9 gene editing system, with four
independent single-guided RNAs targeting the promoter regions of
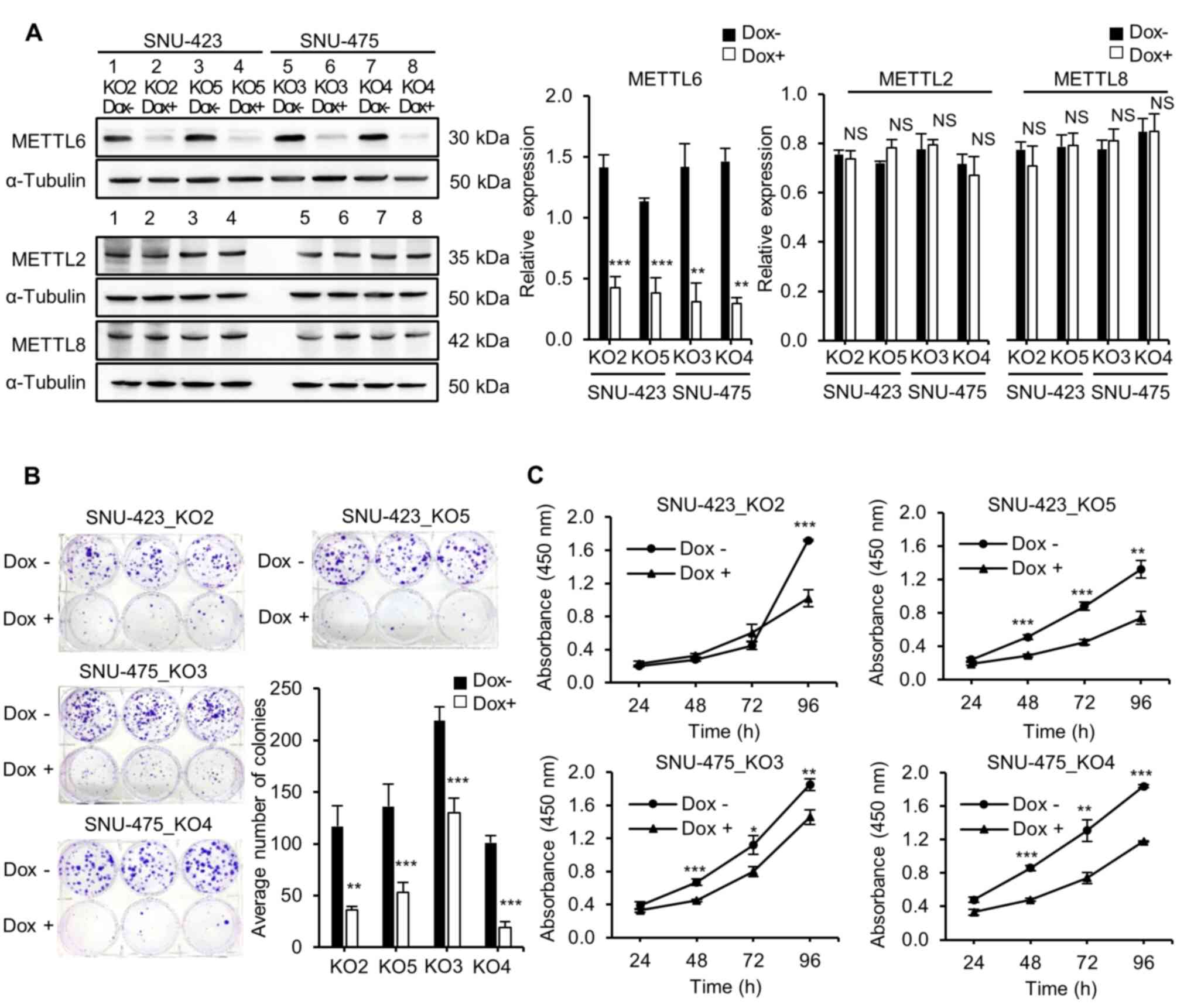
METTL6. Knockout of METTL6 was confirmed using western blot
analysis (Fig. 2A). No noticeable
changes were observed in the protein expression levels of METTL2
and METTL8, which are the other m3C methyltransferases
that exhibit a functional similarity and are closely related to
METTL6, as analyzed by a family tree diagram (33,34). Our bioinformatics analysis
indicated that METTL6 was associated with prognosis;
therefore, we first performed multiple cancer phenotypic assays
using established knockout cell lines. A colony formation assay was
performed to detect the effect of METTL6 expression on the
clonogenicity of HCC cells. As anticipated, the METTL6 knockout
group had significantly fewer and smaller colonies than the METTL6
expressing groups in both SNU-423 and SNU-475 cell lines,
indicating that METTL6 expressing cells had the ability to form
colonies (Fig. 2B).
Next, the role of METTL6 in HCC cells was analyzed
using the CCK-8 to examine cell proliferation. We found that
knockout of METTL6 remarkably inhibited HCC cell viability,
suggesting that METTL6 played a role in cell survival (Fig. 2C). Additionally, we investigated
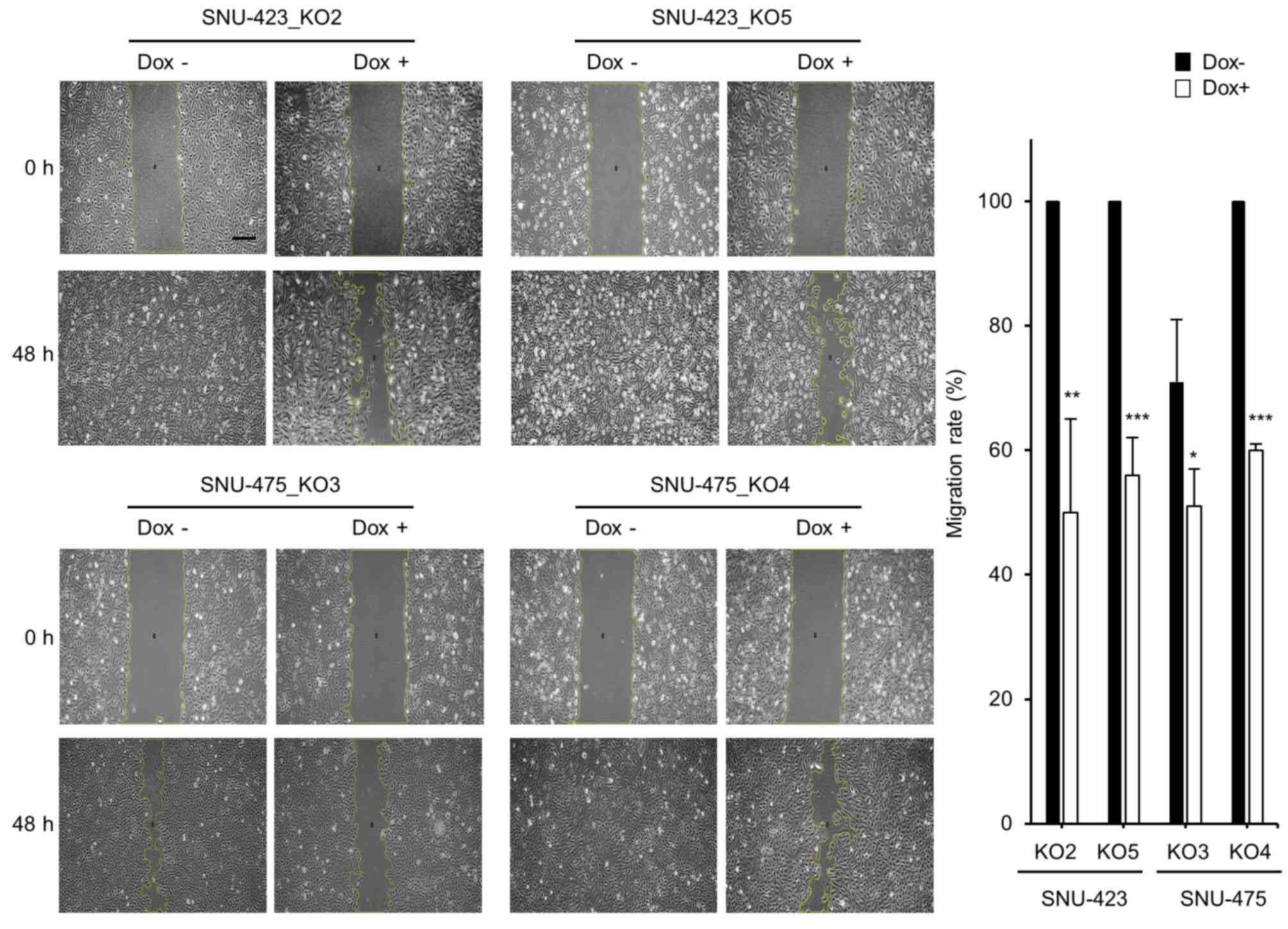
the effect of METTL6 knockout on HCC cell migration. Cell scratch
assays showed that HCC cell migration was significantly suppressed
at 48 h post-scratch in the METTL6 knockout cells (Fig. 3). Collectively, these results
indicate that specific tRNA modifications regulated by METTL6
enhance HCC cell colony formation, proliferation, and migration
abilities.
Loss of METTL6 decreases cell
adhesion-related gene expression levels
To investigate the molecular mechanisms of METTL6
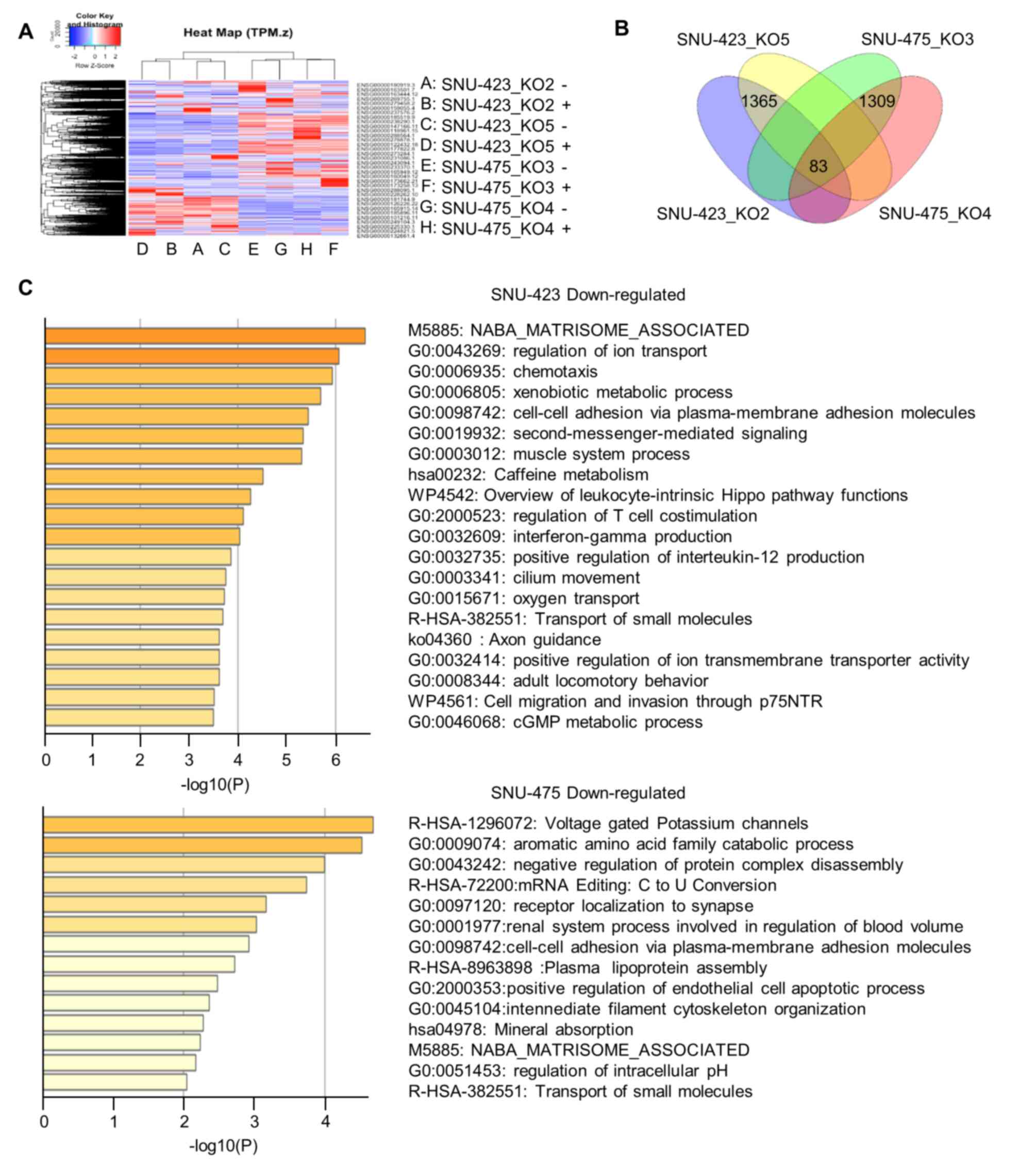
action in HCC, we performed RNA-seq on four METTL6 stable knockout
cells (SNU-423_KO2, SNU-423_KO5, SNU-475_KO3, and SNU-475_KO4). A
heat map and dendrogram summary of the RNA-seq data are shown in
Fig. 4A. In total, we identified
1,365 and 1,309 DEGs in SNU-423 and SNU-475 cells, respectively
(Fig. 4B). GO analysis using
Metascape highlighted that these DEGs were involved in cell-cell
adhesion via plasma-membrane adhesion molecules, extracellular
matrix, cell migration and invasion, cell apoptotic process,
xenobiotic metabolism, and other metabolic processes (Fig. 4C). This indicates that METTL6 may
play an important role in cancer cell biology. In addition, there
was an overlap of 32 upregulated and 51 downregulated genes between
SNU-423 and SNU-475 cells in the different knockout groups
(Fig. 4B). Among the
downregulated genes, we selected a group of cell adhesion proteins
for further investigation, including integrin α-1/β-1 (ITGA1),
claudin-14 molecules (CLDN14), and spondin1 (SPON1). ITGA1 mediates
the distinct interactions between cells and those in the
surrounding extracellular matrix (35); CLDN14 forms tight junctions and
interacts with other CAMs (36)
while SPON1 is another cell adhesion protein that maintains the
extracellular matrix (37). In
addition, these cell adhesion genes have been associated with tumor
progression and metastasis in HCC (38-40). Immunoblotting revealed that the
knockout of METTL6 resulted in a decrease in ITGA1 in SNU-423 cell
lines and CLDN14 in SNU-423 and SNU-475 cell lines (Fig. S2B). Unfortunately, SPON1 protein
expression was not detected in either SNU-423 or SNU-475. RNA-seq
results showed that the average TPM value of ITGA1 in SNU-475 cell
lines was 0.351 while it was 1.61 in SNU-423, which could be one of
the reasons why the protein expression was relatively weak in
SNU-475 cell lines with non-significant decrease after the knockout
of METTL6. In addition, the average TPM value of SPON1 among
SNU-423 and SNU-475 cell lines was 0.13, which indicated that gene
expression levels of SPON1 were the lowest compared with other
proteins. This might explain why SPON1 expression levels were not
detected in these cell lines. Our ChIP-seq analysis demonstrated
that enhancer activity was not significantly different between
control cells and METTL6 KO3 in SNU-475 cells (Fig. S2C). The chromatin binding
activity of the transcription factor CTCF also showed a similar
binding affinity between the control cells and METTL6 KO3 cell
lines (Fig. S2C). These results
suggest that METTL6 does not contribute to the transcriptional
activity but stabilizes the mRNA expression of cell adhesion genes
such as ITGA1 and CLDN14 and increases their protein expression,
probably via post-transcriptional tRNA modifications mediated by
METTL6.
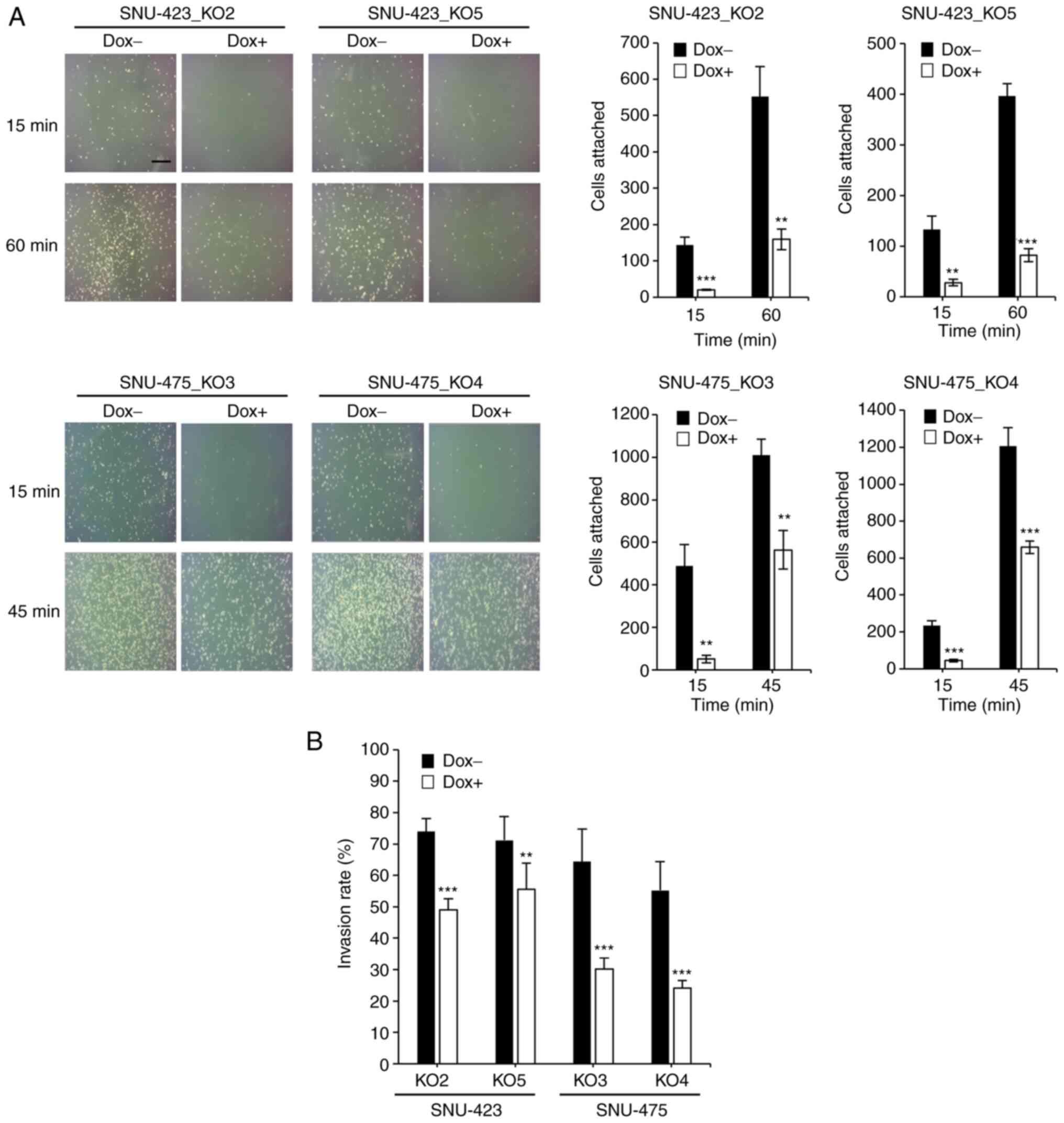
To determine whether METTL6 depletion attenuated
cell adhesion ability and cell invasiveness by altering the
expression levels of adhesion molecules, we performed a cell
adhesion assay, which is often used to evaluate the metastatic
ability of cancer cells. The assay results demonstrated that
knockout of METTL6 dramatically decreased the number of attached
cells after 60 min in SNU-423 and 45 min in SNU-475 cells (Fig. 5A). Furthermore, we conducted a
cell invasion assay and found that the cell invasion ability was
significantly suppressed in METTL6 KO cells (Fig. 5B) compared to that in control
cells. Taken together, RNA-seq analysis, cell adhesion assay and
cell invasion assay results reveal that METTL6 exerts oncogenic
effects by altering the expression of CAMs.
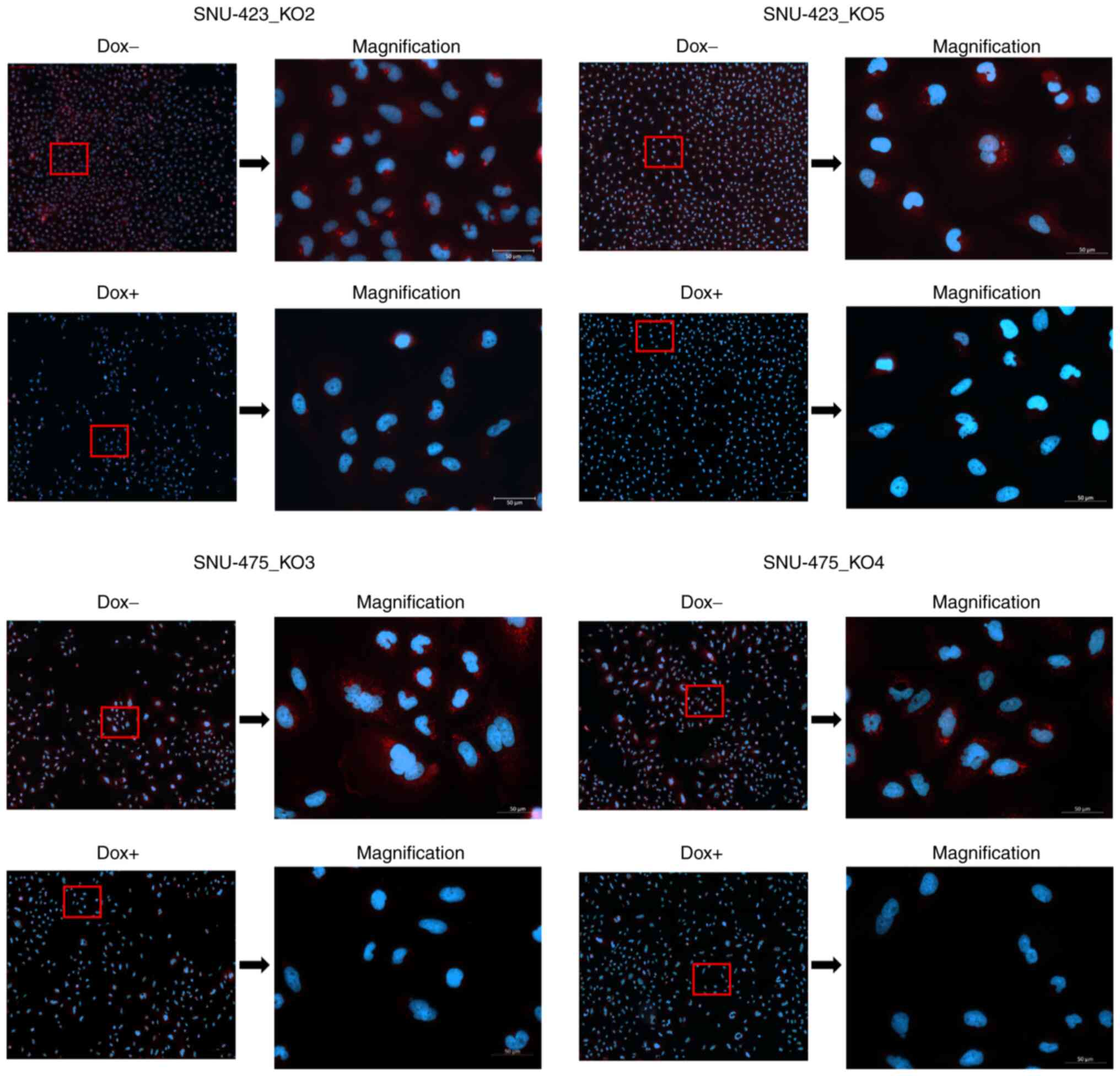
METTL6 is localized in the cytoplasm
Our ChIP-seq results suggested that METTL6 was
directly associated with post-transcriptional regulation; however,
some mRNA expression levels were upregulated and downregulated in
METTL6 KO cells. Therefore, we investigated the subcellular
localization of METTL6. For this, we conducted immunofluorescence
assays and observed an obvious signal reduction in the cytoplasm
after the knockout of METTL6 in SNU-423 and SNU-475 cell
lines, indicating that METTL6 was mainly localized in the cytosol
(Fig. 6). To the best of our
knowledge, this is the first study to confirm that METTL6 is
localized in the cytoplasm. In line with our findings, previous
studies have shown that METTL6 catalyzes the formation of
m3C at C32 (33,41), and another study claimed that
m3C32 modification must occur in the cytoplasm (42).
Discussion
The field of post-transcriptional regulation of gene
expression at the RNA level, and dynamic and reversible
modifications of almost all forms of coding and non-coding RNAs,
including tRNA, have attracted increasing attention. tRNA is an
essential component of protein synthesis. In cancer, deregulation
of tRNA can elevate oncogenic protein translation and fulfill the
energy demand required for cancerous growth. A recent large-scale
analysis revealed an overall overexpression of tRNA levels and
amplification of tRNA modification enzymes across 31 cancer types,
which indicated the dynamic role of tRNA and tRNA modification
proteins in cancer initiation and progression (43). Despite the fact that tRNA
modifications are involved in various cancers, the detailed
characterization of this process and mechanisms of such regulation
are still lacking. This could be attributed to the difficulty of
tRNA quantification due to its abundance, and the intricate
interactions between tRNA and tumor metabolism (44). METTL6 is a tRNA methylation enzyme
that is present in many cancer types; however, we have limited
knowledge about its functions and mechanisms of action in cancer.
Earlier, the enzymatic activity of METTL6 was identified by Xu
et al (33). The knockout
of m3C tRNA-modified enzyme METTL2 showed a 35%
reduction, whereas knockout of METTL6 accounted for 12% reduction
in the total RNA compared to that in the wild-type (WT) tissues.
Moreover, METTL6 is a homolog of Trm141 in S. pombe,
targeting tRNASer; therefore, they identified a novel
target of tRNA and found that m3C32 of
tRNASer(AGA) and tRNASer(GCU) were modified
by METTL6.
RNA modifications of mRNA are spatiotemporally
regulated. For example, METTL14 binds to histone H3 trimethylation
at lysine 36 (H3K36me3), and consequently m6A
modification of mRNA is deposited co-transcriptionally (45). Intriguingly, the METTL3/METTL14
complex co-transcriptionally regulates histone modification. In the
presence of METTL3/METTL14, m6A modified RNA is
recognized by the m6A reader YTHDC1, which recruits
H3K9me2 demethylase KDM3B, and promotes gene expression by
demethylating H3K9me2 (46).
Similar to m6A, m5C and m5C
methyltransferases (RCMTs) contribute to chromatin structure
regulation (47). In our data,
H3K27ac peak levels were identical between METTL6 expressing and
METTL6 depleted cells, suggesting that METTL6 was not associated
with transcriptional regulation. This finding was further supported
by immunocytochemistry in CRISPR/Cas9-mediated knockout cells,
which showed that METTL6 was localized in the cytosol.
As mentioned earlier, more than 170 RNA
modifications have been reported, 90 of which are found in tRNAs
(1). To date, 14 modification
sites have been identified in tRNA (48). It has been reported that
modifications of tRNAs affect the stability of tRNAs and thus the
genetic code of mRNAs can be read accurately (49). Modifications outside the anticodon
region are crucial for tRNA stability and modulating
temperature-sensitive growth (50,51). Additional studies have shown that
mRNA stability is affected by translation, particularly related to
codon stabilization, which is regulated by tRNA anticodon I34
modification by adenosine deaminases (ADATs) (52-54).
The biogenesis and degradation of tRNAs are well
summarized by Motorin and Helm (55). Deregulation of early,
intermediate, and late modifications affected tRNA stability and
degradation. Destabilization of 3D-structured tRNA triggered by
aberrations in RNA modifications involves either the TRAMP pathway
(nuclear tRNA degradation) or rapid tRNA decay (cytoplasmic tRNA
degradation). Widespread tRNA modification regulates tRNA
stability, and hypomodified tRNA is degraded even in bacteria and
archaea (56). Therefore, it is
reasonable to assume that METTTL6 may regulate tRNA stability in
SNU-423 and SNU-475 cell lines and possibly affect mRNA expression
levels, followed by alterations in protein expression.
A tRNA half, which binds with other transcripts and
regulates gene expression as miRNA, may account for a possible
explanation of how METTL6 mediates mRNA expression levels. tRNA
halves are generated by dicers, angiogenins, or other exonucleases
(57,58). Intriguingly, RNA modifications
play important roles in the origin and function of tRNA fragments,
and tRNA modifications promote and protect the expression of tRNA
halves (59). Moreover,
tRNA-derived fragments regulate ribosomal biogenesis by targeting
ribosomal proteins encoding two mRNAs in a mouse model of HCC
(60).
In the present study, we focused on METTL6 and
explored its oncogenic activities in HCC, thereby providing a novel
perspective on its underlying mechanism in cancer progression by
altering the expression levels of membrane proteins. Here, for the
first time, we performed a comprehensive bioinformatics analysis of
RNA modification enzymes and associated proteins in patients with
HCC using a liver cancer dataset and observed that METTL6 was
upregulated in HCC tissues compared to the adjacent non-tumor
tissue samples. High expression levels of METTL6 were associated
with high histological grade and poor survival rates, suggesting
that METTL6 was a potential unfavorable prognostic biomarker in
HCC. To further investigate the functional role of METTL6 in HCC,
we knocked out METTL6 through the CRISPR/Cas9 system in
vitro using five independent gRNAs, and demonstrated that the
depletion of METTL6 significantly suppressed HCC cell growth,
colony formation, cell migration, cell invasion and cell adhesion.
In addition, we used RNA-seq and ChIP-seq to elucidate the
mechanism of METTL6-driven regulation of HCC. In addition to GO and
pathway enrichment analysis, we found that knockout of METTL6
significantly reduced the expression levels of cell adhesion
proteins, including ITGA1, SPON1, and CLDN14, which was further
validated at the protein level.
Previous studies have demonstrated that the
abovementioned cell adhesion proteins are of crucial importance in
HCC and other cancers. Liu et al reported that silencing of
ITGA1 inhibited HCC cell migration and invasion, while upregulation
of ITGA1 enhanced HCC migration and invasion ability in
vitro (38). In addition,
overexpression of ITGA1 has been indicated as a key driver of HCC
lymph gland metastasis (61).
Gharibi et al found that ITGA was increased in pancreatic
cancer and associated with poor prognosis, and that ITGA1 was
necessary for TGFβ/collagen-induced epithelial to mesenchymal
transition and metastasis (62).
Another study suggested that ITGA1 could promote colorectal cancer
cell migration, invasion, and tumorigenicity by activating Ras/Erk
signaling (63). Interestingly,
our RNA-seq results also showed a significant decrease in the
activity of the Ras signaling pathway along with ITGA1, such as
T-cell lymphoma invasion and metastasis-inducing protein 1 (TIAM1)
and GRB2-associated-binding protein 2 (GAB2), while there was an
increase in the expression of kinase suppressor of Ras 2 (KSR2),
which is a Ras/Erk signaling suppressor, after the knockout of
METTL6. Dai et al demonstrated that downregulation of SPON1
inhibited HCC cell proliferation, migration, and invasion (39). Of note, high CLDN14
expression was associated with good prognosis in HCC (P=0.047)
analyzed using GEPIA. However, there are some contradictory
findings regarding the prognostic role of CLDN14 in cancer. One
study showed that CLDN14 was a favorable prognostic biomarker in
HCC (40), and that CLDN14 was a
direct target of EZH2-mediated H3K27me3 and regulated by the
Wnt/β-catenin pathway (40).
RNA-seq analysis in our study indicated that EZH2 expression
was not altered. The expression levels of Wnt family proteins (2B,
3, 5A, 5B, 6, 7B, 10A, and 10B) were altered (downregulated, no
difference, and upregulated). Tryndyak et al reported that
cytosine DNA hypermethylation induced the downregulation of
CLDN14 gene expression in differentiated HepaRG cells
treated with NaAsO2 (64). Another study revealed that the
CLDN14 expression level was negatively correlated with acute
myeloid leukemia (AML) survival rates (65). In breast cancer (BC),
CLDN14 gene expression was upregulated in BC tissues
compared with non-cancer tissues, and a high gene expression of
CLDN14 was associated with a poor overall survival (66). These previous reports suggest that
CLDN14 gene expression, as well as the relationship between
gene expression and patient outcome, are dependent on various
cellular contexts.
The primary challenges of focusing on METTL6 or tRNA
modifications are the lack of m3C recognizing antibody
for RNA immunoprecipitation sequencing to globally address specific
m3C tRNA modification as well as the lack of established
tRNA-focused next-generation sequencing techniques, which means
that solid analyzing pipelines are currently missing. Hence, we
need to continue developing biochemical analytical tools and delve
further into this field for a comprehensive understanding of the
role of tRNA modifications.
One limitation of our study is the difficulty in
revealing how METTL6 interacts with identified cell adhesion
proteins and regulates their role in cancer cell proliferation,
migration, invasion and adhesion. Therefore, further detailed
research and validation such as rescue experiments are required.
The other option is to perform pulldown experiments to pulldown
m3C modified RNA and conduct a high-throughput analysis
(known as MeRIP-seq; methylated RNA immunoprecipitation) to detect
m3C modified tRNA using METTL6-expressing cells (Dox-)
and METTL6-KO cells (Dox+). This may provide a more direct
indication of whether global or specific tRNA modifications by
METTL6 are associated with target gene expression. For validation,
control tRNAs and mutant tRNAs at methylated positions should be
transfected and confirmed by experimentally changing the expression
levels of the methylated tRNAs. In addition, it is needed to
conduct mass spectrometry analysis in advance to determine the
precise methylated position on the RNAs of interest. However, there
are some technical concerns such as the lack of antibodies to
recognize m3C (specificity, sensitivity, and
precipitation efficacy), and/or MeRIP platform for tRNA. Another
limitation of current study is that we only investigated the role
of METTL6 in HCC using cell lines, but did not verify our findings
in vivo. The xenograft mouse models will be established in
future studies to support the findings of the present study.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed in the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
AB, KA, SK, and RH designed the study. AB, KA, SK,
KS, and NI performed the experiments. AB, KA, SK, and HM analyzed
the data and confirmed the accuracy. AB and KA wrote the
manuscript. MK, SS, and RH reviewed, edited, revised critically and
proposed feedback constructive suggestions to improve the quality
of the manuscript. All authors read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors express their gratitude to the past and
present members of the Hamamoto Laboratory. AB is grateful to the
Otsuka Toshimi Scholarship Foundation for their support during her
PhD study.
Funding
This study was supported by JST CREST (grant no. JPMJCR1689),
JSPS Grant-in-Aid for Scientific Research on Innovative Areas
(grant no. JP18H04908), and JSPS KAKENHI (grant no.
JP20K17982).
References
|
1
|
Boccaletto P, Machnicka MA, Purta E,
Piatkowski P, Baginski B, Wirecki TK, de Crécy-Lagard V, Ross R,
Limbach PA, Kotter A, et al: MODOMICS: A database of RNA
modification pathways. 2017 update. Nucleic Acids Res.
46:D303–D307. 2018. View Article : Google Scholar :
|
|
2
|
Barbieri I and Kouzarides T: Role of RNA
modifications in cancer. Nat Rev Cancer. 20:303–322. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Delaunay S and Frye M: RNA modifications
regulating cell fate in cancer. Nat Cell Biol. 21:552–559. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Roundtree IA, Evans ME, Pan T and He C:
Dynamic RNA modifications in gene expression regulation. Cell.
169:1187–1200. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Asada K, Bolatkan A, Takasawa K, Komatsu
M, Kaneko S and Hamamoto R: Critical Roles of
N6-Methyladenosine (m6A) in cancer and virus
infection. Biomolecules. 10:10712020. View Article : Google Scholar
|
|
6
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Herath NI, Leggett BA and MacDonald GA:
Review of genetic and epigenetic alterations in
hepatocarcinogenesis. J Gastroenterol Hepatol. 21:15–21. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nishida N and Goel A: Genetic and
epigenetic signatures in human hepatocellular carcinoma: A
systematic review. Curr Genomics. 12:130–137. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hamamoto R, Furukawa Y, Morita M, Iimura
Y, Silva FP, Li M, Yagyu R and Nakamura Y: SMYD3 encodes a histone
methyltransferase involved in the proliferation of cancer cells.
Nat Cell Biol. 6:731–740. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yagyu R, Hamamoto R, Furukawa Y, Okabe H,
Yamamura T and Nakamura Y: Isolation and characterization of a
novel human gene, VANGL1, as a therapeutic target for
hepatocellular carcinoma. Int J Oncol. 20:1173–1178.
2002.PubMed/NCBI
|
|
11
|
Shigekawa Y, Hayami S, Ueno M, Miyamoto A,
Suzaki N, Kawai M, Hirono S, Okada KI, Hamamoto R and Yamaue H:
Overexpression of KDM5B/JARID1B is associated with poor prognosis
in hepatocellular carcinoma. Oncotarget. 9:34320–34335. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ryu JW, Kim SK, Son MY, Jeon SJ, Oh JH,
Lim JH, Cho S, Jung CR, Hamamoto R, Kim DS and Cho HS: Novel
prognostic marker PRMT1 regulates cell growth via downregulation of
CDKN1A in HCC. Oncotarget. 8:115444–115455. 2017. View Article : Google Scholar
|
|
13
|
Chen M, Wei L, Law CT, Tsang FH, Shen J,
Cheng CL, Tsang LH, Ho DW, Chiu DK, Lee JM, et al: RNA
N6-methyladenosine methyltransferase-like 3 promotes liver cancer
progression through YTHDF2-dependent posttranscriptional silencing
of SOCS2. Hepatology. 67:2254–2270. 2018. View Article : Google Scholar
|
|
14
|
Barbieri I, Tzelepis K, Pandolfini L, Shi
J, Millán-Zambrano G, Robson SC, Aspris D, Migliori V, Bannister
AJ, Han N, et al: Promoter-bound METTL3 maintains myeloid leukaemia
by m6A-dependent translation control. Nature.
552:126–131. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH,
Wang F, Wang TT, Xu QG, Zhou WP and Sun SH: METTL14 suppresses the
metastatic potential of hepatocellular carcinoma by modulating
N6-methyladenosine-dependent primary MicroRNA
processing. Hepatology. 65:529–543. 2017. View Article : Google Scholar
|
|
16
|
Bao J, Yu Y, Chen J, He Y, Chen X, Ren Z,
Xue C, Liu L, Hu Q, Li J, et al: MiR-126 negatively regulates PLK-4
to impact the development of hepatocellular carcinoma via ATR/CHEK1
pathway. Cell Death Dis. 9:10452018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhong L, Liao D, Zhang M, Zeng C, Li X,
Zhang R, Ma H and Kang T: YTHDF2 suppresses cell proliferation and
growth via destabilizing the EGFR mRNA in hepatocellular carcinoma.
Cancer Lett. 442:252–261. 2019. View Article : Google Scholar
|
|
18
|
Gatza ML, Silva GO, Parker JS, Fan C and
Perou CM: An integrated genomics approach identifies drivers of
proliferation in luminal-subtype human breast cancer. Nat Genet.
46:1051–1059. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tan XL, Moyer AM, Fridley BL, Schaid DJ,
Niu N, Batzler AJ, Jenkins GD, Abo RP, Li L, Cunningham JM, et al:
Genetic variation predicting cisplatin cytotoxicity associated with
overall survival in lung cancer patients receiving platinum-based
chemotherapy. Clin Cancer Res. 17:5801–5811. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ignatova VV, Kaiser S, Ho JS, Bing X,
Stolz P, Tan YX, Lee CL, Gay FP, Lastres PR, Gerlini R, et al:
METTL6 is a tRNA m3C methyltransferase that regulates
pluripotency and tumor cell growth. Sci Adv. 6:eaaz45512020.
View Article : Google Scholar
|
|
21
|
Dai X, Jiang W, Ma L, Sun J, Yan X, Qian
J, Wang Y, Shi Y, Ni S and Yao N: A metabolism-related gene
signature for predicting the prognosis and therapeutic responses in
patients with hepatocellular carcinoma. Ann Transl Med. 9:5002021.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lopez-Terrada D, Cheung SW, Finegold MJ
and Knowles BB: Hep G2 is a hepatoblastoma-derived cell line. Hum
Pathol. 40:1512–1515. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nakabayashi H and Taketa K: HuH-7 cell
line established from a highly differentiated human hepatocellular
carcinoma. Okayama Igakkai Zasshi (Journal of Okayama Medical
Association). 124:231–238. 2012. View Article : Google Scholar
|
|
24
|
Yamada T, Abei M, Danjoh I, Shirota R,
Yamashita T, Hyodo I and Nakamura Y: Identification of a unique
hepatocellular carcinoma line, Li-7, with CD13(+) cancer stem cells
hierarchy and population change upon its differentiation during
culture and effects of sorafenib. BMC Cancer. 15:2602015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim S, Bolatkan A, Kaneko S, Ikawa N,
Asada K, Komatsu M, Hayami S, Ojima H, Abe N, Yamaue H and Hamamoto
R: Deregulation of the histone lysine-specific demethylase 1 is
involved in human hepatocellular carcinoma. Biomolecules.
9:8102019. View Article : Google Scholar
|
|
26
|
Schneider CA, Rasband WS and Eliceiri KW:
NIH Image to ImageJ: 25 years of image analysis. Nat Methods.
9:671–675. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou Y, Zhou B, Pache L, Chang M,
Khodabakhshi AH, Tanaseichuk O, Benner C and Chanda SK: Metascape
provides a biologist-oriented resource for the analysis of
systems-level datasets. Nat Commun. 10:15232019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ozawa T, Kaneko S, Szulzewsky F, Qiao Z,
Takadera M, Narita Y, Kondo T, Holland EC, Hamamoto R and Ichimura
K: C11orf95-RELA fusion drives aberrant gene expression through the
unique epigenetic regulation for ependymoma formation. Acta
Neuropathol Commun. 9:362021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Langmead B and Salzberg SL: Fast
gapped-read alignment with Bowtie 2. Nat Methods. 9:357–359. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Robinson JT, Thorvaldsdottir H, Winckler
W, Guttman M, Lander ES, Getz G and Mesirov JP: Integrative
genomics viewer. Nat Biotechnol. 29:24–26. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Anaya J: OncoLnc: Linking TCGA survival
data to mRNAs, miRNAs, and lncRNAs. PeerJ Computer Science.
2:e672016. View Article : Google Scholar
|
|
33
|
Xu L, Liu X, Sheng N, Oo KS, Liang J,
Chionh YH, Xu J, Ye F, Gao YG, Dedon PC and Fu XY: Three distinct
3-methylcytidine (m3C) methyltransferases modify tRNA
and mRNA in mice and humans. J Biol Chem. 292:14695–14703. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Richon VM, Johnston D, Sneeringer CJ, Jin
L, Majer CR, Elliston K, Jerva LF, Scott MP and Copeland RA:
Chemogenetic analysis of human protein methyltransferases. Chem
Biol Drug Des. 78:199–210. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Leitinger B and Hohenester E: Mammalian
collagen receptors. Matrix Biol. 26:146–155. 2007. View Article : Google Scholar
|
|
36
|
Wilcox ER, Burton QL, Naz S, Riazuddin S,
Smith TN, Ploplis B, Belyantseva I, Ben-Yosef T, Liburd NA, Morell
RJ, et al: Mutations in the gene encoding tight junction claudin-14
cause autosomal recessive deafness DFNB29. Cell. 104:165–172. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Klar A, Baldassare M and Jessell TM:
F-spondin: A gene expressed at high levels in the floor plate
encodes a secreted protein that promotes neural cell adhesion and
neurite extension. Cell. 69:95–110. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu X, Tian H, Li H, Ge C, Zhao F, Yao M
and Li J: Derivate Isocorydine (d-ICD) suppresses migration and
invasion of hepatocellular carcinoma cell by downregulating ITGA1
expression. Int J Mol Sci. 18:5142017. View Article : Google Scholar :
|
|
39
|
Dai W, Huang HL, Hu M, Wang SJ, He HJ,
Chen NP and Li MY: microRNA-506 regulates proliferation, migration
and invasion in hepatocellular carcinoma by targeting F-spondin 1
(SPON1). Am J Cancer Res. 5:2697–2707. 2015.PubMed/NCBI
|
|
40
|
Li CP, Cai MY, Jiang LJ, Mai SJ, Chen JW,
Wang FW, Liao YJ, Chen WH, Jin XH, Pei XQ, et al: CLDN14 is
epigenetically silenced by EZH2-mediated H3K27ME3 and is a novel
prognostic biomarker in hepatocellular carcinoma. Carcinogenesis.
37:557–566. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Arimbasseri AG, Iben J, Wei FY, Rijal K,
Tomizawa K, Hafner M and Maraia RJ: Evolving specificity of tRNA
3-methyl-cytidine-32 (m3C32) modification: A subset of tRNAsSer
requires N6-isopentenylation of A37. RNA. 22:1400–1410. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hopper AK and Huang HY: Quality control
pathways for nucleus-encoded eukaryotic tRNA biosynthesis and
subcellular trafficking. Mol Cell Biol. 35:2052–2058. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang Z, Ye Y, Gong J, Ruan H, Liu CJ,
Xiang Y, Cai C, Guo AY, Ling J and Diao L: Global analysis of tRNA
and translation factor expression reveals a dynamic landscape of
translational regulation in human cancers. Commun Biol. 1:2342018.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Santos M, Fidalgo A, Varanda AS, Oliveira
C and Santos MA: tRNA deregulation and its consequences in cancer.
Trends Mol Med. 25:853–865. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Huang H, Weng H, Zhou K, Wu T, Zhao BS,
Sun M, Chen Z, Deng X, Xiao G, Auer F, et al: Histone H3
trimethylation at lysine 36 guides m6A RNA modification
co-transcriptionally. Nature. 567:414–419. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Li Y, Xia L, Tan K, Ye X, Zuo Z, Li M,
Xiao R, Wang Z, Liu X, Deng M, et al: N6-Methyladenosine
co-transcriptionally directs the demethylation of histone H3K9me2.
Nat Genet. 52:870–877. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cheng JX, Chen L, Li Y, Cloe A, Yue M, Wei
J, Watanabe KA, Shammo JM, Anastasi J, Shen QJ, et al: RNA cytosine
methylation and methyltransferases mediate chromatin organization
and 5-azacytidine response and resistance in leukaemia. Nat Commun.
9:11632018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Pereira M, Francisco S, Varanda AS, Santos
M, Santos MA and Soares AR: Impact of tRNA Modifications and
tRNA-modifying enzymes on proteostasis and human disease. Int J Mol
Sci. 19:37382018. View Article : Google Scholar
|
|
49
|
Hoffer ED, Hong S, Sunita S, Maehigashi T,
Gonzalez RL Jnr, Whitford PC and Dunham CM: Structural insights
into mRNA reading frame regulation by tRNA modification and
slippery codon-anticodon pairing. Elife. 9:e518982020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
McLaughlin CS, Grundberg-Manago M, Dondon
J, Michelson AM and Saunders G: Stability of the messenger
RNA-transfer RNA-ribosome complex. J Mol Biol. 32:521–542. 1968.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Alexandrov A, Chernyakov I, Gu W, Hiley
SL, Hughes TR, Grayhack EJ and Phizicky EM: Rapid tRNA decay can
result from lack of nonessential modifications. Mol Cell. 21:87–96.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Carneiro RL, Requiao RD, Rossetto S,
Domitrovic T and Palhano FL: Codon stabilization coefficient as a
metric to gain insights into mRNA stability and codon bias and
their relationships with translation. Nucleic Acids Res.
47:2216–2228. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wu Q, Medina SG, Kushawah G, DeVore ML,
Castellano LA, Hand JM, Wright M and Bazzini AA: Translation
affects mRNA stability in a codon-dependent manner in human cells.
Elife. 8:e453962019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lyu X, Yang Q, Li L, Dang Y, Zhou Z, Chen
S and Liu Y: Adaptation of codon usage to tRNA I34 modification
controls translation kinetics and proteome landscape. PLoS Genet.
16:e10088362020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Motorin Y and Helm M: tRNA stabilization
by modified nucleotides. Biochemistry. 49:4934–4944. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kimura S and Waldor MK: The RNA
degradosome promotes tRNA quality control through clearance of
hypomodified tRNA. Proc Natl Acad Sci USA. 116:1394–1403. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Schimmel P: The emerging complexity of the
tRNA world: Mammalian tRNAs beyond protein synthesis. Nat Rev Mol
Cell Biol. 19:45–58. 2018. View Article : Google Scholar
|
|
58
|
Xie Y, Yao L, Yu X, Ruan Y, Li Z and Guo
J: Action mechanisms and research methods of tRNA-derived small
RNAs. Signal Transduct Target Ther. 5:1092020. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lyons SM, Fay MM and Ivanov P: The role of
RNA modifications in the regulation of tRNA cleavage. FEBS Lett.
592:2828–2844. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kim HK, Fuchs G, Wang S, Wei W, Zhang Y,
Park H, Roy-Chaudhuri B, Li P, Xu J, Chu K, et al: A
transfer-RNA-derived small RNA regulates ribosome biogenesis.
Nature. 552:57–62. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wan J, Wen D, Dong L, Tang J, Liu D, Liu
Y, Tao Z, Gao D, Sun H, Cao Y, et al: Establishment of monoclonal
HCC cell lines with organ site-specific tropisms. BMC Cancer.
15:6782015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Gharibi A, La Kim S, Molnar J, Brambilla
D, Adamian Y, Hoover M, Hong J, Lin J, Wolfenden L and Kelber JA:
ITGA1 is a pre-malignant biomarker that promotes therapy resistance
and metastatic potential in pancreatic cancer. Sci Rep.
7:100602017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Li H, Wang Y, Rong SK, Li L, Chen T, Fan
YY, Wang YF, Yang CR, Yang C, Cho WC and Yang J: Integrin α1
promotes tumorigenicity and progressive capacity of colorectal
cancer. Int J Biol Sci. 16:815–826. 2020. View Article : Google Scholar :
|
|
64
|
Tryndyak VP, Borowa-Mazgaj B, Steward CR,
Beland FA and Pogribny IP: Epigenetic effects of low-level sodium
arsenite exposure on human liver HepaRG cells. Arch Toxicol.
94:3993–4005. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cheng J, Han J and Lin C: A comprehensive
assessment of the prognostic role of cell adhesion molecules in
acute myeloid leukemia. Transl Cancer Res. 9:7605–7618. 2020.
View Article : Google Scholar
|
|
66
|
Yang G, Jian L and Chen Q: Comprehensive
analysis of expression and prognostic value of the claudin family
in human breast cancer. Aging (Albany NY). 13:8777–8796. 2021.
View Article : Google Scholar
|