Introduction
Colorectal cancer (CRC), one of the most common and
aggressive human malignancies, causes numerous cancer-associated
deaths worldwide annually (1,2).
The shift to aerobic glycolytic metabolism in cancer cells has been
widely studied and updated in the well-known 'hallmarks of cancer',
as described by Hanahan and Weinberg (3). However, the current view is that
research on cancer metabolism should not be restricted to cancer
cells (4). The tumor
microenvironment (TME) is composed of several cell types that are
allocated the constant supplies of nutrients required for tumor
survival and progression (5).
Emerging evidence has confirmed that cancer-associated fibroblasts
(CAFs) undergo epigenetic modifications in molecules associated
with glucose metabolism to achieve a symbiotic relationship with
cancer cells (6-8). CAFs with enhanced glycolytic
features subsequently secrete high amounts of pyruvate and lactate,
which are in turn used by cancer cells as new sources of energy
(9). This new concept is referred
to as the 'reverse Warburg effect', and this indicates a potential
therapeutic strategy for cancer (6,10-12). Recently, chip-based 3D co-culture
technology has been applied to simulate the characteristics of the
metabolic coupling and 'reverse Warburg effect' between CRC cells
and fibroblasts in vitro (13).
Green tea is a popular beverage worldwide, and it
has long been described to possess several health benefits
(14). As a safe and the most
powerful ingredient in green tea (15), epigallocatechin-3-gallate (EGCG)
possesses high chemo-preventative properties, and has been proposed
as a possible chemotherapeutic treatment against numerous types of
cancer, including hepatocellular carcinoma (16), CRC (17), breast cancer (18) and B lymphoma (19). It has been concluded that it is
safe for humans to take the amounts equivalent to the EGCG content
in 8-16 cups of green tea once a day for 4 weeks (20). To date, various mechanisms have
been proposed to explain the cancer-preventative effects of EGCG,
including the upregulation of tumor suppressor genes such as p53
(21), and the modulation of cell
signaling pathways, such as nuclear factor-κB (22) and JAK/STAT3 (23). More recently, EGCG has been shown
to inhibit cell growth and induce apoptosis through targeting the
enhanced aerobic glycolytic pathway via directly inhibiting
phosphofructokinase (PFK) activity in cancer cells (18,24).
However, to the best of our knowledge, there are no
previous studies assessing the effects of EGCG on glucose metabolic
alterations involving CAFs in the TME. The aim of the present study
was to investigate the hypothesis that EGCG may intervene in the
metabolic coupling between CAFs and CRC cells through inhibiting
aerobic glycolytic activity in CAFs, thereby suppressing the
malignancy of CRC cells in vitro. The findings obtained
highlight a novel anti-tumor strategy of EGCG, in which it targets
the glucose metabolism of stromal cells in the TME, providing
further insights into elucidating the underlying mechanism of
metabolic coupling between CRC cells and stromal cells.
Materials and methods
Cell culture and reagents
Human colorectal carcinoma cell lines HCT-116
(CCL-247) and HT-29 (HTB-38) were purchased from ATCC and cultured
in McCoy's 5A (Thermo Fisher Scientific, Inc.) supplemented with
10% FBS (Thermo Fisher Scientific, Inc.) and a 1%
penicillin-streptomycin solution (Thermo Fisher Scientific, Inc.).
Human intestinal fibroblasts (HIFs) were obtained from ScienCell
Research Laboratories, Inc. (cat. no. 2920) and cultured in
complete fibroblast medium (cat. no. 2301; ScienCell Research
Laboratories). Cells were cultured at 37°C in a humidified
incubator with 5% CO2 and normal oxygen saturation, and
were used for experiments during the exponential growth phase under
mycoplasma-free conditions. All cell lines used in the present
study were authenticated by short tandem repeat profiling by the
companies they were purchased from, were aliquoted and frozen in
liquid nitrogen immediately upon receipt, and used for the present
experiments within 6 months of thawing. EGCG was purchased from
Funakoshi Co., Ltd. (cat. no. E-5737) and bindarit
(2-methyl-2-[(1-[phenylmethyl]-1H-indazol-3yl)methoxy] propanoic
acid) was supplied by MedChemExpress.
Cell co-culture and conditioned medium
(CM) collection
To induce CAFs, HIFs were co-cultured with HCT-116
or HT-29 cells at a ratio of 1:3 using Falcon® Permeable
supports for six-well plates with 0.4-µm pores (cat. no.
353090; Corning, Inc.) in an indirect contact manner for 3 days,
sharing the same media; DMEM high glucose (4.5 g/l), pyruvate (110
mg/l) (cat. no. 11995065; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS. Subsequently, the cancer cells were
discarded from the co-culture system, and the medium was replaced
with fresh serum-free DMEM for the monoculture of induced CAFs.
After a further 48 h of culture, the supernatant was collected,
centrifuged (500 × g at room temperature for 20 min), and filtered
using a 0.2-µm filter to remove the cell debris. For EGCG or
bindarit treatment, different concentrations (as described in
results section) of reagents were added to fresh DMEM supplemented
with 10% FBS to induce CAFs for a further 24 h after discarding the
cancer cells, and before further replacement of new medium to
generate CM. To generate HIF-CM, HIFs were cultured without cancer
cells, and the same procedure as that described above was employed.
For CAF-siRNA-MCT4-CM and CAF-siRNA-NC-CM, induced CAFs were first
transfected with monocarboxylate transporter (MCT)4 siRNA or Select
Negative Control #1 siRNA respectively, and the remaining steps in
the procedure were the same as those described above. All CM
obtained from the above groups was stored at -80°C. For the further
treatments of cancer cells in the subsequent experiments, CM was
warmed to room temperature and added to fresh medium at a ratio of
1:1.
Immunofluorescence staining
Cells were seeded in the Nunc Lab-Tek II Chamber
Slide system (cat. no. 154526PK; Thermo Fisher Scientific, Inc.)
until they had adhered, washed twice with PBS, and fixed with 4%
paraformaldehyde (cat. no. 163-20145; Fujifilm) at 4°C for 30 min.
After permeabilization with 0.1% Triton X-100 (cat. no. HFH10;
Thermo Fisher Scientific, Inc.) for 5 min at room temperature, all
slides were incubated with 3% bovine serum albumin (BSA) diluted
from a MACS BSA stock solution (cat. no. 130-091-376; Miltenyi
Biotec) in PBS for 60 min at room temperature. Subsequently, the
slides were incubated with an anti-α smooth muscle actin (α-SMA)
antibody (dilution, 1:300; cat. no. no. ab7817; Abcam) or
anti-fibroblast activation protein (anti-FAP) (1:100; cat. no.
ab207178, Abcam) at 4°C overnight. The following day, the slides
were washed three times with PBS and incubated with Alexa Fluor
488-conjugated goat anti-rabbit IgG (H+L) cross-adsorbed secondary
antibody (1:500; cat. no. A-11008, Thermo Fisher Scientific, Inc.)
for FAP or Alexa Fluor 594-conjugated goat anti-mouse IgG (H+L)
cross-adsorbed secondary antibody (1:500; cat. no. A-11005, Thermo
Fisher Scientific, Inc.) for α-SMA for 1 h at 37°C in the dark.
After three washes with PBS protected from light, ProLong™ Gold
Antifade Mount with DAPI (cat. no. P36931; Thermo Fisher
Scientific, Inc.) was used to stain the nuclei for 10 min in the
dark. Finally, the slides were observed and imaged under a
fluorescence microscope (magnification, ×100; BZ-X700; Keyence
Corporation). For negative controls, the primary antibody was
replaced with BSA.
Cell proliferation assay
HCT-116 or HT-29 cells were seeded at a density of
1×104 cells/well into 96-well plates until they had
adhered to the plates. After washing with PBS, the indicated CM was
added to each group for 48 h of culture. Subsequently, the medium
for each group was replaced with fresh medium containing 10% (v/v)
Cell Counting Kit-8 (CCK-8) solution (Dojindo Molecular
Technologies, Inc.), the plates were cultured for a further 2 h,
and the extent of cell proliferation was analyzed by measuring the
absorbance values at 450 nm with a microplate reader (SpectraMax
i3; Molecular Devices, LLC), in accordance with the manufacturer's
instructions. To examine the direct effects of EGCG on cancer cells
and the cytotoxicity of EGCG on normal cells, HCT-116 and HT-29
cells and HIFs were seeded at a density of 5×103
cells/well into 96-well plates and treated with different
concentrations of EGCG. The proliferative status of the cells was
observed every 24 h between days 1 and 3 using CCK-8 solution, as
described above.
Wound healing assay
For wound healing assays, HCT-116 or HT-29 cells
were seeded at a density of 7×105 cells/well into
six-well plates and incubated overnight to form a 90% confluent
monolayer. Subsequently, a 200-µl pipette tip was used to
scratch a wound through the entire center of each well. After
washing with PBS, the cells in each group were cultured with EGCG,
bindarit or the indicated CM in the absence of FBS for 36 h. The
areas of the wounds were observed, and images were captured using a
light microscope (magnification, ×40; DP22-CU; Olympus Corporation)
at 0 and 36 h after scratching. The cell migration rates were
calculated using ImageJ v1.46r software (National Institutes of
Health) according to the following equation: Relative migration
rate=[width (0 h)-width (36 h)]/width (0 h) ×100%.
Migration and invasion assay
A 24-well Transwell system with 8.0-µm pores
(Corning, Inc.) was used for the migration and invasion assays.
Serum-starved cancer cells at a density of 7×104/well
were seeded into the upper chamber in a 100 µl volume
suspension. After cell attachment, the culture medium was changed
to the indicated CM for each group. The final FBS concentration was
5% in the upper chambers and 10% in the lower chambers. After a
subsequent period of incubation (36 h for migration assay; 60 h for
invasion assay), the upper chambers were fixed with 100% methanol
(cat. no. 131-01826; Fujifilm) for 20 min and then stained with 1%
crystal violet (cat. no. 031-04852; Fujifilm) for a further 20 min
at room temperature, and non-migrated cells were removed using
cotton swabs. Subsequently, images were captured, and the number of
remaining cells were calculated and quantified in 5 random fields
of view under a light microscopic (magnification, ×100) equipped
with a digital camera (Olympus Corporation). For the invasion
assays, the upper chambers of the Transwell system were coated with
Matrigel™ (0.5 mg/ml dilatated in DMEM; Corning, Inc.) overnight at
37°C.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted using an RNeasy Mini kit
(Qiagen GmbH, Hilden, Germany), and its concentration was measured
using a spectrophotometer (NanoDrop™ 2000; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Subsequently, 2.5 µg RNA was reverse-transcribed into cDNA
in a total reaction system of 50 µl using an Applied
Biosystems™ High-Capacity cDNA Reverse Transcription kit (Thermo
Fisher Scientific, Inc.) in accordance with the manufacturer's
instructions. Subsequently, an Applied Biosystems StepOnePlus
Real-Time PCR system (Thermo Fisher Scientific, Inc.) was used to
perform TaqMan qPCR using the following thermocycling conditions:
Initial denaturation at 95°C for 3 min; 40 cycles of denaturation
(95°C for 30 sec), annealing (58°C for 30 sec) and extension (72°C
for 45 sec); and final extension at 72°C for 10 min. The following
TaqMan gene expression assays were used: ACTA2 (assay ID,
Hs00426835_g1) FAP (assay ID, Hs00990791_m1), interleukin-6
(IL-6) (assay ID, Hs00985639_m1), PFK (assay ID,
Hs00737347_m1), and SLC16A3 (MCT4) (assay ID,
Hs00358829_m1). A GAPDH assay (assay ID, 4326317E; Thermo
Fisher Scientific, Inc.) was used as the internal control to
normalize the raw data. The 2−∆∆Cq method (25) was used for data analysis, and the
results are presented as the fold changes in the relative mRNA
expression for each experimental group compared with that in the
control group.
MCT4 silencing through siRNA
transfection
Induced CAFs were transfected with 10 pmol/ml
SLC16A3 (MCT4) siRNA (cat. no. 4390824; assay ID: s17417; Thermo
Fisher Scientific, Inc.) or Select Negative Control #1 siRNA (cat.
no. 4390843; Thermo Fisher Scientific, Inc.) with 3 µl/ml
Invitrogen™ Lipofectamine RNAiMax Transfection Reagent (cat. no.
13778075; Thermo Fisher Scientific Inc.) for 12 h according to the
manufacturer's instructions. The efficiency of gene silencing was
verified through performing RT-qPCR and western blotting.
Lactate assay
The supernatant was collected using the same method
as that used for CM after culturing the indicated cells for 6, 12
or 24 h. A lactate assay kit (cat. no. MAK064; Sigma-Aldrich) was
used to detect the concentration of lactate in the supernatant.
Briefly, supernatants and standard samples were diluted with
lactate assay buffer according to the manufacturer's instructions
and added to a 96-well plate. After a 30 min incubation with 50
µl master reaction mix at room temperature, the absorbance
values at 570 nm were measured with a microplate reader, and the
lactate concentrations of samples were calculated according to a
standard curve constructed using standard samples. The relative
secretion of lactate was normalized to the cell number.
Western blot analysis
Freshly prepared RIPA lysis buffer (Thermo Fisher
Scientific, Inc.) containing 1% protease inhibitor cocktail (cat.
no. P8849; Sigma-Aldrich) and 1% phosphatase inhibitor cocktail 3
(cat. no. P0044; Sigma-Aldrich) was used to extract the total
protein, and the protein concentration was measured using a BCA kit
(Thermo Fisher Scientific, Inc.). Equal amounts of protein (10
µg) were separated by SDS-PAGE (10% gels) and transferred to
PVDF membranes with 0.45-µm pores (Sigma-Aldrich; Merck
KGaA). After washing with Tris Buffered Saline with Tween-20 (TBST)
(cat. no. 9997; Cell Signaling Technology, Inc.) for 10 min at room
temperature, 5% skimmed milk was used as the blocking solution to
incubate the membranes under gentle shaking for 90 min at room
temperature. The membranes were washed with TBST 4 times (10 min
each) and then incubated with anti-α-SMA antibody (dilution,
1:2,000; cat. no. ab7817, Abcam), anti-FAP (1:3,000; cat. no.
ab28244; Abcam), anti-PFK (1:20,000; cat. no. ab204131; Abcam),
anti-MCT4 (1:2,000; cat. no. 22787-1-AP; ProteinTech Group, Inc.)
or anti-β-actin (1:2,000; cat. no. 4970; Cell Signaling Technology,
Inc.) as the primary antibodies overnight at 4°C. The following
day, after washing with TBST 4 times (10 min each), anti-rabbit
IgG, HRP-linked (1:3,000; cat. no. 7074; Cell Signaling Technology,
Inc.) and anti-mouse IgG, HRP-linked (1:3,000; cat. no. 7076; Cell
Signaling Technology, Inc.) were used as the secondary antibodies,
according to the species of the primary antibodies, to incubate the
membranes for 1 h at room temperature. Protein signals were
visualized using ECL Prime Western Blotting Detection Reagents
(cat. no. RPN2232; Cytiva).
Statistical analysis
All data are presented as the mean ± standard
deviation of at least three biological repeats. GraphPad Prism v7.0
software (GraphPad Software, Inc.) and ImageJ v1.46r software were
used for the statistical analysis and construction of the graphs.
An unpaired Student's t-test or a Mann-Whitney U test were used for
comparisons between two groups. Differences between multiple groups
were analyzed using an ANOVA followed by a Tukey's post hoc test.
P<0.05 (two-sided) was considered to indicate a statistically
significant difference.
Results
Fibroblasts co-cultured with tumor cells
exhibit a CAF phenotype with enhanced features of aerobic
glycolysis
After co-culture with the HCT-116 and HT-29 CRC cell
lines, HIFs exhibited higher gene expression levels of the CAF
markers ACTA2, FAP and IL-6 (Fig. 1A). The western blotting results
showed that the expression levels of α-SMA and FAP, the two most
widely used markers for the identification of CAFs, were
upregulated in HIFs after co-culture (Fig. 1B). Moreover, the results from the
immunofluorescence staining experiments confirmed the upregulation
of α-SMA and FAP, and a narrower, longer and spindle-shaped
morphology of the cells was observed in the induced CAFs, which was
similar to that commonly exhibited in activated fibroblasts; one of
the characteristic features in CAFs (Fig. 1C). Taken together, these results
confirmed the transformation of the HIFs into a CAF phenotype.
Lactate assays revealed that the extracellular lactate excretion of
CAFs was higher compared with that of HIFs after 12 and 24 h of
culture (Fig. 1D).
Correspondingly, the pH value of the medium was lower in CAFs after
24 h of culture (Fig. 1E).
RT-qPCR and western blotting experiments demonstrated that the
expression of PFK, a critical rate-limiting enzyme of glycolysis,
and MCT4, an important lactate efflux transporter on the cell
membrane, were significantly upregulated in the CAFs (Fig. 1F-H). These findings demonstrated
the enhanced aerobic glycolytic metabolism of the CAFs.
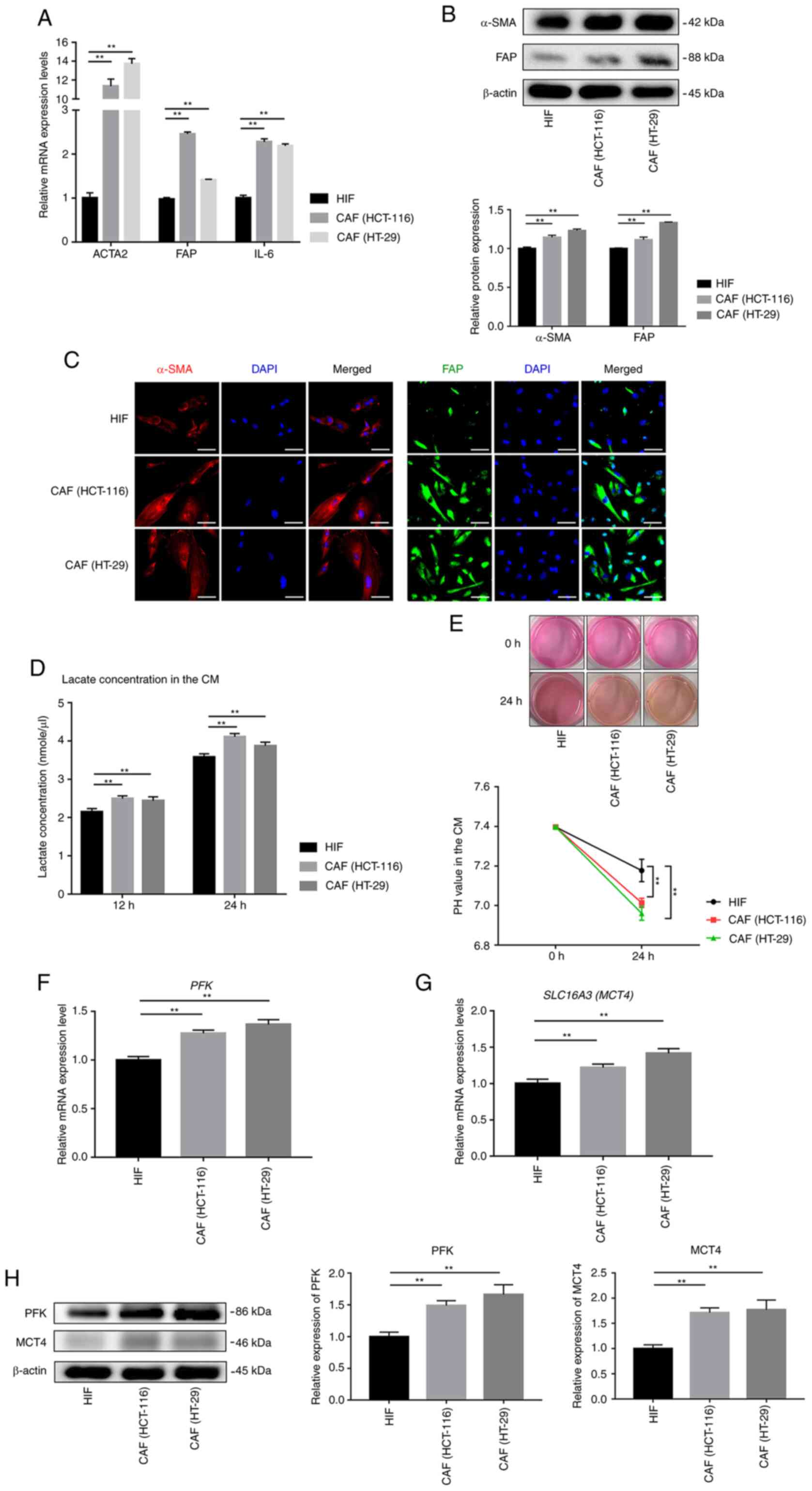 | Figure 1Fibroblasts co-cultured with tumor
cells exhibit a CAF phenotype with enhanced aerobic glycolysis. (A)
After co-culture with HCT-116 or HT-29 CRC cells, the gene
expression levels of CAF markers were detected in HIFs by RT-qPCR
analysis. (B) The expression levels of α-SMA and FAP of HIFs and
CAFs were measured by western blot analysis. (C) Immunofluorescence
staining results, showing the expression levels of α-SMA and FAP in
HIFs and CAFs (scale bar, 50 µm). (D) Extracellular lactate
excretion rates of HIFs and CAFs were evaluated according to the
lactate concentration of CM in lactate assays. (E) Typical images
and pH values of CM in HIF and CAF groups. Gene expression levels
of (F) PFK and (G) MCT4 were detected in HIFs and
CAFs by RT-qPCR analysis. (H) Western blotting results also
confirmed the overexpression of PFK and MCT4 in CAFs compared with
HIFs. **P<0.01. α-SMA, α-smooth muscle actin; ACTA2,
actin α-2; FAP, fibroblast activation protein; HIF, human
intestinal fibroblast; CAFs, cancer-associated fibroblasts; CM,
conditioned medium; RT-qPCR, reverse transcription-quantitative
PCR; PFK, phosphofructokinase; MCT4, monocarboxylate transporter
4. |
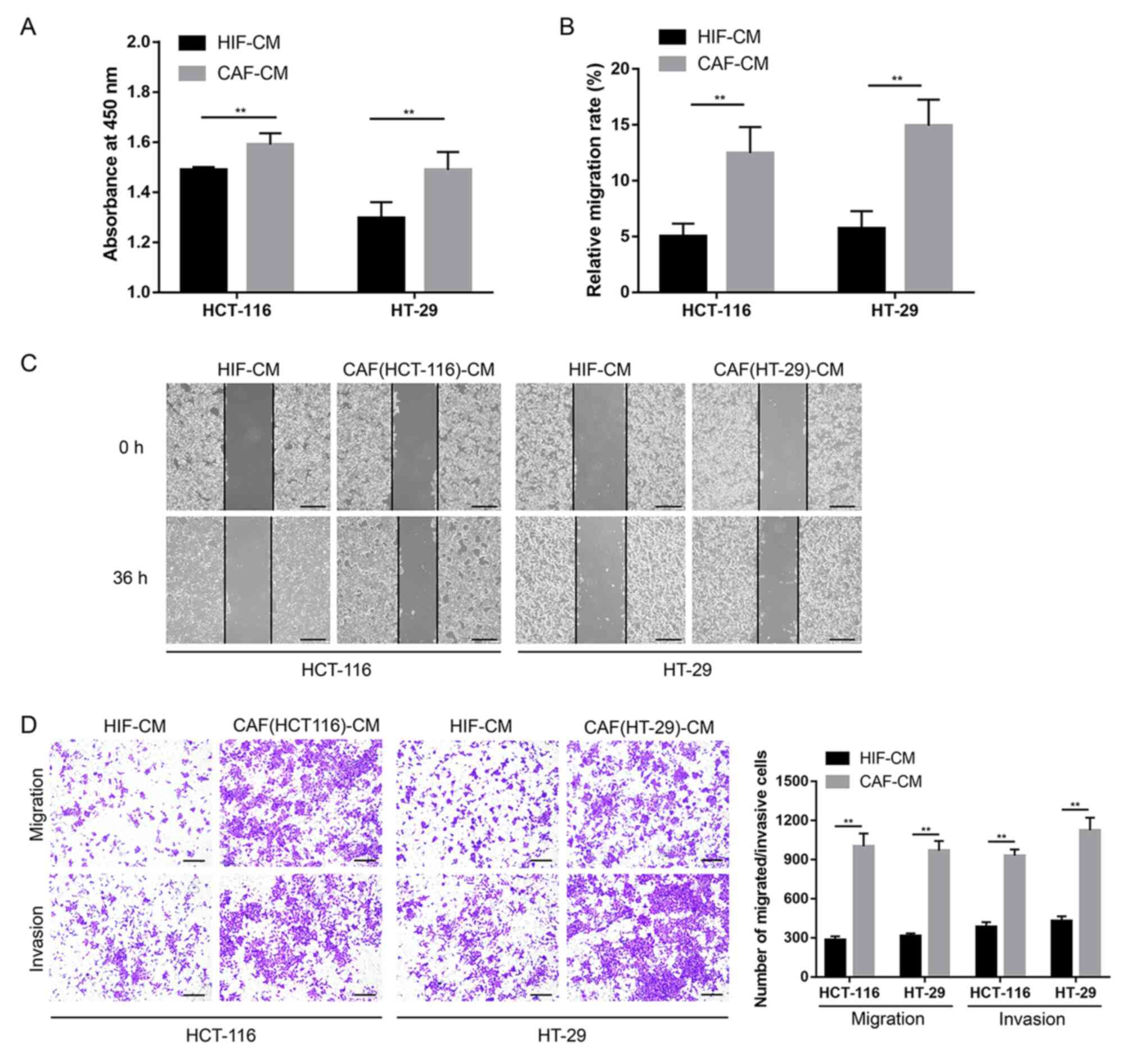
CAFs promote the malignant behavior of
CRC cells in vitro
To examine the effects of CAFs on the cancerous
properties of the CRC cells, CM was collected from the CAFs to
treat the cancer cells. Subsequently, CCK-8 proliferation assays
demonstrated that CAFs were able to support cell proliferation of
the HCT-116 and HT-29 cells compared with HIFs (Fig. 2A). Furthermore, compared with
HIFs, CAFs significantly promoted the migration and invasion of
HCT-116 and HT-29 cells, as shown in the wound healing assays
(Fig. 2B and C) and Transwell
migration and invasion assays (Fig.
2D).
EGCG directly inhibits the proliferation
and migration of CRC cells in vitro
To examine the direct effects of EGCG on tumor
cells, 25, 50 or 100 µM EGCG was used to treat the HCT-116
and HT-29 cells. Proliferation assays demonstrated that EGCG could
inhibit the proliferation of the CRC cells in a dose-dependent
manner (Fig. 3A). The wound
healing assay experiments indicated that treatment with EGCG
elicited a direct suppressive effect on the migration of HCT-116
and HT-29 cells (Fig. 3B).
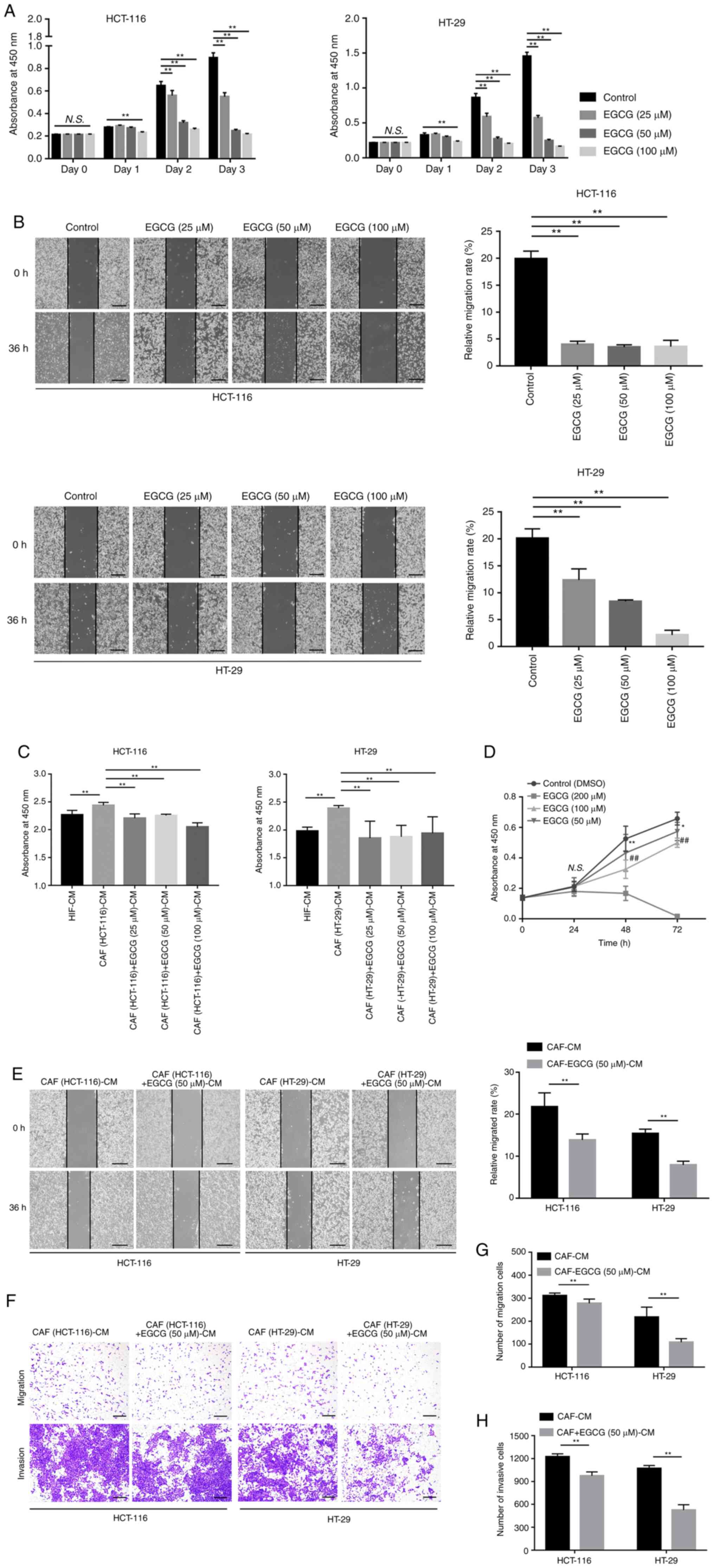 | Figure 3The effects of EGCG treatment on CRC
cells and CAFs. (A) Cell Counting Kit-8 assay results demonstrated
that treatment with 25, 50 and 100 µM EGCG directly
suppressed the proliferation of HCT-116 and HT-29 cells after 1-3
days of culture. (B) Wound healing (scale bar, 400 µm)
indicated the direct effects of different concentrations of EGCG on
the migration of HCT-116 and HT-29 cells. (C) Cell Counting Kit-8
results demonstrated that treatment with 25, 50 and 100 µM
EGCG suppressed the promoting effect of CAFs on the proliferation
of HCT-116 and HT-29 cells after 48 h of culture. (D) Cell Counting
Kit-8 assay results showed the direct effects of different
concentrations and durations of EGCG treatment on the proliferation
of HIFs. *P<0.05 and **P<0.01 vs. 50
µM EGCG; ##P<0.01 vs. 100 µM EGCG. (E)
Wound healing (scale bar, 400 µm) and (F-H) Transwell
migration and invasion assays (scale bar, 200 µm) indicated
that EGCG treatment suppressed the promoting effect of CAFs on the
migration and invasion of HCT-116 and HT-29 cells.
**P<0.01. HIFs, human intestinal fibroblasts; EGCG,
epigallocatechin-3-gallate; CAFs, cancer-associated fibroblasts;
CRC, colorectal cancer; CM, conditioned medium. N.S., not
significant. |
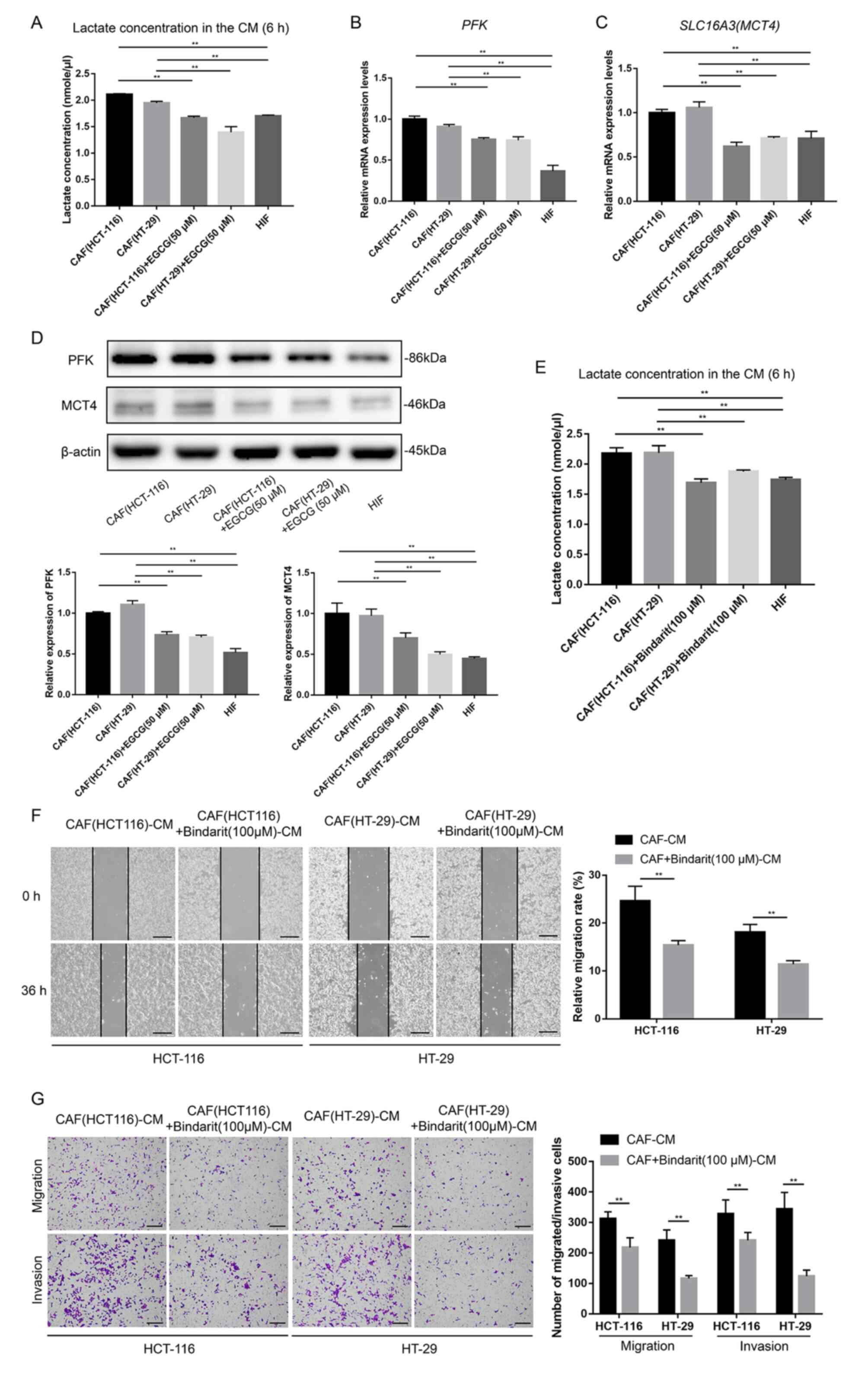
EGCG treatment suppresses the
tumor-promoting capabilities of CAFs
Subsequently, the effects of EGCG on the
malignancy-promoting abilities of CAFs were investigated. EGCG at
concentrations of 0, 25, 50 and 100 µM was used to treat
CAFs for 24 h. After washing with PBS, EGCG-pretreated CAFs were
cultured in serum-free fresh DMEM for a further 48 h to generate
CM. Subsequently, CM from these EGCG-pretreated CAFs was collected
for the further treatment of HCT-116 and HT-29 cells. Proliferation
assays demonstrated that after pretreatment with EGCG, the
capability of CAFs to stimulate cancer cell proliferation was
decreased (Fig. 3C).
The direct effects of EGCG on the survival ability
of HIFs was also investigated. However, treatment with EGCG at
concentrations up to 100 µM for 24 h did not produce any
significant effects on the proliferation of HIFs (Fig. 3D). Therefore, it was concluded
that the safe EGCG treatment conditions for CAFs were
administration of a concentration of 50 µM and a time of 24
h. As shown in Fig. 3E-H, the
ability of CAFs to promote the migration and invasion of HCT-116
and HT-29 cells was also suppressed following EGCG treatment in
this condition.
EGCG treatment inhibits the aerobic
glycolytic activity of CAFs
Previous studies have shown that EGCG is able to
inhibit aerobic glycolysis in tumor cells (14,15), and therefore it was possible to
speculate that EGCG may exert an equivalent effect in CAFs. EGCG
treatment decreased the lactate production of CAFs (Fig. 4A), as well as the expression of
PFK and MCT4 (Fig. 4B-D), which
indicated that the aerobic glycolytic activity of CAFs was
inhibited following treatment with EGCG.
Lactate supplied by CAFs exerts an
important role in tumor-stroma metabolic coupling
To further explore the metabolic coupling between
CAFs and cancer cells, bindarit (an MCT4 inhibitor) was used to
block the lactate supply from the CAFs. Bindarit treatment
decreased the lactate concentration in CM after 6 h of culture of
the CAFs (Fig. 4E). Although
bindarit pretreatment of CAFs did not reveal any obvious effects on
cancer cell proliferation (data not shown), the ability of CAFs to
promote the migration and invasion of HCT-116 and HT-29 cells was
reduced following bindarit treatment (Fig. 4F and G). Furthermore, the direct
effects of bindarit on CRC cells were also examined. As shown in
Fig. S1A and B, treatment with
100 µM bindarit did not reveal any significant effects on
the proliferation and migration of HCT-116 and HT-29 cells.
Furthermore, siRNA experiments were performed to silence MCT4
expression in the CAFs to confirm its important role in the
metabolic coupling process. The RT-qPCR and western blotting
results confirmed the efficiency of MCT4 silencing in CAFs
(Fig. 5A and B). As observed with
the bindarit treatment, the tumor-promoting effects of CAFs were
reduced following MCT4 silencing (Fig. 5C-E). Taken together, the findings
in the present study demonstrated that EGCG exerted an anti-tumor
role in impeding metabolic coupling between the CAFs and CRC cells
(Fig. 5F).
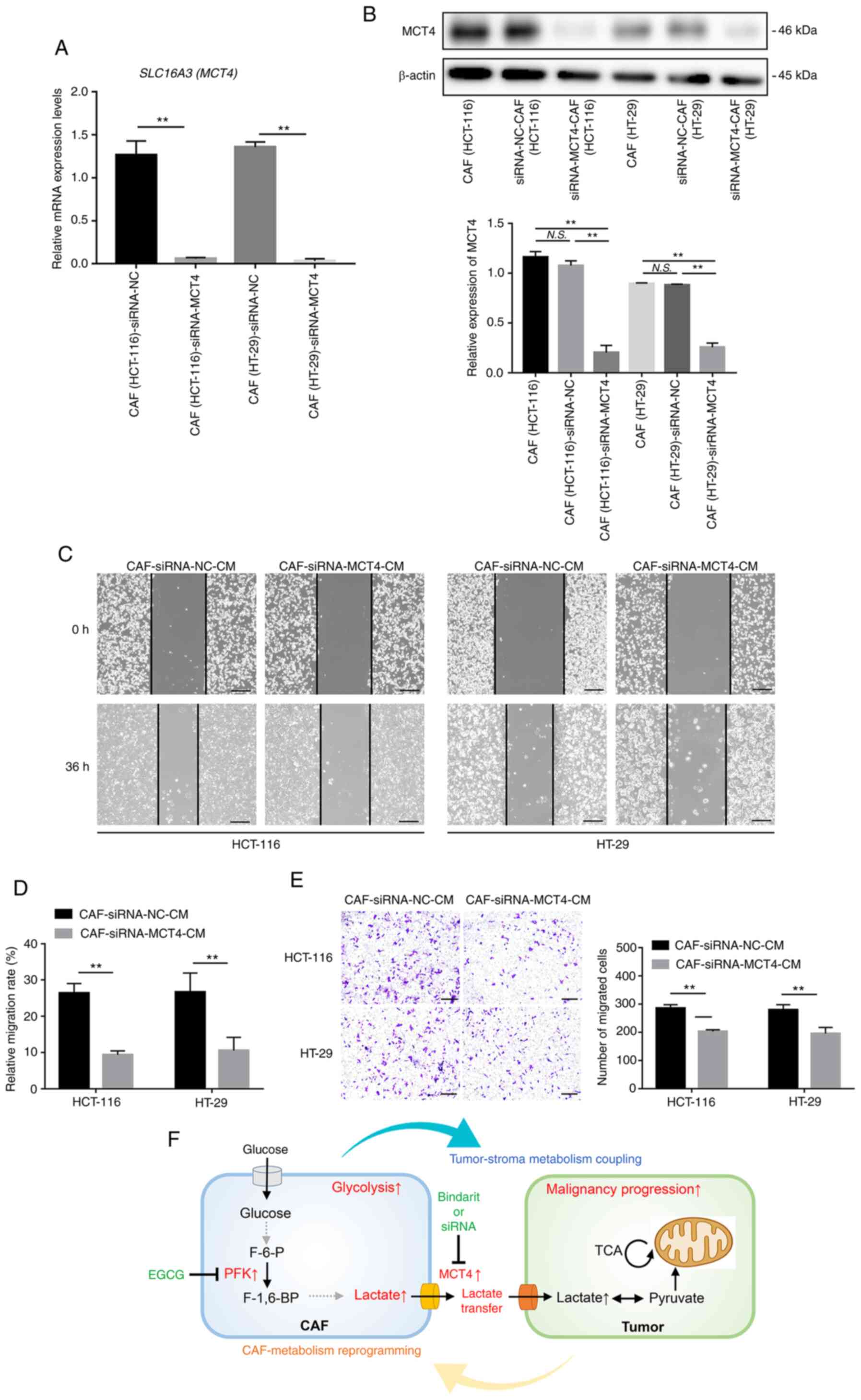 | Figure 5MCT4 silencing in CAFs suppressed its
tumor-promoting effects. (A) Gene expression levels of MCT4 were
detected in siRNA-treated CAFs by RT-qPCR analysis. (B) Western
blotting results confirmed the downregulation of MCT4 protein
levels in CAFs after siRNA treatment. (C and D) Wound healing
(scale bar, 400 µm) and (E) Transwell migration assays
(scale bar, 200 µm) indicated that knockdown of MCT4 in CAFs
suppressed the promoting effect of CAFs on the migration and
invasion of HCT-116 and HT-29 cells. (F) Schematic representation
of EGCG inhibition of the metabolic coupling between CRC and CAFs
via targeting of aerobic glycolysis in CAFs.
**P<0.01. HIFs, human intestinal fibroblasts; EGCG,
epigallocatechin-3-gallate; CAFs, cancer-associated fibroblasts;
CM, conditioned medium; F-6-P, fructose-6-phosphate; F-1,6-BP,
fructose-1,6-bisphosphate; PFK, phosphofructokinase; MCT4,
monocarboxylate transporter 4; tricarboxylic acid cycle, TCA cycle;
NC, negative control; N.S., not significant. |
Discussion
In the present study, it was demonstrated that CAFs
showed enhanced aerobic glycolytic activity, and were able to
promote CRC progression through lactate secretion compared with
normal fibroblasts. Both EGCG treatment and MCT4 inhibition were
shown to reduce the aerobic glycolytic activity of CAFs, thereby
impeding their metabolic coupling with CRC cells and reducing their
promoting effects on tumor progression. The therapeutic application
of EGCG for targeting glucose metabolism in stromal cells in the
TME was therefore been shown to be a safe and effective strategy as
an alternative means of anti-cancer therapy.
As an important stromal cell type in the TME, CAFs
are able to promote tumor progression through various direct or
indirect effects, including paracrine secretion of cytokines
(26,27). Recent research on metabolic
heterogeneity has shifted its focus from cancer cells to the
reprogramming of tumor-stroma metabolic coupling, which results
from the profound crosstalk between CAFs and cancer cells (4,5,9,12,28). This new paradigm has been termed
'two-compartment tumor metabolism' and the 'reverse Warburg effect'
(6,12,29-33), and its existence has been
confirmed in lung cancer (34),
pancreatic cancer (35), oral
squamous cell carcinoma (36,37), breast cancer (7,38),
prostate cancer (39) and
nasopharyngeal cancer (40). In
terms of the specific details, CAFs undergo metabolic
reprogramming, which manifests itself as enhanced aerobic
glycolysis. High amounts of lactic acid secreted by CAFs with an
upregulated expression of MCT4 as an energy substrate are absorbed
by cancer cells, and these participate in the metabolic alteration
of cancer cells, thereby enhancing tumor malignancy. In the present
study, the western blotting results showed that the expression
levels of α-SMA and FAP, the two most widely used markers for
identifying CAFs (41,42), were upregulated in HIFs after
co-culture. Moreover, the results from the immunofluorescence
staining experiments confirmed the upregulation of α-SMA and FAP,
and a narrower, longer and spindle-shaped morphology of the cells
was observed in the induced CAFs that was similar to that commonly
exhibited in activated fibroblasts, as one of their characteristic
features in CAFs (43). The
current consensus is that CAFs are a stromal cell population with
cell-of-origin, phenotypic and functional heterogeneity (43). Although the exact method of
identifying CAFs is still controversial, it is generally
hypothesized that CAFs are derived from activated myofibroblasts in
the TME. The typical spindle-shaped morphology changes compared
with quiescent fibroblasts, and the upregulation of several
specific CAF markers that are widely used, such as α-SMA, FAP and
IL-6, can be used to identify CAFs (41-43). Recently, co-culture with cancer
cells has become a way to obtain and study CAFs in vitro
(44,45). Thus, it was concluded that HIFs
transformed into a CAF phenotype. The induced CAFs exhibited an
enhanced rate of aerobic glycolysis, which was characterized by
upregulation of the glycolytic rate-limiting enzyme PFK and lactate
transporter MCT4, as well as increased lactate production.
Moreover, CM from CAFs had a marked ability to promote the
malignancy of CRC cells in vitro.
EGCG is a safe natural substance, and its direct
anti-tumor properties have been extensively studied (15,20,46). Consistent with a previous study
(17), the results of the present
study also revealed that EGCG was able to directly inhibit the
proliferation and migration of CRC cells. Moreover, it has been
shown for the first time, to the best of our knowledge, that
treatment with EGCG at a concentration of 50 µM (an assured
safe dose) for 24 h led to a reduction in the aerobic glycolytic
capacity of CAFs. Specifically, PFK and MCT4 were downregulated,
and the efflux of lactic acid was reduced upon administering this
dose of EGCG. Interestingly, EGCG-treated CAFs exhibited a reduced
ability to promote CRC malignancy. These findings have expanded the
concept of the anti-tumor mechanism of EGCG, suggesting that it may
be used as a safe anti-cancer adjuvant targeting the TME.
According to the 'two-compartment' metabolic
coupling hypothesis, MCT4 is upregulated in CAFs and contributes to
the excretion of lactic acid, while MCT1 is overexpressed in cancer
to increase lactate uptake. Through this lactic acid shuttle from
CAFs to cancer, lactate becomes an energy source for further
utilization by cancer cells (6,9,28,47,48). From this perspective, targeting of
the MCT family and use of MCT1/MCT4 inhibitors to disrupt the
'lactic acid shuttle' for the 'reverse Warburg effect' has become
an anti-tumor strategy (8,10).
In fact, silencing of MCT4 in CAFs weakens their ability to secrete
lactic acid and its subsequent promoting effect on the invasion of
breast cancer (49). Futagi et
al (50) demonstrated that
bindarit acts as the first potent and highly selective inhibitor of
human MCT4, with a Ki value of 30.2±1.4
µM. In the present study, bindarit was used at a
concentration of 100 µM, which is sufficient for the
inhibition of MCT4. Indeed, using various or more specific
inhibitors of MCT4 may more convincingly confirm this conclusion.
However, novel selective MCT4 inhibitors remain to be
identified/designed. Thus, siRNA technology was also to
specifically silence MCT4 in CAFs in the present study to provide
more experimental support for the conclusion drawn. Similar to the
experiments with EGCG, using an MCT4 inhibitor or silencing MCT4
expression also weakened the tumor-promoting effects of CAF. These
experiments further confirmed that the suppressive effects of EGCG
on CAFs may be attributed to blocking aerobic glycolysis.
Although novel findings have been presented in the
present study, further work is required. Firstly, the lack of
further evidence demonstrating transformation of HIFs into a
CAF-like phenotype is a limitation of the present study. Secondly,
for silencing MCT4, siRNA technology was used, the absence of shRNA
experiments is another limitation of this study. Additionally, the
detailed mechanism through which lactic acid stimulated tumor
progression is still not fully understood. The absorbed lactic acid
could serve as the main source of carbon in tumors and enter the
tricarboxylic acid cycle (51).
It could also directly promote the epithelial-mesenchymal
transition and the expression of angiogenesis-associated genes in
cancer (52). Therefore, in the
context of nutrient metabolism, lactate should be studied further
as a signaling molecule. Furthermore, another study demonstrated
that CAFs were able to directly transfer mitochondria to tumor
cells in order to enhance their oxyphosphate phosphorylation
(38). This phenomenon may
explain why the MCT4 inhibitor did not affect the proliferation of
cancer cells. Metabolic heterogeneity in the TME and the dynamics
of metabolic coupling both require further investigation.
In conclusion, the present study revealed the
metabolic coupling of cancer cells and CAFs in CRC, with the
resultant effects on tumor progression. Knowledge of the anti-tumor
mechanism of EGCG has also been expanded through targeting of the
glycolysis of CAFs in the TME. Collectively, the findings of the
present study should prove to be valuable for further clarifying
tumor-stroma metabolic coupling and proposing novel therapeutic
strategies.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SC, YM and MS designed the experiments. SC, TT, HK
and CT performed the experiments and collected the data. YM, SY and
CT analyzed and interpreted the data. SHC and YW drafted the
manuscript. MN, YM and MS revised the paper critically for
important intellectual content. SHC and MN confirmed the
authenticity of all the raw data. SHC and MN agree to be
accountable for all aspects of the work in ensuring that questions
related to the accuracy or integrity of any part of the work are
appropriately investigated and resolved. All the authors have read
and approved the final version of the manuscript for
publication.
Ethics approval and consent to
participate
This study was approved by The Ethics Committee of
Tokushima University Hospital (TOCMS: 2901-2). This study strictly
followed the ethical guidance of Ministry of Health, Labour and
Welfare in Japan and Tokushima University regulations.
Patient consent for publication
Not applicable.
Competing interests
The conflicts of interest with regard to our study
are as follows: grant support from Taiho Pharmaceutical Co., Ltd,
and Tsumura Co., Ltd.
Acknowledgments
Not applicable.
Funding
This work was supported by the Grants-in-Aid for Scientific
Research (grant nos. 20K08957 to YM), Taiho Pharmaceutical Co.,
Ltd. and Tsumura Pharmaceutical Co., Ltd.
Abbreviations:
|
α-SMA
|
α-smooth muscle actin
|
|
ACTA2
|
actin α-2
|
|
CAF
|
cancer-associated fibroblast
|
|
CM
|
conditioned medium
|
|
CRC
|
colorectal cancer
|
|
EGCG
|
epigallocatechin-3-gallate
|
|
FAP
|
fibroblast activation protein
|
|
HIFs
|
human intestinal fibroblasts
|
|
MCT
|
monocarboxylate transporter
|
|
TBST
|
Tris Buffered Saline with Tween-20
|
|
TME
|
tumor microenvironment
|
|
PFK
|
phosphofructokinase
|
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD, Goding Sauer A,
Fedewa SA, Butterly LF, Anderson JC, Cercek A, Smith RA and Jemal
A: Colorectal Cancer statistics, 2020. CA Cancer J Clin.
70:145–164. 2020. View Article : Google Scholar
|
|
3
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gentric G and Mechta-Grigoriou F: Tumor
cells and cancer-associated fibroblasts: An updated metabolic
perspective. Cancers (Basel). 13:3992021. View Article : Google Scholar
|
|
5
|
Eisenberg L, Eisenberg-Bord M,
Eisenberg-Lerner A and Sagi-Eisenberg R: Metabolic alterations in
the tumor microenvironment and their role in oncogenesis. Cancer
Lett. 484:65–71. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pavlides S, Whitaker-Menezes D,
Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro
MC, Wang C, Fortina P, Addya S, et al: The reverse Warburg effect:
Aerobic glycolysis in cancer associated fibroblasts and the tumor
stroma. Cell Cycle. 8:3984–4001. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Becker LM, O'Connell JT, Vo AP, Cain MP,
Tampe D, Bizarro L, Sugimoto H, McGow AK, Asara JM, Lovisa S, et
al: Epigenetic reprogramming of cancer-associated fibroblasts
deregulates glucose metabolism and facilitates progression of
breast cancer. Cell Rep. 31:1077012020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jung JG and Le A: Targeting metabolic
cross talk between cancer cells and cancer-associated fibroblasts.
Adv Exp Med Biol. 1311:205–214. 2021. View Article : Google Scholar
|
|
9
|
Martinez-Outschoorn UE, Lisanti MP and
Sotgia F: Catabolic cancer-associated fibroblasts transfer energy
and biomass to anabolic cancer cells, fueling tumor growth. Semin
Cancer Biol. 25:47–60. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fu Y, Liu S, Yin S, Niu W, Xiong W, Tan M,
Li G and Zhou M: The reverse Warburg effect is likely to be an
Achilles' heel of cancer that can be exploited for cancer therapy.
Oncotarget. 8:57813–57825. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Martinez-Outschoorn UE, Peiris-Pagés M,
Pestell RG, Sotgia F and Lisanti MP: Cancer metabolism: A
therapeutic perspective. Nat Rev Clin Oncol. 14:11–31. 2017.
View Article : Google Scholar
|
|
12
|
Benny S, Mishra R, Manojkumar MK and
Aneesh TP: From Warburg effect to reverse Warburg effect; the new
horizons of anti-cancer therapy. Med Hypotheses. 144:1102162020.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Keller F, Bruch R, Schneider R,
Meier-Hubberten J, Hafner M and Rudolf R: A scaffold-free 3-D
co-culture mimics the major features of the reverse Warburg effect
in vitro. Cells. 9:19002020. View Article : Google Scholar
|
|
14
|
Jankun J, Selman SH, Swiercz R and
Skrzypczak-Jankun E: Why drinking green tea could prevent cancer.
Nature. 387:5611997. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Aggarwal V, Tuli HS, Tania M, Srivastava
S, Ritzer EE, Pandey A, Aggarwal D, Barwal TS, Jain A, Kaur G, et
al: Molecular mechanisms of action of epigallocatechin gallate in
cancer: Recent trends and advancement. Semin Cancer Biol May.
24:2020Epub ahead of print. View Article : Google Scholar
|
|
16
|
Shen X, Zhang Y, Feng Y, Zhang L, Li J,
Xie YA and Luo X: Epigallocatechin-3-gallate inhibits cell growth,
induces apoptosis and causes S phase arrest in hepatocellular
carcinoma by suppressing the AKT pathway. Int J Oncol. 44:791–796.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Luo KW, Xia J, Cheng BH, Gao HC, Fu LW and
Luo XL: Tea polyphenol EGCG inhibited colorectal-cancer-cell
proliferation and migration via downregulation of STAT3.
Gastroenterol Rep (Oxf). 9:59–70. 2020. View Article : Google Scholar
|
|
18
|
Wei R, Mao L, Xu P, Zheng X, Hackman RM,
Mackenzie GG and Wang Y: Suppressing glucose metabolism with
epigallocatechin-3-gallate (EGCG) reduces breast cancer cell growth
in preclinical models. Food Funct. 9:5682–5696. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang J, Xie Y, Feng Y, Zhang L, Huang X,
Shen X and Luo X: (-)-Epigallocatechingallate induces apoptosis in
B lymphoma cells via caspase-dependent pathway and Bcl-2 family
protein modulation. Int J Oncol. 46:1507–1515. 2015. View Article : Google Scholar :
|
|
20
|
Chow HH, Cai Y, Hakim IA, Crowell JA,
Shahi F, Brooks CA, Dorr RT, Hara Y and Alberts DS:
Pharmacokinetics and safety of green tea polyphenols after
multiple-dose administration of epigallocatechin gallate and
polyphenon E in healthy individuals. Clin Cancer Res. 9:3312–3319.
2003.
|
|
21
|
Kuo PL and Lin CC: Green tea constituent
(-)-epigallocate-chin-3-gallate inhibits Hep G2 cell proliferation
and induces apoptosis through p53-dependent and Fas-mediated
pathways. J Biomed Sci. 10:219–227. 2003.PubMed/NCBI
|
|
22
|
Gupta S, Hastak K, Afaq F, Ahmad N and
Mukhtar H: Essential role of caspases in
epigallocatechin-3-gallate-mediated inhibition of nuclear factor
kappa B and induction of apoptosis. Oncogene. 23:2507–2522. 2004.
View Article : Google Scholar
|
|
23
|
Li S, Xia Y, Chen K, Li J, Liu T, Wang F,
Lu J, Zhou Y and Guo C: Epigallocatechin-3-gallate attenuates
apoptosis and autophagy in concanavalin A-induced hepatitis by
inhibiting BNIP3. Drug Des Devel Ther. 10:631–647. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li S, Wu L, Feng J, Li J, Liu T, Zhang R,
Xu S, Cheng K, Zhou Y, Zhou S, et al: In vitro and in vivo study of
epigallocatechin-3-gallate-induced apoptosis in aerobic glycolytic
hepatocellular carcinoma cells involving inhibition of
phosphofructokinase activity. Sci Rep. 6:284792016. View Article : Google Scholar :
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
26
|
Chen S, Morine Y, Tokuda K, Yamada S,
Saito Y, Nishi M, Ikemoto T and Shimada M: Cancer-associated
fibroblast-induced M2-polarized macrophages promote hepatocellular
carcinoma progression via the plasminogen activator inhibitor-1
pathway. Int J Oncol. 59:592021. View Article : Google Scholar
|
|
27
|
Fiori ME, Di Franco S, Villanova L, Bianca
P, Stassi G and De Maria R: Cancer-associated fibroblasts as
abettors of tumor progression at the crossroads of EMT and therapy
resistance. Mol Cancer. 18:702019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim J and DeBerardinis RJ: Mechanisms and
implications of metabolic heterogeneity in cancer. Cell Metab.
30:434–446. 2019. View Article : Google Scholar :
|
|
29
|
Salem AF, Whitaker-Menezes D, Lin Z,
Martinez-Outschoorn UE, Tanowitz HB, Al-Zoubi MS, Howell A, Pestell
RG, Sotgia F and Lisanti MP: Two-compartment tumor metabolism:
Autophagy in the tumor microenvironment and oxidative mitochondrial
metabolism (OXPHOS) in cancer cells. Cell Cycle. 11:2545–2556.
2012. View Article : Google Scholar :
|
|
30
|
Witkiewicz AK, Whitaker-Menezes D,
Dasgupta A, Philp NJ, Lin Z, Gandara R, Sneddon S,
Martinez-Outschoorn UE, Sotgia F and Lisanti MP: Using the 'reverse
Warburg effect' to identify high-risk breast cancer patients:
Stromal MCT4 predicts poor clinical outcome in triple-negative
breast cancers. Cell Cycle. 11:1108–1117. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pavlides S, Vera I, Gandara R, Sneddon S,
Pestell RG, Mercier I, Martinez-Outschoorn UE, Whitaker-Menezes D,
Howell A, Sotgia F and Lisanti MP: Warburg meets autophagy:
Cancer-associated fibroblasts accelerate tumor growth and
metastasis via oxidative stress, mitophagy, and aerobic glycolysis.
Antioxid Redox Signal. 16:1264–1284. 2012. View Article : Google Scholar :
|
|
32
|
Gonzalez CD, Alvarez S, Ropolo A,
Rosenzvit C, Bagnes MF and Vaccaro MI: Autophagy, Warburg, and
Warburg reverse effects in human cancer. BioMed Res Int.
2014:9267292014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wilde L, Roche M, Domingo-Vidal M, Tanson
K, Philp N, Curry J and Martinez-Outschoorn U: Metabolic coupling
and the Reverse Warburg Effect in cancer: Implications for novel
biomarker and anticancer agent development. Semin Oncol.
44:198–203. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cruz-Bermúdez A, Laza-Briviesca R,
Vicente-Blanco RJ, García-Grande A, Coronado MJ, Laine-Menéndez S,
Alfaro C, Sanchez JC, Franco F, Calvo V, et al: Cancer-associated
fibroblasts modify lung cancer metabolism involving ROS and TGF-β
signaling. Free Radic Biol Med. 130:163–173. 2019. View Article : Google Scholar
|
|
35
|
Shao S, Qin T, Qian W, Yue Y, Xiao Y, Li
X, Zhang D, Wang Z, Ma Q and Lei J: Positive feedback in Cav-1-ROS
signaling in PSCs mediates metabolic coupling between PSCs and
tumour cells. J Cell Mol Med. 24:9397–9408. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang Z, Gao Z, Rajthala S, Sapkota D,
Dongre H, Parajuli H, Suliman S, Das R, Li L, Bindoff LA, et al:
Metabolic reprogramming of normal oral fibroblasts correlated with
increased glycolytic metabolism of oral squamous cell carcinoma and
precedes their activation into carcinoma associated fibroblasts.
Cell Mol Life Sci. 77:1115–1133. 2020. View Article : Google Scholar
|
|
37
|
Zhang X, Dong Y, Zhao M, Ding L, Yang X,
Jing Y, Song Y, Chen S, Hu Q and Ni Y: ITGB2-mediated metabolic
switch in CAFs promotes OSCC proliferation by oxidation of NADH in
mitochondrial oxidative phosphorylation system. Theranostics.
10:12044–12059. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sung JS, Kang CW, Kang S, Jang Y, Chae YC,
Kim BG and Cho NH: ITGB4-mediated metabolic reprogramming of
cancer-associated fibroblasts. Oncogene. 39:664–676. 2020.
View Article : Google Scholar
|
|
39
|
Ippolito L, Morandi A, Taddei ML, Parri M,
Comito G, Iscaro A, Raspollini MR, Magherini F, Rapizzi E,
Masquelier J, et al: Cancer-associated fibroblasts promote prostate
cancer malignancy via metabolic rewiring and mitochondrial
transfer. Oncogene. 38:5339–5355. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wu X, Zhou X, Xu S, Liao C, Chen X, Li B,
Peng J, Li D and Yang L: Extracellular vesicle packaged
LMP1-activated fibroblasts promote tumor progression via autophagy
and stroma-tumor metabolism coupling. Cancer Lett. 478:93–106.
2020. View Article : Google Scholar
|
|
41
|
Deng L, Jiang N, Zeng J, Wang Y and Cui H:
The versatile roles of cancer-associated fibroblasts in colorectal
cancer and therapeutic implications. Front Cell Dev Biol.
9:7332702021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mao X, Xu J, Wang W, Liang C, Hua J, Liu
J, Zhang B, Meng Q, Yu X and Shi S: Crosstalk between
cancer-associated fibroblasts and immune cells in the tumor
microenvironment: New findings and future perspectives. Mol Cancer.
20:1312021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kobayashi H, Enomoto A, Woods SL, Burt AD,
Takahashi M and Worthley DL: Cancer-associated fibroblasts in
gastrointestinal cancer. Nat Rev Gastroenterol Hepatol. 16:282–295.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tan HX, Gong WZ, Zhou K, Xiao ZG, Hou FT,
Huang T, Zhang L, Dong HY, Zhang WL, Liu Y, et al: CXCR4/TGF-β1
mediated hepatic stellate cells differentiation into
carcinoma-associated fibroblasts and promoted liver metastasis of
colon cancer. Cancer Biol Ther. 21:258–268. 2020. View Article : Google Scholar
|
|
45
|
Wu YH, Huang YF, Chang TH, Chen CC, Wu PY,
Huang SC and Chou CY: COL11A1 activates cancer-associated
fibroblasts by modulating TGF-β3 through the NF-κB/IGFBP2 axis in
ovarian cancer cells. Oncogene. 40:4503–4519. 2021. View Article : Google Scholar
|
|
46
|
Park CR, Lee JS, Son CG and Lee NH: A
survey of herbal medicines as tumor microenvironment-modulating
agents. Phytother Res. 35:78–94. 2021. View Article : Google Scholar
|
|
47
|
Whitaker-Menezes D, Martinez-Outschoorn
UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, Birbe RC, Howell
A, Pavlides S, Gandara R, et al: Evidence for a stromal-epithelial
'lactate shuttle' in human tumors: MCT4 is a marker of oxidative
stress in cancer-associated fibroblasts. Cell Cycle. 10:1772–1783.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fiaschi T, Marini A, Giannoni E, Taddei
ML, Gandellini P, De Donatis A, Lanciotti M, Serni S, Cirri P and
Chiarugi P: Reciprocal metabolic reprogramming through lactate
shuttle coordinately influences tumor-stroma interplay. Cancer Res.
72:5130–5140. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sun K, Tang S, Hou Y, Xi L, Chen Y, Yin J,
Peng M, Zhao M, Cui X and Liu M: Oxidized ATM-mediated glycolysis
enhancement in breast cancer-associated fibroblasts contributes to
tumor invasion through lactate as metabolic coupling. EBioMedicine.
41:370–383. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Futagi Y, Kobayashi M, Narumi K, Furugen A
and Iseki K: Identification of a selective inhibitor of human
monocarboxylate transporter 4. Biochem Biophys Res Commun.
495:427–432. 2018. View Article : Google Scholar
|
|
51
|
Hui S, Ghergurovich JM, Morscher RJ, Jang
C, Teng X, Lu W, Esparza LA, Reya T, Le Zhan Yanxiang, Guo J, et
al: Glucose feeds the TCA cycle via circulating lactate. Nature.
551:115–118. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kim BG, Sung JS, Jang Y, Cha YJ, Kang S,
Han HH, Lee JH and Cho NH: Compression-induced expression of
glycolysis genes in CAFs correlates with EMT and angiogenesis gene
expression in breast cancer. Commun Biol. 2:3132019. View Article : Google Scholar :
|