1. Introduction
Metastasis remains the leading cause of mortality in
cancer patients, despite recent advancements in the diagnosis and
treatment of a variety of cancers. It becomes more and more
heterogeneous as cancer progresses, resulting in the production of
aggressive subsets of cancer cells that invade local tissues, lymph
vessels, and circulatory systems before spreading throughout the
body. This aggressive behavior of the original tumor eventually
results in the extensive spread and metastasis of the primary
tumor. Therefore, understanding the mechanism of cancer metastasis
is an important step in determining therapeutic targets that can be
used to slow or stop cancer growth and progression (1). The process of cell transformation
and cancer progression includes gene mutations and epigenetic
changes and the rewiring of cell signals, and the reprogramming of
metabolic pathways. Furthermore, a vast body of literature
published over the past few decades has shown that epigenetic
modifications are implicated in the formation and progression of
the tumor. It has also been said that genetic and epigenetic
changes are closely linked during the development of tumors
(2).
Epigenetic mechanisms of tumor cell growth and gene
expression regulation include DNA methylation, covalent histone
modifications, glycosylation, and ubiquitination. These epigenetic
modifications exhibit unique features and distribution patterns in
different tumor cells. The unique pattern of combinations of these
modifications, collectively referred to as the epigenome, is a
critical determinant of cell fate and gene activity (3). Collectively, the epigenetic state
within cells is tightly controlled to maintain an appropriate state
of differentiation. In cancer, this fine-tuned genomic programming
is disrupted, resulting in unregulated cell proliferation,
defective differentiation, and resistance to apoptosis (4). Epigenetic events, most notable
stability of gene expression and genetic modifications, are not
attributed to any changes in the primary DNA sequence. Here, we
examine the current findings into the contribution of epigenetics
to metastasis, which may guide cell dissemination from the primary
tumor or eventual growth and colonization at distant sites. This
information will allow us to uncover the functional importance of
these epigenetic phenomena and provide informative therapeutic
value for cancers targeting epigenetic changes in metastatic
cells.
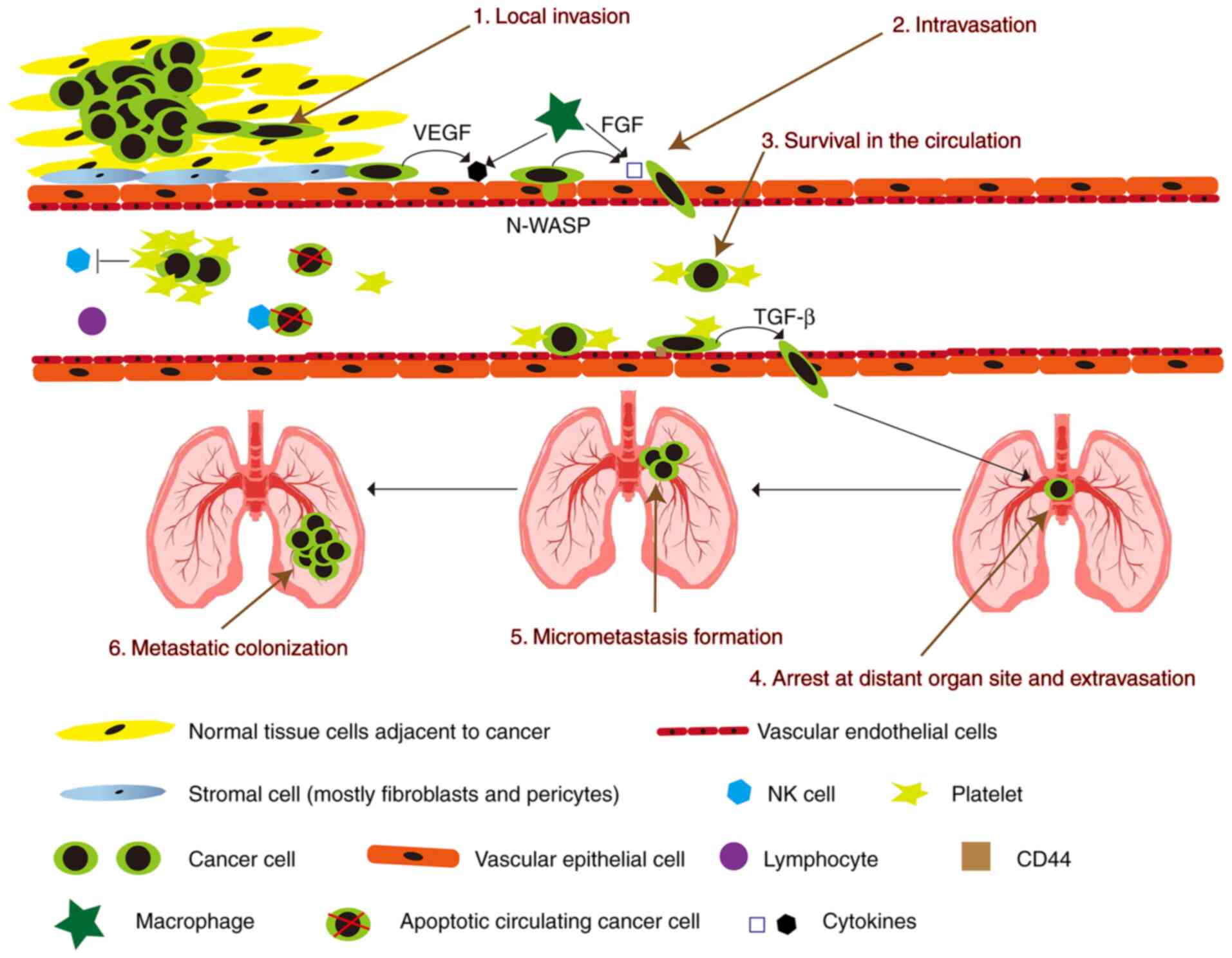
2. Process of cancer metastasis
Metastasis represents a major obstacle in cancer
treatment and is the leading cause of cancer-related deaths. The
process from primary local tumor to distant metastasis is divided
into 6 steps: i) local invasion, ii) intravasation, iii) survival
in the circulation, iv) arrest at the distant organ site and
extravasation, v) micrometastasis formation, and vi) metastatic
colonization (Fig. 1) (5). In order to pass these steps,
different molecular pathways have been shown to be important,
including mitogen-activated protein kinase (MAPK),
phosphatidylinositol 3 kinase (PI3K)/protein kinase B
(Akt)/mammalian target of rapamycin (mTOR), hepatocyte growth
factor (HGF)/mesenchymal-epithelial transition tyrosine kinase
receptor (Met), Wnt/β-catenin, and vascular endothelial growth
factor (VEGF) signal transduction (6). Furthermore, some cytokines secreted
by macrophages can lead to the loss of vascular connections and
increase the permeability of blood vessels when tumor cells invade
blood vessels. Various cellular components in blood vessels will
kill circulating tumor cells, whereas circulating tumor cells can
recruit platelets to resist killing (7).
Cancer cells break away from the original site and
infiltrate the surrounding normal tissues. Morphologically, the
cancer cell transitions from a highly differentiated to an
undifferentiated state (5).
Cancer cells undergo mesenchymal-epithelial transition (EMT) during
migration and invasion. EMT is a cellular process in which cells
lose their epithelial features and acquire mesenchymal ones
(8). The loss of E-cadherin and
its inhibition or attenuation of cell adhesion during EMT are
considered to be a critical step. E-cadherin is a type of
Ca2+-dependent intercellular adhesion molecule in
epithelial tissues. E-cadherin is a single-channel transmembrane
glycoprotein containing five extracellular repeats that mediate
Ca2+-dependent interactions with opposing molecules on
adjacent cells (9). Expression or
the cell surface localization of E-cadherin is frequently lost in
advanced tumors and is associated, at least in some cases, with the
incidence of metastasis and tumor recurrence (10). Loss of E-cadherin expression in
human tumors is most commonly caused by methylation of its
promoter, or upregulation of the transcriptional repressors SNAIL,
SIP1 and zinc finger E-box-binding homeobox 1 (ZEB1), which target
the E-cadherin promoter (11).
Loss of E-cadherin function is a cause of promotion of invasion and
metastasis, primarily through the transformation of epithelial
tumor cells into highly migratory and invasive cells (12). It can combine with β-catenin in
the cytoplasm to form a complex. This complex can be directly
connected to the actin cytoskeleton to maintain the stability of
cell adhesion and cell polarity (13). If the expression of E-cadherin is
blocked, tumor growth factor (TGF)-β, Wnt/β-catenin, Hedgehog,
Notch, and tumor necrosis factor (TNF) signals will induce cancer
cell EMT via Twist/Snail/Slug/ZEB1, causing cancer cells to invade
and metastasize (8,14).
For intravasation, the migration of cancer cells to
blood vessels is caused by factors involved in local invasion-the
secretion of proteases such as matrix metalloproteinase (MMP)-1,
-2, and -9 and the activation of the urokinase-type plasminogen
activator (uPA)/uPA receptor (uPAR) (15). Cancer cells mainly invade
capillaries and venules, and large blood vessels are resistant to
tumor cell invasion. Tissue fibrosis is also resistant to cancer
cell invasion. Cirrhotic organs are not prone to metastasis, and
myofibroblasts from cirrhosis can secrete MMP inhibitors (TIMP). In
addition, the heart and skeletal muscles are also resistant to
metastasis (16).
After cancer cells enter the circulation, they are
prone to anoikis due to their lack of support from the
extracellular matrix (ECM) (17).
It is estimated that less than 1% of circulating tumor cells (CTCs)
that circulate in the blood on a daily basis would survive and have
a possibility of spreading to produce distant metastases (18). When cancer cells are in
circulation, they are attacked by immune cells, which in turn are
assaulted by cancer cells. In order to prevent immune cells from
attacking cancer cells, fibrin and platelets adhere to the surface
of cancer cells (19). Less than
1% of cancer cells found in the blood have a chance of surviving,
and some cancer cells are in a dormant state, and less than 0.01%
of cells with high metastatic potential can emerge from blood
vessels to form metastases (20).
The following are the leading causes of cancer cells
to stop growing. i) Cancer cells are prevented from spreading by
capillary stenosis. Then the cancer cells extravasate through the
endothelial cells and exit the blood vessels to establish
metastases. ii) The cancer cell surface protein interacts with the
microvascular endothelial surface (21). The detailed mechanism of cancer
cell metastasis to the organs is currently unclear, which may be
related to organ microvascular endothelial cells. Studies have
shown that cancer cells only adhere to microvascular endothelial
cells, not large vascular endothelial cells (22). There are some specific molecules
in organ micro-vascular endothelial cells. These molecules include
adhesion molecules, intercellular adhesion molecule-1 (ICAM-1),
selectin and integrin (23), and
endothelial cells secrete chemo-attractants [CXC chemokine receptor
4 (CXCR4), CXC chemokine receptor 12 (CXCR12), and chemokine (C-C
motif) receptor 1 (CCR1)] (24).
Therefore, the specific molecules may be the main reason for cancer
cell arrest. The specificity of metastatic sites for each tumor
entity is also called tissue tropism (25). Tissue tropism helps predict the
future metastatic site through these specific molecules and may
become the target of future anti-metastatic drug therapy (26).
After cell extravasation, CTCs need to create
specific conditions to reach the target organs (27). The steps of cancer cells exiting
the vessel wall are opposite to the direction of their entering the
vessel wall (28). Cancer cells
removed from blood vessels are in an unfavorable environment, and
they have to overcome various difficulties to establish metastases
(29). Primary cancer cells
release growth factors into the circulation (30). These growth factors lead to an
upregulation of vascular endothelial growth factor (VEGF)-A,
placental growth factor (PlGF), transforming growth factor-β
(TGF-β), and inflammatory proteins S100A8/9 at the future
metastatic sites (31). From the
results, the cells that can establish metastases should have the
properties of cancer stem cells (32).
As the final step of the metastatic cascade,
micro-metastases have now spread to distant organs. In specific
niches or in undefined spots, a plethora of genes and signals
support metastatic cell growth and survival (33). Many of these pro-metastatic
stromal mediators ultimately activate stem cell support pathways
(Wnt, TGF-β, BMP, Notch, Stat3), positional and mechanical pathways
(Hedgehog, Hippo), pathways that integrate cell metabolism and
survival (PI3K/AKT, MAPK, HIF), and inflammatory pathways (NF-κB,
Stat1) (34). Cancer kills the
most individuals when it spreads to other parts of the body.
Preventing the formation of new niches may be a new way to treat
metastatic diseases.
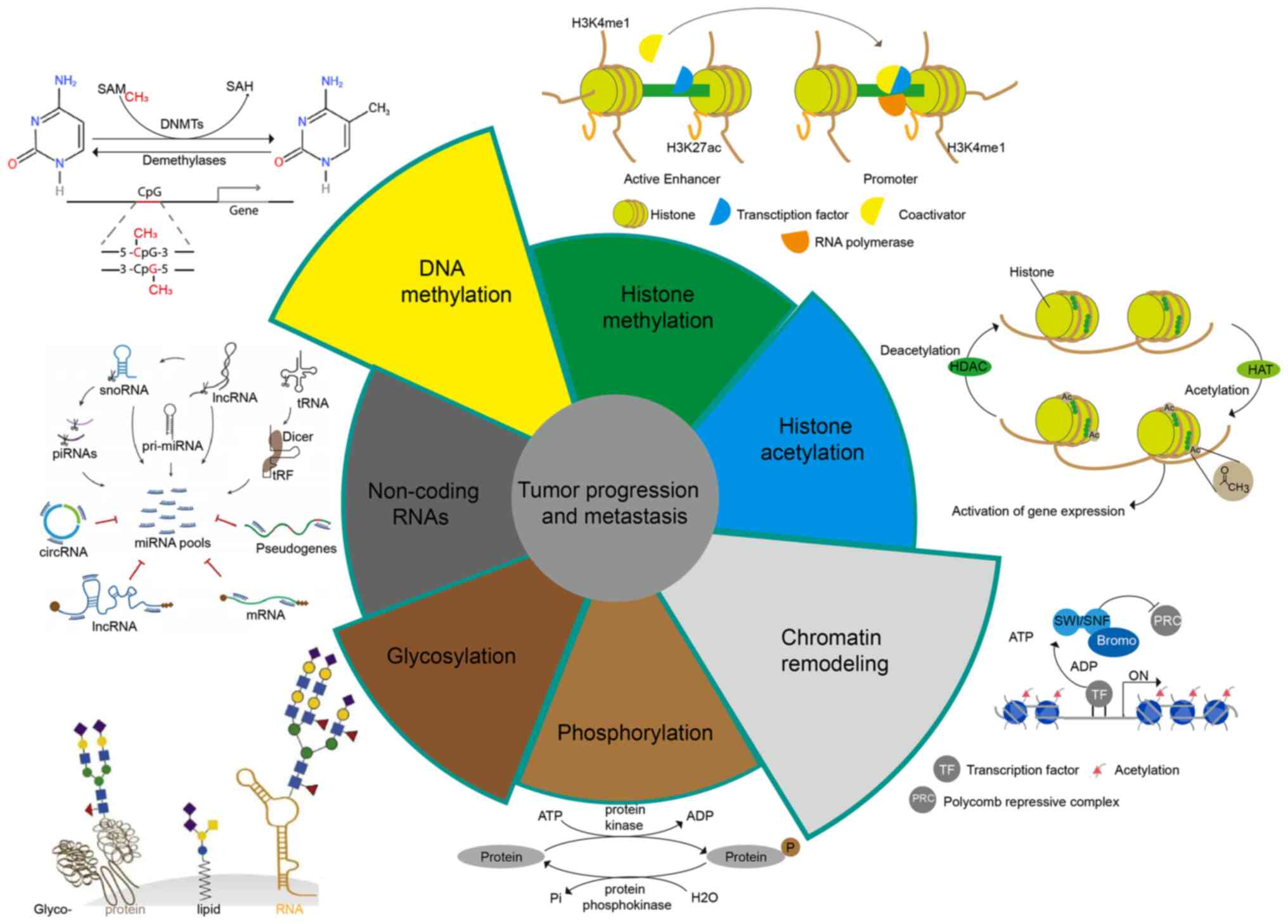
3. Epigenetic in tumor progression and
metastasis
Epigenetic research aims to reveal how the
environment, social status, psychosocial factors, and nutrition
influence the expression of an individual's genetic information
(35). Epigenetics is responsible
for initiating and maintaining epigenetic silencing and regulating
gene expression profiles and is the cornerstone of a range of
cellular processes, including cell differentiation, gene
expression, X chromosome inactivation, embryogenesis, and genomic
imprinting (36). In addition,
epigenetics also plays a significant role in the regulation of
tumor metastasis. We will illustrate the impact of epigenetics on
tumor metastasis from the following aspects (Fig. 2).
Metastasis is regulated by DNA
methylation
DNA methylation is an epigenetic modification first
discovered in humans in the early 1980's and the most intensely
studied in epigenetic regulatory mechanisms (37,38). In a broad sense, DNA methylation
refers to the chemical modification process in which a specific
base in the DNA sequence obtains a methyl group by covalent bonding
with S-adenosyl methionine (SAM) as a methyl donor under the
catalysis of DNA methyltransferase (DNMT). This DNA methylation
modification can occur at the C-5 position of cytosine, the N-6
position of adenine, and the G-7 position of guanine (39). In general, DNA methylation mainly
refers to the methylation process that occurs at the 5th carbon
atom on cytosine in CpG dinucleotides, and the product is called
5-methylcytosine (5-mC), which is the main form of DNA methylation
in eukaryotes such as plants and animals, and the only form of DNA
methylation in mammals found (40). DNA methylation plays an important
role in regulating individual growth, development, gene expression
patterns, and genome stability without changing the DNA sequence,
and this modification is stable during development and cell
proliferation. A large number of studies in recent years have shown
that DNA aberrant methylation is closely related to the occurrence,
development, and carcinogenesis of tumors (41,42).
The DNA methyltransferase (DNMT) family consists of
a group of conserved DNA-modifying enzymes that play central roles
in epigenetic regulation. Five DNMT family members have been found
in mammals: DNMT1, DNMT2, DNMT3a, DNMT3b, and DNMT3L (43). However, only DNMT1, DNMT3a, and
DNMT3b interact to generate the overall cytosine methylation
pattern. These independently encoded proteins are divided into
generating DNA methylases (DNMT3a and DNMT3b) and maintaining DNA
methylases (DNMT1). DNMT2 and DNMT3L are not considered cytosine
methyltransferases (44). DNMT3L,
on the other hand, has been shown to stimulate de novo DNA
methylation via DNMT3a and to mediate transcriptional repression
via interaction with histone deacetylase 1 (45).
The role of DNA methylation in tumors is mainly
manifested in the following aspects. First, cytosine in methylated
CpG island dinucleotides is deaminated to thymine at a high
frequency, causing gene mutation (46). Second, tumor-suppressor genes and
DNA repair genes are silenced due to hypermethylation (47). Third, oncogene methylation levels
are reduced and activated (48);
and fourth, the overall reduction in methylation levels of the
genome causes transposons and repetitive sequences to activate,
resulting in decreased chromosomal stability (49). These factors are important reasons
for the development, metastasis, and progression of tumors, which
eventually lead to the death of patients. The overall DNA
methylation level (i.e., methylation profile) and changes in the
degree of methylation of specific genes can be used as tumor
diagnostic indicators (50).
In normal cells, heterochromatin is hypermethylated
around the central point; however, in many tumors, this mechanism
is disrupted, and DNA methylation in normally inactive regions is
lost. Transposable elements are subsequently reactivated and can
integrate at arbitrary sites in the genome, leading to mutation and
genomic instability (51).
Therefore, DNA methylation plays a vital role in tumor progression
and metastasis. DNMT1 is a maintenance methyltransferase that
methylates cytosines in hemimethylated CpG dinucleotides (52). DNMT1 is required for the
maintenance of trimethylation of lysine 9 at histone H3 in
pericentromeric regions (53),
and DNMT1 can bind to H3K9me3 and H3K27me3 at the promoter sites of
ZEB2 [0.4 kb (3502) site] or Kruppel-like factor 4 (KLF4) [0.4 kb
(3110) site] or Snail to inhibit EMT of prostate cancer (PCa) and
hepatocellular carcinoma (HCC) (54,55). miR-185 and miR-148a can directly
target DNMT1 so that the expression of breast cancer gene 1 (BRCA1)
can be increased to stop the spread of breast cancer and gastric
cancer (GC) (56).
In addition to the inhibitory effect of DNMT1, DNMT1
can also regulate the expression of some genes to promote tumor
metastasis. For example, DNMT1 can bind to the nuclear
transcriptional repressor CpG region of RAD9, which will promote
the transcriptional expression of RAD9 (a gene that
maintains genome integrity, DNA repair, cell cycle checkpoints,
apoptosis, and transcriptional transactivation of specific target
genes) to promote metastasis of PCa cells (57). Osteopontin increases the
expression of DNMT1 to increase the methylation of tumor-suppressor
genes, Ras-associated domain family 1A (RASSF1A), GATA
binding protein 4 (GATA4), cyclin-dependent kinase-like 2
(CDKL2), and death-associated protein kinase (DAPK)
to induce metastasis of liver cancer and esophageal squamous cell
carcinoma (ESCC) (58,59). Moreover, DNMT1 also reduces the
expression of lncRNA-SPRY4-IT1 to promote gastric cancer cell
migration and invasion (60).
However, lncRNA-HNF1A-AS1 can bind to DNMT1 and inhibit its
activation from promoting EMT of lung adenocarcinoma (61). SET and MYND domain-containing
protein 2 (SMYD2) can increase adenomatous polyposis coli 2 (APC2)
methylation by DNMT1 to activate the Wnt/β-catenin pathway, which
promotes colorectal cancer (CRC) EMT (62).
De novo methylation and mammalian development
require DNMT3a and DNMT3b (63).
The target recognition domain (Trd) (residues R 831-f848), the
catalytic loop (residues G 707-k721), and the homodimeric interface
of DNMT3a mediate DNMT3a binding to DNA, and these loops come
together to form a continuous DNA binding surface (64). Recognizing of CpG dinucleotides by
DNMT3a is mediated by catalytic and TRD cycles (65). DNMT3b-mediated transcriptional
repression occurs at CpG island (CGI)-associated promoters and
repeats. The activity of DNMT3b is mainly to promote long-term gene
silencing, which needs to be preserved in certain tissues to
maintain the body's life (66).
In addition to centromeric, pericentromeric, and subtelomeric
repeats, germline genes are also known genomic targets of DNMT3b.
Notably, the maintenance of CGI methylation of certain germ
cell-specific genes in somatic cells depends entirely on DNMT3b
activity (67). DNMT3a and DNMT3b
also play an important role in tumor metastasis. For example,
DNMT3a mutations are more common in acute myeloid leukemia (AML),
and DNMT3A mutations can promote extra-medullary infiltration (EMI)
by upregulating the expression of TWIST1 (68). In addition, DNMT3a also regulates
the expression of Snail and E-cadherin to affect the metastasis of
GC (69). Metastasis-associated
protein 1 (Mta 1) is one of the important chromatin remodeling
factors in eukaryotic cells; it is one of the most upregulated
oncogenes in human tumors and is involved in tumor progression and
metastasis. Mta1 can inhibit the transcription of DNMT3a from
increasing the expression of insulin-like growth factor binding
protein-3 (IGFBP-3), which promotes the metastasis of breast cancer
(70). Smad4 and mastermind-like
transcriptional coactivator 1 (MAML1) are novel direct targets of
miR-34a and miR-133a-3p, but lncRNA-34a can bind to DNMT3a and
inhibit the activity of miR-34a and miR-133a-3p, which increases
Smad4 and MAML1 expression in HCC metastasis (71,72). In addition, DNMT3b also binds to
the promoter region of miR-34, which enhances the expression levels
of hepatocyte nuclear factor 4 γ (HNF4G) and Notch1, which are
downstream targets of miR-34a to promote migration and invasion of
bladder cancer (73).
Furthermore, lncRNA-H19 can directly bind to miR-29b-3p (miR-29b)
and inhibit the expression of DNMT3b in bladder cancer cells. More
importantly, upregulation of lncRNA-H19 was found to antagonize
miR-29b-3p-mediated inhibition of breast cancer cell proliferation,
migration and EMT. On the contrary, lncRNA-H19 knockdown partially
reversed the effect of the miR-29b-3p inhibitor on Dnmt3b and
promoted miR-29b-3p-induced MET (74). DNMT3b also can bind to the nuclear
transcriptional repressor CpG region of RAD9 to promote metastasis
of PCa cells (57).
Demethylation is one method of tumor prevention and
treatment that involves restoring the activity of some key tumor
suppressor or DNA repair genes. At present, DNMT inhibitors are the
most studied, as they can reverse abnormal DNA methylation by
inhibiting DNMT activity (75).
The first phenotype-modifying drug, 5-azacytidine and its analog
5-aza-2′-deoxycytidine (5-aza-CdR), have been approved by the US
FDA to treat preleukemic myelodysplastic syndromes (76). 5-Aza-CdR is an analog of cytosine,
which can be incorporated into the DNA chain during DNA
replication. On the one hand, it can reduce the ability of DNA to
receive methyl groups, and on the other hand, it inhibits DNMT
activity, resulting in the reduction in the DNA methylation level
(77). It has been clinically
shown that 5-aza-CdR can improve the survival rate of some patients
with stage IV small cell lung cancer. However, the drug also has
toxic and side effects that cannot be ignored (for example, the
specificity is not strong, and it cannot be targeted for a specific
tumor-suppressor gene; targeted therapy; high doses of 5-aza-CdR
may induce tumor metastasis), so its clinical application is
greatly limited (78). However,
MCF-7 breast cancer cells were treated with the DNMT inhibitor
5-aza-cytidine to maintain a hypomethylated state. The results
showed that the expression levels of pro-invasive EMT-associated
genes related to the EMT process were upregulated, and the invasive
and metastatic abilities of the cells were enhanced (79). The results of this study are worth
serious deliberation. Although the use of DNMT inhibitors to treat
tumors may inhibit the expression of proto-oncogenes, it may also
increase the risk of tumor cell metastasis and dissemination.
Therefore, we should use these drugs with caution in clinical
practice.
Metastasis regulated by histone
modifications
Histone methylation and
demethylation
Histones protect genetic information, maintain DNA
structure, and regulate gene expression. Histone amino-terminal
(N-terminal) domains protrude from the nucleosome and can interact
with other regulatory proteins and DNA. Histone modifications
include methylation, phosphorylation, acetylation, crotonylation,
ubiquitination, glycosylation, and ADP ribosylation. Imbalances in
histone modifications can lead to tumorigenesis, and loss of
methylation, and acetylation of histone H3 and H4 residues has been
shown to be a marker of tumor cells (80). Histone methylation and
demethylation are usually carried out by histone methyltransferases
(HMTs) and histone demethyltransferase (HDMs). The methylation of
histones is a covalent modification of arginine and lysine.
Arginines can be mono-or di-methylated, while lysines can be mono-,
di-, or tri-methylated (81).
There are three types of enzymes responsible for histone
methylation, lysine-specific SET domain histone methylase,
methylase of amino acid, and methylase of lysine (82). In general, the methylation of
different sites of histone H3 and H4 and the amount of methylation
have great significance for the transcriptional regulation of
genes. Among them, H3K9me3, H3K27me3, and H4K20me2/3 mediate
transcriptional repression, while H3K4me1/2/3, H3K9me1, H3K27me1,
H3K36me1/2/3, and H3k79me1/2/3 mediate transcriptional activation
(83).
According to the different amino acids catalyzed by
histone transferases, HMTs can be divided into two families:
protein lysine methyltransferases (PKMTs) and protein arginine
methyltransferases (PRMTs). The KMT family is further divided into
enzymes that contain SET domains, such as G9a, EZH2, DOT1L,
SUV39H1, CLL8, MLL1, SET8, SETDB1, GLP, and SETD2; the PRMT family
includes PRMT1-9 and CARM1 in mammals. HMTs have a direct
regulatory effect on tumor development and metastasis (84). The enhancer of zeste homolog 2
(EZH2) gene can promote tumor development and is the catalytic
component of the polycomb repressive complex 2 (PRC2). EZH2
utilizes its HMT activity to catalyze the trimethylation of histone
H3 lysine 27, inhibiting the downstream tumor-suppressor genes such
as E-cadherin, transforming growth factor-β (TGF-β) receptor 2
(TGFBR2), P57, and PSP94 (85).
However, EZH2 itself lacks enzymatic activity, and its activity
requires the assistance of the zinc-finger-containing protein
(SUZ12) and the WD40-repeat protein (EED) to maintain the integrity
of the PRC2 complex and the methyltransferase activity of EZH2
(86). EZH2 binds phosphorylated
p38 (p-p38) in breast cancer cells to other core members of PRC2,
EED, and SUZ12. EZH2 overexpression leads to the expression of
p-p38 and activates downstream pathway proteins, which promote
breast cancer motility and metastasis (87). EZH2 overexpression inhibits the
expression of metalloproteinase 2 (MMP-2) and promotes the
proteolytic activity of MMP-2 and -9, which also promotes ovarian
cancer invasion and migration (88). In addition, numerous studies have
shown that EZH2 overexpression promotes the proliferation,
migration, and invasion of cancer cells in lung, bladder, melanoma,
and colorectal cancers (89,90). In addition to the high expression
of EZH2, the low expression of some members of this family promotes
tumor metastasis. For example, loss of SETD2 leads to persistent
activation of AKT through extracellular matrix (ECM) production,
thereby facilitating metastasis of pancreatic ductal adenocarcinoma
(PDAC) (91). More importantly,
loss of SETD2 can activate EZH2 signaling and AMPK signaling to
promote PCa metastasis (92).
KMT1E (also known as SETDB 1) is involved in the epigenetic
silencing of oncogenes and tumor-suppressor genes in cancer cells.
KMT1E, a metastasis suppressor, is strongly downregulated in highly
metastatic lung cancer cells (93). In addition, SET8 (94), G9a (95), GLP (95), DOT1L (95), and MLL1 (96) have also been reported to be
closely associated with tumor metastasis. The PRMT family also has
members involved in tumor metastasis. PRMT1 can directly target the
leukocyte adhesion molecule (ALCAM) to promote the growth and
metastasis of melanoma (97).
PRMT1, PRMT5, PRMT6, and PRMT7 can promote EMT of head and neck
cancer, colorectal cancer, breast cancer, ovarian cancer, lung
adenocarcinoma, and oral cancer cells (98–102).
HDMs also have a variety of domains, including
Jumonji (N/C terminal domains) (83), PHD-finger, Zinc-finger, SWIRM1
(Swi3, Rsc, and Moira domains), and the amine oxidase domain
(103). Lysine-specific
demethylases (KDMs) work in concert with histone lysine methylases
to maintain global histone methylation patterns. The histone
demethylases characterized to date belong to the amine oxidase and
oxygenase superfamilies. The amino oxidase family members degrade
histones through flavin adenine dinucleotide (FAD)-dependent amine
oxidase reaction substrate demethylation (104). The JmjC protein belongs to the
family of oxygenases and demethylates histones in an
α-ketoglutarate and Fe(II) ion-dependent manner (105). Furthermore, they can also be
divided into several families: UTY, KDM1A, KDM1B, KDM2A, KDM2B,
KDM3A, KDM3B, JMJD1C, KDM4A, KDM4B, KDM4C, KDM4D, KDM5A, KDM5B,
KDM5C, KDM5D, KDM6A, and KDM6B (45). Although not extensively studied as
histone methylases, KDMs are also associated with tumor progression
and metastasis. For example, KDM1A (also known as LSD1)-mediated
stabilization of SEPT6 can activate the TGF-β1/SMAD pathway and
VEGF-C/PI3K/AKT signaling pathway, to stability promote the
metastasis of GC, non-small cell lung cancer (NSCLC), and breast
cancer (106–108). At the same time, KDM2A and KDM2B
can downregulate the expression of programmed cell death 4 (PDCD4)
and active the TGF-β1/SMAD and PI3K/AKT/mTOR pathways to stabilize
and promote the metastasis of GC, lung cancer, pancreatic cancer,
and ovarian cancer (109–111).
KDM3A and KDM3B can activate the Wnt/β-catenin pathway and
upregulate c-MYC, lncRNA-MALAT1, and melanoma cell adhesion
molecule (MCAM), and MCAM to promote CRC, Ewing sarcoma, and
neuroblastoma migration and invasion (112,113). In addition to this,
lysine-specific demethylases KDM4A (114) and KDM4B (115) are also closely related to tumor
progression and metastasis.
In the past few years, specific inhibitors of a
large number of different HMTs and HDMs have been developed. EZH2,
DOT1L, PRMT1, PRMT5, LSD1, KDM2B, KDM4D inhibitors have entered the
clinical trial stage. These results demonstrate that HMTs and HDMs
play a critical function in tumor metastasis. Furthermore, we
believe that more targeted drugs for HMTs and HDMs will enter the
clinic in the future, bringing good news to patients.
Histone acetylation and
deacetylation
Protein acetylation refers to the process of adding
acetyl groups to protein lysine residues under the action of an
acetyltransferase. Histone acetylation is a post-translational
modification that mostly occurs at specific lysine residues in the
basic amino acid concentration region at the N-terminal of core
histones and transfers the acetyl group of acetyl-CoA to
sNH3+ of lysine to neutralize a positive charge
(116). Histone acetylation and
deacetylation are usually carried out by histone acetyltransferases
(HATs) and histone deacetylases (HDACs).
HATs can be divided into two families according to
the properties of their substrates, the GNAT family (GCN5-related
N-acetyltransferase family) and the MYST family (MOZ, Ybf2/Sas3,
Sas2, and Tip60). Although both contain acetyl-CoA homologous
sequences, there are differences in their core regions (117). Functionally, the GNAT family is
mainly responsible for the acetylation of lysine sites on histone
H3. In contrast, the MYST family is related to the acetylation of
lysine sites (such as H4K16) on histone H4. According to the
different domains contained, the MYST family can be divided into
the following three types: the subgroup containing the plant
homology domain (including MOZ and MORF), and the subgroup
containing the chromatin domain (including Esa1, dMOF, and Tip60),
and a subgroup containing zinc fingers (HBO1) (118). HATs facilitate the dissociation
of DNA and histone octamers and the relaxation of nucleosome
structure so that various transcription factors and
co-transcription factors can specifically bind to DNA binding sites
and activate gene transcription. Studies have shown that HATs are
involved in tumor growth and metastasis. For example, lysine
acetyltransferase 6A (KAT6A) can activate the Wnt/β-catenin pathway
to promote the growth and metastasis of ovarian cancer (119). Histone acetyltransferase 1
(HAT1) and HBO1 can regulate the expression of TP53 to affect tumor
metastasis (120).
HDACs deacetylate histones, bind tightly to
negatively charged DNA, and make chromatin dense and coiled,
repressing gene transcription (121). HDACs have been identified and
grouped into four classes, including class I consisting of HDAC1,
HDAC2, HDAC3, HDAC8; class II consisting of HDAC4, HDAC5, HDAC6,
HDAC7, HDAC7, HDAC10; class III consisting of SIRT1, SIRT2, SIRT3,
SIRT4, SIRT5, SIRT6, SIRT7; and class IV consisting of HDAC11
(122). Although HDACs can
promote tumor metastasis, sometimes, some HDACs can also inhibit
tumor metastasis. For example, HDAC1, HDAC2, HDAC4, HDAC5, and
HDAC6 are required for both proliferation and metastasis of
melanoma, pancreatic cancer, nasopharyngeal carcinoma, and
hepatocellular carcinoma by downregulating E-cadherin (123–126). IRT7 is transcriptionally
repressed by HDAC8, a novel cofactor of the SMAD3/4 complex,
through local chromatin remodeling, further activating TGF-β
signaling and causing lung cancer metastasis (127). SIRT1 can promote the expression
of ZEB1 to induce EMT of osteosarcoma (128). Overexpression of SIRT6 not only
enhances the phosphorylation of extracellular signal-regulated
kinase 1/2 (p-ERK 1/2) but also activates MMP 9 to promote tumor
cell migration and invasion (129). SIRT6 also suppresses cell
metastasis of pancreatic ductal adenocarcinoma (PDAC) by regulating
the expression of HMGA 2, IGF2BP1, and IGF2BP3 (130). In addition, SIRT3 can activate
FOXO3a and inhibit the wnt/β-catenin pathway, thereby inhibiting
EMT of prostate cancer cells (131). SIRT4 inhibits Drp1
phosphorylation through interaction with fis-1, and suppresses
MEK/ERK activity to inhibit NSCLC cell invasion and migration
(132). SIRT7 is a key regulator
of TGF-β signaling and a suppressor of breast cancer metastasis,
and its deletion promotes the metastasis of breast cancer cells
(133).
Several HDAC inhibitors (HDACi) with different
chemical structures have been purified from natural extracts or
chemical synthesis. To date, there are at least nine different
HDACi, including Saha, LAQ 824, FK 228, MS-275, CI-994, PXD 101,
valproic acid, pyroxamine, and sodium butyrate, currently in use
alone or in combination with therapy for blood disorders and solid
tumors (134,135). HDACi selectively kills tumor
cells and has little toxicity to normal cells. The basis for the
selective toxicity of HDACi is unclear but may be related to the
HDAC overexpression observed in cancer cells (2). HDACi has the ability to effectively
activate multiple molecular pathways and mediate its antitumor
effect (136,137). The antitumor effect of HDACi
apparently depends on inhibiting the proliferation, survival,
migration, invasion, metastasis, and angiogenesis of cancer cells
by regulating gene transcription (138). In conclusion, the molecular
basis for the tumor-selective cytotoxicity of HDACi has not been
extensively studied; therefore, the molecular basis requires
further in-depth validation.
Metastasis regulated by ubiquitination
and deubiquitination
Ubiquitination refers to the process in which the
ubiquitin (a small 76-residue regulatory protein ubiquitously
expressed in eukaryotes) molecule classifies proteins in cells
under the action of a series of special enzymes, selects target
protein molecules from them, and modifies explicitly the target
protein (139). These special
enzymes include ubiquitin-activating enzymes (E1),
ubiquitin-conjugating enzymes (E2), ubiquitin-protein ligase (E3),
and degrading enzymes (140).
Substrates can be modified with single ubiquitin
(monoubiquitination) or polymeric Ub chains. Depending on which
internal lysine (K6, K11, K27, K29, K33, K48, K63), or the
N-terminal methionine residue of the Ub (m1, linear or
head-to-tail) can be used to link to the far Ub chain type of end
(141). Deubiquitinating enzymes
(Dubs) balance ubiquitin chain growth by removing ubiquitin. The
synergistic effect of Dubs recognition and Ub hydrolysis creates a
dynamic network that controls the distribution of different
ubiquitin signals, which in turn regulates numerous biological
processes within the cancer cell (142). Approximately 103 Dubs have been
recognized in the human genome, and they can be divided into six
families according to their sequence and the sequence of conserved
regions: USPs (ubiquitin-specific proteases) such as USP2, USP6,
USP7, USP8, USP11, USP15, USP16, USP21, USP 28, USP35; UCHs
(ubiquitin C-terminal hydrolysis enzymes) such as UCH-L1, UCH-L3,
UCH-L5; MJDs (Machado-Joshphin domain-containing proteases) such as
JosD1, JosD1; OUTs (ovarian cancer proteases) such as A20, OTUB2,
TRABIO; SENPs (motif-interacting with ubiquitin-containing novel
DUB family), such as SENP1, SENP2, SENP8; JAMMs (JAB1, MPN, MOV34
family) such as AMSH (143).
Ubiquitination and deubiquitination play an
essential role in protein localization, metabolism, function,
regulation, and degradation. At the same time, it regulates nearly
all life activities, including cell cycle, proliferation,
apoptosis, differentiation, metastasis, gene expression,
transcriptional regulation, signal transmission, damage repair,
inflammation, and immunity (144). For example, E3 ubiquitin ligase
RNF126 specifically regulates PTEN stability and then induces cell
proliferation and metastasis of bladder cancer by upregulating the
EGFR/PI3K/AKT signaling pathway (145). STAMBP is a deubiquitinase (Dubs)
family member in the Jab1/MPN family of metalloenzymes that
specifically cleaves K63-linked polyubiquitinated chains from
substrates to promote the stabilization of EGFR and the active AMPK
signaling pathway to induce cell metastasis of lung adenocarcinoma
(146). RING finger-domain E3
ubiquitin ligase RBBP6-mediated ubiquitination and degradation of
IκBα enhances p65 nuclear translocation, triggering activation of
the NF-κB pathway, which in turn induces EMT of colorectal cancer
(147). In addition, some
deubiquitinating enzymes can also play a role in promoting tumor
metastasis. USP5 can induce EMT of non-small cell lung cancer
(NSCLC) by activating the Wnt/β-catenin pathway (148). USP10 directly interacts with
SMAD4 and stabilizes it to promote HCC proliferation and metastasis
(149). USP21 can deubiquitinate
and stabilize EZH2 to induce cell proliferation and metastasis of
bladder cancer (150). USP11
promotes colorectal cancer growth and metastasis by stabilizing
PPP1CA, activating the ERK/MAPK pathway, and insulin-like growth
factor 2 mRNA binding protein 3 (IGF2BP3) signaling (151), and interacting with nuclear
factor 90 (NF90) to promote HCC proliferation and metastasis
(152). OTU domain-containing
protein 3 (OTUD3), a deubiquitinating enzyme, also promotes HCC
proliferation and metastasis by regulating α-actinin 4 (ACTN4)
(153). The deubiquitinating
enzyme PSMD14 directly interacts with Snail and stabilizes it to
promote cell migration and tumor metastasis of esophageal squamous
cell carcinoma (154). In
addition to their promoting effects, ubiquitinases and
deubiquitinases can also inhibit tumor metastasis. For instance, E3
ligase zinc finger protein 91 (ZFP91) can regulate PKM splicing to
inhibit HCC metastasis (155).
Ubiquitin-conjugating enzyme variant proteins (Ube2v) and E3
ubiquitin ligase SMURF2 can degrade the protein level of SIRT1,
thereby suppressing cell proliferation and metastasis of CRC
(156,157). BTRC, an E3 ubiquitin ligase, can
degrade the protein level of Twist1 to suppress cell proliferation
and metastasis of GC (158).
The drug development of E3 ligase has been a very
challenging research hotspot in recent years. At present, antitumor
drugs targeting E3 ligase are mainly divided into four categories
according to their mechanism of action: E3 ligase targeted
inhibitors, E3 ligase targeted agonists, proteolytic targeting
chimeras (PROTACs), and molecular glues (159). Considering the complex and
extensive life activities regulated by ubiquitination, blocking or
activating E3 ligase to treat tumors may have adverse effects on
other everyday life metabolic activities. Therefore, exploring and
solving this problem remains a considerable challenge. Moreover,
most ubiquitination inhibitors found to be beneficial in
preclinical studies have shown poor results in clinical trials.
This discrepancy may be because we do not know enough about the
target protein's structural analysis and medicinal chemistry, which
requires technological advances.
Similarly, a large number of Dubs play an important
role in the development of several stages of cancer development.
The potential to affect processes such as signal transduction,
proliferation, and apoptosis by affecting ubiquitination and
proteasomal degradation of key regulators is promising and
exciting. Dubs are more likely candidates than E3 ligases, which
lack well-defined catalytic residues (160). For example, the first highly
selective proteasome-bound USP14 inhibitor
uu1(ubiquitin-7-amino-4-methylcoumarin, ub-amc) was identified by
high-throughput screening of Ub-amc (161). P5091
(1-[5-[(2,3-dichlorophenyl)thio]-4-nitro-2-thienyl]-ethanone) was
discovered to be a specific inhibitor of USP7 for the first time.
This compound inhibits the stabilizing effect of USP7 on Hdm2 and
exerts a pro-apoptotic effect by stabilizing p21 and p53 (162). Betulinic acid (BA)
[(3β)-3-hydroxy-lup-20(29)-en-28-oc acid)] is a naturally
occurring plant-derived compound with proapoptotic and highly
specific effects on cancer cells. BA was identified as a broad
inhibitor of deubiquitination that induces apoptosis by releasing
mitochondrial proteins and leads to the downregulation of
angiogenic markers (163).
Overall, USPs are a complex system, and not all components have
been adequately evaluated. It is well known that the UPSs are
involved in almost all tumor cell processes. There are many
important clinical implications to new ways to target UPS in cancer
therapy, and we expect that UPS-based therapy will play a
significant role on a clinical level in the near future.
Metastasis regulated by
glycosylation
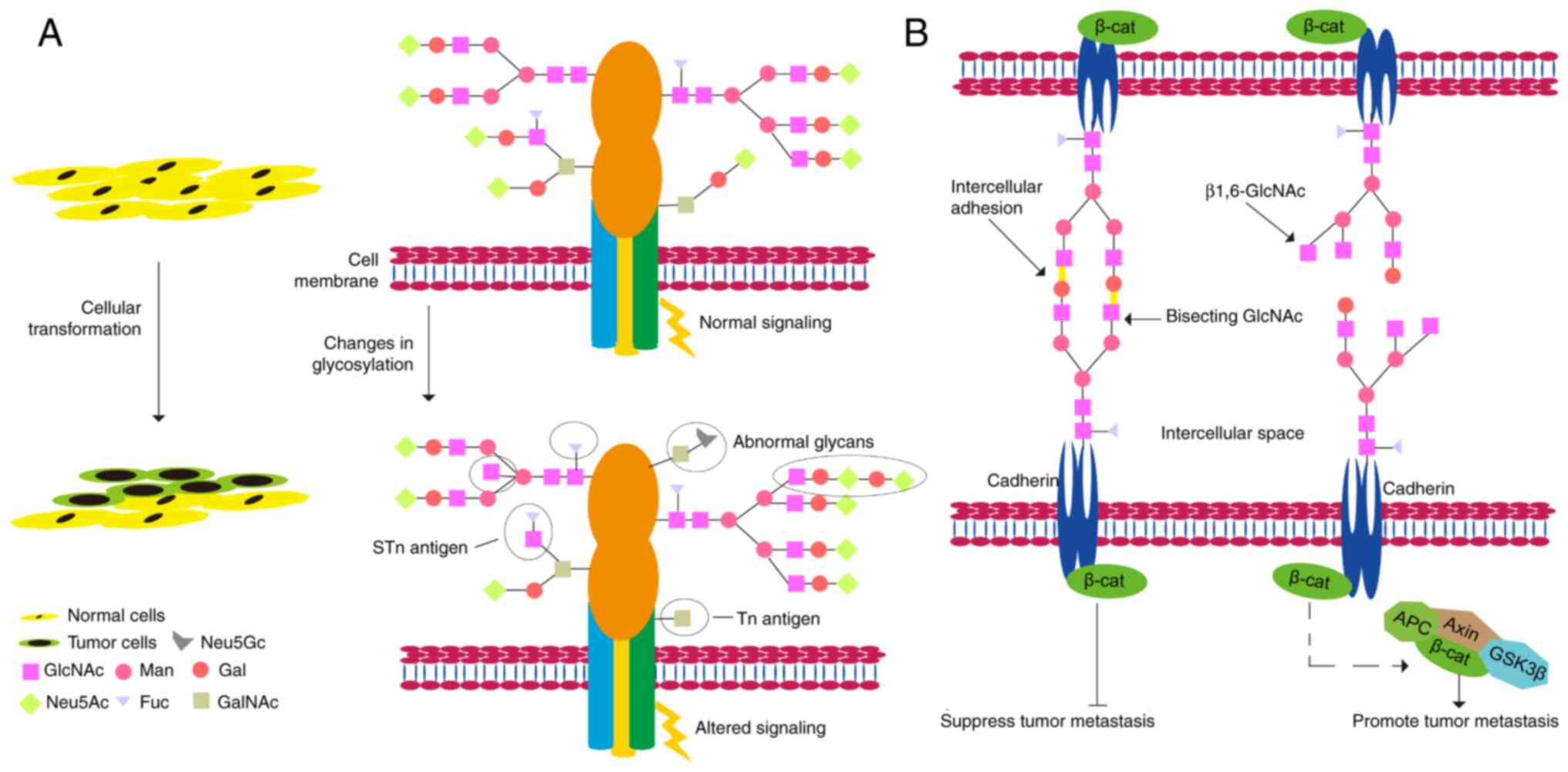
Abnormal glycosylation plays a vital role in the key
pathological processes of tumorigenesis and development (Fig. 3) (164). Glycans play important roles in
tumor cell signaling, tumor cell separation and invasion,
cell-matrix interaction, angiogenesis, metastasis, and immune
regulation, and abnormal glycosylation is often referred to as a
'signature of cancer' (165).
The synthesis of N-linked sugar chains starts in the endoplasmic
reticulum and is completed in the Golgi apparatus. Most of the
mannose in the original sugar chain is excised, but various
glycosyltransferases add different types of sugar molecules, in
turn, to form oligosaccharide chains with different structures
(166). The spatial structure of
a glycoprotein determines which glycosyltransferase it can bind to
undergo specific glycosylation modifications (167). Many glycoproteins have both
N-linked sugar chains and O-linked sugar chains (168). The O-linked glycosylation is
carried out in the Golgi apparatus; usually, the first linked sugar
unit is N-acetylgalactose, and the linked sites are the hydroxyl
groups of Ser, Thr, and Hyp, and then the sugar groups are
successively transferred to it to form an oligosaccharide chain;
the donor of sugar is also a nucleoside sugar, such as
UDP-galactose (169). As a
result of glycosylation, different proteins are marked differently,
changing the polypeptide's conformation and increasing the
protein's stability (170).
Metastatic tumor cells must undergo a series of important events,
including EMT, detachment from the primary tumor mass, adherence to
ECM proteins, migration and degradation of ECM proteins, invasion
of adjacent tissues, penetration of lymphatic or blood vessels,
spread to different parts of the body, and outflow from blood
vessels to form metastatic tumors (171). In humans, there are more than
2,000 proteins that contain an amino acid motif suitable for
N-glycosylation. They are either membrane-bound or secreted but by
no means cytoplasmic or nuclear. Therefore, glycoproteins are
critical for tumor metastasis (172). Examples of N-glycans are
secreted proteinases such as kallikreins, carboxypeptidase E,
cathepsins, and others; adhesion proteins including members of the
immunoglobulin superfamily (ALCAM, ICAM1, BCAM, and others),
cadherins; Wnt family members; c-Kit, TIMP1, tetraspanins,
clusterin, and others; ECM molecules such as fibronectin, laminin,
and others (173).
N-glycosylated cadherins have substantial effects on their
functions as adhesion molecules, signaling proteins, and tumor
suppressors (174). Human
E-cadherin displays four potential N-glycosylation sites (175). Moreover, alterations in the
expression profiles of E-cadherin-linked N-glycans are associated
with malignant and invasive phenotypes and poor survival in cancer
patients (176). Another
important part of N-glycosylation is that it helps keep N-cadherin
stable. It also plays a role in preventing cell-cell adhesion and
promoting GBM cell migration (177).
The ECM is the acellular part of tissue composed of
collagen, glycoproteins, proteoglycans, and crevices. Dynamic
changes in cell-ECM interactions, including those coordinated by
glycans, are critical for tumor cells to acquire migratory and
invasive capabilities (178).
The integrin family encompasses the primary surface receptors
involved in the adhesion of cells to the ECM elements. N-linked
glycans modulate integrin function regulating the migration
capacity of tumor cells (179).
ST6GAL1 (ST6 β-galactoside α-2,6-sialyltransferase 1), which
catalyzes the transfer of sialic acids to terminal galactose
residues of N-glycans, is upregulated, which can increase the
migration and invasion of human CRC cells (180).
Similarly, ST6GAL1 can increase adhesion to ECM
structures and increase the invasiveness of breast cancer (181). N-glycosylation of
N-acetylglucosaminyltransferase V enhances CD 147/basgin
interaction with integrin β1 to promote liver cancer metastasis
(182). Cluster of
differentiation 147 (CD147) is an extracellular matrix
metalloproteinase inducer, and modification of N-glycosylation of
Asn152 on CD147 strongly promotes invasion and migration of HCC
(183). Of course, in addition
to the promoting effect, N-glycosylation also has an inhibitory
effect on the metastasis of tumor cells. For example, glycosylation
of Asn-144 of Golgi protein 73 (GP73) inhibits HCC metastasis
(184).
O-glycosylation has the same effect on tumor cell
metastasis as N-glycosylation. For instance, GalNAc-type
O-glycosylation can modify TGF-β to facilitate breast cancer cell
migration and invasion via the EMT process (184). N-ac
etylgalactosaminyltransferase 6 (GALNT6), an enzyme that mediates
the initial step of mucin-type O-glycosylation, enhances
O-glycosylation of α2-macroglobulin (α2M) and activates the
downstream PI3K/Akt signaling pathway to promote migration and
invasion of breast cancer and ovarian cancer (185,186). However,
N-acetyl-galactosaminotransfe rase 2 (GALNT2), an enzyme that
initiates O-glycosylation of mucins, inhibits metastasis of gastric
adenocarcinoma by reducing EGFR-AKT signaling (187). Osteopontin (OPN) is a
multiphosphorylated extracellular glycoprotein. O-terminal
glycosylation of OPN can increase cell adhesion, thereby inhibiting
tumor cell metastasis (188).
As glycosylation is implicated in every link from
tumor occurrence to metastasis, antitumor medication development
for glycosylation, such as cancer-related glycoforms and
glycosyltransferases related to synthetic sugars, which are all
potential therapeutic targets is urgently needed (189). Glycolectin interactions are
central axes of multiple aspects of cancer progression, such as
immune evasion, cell proliferation, invasion, and extravasation.
Blocking this interaction is a promising new strategy for
single-agent and combination therapy (190). Finally, we emphasize that in
addition to the need to develop new cancer drug strategies
targeting glycosylation, it is worth exploring efficient cancer
delivery systems to avoid side effects. We believe that exploring
specific glycosylation targets may be the beginning of a new era in
cancer therapy.
Metastasis regulated by
phosphorylation and dephosphorylation
Phosphorylation and dephosphorylation are some of
the most prevalent chemical modifications in cells and are
catalyzed by phosphorylase and phosphatase, respectively (191). Protein phosphorylation is a
ubiquitous regulatory mechanism in organisms. It plays an important
role in various aspects of every organism, such as gene
transcription, expression, cell proliferation, differentiation,
apoptosis, signal transduction, immune regulation, tumor
occurrence, and transfer (192).
Phosphorylation refers to the addition of a phosphate
(PO4) group to a protein or other types of molecules,
which can also be defined as 'introducing a phosphate group into an
organic molecule' (193).
Protein phosphorylation can be divided into four categories
according to the amino acid residues that are phosphorylated:
phosphorylation of serine, threonine, and tyrosine constitutes
O-phosphorylation; phosphorylation of histidine, arginine, and
lysine constitutes N-phosphorylation; phosphorylation of aspartate
and glutamate constitutes S-phosphorylation; phosphorylation of
cysteine constitutes acyl phosphorylation (192). More than 90% of the proteins
encoded by the human genome are phosphorylated. Therefore,
phosphorylated or dephosphorylated proteins can regulate tumor
growth and metastasis.
For example, phosphorylation of cyclase-associated
protein 1 (CAP1) can induce EMT of lung cancer (194). Longevity assurance homolog 2 of
yeast LAG1 (LASS2), a novel tumor-suppressor gene, is thought to be
a ceramide synthase that synthesizes very long acyl-chain
ceramides. Therefore, phosphorylated LASS2 inhibits Wnt/β-catenin
signaling to reduce prostate cancer growth and metastasis (195). Protein phosphatase 2A
(PP2A)-induced dephosphorylation of girdin is involved in
inhibiting breast cancer cell migration (196).
Signaling pathways regulated by protein kinases are
involved in the occurrence and development of almost all types of
cancer. Therefore, the study of kinase-mediated signaling pathways
and the possibility of blocking them with targeted therapy may have
important clinical therapeutic implications, especially since many
of these proteins are oncogenes (197). The signaling networks through
which protein kinases operate are very complex, but we believe that
understanding the regulatory functions of kinases may be an
effective means of identifying more effective cancer treatments
(198). Many drug kinase
inhibitors, such as imatinib mesylate (199), crizotinib (200), vemurafenib, and cobimetinib
(201), are already on the
market; nevertheless, their efficacy is often reduced due to the
development of complex resistance mechanisms (202). In short, protein phosphatase may
be a new drug target for treating malignant tumors in the
future.
Metastasis regulated by chromatin
remodeling
Chromatin remodeling refers to the molecular
mechanism of changes in the packaging state of chromatin, histones
in nucleosomes, and corresponding DNA molecules during the
replication and recombination of gene expression (203). Chromatin remodeling can lead to
changes in the position and structure of nucleosomes, causing
chromatin changes. ATP-dependent chromatin remodelers reposition
nucleosomes, alter nucleosome structure, and covalently modify
histones (204). In the process
of nucleosome remodeling, the role of the remodeling factor complex
(ATPase activity) is crucial, including the SWI/SNF family, ISWI
family, CHD family, and INO80 family (205). These shared properties enable
nucleosome engagement, selection, and remodeling. Each ATPase
family catalyzes a different remodeling activity which can include
incremental nucleosome sliding on DNA in cis-ribosomes; DNA
loops generated on the surface of nucleosomes; histone H2A/H2B
dimers cleared; histone VIII aggregates are cleared, or histone
octapeptide subunits are exchanged within the nucleosome to alter
their composition (206). Each
of these activities alters the accessibility of DNA in chromatin to
DNA-binding factors, which in turn regulate fundamental nuclear
processes such as transcription, DNA replication, and DNA
repair.
The chromatin remodeling activity of ATPases
usually occurs in a large multi-subunit complex, but they are
called single ATPase subunits in rare cases. Complexes in the
SWI/SNF family include the large multi-subunit BAF, PBAF, and WINAC
complexes, which are co-regulators of transcription and which also
contribute to DNA damage repair (207). Like SWI/SNF, members of the INO
80 family are large multi-subunit complexes that regulate
transcription, but their roles in DNA damage repair are more
pronounced (208). These
complexes are unique among ATP-dependent remodeling complexes in
that they catalyze the exchange of histones from the nucleosomal
structure. Complexes with optimal function include SRCAP and Tip
60/P 400, which exchange H2A/H2B histone dimers found in standard
nucleosomes for variant H2A.Z/H2B dimers (209,210). The CHD family contains nine
different ATPases and is the largest in the remodeling family. The
most characteristic complex in this family is NURD (211). NURD contains both ATP-dependent
chromatin remodeling and HDAC activities (211). MBD2 is a unique subunit of the
NURD complexes and can bind 5mC DNA (212). Once recruited by 5mC-enriched
DNA, the MBD2-NURD complex inhibits gene expression through its
remodeling and histone deacetylase activity. MBD3 can replace the
MBD2 subunit, dissociating NURD from 5mC and promoting its function
as a transcriptional activator (213). Most imitation switch (ISWI)
complexes are relatively small, consisting of only two or three
subunits. Each of these complexes contains a large subunit with
several histone-binding domains (including PHD and bromodomains)
and an ISWI family of ATPases (214). These complexes have multiple
functions, including spacing of nucleosomes after DNA replication
(CHRAC, ACF), RNA polymerase elongation (RSF), as co-regulators of
transcription (CERF, NURF, NoRC, b-WICH) and regulation of DNA
damage repair (WICH) (215).
All proteins involved in chromatin remodeling also
share five basic properties: a) domains that recognize covalent
histone modifications; b) an affinity for the nucleosome, beyond
DNA itself; c) domains and/or proteins that regulate the ATPase
domain; d) a similar DNA-dependent ATPase domain, required for
remodeling and serving as a DNA-translocating motor to break
histone-DNA contacts; and e) domains and/or proteins for
interaction with other chromatin or transcription factors (205).
The role of chromatin remodeling in cancer
metastasis is well-studied (216). For example, chromatin remodeling
protein BRG1, a core component of the mammalian chromatin
remodeling complex, regulates the transcription of long-chain fatty
acid elongase 3 (Elovl3), which promotes cell metastasis of PCa
(217). MORC family CW-type zinc
finger 2 (MORC2) is a newly discovered chromatin remodeling protein
that promotes breast cancer invasion and metastasis through
interacting with catenin delta 1 (CTNND1, also known as
p120-catenin, was originally identified as a substrate of the
oncogenic tyrosine kinase Src and subsequently defined as a
component of the adherens junction complex that includes E-cadherin
and α-, β-, γ-catenins) (218).
However, chromatin remodeling factor ARID2 is one subunit of the
chromatin-remodeling SWI/SNF complex and inhibits EMT of HCC cells
by recruiting DNMT1 to Snail promoter, which increases promoter
methylation and inhibits Snail transcription [55]. ARID1A, a
SWI/SNF chromatin remodeling complex subunit, inhibits cancer cell
migration, invasion, and metastasis by downregulating β-catenin
signaling (219).
Signaling pathways regulated by chromatin
remodeling proteins are involved in the occurrence and development
of almost all types of cancer. Changes in chromatin structure have
profound effects on gene expression during normal cellular
homeostasis and malignant transformation. Therefore, screening
small molecules targeting recombinant chromatin proteins has
important clinical implications.
Metastasis regulated by non-coding
RNAs
Non-coding RNAs (ncRNAs) refer to RNAs that do not
encode proteins. These include RNAs with known functions such as
rRNA, tRNA, circular RNA, snRNA, snoRNA, and microRNA and RNAs with
unknown functions (220). The
common feature of these RNAs is that they can all be transcribed
from the genome but not translated into proteins to perform their
respective biological functions at the RNA level. Non-coding RNAs
can be divided into three categories in terms of length: less than
50 nt, including microRNAs, siRNAs, piRNAs; 50–500 nt, including
rRNAs, tRNAs, snRNAs, snoRNAs, siRNAs, srpRNAs; greater than 500
nt, including long non-coding RNAs, long non-coding RNAs without
polyA tails (221).
MicroRNA is a class of 21–23 nt small RNAs, its
precursor is approximately 70–100 nt, forming a standard stem
structure, and after processing, it becomes a 21–23 nt
single-stranded RNA. The mechanism of action of a microRNA is to
complement mRNA, silencing or degrading mRNA (222). snRNA is short for small nuclear
RNA. Its function is to combine with protein factors to form small
nuclear ribonucleoprotein particles (snRNPs) and perform the
function of splicing mRNA. There are mainly five types of snRNAs:
U1, U2, U4, U5, and U6 (223).
snoRNAs are the first small RNAs discovered in the nucleolus,
called small nucleolar RNAs, and their biological functions were
initially discovered to modify rRNA. Most small nucleolar RNAs can
be divided into two categories. One is C Dbox snoRNA, which is
methylated on RNA bases. It contains 4 Boxes: Box C, Box D, BoxC'
and BoxD'. The other type is the H/ACA box. This type of snoRNA
carries out methyluracilylation modification to the base of RNA.
Its characteristic is to form a double stem and add a loop area in
the middle, among which boxH in the middle loop area (224). Long non-coding RNAs (lncRNAs)
are longer than 200 bp and are transcribed from independent
promoters by RNA Pol II (225).
Its sequence conservation is not high, and its expression abundance
is low, showing strong specificity in tissues and cells. lncRNAs
are involved in various critical regulatory processes such as X
chromosome silencing, genomic imprinting, chromatin modification,
transcriptional activation, transcriptional interference, and
intranuclear transport (226).
Unlike known linear RNAs, circular RNAs (circRNAs) form a
covalently closed continuous loop. In circRNAs, the 3' and 5'ends
of the RNA molecule are usually joined together. This property
confers several properties on circRNAs, many of which have only
recently been identified. Many circRNAs are derived from
protein-coding genes, but some studies have shown that some
circRNAs can encode proteins (227). Because circRNAs were previously
found to have no protein-coding function, they were classified as
non-coding RNAs. Recently, some circRNAs have shown potential as
gene regulators (228).
There are many studies showing that non-coding RNAs
can regulate tumor growth and metastasis. Over the past decade, the
regulatory roles of lncRNAs and miRNAs in various biological
processes have emerged. The regulatory mechanisms of lncRNAs and
miRNAs are diverse, relying primarily on their localization and
interacting proteins or RNAs (229,230). For many metastasis-related
miRNAs, target genes with established roles in tumor cell invasion,
migration, and metastasis have been identified, such as MMPs, HER
receptors, BMPs, PTEN, ZEB1, ZEB2, or E-cadherin (231). Some metastasis-related genes are
directly or indirectly regulated by multiple miRNAs simultaneously.
For example, members of the miR-200 family (miR-141, miR-200a,
miR-200b, miR-200c, miR-429) have been shown to regulate the
epithelial properties of cells by silencing Zeb proteins (232), and ZEB1/ZEB2 are also affected
by regulation of miR-205 and or miR-192 (233).
lncRNAs are abnormally regulated in many cancers
and are associated with tumor metastasis. lncRNAs can be used as
new biomarkers for tumor diagnosis and treatment. Using lncRNA
arrays or RNA-seq analysis, many lncRNAs have been found to be
markers of tumor prognosis (234). More and more studies have shown
that lncRNAs are potent regulators of EMT during tumor metastasis
by controlling key molecules in several intracellular signaling
pathways, including TGF-β/SMAD, Wnt/β-catenin, Notch, MEK/ERK,
PI3K/AKT, and JAK/STAT, notably Snail, ZEB and TWIST, which inhibit
the expression of epithelial state-associated genes while
simultaneously activating the expression of mesenchymal
state-associated genes and concomitantly (235–238).
Since miRNAs play critical roles in tumorigenesis
and metastasis, they are likely to be tumor-suppressor targets or
therapeutic drugs. In the case of abnormal upregulation, miRNAs can
be silenced by directly targeting miRNAs involved in tumor
pathogenesis by 'anti-miRNA' molecules. Conversely, when miRNAs
aberrantly downregulate mRNAs that directly target genes involved
in tumor pathogenesis, so-called miRNA mimics or similar drugs can
be used as drugs to replace pathologically downregulated miRNAs
(231). Furthermore, to date,
research on lncRNAs and metastasis has mainly focused on different
organ-specific metastases. However, the reality is that many
patients suffer from or are at risk for multi-organ metastases. In
addition, the function of lncRNAs can be altered through epigenetic
modification and microenvironmental shift (239). This complexity makes it
difficult to fully understand the specific molecular mechanisms of
tumor metastasis mediated by lncRNAs (240). Because lncRNAs are mediators of
tumor metastasis, targeting them as a common therapeutic target in
different types of tumors may be important for future research.
4. Conclusion
Epigenetic regulation and metastasis play unique
roles in the occurrence and development of tumors, and related
enzymes and regulatory factors are potential drug targets for
cancer treatment. Although no single epigenetic regulator is
mutated in tumor metastasis, there is growing evidence that
metastatic tissue has a distinct epigenetic status compared to the
primary tumor tissue for use in cancer therapy and diagnosis
(241). Due to the nature of
epigenetic modifications, modulating these changes and
understanding the role of epigenetics in EMT and metastasis will
provide new insights into our understanding of tumor progression
and metastasis. Because epigenetic modifications are reversible, a
thorough understanding of their function in EMT provides us with
new insights into tumor progression and metastasis. In addition, it
can further facilitate the development of human cancer diagnostic
and therapeutic strategies. Epigenetic alterations associated with
EMT are considered clinically as a potential biomarker. Given the
complexity of epigenetic transformation, we also need to pay
special attention to the epigenetic characteristics of the target
protein or RNA when using targeted drugs, which means that our
drugs need to be more cautious. Nevertheless, we believe that a
detailed understanding of the epigenetic aspects of EMT regulation
and metastasis will undoubtedly be an excellent way to develop
prognostic cancer biomarkers. Moreover, the development of specific
inhibitors of enzymatic proteins designed by epigenetic
modification will also be new hope for treating tumor
metastasis.
Availability of data and materials
All information provided in this review is
documented by relevant references. All references are open-ended
articles.
Authors' contributions
HC and YC contributed to the conception and design
of the review. TT and PS organized the database and wrote the first
draft of the manuscript. MNA, YW and JX wrote sections of the
manuscript. All authors contributed to manuscript revision, read,
and approved the submitted version.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing
interests.
Acknowledgements
Not applicable.
Funding
This research was supported by the Natural Science Foundation of
Chongqing (cstc2019jcyjzdxmX0033), and the Graduate Research and
Innovation Project of Chongqing of China (no. 2019CYB19117).
References
|
1
|
Rodenhiser DI: Epigenetic contributions to
cancer metastasis. Clin Exp Metastasis. 26:5–18. 2009. View Article : Google Scholar
|
|
2
|
Timp W and Feinberg AP: Cancer as a
dysregulated epigenome allowing cellular growth advantage at the
expense of the host. Nat Rev Cancer. 13:497–510. 2013. View Article : Google Scholar
|
|
3
|
Dario LS, Rosa MA, Mariela E, Roberto G
and Caterina C: Chromatin remodeling agents for cancer therapy. Rev
Recent Clin Trials. 3:192–203. 2008. View Article : Google Scholar
|
|
4
|
Werner RJ, Kelly A and DIssa JJ:
Epigenetics and precision oncology. Cancer J. 23:262–269. 2017.
View Article : Google Scholar
|
|
5
|
Guan X: Cancer metastases: Challenges and
opportunities. Acta Pharm Sin B. 5:402–418. 2015. View Article : Google Scholar
|
|
6
|
Pachmayr E, Treese C and Stein U:
Underlying mechanisms for distant metastasis-molecular biology.
Visc Med. 33:11–20. 2017. View Article : Google Scholar
|
|
7
|
Micalizzi DS, Maheswaran S and Haber DA: A
conduit to metastasis: Circulating tumor cell biology. Genes Dev.
31:1827–1840. 2017. View Article : Google Scholar
|
|
8
|
Pastushenko I and Blanpain C: EMT
transition states during tumor progression and metastasis. Trends
Cell Biol. 29:212–226. 2019. View Article : Google Scholar
|
|
9
|
van Roy F and Berx G: The cell-cell
adhesion molecule E-cadherin. Cell Mol Life Sci. 65:3756–3788.
2008. View Article : Google Scholar
|
|
10
|
Birchmeier W and Behrens J: Cadherin
expression in carcinomas: Role in the formation of cell junctions
and the prevention of invasiveness. Biochim Biophys Acta.
1198:11–26. 1994.
|
|
11
|
Berx G and van Roy F: Involvement of
members of the cadherin superfamily in cancer. Cold Spring Harb
Perspect Biol. 1:a0031292009. View Article : Google Scholar
|
|
12
|
Derksen PW, Liu X, Saridin F, van der
Gulden H, Zevenhoven J, Evers B, van Beijnum JR, Griffioen AW, Vink
J, Krimpenfort P, et al: Somatic inactivation of E-cadherin and p53
in mice leads to meta-static lobular mammary carcinoma through
induction of anoikis resistance and angiogenesis. Cancer Cell.
10:437–449. 2006. View Article : Google Scholar
|
|
13
|
Wong SHM, Fang CM, Chuah LH, Leong CO and
Ngai SC: E-cadherin: Its dysregulation in carcinogenesis and
clinical implications. Crit Rev Oncol Hematol. 121:11–22. 2018.
View Article : Google Scholar
|
|
14
|
Odero-Marah V, Hawsawi O, Henderson V and
Sweeney J: Epithelial-mesenchymal transition (EMT) and prostate
cancer. Adv Exp Med Biol. 1095:101–110. 2018. View Article : Google Scholar
|
|
15
|
Chiang SP, Cabrera RM and Segall JE: Tumor
cell intravasation. Am J Physiol Cell Physiol. 311:C1–C14. 2016.
View Article : Google Scholar
|
|
16
|
Hamilton G and Rath B:
Mesenchymal-epithelial transition and circulating tumor cells in
small cell lung cancer. Adv Exp Med Biol. 994:229–245. 2017.
View Article : Google Scholar
|
|
17
|
Zhao B, Li L, Wang L, Wang CY, Yu J and
Guan KL: Cell detachment activates the Hippo pathway via
cytoskeleton reorganization to induce anoikis. Genes Dev. 26:54–68.
2012. View Article : Google Scholar
|
|
18
|
Pantel K and Speicher MR: The biology of
circulating tumor cells. Oncogene. 35:1216–1224. 2016. View Article : Google Scholar
|
|
19
|
Joyce JA and Pollard JW:
Microenvironmental regulation of metastasis. Nat Rev Cancer.
9:239–252. 2009. View Article : Google Scholar
|
|
20
|
Paoletti C and Hayes DF: Circulating tumor
cells. Adv Exp Med Biol. 882:235–258. 2016. View Article : Google Scholar
|
|
21
|
Haemmerle M, Stone RL, Menter DG,
Afshar-Kharghan V and Sood AK: The platelet lifeline to cancer:
Challenges and opportunities. Cancer Cell. 33:965–983. 2018.
View Article : Google Scholar
|
|
22
|
Fu BM: Tumor metastasis in the
microcirculation. Adv Exp Med Biol. 1097:201–218. 2018. View Article : Google Scholar
|
|
23
|
Bui TM, Wiesolek HL and Sumagin R: ICAM-1:
A master regulator of cellular responses in inflammation, injury
resolution, and tumorigenesis. J Leukoc Biol. 108:787–799. 2020.
View Article : Google Scholar
|
|
24
|
Sarvaiya PJ, Guo D, Ulasov I, Gabikian P
and Lesniak MS: Chemokines in tumor progression and metastasis.
Oncotarget. 4:2171–2185. 2013. View Article : Google Scholar
|
|
25
|
Mielgo A and Schmid MC: Liver Tropism in
Cancer: The hepatic metastatic niche. Cold Spring Harb Perspect
Med. 10:a0372592020. View Article : Google Scholar
|
|
26
|
Walker S, Busatto S, Pham A, Tian M, Suh
A, Carson K, Quintero A, Lafrence M, Malik H, Santana MX and
Wolfram J: Extracellular vesicle-based drug delivery systems for
cancer treatment. Theranostics. 9:8001–8017. 2019. View Article : Google Scholar
|
|
27
|
Pramani KA, Jones S, Gao Y, Sweet C,
Vangara A, Begum S and Ray PC: Multifunctional hybrid graphene
oxide for circulating tumor cell isolation and analysis. Adv Drug
Deliv Rev. 125:21–35. 2018. View Article : Google Scholar
|
|
28
|
Dabagh M and Randles A: Role of deformable
cancer cells on wall shear stress-associated-VEGF secretion by
endothelium in microvasculature. PLoS One. 14:e02114182019.
View Article : Google Scholar
|
|
29
|
Hsu SK, Chiu CC, Dahms HU, Chou CK, Cheng
CM, Chang WT, Cheng KC, Wang HD and Lin IL: Unfolded protein
response (UPR) in survival, dormancy, immunosuppression,
metastasis, and treatments of cancer cells. Int J Mol Sci.
20:25182019. View Article : Google Scholar
|
|
30
|
Hu X, Zang X and Lv Y: Detection of
circulating tumor cells: Advances and critical concerns. Oncol
Lett. 21:4222021. View Article : Google Scholar
|
|
31
|
Kaplan RN, Riba RD, Zacharoulis S, Bramley
AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, et
al: VEGFR1-positive haematopoietic bone marrow progenitors initiate
the pre-metastatic niche. Nature. 438:820–827. 2005. View Article : Google Scholar
|
|
32
|
Liu T, Xu H, Huang M, Ma W, Saxena D,
Lustig RA, Alonso-Basanta M, Zhang Z, O'Rourke DM, Zhang L, et al:
Circulating glioma cells exhibit stem cell-like properties. Cancer
Res. 78:6632–6642. 2018.
|
|
33
|
Malanchi I, Santamaria-Martínez A, Susanto
E, Peng H, Lehr HA, Delaloye JF and Huelsken J: Interactions
between cancer stem cells and their niche govern metastatic
colonization. Nature. 481:85–89. 2011. View Article : Google Scholar
|
|
34
|
Oskarsson T, Batlle E and Massagué J:
Metastatic stem cells: Sources, niches, and vital pathways. Cell
Stem Cell. 14:306–321. 2014. View Article : Google Scholar
|
|
35
|
Kaminsky ZA, Tang T, Wang SC, Ptak C, Oh
GH, Wong AH, Feldcamp LA, Virtanen C, Halfvarson J, Tysk C, et al:
DNA methylation profiles in monozygotic and dizygotic twins. Nat
Genet. 41:240–245. 2009. View
Article : Google Scholar
|
|
36
|
Riggs AD: X inactivation, differentiation,
and DNA methylation. Cytogenet Cell Genet. 14:9–25. 1975.
View Article : Google Scholar
|
|
37
|
Cooper DN: Eukaryotic DNA methylation.
Human Genet. 64:315–333. 1983. View Article : Google Scholar
|
|
38
|
Compere SJ and Palmiter RD: DNA
methylation controls the inducibility of the mouse
metallothionein-I gene lymphoid cells. Cell. 25:233–240. 1981.
View Article : Google Scholar
|
|
39
|
Dong Z, Pu L and Cui H: Mitoepigenetics
and its emerging roles in cancer. Front Cell Dev Biol. 8:42020.
View Article : Google Scholar
|
|
40
|
Moore LD, Le T and Fan G: DNA methylation
and its basic function. Neuropsychopharmacology. 38:23–38. 2013.
View Article : Google Scholar
|
|
41
|
Morgan AE, Davies TJ and Mc Auley MT: The
role of DNA methylation in ageing and cancer. Proc Nutr Soc.
77:412–422. 2018. View Article : Google Scholar
|
|
42
|
Zhao H, Yang L and Cui H: SIRT1 regulates
autophagy and diploidization in parthenogenetic haploid embryonic
stem cells. Biochem Biophys Res Commun. 464:1163–1170. 2015.
View Article : Google Scholar
|
|
43
|
Lyko F: The DNA methyltransferase family:
A versatile toolkit for epigenetic regulation. Nat Rev Genet.
19:81–92. 2018. View Article : Google Scholar
|
|
44
|
Kausar S, Abbas MN and Cui H: A review on
the DNA methyltransferase family of insects: Aspect and prospects.
Int J Biol Macromol. 186:289–302. 2021. View Article : Google Scholar
|
|
45
|
Dong Z and Cui H: Epigenetic modulation of
metabolism in glioblastoma. Semin Cancer Biol. 57:45–51. 2019.
View Article : Google Scholar
|
|
46
|
Anteneh H, Fang J and Song J: Structural
basis for impairment of DNA methylation by the DNMT3A R882H
mutation. Nat Commu. 11:22942020. View Article : Google Scholar
|
|
47
|
Hayashi K, Hishikawa A and Itoh H: DNA
damage repair and DNA methylation in the kidney. Am J Nephrol.
50:81–91. 2019. View Article : Google Scholar
|
|
48
|
de Araújo ÉS, Pramio DT, Kashiwabara AY,
Pennacchi PC, Maria-Engler SS, Achatz MI, Campos AH, Duprat JP,
Rosenberg C, Carraro DM and Krepischi AC: DNA methylation levels of
melanoma risk genes are associated with clinical characteristics of
melanoma patients. Biomed Res Int. 2015:3764232015. View Article : Google Scholar
|
|
49
|
Farhadova S, Gomez-Velazquez M and Feil R:
Stability and lability of parental methylation imprints in
development and disease. Genes (Basel). 10:9992019. View Article : Google Scholar
|
|
50
|
Horvath S and Raj K: DNA methylation-based
biomarkers and the epigenetic clock theory of ageing. Nat Rev
Genet. 19:371–384. 2018. View Article : Google Scholar
|
|
51
|
Wu A, Cremaschi P, Wetterskog D, Conteduca
V, Franceschini GM, Kleftogiannis D, Jayaram A, Sandhu S, Wong SQ,
Benelli M, et al: Genome-wide plasma DNA methylation features of
metastatic prostate cancer. J Clin Invest. 130:1991–2000. 2020.
View Article : Google Scholar
|
|
52
|
Hermann A, Goyal R and Jeltsch A: The
Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA
processively with high preference for hemimethylated target sites.
J Biol Chem. 279:48350–48359. 2004. View Article : Google Scholar
|
|
53
|
Espada J, Ballestar E, Fraga MF,
Villar-Garea A, Juarranz A, Stockert JC, Robertson KD, Fuks F and
Esteller M: Human DNA methyltransferase 1 is required for
maintenance of the histone H3 modification pattern. J Biol Chem.
279:37175–37184. 2004. View Article : Google Scholar
|
|
54
|
Lee E, Wang J, Yumoto K, Jung Y, Cackowski
FC, Decker AM, Li Y, Franceschi RT, Pienta KJ and Taichman RS:
DNMT1 regulates epithelial-mesenchymal transition and cancer stem
cells, which promotes prostate cancer metastasis. Neoplasia.
18:553–566. 2016. View Article : Google Scholar
|
|
55
|
Jiang H, Cao HJ, Ma N, Bao WD, Wang JJ,
Chen TW, Zhang EB, Yuan YM, Ni QZ, Zhang FK, et al: Chromatin
remodeling factor ARID2 suppresses hepatocellular carcinoma
metastasis via DNMT1-Snail axis. Proc Natl Acad Sci USA.
117:4770–4780. 2020. View Article : Google Scholar
|
|
56
|
Tang H, Liu P, Yang L and Xie X, Ye F, Wu
M, Liu X, Chen B, Zhang L and Xie X: miR-185 suppresses tumor
proliferation by directly targeting E2F6 and DNMT1 and indirectly
upregulating BRCA1 in triple-negative breast cancer. Mol Cancer
Ther. 13:3185–3197. 2014. View Article : Google Scholar
|
|
57
|
Zhu A, Hopkins KM, Friedman RA, Bernstock
JD, Broustas CG and Lieberman HB: DNMT1 and DNMT3B regulate
tumorigenicity of human prostate cancer cells by controlling RAD9
expression through targeted methylation. Carcinogenesis.
42:220–231. 2021. View Article : Google Scholar
|
|
58
|
Gao X, Sheng Y, Yang J, Wang C, Zhang R,
Zhu Y, Zhang Z, Zhang K, Yan S, Sun H, et al: Osteopontin alters
DNA methylation through up-regulating DNMT1 and sensitizes
CD133+/CD44+ cancer stem cells to 5
azacytidine in hepatocellular carcinoma. J Exp Clin Cancer Res.
37:1792018. View Article : Google Scholar
|
|
59
|
Bai J, Zhang X, Hu K, Liu B, Wang H, Li A,
Lin F, Zhang L, Sun X, Du Z and Song J: Silencing DNA
methyltransferase 1 (DNMT1) inhibits proliferation, metastasis and
invasion in ESCC by suppressing methylation of RASSF1A and DAPK.
Oncotarget. 7:44129–44141. 2016. View Article : Google Scholar
|
|
60
|
Xie M, Nie FQ, Sun M, Xia R, Liu YW, Zhou
P, De W and Liu XH: Decreased long noncoding RNA SPRY4-IT1
contributing to gastric cancer cell metastasis partly via affecting
epithelial-mesenchymal transition. J Transl Med. 13:2502015.
View Article : Google Scholar
|
|
61
|
Wu Y, Liu H, Shi X, Yao Y, Yang W and Song
Y: The long non-coding RNA HNF1A-AS1 regulates proliferation and
metastasis in lung adenocarcinoma. Oncotarget. 6:9160–9172. 2015.
View Article : Google Scholar
|
|
62
|
Meng F, Liu X, Lin C, Xu L, Liu J, Zhang
P, Zhang X, Song J, Yan Y, Ren Z and Zhang Y: SMYD2 suppresses APC2
expression to activate the Wnt/β-catenin pathway and promotes
epithelial-mesenchymal transition in colorectal cancer. Am J Cancer
Res. 10:997–1011. 2020.
|
|
63
|
Okano M, Bell DW, Haber DA and Li E: DNA
methyltransferases Dnmt3a and Dnmt3b are essential for de novo
methylation and mammalian development. Cell. 99:247–257. 1999.
View Article : Google Scholar
|
|
64
|
Chédin F: The DNMT3 family of mammalian de
novo DNA methyltransferases. Prog Mol Biol Transl Sci. 101:255–285.
2011. View Article : Google Scholar
|
|
65
|
Zhang ZM, Lu R, Wang P, Yu Y, Chen D, Gao
L, Liu S, Ji D, Rothbart SB, Wang Y, et al: Structural basis for
DNMT3A-mediated de novo DNA methylation. Nature. 554:387–391. 2018.
View Article : Google Scholar
|
|
66
|
Walton EL, Francastel C and Velasco G:
Maintenance of DNA methylation: Dnmt3b joins the dance.
Epigenetics. 6:1373–1377. 2011. View Article : Google Scholar
|
|
67
|
Walton EL, Francastel C and Velasco G:
Dnmt3b prefers germ line genes and centromeric regions: Lessons
from the ICF syndrome and cancer and implications for diseases.
Biology. 3:578–605. 2014. View Article : Google Scholar
|
|
68
|
Xu J, Zhang W, Yan XJ, Lin XQ, Li W, Mi
JQ, Li JM, Zhu J, Chen Z and Chen SJ: DNMT3A mutation leads to
leukemic extramedullary infiltration mediated by TWIST1. J Hematol
Oncol. 9:1062016. View Article : Google Scholar
|
|
69
|
Cui H, Hu Y, Guo D, Zhang A, Gu Y, Zhang
S, Zhao C, Gong P, Shen X, Li Y, et al: DNA methyltransferase 3A
isoform b contributes to repressing E-cadherin through cooperation
of DNA methylation and H3K27/H3K9 methylation in EMT-related
metastasis of gastric cancer. Oncogene. 37:4358–4371. 2018.
View Article : Google Scholar
|
|
70
|
Deivendran S, Marzook H, Santhoshkumar TR,
Kumar R and Pillai MR: Metastasis-associated protein 1 is an
upstream regulator of DNMT3a and stimulator of insulin-growth
factor binding protein-3 in breast cancer. Sci Rep. 7:442252017.
View Article : Google Scholar
|
|
71
|
Zhang L, Niu H, Ma J, Yuan BY, Chen YH,
Zhuang Y, Chen GW, Zeng ZC and Xiang ZL: The molecular mechanism of
lncRNA34a-mediated regulation of bone metastasis in hepatocellular
carcinoma. Mol Cancer. 18:1202019. View Article : Google Scholar
|
|
72
|
Shi W, Tang T, Li X, Deng S, Li R, Wang Y,
Wang Y, Xia T, Zhang Y, Zen K, et al: Methylation-mediated
silencing of miR-133a-3p promotes breast cancer cell migration and
stemness via miR-133a-3p/MAML1/DNMT3A positive feedback loop. J Exp
Clin Cancer Res. 38:4292019. View Article : Google Scholar
|
|
73
|
Xu K, Chen B, Li B, Li C, Zhang Y, Jiang N
and Lang B: DNMT3B silencing suppresses migration and invasion by
epigenetically promoting miR-34a in bladder cancer. Aging.
12:23668–23683. 2020. View Article : Google Scholar
|
|
74
|
Lv M, Zhong Z, Huang M, Tian Q, Jiang R
and Chen J: lncRNA H19 regulates epithelial-mesenchymal transition
and metastasis of bladder cancer by miR-29b-3p as competing
endogenous RNA. Biochimica et biophysica acta. Biochim Biophys Acta
Mol Cell Res. 1864:1887–1899. 2017. View Article : Google Scholar
|
|
75
|
Takeshima H, Niwa T, Yamashita S,
Takamura-Enya T, Iida N, Wakabayashi M, Nanjo S, Abe M, Sugiyama T,
Kim YJ and Ushijima T: TET repression and increased DNMT activity
synergistically induce aberrant DNA methylation. J Clin Invest.
130:5370–5379. 2020. View Article : Google Scholar
|
|
76
|
Ning B, Liu G, Liu Y, Su X, Anderson GJ,
Zheng X, Chang Y, Guo M, Liu Y, Zhao Y and Nie G:
5-aza-2'-deoxycytidine activates iron uptake and heme biosynthesis
by increasing c-Myc nuclear localization and binding to the E-boxes
of transferrin receptor 1 (TfR1) and ferrochelatase (Fech) genes. J
Biol Chemistry. 286:37196–37206. 2011. View Article : Google Scholar
|
|
77
|
Schmelz K, Sattler N, Wagner M, Lübbert M,
Dörken B and Tamm I: Induction of gene expression by
5-Aza-2'-deoxycytidine in acute myeloid leukemia (AML) and
myelodysplastic syndrome (MDS) but not epithelial cells by
DNA-methylation-dependent and -independent mechanisms. Leukemia.
19:103–111. 2005. View Article : Google Scholar
|
|
78
|
Tong HY and Lin MF: Effect of
5-aza-2'-deoxycytidine on cell of high-risk patients with
myelodysplastic syndrome in vitro. Zhongguo Shi Yan Xue Ye Xue Za
Zhi. 12:467–471. 2004.In Chinese.
|
|
79
|
Gagnon J, Shaker S, Primeau M, Hurtubise A
and Momparler RL: Interaction of 5-aza-2'-deoxycytidine and
depsipeptide on anti-neoplastic activity and activation of
14-3-3sigma, E-cadherin and tissue inhibitor of metalloproteinase 3
expression in human breast carcinoma cells. Anticancer Drugs.
14:193–202. 2003. View Article : Google Scholar
|
|
80
|
Jambhekar A, Dhall A and Shi Y: Roles and
regulation of histone methylation in animal development. Nat Rev
Mol Cell Biol. 20:625–641. 2019. View Article : Google Scholar
|
|
81
|
Zhao E, Ding J, Xia Y, Liu M, Ye B, Choi
JH, Yan C, Dong Z, Huang S, Zha Y, et al: KDM4C and ATF4 cooperate
in transcriptional control of amino acid metabolism. Cell Rep.
14:506–519. 2016. View Article : Google Scholar
|
|
82
|
Hyun K, Jeon J, Park K and Kim J: Writing,
erasing and reading histone lysine methylations. Exp Mol Med.
49:e3242017. View Article : Google Scholar
|
|
83
|
Tsukada Y, Fang J, Erdjument-Bromage H,
Warren ME, Borchers CH, Tempst P and Zhang Y: Histone demethylation
by a family of JmjC domain-containing proteins. Nature.
439:811–816. 2006. View Article : Google Scholar
|
|
84
|
Skucha A, Ebner J and Grebien F: Roles of
SETD2 in Leukemia-Transcription, DNA-Damage, and Beyond. Int J Mol
Sci. 20:10292019. View Article : Google Scholar
|
|
85
|
Cao R, Wang L, Wang H, Xia L,
Erdjument-Bromage H, Tempst P, Jones RS and Zhang Y: Role of
histone H3 lysine 27 methylation in Polycomb-group silencing.
Science. 298:1039–1043. 2002. View Article : Google Scholar
|
|
86
|
Montgomery ND, Yee D, Chen A, Kalantry S,
Chamberlain SJ, Otte AP and Magnuson T: The murine polycomb group
protein Eed is required for global histone H3 lysine-27
methylation. Curr Biol. 15:942–947. 2005. View Article : Google Scholar
|
|
87
|
Moore HM, Gonzalez ME, Toy KA,
Cimino-Mathews A, Argani P and Kleer CG: EZH2 inhibition decreases
p38 signaling and suppresses breast cancer motility and metastasis.
Breast Cancer Res Treat. 138:741–752. 2013. View Article : Google Scholar
|
|
88
|
Yi X, Guo J, Guo J, Sun S, Yang P, Wang J,
Li Y, Xie L, Cai J and Wang Z: EZH2-mediated epigenetic silencing
of TIMP2 promotes ovarian cancer migration and invasion. Sci Rep.
7:35682017. View Article : Google Scholar
|
|
89
|
Mahmoud F, Shields B, Makhoul I, Hutchins
LF, Shalin SC and Tackett AJ: Role of EZH2 histone methyltrasferase
in melanoma progression and metastasis. Cancer Biol Ther.
17:579–591. 2016. View Article : Google Scholar
|
|
90
|
Lo Sardo F, Pulito C, Sacconi A, Korita E,
Sudol M, Strano S and Blandino G: YAP/TAZ and EZH2 synergize to
impair tumor suppressor activity of TGFBR2 in non-small cell lung
cancer. Cancer Lett. 500:51–63. 2021. View Article : Google Scholar
|
|
91
|
Niu N, Lu P, Yang Y, He R, Zhang L, Shi J,
Wu J, Yang M, Zhang ZG, Wang LW, et al: Loss of Setd2 promotes
Kras-induced acinar-to-ductal metaplasia and epithelia-mesenchymal
transition during pancreatic carcinogenesis. Gut. 69:715–726. 2020.
View Article : Google Scholar
|
|
92
|
Yuan H, Han Y, Wang X, Li N, Liu Q, Yin Y,
Wang H, Pan L, Li L, Song K, et al: SETD2 restricts prostate cancer
metastasis by integrating EZH2 and AMPK signaling pathways. Cancer
Cell. 38:350–365.e7. 2020. View Article : Google Scholar
|
|
93
|
Wu PC, Lu JW, Yang JY, Lin IH, Ou DL, Lin
YH, Chou KH, Huang WF, Wang WP, Huang YL, et al: H3K9 histone
methyl-transferase, KMT1E/SETDB1, cooperates with the SMAD2/3
pathway to suppress lung cancer metastasis. Cancer Res.
74:7333–7343. 2014. View Article : Google Scholar
|
|
94
|
Luan X and Wang Y: Long non-coding RNA
XLOC_006390 promotes cervical cancer proliferation and metastasis
through the regulation of SET domain containing 8. Oncol Rep.
38:159–166. 2017. View Article : Google Scholar
|
|
95
|
Kang J, Shin SH, Yoon H, Huh J, Shin HW,
Chun YS and Park JW: FIH Is an oxygen sensor in ovarian cancer for
G9a/GLP-Driven epigenetic regulation of metastasis-related genes.
Cancer Res. 78:1184–1199. 2018. View Article : Google Scholar
|
|
96
|
Qiang R, Cai N, Wang X, Wang L, Cui K,
Wang X and Li X: MLL1 promotes cervical carcinoma cell
tumorigenesis and metastasis through interaction with β-catenin.
OncoTargets Ther. 9:6631–6640. 2016. View Article : Google Scholar
|
|
97
|
Li L, Zhang Z, Ma T and Huo R: PRMT1
regulates tumor growth and metastasis of human melanoma via
targeting ALCAM. Mol Med Rep. 14:521–528. 2016. View Article : Google Scholar
|
|
98
|
Chuang CY, Chang CP, Lee YJ, Lin WL, Chang
WW, Wu JS, Cheng YW, Lee H and Li C: PRMT1 expression is elevated
in head and neck cancer and inhibition of protein arginine
methylation by adenosine dialdehyde or PRMT1 knockdown
downregulates proliferation and migration of oral cancer cells.
Oncol Rep. 38:1115–1123. 2017. View Article : Google Scholar
|
|
99
|
Yin XK, Wang YL, Wang F, Feng WX, Bai SM,
Zhao WW, Feng LL, Wei MB, Qin CL, Wang F, et al: PRMT1 enhances
oncogenic arginine methylation of NONO in colorectal cancer.
Oncogene. 40:1375–1389. 2021. View Article : Google Scholar
|
|
100
|
Yao R, Jiang H, Ma Y, Wang L, Wang L, Du
J, Hou P, Gao Y, Zhao L, Wang G, et al: PRMT7 induces
epithelial-to-mesenchymal transition and promotes metastasis in
breast cancer. Cancer Res. 74:5656–5667. 2014. View Article : Google Scholar
|
|
101
|
Bao X, Zhao S, Liu T, Liu Y, Liu Y and
Yang X: Overexpression of PRMT5 promotes tumor cell growth and is
associated with poor disease prognosis in epithelial ovarian
cancer. J Histochem Cytochem. 61:206–217. 2013. View Article : Google Scholar
|
|
102
|
Tang J, Meng Q, Shi R and Xu Y: PRMT6
serves an oncogenic role in lung adenocarcinoma via regulating p18.
Mol Med Rep. 22:3161–3172. 2020.
|
|
103
|
Allis CD, Berger SL, Cote J, Dent S,
Jenuwien T, Kouzarides T, Pillus L, Reinberg D, Shi Y, Shiekhattar
R, et al: New nomenclature for chromatin-modifying enzymes. Cell.
131:633–636. 2007. View Article : Google Scholar
|
|
104
|
Schneider J and Shilatifard A: Histone
demethylation by hydroxylation: Chemistry in action. ACS Chem Biol.
1:75–81. 2006. View Article : Google Scholar
|
|
105
|
Varier RA and Timmers HT: Histone lysine
methylation and demethylation pathways in cancer. Biochim Biophys
Acta. 1815:75–89. 2011.
|
|
106
|
Hong Y, Li X and Zhu J: LSD1-mediated
stabilization of SEPT6 protein activates the TGF-β1 pathway and
regulates non-small-cell lung cancer metastasis. Cancer Gene Ther.
29:189–201. 2022. View Article : Google Scholar
|
|
107
|
Liu J, Feng J, Li L, Lin L, Ji J, Lin C,
Liu L, Zhang N, Duan D, Li Z, et al: Arginine methylation-dependent
LSD1 stability promotes invasion and metastasis of breast cancer.
EMBO Rep. 21:e485972020. View Article : Google Scholar
|
|
108
|
Pan HM, Lang WY, Yao LJ, Wang Y and Li XL:
shRNA-interfering LSD1 inhibits proliferation and invasion of
gastric cancer cells via VEGF-C/PI3K/AKT signaling pathway. World J
Gastrointest Oncol. 11:622–633. 2019. View Article : Google Scholar
|
|
109
|
Huang Y, Liu Y, Yu L, Chen J, Hou J, Cui
L, Ma D and Lu W: Histone demethylase KDM2A promotes tumor cell
growth and migration in gastric cancer. Tumour Biol. 36:271–278.
2015. View Article : Google Scholar
|
|
110
|
Wanna-Udom S, Terashima M, Suphakhong K,
Ishimura A, Takino T and Suzuki T: KDM2B is involved in the
epigenetic regulation of TGF-β-induced epithelial-mesenchymal
transition in lung and pancreatic cancer cell lines. J Biol Chem.
296:1002132021. View Article : Google Scholar
|
|
111
|
Lu DH, Yang J, Gao LK, Min J, Tang JM, Hu
M, Li Y, Li ST, Chen J and Hong L: Lysine demethylase 2A promotes
the progression of ovarian cancer by regulating the PI3K pathway
and reversing epithelial-mesenchymal transition. Oncol Rep.
41:917–927. 2019.
|
|
112
|
Tee AE, Ling D, Nelson C, Atmadibrata B,
Dinger ME, Xu N, Mizukami T, Liu PY, Liu B, Cheung B, et al: The
histone demethylase JMJD1A induces cell migration and invasion by
up-regulating the expression of the long noncoding RNA MALAT1.
Oncotarget. 5:1793–1804. 2014. View Article : Google Scholar
|
|
113
|
Sechler M, Parrish JK, Birks DK and
Jedlicka P: The histone demethylase KDM3A, and its downstream
target MCAM, promote Ewing Sarcoma cell migration and metastasis.
Oncogene. 36:4150–4160. 2017. View Article : Google Scholar
|
|
114
|
Sun S, Yang F, Zhu Y and Zhang S: KDM4A
promotes the growth of non-small cell lung cancer by mediating the
expression of Myc via DLX5 through the Wnt/β-catenin signaling
pathway. Life Sci. 262:1185082020. View Article : Google Scholar
|
|
115
|
Li S, Wu L, Wang Q, Li Y and Wang X: KDM4B
promotes epithelial-mesenchymal transition through up-regulation of
ZEB1 in pancreatic cancer. Acta Biochim Biophys Sin (Shanghai).
47:997–1004. 2015.
|
|
116
|
Shen Y, Wei W and Zhou DX: Histone
acetylation enzymes coordinate metabolism and gene expression.
Trends Plant Sci. 20:614–621. 2015. View Article : Google Scholar
|
|
117
|
Wang Y, Miao X, Liu Y, Li F, Liu Q, Sun J
and Cai L: Dysregulation of histone acetyltransferases and
deacetylases in cardiovascular diseases. Oxid Med Cell Longev.
2014:6419792014. View Article : Google Scholar
|
|
118
|
Gujral P, Mahajan V, Lissaman AC and
Ponnampalam AP: Histone acetylation and the role of histone
deacetylases in normal cyclic endometrium. Reprod Biol Endocrinol.
18:842020. View Article : Google Scholar
|
|
119
|
Liu W, Zhan Z, Zhang M, Sun B, Shi Q, Luo
F, Zhang M, Zhang W, Hou Y, Xiao X, et al: KAT6A, a novel regulator
of β-catenin, promotes tumorigenicity and chemoresistance in
ovarian cancer by acetylating COP1. Theranostics. 11:6278–6292.
2021. View Article : Google Scholar
|
|
120
|
Santos GC Jr, da Silva AP, Feldman L,
Ventura GM, Vassetzky Y and de Moura Gallo CV: Epigenetic
modifications, chromatin distribution and TP53 transcription in a
model of breast cancer progression. J Cell Biochem. 116:533–541.
2015. View Article : Google Scholar
|
|
121
|
Legube G and Trouche D: Regulating histone
acetyltransferases and deacetylases. EMBO Rep. 4:944–947. 2003.
View Article : Google Scholar
|
|
122
|
Parra M and Verdin E: Regulatory signal
transduction pathways for class IIa histone deacetylases. Curr Opin
Pharmacol. 10:454–460. 2010. View Article : Google Scholar
|
|
123
|
Liu J, Gu J, Feng Z, Yang Y, Zhu N, Lu W
and Qi F: Both HDAC5 and HDAC6 are required for the proliferation
and metastasis of melanoma cells. J Transl Med. 14:72016.
View Article : Google Scholar
|
|
124
|
Dong L, Dong Q, Chen Y, Li Y, Zhang B,
Zhou F, Lyu X, Chen GG, Lai P, Kung HF and He ML: Novel
HDAC5-interacting motifs of Tbx3 are essential for the suppression
of E-cadherin expression and for the promotion of metastasis in
hepatocellular carcinoma. Signal Transduct Target Ther. 3:222018.
View Article : Google Scholar
|
|
125
|
von Burstin J, Eser S, Paul MC, Seidler B,
Brandl M, Messer M, von Werder A, Schmidt A, Mages J, Pagel P, et
al: E-cadherin regulates metastasis of pancreatic cancer in vivo
and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex.
Gastroenterology. 137:361–371. e1–e5. 2009. View Article : Google Scholar
|
|
126
|
Cheng C, Yang J, Li SW, Huang G, Li C, Min
WP and Sang Y: HDAC4 promotes nasopharyngeal carcinoma progression
and serves as a therapeutic target. Cell Death Dis. 12:1372021.
View Article : Google Scholar
|
|
127
|
Tang X, Li G, Su F, Cai Y, Shi L, Meng Y,
Liu Z, Sun J, Wang M, Qian M, et al: HDAC8 cooperates with SMAD3/4
complex to suppress SIRT7 and promote cell survival and migration.
Nucleic Acids Res. 48:2912–2923. 2020. View Article : Google Scholar
|
|
128
|
Yu XJ, Guo XZ, Li C, Chong Y, Song TN,
Pang JF and Shao M: SIRT1-ZEB1-positive feedback promotes
epithelial-mesenchymal transition process and metastasis of
osteosarcoma. J Cell Biochem. 120:3727–3735. 2019. View Article : Google Scholar
|
|
129
|
Bai L, Lin G, Sun L, Liu Y, Huang X, Cao
C, Guo Y and Xie C: Upregulation of SIRT6 predicts poor prognosis
and promotes metastasis of non-small cell lung cancer via the
ERK1/2/MMP9 pathway. Oncotarget. 7:40377–40386. 2016. View Article : Google Scholar
|
|
130
|
Kugel S, Sebastián C, Fitamant J, Ross KN,
Saha SK, Jain E, Gladden A, Arora KS, Kato Y, Rivera MN, et al:
SIRT6 suppresses pancreatic cancer through control of Lin28b. Cell.
165:1401–1415. 2016. View Article : Google Scholar
|
|
131
|
Li R, Quan Y and Xia W: SIRT3 inhibits
prostate cancer metastasis through regulation of FOXO3A by
suppressing Wnt/β-catenin pathway. Exp Cell Res. 364:143–151. 2018.
View Article : Google Scholar
|
|
132
|
Fu L, Dong Q, He J, Wang X, Xing J, Wang
E, Qiu X and Li Q: SIRT4 inhibits malignancy progression of NSCLCs,
through mitochondrial dynamics mediated by the ERK-Drp1 pathway.
Oncogene. 36:2724–2736. 2017. View Article : Google Scholar
|
|
133
|
Tang X, Shi L, Xie N, Liu Z, Qian M, Meng
F, Xu Q, Zhou M, Cao X, Zhu WG and Liu B: SIRT7 antagonizes TGF-β
signaling and inhibits breast cancer metastasis. Nat Commun.
8:3182017. View Article : Google Scholar
|
|
134
|
Sun Y, Sun Y, Yue S, Wang Y and Lu F:
Histone deacetylase inhibitors in cancer therapy. Curr Top Med
Chem. 18:2420–2428. 2018. View Article : Google Scholar
|
|
135
|
Kelly WK and Marks PA: Drug insight:
Histone deacetylase inhibitors-development of the new targeted
anticancer agent suberoylanilide hydroxamic acid. Nat Clin Pract
Oncol. 2:150–157. 2005. View Article : Google Scholar
|
|
136
|
Greenberg VL, Williams JM, Cogswell JP,
Mendenhall M and Zimmer SG: Histone deacetylase inhibitors promote
apoptosis and differential cell cycle arrest in anaplastic thyroid
cancer cells. Thyroid. 11:315–325. 2001. View Article : Google Scholar
|
|
137
|
Nishida K, Komiyama T, Miyazawa S, Shen
ZN, Furumatsu T, Doi H, Yoshida A, Yamana J, Yamamura M, Ninomiya
Y, et al: Histone deacetylase inhibitor suppression of
autoanti-body-mediated arthritis in mice via regulation of p16INK4a
and p21(WAF1/Cip1) expression. Arthritis Rheum. 50:3365–3376. 2004.
View Article : Google Scholar
|
|
138
|
Deroanne CF, Bonjean K, Servotte S, Devy
L, Colige A, Clausse N, Blacher S, Verdin E, Foidart JM, Nusgens BV
and Castronovo V: Histone deacetylases inhibitors as
anti-angiogenic agents altering vascular endothelial growth factor
signaling. Oncogene. 21:427–436. 2002. View Article : Google Scholar
|
|
139
|
Dikic I: Proteasomal and autophagic
degradation systems. Ann Rev Biochem. 86:193–224. 2017. View Article : Google Scholar
|
|
140
|
Nandi D, Tahiliani P, Kumar A and Chandu
D: The ubiq-uitin-proteasome system. J Biosci. 31:137–155. 2006.
View Article : Google Scholar
|
|
141
|
Ikeda F and Dikic I: Atypical ubiquitin
chains: New molecular signals. 'Protein Modifications: Beyond the
Usual Suspects' review series. EMBO Rep. 9:536–542. 2008.
View Article : Google Scholar
|
|
142
|
Mevissen TET and Komander D: Mechanisms of
deubiquitinase specificity and regulation. Ann Rev Biochem.
86:159–192. 2017. View Article : Google Scholar
|
|
143
|
Snyder NA and Silva GM: Deubiquitinating
enzymes (DUBs): Regulation, homeostasis, and oxidative stress
response. J Biol Chem. 297:1010772021. View Article : Google Scholar
|
|
144
|
van Wijk SJ, Fulda S, Dikic I and
Heilemann M: Visualizing ubiquitination in mammalian cells. EMBO
Rep. 20:e465202019. View Article : Google Scholar
|
|
145
|
Xu H, Ju L, Xiong Y, Yu M, Zhou F, Qian K,
Wang G, Xiao Y and Wang X: E3 ubiquitin ligase RNF126 affects
bladder cancer progression through regulation of PTEN stability.
Cell Death Dis. 12:2392021. View Article : Google Scholar
|
|
146
|
Xu H, Yang X, Xuan X, Wu D, Zhang J, Xu X,
Zhao Y, Ma C and Li D: STAMBP promotes lung adenocarcinoma
metastasis by regulating the EGFR/MAPK signaling pathway.
Neoplasia. 23:607–623. 2021. View Article : Google Scholar
|
|
147
|
Xiao C, Wu G, Zhou Z, Zhang X, Wang Y,
Song G, Ding E, Sun X, Zhong L, Li S, et al: RBBP6, a RING
finger-domain E3 ubiquitin ligase, induces epithelial-mesenchymal
transition and promotes metastasis of colorectal cancer. Cell Death
Dis. 10:8332019. View Article : Google Scholar
|
|
148
|
Xue S, Wu W, Wang Z, Lu G, Sun J, Jin X,
Xie L, Wang X, Tan C, Wang Z, et al: USP5 Promotes metastasis in
non-small cell lung cancer by inducing epithelial-mesenchymal
transition via Wnt/β-catenin pathway. Front Pharmacol. 11:6682020.
View Article : Google Scholar
|
|
149
|
Yuan T, Chen Z, Yan F, Qian M, Luo H, Ye
S, Cao J, Ying M, Dai X, Gai R, et al: Deubiquitinating enzyme
USP10 promotes hepatocellular carcinoma metastasis through
deubiquitinating and stabilizing Smad4 protein. Mol Oncol.
14:197–210. 2020. View Article : Google Scholar
|
|
150
|
Chen Y, Zhou B and Chen D: USP21 promotes
cell proliferation and metastasis through suppressing EZH2
ubiquitination in bladder carcinoma. Onco Targets Ther. 10:681–689.
2017. View Article : Google Scholar
|
|
151
|
Sun H, Ou B, Zhao S, Liu X, Song L, Liu X,
Wang R and Peng Z: USP11 promotes growth and metastasis of
colorectal cancer via PPP1CA-mediated activation of ERK/MAPK
signaling pathway. EBioMedicine. 48:236–247. 2019. View Article : Google Scholar
|
|
152
|
Zhang C, Xie C, Wang X, Huang Y, Gao S, Lu
J, Lu Y and Zhang S: Aberrant USP11 expression regulates NF90 to
promote proliferation and metastasis in hepatocellular carcinoma.
Am J Cancer Res. 10:1416–1428. 2020.
|
|
153
|
Xie P, Chen Y, Zhang H, Zhou G, Chao Q,
Wang J, Liu Y, Fang J, Xie J, Zhen J, et al: The deubiquitinase
OTUD3 stabilizes ACTN4 to drive growth and metastasis of
hepatocellular carcinoma. Aging. 13:19317–19338. 2021. View Article : Google Scholar
|
|
154
|
Zhu R, Liu Y, Zhou H, Li L, Li Y, Ding F,
Cao X and Liu Z: Deubiquitinating enzyme PSMD14 promotes tumor
metastasis through stabilizing SNAIL in human esophageal squamous
cell carcinoma. Cancer Lett. 418:125–134. 2018. View Article : Google Scholar
|
|
155
|
Chen D, Wang Y, Lu R, Jiang X, Chen X,
Meng N, Chen M, Xie S and Yan GR: E3 ligase ZFP91 inhibits
Hepatocellular Carcinoma Metabolism Reprogramming by regulating PKM
splicing. Theranostics. 10:8558–8572. 2020. View Article : Google Scholar
|
|
156
|
Yu L, Dong L, Li H, Liu Z, Luo Z, Duan G,
Dai X and Lin Z: Ubiquitination-mediated degradation of SIRT1 by
SMURF2 suppresses CRC cell proliferation and tumorigenesis.
Oncogene. 39:4450–4464. 2020. View Article : Google Scholar
|
|
157
|
Shen T, Cai LD, Liu YH, Li S, Gan WJ, Li
XM, Wang JR, Guo PD, Zhou Q, Lu XX, et al: Ube2v1-mediated
ubiquitination and degradation of Sirt1 promotes metastasis of
colorectal cancer by epigenetically suppressing autophagy. J
Hematol Oncol. 11:952018. View Article : Google Scholar
|
|
158
|
Guo W, You X, Xu D, Zhang Y, Wang Z, Man
K, Wang Z and Chen Y: PAQR3 enhances Twist1 degradation to suppress
epithelial-mesenchymal transition and metastasis of gastric cancer
cells. Carcinogenesis. 37:397–407. 2016. View Article : Google Scholar
|
|
159
|
Ye P, Chi X, Cha JH, Luo S, Yang G, Yan X
and Yang WH: Potential of E3 ubiquitin ligases in cancer immunity:
Opportunities and challenges. Cells. 10:33092021. View Article : Google Scholar
|
|
160
|
Wei R, Liu X, Yu W, Yang T, Cai W, Liu J,
Huang X, Xu GT, Zhao S, Yang J and Liu S: Deubiquitinases in
cancer. Oncotarget. 6:12872–12889. 2015. View Article : Google Scholar
|
|
161
|
Lee BH, Lee MJ, Park S, Oh DC, Elsasser S,
Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP, et al: Enhancement
of proteasome activity by a small-molecule inhibitor of USP14.
Nature. 467:179–184. 2010. View Article : Google Scholar
|
|
162
|
Chauhan D, Tian Z, Nicholson B, Kumar KG,
Zhou B, Carrasco R, McDermott JL, Leach CA, Fulcinniti M, Kodrasov
MP, et al: A small molecule inhibitor of ubiquitin-specific
protease-7 induces apoptosis in multiple myeloma cells and
overcomes bortezomib resistance. Cancer Cell. 22:345–358. 2012.
View Article : Google Scholar
|
|
163
|
Reiner T, Parrondo R, de Las Pozas A,
Palenzuela D and Perez-Stable C: Betulinic acid selectively
increases protein degradation and enhances prostate cancer-specific
apoptosis: Possible role for inhibition of deubiquitinase activity.
PLoS One. 8:e562342013. View Article : Google Scholar
|
|
164
|
Stowell SR, Ju T and Cummings RD: Protein
glycosylation in cancer. Ann Rev Pathol. 10:473–510. 2015.
View Article : Google Scholar
|
|
165
|
Eichler J: Protein glycosylation. Curr
Bio. 29:R229–R231. 2019. View Article : Google Scholar
|
|
166
|
Mammadova-Bach E, Jaeken J, Gudermann T
and Braun A: Platelets and defective N-glycosylation. Int J Mol
Sci. 21:56302020. View Article : Google Scholar
|
|
167
|
Clerc F, Reiding KR, Jansen BC, Kammeijer
GS, Bondt A and Wuhrer M: Human plasma protein N-glycosylation.
Glycoconj J. 33:309–343. 2016. View Article : Google Scholar
|
|
168
|
Shajahan A, Supekar NT, Gleinich AS and
Azadi P: Deducing the N-and O-glycosylation profile of the spike
protein of novel coronavirus SARS-CoV-2. Glycobiology. 30:981–988.
2020. View Article : Google Scholar
|
|
169
|
Magalhães A, Duarte HO and Reis CA: The
role of O-glycosylation in human disease. Mol Aspects Med.
79:10096420210. View Article : Google Scholar
|
|
170
|
Van den Steen P, Rudd PM, Dwek RA and
Opdenakker G: Concepts and principles of O-linked glycosylation.
Crit Rev Biochem Mol Biol. 33:151–208. 1998. View Article : Google Scholar
|
|
171
|
Chiang AC and Massagué J: Molecular basis
of metastasis. N Engl J Med. 359:2814–2823. 2008. View Article : Google Scholar
|
|
172
|
Läubli H and Borsig L: Altered cell
adhesion and glycosylation promote cancer immune suppression and
metastasis. Front Immunol. 10:21202019. View Article : Google Scholar
|
|
173
|
Oliveira-Ferrer L, Legler K and
Milde-Langosch K: Role of protein glycosylation in cancer
metastasis. Semin Cancer Biol. 44:141–152. 2017. View Article : Google Scholar
|
|
174
|
Sengupta PK, Bouchie MP, Nita-Lazar M,
Yang HY and Kukuruzinska MA: Coordinate regulation of
N-glycosylation gene DPAGT1, canonical Wnt signaling and E-cadherin
adhesion. J Cell Sci. 126:484–496. 2013. View Article : Google Scholar
|
|
175
|
Zhao H, Liang Y, Xu Z, Wang L, Zhou F, Li
Z, Jin J, Yang Y, Fang Z, Hu Y, et al: N-glycosylation affects the
adhesive function of E-Cadherin through modifying the composition
of adherens junctions (AJs) in human breast carcinoma cell line
MDA-MB-435. J Cell Biochem. 104:162–175. 2008. View Article : Google Scholar
|
|
176
|
Pinho SS, Reis CA, Paredes J, Magalhães
AM, Ferreira AC, Figueiredo J, Xiaogang W, Carneiro F, Gärtner F
and Seruca R: The role of N-acetylglucosaminyltransferase III and V
in the post-transcriptional modifications of E-cadherin. Hum Mol
Genet. 18:2599–2608. 2009. View Article : Google Scholar
|
|
177
|
Xu Y, Chang R, Xu F, Gao Y, Yang F, Wang
C, Xiao J, Su Z, Bi Y, Wang L and Zha X: N-Glycosylation at Asn 402
Stabilizes N-cadherin and promotes cell-cell adhesion of glioma
cells. J Cell Biochem. 118:1423–1431. 2017. View Article : Google Scholar
|
|
178
|
Binder MJ, McCoombe S, Williams ED,
McCulloch DR and Ward AC: The extracellular matrix in cancer
progression: Role of hyalectan proteoglycans and ADAMTS enzymes.
Cancer Lett. 385:55–64. 2017. View Article : Google Scholar
|
|
179
|
Lagana A, Goetz JG, Cheung P, Raz A,
Dennis JW and Nabi IR: Galectin binding to Mgat5-modified N-glycans
regulates fibronectin matrix remodeling in tumor cells. Mol Cell
Biol. 26:3181–3193. 2006. View Article : Google Scholar
|
|
180
|
Park JJ and Lee M: Increasing the α 2,6
sialylation of glyco-proteins may contribute to metastatic spread
and therapeutic resistance in colorectal cancer. Gut Liver.
7:629–641. 2013. View Article : Google Scholar
|
|
181
|
Suzuki O, Abe M and Hashimoto Y:
Sialylation by β-galactoside α-2,6-sialyltransferase and N-glycans
regulate cell adhesion and invasion in human anaplastic large cell
lymphoma. Int J Oncol. 46:973–980. 2015. View Article : Google Scholar
|
|
182
|
Cui J, Huang W, Wu B, Jin J, Jing L, Shi
WP, Liu ZY, Yuan L, Luo D, Li L, et al: N-glycosylation by
N-acetylglucosaminyltra nsferase V enhances the interaction of
CD147/basigin with integrin β1 and promotes HCC metastasis. J
Pathol. 245:41–52. 2018. View Article : Google Scholar
|
|
183
|
Li JH, Huang W, Lin P, Wu B, Fu ZG, Shen
HM, Jing L, Liu ZY, Zhou Y, Meng Y, et al: N-linked glycosylation
at Asn152 on CD147 affects protein folding and stability: Promoting
tumour metastasis in hepatocellular carcinoma. Sci Rep.
6:352102016. View Article : Google Scholar
|
|
184
|
Jiang K, Li W, Zhang Q, Yan G, Guo K,
Zhang S and Liu Y: GP73 N-glycosylation at Asn144 reduces
hepatocellular carcinoma cell motility and invasiveness.
Oncotarget. 7:23530–23541. 2016. View Article : Google Scholar
|
|
185
|
Lin TC, Chen ST, Huang MC, Huang J, Hsu
CL, Juan HF, Lin HH and Chen CH: GALNT6 expression enhances
aggressive phenotypes of ovarian cancer cells by regulating EGFR
activity. Oncotarget. 8:42588–42601. 2017. View Article : Google Scholar
|
|
186
|
Liu C, Li Z, Xu L, Shi Y, Zhang X, Shi S,
Hou K, Fan Y, Li C, Wang X, et al: GALNT6 promotes breast cancer
metastasis by increasing mucin-type O-glycosylation of α2M. Aging.
12:11794–11811. 2020. View Article : Google Scholar
|
|
187
|
Hu WT, Yeh CC, Liu SY, Huang MC and Lai
IR: The O-glycosylating enzyme GALNT2 suppresses the malignancy of
gastric adenocarcinoma by reducing EGFR activities. Am J Cancer
Res. 8:1739–1751. 2018.
|
|
188
|
Kariya Y, Kanno M, Matsumoto-Morita K,
Konno M, Yamaguchi Y and Hashimoto Y: Osteopontin O-glycosylation
contributes to its phosphorylation and cell-adhesion properties.
Biochem J. 463:93–102. 2014. View Article : Google Scholar
|
|
189
|
Ponath P, Menezes D, Pan C, Chen B, Oyasu
M, Strachan D, LeBlanc H, Sun H, Wang XT, Rangan VS, et al: A
novel, fully human Anti-fucosyl-GM1 antibody demonstrates potent in
vitro and in vivo antitumor activity in preclinical models of small
cell lung cancer. Clin Cancer Res. 24:5178–5189. 2018. View Article : Google Scholar
|
|
190
|
Festuccia C, Mancini A, Gravina GL,
Colapietro A, Vetuschi A, Pompili S, Ventura L, Delle Monache S,
Iorio R, Del Fattore A, et al: Dual CXCR4 and E-selectin inhibitor,
GMI-1359, shows Anti-bone metastatic effects and synergizes with
docetaxel in prostate cancer cell intraosseous growth. Cells.
9:322019. View Article : Google Scholar
|
|
191
|
Taracha A, Kotarba G and Wilanowski T:
Methods of analysis of protein phosphorylation. Postepy Biochem.
63:137–142. 2017.In Polish.
|
|
192
|
Tokuda M and Hatase O: Regulation of
neuronal plasticity in the central nervous system by
phosphorylation and dephosphorylation. Mol Neurobiol. 17:137–156.
1998. View Article : Google Scholar
|
|
193
|
Csolle MP, Ooms LM, Papa A and Mitchell
CA: PTEN and other PtdIns(3,4,5)P3 lipid phosphatases in
breast cancer. Int J Mol Sci. 21:91892020. View Article : Google Scholar
|
|
194
|
Zeng J, Li X, Liang L, Duan H, Xie S and
Wang C: Phosphorylation of CAP1 regulates lung cancer
proliferation, migration, and invasion. J Cancer Res Clin Oncol.
148:137–153. 2022. View Article : Google Scholar
|
|
195
|
Zhang K, Wu R, Mei F, Zhou Y, He L, Liu Y,
Zhao X, You J, Liu B, Meng Q and Pei F: Phosphorylated LASS2
inhibits prostate carcinogenesis via negative regulation of
Wnt/β-catenin signaling. J Cell Biochem. Apr 14–2021.Epub ahead of
print. View Article : Google Scholar
|
|
196
|
Li J, Enomoto A, Weng L, Sun L and
Takahashi M: Dephosphorylation of Girdin by PP2A inhibits breast
cancer metastasis. Biochem Biophys Res Commun. 513:28–34. 2019.
View Article : Google Scholar
|
|
197
|
Hainaut P and Plymoth A: Targeting the
hallmarks of cancer: Towards a rational approach to next-generation
cancer therapy. Curr Opin Oncol. 25:50–51. 2013. View Article : Google Scholar
|
|
198
|
Elkabets M, Vora S, Juric D, Morse N,
Mino-Kenudson M, Muranen T, Tao J, Campos AB, Rodon J, Ibrahim YH,
et al: mTORC1 inhibition is required for sensitivity to PI3K p110α
inhibitors in PIK3CA-mutant breast cancer. Sci Transl Med.
5:196ra992013. View Article : Google Scholar
|
|
199
|
Druker BJ: Imatinib mesylate in the
treatment of chronic myeloid leukaemia. Expert Opin Pharmacother.
4:963–971. 2003. View Article : Google Scholar
|
|
200
|
Ulivi P, Chiadini E, Dazzi C, Dubini A,
Costantini M, Medri L, Puccetti M, Capelli L, Calistri D, Verlicchi
A, et al: Nonsquamous, non-small-cell lung cancer patients who
carry a double mutation of EGFR, EML4-ALK or KRAS: Frequency,
clinical-pathological characteristics, and response to therapy.
Clin Lung Cancer. 17:384–390. 2016. View Article : Google Scholar
|
|
201
|
Larkin J, Ascierto PA, Dréno B, Atkinson
V, Liszkay G, Maio M, Mandalà M, Demidov L, Stroyakovskiy D, Thomas
L, et al: Combined vemurafenib and cobimetinib in BRAF-mutated
melanoma. N Engl J Med. 371:1867–1876. 2014. View Article : Google Scholar
|
|
202
|
Murray BW and Miller N: Durability of
Kinase-directed therapies-A Network perspective on response and
resistance. Mol Cancer Ther. 14:1975–1984. 2015. View Article : Google Scholar
|
|
203
|
Bagert JD, Mitchener MM, Patriotis AL, Dul
BE, Wojcik F, Nacev BA, Feng L, Allis CD and Muir TW: Oncohistone
mutations enhance chromatin remodeling and alter cell fates. Nat
Chem Biol. 17:403–411. 2021. View Article : Google Scholar
|
|
204
|
Dawson MA and Kouzarides T: Cancer
epigenetics: From mechanism to therapy. Cell. 150:12–27. 2012.
View Article : Google Scholar
|
|
205
|
Clapier CR and Cairns BR: The biology of
chromatin remodeling complexes. Ann Rev Biochem. 78:273–304. 2009.
View Article : Google Scholar
|
|
206
|
Becker PB and Workman JL: Nucleosome
remodeling and epigenetics. Cold Spring Harbor Perspect Biol.
5:a0179052013. View Article : Google Scholar
|
|
207
|
Wang W, Côté J, Xue Y, Zhou S, Khavari PA,
Biggar SR, Muchardt C, Kalpana GV, Goff SP, Yaniv M, et al:
Purification and biochemical heterogeneity of the mammalian SWI-SNF
complex. EMBO J. 15:5370–5382. 1996. View Article : Google Scholar
|
|
208
|
Morrison AJ and Shen X: Chromatin
remodelling beyond transcription: The INO80 and SWR1 complexes. Nat
Rev Mol Cell Biol. 10:373–384. 2009. View Article : Google Scholar
|
|
209
|
Gévry N, Chan HM, Laflamme L, Livingston
DM and Gaudreau L: p21 transcription is regulated by differential
localization of histone H2A.Z. Genes Dev. 21:1869–1881. 2007.
View Article : Google Scholar
|
|
210
|
Wong MM, Cox LK and Chrivia JC: The
chromatin remodeling protein, SRCAP, is critical for deposition of
the histone variant H2A.Z at promoters. J Biol Chem.
282:26132–26139. 2007. View Article : Google Scholar
|
|
211
|
Tong JK, Hassig CA, Schnitzler GR,
Kingston RE and Schreiber SL: Chromatin deacetylation by an
ATP-dependent nucleosome remodelling complex. Nature. 395:917–921.
1998. View Article : Google Scholar
|
|
212
|
Hendrich B and Bird A: Identification and
characterization of a family of mammalian methyl-CpG binding
proteins. Mol Cell Biol. 18:6538–6547. 1998. View Article : Google Scholar
|
|
213
|
Günther K, Rust M, Leers J, Boettger T,
Scharfe M, Jarek M, Bartkuhn M and Renkawitz R: Differential roles
for MBD2 and MBD3 at methylated CpG islands, active promoters and
binding to exon sequences. Nucleic Acids Res. 41:3010–3021. 2013.
View Article : Google Scholar
|
|
214
|
Poot RA, Bozhenok L, van den Berg DL,
Steffensen S, Ferreira F, Grimaldi M, Gilbert N, Ferreira J and
Varga-Weisz PD: The Williams syndrome transcription factor
interacts with PCNA to target chromatin remodelling by ISWI to
replication foci. Nat Cell Biol. 6:1236–1244. 2004. View Article : Google Scholar
|
|
215
|
Cavellán E, Asp P, Percipalle P and
Farrants AK: The WSTF-SNF2h chromatin remodeling complex interacts
with several nuclear proteins in transcription. J Biol Chem.
281:16264–16271. 2006. View Article : Google Scholar
|
|
216
|
Centore RC, Sandoval GJ, Soares LMM,
Kadoch C and Chan HM: Mammalian SWI/SNF chromatin remodeling
complexes: Emerging mechanisms and therapeutic strategies. Trends
Genet. 36:936–950. 2020. View Article : Google Scholar
|
|
217
|
Yang Y, Liu L, Li M, Cheng X, Fang M, Zeng
Q and Xu Y: The chromatin remodeling protein BRG1 links ELOVL3
transactivation to prostate cancer metastasis. Biochim Biophys Acta
Gene Regul Mech. 1862:834–845. 2019. View Article : Google Scholar
|
|
218
|
Liao XH, Zhang Y, Dong WJ, Shao ZM and Li
DQ: Chromatin remodeling protein MORC2 promotes breast cancer
invasion and metastasis through a PRD domain-mediated interaction
with CTNND1. Oncotarget. 8:97941–97954. 2017. View Article : Google Scholar
|
|
219
|
Wang J, Yan HB, Zhang Q, Liu WY, Jiang YH,
Peng G, Wu FZ, Liu X, Yang PY and Liu F: Enhancement of E-cadherin
expression and processing and driving of cancer cell metastasis by
ARID1A deficiency. Oncogene. 40:5468–5481. 2021. View Article : Google Scholar
|
|
220
|
Hombach S and Kretz M: Non-coding RNAs:
Classification, biology and functioning. Adv Exp Med Biol.
937:3–17. 2016. View Article : Google Scholar
|
|
221
|
Zhang P, Wu W, Chen Q and Chen M:
Non-coding RNAs and their integrated networks. J Integr Bioinform.
16:201900272019. View Article : Google Scholar
|
|
222
|
Lu TX and Rothenberg ME: MicroRNA. J
Allergy Clin Immunol. 141:1202–1207. 2018. View Article : Google Scholar
|
|
223
|
Ro-Choi TS: Nuclear snRNA and nuclear
function (discovery of 5'cap structures in RNA). Crit Rev Eukaryot
Gene Expr. 9:107–158. 1999. View Article : Google Scholar
|
|
224
|
Deryusheva S, Talross GJS and Gall JG:
SnoRNA guide activities: Real and ambiguous. RNA. 27:1363–1373.
2021. View Article : Google Scholar
|
|
225
|
Ali T and Grote P: Beyond the
RNA-dependent function of lncRNA genes. Elife. 9:e605832020.
View Article : Google Scholar
|
|
226
|
Bridges MC, Daulagala AC and Kourtidis A:
LNCcation: lncRNA localization and function. J Cell Biol.
220:e2020090452021. View Article : Google Scholar
|
|
227
|
Yang Q, Li F, He AT and Yang BB: Circular
RNAs: Expression, localization, and therapeutic potentials. Mol
Ther. 29:1683–1702. 2021. View Article : Google Scholar
|
|
228
|
Kristensen LS, Andersen MS, Stagsted LVW,
Ebbesen KK, Hansen TB and Kjems J: The biogenesis, biology and
characterization of circular RNAs. Nat Rev Genet. 20:675–691. 2019.
View Article : Google Scholar
|
|
229
|
Chen B, Li Q, Zhou Y, Wang X, Zhang Q,
Wang Y, Zhuang H, Jiang X and Xiong W: The long coding RNA
AFAP1-AS1 promotes tumor cell growth and invasion in pancreatic
cancer through upregulating the IGF1R oncogene via sequestration of
miR-133a. Cell Cycle. 17:1949–1966. 2018. View Article : Google Scholar
|
|
230
|
Petri BJ and Klinge CM: Regulation of
breast cancer metastasis signaling by miRNAs. Cancer Metastasis
Rev. 39:837–886. 2020. View Article : Google Scholar
|
|
231
|
Aigner A: MicroRNAs (miRNAs) in cancer
invasion and metastasis: Therapeutic approaches based on
metastasis-related miRNAs. J Mol Med. 89:445–457. 2011. View Article : Google Scholar
|
|
232
|
Feng X, Wang Z, Fillmore R and Xi Y:
MiR-200, a new star miRNA in human cancer. Cancer Lett.
344:166–173. 2014. View Article : Google Scholar
|
|
233
|
Krupa A, Jenkins R, Luo DD, Lewis A,
Phillips A and Fraser D: Loss of MicroRNA-192 promotes fibrogenesis
in diabetic nephropathy. J Am So Nephrol. 21:438–447. 2010.
View Article : Google Scholar
|
|
234
|
Li J, Meng H, Bai Y and Wang K: Regulation
of lncRNA and its role in cancer metastasis. Oncol Res. 23:205–217.
2016. View Article : Google Scholar
|
|
235
|
Hao Y, Baker D and Ten Dijke P:
TGF-β-mediated epithelial-mesenchymal transition and cancer
metastasis. Int J Mol Sci. 20:27672019. View Article : Google Scholar
|
|
236
|
Bray SJ: Notch signalling in context. Nat
Rev Mol Cell Biol. 17:722–735. 2016. View Article : Google Scholar
|
|
237
|
Browning L, Patel MR, Horvath EB, Tawara K
and Jorcyk CL: IL-6 and ovarian cancer: Inflammatory cytokines in
promotion of metastasis. Cancer Manage Res. 10:6685–6693. 2018.
View Article : Google Scholar
|
|
238
|
Yang S, Liu Y, Li MY, Ng CSH, Yang SL,
Wang S, Zou C, Dong Y, Du J, Long X, et al: FOXP3 promotes tumor
growth and metastasis by activating Wnt/β-catenin signaling pathway
and EMT in non-small cell lung cancer. Mol Cancer. 16:1242017.
View Article : Google Scholar
|
|
239
|
Li Y, Egranov SD, Yang L and Lin C:
Molecular mechanisms of long noncoding RNAs-mediated cancer
metastasis. Genes Chromosomes Cancer. 58:200–207. 2019. View Article : Google Scholar
|
|
240
|
Fang C, Wang L, Gong C, Wu W, Yao C and
Zhu S: Long non-coding RNAs: How to regulate the metastasis of
non-small-cell lung cancer. J Cell Mol Med. 24:3282–3291. 2020.
View Article : Google Scholar
|
|
241
|
Roe JS, Hwang CI, Somerville TDD, Milazzo
JP, Lee EJ, Da Silva B, Maiorino L, Tiriac H, Young CM, Miyabayashi
K, et al: Enhancer reprogramming promotes pancreatic cancer
metastasis. Cell. 170:875–888.e20. 2017. View Article : Google Scholar
|