Introduction
The 5-year survival rate of patients with liver
cancer (20%) is among the lowest compared with other types of
cancer (1). Hepatocellular
carcinoma (HCC) and cholangiocellular carcinoma (CCC) are two of
the most important types of primary liver cancers, with HCC being
the most common, which accounts for 70-90% of all types of liver
cancer (2,3). By contrast, CCC accounts for ~10% of
all liver malignancies (2,3).
Although CCC is comparatively rare, they are highly aggressive and
are typically characterised by poor 5-year survival rates,
specifically 5-15% (2,3). The standard systemic therapeutic
strategy for HCC is by using the multi-kinase inhibitor sorafenib,
which remains unchanged over the last decade (4-7).
However, adverse side effects, coupled with the increasing
incidence of resistance, are posing significant therapeutic
obstacles that needs to be overcome for the effective treatment of
HCC (4-7). In addition, currently available
treatment options for CCC are even more limited. At early stages
surgical tumour resection is a curative option, but only palliative
measures are available for advanced and metastatic CCC (8,9).
Therefore, treatment possibilities remain limited, which induce
severe side effects and only marginally increase the survival time
(8,9). There is an urgent demand for novel
treatment options for patients with advanced HCC or CCC.
One potential approach of tackling the therapeutic
resistance and metabolic evasiveness of cancer cells is the
development of 'chimeric inhibitors', which has been garnering
interest over recent years (10,11). Using the advent of molecular
hybridisation, two distinct drug pharmacophores can be merged into
a single molecule, which can simultaneously attack different
cellular and molecular targets (10,11). Due to their extensively researched
structure-activity relationships, histone deacetylase inhibitors
(HDACi) are at the centre of this chimeric drug approach. In recent
years, chimeric agents featuring HDACi pharmacophores linked to
either protein kinase inhibitors, modulators of the DNA structure
or to moieties that can interfere with the cancer cell
cytoskeleton, have been developed (11).
HDACs are amidohydrolases that serve a pivotal role
in cellular chromatin remodelling (12). They have been previously
implicated in the epigenetic regulation of cell metabolism,
proliferation and differentiation of various solid cancers, such as
urothelial, cervical, myeloma, ovarian and lung cancer, where they
have already been tested in early-stage clinical trials (12,13). In several malignancies, including
liver cancer, HDACs were found to be overexpressed, where their
activity was also correspondingly enhanced (14). In turn, they stifle the expression
of tumour suppressor genes, leading to uncontrolled cell division
and insensitivity to cell repair mechanisms and suppression of
apoptosis (14). Therefore, HDACi
are regarded to be promising novel compounds for the therapy of a
number of cancers, including liver cancer (14-16). In particular, vorinostat was the
first HDACi to be clinically approved for the treatment of T-cell
lymphoma (16-18).
Recently, combined treatment with the new HDACi
resminostat and the established liver cancer drug sorafenib was
reported to effectively counteract HCC growth and progression
(19). Similarly, a synergistic
inhibitory effect on the proliferation of HT-29 and HCT116 colon
cancer cells has been reported for the combination of HDACi and
microtubule disrupting agents (MDAs) (20). MDAs trigger the upregulation of
p53 and enhance post-translational modifications on p53, such as
phosphorylation and acetylation, in non-small lung cancer cells
(21). In this regard,
potentiation of apoptosis mediated by the combined treatment of the
o-phenylenediamine-based HDACi MS-275 (entinostat) with the
MDA taxol combretastatin (CA-4) was previously reported in MCF7
(breast cancer) and HCT (colon cancer) cell lines (22). Treatment with this drug
combination led to a pronounced inhibition of tubulin
polymerisation to impede neovascularisation and disrupt the tumour
vasculature (23). These previous
findings aforementioned therefore argue for the acceleration in the
development of therapeutic agents that can simultaneously address ≥
one target. Drugs that can accomplish this by the means of several
covalently-linked pharmacophores are called chimeric drugs
(24). Recently, the chimeric
inhibitor animacroxam, which induces both HDAC inhibition and
cytoskeleton-interfering effects, has been shown to be particularly
efficacious against testicular germ cell cancer (15).
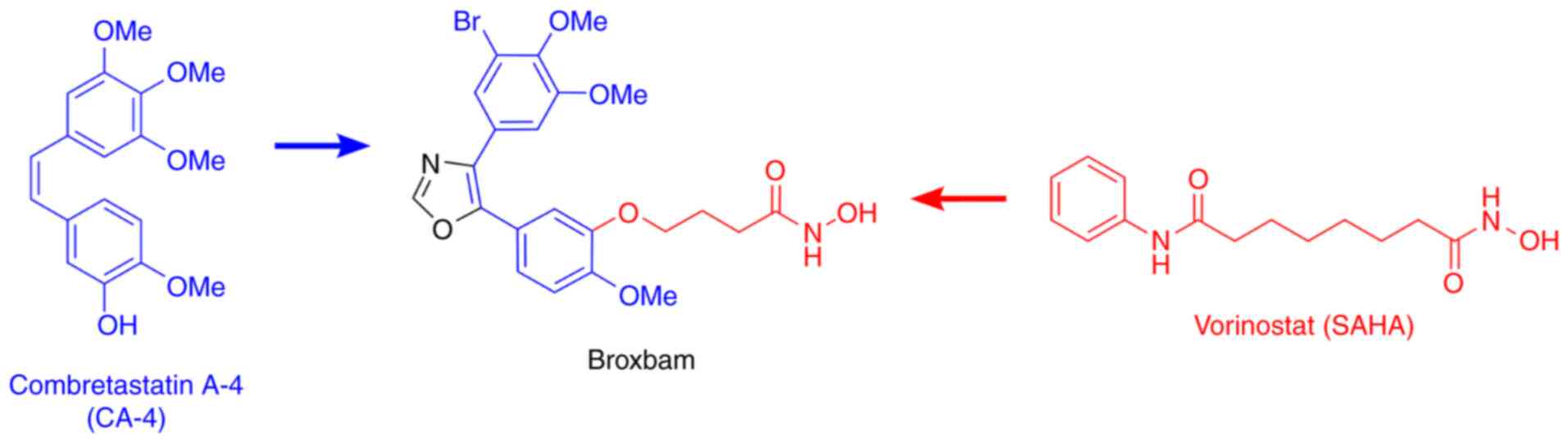
Broxbam is a promising chimeric HDACi inhibitor. The
pleiotropic effects of broxbam have been previously reported in the
518A2 melanoma cell line, where it was demonstrated to inhibit
tubulin polymerisation and HDAC activity, disrupt the cytoskeleton
and induce cell cycle arrest (20). Broxbam contains the structural
hydroxamate motif of 1st generation HDACi that can also be found on
vorinostat in addition to the trimethoxyphenyl motif commonly found
on cytostatic MDAs, such as combretastatin A-4 (CA-4) (23,25). In particular, the oxazole bridge
of the trimethoxyphenyl motif stabilizes the
cis-configuration of the alkene (Fig. 1), which is essential for the
attachment to the colchicine-binding site of tubulin heterodimers
(26). It was recently shown that
broxbam can exert antiproliferative effects in melanoma and
colorectal cancer cell models (20). However, its potential suitability
and efficacy against liver cancer remain poorly understood.
Therefore, the present study aims to investigate the effects of
broxbam on the physiology of liver cancer cells and the under-lying
molecular mechanism of action using both in vivo and in
vitro models.
Materials and methods
Compounds
Broxbam was synthesized according to literature
(20). In brief, Van Leusen
reaction of the 3-bromo-4,5-dimethoxyphenyl toluenesulfonylmethyl
isocyanide reagent with ethyl 4-(1-methoxy-4-formyl-2-phenoxy)
butyrate led to the ester-functionalised oxazole intermediate,
which was reacted with hydroxylamine to form the target compound
broxbam. Stock solutions (20 µM) of broxbam and vorinostat
(cat. no. V-8477; LC Laboratories), were prepared in DMSO (Thermo
Fisher Scientific, Inc.) stored at −20°C and diluted to the final
concentration in fresh media before each experiment. In all
experiments, the final DMSO concentration was <0.2%.
Cell culture
HepG2 (hepatoma; DSMZ no. ACC 180), Huh7
(hepatocellular carcinoma; RRID, CVCL_0336), TFK1
[cholangiocellular carcinoma; Deutsche Sammlung von Mikroorganismen
und Zellkulturen (DMSZ) no. ACC 344] and EGI1 (cholangiocellular
carcinoma; DSMZ no. ACC 385) cells, in addition to the
non-transformed hepatocyte cell line non-transformed hepatocyte
cell line AML12 (cat. no. CRL-2254) (27) were purchased from ATCC Company and
stored for long time in liquid nitrogen in an in-house repository.
Only cells in the early passages (<30 passages) were used for
the study. The liver cancer cell lines are representative for the
established hepatocellular carcinoma, hepatoblastoma and
cholangiocarcinoma cell models for the in vitro research of
liver cancer (28-32). The murine non-transformed
hepatocyte cell line AML-12 (33,34) was used instead of a
non-transformed human hepatocyte cell model, which was not
available for the present study. However, AML-12 cells represent a
widely applied non-transformed hepatocyte cell model (33,34).
The cells were maintained in RPMI 1640 medium
containing 10% FBS and 100 U/ml penicillin and strepto-mycin (all
from Gibco, Thermo Fisher Scientific, Inc.) and cultured at 37°C
and 5% CO2 in a humidified atmosphere unless stated
otherwise. Cell lines were serially passaged after trypsinisation,
using 0.05% trypsin/0.02% EDTA solution (Bio & SELL GmbH). Only
mycoplasma-free cultures were used and potential contamination was
routinely monitored. Glucose and lactate levels from the cell
culture supernatants were measured using a blood gas analyser
(ABL800 Flex; Radiometer GmbH).
Small-interfering (si)RNA
transfection
For the siRNA-mediated knockdown of HDAC6, Huh7
cells were seeded into six-well plates and cultured until they
reached 40-50% confluency. Cells were then transfected with siRNAs
(75 nM; ON-TARGET plus SMART pool human HDAC6, cat. no.
L-003499-00-0010 or ON-TARGET plus non-targeting control pool, cat.
no. D-001810-10-05; PerkinElmer, Inc.; https://horizondiscovery.com/en/gene-modulation/knockdown/sirna/products/on-target-plus-sirna-reagents?nodeid=entrezgene-10013&catalognumber=L-003499-00-0010)
using the transfection reagent DharmaFECT1 (cat. no. T-2005-01)
according to the protocol provided by Dharmacon; PerkinElmer, Inc.
In the present study, SMART pool siRNAs targeting human HDAC6 and
the non-targeting control containing a mixture of four
oligonucleotides were used. The target sequences for human HDAC6
are as follows: Sequence (Seq) 1, 5′-GGG AGG UUC UUG UGA GAU C-3′;
Seq2, 5′-GGA GGG UCC UUA UCG UAG A-3′; Seq3, 5′-GCA GUU AAA UGA AUU
CCA U-3′ and Seq4, 5′-GUU CAC AGC CUA GAA UAU A-3′. Non-targeting
control sequences are as follows: Seq1, 5′-UGG UUU ACA UGU CGA CUA
A-3′; Seq2, 5′-UGG UUU ACA UGU UGU GUG A-3′; Seq3, 5′-UGG UUU ACA
UGU UUU CUG A-3′ and Seq4, 5′-UGG UUU ACA UGU UUU CCU A-3′. After
48 h of transfection, cells were harvested and analysed by western
blotting and reverse-transcription-quantitative PCR (RT-qPCR).
Crystal violet assay
Changes in cell numbers associated with drug
treatment were monitored using crystal violet staining as
previously described (35). In
total, 1,500-3,000 cells/well were first seeded into 96-well plates
and allow to detach for 72 h at 37°C, 5% CO2 and 95%
humidity. The cells were then treated with either broxbam or
vorinostat in the following concentrations: 0.1, 0.2, 0.4, 0.8,
1.6, 3.2, 6.4 and 10.0 µM for 24, 48 and 72 h at 37°C, 5%
CO2 and 95% humidity, fixed with 1% glutaraldehyde for
30 min at room temperature and stained with 0.1% crystal violet
(Sigma-Aldrich; Merck KGaA) for 30 min at room temperature. Water
rinsing was then performed to remove any unbound dye, before 0.2%
Trition-X100 was added to solubilise the bound crystal violet and
the absorbance at 570 nm was measured using a microplate reader
(Dynex Technologies). The light absorbance was assumed in linear
proportion to the number of cells.
Real-time inhibition of cell
proliferation
The measurement of cell proliferation in real-time
was performed as previously described (15). Briefly, HepG2, Huh7
(1.0×104 cells/well), TFK1 (5×103 cells/well)
and EGI1 (3×103 cells/well) cells were seeded into in
eight-well E-plates (ACEA Biosciences, Inc.) and maintained under
normal cell culture conditions for 24 h. After attachment of the
cells, they were treated with concentrations of broxbam (0.1, 0.25,
0.5 and 1.0 µM) for ≤70 h at 37°C, 5% CO2 and 95%
humidity. An impedance-based iCEL-Ligence system (RTCA Software Vs
2.1.0, ACEA Biosciences) was used to monitor the real-time
proliferation of viable cells in the eight-well plates at 37°C, 5%
CO2 and 95% humidity, every 15 min for 70 h. Cell
proliferation was recorded as a unitless parameter called the 'cell
index', which was defined as (Rtn-Rt0)/4.6
Ohm, with Rtn representing the measured resistance at
time point n and Rt0 representing the background
resistance measured at time point T0
Lactate dehydrogenase (LDH) cytotoxicity
assay
Cells were seeded into 96-well microtiter plates at
a density of 8×103 cells/well and treated with
increasing concentrations (0.1, 0.3, 0.6, 1.5, 3.0 and 10.0
µM) of broxbam for 24 h at 37°C, 5% CO2 and 95%
humidity. Cytoplasmic LDH release into the cell culture medium was
measured using a colorimetric assay kit (cat. no. 11644793001;
Sigma-Aldrich; Merck KGaA) according to the manufacturers'
protocols (36).
Induction of chorioallantoic membrane
(CAM) tumours
In total 2×106 HepG2 cells were
resuspended in 10 µl RPMI medium containing 10% FBS and 100
U/ml penicillin and streptomycin (all from Gibco; Thermo Fisher
Scientific, Inc.) and 10 µl Matrigel (BD Biosciences),
before the cell suspension was applied into a silicone ring 5 mm in
diameter onto the CAM of fertilised white Leghorn chicken eggs
(Gallus domesticus) at day 8 of their embryonic development.
Fertilized eggs were obtained from Valo Biomedia GmbH (Cuxhaven,
Germany and the embryonic development was induced by incubating the
eggs at 37.5°C and 80% humidity as described earlier (37). The tumourbearing chicken eggs were
incubated for 24 h at 37.5°C to stimulate tumour formation,
followed by the topical application of 20 µl PBS containing
three concentrations of broxbam (1.2, 3.0 and 5.0 µM). After
an incubation period of 72 h at 37.5°C and 80% humidity, the
tumours were excised and carefully weighed to determine their
mass.
Apoptosis detection
Measurement of caspase-3 activity
HepG2 and Huh7 cells were incubated (37°C, 5%
CO2 and 95% humidity) for 48 h in RPMI growth medium
(Gibco; Thermo Fisher Scientific, Inc.) containing the respective
concentrations of test compounds (0.1, 0.3, 0.6 and 1.2 µM).
The cells were rinsed twice with PBS. All cells were lysed with 500
µl lysis buffer (10 mM Tris-HCl, 10 mM
NaH2PO4/Na2HPO4, 130 mM
NaCl, 1% Triton X-100 and 10 mM NaPPi, pH 7.5) per 100 cells.
Protein concentration was determined using a BCA protein assay kit
(Pierce; Thermo Fisher Scientific, Inc.). The activity of caspase-3
was measured using the fluorogenic substrate AC-DEVD-AMC (cat. no.
14987; Caymen Chemical Company). In brief, the cell lysate was
adjusted to a protein concentration of 500 mg/ml before 100
µl of this cell lysate were mixed with 100 µl
substrate solution (20 µg/ml caspase-3 substrate
AC-DEVD-AMC, 20 mM HEPES, 10% glycerol and 2 mM DTT, pH 7.5;
Sigma-Merck; Merck KGaA). The samples were then incubated for 1 h
at 37°C. The fluorescence of the substrate, cleaved by caspase-3,
(excitation wavelength=380 nm, emission wavelength=460 nm) was
measured using a Varioskan Flash fluorometer (Thermo Fisher
Scientific, Inc.).
Detection of changes in the mitochondrial membrane
potential (MMP HepG2 and Huh7 cells were seeded at a density of
8×103 cells/well in 96-well plates and maintained for 72
h at 37°C, 5% CO2 and 95% humidity until they were
treated with 0.6 µM broxbam or 2.0 µM vorinostat for
3, 6 and 18 h at 37°C, 5% CO2 and 95% humidity. To
measure MMP, cells were stained for 15 min in the dark at 37°C
using the JC-1 dye (1 mg/ml;
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyanine
iodide; Molecular Probes; Thermo Fisher Scientific, Inc.). The
cells were analysed using a Varioscan Flash fluorometer (Thermo
Fisher Scientific, Inc.) at excitation wavelengths of 485 nm and
emission wavelengths of 535 nm. Signals in the orange region of the
fluorescence can be detected when JC-1 aggregates occur because of
a negative MMP, which indicates healthy mitochondria. In the event
of a positive MMP, which results from mitochondrial damage, JC-1
occurs in its monomeric form giving rise to fluorescence in the
green wavelength region (38).
Accordingly, measurement of the ratio of orange to green
fluorescence signal intensities allows the determination of changes
in the MMP.
Western blotting
Whole cell extracts were prepared after harvesting
substance-treated cells. HepG2 and Huh7 cells were incubated (37°C,
5% CO2 and 95% humidity) for 24 h in RPMI growth medium
(Gibco; Thermo Fisher Scientific, Inc.) containing the respective
concentrations of test compounds (BB: 0.6 and 1.2 µM; Vs,
4.0 µM). Lysis was performed by using lysis buffer (0.1%
SDS, 0.5% sodium deoxycholic acid, 1% Nonidet P-40, 0.1 mM PMSF, 1
mg/ml aprotinin and 1 mg/ml pepstatin A1; all from Sigma Aldrich;
Merck KGaA). Protein contents of samples were determined using a
BCA protein assay kit and samples containing 30 µg protein
subjected to 7.5% or 12% SDS-PAGE. Proteins were then transferred
onto PVDF membranes by electroblotting for 1.5 h. Membranes were
blocked for 1 h using 5% skimmed milk powder solution followed by
incubation at 4°C overnight with primary antibodies. The following
antibodies were used: Glucose transporter (GLUT) 2 (1:1,000; cat.
no. 071402 MilliporeSigma), p53 and phosphorylated (p-) p53
(1:1,000; cat. nos. 9282 and 9286, respectively; Cell Signalling
Technology, Inc.), HDAC1, HDAC2, HDAC4 and HDAC6 (1:1,000; cat.
nos. 5356, 5113, 7628 and 7558, respectively; Cell Signalling
Technology, Inc.). Detection of tubulin (anti-Tubb2B, cat. no.
TA337744; Origene Technologies, Inc.) served as a loading control.
Membranes were washed with 0.1% Tween in PBS and incubated with
HRP-coupled anti-IgG antibody (1:10,000; cat. nos. NA934 and NA931,
Amersham; Cytiva) for 1 h at room temperature. Protein signals were
visualised using enhanced chemiluminescent detection kit (Amersham;
Cytiva) and a Fusion SL camera (Vilber Lourmat Deutschland GmbH).
For quantification, ImageJ (Vs 1.53j, National Institutes of
Health) was used and the density of the protein bands was
normalised to tubulin as a loading control.
RT-qPCR
Cellular RNA of untreated HepG2 and Huh7 cells were
extracted using GeneMATRIX Universal RNA Purification Kit (Roboklon
GmbH) according to the manufacturers' protocols, followed by
treatment with 1 U DNAse I (Gibco; Thermo Fisher Scientific, Inc.)
per µg RNA for the elimination of possible DNA
contaminations. Reverse transcription into cDNA and qPCR were
performed using GoTaq® 1-Step RT-qPCR System (Promega
Corporation) on a QuantStudio 5 Real-Time PCR System (Thermo Fisher
Scientific, Inc.). The mixture for each sample/primer mix had a
final volume of 10 µl containing 20 ng RNA and 250 nM of
each primer. Melt curve controls were run to ensure primer
specificity. The parameters for reverse transcription were as
follows: 37°C for 15 min, 95°C for 10 min. Primer sequences are
listed in Table SI. The obtained
cDNA was analysed by using GoTaq® 1-Step RT-qPCR System
(Promega Corporation) and qPCR was run on a StepOne-Cycler (Thermo
Fisher Scientific, Inc.) for 40 cycles of amplification (initial
denaturation for 5 min at 95°C; followed by denaturation at 95°C
for 15 sec, annealing at 60°C for 20 sec, elongation at 72°C for 45
sec for 40 cycles). All samples were run in tripli-cate. The
relative target gene expression was calculated using the
2−ΔΔCq method using GAPDH, β-actin or 18S rRNA as an
internal control (39).
Inhibition of HDAC activity
The ability of broxbam to inhibit HDAC1, -2, -4 and
was measured using a cell free fluorogenic HDAC Assay (cat. nos.
50061, 50062, 50064, 50076 BPS Bioscience, Inc.). The HDAC activity
was measured according to the protocols of the manufacturer.
Briefly, purified human recombinant HDAC enzymes contained with the
assay kit and fluorogenic HDAC substrates were used to measure HDAC
activity. In total, 50 µl assay buffer containing 1
µg/µl BSA, the human recombinant HDAC enzyme, the
test compound and the corresponding HDAC substrate was added into a
black 96-well assay plate. The reaction in each well was incubated
at 37°C for 30 min, followed by the addition of 50 µl HDAC
developer reagent and incubation at room temperature for 15 min.
Fluorescence intensity was measured using a Varioskan Flash
Fluorometer (Thermo Fisher Scientific, Inc.) using an excitation
wavelength of 380 nm and an emission wavelength of 460 nm.
Immunofluorescence staining of the
cytoskeleton
HepG2 cells were seeded onto glass coverslips at a
density of 1×105 and were allowed to attach and
proliferate for 24 h (37°C, 5% CO2 and 95% humidity).
Cells were treated with broxbam (0.6 µM) and vorinostat (4
µM) whereas a corresponding volume of DMSO was used as a
negative control. Treated cells were incubated for additional 24 h
(37°C, 5% CO2 and 95% humidity) and fixed with 4%
formaldehyde in PBS for 20 min at room temperature. The samples
were then blocked and permeabilised (1% BSA and 0.1% Triton X-100
in PBS) for 30 min at room temperature. For the immunostaining of
the microtubule structures, the cells were incubated for 1 h at
37°C in the dark with mouse primary anti-human α-tubulin monoclonal
antibody (1:500; cat. no. T6199; Sigma Aldrich; Merck KGaA),
followed by treatment with a AlexaFluor®-conjugated 546
goat anti-mouse secondary antibody (1:500; cat. no A-21133; Thermo
Fisher Scientific, Inc.) for 1 h at room temperature in the dark.
Cellular actin filaments were stained for 1 h at 37°C in the dark
using phalloidin (1:1,000, cat. No A12379
AlexaFluor®-conjugated 488; Invitrogen; Thermo Fisher
Scientific, Inc.). Coverslips were mounted in DAPI containing
Mowiol (Mowiol 4-88; 1 µg/ml DAPI; Carl Roth GmbH) for
additional nuclei counterstaining. Fluorescence microscopic imaging
(magnification, ×60; Spinning Disk Confocal microscope; Nikon
Corporation) was performed at the Charité Advanced Medical
BioImaging Core Facility (Berlin, Germany).
Scratch wound healing assay
HepG2 cells were allowed to proliferate to
confluence in six-well plates. Using a 10 µl pipette tip,
the cell monolayer was scratched once horizontally and vertically.
Wells were rinsed with PBS and fresh medium was added, containing
two concentrations of broxbam (0.6 and 1.2 µM), whereas a
corresponding volume of DMSO was used for a control. The cell
monolayers were incubated for 24 h (37°C, 5% CO2 and 95%
humidity), followed by photographic documentation using a digital
camera (Kappa Optronics GmbH). Migration of cells was quantified
using the TScratch software (version 1.0; CSElab). Migration values
were normalised to control, which was set as 100%. The experiments
were performed under normal FBS conditions (10%), since HepG2 cells
could not tolerate serum deprivation and reacted with immediate and
pronounced cell detachment. In addition, serum deprivation is a
procedure that is preferable necessary for long-term observations
(>24-48 h), where cell proliferation instead of migration
becomes the predominant factor for wound healing (40,41).
Zebrafish angiogenesis assay
To examine the angiogenesis in vivo,
experiments with zebrafish embryos were performed, which represents
a viable model for the evaluation of angiogenic effects of small
molecules (42). In the
developing zebrafish embryo, the sub-intestinal veins (SIVs)
represent a characteristic blood vessel system that can be easily
visualised (42). Eggs from the
transgenic Tg(fli1:EGFP)y1 mutant Casper zebrafish stem
were obtained 2-5 h after egg deposition connected to spawning
process from the animal breeding facility of the University of
Bayreuth, which has the permission to keep and breed vertebrates
and cephalopods for experimental purposes according to § 11 German
animal Welfare Law (permission no. OBK/A 2; Bayreuth, Germany).
Zebrafish are kept at 20-26°C in water pH 6.0-8.0 under standard
atmosphere.
After the egg deposition, the spawn fish eggs (1 day
post fertilisation) were collected, rinsed, and transferred into a
petri dish filled with E3-medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM
CaCl2, 0.33 mM MgSO4 and 0.01% methylene blue
in ddH2O; pH 7.2) and incubated for 24 h at 28°C.
Healthy embryos were then dechorionated, which means the removal of
the egg shell, and five embryos per well were transferred into
six-well plates in 5 ml E3-medium. In total, 100-fold pre-dilutions
of test compounds (final concentrations were 1 µM, 750 and
500 nM) in ddH2O were prepared and added to the
E3-medium at 50.5 µl per well, followed by incubation for 48
h at 28°C. After the addition of 50 µl 0.04% tricaine
solution (ethyl-3-aminobenzoate-methanesulfonate; Sigma Aldrich;
Merck KGaA) to the E3-medium in the six-well plates, the embryos (3
days post fertilisation) were anaesthetised (5 min at room
temperature) to a final tricaine concentration of 0.0004% before
the blood vessel system was visualised and documented by
fluorescence microscopy with up to two-fold magnification
(excitation wavelength, 488 nm; emission wavelength, 510 nm; Leica
MZ10F fluorescence microscope; Leica Microsystems GmbH; AxioCam l
cm; Carl Zeiss AG). The area of SIVs was measured using Image J
version 1.52a. At the end of the experiments, 1 ml anaesthetic
tricaine (tricaine mesylate; 5 g/l) was added to 5 ml E3-media to
kill the zebrafish larvae (3 days post fertilisation) with an
overdose of the anaesthetic, at a final concentration of 833
mg/l.
Statistical analysis
Statistical analysis of the data was performed using
GraphPad Prism 8 software for Windows (GraphPad Software, Inc.). If
not indicated otherwise, data were presented as the mean ± standard
error of the mean. Unpaired Student's t-tests were used for
two-group comparisons. To test for statistical significance between
> two groups, one-way ANOVA coupled with Tukey's post hoc tests
was used. P<0.05 were considered to indicate a statistically
significant difference. All experiments were performed in
triplicate unless otherwise stated. The exact number of independent
repetitions for each assay is provided in the figure legends.
Results
Inhibition of cell proliferation and
cytotoxicity
It was previously reported that broxbam exerts
selective anti-tumour effects at half maximal inhibitory
concentrations (IC50) in the low µM to two-digit
nM range (20). In the present
study, the potential effects of broxbam in liver cancer cell models
were evaluated. Table I
summarises the IC50 values calculated for broxbam and
vorinostat in liver cancer cell lines HCC and CCC and
non-transformed hepatocytes. Notably, broxbam exhibited higher
antiproliferative potency in all liver cancer cell lines compared
with non-transformed hepatocytes, which required three-times higher
IC50 concentrations (Table
I). This suggests the selectivity of broxbam towards liver
cancer cells. In addition, broxbam exerted higher antiproliferative
capabilities compared with the clinically relevant HDACi vorinostat
in all liver cancer cell models test. Unlike broxbam, vorinostat
did not appear to display cancer specificity, since its
IC50 values did not differ between the liver cancer cell
lines and the non-transformed AML-12 cells (Table I).
 | Table IGrowth inhibitory concentrations
(IC50) of compounds applied to liver cancer cell lines
HepG2 and Huh7, CCC cell lines TFK-1 and EGI-1 and mouse
hepatocytes (AML12)a. |
Table I
Growth inhibitory concentrations
(IC50) of compounds applied to liver cancer cell lines
HepG2 and Huh7, CCC cell lines TFK-1 and EGI-1 and mouse
hepatocytes (AML12)a.
| Origin | Cell line | Broxbam
(µM) | Vorinostat
(µM) |
|---|
| Liver cancer | HepG2 | 0.6±0.2 | 2.1±0.4 |
| Huh7 | 0.6±0.1 | 1.8±0.5 |
| CCC | TFK-1 | 0.6±0.1 | 1.4±0.1 |
| EGI-1 | 0.6±0.2 | 3.2±0.1 |
| Non-transformed
mouse hepatocytes | AML12 | 1.9±0.2 | 1.7±0.2 |
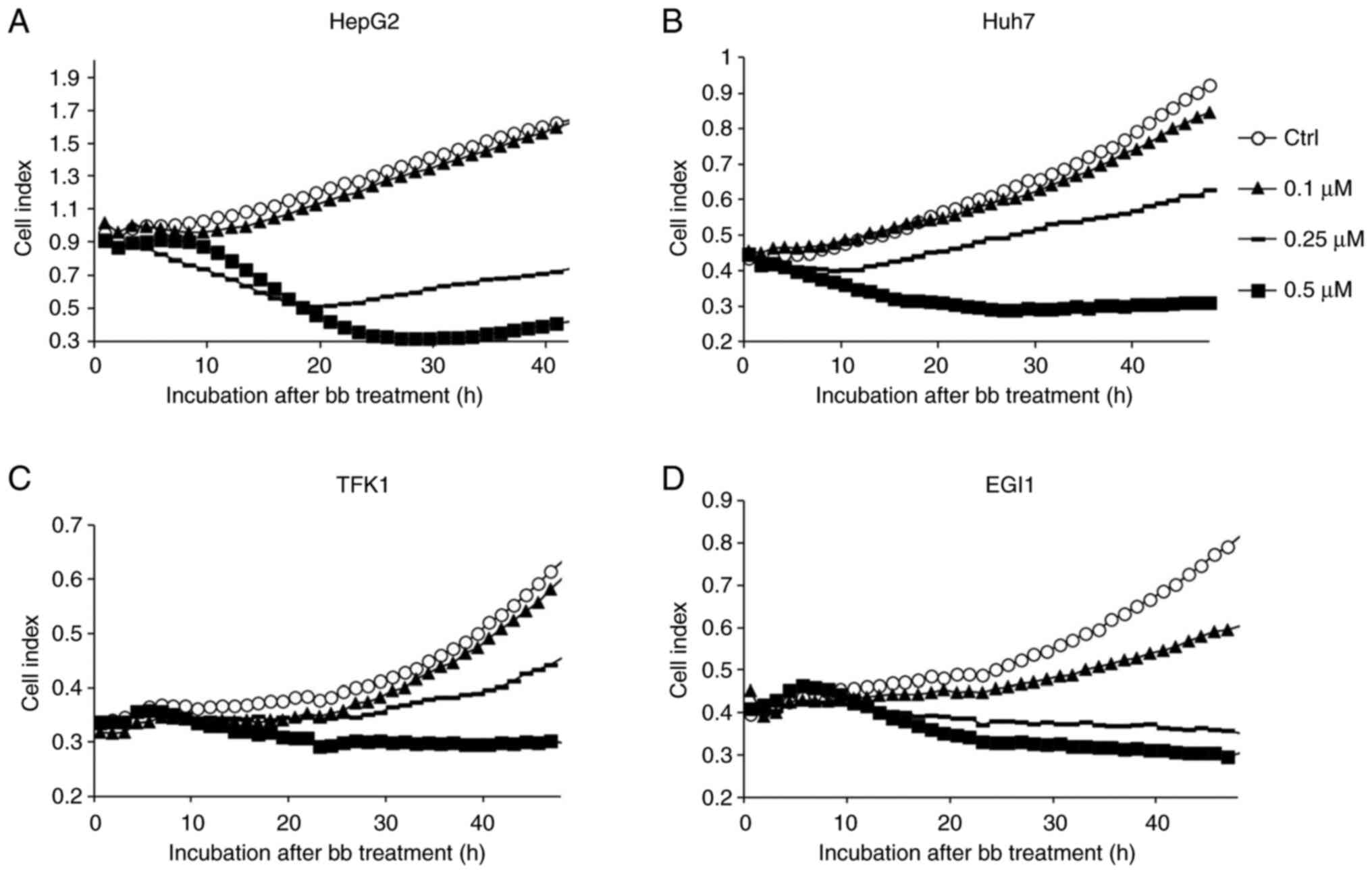
Using the iCELLigence real-time cell viability and
proliferation analysis system, the kinetic profile and onset of the
inhibition of proliferation mediated by broxbam were deter-mined.
In case of the HepG2 and Huh7 cell lines, a distinct reduction in
cell proliferation was observed at as early as 12 h after the
application of broxbam in a dose-dependent manner (Fig. 2A and B). In CCC cell lines, this
dose-dependent inhibitory effect was also detected, but instead
becoming more pronounced 24 h after treatment (Fig. 2C and D). To exclude the
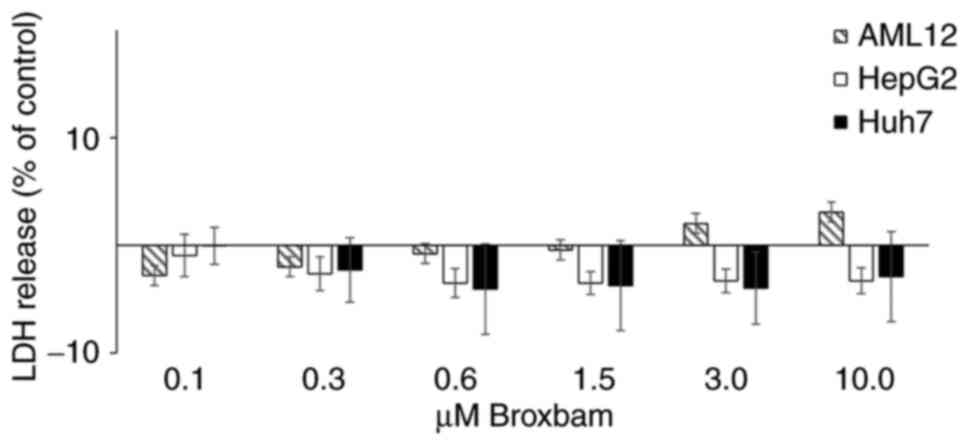
contribution of unspecific cytotoxicity to these antiproliferative
effects exerted by broxbam, the release of lactate dehydrogenase
(LDH) from the cytosol into the cell culture supernatant of HepG2,
Huh7 and AML12 cells was measured (Fig. 3). Increased LDH release would
indicate the presence of nonspecific and necrotic cell death due to
treatment-induced damage of cell membranes (36). However, treatment of HepG2 or Huh7
cells with rising concentrations of broxbam (0.1-10 µM) did
not lead to significant increases in LDH release after 24 h
(Fig. 3). The same effects were
also observed in non-transformed hepatocytes (AML12) after broxbam
treatment (Fig. 3). This suggests
that broxbam does not compromise cell membrane integrity and that
it exerted no immediate cytotoxic effects even at concentrations as
high as 10 µM.
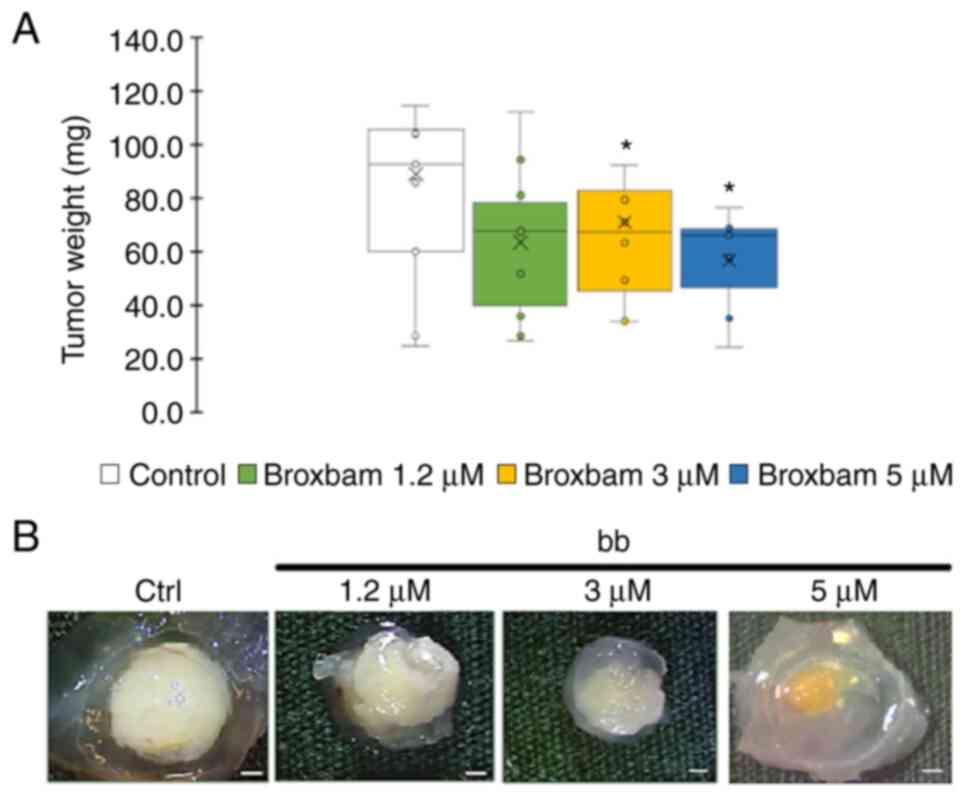
In vivo tumour growth reduction
The potential antineo-plastic effects of broxbam in
liver cancer tumours was next tested using in vivo chick
embryo experiments, specifically by applying the modified
chorioallantoic membrane (CAM) assay. HepG2-derived microtumours
from an initial volume of 20 µl were grown on the CAM of
fertilised chicken eggs for 24 h. Subsequently, the microtumours
were treated for 72 h with either PBS (control) or rising
concentrations of broxbam (1.2-5.0 µM). At the end of the
experiment, tumour masses were excised and weighed. Compared to
PBS-treated controls, broxbam (3 and 5 µM) induced a
significant reduction in liver cancer microtumour growth (Fig. 4).
To further characterise the underlying signalling
and metabolic events underlying these antitumor effects of broxbam
observed in the present study, its potential effects on glucose
metabolism were next assessed. Cancer cells preferentially use
glycolysis to consume glucose as the substrate for energy
metabolism, in a process known as the Warburg effect (43). Therefore, the impact of broxbam on
the expression of GLUT2, which is crucial for hepatic glucose
uptake (44), was investigated.
In addition, the glycolytic activity of HepG2 and Huh7 cells was
also assessed by measuring the consumption of glucose, production
of lactate and its release into the supernatant of the cell culture
medium. Protein expression levels of GLUT2 were not affected by
broxbam, though the HDACi vorinostat markedly reduced the
expression of GLUT2 compared to broxbam treated group in both HepG2
and Huh7 cells (Fig. S1). This
finding suggests that vorinostat, but not broxbam, can inhibit
cellular glucose uptake by downregulating GLUT2 expression. By
contrast, the glycolytic activity of broxbam-treated liver cancer
cells was found to be decreased significantly. Compared with that
in the untreated controls, the remaining glucose in the cell
culture supernatant of broxbam-treated HepG2 cells was markedly
higher whereas the production and release of lactate was decreased
(Fig. S1). This lower glucose
consumption and reduced lactate production indicate reduced
glycolytic activity in broxbam-treated cells. In addition, no such
reduction in glycolytic activity could be observed in HepG2 cells
following vorinostat treatment, suggesting no effects were mediated
by vorinostat on glucose uptake or lactate production and
release.
Proapoptotic mode of action
To further elucidate the mechanisms by which broxbam
exerted antitumour effects on the liver cancer cells the extent of
apoptosis induction was next measured. Apoptosis induction
considered to be one of the primary objectives of targeted cancer
therapy since it results in the specific and inflammation-free
elimination of cancer cells. Apoptosis induction occurs through two
distinct mechanisms, either by the activation of cellular death
receptors or through the mitochondria-driven activation of
caspase-3 (45). Broxbam was
found to induce mitochondria-driven apoptosis in liver cancer
cells, as evidenced by fluorometric changes in the MMP of broxbam
treated cells (Fig. 5A). An
increase in the green/red ratio indicates a reduction in the MMP
and therefore mitochondrial damage, which may have served as a
trigger for the activation of the apoptosis-associated tumour
suppressor protein p53. A significant increase in the green/red
ratio following treatment with the IC50 concentration of
broxbam at 0.6 µM was observed after 3 and 6 h (Fig. 5A).
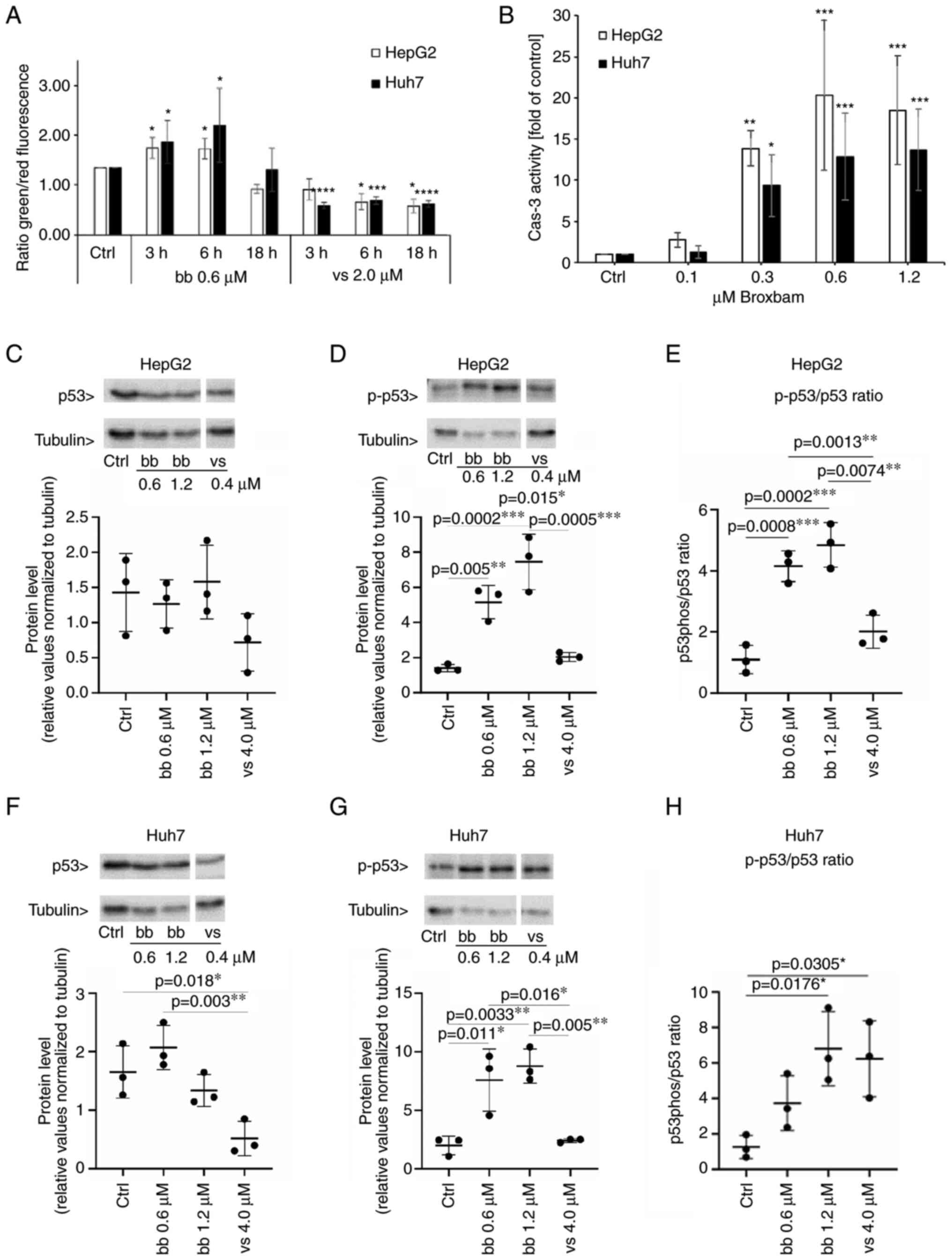 | Figure 5Detection of proapoptotic events in
liver cancer cells. (A) Influence of bb and vs on the loss of
mitochondrial membrane potential of liver cancer cell lines HepG2
and Huh7. Indicated values were measured by fluorometric
measurement of JC-1-stained cells. The effect of IC50
concentrations of bb and vs was monitored after 3, 6 and 18 h.
Mitochondrial accumulation of JC-1 results in red fluorescence
whereas loss of MMP results in monomeric JC-1, which emits green
fluorescence. Data are presented as the ratio of green/red
fluorescence of cells. Data are presented as the mean ± SEM from
three experiments. One-way ANOVA with Tukey's post hoc test was
used to test for significance. *P<0.05,
***P<0.001 and ****P<0.0001 vs. Ctrl.
(B) Caspase-3 activity in HepG2and Huh7 cells upon treatment with
bb for 24 h. Data are presented as the mean ± SEM percentage of
untreated controls from four experiments. One-way ANOVA with
Tukey's post hoc test was used to test for significance.
*P<0.05, **P<0.01 and
***P<0.001 vs. ctrl. (C-H) HepG2 and Huh7 cells were
treated either with solvent (Ctrl), bb at IC50 (0.6
µM), bb at two-fold IC50 (1.2 µM) or vs.
at two-fold IC50 (4.0 µM). Western blotting results of (C)
p53 expression and (D) phosphorylation levels in HepG2 cells. (E)
p-p53/p53 ratio was also quantified in HepG2 cells. Western
blotting results of (F) p53 expression and (G) phosphorylation
levels in Huh7 cells. (H) p-p53/p53 ratio was also quantified in
Huh7 cells. Corresponding representative western blotting images
are shown in Fig. S2. n=3 per
group. One-way ANOVA with Tukey's post hoc test was used to test
for significance. Bb, broxbam; vs, vorinostat; Ctrl, control; Cas
3, caspase 3; p-, phosphorylated. |
By contrast, treatment with vorinostat at its
IC50 concentration of 2 µM, did not lead to an
apoptosis-indicating increase in the green/red ratio. Instead, a
significant reduction was observed, suggesting that vorinostat was
not likely to induce mitochondria-driven apoptosis (Fig. 5A).
In following experiments broxbam treatment was shown
to induce a dose-dependent increase in the phosphorylation of p53
of HepG2 and Huh7 cells (Fig.
5C-H), consistent with the aforementioned data on the
broxbam-induced changes in MMP for the initiation of
mitochondria-driven apoptosis (Fig.
5A). In addition, no increase in p53 phosphorylation in HepG2
(Fig. 5D) or Huh7 (Fig. 5G) cells could be observed after
vorinostat treatment, which is in accordance with the failure of
vorinostat to decrease the MMP (Fig.
5A).
Mitochondria serve a pivotal role in the regulation
of apoptotic caspase activation. The caspase cascade lies
down-stream of the loss of MMP and activation of p53 (46-48). This cascade eventually leads to
the activation of the executioner caspase-3 (49). Therefore, Caspase-3 activity was
next assessed using a fluorogenic caspase-3 assay, which functions
by the specific recognition of the enzymatic activity of
phosphorylated caspase-3 (15).
After broxbam treatment, HepG2 and Huh7 cells exhibited a
dose-dependent increase in apoptotic caspase-3 activity compared
with that in the control group (Fig.
5B). Of note, maximal capsase-3 activity could already be
observed at the concentration of 0.6 µM broxbam (Fig. 5B), which corresponded to the
IC50 values of broxbam in these cells (Table I).
HDAC expression and activity
To clarify if HDAC inhibition forms part of the
antitumour mechanism of broxbam, its effects on HDAC expression and
activity were investigated. RT-qPCR was used to measure the
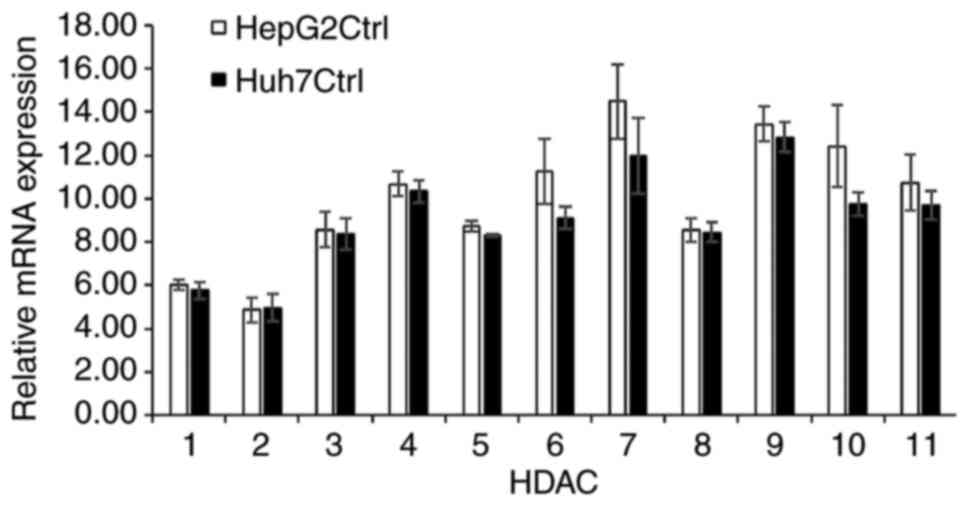
expression levels of the 11 subtypes of HDACs (HDACs 1-11) in the
HepG2 and Huh7 cells. Although the mRNA expression levels of HDAC1
and HDAC2 were considerably lower compared with those of the other
HDAC subtypes, all 11 HDACs tested were found to be expressed in
both cell lines (Fig. 6).
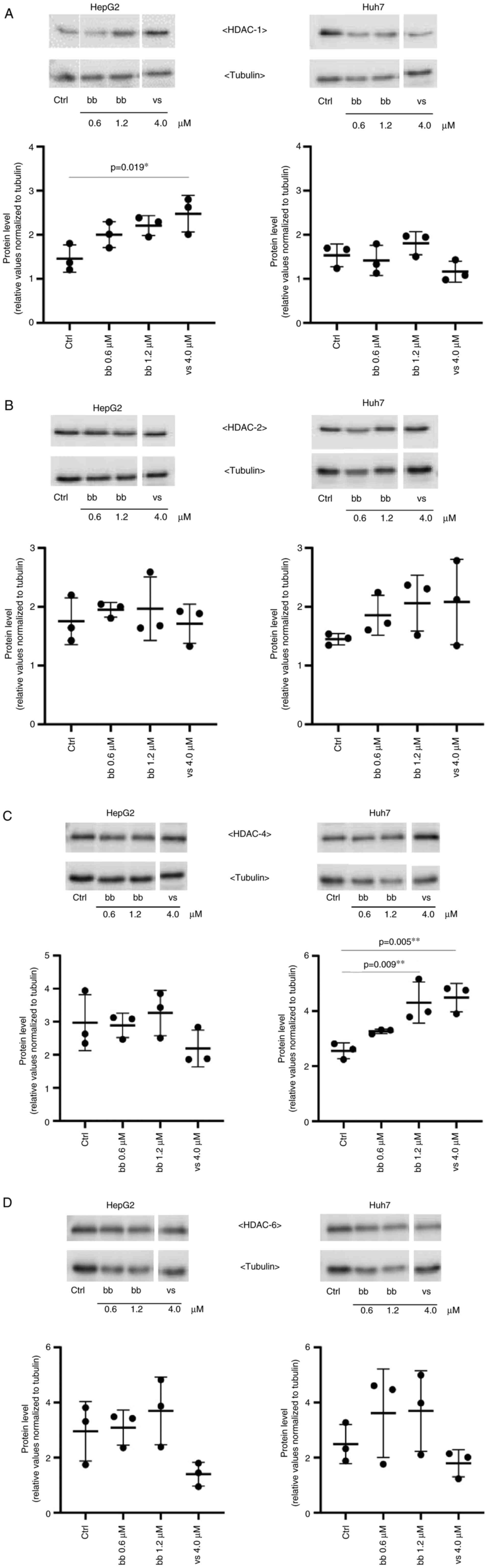
Subsequently, the potential treatment-induced
changes in the protein expression levels of HDAC1, -2, -4 and -6
were assessed by western blotting and densitometric analysis. HDAC1
and HDAC2 belongs to class I HDACs and are local-ized in the
nucleus of the cell (50). HDAC4
and HDAC6 are class II HDACs and both can be found in the cytoplasm
as well as in the nucleus of a cell (50). These HDACs were previously found
to be overexpressed in human pancreatic adenocarcinoma and
hepatocellular carcinoma (50,51). Exposure of HepG2 and Huh7 cells to
0.6 µM and 1.2 µM broxbam, concentrations
corresponding to their respective IC50 and two-fold
IC50 values, did not lead to significant changes in any
of the HDAC subtypes (Fig. 7). An
exception was the significant increase in HDAC4 expression in Huh7
cells treated with 1.2 µM broxbam compared with that in the
control group (Fig. 7C). This
finding suggests that broxbam functions as an inhibitor of HDAC
activity instead of being a regulator of HDAC expression (Fig. 7).
The activities of HDACs -1, -2, -4 and -6 following
treatment with IC50 and two-fold IC50
concentrations of broxbam and vorinostat were assessed using a
cell-free enzymatic assay system. HDAC subtype-specific inhibition
of the cleavage of fluorogenic peptide substrates (Fig. 8A) was next detected and compared
with the untreated control group, which was set to 100%. Broxbam
exerted an attenuating effect on the activity of HDAC1 and -6, but
did not affect the activities of HDACs -2 and -4. Vorinostat
mediated potent inhibitory effects on the HDACs -1 and -2, which
exceeded that of broxbam. Therefore, broxbam and vorinostat
inhibited the individual HDAC isoforms with distinct profiles.
However, both compounds were concluded inhibit HDAC1, with broxbam
showing specificity towards HDAC6 whereas vorinostat showed
specificity towards HDAC2. Due to this potent inhibitory effect of
broxbam against HDAC6, the present study next examined its
potential dose dependency over the concentration range from 0.6 to
10 µM, which revealed 90% inhibition of HDAC6 activity at 10
µM, the highest broxbam concentration tested (Fig. 8B).
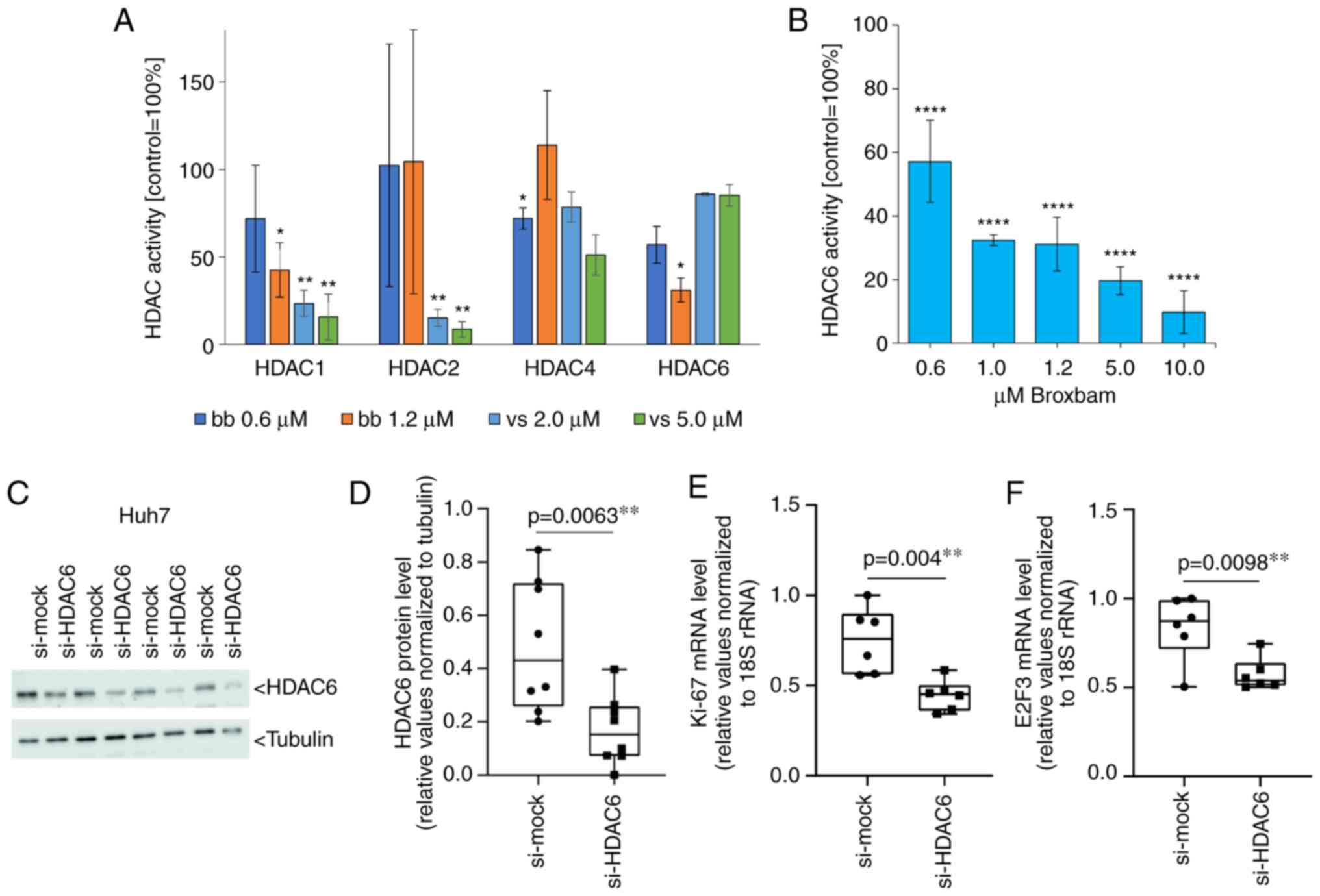 | Figure 8Effects of inhibiting the selective
HDACs of broxbam in Huh-7 cells. (A) Activities of HDAC1, -2, -4
and -6 after treatment with bb or vs at concen-trations
corresponding to their respective IC50 and two-fold
IC50. Data are presented as the means ± SEM percentage
of untreated control, which was set to 100%, from three
experiments. *P<0.05, **P<0.01 vs.
Ctrl. One-way ANOVA with Tukey's post hoc test was used to test for
significance (B) Dose-dependent inhibitory effect of bb on the
activity of HDAC6 relative to control, represented as the means ±
SEM from three independent experiments. ****P<0.0001
vs. Ctrl. One-way ANOVA with Tukey's post hoc test was used to test
for significance. (C) Western blot analysis and (D) quantification
of siRNA-mediated knockdown of HDAC6 protein expression in Huh7
cells transfected with control (si-mock) or HDAC6-specific
(si-HDAC6) siRNAs for 48 h. Each set of si-mock and si-HDAC6 lanes
represents one transfection experiment. Tubulin was used as the
loading control. Analysis of (E) Ki-67 and (F) E2F3 mRNA expression
by reverse transcription-quantitative PCR. The mRNA expression
levels were normalised to that of 18S rRNA. Data was presented as
the mean ± SEM from six independent experiments. One-way ANOVA with
Tukey's post hoc test was used to test for significance. HDAC,
histone deacetylase; bb, broxbam; vs, vorinostat; si, small
interfering. |
To investigate the contribution of HDAC6 inhibition
towards the antiproliferative effects of broxbam in liver cancer
cells (Fig. 2), HDAC6 knockdown
was performed using siRNA. After 48 h of transfection with
HDAC6-specific siRNA, the expression of HDAC6 protein in Huh7 cells
was significantly decreased compared with that in cells transfected
with the si-mock (Fig. 8C and D).
Furthermore, the expression of typical markers of cell
proliferation Ki-67 and E2F3 (52,53), were also found to be decreased
significantly compared with that in cells transfected with the
si-mock (Fig. 8E and F)
suggesting a distinct contribution of HDAC6 inhibition towards the
antiproliferative (Figs. 1 and
2) and antineoplastic (Fig. 4) effects of broxbam in liver
cancer.
Dynamics of the cytoskeleton and cellular
migration
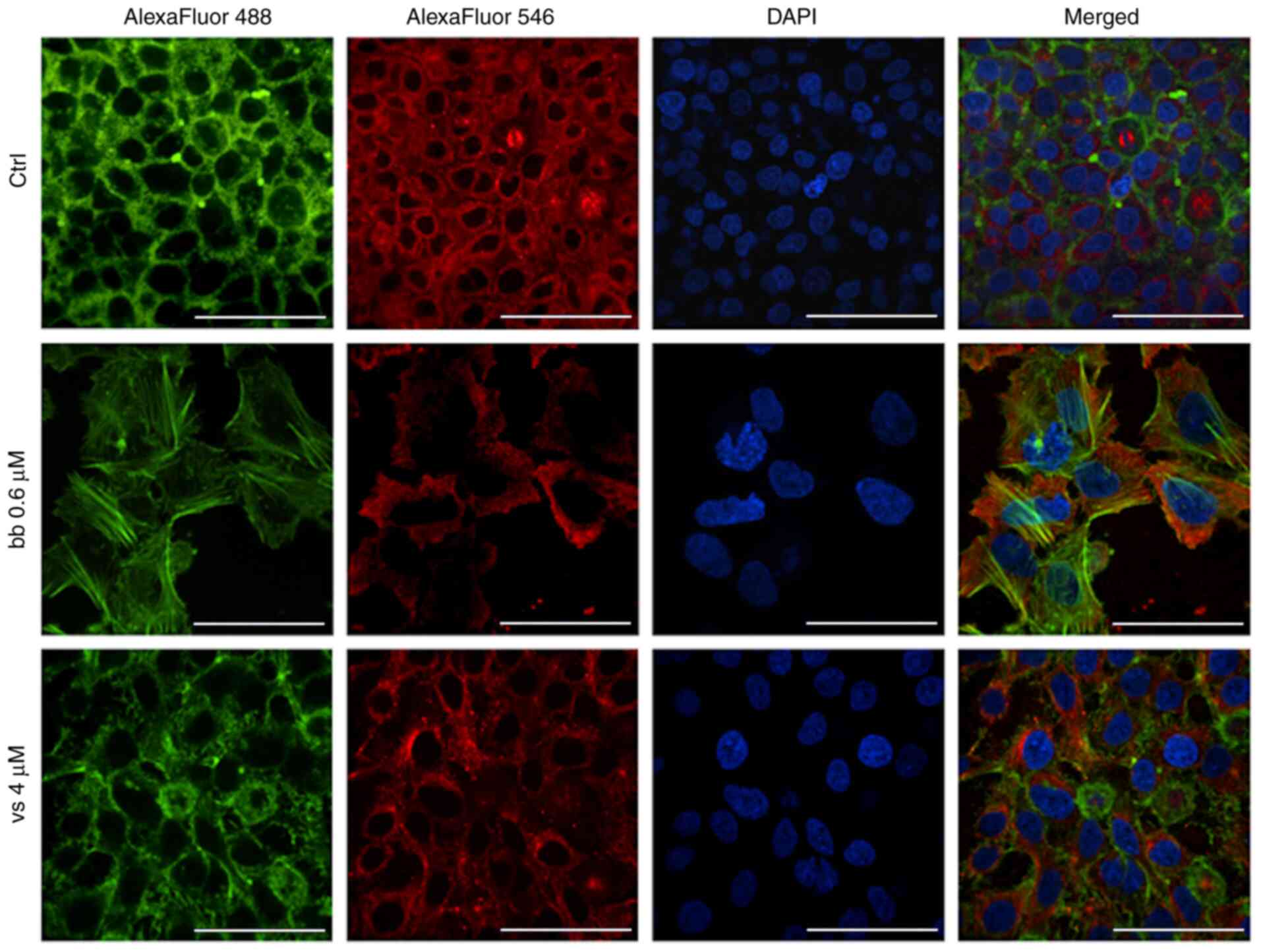
Since broxbam contains the trimethoxystilbene motif
of the vascular disrupting natural product CA-4 within its
structure (23,25,26), it was next examined for its
possible effects on the tubulin cytoskeleton and associated
processes. Its inhibitory effects on the polymerisation of purified
tubulin in vitro have already been previously reported
(20). As HDAC6 has been found to
function as a tubulin deacetylase to interfere with
microtubule-dependent cell motility (54), the effects of broxbam on the
microtubule and F-actin cytoskeleton of HepG2 cells were
investigated using immunofluorescence staining. The formation of
F-actin stress fibres and focal adhesions was observed following
treatment with broxbam at its IC50 concentration
(Fig. 9). Furthermore, the
microtubule cytoskeleton appeared to be markedly altered compared
with that in the control group. In general, broxbam-treated cells
were considerably enlarged, especially their nuclei (Fig. 9). These effects were more
prominent compared with those mediated by vorinostat, even after
treatment with vorinostat at a concentration of two-fold
IC50 (Fig. 9).
Cytoskeletal components, such as microtubules and
actin fibres, are critical for cell migration (55,56). To investigate the effect of
broxbam on cell migration, a scratch wound healing assay was
performed. An observation period of 24 h was chosen to minimise the
possibility of the migratory process being distorted by cell
proliferation. The average doubling time of HepG2 cells is ~48 h,
suggesting that HepG2 cell proliferation is unlikely to contribute
significantly to wound closing (57). Broxbam treatment led to a
significant reduction in gap closing by the HepG2 cell monolayer
compared with that in the untreated control group (Fig. 10). In addition, broxbam exerted a
potent antimigratory effect even when applied at its
IC50 concentration of 0.6 µM (Fig. 10B).
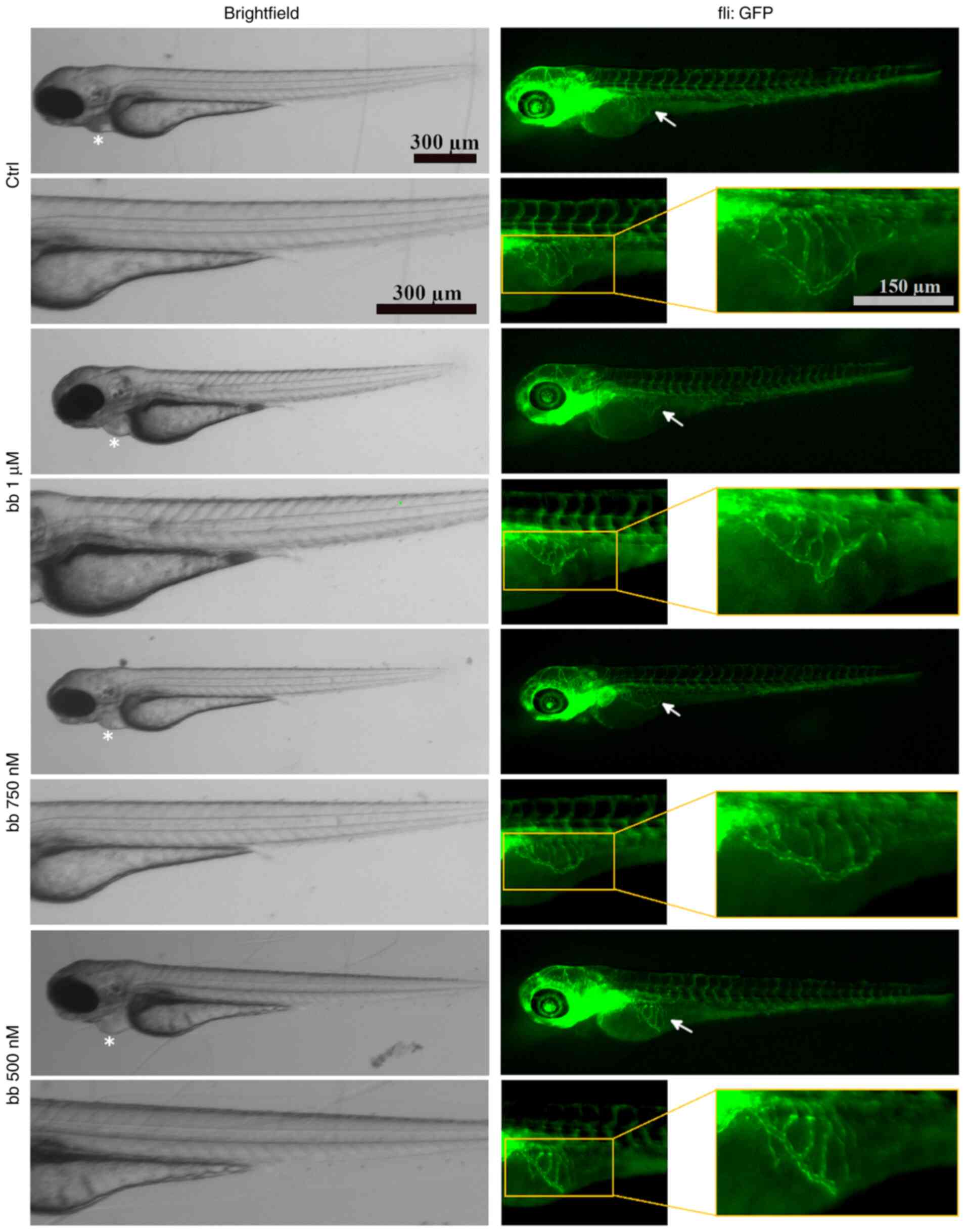
Antiangiogenic effects in vivo
A previous study validated the properties of the
two structural motifs on broxbam, which are HDAC-inhibition
mediated by the hydroxamic acid pharmacophore and interference with
cytoskeletal dynamics by its trimethoxystilbene motif (20). There is a possibility that
inhibition of tubulin polymerisation and HDAC activity by broxbam
may also translate into an antiangiogenic effect. Due to the
previously known antiangiogenic effects of CA-4 (23) and the well-established association
between HDACs as modulators of hypoxia-induced factor-1α and
vascular endothelial growth factor (VEGF) expression (58), it was hypothesised that broxbam
may also exert antiangiogenic properties. Therefore, in vivo
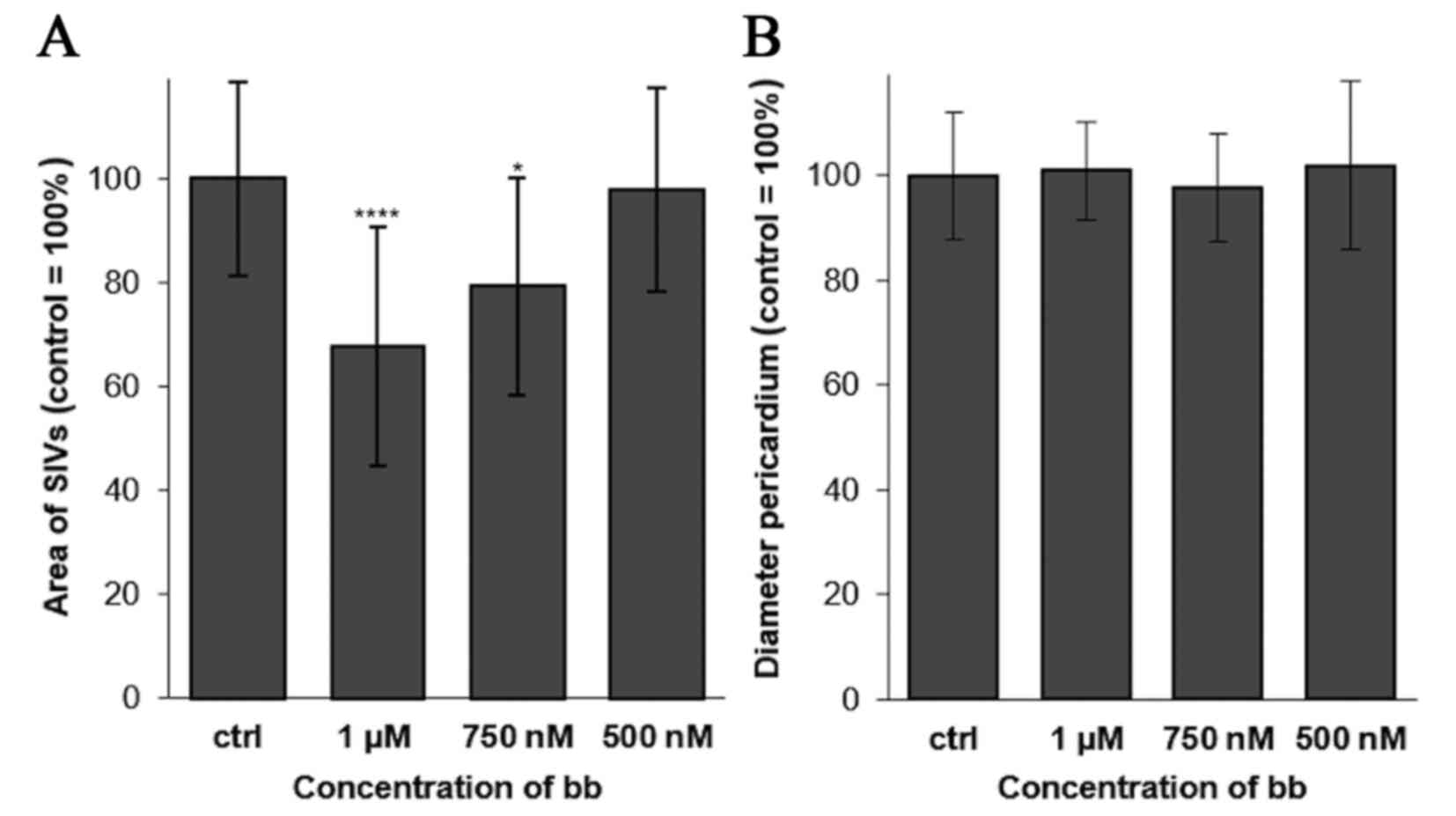
zebrafish angiogenesis assays were performed (42). Broxbam mediated a dose-dependent
antiangiogenic effect in reducing the area of the sub-intestinal
veins (SIV) in developing zebrafish embryos, which became visible
at concentrations equating to its IC50 value (Figs. 11 and 12). Signs of cardiotoxicity, such as
dilation of the pericardial sac, which is a known unwanted side
effect of antiangiogenic substances, such as CA-4 (59), could not be observed (Figs. 11 and 12).
Discussion
Broxbam was recently reported to be a potent
anticancer agent against various non-liver cancer cell models
(20). In the present study, it
was shown to exert antiproliferative properties in HCC and CCC
liver cancer cell models. This novel chimeric compound, which
consists of a HDAC-inhibitory hydroxamic acid pharmacophore and a
cytoskeleton-interfering trimethoxystilbene motif, inhibited the
proliferation of liver cancer cells at near-nanomolar
concentrations (IC50, ~0.6 µM). This exceeded the
antiproliferative potency of the clinically established HDACi
vorinostat (IC50, 1.4-3.2 µM). Unlike vorinostat,
broxbam exerted preferential specificity for cancer cells over
non-transformed murine hepatocytes. Real-time kinetic investigation
of the antiproliferative effects of broxbam revealed an early onset
of this effect, at 12-24 h, after drug application. The prohibitive
effects of broxbam on liver cancer cells was found not likely to be
due to unspecific cytotoxicity, but instead likely to be due to the
targeted and orchestrated modulation of cancer cell-specific
signalling pathways. This in turn lead to antiproliferative,
anti-angiogenic, anti-migratory and apoptotic effects, in addition
to the breakdown of cellular energy metabolism. The decrease in the
glycolytic activity of liver cancer cells after broxbam treatment
may be of therapeutic importance in terms of the glucose addiction
of cancer cells, which is required for the maintenance of
metabolism (the Warburg effect) (43).
Broxbam was found to induce the apoptosis of liver
cancer cells through the mitochondrial-driven apoptosis pathway. A
protein of importance for mitochondrial apoptosis is the p53 tumour
suppressor protein, the phosphorylation of which is associated with
the disruption of mitochondrial function and subsequent cell death
(48). Phosphorylation of p53
induces the expression of p53 regulated apoptosis inducing protein
1, which is localised in the mitochondria and leads to the
disruption of MMP en route to triggering apoptosis (60,61). Consistent with this mechanism, the
present study found that broxbam induced p53 phosphorylation,
reduced MMP and subsequently activated the apoptosis-specific
effector caspase-3. The activation of caspase-3 directly
contributes to core apoptotic processes, such as the dismantling of
cell components and formation of apoptotic bodies therefore, it
represents a direct marker for the activation of apoptosis
(62). The extent of apoptosis
mediated by broxbam was studied in the present study in two liver
cancer cell lines with distinct p53 statuses. HepG2 cells
represents a p53-wild-type cell line whereas Huh7 cells harbour a
mutated codon in the p53 gene leading to the overexpression of the
functionally-repressed p53 (63).
Caspase-3 activation by broxbam was more pronounced in the
p53-native HepG2 cells compared with that in the p53-mutated Huh7
cells. However, broxbam promoted p53 phosphorylation in both cell
models without altering the expression of the total p53 protein. By
contrast, vorinostat did not alter p53 phosphorylation in either of
the cell models, whilst significantly downregulating p53 expression
in Huh7 cells. It is tempting to speculate that this may be the
result of differences in the HDAC subtype specificity between
broxbam and vorinostat, especially in their inhibitory activities
towards HDAC6 activity. However, further research is required to
clarify this putative association. It is noteworthy that broxbam
can induce p53 phosphorylation both in cells with wild-type and
mutant p53, whilst vorinostat could not induce p53
phosphorylation.
Evaluation of the effects of broxbam and vorinostat
on HDAC revealed that neither of the compounds significantly
influenced the protein expression of the HDAC subtypes -1, -2, -4
and -6, instead inhibiting their HDAC enzyme activity.
Subtype-specific profiling revealed pronounced differences in the
ability of broxbam and vorinostat to inhibit HDAC6. Whilst broxbam
potently inhibited HDAC6 activity (>90%) at a concentration of
10 µM, vorinostat could only mediate a comparatively weak
inhibition of 20%.
HDAC6 is a unique member of the HDAC family not
only for its role in histone acetylation and deacetylation, but
also in targeting several non-histone substrates, such as cortactin
and heat shock protein 90, thereby regulating cell proliferation,
metastasis, invasion, and mitosis in tumours of SKOV3 human ovarian
cancer cells (64). Accordingly,
it has been previously demonstrated that HDAC6 inhibition can lead
to the inhibition of cell proliferation, apoptosis, reduction in
migration and motility of fibroblasts, SKOV3 ovarian cancer, SKBR3
breast carcinoma, and MCF7 breast cancer cell lines (65). This is consistent with
observations in the present study with regards to the
broxbam-mediated HDAC6 inhibition of liver cancer cells.
HDAC6-specific siRNA silencing in Huh7 cells, which mimicked the
effects of HDAC6 inhibition exerted by broxbam, led to a
significant downregulation of proliferation markers Ki67 and E2F3.
α-Tubulin is another non-histone substrate that can be deacetylated
by HDAC6 (54,56,66). HDAC6-dependent deacetylation of
tubulin has been reported to result in potent effects on
cytoskeletal dynamics and cell migration (54,66,67). HDAC6 has also been previously
associated with the function of microtubule dynamics and regulation
of microtubule-dependent cell migration (54). In the present study,
broxbam-treated liver cancer cells displayed profound alterations
in their cytoskeleton components F-actin and microtubules. After
microtubules become disrupted, f-actin stress fibres and focal
adhesions are formed in response (68,69). The cytoskeletal effects of broxbam
exceeded those mediated by vorinostat, which corresponded with the
weaker inhibitory effects of vorinostat on HDAC6 activity.
Broxbam was also found to exert antiangiogenic
effects in vivo, as shown by the suppression of SIV growth
in the zebrafish embryos. This effect was visible already at nM
concentrations, resembling the antiproliferative IC50
values of broxbam in liver cancer cells. Notably, the
antiangiogenic effects of broxbam were not accompanied by signs of
cardiotoxicity or impairments in the development of zebrafish
embryos, which are known to be highly sensitive to toxic stress
(70). The antiangiogenic effects
of broxbam were likely to be due to a combination of mechanisms,
with contributions from both pharmacophores contained within the
structure of this chimeric compound. In this respect, HDACis are
known to inhibit VEGF-induced angiogenesis in vitro as well
as in vivo (71), such
that CA-4-driven anti-angiogenesis has also been linked to the
inhibition of VEGF (72).
However, tumour vasculature disrupting agents, especially those
derived from CA4, have been shown to confer a high toxicity risk
(59). Therefore, although
broxbam contains a CA4-derived pharmacophore, it did not mediate
toxic effects. This suggests that the effects of this novel
chimeric compound may instead arise from its inhibitory effects on
HDACs. The present study revealed that HDAC6-specific knockdown
resulted in the downregulation of E2F3 expression in Huh7 cells.
E2F3 is regarded to be a key factor for endothelial cell
proliferation and angiogenesis (73). Therefore, it is conceivable that
the pronounced HDAC6 inhibition of broxbam substantially
contributed to its antiangiogenic effects due to the suppression of
endothelial E2F3 expression. Further study is warranted to verify
this hypothesis. The effects of broxbam on liver cancer cells
revealed in the present study are summarized in Fig. 13.
Currently available treatment options for advanced
and inoperable liver cancer are restricted to multi-kinase
inhibitors, such as sorafenib or regorafenib (74). Although the therapeutic
concentration of sorafenib is ~8 µM (75), lower concentrations of broxbam was
able to induce pronounced antitumor effects in the in vivo
experiments with liver cancer xenografts on the CAM of fertilised
chicken eggs. In addition to the therapeutic effects mediated by
this low dosage of broxbam, which prob-ably does not induce
pronounced side effects, it also exhibited pronounced
antineoplastic effects. This renders broxbam to be the experimental
compound for alternative approaches for clinical liver cancer
treatment.
The combination of HDACis and sorafenib has
previously been demonstrated to exert enhanced anticancer activity
and may therefore be an advantageous approach compared with
sorafenib monotherapy (51,76,77). Freese et al previously
reported that HDACi sensitised HCC cells to sorafenib treatment
(78), which may be due to the
overexpression of HDACs in HCCs. In a recent review, Garmpis et
al (51) highlighted the
potential suitability of HDACis as anticancer agents and their
application in combinatorial therapies for the enhanced treatment
of HCC. Although the preclinical data of HDACi-based combination
therapies are promising, these authors also reported results from a
critical phase 1 clinical trial in which sorafenib was tested in
combination with vorinostat in patients with liver cancer. Although
some patients showed good tolerability and stable disease, this
treatment also led to adverse toxicities in the majority of
patients, which necessitated dose modifications and prevented
progression towards phase 2 clinical trial (79). In the present study, results from
the preclinical in vitro and in vivo studies suggest
the suitability and tolerability of this chimeric
hydroxamate-trimethoxystilbene conjugate for liver cancer
treatment. The fact that signs of toxicity were observed in neither
zebrafish embryos nor in the CAM, couple with the findings that the
chicken embryos developed and survived, suggest that broxbam may
exert fewer toxic effects compared with other HDACis, including
vorinostat. Future studies involving the effects of combination
therapies with broxbam and sorafenib should clarify this important
issue. In addition, to address the suitability of broxbam for
personalised therapeutic approaches, investigation of
patient-derived primary liver cancer cells should be executed.
To conclude, the present study evaluated the
recently-introduced chimeric inhibitor broxbam (20) for its potential anticancer
activity in liver cancer cells and the underlying molecular
mechanism. Additionally, its possible antitumoral and
antiangiogenic effects were examined in in vivo settings by
performing in chick embryo and zebrafish experiments. On the basis
of the presented data, broxbam is implicated to be a promising
chimeric HDACi that should be evaluated further as a potential
compound for liver cancer treatment, either as a monotherapy or in
combination with other clinically-relevant HCC therapeutics, such
as sorafenib.
Supplementary Data
Availability of data and materials
The datasets used and/or analysed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
SIB: Investigation, zebrafish angiogenesis assay
and data analysis. AD: Investigation, crystal violet assay,
real-time inhibition of cell proliferation, lactate dehydrogenase
(LDH) cytotoxicity assay, western blotting, reverse
transcription-quantitative PCR (RT-qPCR), apoptosis detection, HDAC
activity assay, immunofluorescence assay and scratch wound healing
assay. GTA: Investigation, small-interfering (si)RNA assay, western
blotting and RT-qPCR. MF: Western blotting and RT-qPCR. AK:
Investigation, crystal violet staining and LDH-Assay. LK:
Investigation, crystal violet staining and LDH-Assay. BB:
Conceptualisation, proof reading, validation. RS:
Conceptualisation, validation and language proof reading. BN:
Conceptualisation and CAM Assay. MH: Conceptualisation and
validation and proof reading. All authors have read and agreed to
the published version of the manuscript. SIB, BN, MF and AK confirm
the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was funded by the Else
Kröner-Fresenius-Stiftung Grant No. 2016 A47 (to AD).
Abbreviations:
|
CA-4
|
combretastatin A-4
|
|
CAM
|
chorioallantoic membrane
|
|
CCC
|
cholangiocellular carcinoma
|
|
GLUT2
|
glucose transporter 2
|
|
HCC
|
hepatocellular carcinoma
|
|
HDAC
|
histone deacetylase
|
|
HDACi
|
histone deacetylase inhibitor
|
|
IC50
|
mean inhibitory concentration
|
|
LDH
|
lactate dehydrogenase
|
|
MMP
|
mitochondrial membrane potential
|
|
SIV
|
subintestinal veins
|
References
|
1
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2021. CA Cancer J Clin. 71:7–33. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mosconi S, Beretta GD, Labianca R, Zampino
MG, Gatta G and Heinemann V: Cholangiocarcinoma. Crit Rev Oncol
Hematol. 69:259–270. 2009. View Article : Google Scholar
|
|
3
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vogel A, Cervantes A, Chau I, Daniele B,
Llovet JM, Meyer T, Nault JC, Neumann U, Ricke J, Sangro B, et al:
Hepatocellular carcinoma: ESMO clinical practice guidelines for
diagnosis, treatment and follow-up. Ann Oncol. 29(Suppl 4):
iv238–iv255. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lim H, Ramjeesingh R, Liu D, Tam VC, Knox
JJ, Card PB and Meyers BM: Optimizing survival and the changing
landscape of targeted therapy for intermediate and advanced
hepatocellular carcinoma: A systematic review. J Natl Cancer Inst.
113:123–136. 2021. View Article : Google Scholar :
|
|
6
|
Li Y, Gao ZH and Qu XJ: The adverse
effects of sorafenib in patients with advanced cancers. Basic Clin
Pharmacol Toxicol. 116:216–221. 2015. View Article : Google Scholar
|
|
7
|
Tang W, Chen Z, Zhang W, Cheng Y, Zhang B,
Wu F, Wang Q, Wang S, Rong D, Reiter FP, et al: The mechanisms of
sorafenib resistance in hepatocellular carcinoma: Theoretical basis
and therapeutic aspects. Sig Transduct Target Ther. 5:872020.
View Article : Google Scholar
|
|
8
|
Doherty B, Nambudiri VE and Palmer WC:
Update on the diagnosis and treatment of cholangiocarcinoma. Curr
Gastroenterol Rep. 19:22017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Morise Z, Sugioka A, Tokoro T, Tanahashi
Y, Okabe Y, Kagawa T and Takeura C: Surgery and chemotherapy for
intrahepatic cholangiocarcinoma. World J Hepatol. 2:58–64. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Goehringer N, Biersack B, Peng Y, Schobert
R, Herling M, Ma A, Nitzsche B and Höpfner M: Anticancer activity
and mechanisms of action of new chimeric EGFR/HDAC-inhibitors. Int
J Mol Sci. 22:84322021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Biersack B, Polat S and Höpfner M:
Anticancer properties of chimeric HDAC and kinase inhibitors. Semin
Cancer Biol. Nov 12–2020.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park SY and Kim JS: A short guide to
histone deacetylases including recent progress on class II enzymes.
Exp Mol Med. 52:204–212. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kaletsch A, Pinkerneil M, Hoffmann MJ,
Jaguva Vasudevan AA, Wang C, Hansen FK, Wiek C, Hanenberg H,
Gertzen C, Gohlke H, et al: Effects of novel HDAC inhibitors on
urothelial carcinoma cells. Clin Epigenetics. 10:1002018.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gong D, Zeng Z, Yi F and Wu J: Inhibition
of histone deacetylase 11 promotes human liver cancer cell
apoptosis. Am J Transl Res. 11:983–990. 2019.PubMed/NCBI
|
|
15
|
Steinemann G, Dittmer A, Kuzyniak W,
Hoffmann B, Schrader M, Schobert R, Biersack B, Nitzsche B and
Höpfner M: Animacroxam, a novel dual-mode compound targeting
histone deacetylases and cytoskeletal integrity of testicular germ
cell cancer cells. Mol Cancer Ther. 16:2364–2374. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Manal M, Chandrasekar MJN, Gomathi Priya J
and Nanjan MJ: Inhibitors of histone deacetylase as antitumor
agents: A critical review. Bioorg Chem. 67:18–42. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Juan LJ, Shia WJ, Chen MH, Yang WM, Seto
E, Lin YS and Wu CW: Histone deacetylases specifically
down-regulate p53-dependent gene activation. J Biol Chem.
275:20436–20443. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mann BS, Johnson JR, Cohen MH, Justice R
and Pazdur R: FDA approval summary: Vorinostat for treatment of
advanced primary cutaneous T-cell lymphoma. Oncologist.
12:1247–1252. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Streubel G, Schrepfer S, Kallus H,
Parnitzke U, Wulff T, Hermann F, Borgmann M and Hamm S: Histone
deacetylase inhibitor resminostat in combination with sorafenib
counteracts platelet-mediated protumoral effects in hepatocellular
carcinoma. Sci Rep. 11:95872021. View Article : Google Scholar
|
|
20
|
Schmitt F, Gosch LC, Dittmer A, Rothemund
M, Mueller T, Schobert R, Biersack B, Volkamer A and Höpfner M:
Oxazole-bridged combretastatin A-4 derivatives with tethered
hydroxamic acids: Structure-activity relations of new inhibitors of
HDAC and/or tubulin function. Int J Mol Sci. 20:3832019. View Article : Google Scholar
|
|
21
|
Méndez-Callejas GM, Leone S, Tanzarella C
and Antoccia A: Combretastatin A-4 induces p53
mitochondrial-relocalisation independent-apoptosis in non-small
lung cancer cells. Cell Biol Int. 38:296–308. 2014. View Article : Google Scholar
|
|
22
|
Kim JH, Yoon EK, Chung HJ, Park SY, Hong
KM, Lee CH, Lee YS, Choi K, Yang Y, Kim K and Kim IH: p53
acetylation enhances Taxol-induced apoptosis in human cancer cells.
Apoptosis. 18:110–120. 2013. View Article : Google Scholar
|
|
23
|
Tron GC, Pirali T, Sorba G, Pagliai F,
Busacca S and Genazzani AA: Medicinal chemistry of combretastatin
A4: present and future directions. J Med Chem. 49:3033–3044. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hesham HM, Lasheen DS and Abouzid KAM:
Chimeric HDAC inhibitors: Comprehensive review on the HDAC-based
strategies developed to combat cancer. Med Res Rev. 38:2058–2109.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Griggs J, Metcalfe JC and Hesketh R:
Targeting tumour vasculature: The development of combretastatin A4.
Lancet Oncol. 2:82–87. 2001. View Article : Google Scholar
|
|
26
|
Gaspari R, Prota AE, Bargsten K, Cavalli A
and Steinmetz MO: Structural basis of cis-and trans-combretastatin
binding to tubulin. Chem. 2:102–113. 2017. View Article : Google Scholar
|
|
27
|
Schaller E, Ma A, Gosch LC, Klefenz A,
Schaller D, Goehringer N, Kaps L, Schuppan D, Volkamer A, Schobert
R, et al: New 3-Aryl-2-(2-thienyl)acrylonitriles with high activity
against hepatoma cells. Int J Mol Sci. 22:22432021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nwosu ZC, Battello N, Rothley M, Piorońska
W, Sitek B, Ebert MP, Hofmann U, Sleeman J, Wölfl S, Meyer C, et
al: Liver cancer cell lines distinctly mimic the metabolic gene
expression pattern of the corresponding human tumours. J Exp Clin
Cancer Res. 37:2112018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kasai F, Hirayama N, Ozawa M, Satoh M and
Kohara A: HuH-7 reference genome profile: Complex karyotype
composed of massive loss of heterozygosity. Hum Cell. 31:261–267.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nakabayashi H, Taketa K, Miyano K, Yamane
T and Sato J: Growth of human hepatoma cells lines with
differentiated functions in chemically defined medium. Cancer Res.
42:3858–3863. 1982.PubMed/NCBI
|
|
31
|
Monteil M, Migianu-Griffoni E,
Sainte-Catherine O, Di Benedetto M and Lecouvey M: Bisphosphonate
prodrugs: Synthesis and biological evaluation in HuH7
hepatocarcinoma cells. Eur J Med Chem. 77:56–64. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xu L, Hausmann M, Dietmaier W, Kellermeier
S, Pesch T, Stieber-Gunckel M, Lippert E, Klebl F and Rogler G:
Expression of growth factor receptors and targeting of EGFR in
cholangio-carcinoma cell lines. BMC Cancer. 10:3022010. View Article : Google Scholar
|
|
33
|
Jo JR, An S, Ghosh S, Nedumaran B and Kim
YD: Growth hormone promotes hepatic gluconeogenesis by enhancing
BTG2-YY1 signaling pathway. Sci Rep. 11:189992021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kuete V, Sandjo LP, Ouete JLN, Fouotsa H,
Wiench B and Efferth T: Cytotoxicity and modes of action of three
naturally occurring xanthones (8-hydroxycudraxanthone G, morusignin
I and cudraxanthone I) against sensitive and multidrug-resistant
cancer cell lines. Phytomedicine. 21:315–322. 2014. View Article : Google Scholar
|
|
35
|
Gillies RJ, Didier N and Denton M:
Determination of cell number in monolayer cultures. Anal Biochem.
159:109–113. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Korzeniewski C and Callewaert DM: An
enzyme-release assay for natural cytotoxicity. J Immunol Methods.
64:313–320. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nitzsche B, Gloesenkamp C, Schrader M,
Ocker M, Preissner R, Lein M, Zakrzewicz A, Hoffmann B and Höpfner
M: Novel compounds with antiangiogenic and antiproliferative
potency for growth control of testicular germ cell tumours. Br J
Cancer. 103:18–28. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sivandzade F, Bhalerao A and Cucullo L:
Analysis of the mito-chondrial membrane potential using the
cationic JC-1 dye as a sensitive fluorescent probe. Bio Protoc.
9:e31282019. View Article : Google Scholar
|
|
39
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
40
|
Bobadilla AV, Arévalo J, Sarró E, Byrne
HM, Maini PK, Carraro T, Balocco S, Meseguer A and Alarcón T: In
vitro cell migration quantification method for scratch assays. J R
Soc Interface. 16:201807092019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Almeida VM, Bezerra MA Jr, Nascimento JC
and Amorim LMF: Anticancer drug screening: Standardization of in
vitro wound healing assay. J Bras Patol Med Lab. 55:606–619. 2019.
View Article : Google Scholar
|
|
42
|
Serbedzija GN, Flynn E and Willett CE:
Zebrafish angiogenesis: A new model for drug screening.
Angiogenesis. 3:353–359. 1999. View Article : Google Scholar
|
|
43
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Thorens B: GLUT2, glucose sensing and
glucose homeostasis. Diabetologia. 58:221–232. 2015. View Article : Google Scholar
|
|
45
|
Kaufmann SH and Earnshaw WC: Induction of
apoptosis by cancer chemotherapy. Exp Cell Res. 256:42–49. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Reers M, Smiley ST, Mottola-Hartshorn C,
Chen A, Lin M and Chen LB: Mitochondrial membrane potential
monitored by JC-1 dye. Methods Enzymol. 260:406–417. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kim R, Emi M and Tanabe K: Role of
mitochondria as the gardens of cell death. Cancer Chemother
Pharmacol. 57:545–553. 2006. View Article : Google Scholar
|
|
48
|
Fridman JS and Lowe SW: Control of
apoptosis by p53. Oncogene. 22:9030–9040. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kim R, Tanabe K, Uchida Y, Emi M, Inoue H
and Toge T: Current status of the molecular mechanisms of
anticancer drug-induced apoptosis. The contribution of
molecular-level analysis to cancer chemotherapy. Cancer Chemother
Pharmacol. 50:343–352. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Giaginis C, Damaskos C, Koutsounas I,
Zizi-Serbetzoglou A, Tsoukalas N, Patsouris E, Kouraklis G and
Theocharis S: Histone deacetylase (HDAC)-1, -2, -4 and -6
expression in human pancreatic adenocarcinoma: Associations with
clinicopathological parameters, tumor proliferative capacity and
patients' survival. BMC Gastroenterol. 15:1482015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Garmpis N, Damaskos C, Garmpi A,
Georgakopoulou VE, Sarantis P, Antoniou EA, Karamouzis MV, Nonni A,
Schizas D, Diamantis E, et al: Histone deacetylase inhibitors in
the treatment of hepatocellular carcinoma: Current evidence and
future opportunities. J Pers Med. 11:2232021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Scholzen T and Gerdes J: The Ki-67
protein: From the known and the unknown. J Cell Physiol.
182:311–322. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kim HR, Rahman FU, Kim KS, Kim EK, Cho SM,
Lee K, Moon OS, Seo YW, Yoon WK, Won YS, et al: Critical roles of
E2F3 in growth and musculo-skeletal phenotype in mice. Int J Med
Sci. 16:1557–1563. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hubbert C, Guardiola A, Shao R, Kawaguchi
Y, Ito A, Nixon A, Yoshida M, Wang XF and Yao TP: HDAC6 is a
microtubule-associated deacetylase. Nature. 417:455–458. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Garcin C and Straube A: Microtubules in
cell migration. Essays Biochem. 63:509–520. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Schaks M, Giannone G and Rottner K: Actin
dynamics in cell migration. Essays Biochem. 63:483–495. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Norouzzadeh M, Kalikias Y, Mohammadpour Z,
Sharifi L and Mahmoudi M: Determining population doubling time and
the appropriate number of HepG2 cells for culturing in 6-well
plate. IJSBAR. 10:299–303. 2016.
|
|
58
|
Deng B, Luo Q, Halim A, Liu Q, Zhang B and
Song G: The antiangiogenesis role of histone deacetylase
inhibitors: Their potential application to tumor therapy and tissue
repair. DNA Cell Biol. 39:167–176. 2020. View Article : Google Scholar
|
|
59
|
Tomaszewska B, Muzolf M, Grabysa R and
Bodnar L: Cardiotoxicity of antiangiogenic drugs: Causes and
mechanisms. OncoReview. 11:12–18. 2021. View Article : Google Scholar
|
|
60
|
Oda K, Arakawa H, Tanaka T, Matsuda K,
Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y and
Taya Y: p53AIP1, a potential mediator of p53-dependent apoptosis,
and its regulation by Ser-46-phosphorylated p53. Cell. 102:849–862.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Matsuda K, Yoshida K, Taya Y, Nakamura K,
Nakamura Y and Arakawa H: p53AIP1 regulates the mitochondrial
apoptotic pathway. Cancer Res. 62:2883–2889. 2002.PubMed/NCBI
|
|
62
|
Porter AG and Jänicke RU: Emerging roles
of caspase-3 in apoptosis. Cell Death Differ. 6:99–104. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lee YR and Park SY: P53 expression in
hepatocellular carcinoma: Influence on the radiotherapeutic
response of the hepatocellular carcinoma. Clin Mol Hepatol.
21:230–231. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Li T, Zhang C, Hassan S, Liu X, Song F,
Chen K, Zhang W and Yang J: Histone deacetylase 6 in cancer. J
Hematol Oncol. 11:1112018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Aldana-Masangkay GI and Sakamoto KM: The
role of HDAC6 in cancer. J Biomed Biotechnol. 2011:8758242011.
View Article : Google Scholar
|
|
66
|
Gao Y, Hubbert CC, Lu J, Lee YS, Lee JY
and Yao TP: Histone deacetylase 6 regulates growth factor-induced
actin remodeling and endocytosis. Mol Cell Biol. 27:8637–8647.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Tran AD-A, Marmo TP, Salam AA, Che S,
Finkelstein E, Kabarriti R, Xenias HS, Mazitschek R, Hubbert C,
Kawaguchi Y, et al: HDAC6 deacetylation of tubulin modulates
dynamics of cellular adhesions. J Cell Sci. 120:1469–1479. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Enomoto T: Microtubule disruption induces
the formation of actin stress fibers and focal adhesions in
cultured cells: Possible involvement of the rho signal cascade.
Cell Struct Funct. 21:317–326. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Liu BP, Chrzanowska-Wodnicka M and
Burridge K: Microtubule depolymerization induces stress fibers,
focal adhesions, and DNA synthesis via the GTP-binding protein Rho.
Cell Adhes Commun. 5:249–255. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zhang C, Willett C and Fremgen T:
Zebrafish: An animal model for toxicological studies. Curr Protoc
Toxicol. 17:1.7.1–1.7. 2003.
|
|
71
|
Deroanne CF, Bonjean K, Servotte S, Devy
L, Colige A, Clausse N, Blacher S, Verdin E, Foidart JM, Nusgens BV
and Castronovo V: Histone deacetylases inhibitors as
anti-angiogenic agents altering vascular endothelial growth factor
signaling. Oncogene. 21:427–436. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Su M, Huang J, Liu S, Xiao Y, Qin X, Liu
J, Pi C, Luo T, Li J, Chen X and Luo Z: The anti-angiogenic effect
and novel mechanisms of action of Combretastatin A-4. Sci Rep.
6:281392016. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zhou J, Cheng M, Wu M, Boriboun C, Jujo K,
Xu S, Zhao TC, Tang YL, Kishore R and Qin G: Contrasting roles of
E2F2 and E2F3 in endothelial cell growth and ischemic angiogenesis.
J Mol Cell Cardiol. 60:68–71. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Heo YA and Syed YY: Regorafenib: A review
in hepatocellular carcinoma. Drugs. 78:951–958. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Labeur TA, Hofsink Q, Takkenberg RB, van
Delden OM, Mathôt RAA, Schinner R, Malfertheiner P, Amthauer H,
Schütte K, Basu B, et al: The value of sorafenib trough levels in
patients with advanced hepatocellular carcinoma-a substudy of the
SORAMIC trial. Acta Oncol. 59:1028–1035. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Yuan H, Li AJ, Ma SL, Cui LJ, Wu B, Yin L
and Wu MC: Inhibition of autophagy significantly enhances
combination therapy with sorafenib and HDAC inhibitors for human
hepa-toma cells. World J Gastroenterol. 20:4953–4962. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Bitzer M, Horger M, Giannini EG, Ganten
TM, Wörns MA, Siveke JT, Dollinger MM, Gerken G, Scheulen ME, Wege
H, et al: Resminostat plus sorafenib as second-line therapy of
advanced hepatocellular carcinoma-The SHELTER study. J Hepatol.
65:280–288. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Freese K, Seitz T, Dietrich P, Lee SML,
Thasler WE, Bosserhoff A and Hellerbrand C: Histone deacetylase
expressions in hepato-cellular carcinoma and functional effects of
histone deacetylase inhibitors on liver cancer cells in vitro.
Cancers (Basel). 11:15872019. View Article : Google Scholar
|
|
79
|
Gordon SW, McGuire WP III, Shafer DA,
Sterling RK, Lee HM, Matherly SC, Roberts JD, Bose P, Tombes MB,
Shrader EE, et al: Phase I study of sorafenib and vorinostat in
advanced hepatocellular carcinoma. Am J Clin Oncol. 42:649–654.
2019. View Article : Google Scholar : PubMed/NCBI
|