Introduction
According to a status report on the global burden of
cancer, lung cancer is the most commonly diagnosed cancer,
representing 11.6% of the total cases, and the leading cause of
cancer-related death (1). With
recent advances in the field of cancer immunotherapy, various
highly effective therapies, particularly immune checkpoint
inhibitors (ICIs) have been developed to enhance anti-tumor
immunity, which have demonstrated a survival benefit for a subset
of patients with lung cancer (2,3).
However, despite observable clinical improvement, the majority of
patients fail to respond to ICI therapy due to drug resistance,
resulting both from tumor cell-intrinsic mechanisms and other
factors relating to the tumor microenvironment (TME) (4,5).
Thus, new immunotherapeutic approaches are urgently needed for
patients with lung cancer.
Toll-like receptor (TLR) agonists are immune
adjuvants used in antiviral and anti-bacterial treatment that can
promote T helper (Th) 1 immune response (6,7).
Among the 13 known TLRs, TLR7 and TLR8, are mainly expressed on the
endosomal membranes (8) of cells
of the innate immune system, including dendritic cells (DCs),
macrophages and natural killer (NK) cells (9). Resiquimod (R848), an antiviral
imidazoquinoline derivative, stimulates immune cells through TLR7
and/or TLR8 in a MyD88-dependent manner. Nevertheless, R848
selectively acts on TLR7 in mice (10,11) and has been demonstrated to induce
Th1 immune responses following activation of antigen-presenting
cells, leading to the production of numerous pro-inflammatory
cytokines including IL-12 and IFN-γ. R848 exhibits a robust
antitumor effect in several tumor models, such as dermatological
malignancies, cutaneous T-cell lymphoma and breast cancer (12-14). Moreover, recent studies focusing
on macrophages have highlighted the therapeutic efficacy of R848,
either alone or in combination with other drugs, in lung cancer
(15,16). However, the mechanism of R848's
efficacy in lung cancer is not completely clear.
In the present study, it was initially confirmed
that R848 targets TLR7 expression in the immune cells of lung
tumor-bearing mice and the effects of the aforementioned compound
in subcutaneous (s.c.) and metastatic models of lung cancer were
evaluated. The volumes of the s.c. solid tumors, the survival of
tumor-bearing mice and tumor metastasis were examined. Furthermore,
the ability of R848 to stimulate host immunity in tumor-bearing
mice was assessed. It was also determined whether R848 would alter
the composition of immune cells in the TME. The present findings
may provide insight into the future use of R848-based immunotherapy
for lung cancer.
Materials and methods
Mice and cell lines
A total of 151, male, 6-8 weeks-old C57BL/6 mice
(~20 g) were purchased from Beijing Tengxin Company and housed in
individually ventilated cages. OT1 mice (stock no. 003831) and TLR7
knock-out mice (stock no. 008380) were purchased from the Jackson
Laboratory Company and kept in a specific-pathogen-free animal
facility (temperature, 22±2 degrees; 12-h light cycle; air changed
16 times/h; humidity, 55±10%) with free access to water and food.
Mice were anesthetized by intraperitoneal (i.p.) injection with 1%
pentobarbital sodium (50 mg/kg), then sacrificed by cervical
dislocation. Death was confirmed by cessation of the heartbeat and
breathing. The animal study was approved (approval no. 2022-152) by
the ethics committee of The First Affiliated Hospital of Chongqing
Medical University (Chongqing, China). The Lewis lung carcinoma
(LLC) cell line was provided by Professor Haixia Long (Department
of Oncology, Xinqiao Hospital, The Army Medical University), and
maintained in Dulbecco's modified Eagle's medium (DMEM)
supplemented with 10% FBS and 1% penicillin-streptomycin (all from
Gibco; Thermo Fisher Scientific, Inc.) in a humidified incubator at
37°C with 5% CO2/95% air. The B16 F10 cell line was
provided by Professor Haixia Long and was cultured in RPMI-1640
(Hyclone; Cytiva) supplemented with 10% FBS and 1%
penicillin-streptomycin at 37°C.
Reagents and antibodies
For in vivo administration, R848
(MedChemExpress) was dissolved in DMSO and frozen in −80°C.
Live/Dead viability kits (cat. no. L34958) and anti-TLR7 antibodies
(cat. no. MA5-16249) were purchased from Invitrogen; Thermo Fisher
Scientific, Inc. and diluted at 1:200 for use. For flow cytometry,
antibodies specific for CD45 (cat. no. 103108), CD3 (cat. no.
100218), CD8 (cat. no. 100714), CD4 (cat. no. 100422), CD11c (cat.
no. 100422), CD11b (cat. no. 101230), CD49b (cat. no. 108910), CD19
(cat. no. 115520), Ly6G (cat. no. 127607), Ly6C (cat. no. 128016),
CD80 (cat. no. 104705), CD86 (cat. no. 105014), MHC-II (cat. no.
116407), PD-1(cat. no. 135224), FoxP3 (cat. no. 126404), F4/80
(cat. no. 123116) and IFN-γ (cat. no. 505826) were purchased from
Biolegend, Inc. and used for staining following the manufacturer's
protocol. Fixation/Permeabilization kit (with BD Golgistop™ (cat.
no. 554715; BD Biosciences) was used for intracellular staining.
The Foxp3/Transcription Factor Staining Buffer Set (cat. no.
00-5523; Invitrogen; Thermo Fisher Scientific, Inc.) was used for
intranuclear Foxp3 staining. The anti-asialo GM1 rabbit polyclonal
antibody (cat. no. 986-10001) was purchased from FUJIFILM Wako Pure
Chemical Corporation. Anti-mouse CD8 (clone 2.43; cat. no. BE0061)
and anti-mouse CD4 (clone YTS191; cat. no. BE0119) antibodies were
purchased from Bio X Cell and used following the indicated
concentrations for cells depletion.
Cell culture
Primary bone marrow-derived DCs isolated from the
femur and tibia of C57BL/6 mice were cultured with 10 ng/ml IL-4
and GM-CSF (Novoprotein) at 37°C for 4 days. Immature DCs were then
collected and purified with Histodenz (Sigma-Aldrich; Merck KGaA).
DC suspensions with a purity >85% with CD11C and MHC-II
double-staining were used for subsequent assays. NK cells were
positively selected from the spleen (SPL) of C57BL/6 mice using the
Mouse CD49b Positive Selection kit (cat. no. 18755, Stemcell
Technologies, Inc.). CD8+ T cells were negatively
isolated from SPL and lymph nodes (LN) from OT-1 mice according to
the manufacturer's protocol (cat. no. 19835A; Stemcell
Technologies, Inc.). The isolated CD8+ T cells
pre-labeled with 5,6-carboxyfluorescein diacetate succinimidyl
ester (eBioscience; Thermo Fisher Scientific, Inc.) were added
after DCs co-cultured with NK cells in the presence of R848 for 24
h. Following 3 days of co-culture, IFN-γ+
CD8+ T and CD8+ T proliferation was analyzed
by flow cytometry.
Proliferation and apoptosis assay
Proliferation was measured using the Cell Titer
96® AQueous One Solution Cell Proliferation Assay
(Promega Corporation). 1×106 LLC cells were seeded in a
100-µl volume in a 96-well plate and stimulated with R848 at
1, 5 or 10 µg/ml for 24 or 48 h. Each well was plated in
triplicate. A 20-µl volume of Cell Titer 96®
AQueous One Solution Reagent was then added into each well. The
plate was then incubated at 37°C for 1-4 h in a humidified 5%
CO2 atmosphere. The absorbance at 490 nm was then
measured using a 96-well plate reader.
To measure apoptosis, LLC cells were stimulated
under the same conditions, then stained using a PE Annexin V
Apoptosis Detection kit (cat. no. 559763; BD Biosciences) for 20
min at room temperature (RT) in the dark according to the
manufacturer's protocol.
Preparation of single-cell suspensions
and flow cytometry
Following sacrifice, tumors, SPL, LN and femur of
tumor-bearing mice were isolated. Tumors were immersed in DMEM on
ice, then minced and enzymatically digested in DMEM containing 1.0
mg/ml collagenase IV and 50 U/ml DNAse I for 1 h at 37°C. The
digested tumors were then ground, and the cells were filtered
through 70- and 40-µM cell strainers. The other samples,
such as SPL and LN, were collected in cold PBS with 2% FBS, gently
ground, then filtered through 70-µM cell strainers. The
cells were then centrifuged at 500 × g for 5 min at 4°C, then
resuspended and immediately stained for flow cytometry at 4°C for
30 min.
For surface staining, the cells were firstly blocked
with CD16/32 antibody (1:200; cat. no. 101320; Biolegend, Inc.) for
20 min at 4°C, then stained with live/dead kits and streaming
antibodies for 20 min. For intracellular staining, 1×106
cultured cells were added to GolgiStop™ buffer 4-6 h before
staining. Intracellular Foxp3 was conducted using a
Foxp3/Transcription Factor Staining Buffer Set according to the
manufacturer's protocol. Lastly, the samples were tested using a
Beckman Gallios flow cytometer (Beckman Coulter, Inc.) and analyzed
with FlowJo version 7.6.1 (FlowJo LLC).
Systemic immune activation
In vivo, 3 mg/kg R848 (17) was intravenously injected into mice
with a s.c. tumor on day 7; serum, LN and SPL were collected at 0,
6, 24 and 48 h. TLR7 expression on DCs, macrophages,
myeloid-derived suppressor cells (MDSCs), NK cells, CD4+
T and CD8+ T cells, CD80 and CD86 expression on DCs, and
CD69 expression on NK cells, CD4+ T cells and
CD8+ T cells were analyzed by flow cytometry. Levels of
cytokines in the serum of R848-treated mice at 0, 6, 24 and 48 h
were determined using a Multiple mouse cytokine detection kit (cat.
no. B299045; Biolegend, Inc.) following the manufacturer's
protocol.
S.c. lung cancer model and treatment
A total of 1×106 LLC cells in 100
µl PBS were injected subcutaneously into the right flank of
C57BL/6 mice. At the same day following inoculation, the mice were
injected i.p. with 20 µg R848 every other day till endpoint.
The control group received 100 µl PBS. After the formation
of palpable tumors, tumor growth was evaluated by measuring the
length and width using electronic calipers every 3 days. The
results are expressed as volumes, which were calculated according
to the following formula: 0.52 × (length) × (width)2.
Immune cells in the TME were detected after continuous R848
treatment for 10 times.
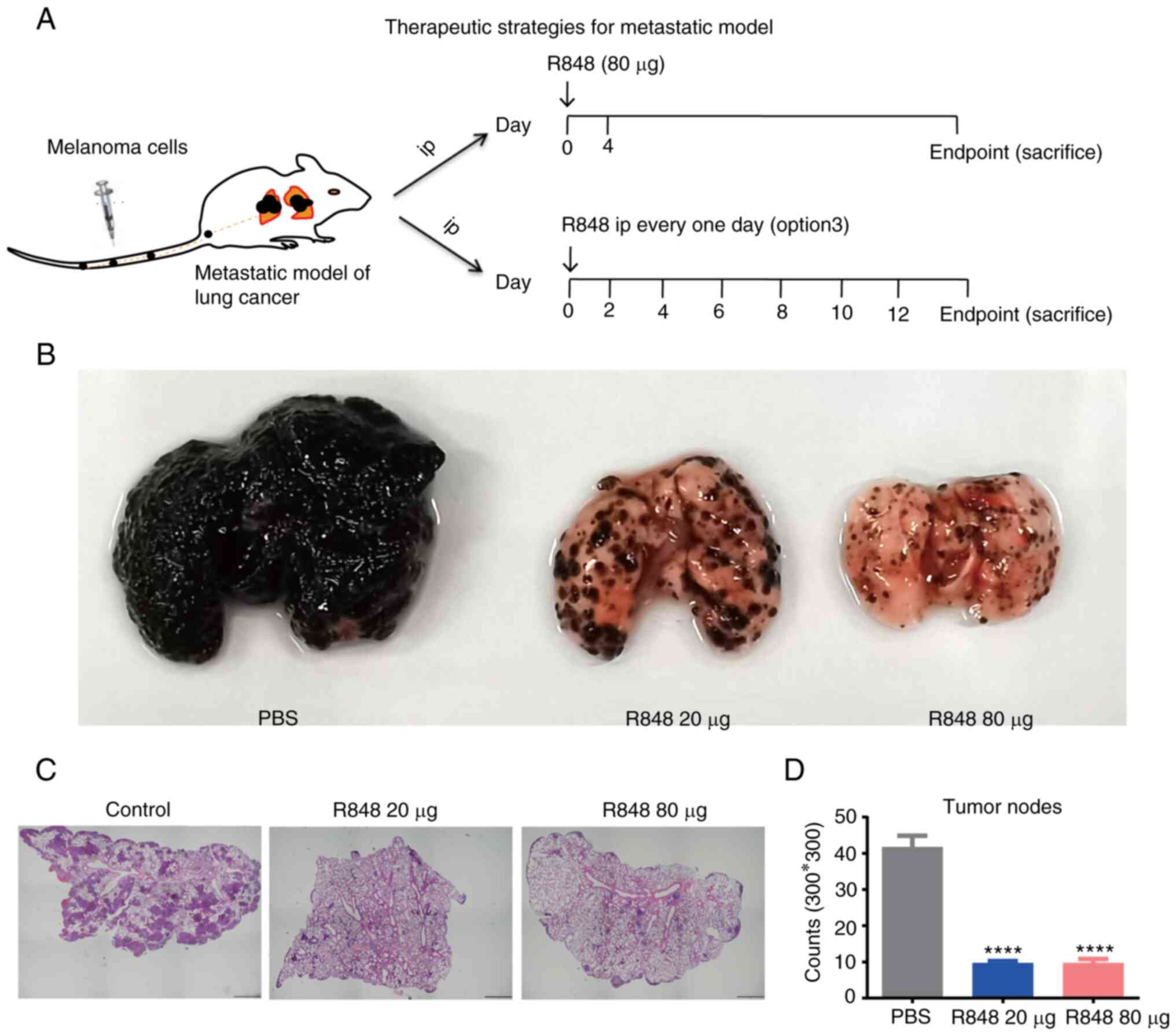
Metastatic lung cancer model
A total of 1×106 B16/F10 melanoma cells
were cultured in complete RPMI-1640 medium, then injected
intravenously into C57BL/6 mice. On day 0 after inoculation,
treatment with 20 µg R848 i.p. was initiated and continued
every other day till endpoint. In addition, 80 µg R848 in
100 µl PBS therapy was started on day 0 and injected twice
with a 4-day interval between the two injections. The numbers of
metastatic tumor nodules in the lung were recorded from day 14. The
left lung with metastatic tumor nodules was fixed in 4%
paraformaldehyde at RT for 24 h, embedded, then sectioned at the
maximum cross-section for further analysis. The 5 mm-thick slides
were stained with hematoxylin and eosin. The tumor nodules were
examined and enumerated under a fluorescence microscope. A total of
1×106 LLC cells in 100 µl PBS were injected
intravenously into C57BL/6 mice. On day 21 after inoculation, tumor
nodes in the lung were extracted for TLR7 gene quantification.
Depletion experiment
Anti-asialo GM1 rabbit polyclonal antibody (50
µl in 100 µl PBS) was administered by i.p. injection
at day -1 every 3 days till the endpoint for NK cell depletion.
Anti-CD4 and anti-CD8 in vivo depletion antibodies were used
at 250 µg/injection, injected daily for the first 2 days,
then twice-weekly until the end of the experiment as previously
described (18). It was confirmed
that >90% CD8+ T, NK and CD4+ T cells were
depleted.
RNA extraction, cDNA synthesis and
reverse transcriptionquantitative (RT-q) PCR
Freshly isolated tumors were immediately frozen and
ground to powder. Total RNA from LLC powder was isolated using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. The RNA was
reverse-transcribed to cDNA using a Reverse Transcription kit
(Promega Corporation) following the manufacturer's protocol. The
following primer sequences were used: mouse TLR7 forward, 5′-GGT
CCA AAG CCA ATG TGT GT-3′ and reverse, 5′-GGA TGG CAG ATC CTG TGG
TA-3′; and β-actin forward, 5′-CTA GGC CAC AGA ATT GAA AGA TCT-3′
and reverse, 5′-GTA GGT GGA AAT TCT AGC ATC ATC C-3′. qPCR was
performed on a Bio-Rad Fast RT-PCR system using GoTaq®
Green Master Mix (Promega Corporation). The qPCR conditions were as
follows: Initial denaturation at 95°C for 15 sec; 40 cycles of 95°C
for 10 sec, 60°C annealing for 20 sec and extension at 72°C for 20
sec. All samples were set up in triplicate. The raw cycle threshold
(Ct) values obtained for β-actin mRNA were deducted from those of
TLR7 to obtain the ΔCt values. Analysis of relative gene expression
data was performed using qPCR and the 2−ΔΔCq method
(19).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism software version 6 (GraphPad Software, Inc.). Comparisons
between groups were carried out using unpaired Student's-test.
Tumor growth speed was evaluated with Linear regression test.
Survival analysis was performed by log-rank test. For all
comparisons, P<0.05 was considered to indicate a statistically
significant difference.
Results
TLR7 is predominantly expressed in LLC
s.c. tumorinfiltrating lymphocytes (TiLs)
R848 selectively targets TLR7 in mice (20). Therefore, prior to the initiation
of R848 treatment for murine lung cancer, TLR7 expression was
examined in mice bearing lung tumors. qPCR results suggested that
TLR7 mRNA was highly expressed both in LLC metastatic and s.c.
tumors, compared with LLC cells (Fig.
1A). Moreover, TLR7 was expressed at high levels on
CD45+ leukocytes in both LLC metastatic and s.c. tumors,
but not in CD45− cells (Fig. 1B and C). The stimulation of TLR7
mainly located on innate cells initiates adaptive immunity by TLR7
agonists, which lay the basis of R848 for its antiviral and
antitumor effect (8,21). Therefore, the dynamic change of
TLR7 on leukocytes was investigated in the TME, LN, SPL and bone
marrow (BM) from mice with LLC s.c. tumors. The proportion of
TLR7+ TiL increased from day 0 to day 22, decreased in
the later period from day 22 to day 30 of s.c. lung tumors
(Fig. 1D). However, there was no
significant difference in the frequency TLR7+ leukocytes
in the BM, LN and SPL (Fig. 1D).
Next, TLR7 distribution was compared among various immune cells
from SPL, LN and tumor tissue samples from LLC s.c. tumor-bearing
mice. The results demonstrated that TLR7 on various immune cells
was dynamically expressed. Indeed, TLR7 expression increased from
day 8 to 22, then declined during days 22-30 (Fig. 1E and F). However, similar to
TLR7+ TiLs, TLR7 expression on various immune cells was
stable from day 8 to day 30 in LN and SPL (Fig. 1G and H). The upregulation of
TLR7+ on immune cells could result from tumor antigen
stimulation in the TME. TLR7 expression levels on immune cells,
including macrophages, NK, CD4+ T and CD8+ T
cells were relatively stable in LN and SPL, with the exception of
LN DCs, where TLR7 expression increased gradually from day 22
(Fig. 1I), which we hypothesize
may be caused by tumor cell invasion in the LN. These findings on
the dynamic patterns of expression of TLR7 may help in the design
of R848 therapeutic regimens for lung cancer.
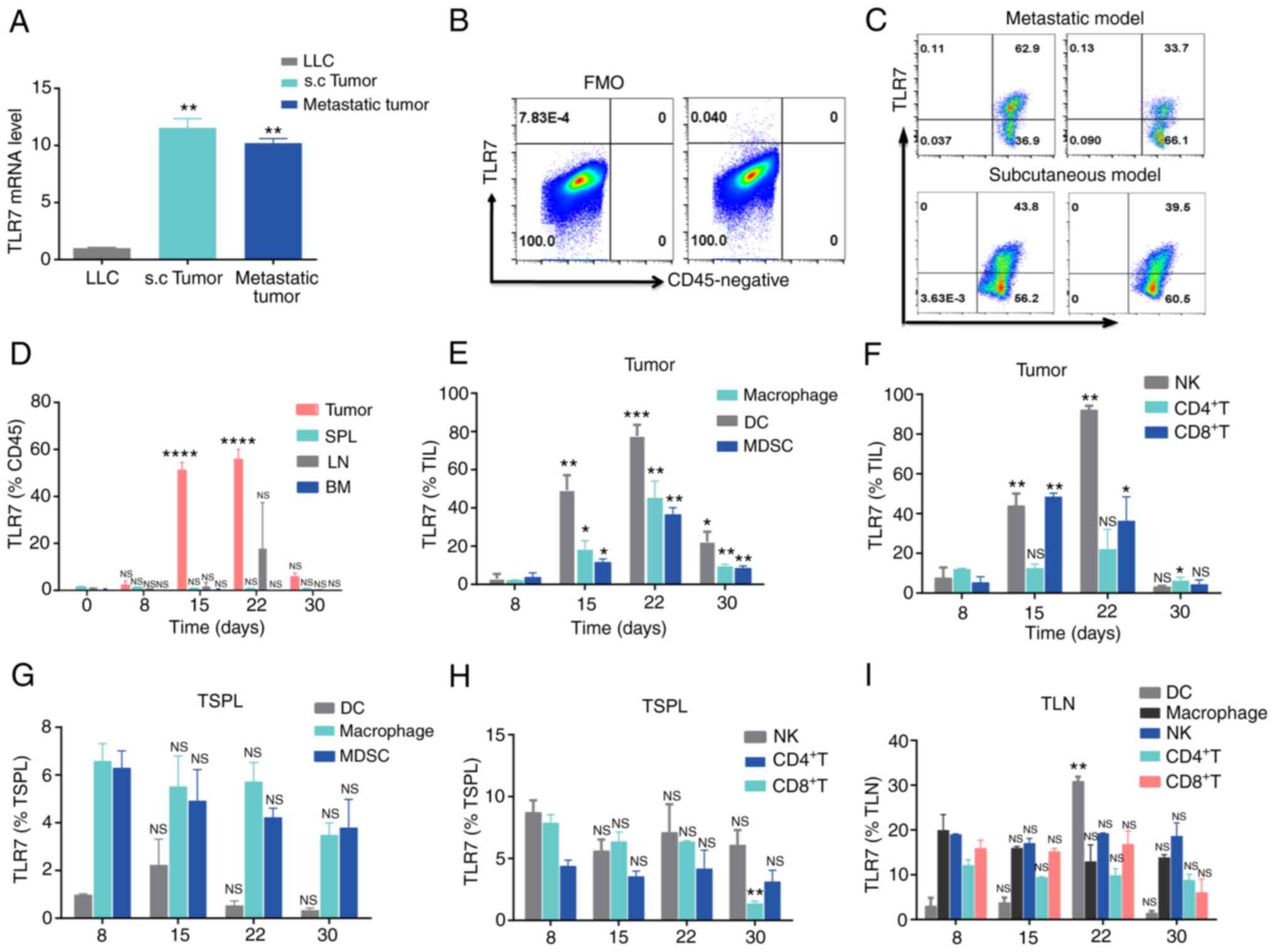 | Figure 1Dynamic TLR7 expression in immune
cells from LLC tumor-bearing mice. (A) TLR7 mRNA abundance in
parental LLC, s.c. and metastatic tumors in C57BL/6
mice.**P<0.01 compared with LLC. (B and C)
Representative flow cytometry plots of TLR7 expression in (B)
CD45− and (C) CD45+ cells from LLC s.c. and
metastatic tumors. (D) Histogram representing
TLR7+CD45+ cells in bone marrow, SPL, LN and
tumor samples at day 0, 8, 15, 22 and 30 in mice with LLC s.c.
tumors (n=3). (E and F) TLR7 expression on (E) DCs, macrophages,
MDSCs and (F) NK, CD4+ T and CD8+ T cells
from LLC s.c. tumors at day 8, 15, 22 and 30 (n=3). (G-I) Dynamic
changes in TLR7 expression on various immune cells from the SPL and
LN of LLC tumor-bearing mice (n=3). The data are presented as the
mean ± SEM. *P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001 compared
with day 0. TLR, Toll-like receptor; LLC, Lewis lung carcinoma;
s.c., subcutaneous; SPL, spleen; LN, lymph nodes; DCs, dendritic
cells; MDSCs, myeloid-derived suppressor cells; NK, natural killer;
NS, not significant. |
Administration of R848 initially targets
DCs and induces systemic immune activation
Previous studies have demonstrated that R848
promotes the maturation of DCs, including the upregulation of
co-stimulatory molecules and production of IL-12 and enhances
adaptive immunity (22,23). It was next sought to determine the
ability of R848 to enhance systemic immune activation in the LLC
s.c. tumor model. Notably, systemic administration of R848
significantly upregulated TLR7 in DCs, but not in macrophages,
MDSCs, NK, CD4+ T and CD8+ T cells from SPL
and LN (Fig. 2A). Compared with
CD8+ T, CD4+ T, NK and MDSCs, TLR7 expression
was relatively higher on macrophages, although no significant
difference was observed following R848 injection (Fig. 2A). R848 administration
significantly unregulated the co-stimulatory markers CD80 (Fig. 2B) and CD86 (Fig. 2C) of DCs in LN and SPL. TLR7
expression in MDSCs from LN was not evaluated, as very few of these
cells were detected. Although no significant changes in TLR7
expression were observed in NK and CD8+ T cells, these
cells displayed an activated phenotype (NK cells: CD69;
CD8+ T cells: CD69+) in LN and SPL (Fig. 2D). Increased CD69 expression on
CD4+ T cells was observed after R848 administration,
although this was not statistically significant. Furthermore,
increased serum levels of Th1-associated cytokines, including IL-2,
IFN-γ and TNF-α (24,25), were observed following R848
injection (Fig. 2E). These
findings suggested that R848 initially activates DCs and further
induces activation of the immune system.
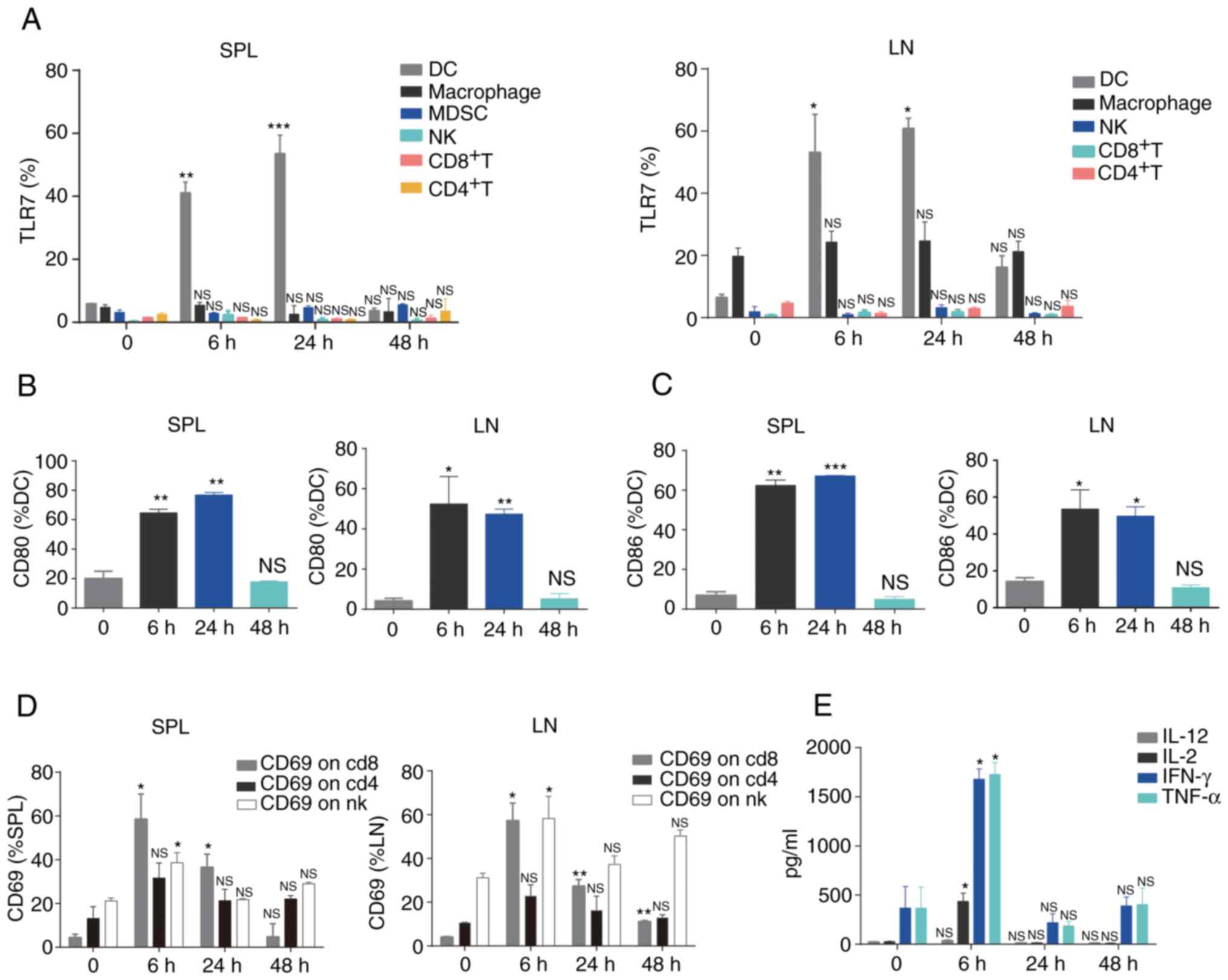 | Figure 2Administration of R848 promotes
systemic immune activation. (A) TLR7 expression on DCs,
macrophages, MDSCs, NK, CD4+ T and CD8+ T
cells was analyzed by flow cytometry (n=3). (B) CD80 and (C) CD86
expression on DCs was detected by flow cytometry (n=3). (D) CD69
expression was assessed in NK, CD4+ T and
CD8+ T cells (n=3). (E) Levels of cytokines were
evaluated in the serum (n=3). *P<0.05,
**P<0.01 and ***P<0.001 compared with
PBS. TLR, Toll-like receptor; DCs, dendritic cells; MDSCs,
myeloid-derived suppressor cells; NK, natural killer; NS, not
significant. |
R848 inhibits tumor growth and prolongs
survival in the s.c. lung cancer model
R848 has been used in the treatment of breast
cancer, lymphoma and pancreatic ductal adenocarcinoma (17,26). Since TLR7 expression was
demonstrated in a s.c. lung cancer model and it was confirmed that
R848 could stimulate systemic immunity, it was hypothesized that
R848 treatment could act on TLR7+ leukocytes, which
would reduce tumor burden in mice. Firstly, 20 µg (27) R848 s.c. (option 1) or i.p. (option
2) administration in lung tumor-bearing mice bearing from day 7,
twice a week until the experimental endpoint was reached, and
tumors were measured every 3 days once palpable (Fig. 3A). The i.p. or s.c. administration
of R848 reduced tumor growth compared with the control group;
however, this was statistically significant only in the i.p. group
(Fig. 3B). There were no
differences in the survival rates of the R848 group (i.p. or s.c.)
and the PBS control group (Fig.
3C). Since i.p. administration showed a significant inhibitory
effect on tumor growth, whereas s.c. did not, this route of
administration was used in subsequent experiments. A more intensive
scheme (option 3), in which R848 (20 µg) treatment was
initiated at day 0 and repeated every other day (Fig. 3D) also significantly prolonged the
survival time (Fig. 3E) and
effectively reduced tumor growth (Fig. 3F and G). These results
demonstrated that R848 inhibits lung tumor growth and prolongs the
survival of LLC tumor-bearing mice.
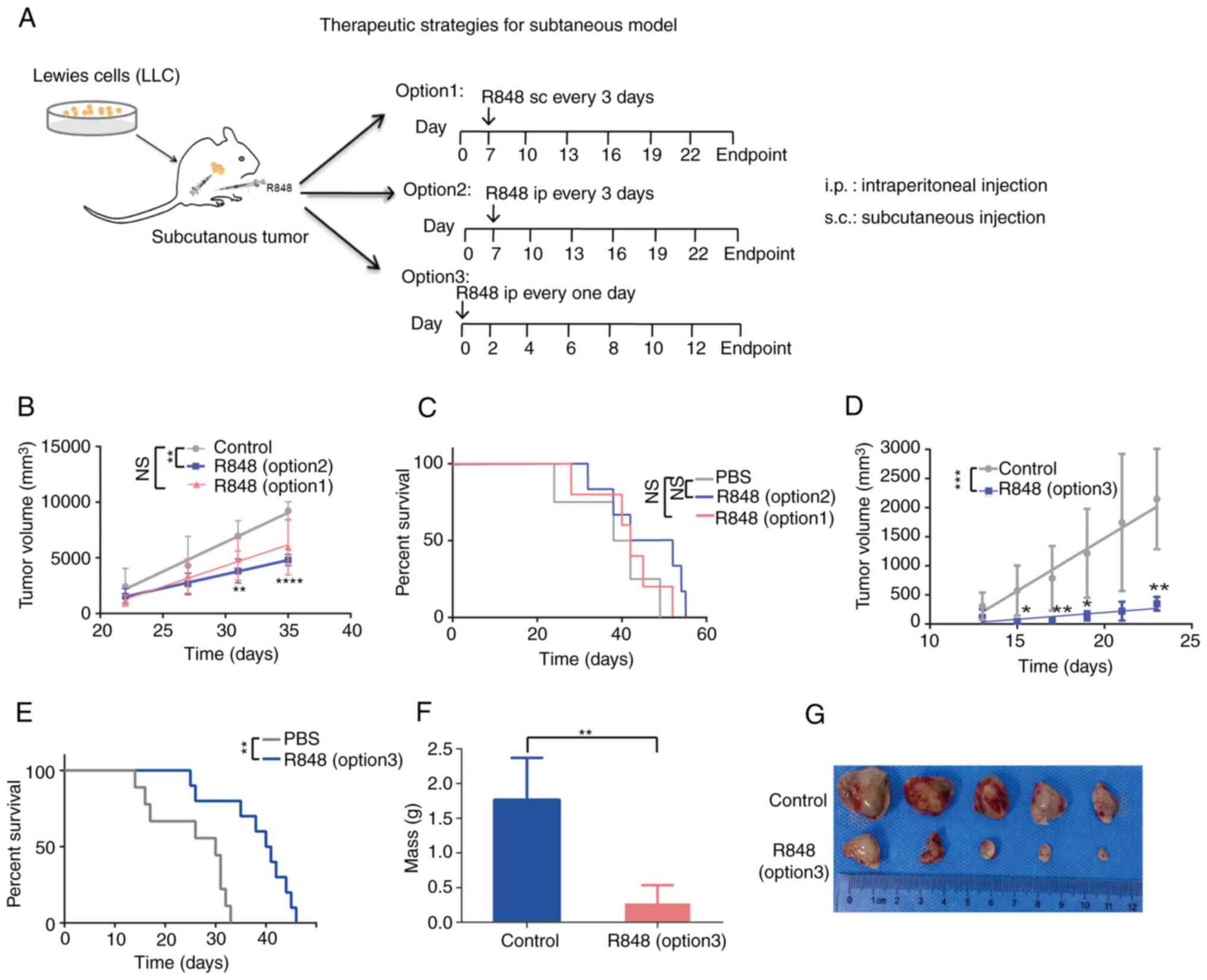 | Figure 3R848 inhibits tumor growth and
prolongs the survival of mice with s.c. lung tumors. (A) R848
treatment regimen. A total of 1×106 LLC cells were
injected s.c. into 6-8 weeks-old C57BL/6 mice. Starting at day 7
post-inoculation, 20 µg R848 in 100 µl PBS was
injected s.c. (option 1) or i.p. (option 2) every 3 days until the
experimental endpoint. The control received 100 µl PBS. In a
separate group, 20 µg R848 injected i.p. at day 0, then
every other day (option 3). (B) Tumor growth curves were generated
for the R848 (20 µg i.p. or s.c.) and the control groups.
(C) The survival of tumor-bearing mice in the R848 (20 µg,
i.p. or s.c.) and control groups was demonstrated. (D) Tumor growth
curves were generated for the R848 (option 3, 20 µg, i.p.)
and control groups. (E) Kaplan-Meier curves of the survival of
tumor-bearing mice in the R848 (option 3, 20 µg, i.p.) and
control groups. (F and G) Tumor weight (F) and picture of tumors
(G) were demonstrated in the R848 (option 3, 20 µg, i.p.)
and control groups (n=8-10). *P<0.05,
**P<0.01, ***P<0.001 and
****P<0.0001 compared with the control group. R848,
resiquimod; s.c., subcutaneous; LLC, Lewis lung carcinoma; i.p.,
intraperitoneal. |
R848 prevents melanoma cell metastasis to
the lung
To evaluate whether R848 can prevent metastasis to
the lung, the murine B16 melanoma metastasis model was used. A
total of 1×106 B16 F10 cells were injected via the tail
vein into C57BL/6 mice, and R848 therapy was initiated from day 0.
As a high dose of R848 (80 µg) may induce toxicity when
applied every other day, 20 µg R848 (i.p.) was administered
every other day (option 3) until the experimental endpoint or 80
µg (i.p.) R848 twice with a 4-day interval (Fig. 4A). The results demonstrated that
either 20 or 80 µg R848 treatment effectively inhibited the
metastasis of melanoma cells to the lung (Fig. 4B-D). During the experiment, B16
F10 metastatic nodes were not observed on the liver, chest wall, or
abdominal cavity without checking of brain or bones.
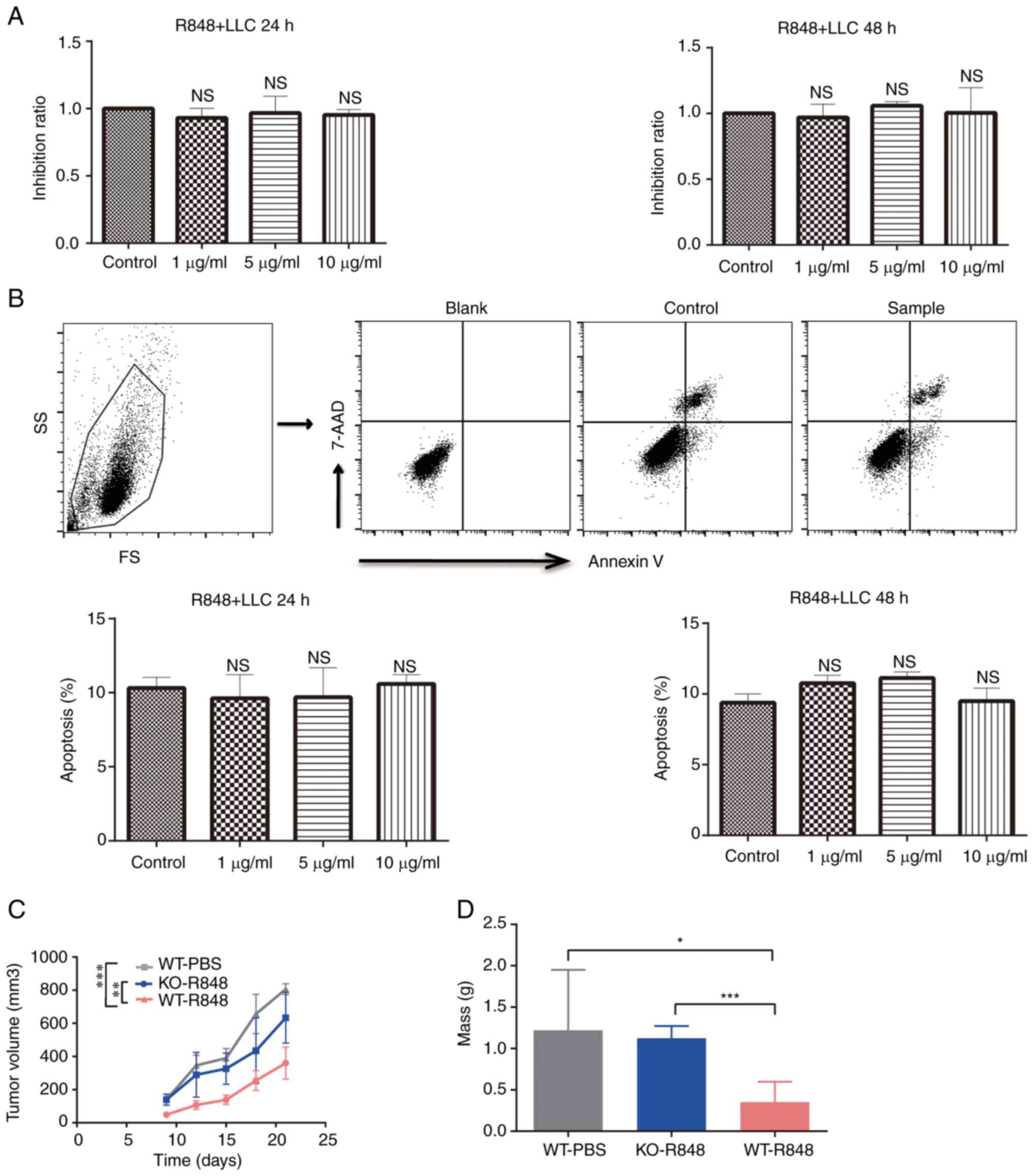
R848 exerts no direct effect on the
proliferation and apoptosis of LLC cells
To investigate the mechanism underlying the
antitumor effects of R848, LLC cells were treated with R848 in
vitro at different concentrations. Cell proliferation was not
affected by R848 treatment (Fig.
5A). Similarly, no significant effect was observed on LLC cell
apoptosis (Fig. 5B). Moreover,
R848 inhibited tumor burden in wild-type but not TLR7 knock-out
(TLR7-KO) mice (Fig. 5C). Indeed,
the tumor mass and volume at the endpoint confirmed that R848 did
not significantly inhibit the growth of s.c. LLC tumors in TLR7-KO
mice (Fig. 5D). These results
indicated that R848 served an antitumor role that is dependent on
TLR7 expression.
R848 modulates the TME
Since R848 did not affect LLC cells directly, but
induced the activation of DCs, CD8+ T cells and NK
cells, it was sought to determine whether R848 could modulate the
TME, as another underlying mechanism for its antitumor effect.
Tumor-infiltrating immune cell populations were detected in R848-
and PBS-treated s.c. LLC tumors. The gating strategy is shown in
Fig. S1. The total frequency of
CD45+ leukocytes was increased in R848-treated tumors
compared with the controls (Fig.
6A; 19.1±0.3480 vs. 13±1.8% of live cells; P<0.05).
CD3+ T cell (4.06±0.068 vs. 2.02±0.22% of
CD45+ cells; P<0.01) and CD8+ T cell
(64.5±0.74 vs. 55.8±0.45% of CD8+ T cells; P<0.01)
frequency increased following R848 treatment (Fig. 6B and C), while no significant
difference was observed in CD4+ T cells (Fig. 6D). The frequency of
FoxP3+ regulatory T (Treg) cells decreased following
R848 treated (11.5±0.45 vs. 34.36±3.66% of CD4+ T cells;
P<0.05) (Fig. 6E). However, a
significant downregulation of PD-1 on CD8+ T cells was
not observed. The proportion of NK cells significantly increased
following R848 treatment (0.98±0.12 vs. 0.53±0.05% of
CD45+ cells, P<0.05) (Fig. 6K). No significant difference was
observed in the B cell population (Fig. 6H). Macrophages
(CD11b+F4/80+) appeared enriched after
treatment with R848, although this was not statistically
significant (Fig. 6G). However,
the R848 treatment promoted M2 polarization towards the M1 subtype,
as evidenced by increased MHC-II expression (Fig. 6L) and lower CD206 expression
(Fig. 6M). The frequency of
tumor-infiltrating DCs (CD11b+CD11C+)
significantly increased (57.72±1.45 vs. 42.3±3.33% of
CD45+ cells; P<0.01) following treatment with R848
(Fig. 6F). In addition, the
frequency of neutrophils
(CD11b+Ly6G+Ly6C−, granulocytic
-MDSC) increased following treatment with R848 (23.6±2.52 vs.
7.63±1.62% of CD45+ cells; P<0.05) (Fig. 6J). There was a trend towards
reduced monocyte frequency
(CD11b+Ly6G−Ly6C+, monocytic-MDSC)
following R848 treatment, although this was not statistically
significant (Fig. 6I). These
findings suggested that R848 modulates the distribution of immune
cells in the TME of s.c. LLC tumors.
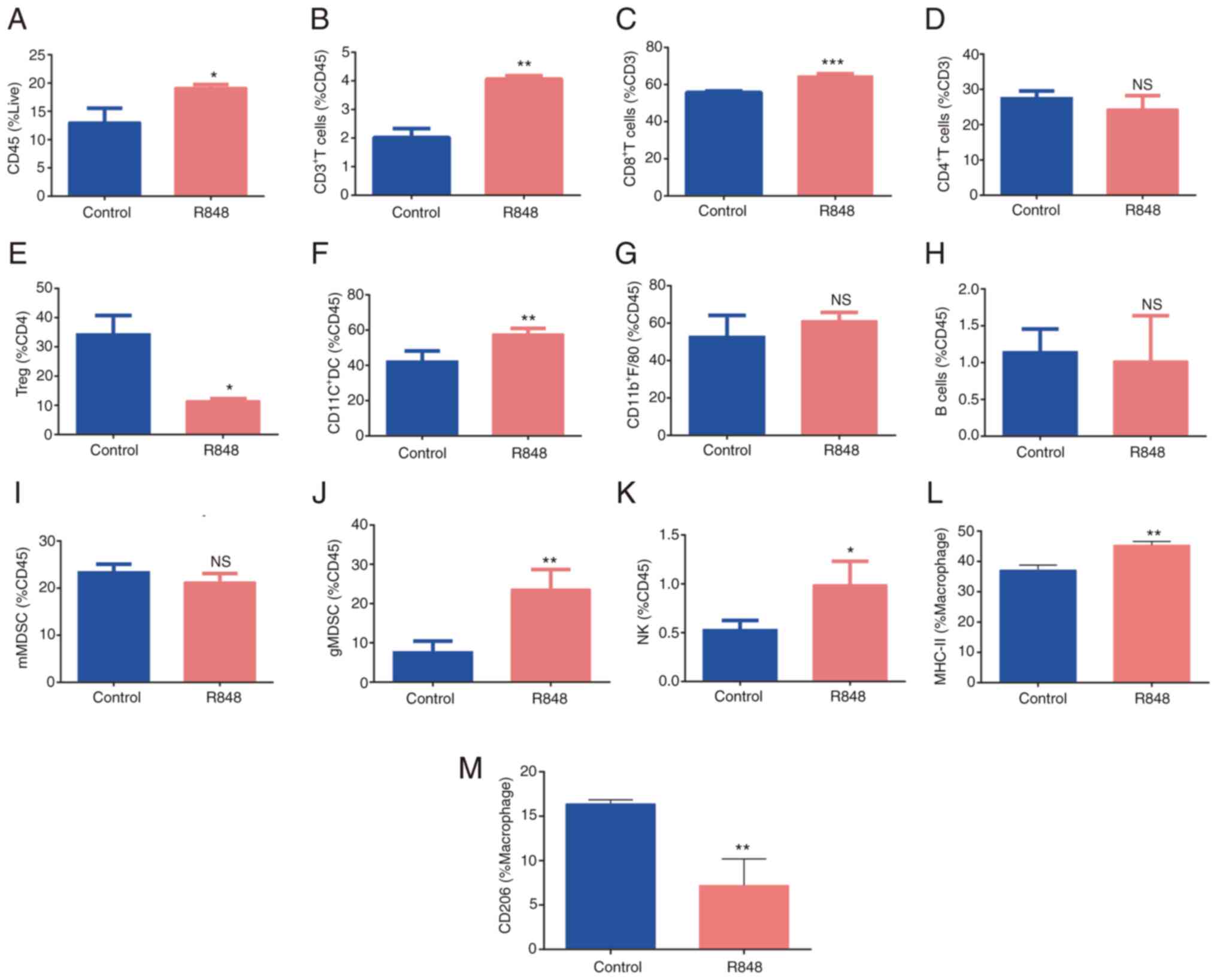 | Figure 6R848 alters the components of immune
cells in tumor microenvironment. (A) Distribution of
CD45+ cells, (B) total T cells or CD3+ T
cells, (C) CD8+ T cells, (D) CD4+ T cells,
(E) Tregs, (F) DCs, (G) macrophages, (H) B cells, (I) mMDSCs, (J)
gMDSCs and (K) NK cells in R848- and PBS-treated subcutaneous Lewis
lung carcinoma tumors (n=3-5). (L) MHC-II (M1) and (M) CD206 (M2)
expression on macrophages (n=3-5). *P<0.05,
**P<0.01 and ***P<0.001 compared with
the PBS control group. R848, resiquimod; Tregs, T regulatory cells;
DCs, dendritic cells; MDSCs, myeloid-derived suppressor cells; NK,
natural killer. |
NK cells and CD8+T cells takes
greatly part in R848-mediated therapeutic efficacy in lung
cancer
Based on the activation and alteration of immune
cells in host and TME of mice with s.c. LLC tumors following R848
treatment, the key immune cells involved in R848-mediated
therapeutic efficacy in murine lung cancer were further explored.
It has been documented that R848 enhances antitumor efficacy in
murine lymphoma models via NK and CD4+ T cells (17). In the present study, the reduction
in tumor burden by R848 was partly abrogated following NK cell
depletion (Fig. S2A). On one
hand, NK cells have been reported to be important immune cells for
cancer immunotherapy due to their cytotoxicity against tumor cells;
on the other hand, NK cells have been revealed to help TLR7
agonist-activated DCs to induce CD8+ T cell immunity
(28,29). Consistently, R848-stimulated DCs
induced CD8+ T cell proliferation and increased
IFN-γ+CD8+ T cell frequencies in the presence
of NK cells in the present study (Fig. S2B and C). R848 treatment with
CD8+ T cell depletion reduced mouse survival and weight,
which indicated that CD8+ T cells are likely to be a
significant factor in R848-mediated lung cancer resistance
(Fig. S2D). However, it was
demonstrated in the present that the antitumor role of R848 in lung
cancer is negatively associated with CD4+ T cells
including tumor volume, mice weight and survival, which is
consistent with the low frequency of CD4+ T cells in the
TME after R848 treatment (Fig. S2A,
D and E). Collectively, these results indicated that NK cells
and CD8+ T cells are closely involved in therapeutic
effecacy of R848 muring lung cancer.
Discussion
A growing number of immunotherapeutic drugs such as
cytokines, tumor vaccines, immune-stimulatory small molecules and
checkpoint inhibitors have exhibited considerable benefits in
patients with lung cancer (30,31). Several studies have also
highlighted the potential benefit of TLR7 agonists as antitumor
agents (7,32). In the present study, it was
demonstrated that the TLR7 agonist, R848, effectively inhibited
murine LLC s.c. tumor progression and metastasis and improved the
survival of LLC tumor-bearing mice. The antitumor effect elicited
by R848 could be attributed to TLR7-induced immunity, which was
characterized by activation of DCs, NK cells and CD8+ T
cells in lymphoid organs and immunomodulation of the TME.
TLR7 is a receptor of the innate immune system that
recognizes highly conserved molecules expressed by pathogens
(33). Upon binding single
stranded RNA or other agonists, TLR7 signaling results in the
production of pro-inflammatory cytokines, T cell proliferation, and
induction of adaptive immunity (34). In the present study, dynamic TLR7
expression was observed in lymphocytes from LLC tumors. In
addition, increased frequency of TLR7+ DCs was also
observed following R848 injection, which suggests that DCs are the
primary target cells of R848 in murine s.c. LLC model. In addition,
various immune cells play collaborating roles in antitumor
responses to lung cancer. Although the use of R848 for the
treatment of lung cancer has already been reported in certain
studies (15,35), the present findings provided
detailed, novel insight into the dynamics of TLR7 expression in
lung tumor-bearing mice. Furthermore, these findings are necessary,
as they provide a rationale for the use of R848 for treatment and
may guide decisions on therapy duration. Indeed, the findings of
the present study suggested that treatment with R848 at the
early/mid stage of lung cancer may be more beneficial than at later
stages.
Recent studies have reported the use of R848 in
combination with chemotherapy or nanoparticles, which had
therapeutic benefits in lung cancer (15,36). Consistent with these findings, a
reduction in tumor burden and improvements in survival were
observed in s.c. LLC model following i.p. injection of R848 in the
present study. Furthermore, it was revealed that R848 treatment
effectively prevented melanoma cells from metastasizing to the
lung. In the present study, a comparison of therapeutic strategies
was also made, including route of administration, dose and duration
choice. The results indicated that i.p. injection was superior to
s.c. administration. Abdominal blood flow is more abundant than in
subcutaneous tissue, thus, it was hypothesized that the absorption
of R848 i.p. may be more effective than the s.c. route. Since TLR7
was highly expressed in TiLs, R848 was initially used
intratumorally. However, frequent injection of R848 directly into
the tumor mass reduced natural tumor progression (data not shown).
R848 encapsulated with nanoparticles (37) or combined with materials targeting
TiLs may be worth exploring in future studies, as the present study
provided evidence that TLR7 is highly expressed in TiLs.
An important factor to consider for cancer treatment
is the complex immunity in the TME. Immune cells within the TME can
have dual roles, either promoting or inhibiting tumor growth.
Immune cells that promote tumor progression include MDSCs, Tregs
and M2-like macrophages. By contrast, DCs, neutrophils, NK, B and
effector T cells in the TME have antitumor effects (38,39). Changes were observed in the
distribution of these immune populations within s.c. LLC tumors
from mice treated with R848, including increased DCs, NK cells,
neutrophils (g-MDSC) and CD8+ T-cells, and decreased
Foxp3+ Tregs. These findings provided a rationale for
the use of R848 to improve therapeutic efficacy in lung tumors.
Although significant changes were not observed in the total
frequency of macrophages, the proportion of M2-like macrophages was
reduced and that of M1-like macrophages was increased, which is
consistent with a previous study demonstrating that R848-loaded
nanoparticles target the polarization of tumor-associated
macrophages to enhance cancer immunotherapy (40). CD8+ T lymphocytes play
a leading role in tumor immunity via their capacity to eliminate
malignant cells upon recognition by the T cell receptor of specific
antigenic peptides presented on the surface of cancer cells
(41). The expression of immune
checkpoint molecules, notably PD-1, is associated with poor
prognosis and reduced therapeutic effect. Furthermore,
CD8+ T cells with reduced PD-1 expression mediated
greater antitumor results in vivo (42). Continuous R848 treatment-induced
CD8+ T cell infiltration and showed PD-1 downregulation
on CD8+ T cells, although this was not statistically
significant. These findings, if confirmed, may provide a rationale
for the use R848 in combination with PD-1 inhibitors.
The role of TLR expression in cancer cells is not
fully understood and has been associated with either good or poor
outcomes (43). Previous studies
reported that TLR7 activation on immune cells results in
therapeutic benefits, whereas TLR7 expression on lung tumor cells
promotes tumor progression (44,45). Based on the absence of tumor
reduction in TLR7-deficient hosts, it is proposed that the
beneficial effects of R848 require a host, rather than tumor TLR7
expression. Future studies investigating the efficacy of R848 on
tumors expressing TLR7 are warranted.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JCZ, GSW and YX conceived and designed the study.
JCZ and HY performed the experiments with the guidence of YX. GSW,
THM, YX and YLL performed development of writing, review and
revision of the manuscript. YLL and THM supervised the study. YLL
and THM acquired funding. All authors made contributions to the
article. JCZ and YX confirm the authenticity of all the raw data.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The animal study was approved (approval no.
2022-152) by the ethics committee of The First Affiliated Hospital
of Chongqing Medical University (Chongqing, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by The Fifth funding for young
and middle-aged medical high talents of Chongqing (grant no.
2019GDRC028).
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar
|
|
2
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2021. CA Cancer J Clin. 71:7–33. 2021.
View Article : Google Scholar
|
|
3
|
Chen R, Manochakian R, James L, Azzouqa
AG, Shi H, Zhang Y, Zhao Y, Zhou K and Lou Y: Emerging therapeutic
agents for advanced non-small cell lung cancer. J Hematol Oncol.
13:582020. View Article : Google Scholar
|
|
4
|
Sharma P, Wagner K, Wolchok JD and Allison
JP: Novel cancer immunotherapy agents with survival benefit: Recent
successes and next steps. Nat Rev Cancer. 11:805–812. 2011.
View Article : Google Scholar
|
|
5
|
Horvath L, Thienpont B, Zhao L, Wolf D and
Pircher A: Overcoming immunotherapy resistance in non-small cell
lung cancer (NSCLC)-novel approaches and future outlook. Mol
Cancer. 19:1412020. View Article : Google Scholar
|
|
6
|
Vasilakos JP and Tomai MA: The use of
toll-like receptor 7/8 agonists as vaccine adjuvants. Expert Rev
Vaccines. 12:809–819. 2013. View Article : Google Scholar
|
|
7
|
Adams S: Toll-like receptor agonists in
cancer therapy. Immunotherapy. 1:949–964. 2009. View Article : Google Scholar
|
|
8
|
Urban-Wojciuk Z, Khan MM, Oyler BL,
Fahraeus R, Marek-Trzonkowska N, Nita-Lazar A, Hupp TR and Goodlett
DR: The role of TLRs in anti-cancer immunity and tumor rejection.
Front Immunol. 10:23882019. View Article : Google Scholar
|
|
9
|
Lim KH: TLR7 and TLR8, Resiquimod, and
852A. Cancer Therapeutic Targets. Marshall J: Springer; New York,
NY: pp. 1–8. 2013
|
|
10
|
Schön MP and Schön M: TLR7 and TLR8 as
targets in cancer therapy. Oncogene. 27:190–199. 2008. View Article : Google Scholar
|
|
11
|
Zhou Z, Yu X, Zhang J, Tian Z and Zhang C:
TLR7/8 agonists promote NK-DC cross-talk to enhance NK cell
anti-tumor effects in hepatocellular carcinoma. Cancer Lett.
369:298–306. 2015. View Article : Google Scholar
|
|
12
|
Yin T, He S and Wang Y: Toll-like receptor
7/8 agonist, R848, exhibits antitumoral effects in a breast cancer
model. Mol Med Rep. 12:3515–3520. 2015. View Article : Google Scholar
|
|
13
|
Rook AH, Gelfand JM, Wysocka M, Troxel AB,
Benoit B, Surber C, Elenitsas R, Buchanan MA, Leahy DS, Watanabe R,
et al: Topical resiquimod can induce disease regression and enhance
T-cell effector functions in cutaneous T-cell lymphoma. Blood.
126:1452–1461. 2015. View Article : Google Scholar
|
|
14
|
Killock D: Haematological cancer:
Resiquimod-a topical CTCL therapy. Nat Rev Clin Oncol. 12:5632015.
View Article : Google Scholar
|
|
15
|
Koh J, Kim S, Lee SN, Kim SY, Kim JE, Lee
KY, Kim MS, Heo JY, Park YM, Ku BM, et al: Therapeutic efficacy of
cancer vaccine adjuvanted with nanoemulsion loaded with TLR7/8
agonist in lung cancer model. Nanomedicine. 37:1024152021.
View Article : Google Scholar
|
|
16
|
Li F, Zheng X, Wang X, Xu J and Zhang Q:
Macrophage polarization synergizes with oxaliplatin in lung cancer
immunotherapy via enhanced tumor cell phagocytosis. Transl Oncol.
14:1012022021. View Article : Google Scholar
|
|
17
|
Cheadle EJ, Lipowska-Bhalla G, Dovedi SJ,
Fagnano E, Klein C, Honeychurch J and Illidge TM: A TLR7 agonist
enhances the antitumor efficacy of obinutuzumab in murine lymphoma
models via NK cells and CD4 T cells. Leukemia. 31:1611–1621. 2017.
View Article : Google Scholar
|
|
18
|
Gallotta M, Assi H, Degagne E, Kannan SK,
Coffman RL and Guiducci C: Inhaled TLR9 agonist renders lung tumors
permissive to PD-1 blockade by promoting optimal CD4+ and CD8+ T
cell interplay. Cancer Res. 78:4943–4956. 2018. View Article : Google Scholar
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Method. 25:402–408. 2001.
View Article : Google Scholar
|
|
20
|
Alam MM, Yang D, Trivett A, Meyer TJ and
Oppenheim JJ: HMGN1 and R848 synergistically activate dendritic
cells using multiple signaling pathways. Front Immunol. 9:29822018.
View Article : Google Scholar
|
|
21
|
Frega G, Wu Q, Le Naour J, Vacchelli E,
Galluzzi L, Kroemer G and Kepp O: Trial watch: Experimental
TLR7/TLR8 agonists for oncological indications. Oncoimmunology.
9:17960022020. View Article : Google Scholar
|
|
22
|
Lu R, Groer C, Kleindl PA, Moulder KR,
Huang A, Hunt JR, Cai S, Aires DJ, Berkland C and Forrest ML:
Formulation and preclinical evaluation of a toll-like receptor 7/8
agonist as an anti-tumoral immunomodulator. J Control Release.
306:165–176. 2019. View Article : Google Scholar
|
|
23
|
Cen X, Liu S and Cheng K: The role of
toll-like receptor in inflammation and tumor immunity. Front
Pharmacol. 9:8782018. View Article : Google Scholar
|
|
24
|
Akira S, Takeda K and Kaisho T: Toll-like
receptors: Critical proteins linking innate and acquired immunity.
Nat Immunol. 2:675–680. 2001. View
Article : Google Scholar
|
|
25
|
Moingeon P, Haensler J and Lindberg A:
Towards the rational design of Th1 adjuvants. Vaccine.
19:4363–4372. 2001. View Article : Google Scholar
|
|
26
|
Michaelis KA, Norgard MA, Zhu X, Levasseur
PR, Sivagnanam S, Liudahl SM, Burfeind KG, Olson B, Pelz KR, Ramos
DMA, et al: The TLR7/8 agonist R848 remodels tumor and host
responses to promote survival in pancreatic cancer. Nat Commun.
10:46822019. View Article : Google Scholar
|
|
27
|
Hosoya T, Sato-Kaneko F, Ahmadi A, Yao S,
Lao F, Kitaura K, Matsutani T, Carson DA and Hayashi T: Induction
of oligoclonal CD8 T cell responses against pulmonary metastatic
cancer by a phospholipid-conjugated TLR7 agonist. Proc Natl Acad
Sci USA. 115:E6836–E6844. 2018. View Article : Google Scholar
|
|
28
|
Karimi K, Boudreau J, Fraser K, Liu H,
Delanghe J, Gauldie J, Xing Z, Bramson JL and Wan Y: Enhanced
antitumor immunity elicited by dendritic cell vaccines is a result
of their ability to engage both CTL and IFN gamma-producing NK
cells. Mol Ther. 16:411–418. 2008. View Article : Google Scholar
|
|
29
|
Stojanovic A and Cerwenka A: Natural
killer cells and solid tumors. J Innate Immun. 3:355–364. 2011.
View Article : Google Scholar
|
|
30
|
Gotwals P, Cameron S, Cipolletta D,
Cremasco V, Crystal A, Hewes B, Mueller B, Quaratino S,
Sabatos-Peyton C, Petruzzelli L, et al: Prospects for combining
targeted and conventional cancer therapy with immunotherapy. Nat
Rev Cancer. 17:286–301. 2017. View Article : Google Scholar
|
|
31
|
Meador CB and Hata AN: Acquired resistance
to targeted therapies in NSCLC: Updates and evolving insights.
Pharmacol Ther. 210:1075222020. View Article : Google Scholar
|
|
32
|
Wang D, Precopio M, Lan T, Yu D, Tang JX,
Kandimalla ER and Agrawal S: Antitumor activity and immune response
induction of a dual agonist of toll-like receptors 7-8. Mol Cancer
Ther. 9:1788–1797. 2010. View Article : Google Scholar
|
|
33
|
Pandey S, Kawai T and Akira S: Microbial
sensing by toll-like receptors and intracellular nucleic acid
sensors. Cold Spring Harb Perspect Biol. 7:a0162462014. View Article : Google Scholar
|
|
34
|
Petes C, Odoardi N and Gee K: The toll for
trafficking: Toll-like receptor 7 Delivery to the endosome. Front
Immunol. 8:10752017. View Article : Google Scholar
|
|
35
|
Smith AA, Gale EC, Roth GA, Maikawa CL,
Correa S, Yu AC and Appel EA: Nanoparticles presenting potent
TLR7/8 agonists enhance Anti-PD-L1 immunotherapy in cancer
treatment. Biomacromolecules. 21:3704–3712. 2020. View Article : Google Scholar
|
|
36
|
Bahmani B, Gong H, Luk BT, Haushalter KJ,
DeTeresa E, Previti M, Zhou J, Gao W, Bui JD, Zhang L, et al:
Intratumoral immunotherapy using platelet-cloaked nanoparticles
enhances antitumor immunity in solid tumors. Nat Commun.
12:19992021. View Article : Google Scholar
|
|
37
|
Rodell CB, Ahmed MS, Garris CS, Pittet MJ
and Weissleder R: Development of adamantane-conjugated TLR7/8
agonists for supramolecular delivery and cancer immunotherapy.
Theranostics. 9:8426–8436. 2019. View Article : Google Scholar
|
|
38
|
Goliwas K, Deshane J, Elmets C and Athar
M: Moving immune therapy forward targeting TME. Physiol Rev.
101:417–425. 2021. View Article : Google Scholar
|
|
39
|
Taube JM, Galon J, Sholl LM, Rodig SJ,
Cottrell TR, Giraldo NA, Baras AS, Patel SS, Anders RA, Rimm DL and
Cimino-Mathews A: Implications of the tumor immune microenvironment
for staging and therapeutics. Mod Pathol. 31:214–234. 2018.
View Article : Google Scholar
|
|
40
|
Rodell CB, Arlauckas SP, Cuccarese MF,
Garris CS, Li R, Ahmed MS, Kohler RH, Pittet MJ and Weissleder R:
TLR7/8-agonist-loaded nanoparticles promote the polarization of
tumour-associated macrophages to enhance cancer immunotherapy. Nat
Biomed Eng. 2:578–588. 2018. View Article : Google Scholar
|
|
41
|
Durgeau A, Virk Y, Corgnac S and
Mami-Chouaib F: Recent advances in targeting CD8 T-cell immunity
for more effective cancer immunotherapy. Front Immunol. 9:142018.
View Article : Google Scholar
|
|
42
|
Zahm CD, Colluru VT, McIlwain SJ, Ong IM
and McNeel DG: TLR stimulation during T-cell activation lowers PD-1
expression on CD8(+) T cells. Cancer Immunol Res. 6:1364–1374.
2018. View Article : Google Scholar
|
|
43
|
Dajon M, Iribarren K and Cremer I: Dual
roles of TLR7 in the lung cancer microenvironment. Oncoimmunology.
4:e9916152015. View Article : Google Scholar
|
|
44
|
Chatterjee S, Crozet L, Damotte D,
Iribarren K, Schramm C, Alifano M, Lupo A, Cherfils-Vicini J, Goc
J, Katsahian S, et al: TLR7 promotes tumor progression,
chemotherapy resistance, and poor clinical outcomes in non-small
cell lung cancer. Cancer Res. 74:5008–5018. 2014. View Article : Google Scholar
|
|
45
|
Cherfils-Vicini J, Platonova S, Gillard M,
Laurans L, Validire P, Caliandro R, Magdeleinat P, Mami-Chouaib F,
Dieu-Nosjean MC, Fridman WH, et al: Triggering of TLR7 and TLR8
expressed by human lung cancer cells induces cell survival and
chemoresistance. J Clin Invest. 120:1285–1297. 2010. View Article : Google Scholar
|