Introduction
Leukemia is a life-threatening hematopoietic
malignancy characterized by the malignant proliferation of
hematopoietic stem cells and dysregulation of regulated cell death
(RCD) (1-3). The age-standardized 5-year relative
survival rate of Chinese patients with leukemia increased from
19.6% in 2003-2005 to 25.4% in 2012-2015 owing to progression in
pathogenesis and therapeutic strategies for leukemia (4). However, side effects and drug
resistance are inevitable after the long-term use of
chemotherapeutic drugs (5). Thus,
new antileukemic drugs need to be discovered.
A delicate balance between RCD and cell
proliferation is essential to maintain a healthy cell population;
however, this balance is out of control in leukemia cell
populations. Conventional drugs, such as cytarabine and
daunorubicin, are used to treat leukemia via the induction of DNA
damage and cell cycle arrest (6).
The novel oral drug venetoclax, which targets the apoptotic
pathway, has been approved by the Food and Drug Administration for
acute myeloid leukemia (7,8).
Pyroptosis is a novel form of RCD characterized by
cell swelling, plasma membrane rupture, and DNA condensation and
fragmentation (9,10). Gasdermins play a pivotal role in
pyroptosis. Apart from APJK (DFNB59), all gasdermins (GSDMA-E)
share the gasdermin N-terminal effector domain (11-13).
The N-terminal effector domain is activated by the
caspase-dependent cleavage of GSDMD or GSDME and exhibits
pore-forming, intrinsic cytotoxic, and antibacterial properties
(11,12). Activated caspase-1 and
caspase-4/5/11 are responsible for canonical and non-canonical
GSDMD-related pyroptosis, respectively (14,15).
Activated caspase-3 that is induced by tumor necrosis factor
(TNF)-α or DNA-damaging chemotherapeutic drugs are responsible for
GSDME-related pyroptosis (10,13,16).
Preclinical studies involving GSDMD-related pyroptosis induced by
DPP8/9 inhibitors and GSDME-related pyroptosis induced by
pyridoxine, aimed at the treatment of acute myeloid leukemia, have
indicated that pyroptosis is a new target for the management of
leukemia (17-19). Apoptosis, characterized by
cytoplasmic shrinkage and small apoptotic bodies, also commonly
known as type I cell death, is a classical form of RCD mediated by
the activation of caspase-3 (10).
As caspase-3 is activated in both pyroptosis and apoptosis, the
final cell destination is dependent on the expression levels of
GSDME (20). It is generally
accepted that GSDME-positive cells undergo pyroptosis, whereas
GSDME-negative cells develop apoptotic responses (13,16).
Autophagy, commonly known as type II cell death, is a core
molecular process that maintains cellular and organismal
homeostasis (21). Notably,
increasing evidence suggests the existence of multilevel crosstalk
between different types of RCD (20,22-24).
Liproxstatin-1 (Lip-1) is a small molecule with a
variety of biological properties, including antibacterial and
anti-ferroptosis activities (25,26).
However, the antileukemic effect of Lip-1 has not yet been
reported. The leukemia cell line K562, derived from a patient with
chronic myeloid leukemia during the blast crisis in the 1970s
(27), is a good cellular model
for in vitro research. K562 cells express low levels of
GSDME (13), suggesting that K562
cells will develop apoptotic responses prior to secondary necrotic
responses following treatment with certain chemotherapeutic drugs
and cleavage of caspase-3. Interestingly, Lip-1-treated K562 cells
developed caspase-3/GSDME-dependent secondary pyroptosis after
apoptosis. Moreover, the results indicated that endoplasmic
reticulum (ER) stress, autophagy, and cell cycle arrest were
involved in the modulation of K562 cell proliferation. Taken
together, Lip-1 may provide new hope for the treatment of
leukemia.
Materials and methods
Antibodies and reagents
Lip-1 (cat. no. S7699) was purchased from Selleck
Chemicals. Z-DEVD-FMK (cat. no. HY-12466) was purchased from
MedChemExpress. RPMI-1640 medium (cat. no. 11835-030) was purchased
from Thermo Fisher Scientific, Inc., fetal bovine serum (cat. no.
900-108) was purchased from Gemini Bio Products, and
penicillin-streptomycin (product no. V900929) was purchased from
Sigma-Aldrich; Merck KGaA. CryoStor CS10 (cat. no. 07930) was
obtained from StemCell Technologies, Inc. Dimethyl sulfoxide and
Dulbecco's phosphate-buffered saline (PBS) were purchased from
Dalian MeilunBio Biotechnology Co., Ltd. QuickBlock Blocking Buffer
for western blotting was purchased from Beyotime Institute of
Biotechnology. Thermo Scientific PageRular Prestained Protein
Ladder (cat. no. 26616) was purchased from Thermo Fisher
Scientific, Inc. SH-SY5Y whole cell lysate used as the positive
control for GSDME-N-terminal in western blotting was gifted by
Abcam.
The primary antibodies used were as follows: C/EBP
homologous (CHOP) antibody (cat. no. 15204-1-AP) and PUMA antibody
(cat. no. 55120-1-AP) obtained from ProteinTech Group, Inc.; LC3A/B
(D3U4C) antibody (product no. 12741) and cleaved caspase-3 (ASP175)
antibody (product no. 9661) purchased from Cell Signaling
Technology, Inc.; CDKN1A/p21CIP1 antibody (cat. no. A19094)
obtained from ABclonal Biotech Co., Ltd.;
anti-DFNA5/GSDME-N-terminal antibody (product code ab215191)
obtained from Abcam; GSDME antibody-N-terminal (cat. no. AF4016)
from Affinity Biosciences, Ltd.; and β-tubulin antibody (cat. no.
M30109), BCL2 associated X (BAX) antibody (product no. T40051),
caspase-3 antibody (product no. T40044), PARP antibody (product no.
T40050), and actin (2P2) antibody (product no. M20011) were gifted
by Abmart Pharmaceutical Technology Co., Ltd. The β-tubulin
antibody served as a loading control. The secondary antibody, goat
anti-rabbit mouse IgG-HRP (product no. M21003) was gifted by
Abmart. Cy3-conjugated goat anti-mouse IgG (H+L) (product no.
GB21301) and FITC-conjugated goat anti-rabbit IgG (H+L) (product
no. GB22303) were purchased from Wuhan Servicebio Technology Co.,
Ltd.
Cell culture
The K562 cell line (human leukemia cell line) (cat.
no. CL-0130) and SH-SY5Y cell line (cat. no. CL-0208) were
purchased from Procell Life Science & Technology Co., Ltd. K562
and SH-SY5Y cells were cultured in RPMI-1640 medium containing 10%
(v/v) fetal bovine serum, penicillin (100 U/ml), and streptomycin
(100 µg/ml). Cells were grown at 37°C in a 5% CO2
incubator. The K562 cell line tested mycoplasma-negative using a
GMyc-PCR Mycoplasma Test Kit (Shanghai Yeasen Biotechnology, Inc.),
according to the manufacturer's instructions. Cell identities of
K562 and SH-SY5Y cell lines were authenticated by Genetic Testing
Biotechnology Corp. using short tandem repeat profiling.
Cell viability assay
Exponentially growing K562 cells were seeded at a
density of 1x104 cells/well in 100 µl of RPMI-1640
medium in 96-well plates. Lip-1 was dissolved in dimethyl
sulfoxide, and cells were treated with various concentrations (2,
4, 10, 20, or 40 µM) of Lip-1 for 24 or 48 h. Subsequently, 10 µl
of CCK-8 solution (Dalian MeilunBio Biotechnology Co., Ltd.) was
added to each well, and the 96-well plate was incubated at 37°C for
3 h in the dark. The absorbance (A) was measured at 450 nm using a
Varioskan LUX microplate reader (Life Technologies; Thermo Fisher
Scientific, Inc.). Cell viability was calculated using the
following formula: Viability
(%)=[(Adrug-Ablank)/(Acontrol-Ablank)]
x100%. The half-maximal inhibitory concentration was calculated
using GraphPad Prism 7 (GraphPad Software, Inc.). In other
experiments, K562 cells were pretreated with the caspase-3-specific
inhibitor z-DEVD-FMK at a concentration of 200 µM for 3 h, and
further treated with 20 µM Lip-1 for 12 h. The CCK-8 assay was
performed to determine cell viability.
Microscopy
To investigate the morphology of apoptotic cells,
K562 cells were seeded in a 6-well plate (2x105
cells/ml) and subjected to Lip-1 treatment at a concentration of 10
or 20 µM for 24 h. Dimethyl sulfoxide at a dilution of 5/10,000
served as the vehicle control. Static bright-field images were
captured using an IX71 microscope (Olympus Corporation).
Scanning electron microscopy
To further investigate the morphological changes in
Lip-1-induced K562 cells, scanning electron microscopy was
performed on K562 cells seeded in 6-well plates (2x105
cells/ml) and treated with 10 or 20 µM Lip-1 for 24 h. The cells
were then washed with PBS, double-fixed in 2.5% glutaraldehyde and
1% osmic acid at room temperature for 2 h, dehydrated using ethanol
solutions, and dried with a critical point dryer. Finally, the
specimens were coated with gold using carbon stickers for 30 sec
and visualized under a Hitachi SU8100 scanning electron microscope
(Hitachi, Ltd.).
Transmission electron microscopy
To examine the ultrastructural damage in
Lip-1-induced K562 cells, transmission electron microscopy was
performed on K562 cells, and the cells at a density of
2x105 cells/ml were treated with 10 or 20 µM Lip-1. The
cells were then washed with PBS, double-fixed in 2.5%
glutaraldehyde and 1% osmic acid at room temperature for 2 h,
dehydrated using a series of ethanol solutions, and embedded at
60°C for 48 h in SPI-PON 812 resin (SPI; Structure Probe, Inc.).
Finally, after polymerization, ultrathin sectioning (resin blocks
were cut to 60-80 nm thickness using an ultramicrotome), and
staining procedures (2% uranium acetate saturated alcohol solution
staining in the dark for 8 min, 2.6% lead citrate staining for 9
min) the cuprum grids were placed into the grid board and dried at
room temperature overnight. The specimens were visualized using a
Hitachi HT7800 transmission electron microscope (Hitachi,
Ltd.).
DNA fragmentation evaluation
K562 cells at a density of 2x105 cells/ml
were cultured in a 6-well plate, followed by treatment with 10 or
20 µM Lip-1 for 24 h. K562 cells were fixed in 4% paraformaldehyde
at room temperature for 2 h. DNA fragmentation evaluation assay was
performed using the FITC TUNEL Cell Apoptosis Detection Kit (Wuhan
Servicebio Technology Co, Ltd.), according to the manufacturer's
instructions, and the specimens were stained with DAPI at room
temperature avoiding light for 10 min. For the positive control,
samples were treated with 2 U/ml DNase I (Wuhan Servicebio
Technology Co, Ltd.) for 10 min. Whole-mounted specimens were
imaged using a fluorescence microscope (Nikon Corporation). DNA
fragmentation was measured as follows: Four randomly selected
fields per section, equivalent to at least 600 cells, were manually
analyzed using ImageJ software V 1.8.0 (National Institutes of
Health).
Immunofluorescence and confocal
microscopy
To study the effect of Lip-1 on autophagosome
formation, an immunofluorescence assay was performed on K562 cells
seeded in 6-well plates and treated with 10 or 20 µM Lip-1 for 24
h. Following treatment, the cells were fixed with 4%
paraformaldehyde at room temperature for 2 h, permeabilized with
0.1% Triton X-100 (Sigma-Aldrich: Merck KGaA), blocked with 3%
bovine serum albumin at room temperature for 30 min, and incubated
with primary antibodies (LC3A/B and actin) at a dilution of 1:300
overnight at 4°C. Next, the cells were incubated with
Cy3-conjugated goat anti-mouse IgG (H+L) and FITC-conjugated goat
anti-rabbit IgG (H+L) at a dilution of 1:500 at room temperature
avoiding light for 50 min, and the nuclei were counterstained with
DAPI at room temperature avoiding light for 10 min. Specimens were
visualized using a confocal laser-scanning microscope (Nikon
Corporation). The ratio of LC3-positive cells was measured as
follows: Two randomly selected fields per section, equivalent to at
least 100 cells, were manually analyzed using ImageJ software V
1.8.0.
RNA sequencing and differential
expression analysis
K562 cells were treated with 10 or 20 µM Lip-1 for
24 h. Total RNA was isolated using TRIzol reagent (Invitrogen;
Thermo Fisher Scientific, Inc.), and then sent to Genesky Bio-Tech
Co., Ltd. for RNA sequencing (RNA-seq) and differential expression
analysis. Briefly, libraries were constructed using the TruSeq RNA
Sample Preparation Kit v2 (Illumina, Inc.) and purified using the
Agencourt SPRIselect Reagent Kit (Beckman Coulter, Inc.).
Sequencing data were analyzed using StringTie (28), to determine the expression level of
each gene. Following DESeq2 (29),
genes with |log2(fold-change)| >1 and adjusted
P<0.05 were regarded as differentially expressed genes (DEGs).
Volcano plots were constructed based on DEGs. Gene Ontology
(30) and Kyoto Encyclopedia of
Genes and Genomes (31) analyses
were performed to assess gene enrichment and functional annotation
using clusterProfiler (32). Raw
sequencing data were submitted to GEO (GSE205036).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from K562 cells using TRIzol
reagent, and complementary DNA was produced using the PrimeScript
RT Reagent Kit (Takara Bio, Inc.), according to the manufacturer's
instructions. RT-qPCR was conducted on an Applied Biosystems 7500
Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific,
Inc.) using TB Green Premix Ex Taq II (Takara Bio, Inc.). Relative
gene expression was calculated using the 2-ΔΔCq method
(33) and normalized to that of
RPLP0. Human RPLP0 endogenous reference gene primers were obtained
from Sangon Biotech Co., Ltd. The sequences of the primers used for
RT-qPCR were as follows: RPLP0 forward, 5'-GCA GCA TCT ACA ACC CTG
AAG-3' and reverse, 5'-CAC TGG CAA CAT TGC GGA C-3'; DDIT3 forward,
5'-GGA AAC AGA GTG GTC ATT CCC-3' and reverse, 5'-CTG CTT GAG CCG
TTC ATT CTC-3'; BBC3 forward, 5'-GAC CTC AAC GCA CAG TAC GAG-3' and
reverse, 5'-AGG AGT CCC ATG ATG AGA TTG T-3'; MAP1LC3B forward,
5'-GAG AAG ACC TTC AAG CAG CG-3' and reverse, 5'-TAT CAC CGG GAT
TTT GGT TG-3'; CDKN1A forward, 5'-TGT CCG TCA GAA CCC ATG C-3' and
reverse, 5'-AAA GTC GAA GTT CCA TCG CTC-3'; TNF forward, 5'-GAG GCC
AAG CCC TGG TAT G-3' and reverse, 5'-CGG GCC GAT TGA TCT CAG C-3';
and GSDME forward, 5'-TGC CTA CGG TGT CAT TGA GTT-3' and reverse,
5'-TCT GGC ATG TCT ATG AAT GCA AA-3'.
Cell cycle assay
Cell cycle analysis was conducted using a Cell Cycle
Staining Kit [Hangzhou Multi Sciences (Lianke) Biotech Co., Ltd.],
according to the manufacturer's instructions. Briefly, K562 cells
treated with or without Lip-1 were harvested, washed twice with
PBS, and treated with 1 ml DNA staining solution and 10 µl
permeabilization solution at 4°C in the dark for 30 min. Finally,
single-cell suspensions were analyzed using a BD FACSCalibur flow
cytometer (BD Biosciences). Data were analyzed using FlowJo
software v10.0.8 (BD Biosciences).
Annexin V-FITC/PI double staining
assay
The integrity of the cell membrane was evaluated
using an Annexin V-FITC/PI Apoptosis Kit [Hangzhou Multi Sciences
(Lianke) Biotech Co., Ltd.], according to the manufacturer's
instructions. Briefly, K562 cells treated with 10 or 20 µM Lip-1
for 24 h were harvested, washed twice with binding buffer, and
stained with Annexin V-FITC and PI at 4°C in the dark for 10 min.
Finally, the cells were analyzed using a BD FACSCalibur flow
cytometer, and the data were analyzed using FlowJo software
v10.0.8.
Western blotting
Total protein was extracted from the cells using
RIPA lysis buffer (Dalian MeilunBio Biotechnology Co., Ltd.). After
measuring the protein concentration using a BCA Protein Assay Kit
(Beyotime Institute of Biotechnology), 15 µg of total protein was
loaded onto 12.5% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis gels, processed for electrophoresis, transferred to
a polyvinylidene fluoride membrane (Merck KGaA). The polyvinylidene
fluoride membranes were blocked with QuickBlock™ Blocking Buffer
(cat. no. P0252; Beyotime Institute of Biotechnology) at room
temperature for 15 min. Membranes were incubated with C/EBP
homologous (CHOP) antibody (1:1,000 dilution), PUMA antibody
(1:1,000 dilution), LC3A/B (D3U4C) antibody (1:1,000 dilution),
cleaved caspase-3 (ASP175) antibody (1:1,000 dilution),
CDKN1A/p21CIP1 antibody (1:1,000 dilution),
anti-DFNA5/GSDME-N-terminal antibody (1:1,000 dilution), GSDME
antibody-N-terminal (1:1,000 dilution), β-tubulin antibody (1:8,000
dilution), BCL2 associated X (BAX) antibody (1:1,000 dilution),
caspase-3 antibody (1:1,000 dilution) and PARP antibody (1:1,000
dilution) at 4°C for 12 h. Next, the membranes were incubated with
goat anti-rabbit mouse IgG-HRP secondary antibody at room
temperature for 1 h. The membranes were treated with
Meilunbio® FGSuper Sensitive ECL Luminescence Reagent
(cat. no. MA0186; Dalian MeilunBio Biotechnology Co., Ltd.),
according to the manufacturer's instructions, and protein bands
were scanned using GelDoc XR (Image Lab; Bio-Rad Laboratories,
Inc.). Densitometric measurements of the protein bands were
performed using ImageJ software v1.8.0.
Caspase-3 and caspase-1 activity
assessment
Caspase-3 and caspase-1 activities were determined
using Colorimetric Assay Kits (Dalian MeilunBio Biotechnology Co.,
Ltd.), which utilized two types of substrates: Ac-DEVD-pNA for
caspase-3 and Ac-YVAD-pNA for caspase-1. Briefly, K562 cells at a
density of 2x105 cells/ml were cultured in 6-well
plates, followed by treatment with 10 or 20 µM Lip-1 for 12 or 24
h. The cells were lysed in lysis buffer at 4°C for 15 min. Protein
concentration was measured using a Bradford Assay Kit (Dalian
MeilunBio Biotechnology Co., Ltd.). Following incubation, caspase-3
and caspase-1 activities were determined by measuring the changes
in absorbance at 405 nm using a microplate reader.
Enzyme-linked immunosorbent assay
(ELISA)
K562 cells at a density of 1x106 cells/ml
were cultured in 6-well plates and treated with 10 or 20 µM Lip-1
for 24 h. Cell culture supernatants were collected, and cell
lysates were harvested via centrifugation at 600 x g at 4°C for 5
min and lysed with lysis buffer at 4°C for 15 min. Protein
concentration was determined using a Bradford Assay Kit. The levels
of human TNF-α in the cell culture supernatants and cell lysates
were detected using an ELISA Kit (cat. no. RK00030; ABclonal
Biotech Co., Ltd.), according to the manufacturer's instructions.
The relative TNF-α concentration in the cell lysates was calculated
as follows: Relative TNF-α (pg/µg)=[(TNF-α concentration)/(protein
concentration)]. The minimum detectable concentration was 6.9
pg/ml.
Statistical analysis
Quantitative variables are presented as the mean ±
SD. SPSS 25.0 (IBM Corp.) was used for statistical analysis.
Statistical analysis was performed using unpaired two-tailed
Student's t-test for comparison of two groups and one-way
ANOVA followed by Dunnett's post hoc test for comparison of three
groups. The Benjamini-Hochberg adjusted P-value was used for
sequencing data. P<0.05 was considered to indicate a
statistically significant difference.
Results
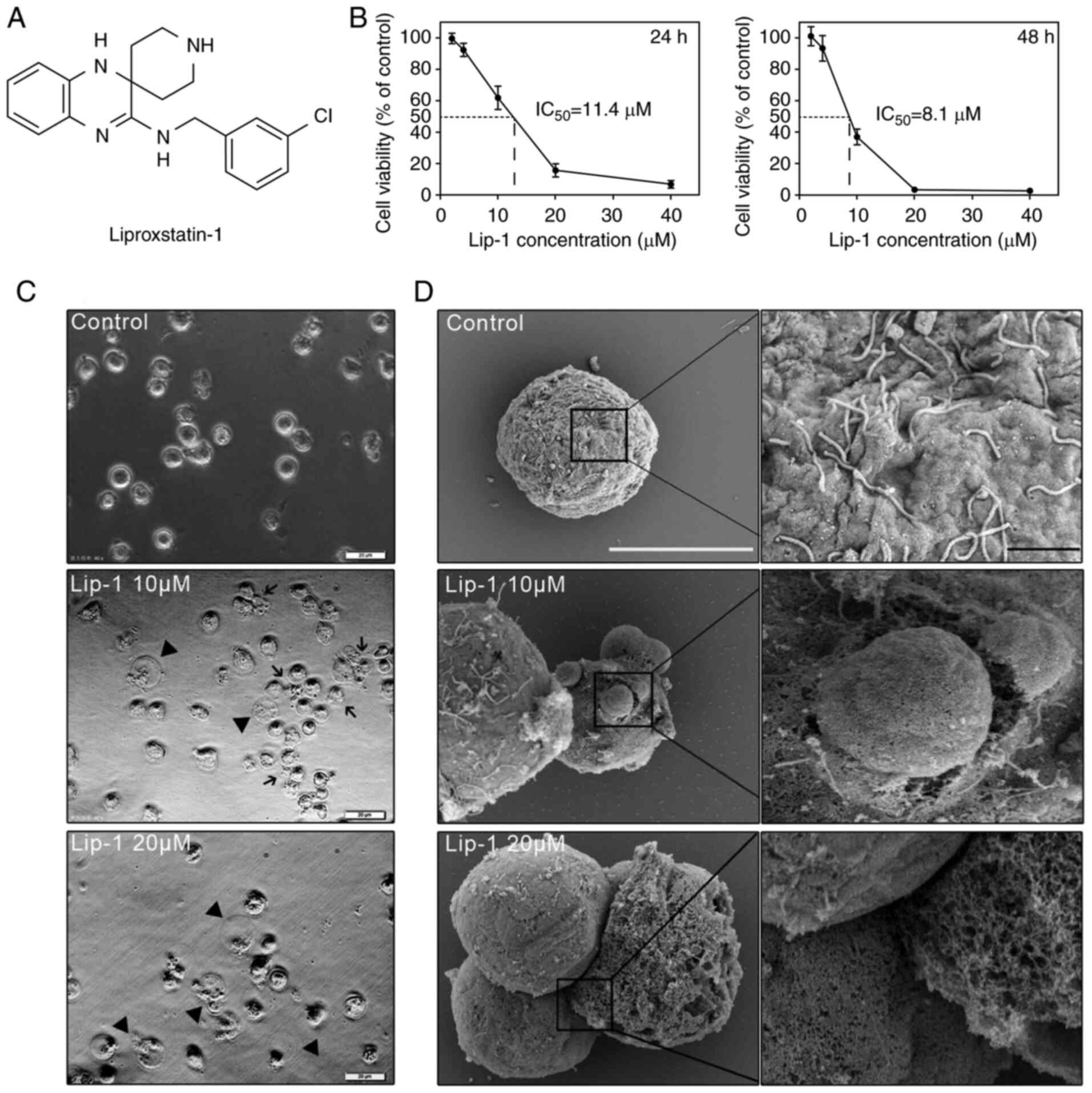
Lip-1 inhibits viability of K562 leukemia
cells in a dose-dependent and time-dependent manner
To verify ferroptosis induction in K562 leukemia
cells by some agents, K562 cells were pre-treated with Lip-1 (an
inhibitor of ferroptosis; Fig.
1A). In the Lip-1-treated group, cell death was observed under
a microscope, and the result revealed that the viability of K562
cells decreased. The antileukemic effect of Lip-1 has yet to be
reported. Thus, K562 cells were treated with a series of Lip-1
concentrations (0, 2, 4, 10, 20, and 40 µM) for different lengths
of time (24 and 48 h) and K562 cell viability was assessed using
the CCK-8 assay. The inhibitory effect of Lip-1 on cell viability
was dose-dependent and time-dependent; the 24and 48-h half-maximal
inhibitory concentrations of Lip-1 in K562 cells were 11.4 and 8.1
µM, respectively (Fig. 1B). Based
on CCK-8 assay data, K562 cells treated with 10 and 20 µM Lip-1 for
24 h were regarded as the experimental groups, and these groups
were used for subsequent experiments.
Lip-1 induces ultrastructural changes in
K562 cells
Because Lip-1-induced cell death remains largely
unknown, the ultrastructural changes in K562 cells were examined.
Excessive cell swelling was observed in Lip-1-treated K562 cells in
phase-contrast images (Fig. 1C).
With higher magnification via SEM, it was observed that untreated
K562 cells had intact plasma membranes with abundant microvilli
whereas 10 µM Lip-1-treated K562 cells had large membrane bubbles
with breakage; furthermore, loss of microvilli and membrane pores
of varying sizes were observed in K562 cells after 20 µM Lip-1
treatment (Fig. 1D). Additionally,
TEM images depicted the rupture of the plasma membrane, an expanded
rough endoplasmic reticulum (RER), and the increased number of
autolysosomes (Fig. 2A-C).
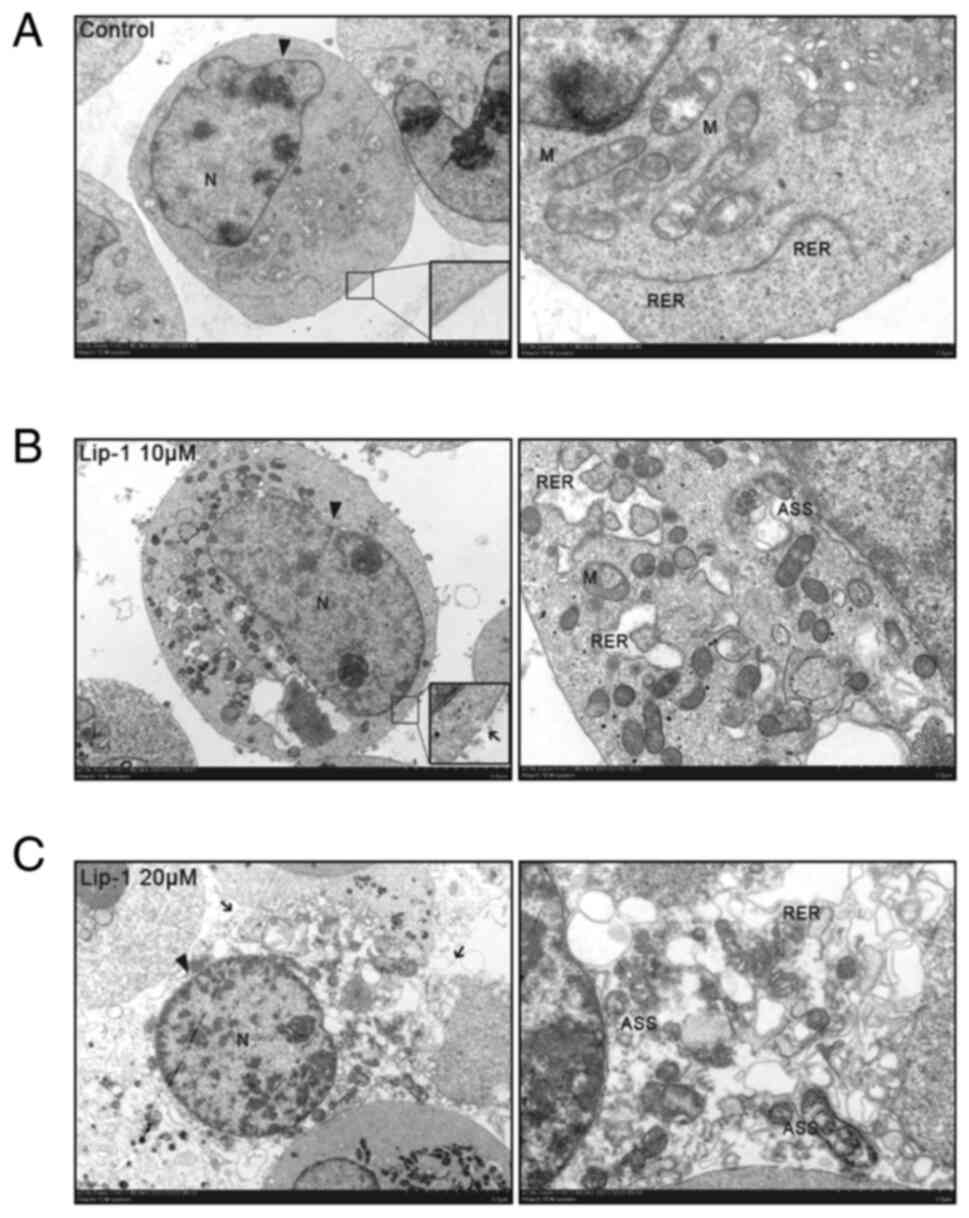 | Figure 2Representative transmission electron
micrographs of K562 cells treated with Lip-1. (A) Untreated K562
cells characterized by unexpanded RER with the plasma membrane
intact. (B) Expanded RER, ruptured plasma membrane, and ASS
observed in K562 cells treated with 10 µM Lip-1 for 24 h. (C)
Post-treatment with 20 µM Lip-1 for 24 h. K562 cells were
characterized by swelling, ruptured plasma membrane, expanded RER,
and an abundance of ASS. N, nuclear; M, mitochondrion. Arrowheads
indicate the nuclear membrane; arrows indicate the ruptured plasma
membrane. Scale bars, 5 and 2 µm for x2,500 and x7,000 images,
respectively. Lip-1, liproxstatin-1; RER, rough endoplasmic
reticulum; ASS, autolysosomes. |
RNA-seq of the effects of Lip-1 on K562
cells
To explore the molecular mechanisms underlying
Lip-1-induced cell death, RNA-seq was performed on K562 cells
treated with 0, 10, 20 µM Lip-1. To confirm the type of cell death,
DEGs were annotated via GO analysis, and the main biological
processes regulated by Lip-1, including G1/S transition of miotic
cell cycle, the extrinsic apoptotic signaling pathway, and the
intrinsic apoptotic signaling pathway (Fig. 3A and B) were identified. By
analyzing sequencing data, DEGs were distinguished and volcano
plots were constructed (Fig. 3C and
D). In addition, KEGG analysis indicated that signaling
pathways that are fundamental for the modulation of cell
proliferation and death, including autophagy and apoptosis, were
deeply affected by Lip-1 treatment (Fig. 3E and F). Collectively, these
results demonstrated that cell-cycle arrest, apoptosis, and
autophagy are involved in Lip-1-induced cell death at the
transcriptome level.
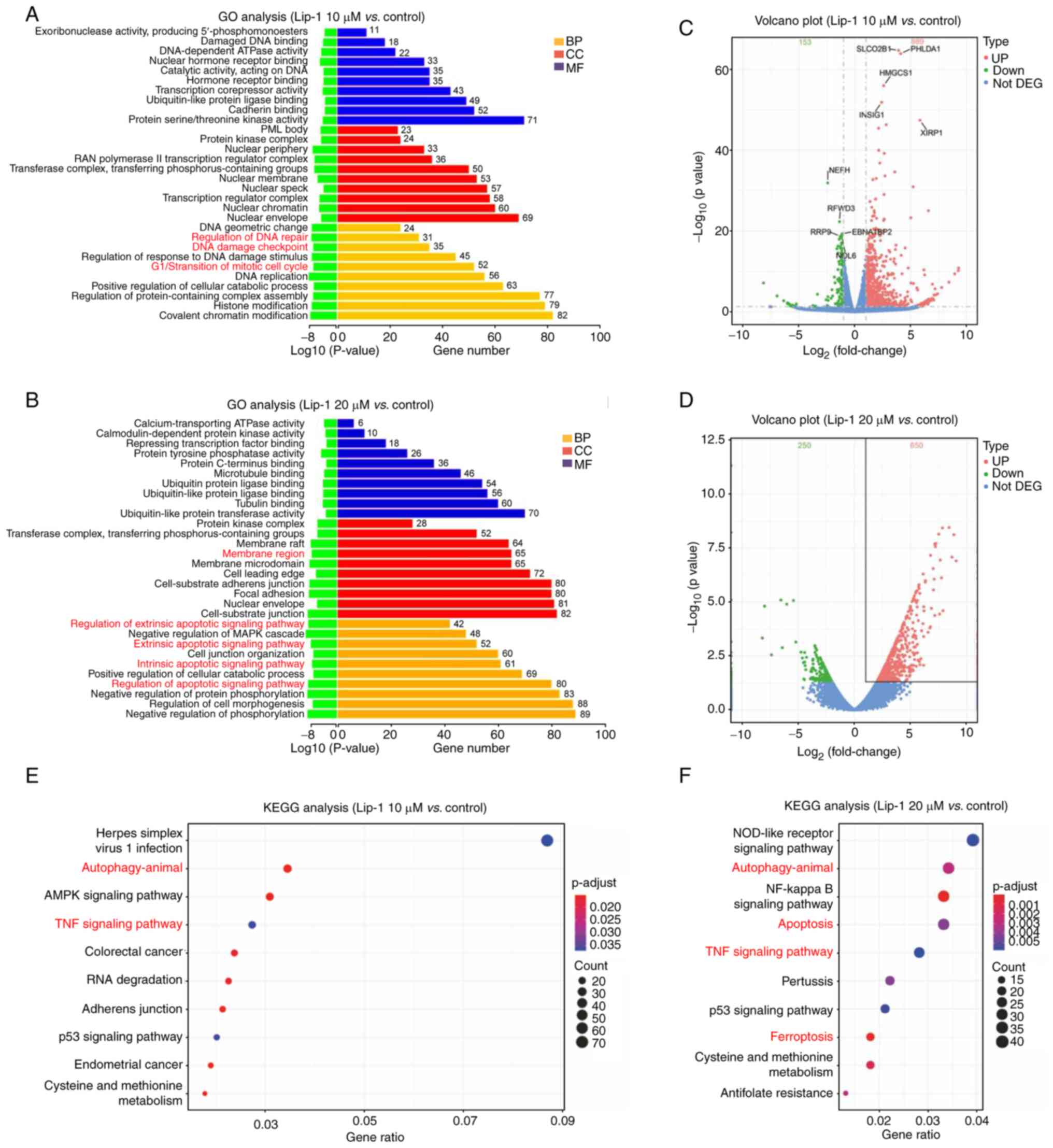 | Figure 3RNA sequencing analysis of the effect
of Lip-1 on the expression profile of K562 cells. (A and B) Gene
Ontology annotations of DEGs were summarized according to
biological process, cellular component, and molecular function. (C
and D) Volcano plots were used to visualize DEGs between the
Lip-1-treated and control groups. (E and F) Top 10 enriched Kyoto
Encyclopedia of Genes and Genomes pathways in 10 and 20 µM
Lip-1-treated K562 cells compared to the control group. p.adjust,
Benjamini-Hochberg adjusted P-value. Lip-1, liproxstatin-1; GO,
Gene Ontology; DEGs, differentially expressed genes; BP, biological
process; CC, cellular component; MF, molecular function; KEGG,
Kyoto Encyclopedia of Genes and Genomes. |
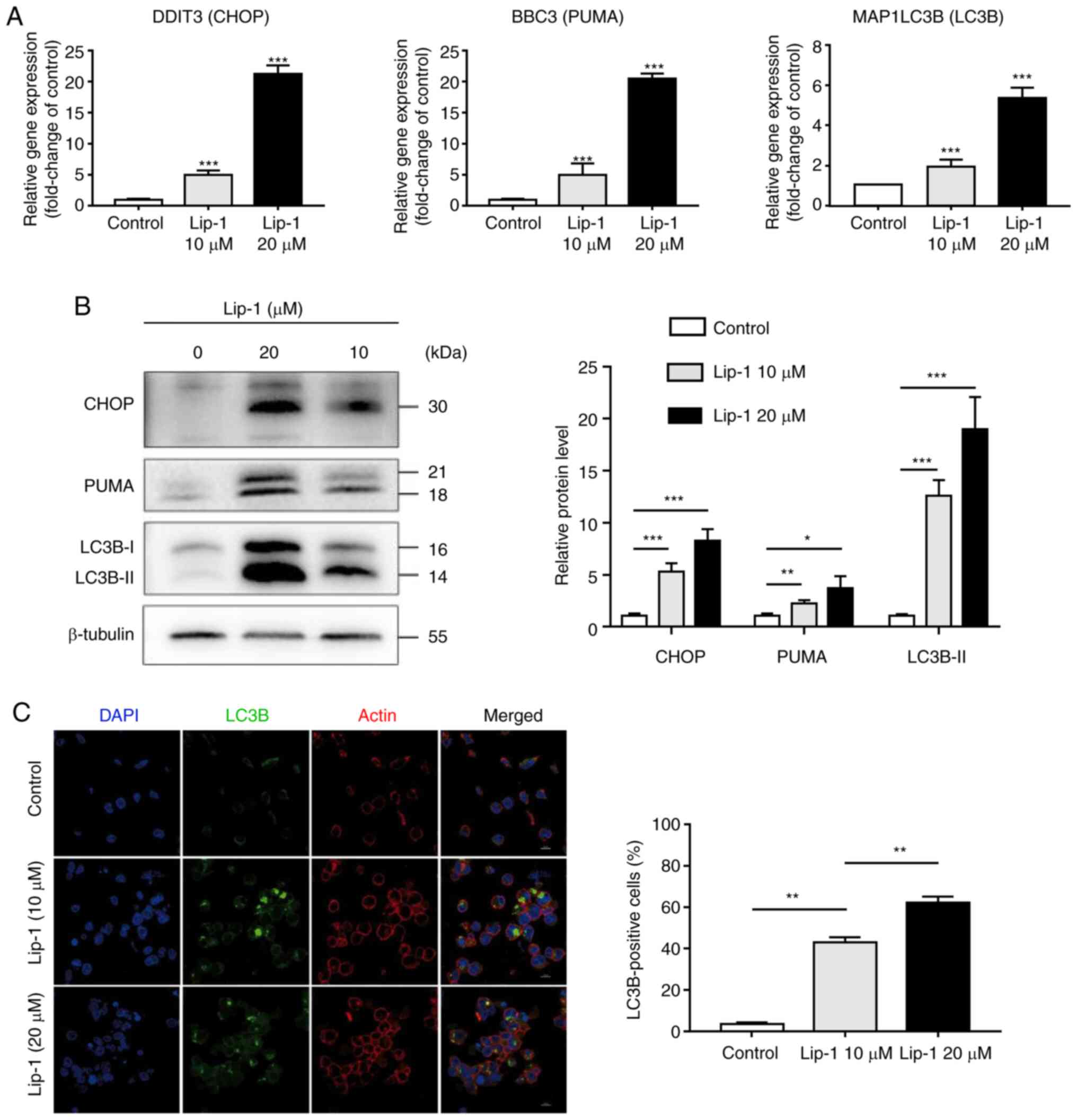
ER stress and autophagy are involved in
Lip-1 induced K562 cells
To identify whether the expanded ER was related to
ER stress, the expression levels of CHOP and PUMA, both of which
are associated with ER stress-mediated apoptosis, were examined
(34). As expected, transcriptome
and protein levels of CHOP and PUMA were elevated in Lip-1-treated
K562 cells (Fig. 4A and B). To
explore whether Lip-1 induced autophagy in K562 cells, RT-qPCR and
western blotting were performed; it was determined that LC3B gene
expression was significantly increased in Lip-1-treated cells than
in untreated cells and the expression of LC3B-Ⅱ, a hallmark product
of autophagy, was markedly increased after 24-h treatment of K562
cells with 10 or 20 µM Lip-1 (Fig. 4A
and B). To visualize the formation of autophagosomes,
immunofluorescence staining was performed; using a confocal laser
scanning microscope, it was observed that the number of LC3 puncta
was increased in Lip-1-treated cells (Fig. 4C). These results collectively
indicated that treatment with Lip-1 potently induced ER stress and
autophagy in K562 cells.
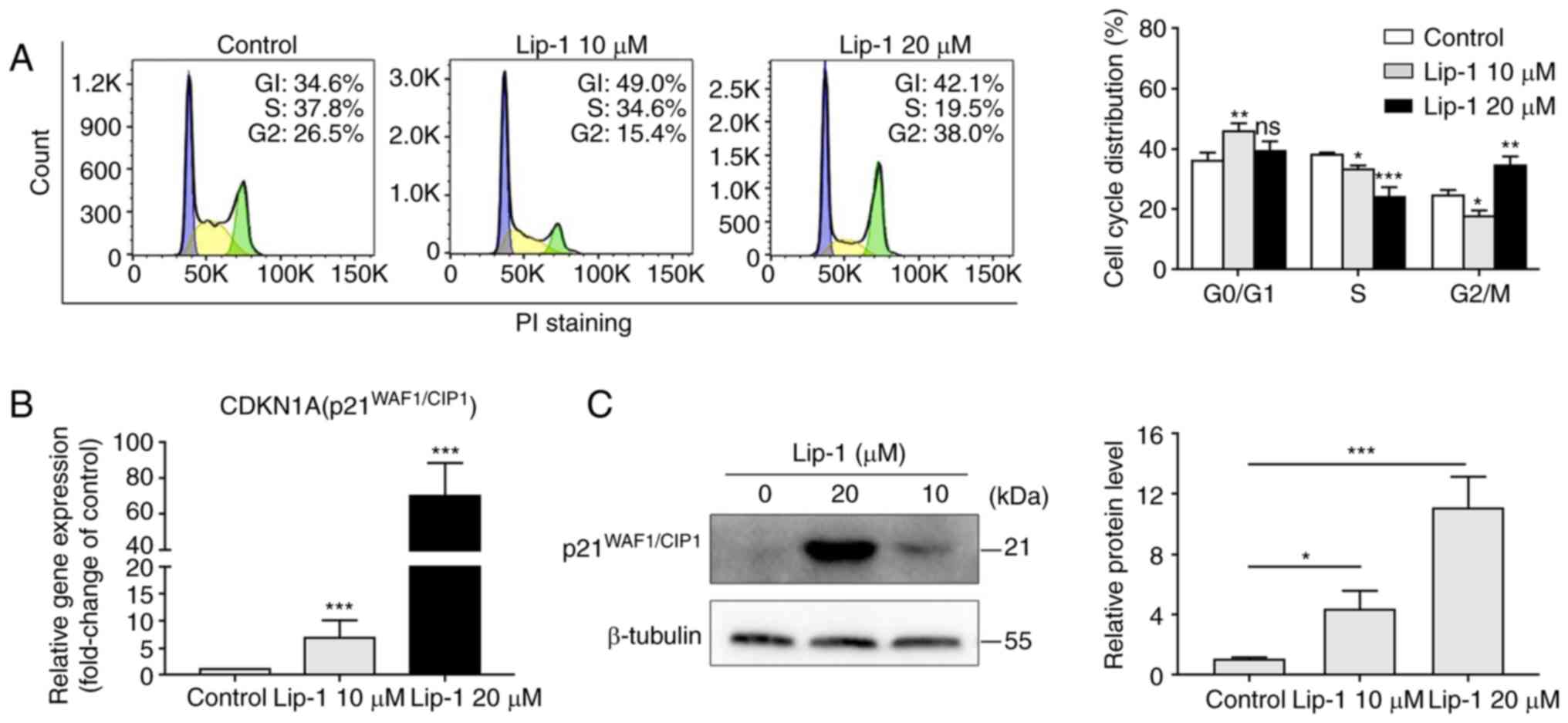
Lip-1 induces
p21WAF1/CIP1-mediated cell cycle arrest in K562
cells
To investigate the inhibitory effect of cell
proliferation, flow cytometry was performed to analyze the
cell-cycle distribution. As revealed in Fig. 5A, the proportion of cells in the S
phase decreased in a concentration-dependent manner, but the
opposite changes were noted in the proportion of cells in the G2/M
phase upon treatment with 10 or 20 µM Lip-1 for 24 h.
Flow cytometric results indicated that Lip-1
arrested the cell cycle at the G1 phase in both experimental
groups, but cell cycle at the G2 or M phase was only observed in
the group treated with a higher concentration of Lip-1.
p21WAF1/CIP1, a universal inhibitor of cyclin-dependent
kinases (35,36), was upregulated in K562 cells
treated with Lip-1 compared with that in untreated cells at the
transcriptome level (Fig. 5B).
Western blot analysis demonstrated that p21WAF1/CIP was
highly expressed in Lip-1-treated K562 cells but expressed at a
very low level in normal K562 cells (Fig. 5C). Taken together, the data
indicated that Lip-1 markedly upregulated p21WAF1/CIP
expression and induced cell cycle arrest.
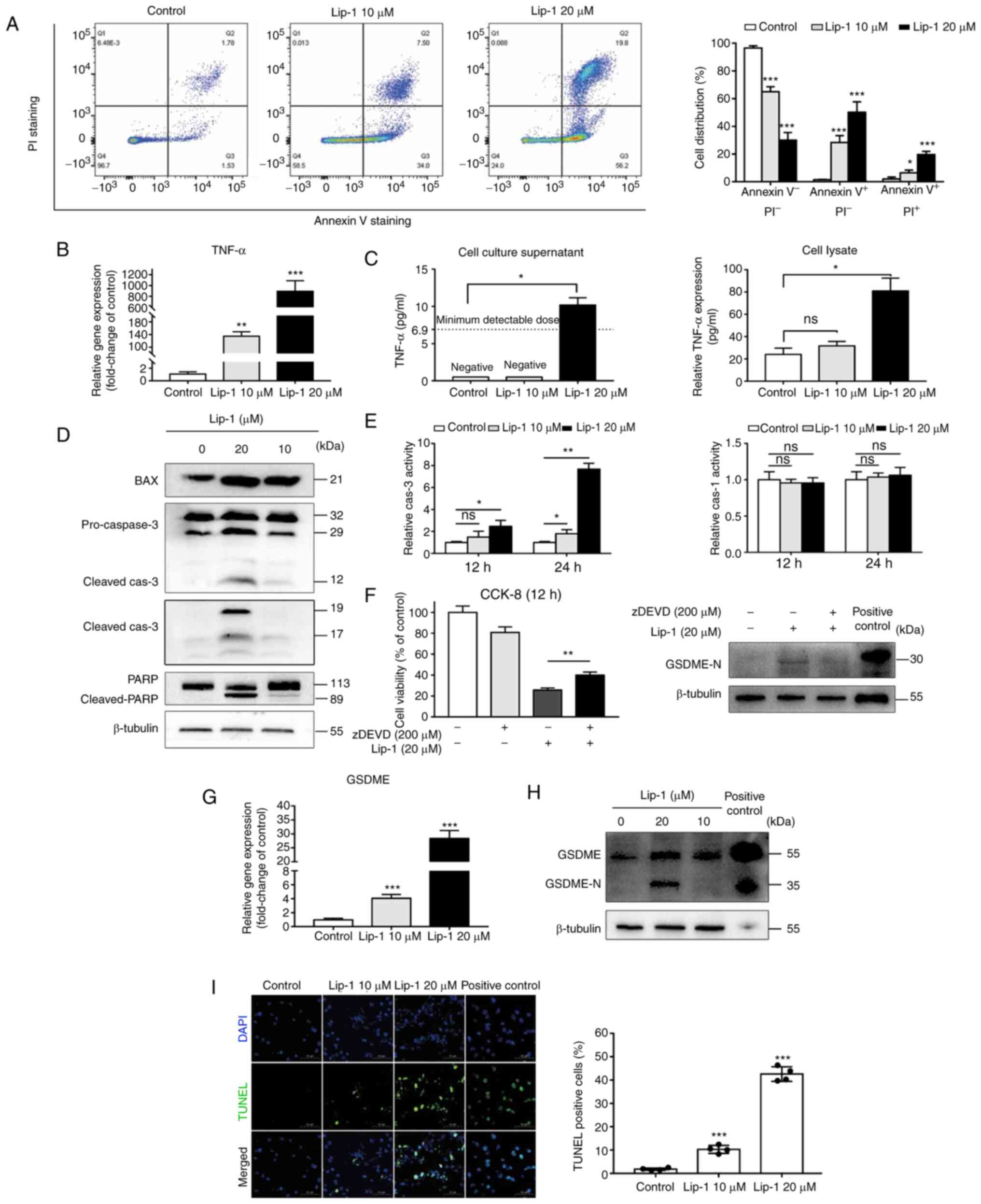
Lip-1 induces apoptosis,
caspase-3/GSDME-dependent secondary pyroptosis, and DNA damage in
K562 cells
To investigate the function of the plasma membrane,
an Annexin Ⅴ-FITC/PI double staining assay was performed. Because
PI cannot penetrate an unimpaired plasma membrane, the increase in
the PI-positive cell population indicated that Lip-1 damaged the
plasma membrane (Fig. 6A). TNF-α
can activate the extrinsic apoptotic signaling pathway as a type of
death ligand (37). Thus, it was
determined that the gene expression of TNF-α was significantly
increased in Lip-1-treated cells compared to that in untreated
cells (Fig. 6B). Subsequently,
ELISA was used to assess TNF-α protein levels in cell supernatants
and lysates. Limited by the sensitivity of the ELISA kit used, the
TNF-α concentration was undetectable in the cell supernatant in the
control group and in the lower concentration experimental group
(Lip-1 10 µM); however, the TNF-α concentration in another
experimental group (Lip-1, 20 µM) was 10.2 pg/ml on average
(Fig. 6C, left panel). The
concentration of TNF-α in cell lysates was greater in the higher
concentration experimental group (Lip-1, 20 µM) than in the control
group; the same was true for supernatants (Fig. 6C, right panel). It was also found
that the protein level of BAX was upregulated in Lip-1-treated K562
cells (Fig. 6D), indicating that
the intrinsic apoptotic signaling pathway was involved in
Lip-1-induced cell death.
Caspase-3 is an executor in both intrinsic and
extrinsic apoptosis, and it can be cleaved into different subunits
to execute biological functions (10,38,39).
Western blot analysis revealed that caspase-3 was activated in
Lip-1-treated cells in a dose-dependent manner (Fig. 6D). Accompanied by caspase-3
activation, PARP, as a substrate of caspase-3 (40), was cleaved into an 89-kDa fragment
(Fig. 6D). The colorimetric assay
was performed to validate and quantify the activation of caspase-3
(Fig. 6E, left panel). Although
caspase-3 is a hallmark of apoptosis, in this scenario, the
morphology and appearance of the dying cells treated with 20 µM
Lip-1 were not consistent with typical apoptotic cells.
Furthermore, caspase-1 was not activated in Lip-1-treated K562
cells (Fig. 6E, right panel). Some
dying cells were rescued upon using the caspase-3-specific
inhibitor z-DEVD-FMK, and the protein level of N-terminal GSDME was
reduced after z-DEVD-FMK treatment (Fig. 6F). RT-qPCR results demonstrated
that the gene expression of GSDME was upregulated in both
experimental groups compared to that in the control group (Fig. 6G). However, the protein level of
the cleaved N-terminal GSDME, which plays a key role in cell
pyroptosis, only increased in K562 cells treated with 20 µM Lip-1
for 24 h (Fig. 6H). In addition,
it was determined that K562 cells expressed little GSDME compared
to positive control SH-SY5Y cells, based on the integrated density
of the loading control (Fig. 6H).
These results indicated that Lip-1 treatment potently induced cell
apoptosis and secondary pyroptosis and that the induction of
secondary pyroptosis in K562 cells was caspase-3/GSDME-dependent
and not caspase-1/GSDMD-dependent.
Finally, the terminal deoxynucleotidyl transferase
dUTP nick-end labelling (TUNEL) assay was performed to evaluate DNA
fragmentation. It was determined that the number of TUNEL-positive
cells increased in a concentration-dependent manner (Fig. 6I). Therefore, Lip-1 is considered
to be a type of DNA-damaging drug.
Discussion
Lip-1, a spiroquinoxalinamine derivative, was first
discovered in a library of small molecules in 2014 (25), and has since been used as a
ferroptosis inhibitor. Lip-1 physically disrupts the outer membrane
of gram-negative bacteria (26).
However, the anticancer effects of Lip-1 have not yet been
reported. In the present study, it was determined that Lip-1
inhibits K562 leukemia cell proliferation by inducing cell cycle
arrest, apoptosis, and secondary pyroptosis, and it is considered
that the apoptosis-to-pyroptosis switch requires relatively high
levels of GSDME and caspase-3 activation.
GSDME belongs to the gasdermin family, and cleavage
of GSDME by caspase-3 during apoptosis mediates progression to
secondary pyroptosis (16).
Caspase-3 plays a key role in apoptosis and can be induced via
extrinsic or intrinsic apoptotic signaling pathways. Extrinsic
apoptosis is initiated by death receptors, including TNFR1/2 and
TRAILR1/2. Once ligand binding occurs, such as TNF-α binding to
TNFR1, the adaptor protein Fas-associated death domain protein is
recruited, followed by initiator caspase (caspase-8/-10) and
downstream executioner caspase activation (mainly caspase-3/-7)
(10,41). Intrinsic apoptosis is initiated by
a variety of microenvironmental perturbations, such as DNA damage,
ER stress, and mitotic defects (10). Subsequently, mitochondrial outer
membrane permeabilization results in the release of cytochrome
c, which activates caspase-9 (an initiator caspase) and
downstream caspase-3 (an executioner caspase) (20,41,42).
Proapoptotic (BAX/BIM/PUMA) and antiapoptotic
(BCL2/BCL-XL/BCL-W) members of the BCL2 family are
involved in the release of cytochrome c (10). Of note, ER stress is a common
cellular process that responds to microenvironmental perturbations.
If ER-stressed cells cannot restore their capacity for protein
folding, they commit to self-destruction via the induction of CHOP,
which inhibits the expression of BCL2 and upregulates BIM (43). Herein, it was determined that both
extrinsic and intrinsic apoptotic signaling pathways are involved
in Lip-1-induced cell death via RNA-seq. The expression of BAX and
TNF-α, which represent intrinsic and extrinsic apoptosis,
respectively, were further investigated. BAX was upregulated in
both experimental groups, whereas TNF-α overexpression was observed
only in the experimental group treated with higher concentrations
of Lip-1. Using several techniques, it was determined that
caspase-3 was activated in a doseand time-dependent manner.
However, apoptotic cells did not exhibit a typical apoptotic
appearance. By contrast, cell swelling, membrane rupture, and ER
expansion was observed in Lip-1-treated K562 cells. The plasma
membrane remains intact in apoptotic cells until the very late
stage of apoptosis, and apoptotic cells are characterized by cell
shrinkage, not swelling (44). The
morphology of Lip-1-treated K562 cells was very different from that
of apoptotic cells; therefore, it was hypothesized that necrosis or
pyroptosis may occur in Lip-1-induced cell death. Cleaved
N-terminal GSDME production was then assessed and it was observed
that Lip-1-induced apoptosis mediated progression to secondary
pyroptosis. Additionally, the expanded ER and increased expression
of CHOP and PUMA suggested that ER stress plays a role in
Lip-1-induced cell death. In the past few decades, researchers have
designed numerous drugs targeting the apoptotic pathway, including
BH3 mimetics, stapled peptides, and IAP inhibitors, some of which
have achieved favorable clinical results (41). The use of venetoclax (a novel
selective BCL2 inhibitor) is successful; however, the possibility
of primary or acquired drug resistance should not be ignored
(45,46). Pyroptosis has several advantages
over apoptosis, such as induction of faster cell death and
activation of antitumor immunity (16,19),
indicating that targeting the pyroptosis pathway is a better
therapeutic strategy than targeting the apoptotic pathway.
Therefore, theoretically, venetoclax-resistant cells could respond
to Lip-1 (a pyroptosis inducer).
Various types of RCD occur in distinct cells treated
concurrently with a specific drug in vitro. End-stage
apoptosis is usually followed by rupture of the plasma membrane and
the development of a necrotic morphotype (secondary necrosis)
(10,47). Guzik et al (48) elucidated that neutrophil exposure
to cigarette smoke extract leads to atypical cell death with
features shared among autophagy, apoptosis, and necrosis. In
another study (49), necrotic
cells were often observed concurrently with apoptotic cells, when
the cells were treated with parthenolide at the same concentration.
In the present study, both apoptotic and pyroptotic cells were
observed. Consistent with the results from earlier studies
(13,16), cells with high levels of GSDME
developed secondary pyroptosis following apoptosis. Therefore, it
is considered that the dying cells were undergoing apoptosis and
that apoptosis and pyroptosis in Lip-1-treated K562 cells occurred
sequentially.
Different types of RCD occasionally share the same
upstream molecules; accordingly, crosstalk between RCD pathways is
inevitable. Ferroptosis is an iron-dependent form of RCD, and
differs from apoptosis by excessive lipid peroxidation, lack of
glutathione, and inactivation of caspase-3 (50,51).
However, a recent study demonstrated that the ER stress-mediated
CHOP-PUMA pathway was associated with the interaction between
apoptosis and ferroptosis (52).
Moreover, p53 is a key factor that mediates the crosstalk between
apoptosis and ferroptosis (53-55).
Caspase-3 plays a central role in both apoptosis and pyroptosis,
and whether cells undergo pyroptosis depends on the expression of
GSDME, a downstream molecule of caspase-3 (13). A recent study by Rogers et
al (56) demonstrated that
cleaved N-terminal GSDME targets the mitochondria to release
cytochrome c and activate caspase-3. The interaction between
apoptosis and pyroptosis is delicate and complex. TNF-α acts as a
death ligand in the extrinsic apoptotic pathway and initiates
caspase-8 activation (10). TNF-α
is also a crucial signaling molecule in the necroptotic pathway;
however, in necroptosis, the activity of caspase-8 is suppressed,
which eventually results in the expression of RIPK1/3, exerting a
series of autophosphorylation or transphosphorylation effects
(10,57,58).
In the present study, it was determined that TNF-α and activated
caspase-3 were highly expressed in Lip-1-treated K562 cells, and
that the caspase-3-specific inhibitor z-DEVD-FMK could not
completely reverse cell death. Therefore, it was hypothesized that
another type of RCD may be involved in Lip-1-induced cell death.
Cell cycle progression and division are regulated by cyclins,
cyclin-dependent kinases, and cyclin-dependent kinase inhibitors.
There are three main checkpoints: G1, G2/M, and metaphase (59). Non-functional checkpoints result in
uncontrolled cell proliferation (60), and unscheduled cell division is a
hallmark of malignant tumors. In the past few decades, scientists
have developed adequate cell cycle therapeutics. Palbociclib, a
CDK4/6 inhibitor, has been used to treat breast cancer (61). Cytarabine, a first-line drug for
the treatment of leukemia, kills leukemic cells by blocking the
cell cycle at the G1 phase (6). In
the present study, it was determined that the G1/S transition of
the mitotic cell cycle was associated with Lip-1-induced cell death
via RNA-seq. Flow cytometry revealed that Lip-1 arrested the cell
cycle at the G1 phase, and even at the G2/M phase. Moreover,
p21WAF1/CIP1, a member of the CIP/KIP family, was
upregulated in Lip-1-treated K562 cells. As a type of
cyclin-dependent kinase inhibitor, p21WAF1/CIP1 can
block the cell cycle at different phases by inhibiting numerous
types of cyclin/cyclin-dependent kinase complexes, including cyclin
A/E-CDK2, cyclin A-CDK1, and cyclin B-CDK1 (61,62).
Thus, Lip-1 is a potential cell cycle therapeutic agent.
There are some limitations to the present study.
First, numerous cancers express low levels of GSDME owing to
aberrant promoter hypermethylation, and demethylation treatment
increases cellular susceptibility to apoptosis (63-65).
In a previous study, it was revealed that the expression of GSDME
in K562 cells is very low, and cells develop secondary necrosis
after apoptosis when lacking sufficient GSDME (13). Upon treatment with Lip-1, GSDME
expression increased, and it is considered that K562 cells
underwent secondary pyroptosis, not necrosis, after apoptosis.
Thus, whether Lip-1 has demethylation effects requires further
investigation. Second, although autophagy is considered a survival
process to maintain cellular homeostasis, autophagy-dependent cell
death is a novel type of RCD (10,21).
It was determined that autophagy is involved in Lip-1-induced cell
death; however, whether it is beneficial or harmful to cell
survival remains unknown. Third, p21WAF1/CIP1 was
upregulated in K562 cells; however, how p21WAF1/CIP1
1delicately modulated the cell cycle remains unclear. Thus, p21
knockdown or knockout should be performed in future studies.
Finally, GSDME is highly expressed in various normal tissues
(63,66), and high levels of GSDME increase
the side effects of chemotherapeutic drugs (13,19).
The efficacy and side effects of Lip-1 treatment in vivo
were not determined; therefore, it is difficult to elucidate the
role of Lip-1 in leukemogenesis. The underlying mechanisms should
be studied further.
In conclusion, it was revealed that Lip-1 inhibits
K562 cell proliferation in vitro. To the best of our
knowledge, the present study is the first to report that Lip-1
treatment induces cell cycle arrest, apoptosis, and
caspase-3/GSDME-dependent secondary pyroptosis in K562 leukemia
cells. Moreover, caspase-3 activation and relatively high levels of
GSDME were revealed to be responsible for inducing secondary
pyroptosis. Additionally, ER stress and autophagy were involved in
Lip-1-induced cell death. These findings provide new insights into
Lip-1 as a promising candidate for the treatment of leukemia. This
research would be the base for further studies.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
HQD contributed to the design of the study. HQD,
SJL, PC, YLX, YD, NS, and DHD contributed to the acquisition,
analysis, or interpretation of the data. HQD and SJL drafted the
manuscript, and PC revised it critically for important intellectual
content. HQD and PC confirm the authenticity of all the raw data.
All authors read and approved the final version to be published and
agree to be accountable for all aspects of the work in ensuring
that questions related to the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
We would like to thank Professor Jiao Lan and Mr
Ming-Zheng Mo (Experiment Center, The People's Hospital of Guangxi
Zhuang Autonomous Region, Nanning, China), for their assistance
with flow cytometry. We would also like to thank Mr Jian-Long Dong
and Mrs Gui-Xiang Li for their English language assistance and Mr
Yi-Qiang Wang for supplying some of the antibodies.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 81660025). The funders had no role
in the study design, data collection and analysis, interpretation
of the data, writing of the report, or the decision to submit this
article for publication.
References
|
1
|
Bonnet D and Dick JE: Human acute myeloid
leukemia is organized as a hierarchy that originates from a
primitive hematopoietic cell. Nat Med. 3:730–737. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Steinwascher S, Nugues AL, Schoeneberger H
and Fulda S: Identification of a novel synergistic induction of
cell death by Smac mimetic and HDAC inhibitors in acute myeloid
leukemia cells. Cancer Lett. 366:32–43. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tamm I, Kornblau SM, Segall H, Krajewski
S, Welsh K, Kitada S, Scudiero DA, Tudor G, Qui YH, Monks A, et al:
Expression and prognostic significance of IAP-family genes in human
cancers and myeloid leukemias. Clin Cancer Res. 6:1796–1803.
2000.PubMed/NCBI
|
|
4
|
Zeng H, Chen W, Zheng R, Zhang S, Ji JS,
Zou X, Xia C, Sun K, Yang Z, Li H, et al: Changing cancer survival
in China during 2003-15: A pooled analysis of 17 population-based
cancer registries. Lancet Glob Health. 6:e555–e567. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Long L, Assaraf YG, Lei ZN, Peng H, Yang
L, Chen ZS and Ren S: Genetic biomarkers of drug resistance: A
compass of prognosis and targeted therapy in acute myeloid
leukemia. Drug Resist Updat. 52:1007032020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Murphy T and Yee KWL: Cytarabine and
daunorubicin for the treatment of acute myeloid leukemia. Expert
Opin Pharmacother. 18:1765–1780. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
DiNardo CD, Pratz K, Pullarkat V, Jonas
BA, Arellano M, Becker PS, Frankfurt O, Konopleva M, Wei AH,
Kantarjian HM, et al: Venetoclax combined with decitabine or
azacitidine in treatment-naive, elderly patients with acute myeloid
leukemia. Blood. 133:7–17. 2019. View Article : Google Scholar :
|
|
8
|
Carter JL, Hege K, Yang J, Kalpage HA, Su
Y, Edwards H, Hüttemann M, Taub JW and Ge Y: Targeting multiple
signaling pathways: The new approach to acute myeloid leukemia
therapy. Signal Transduct Target Ther. 5:2882020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xia X, Wang X, Cheng Z, Qin W, Lei L,
Jiang J and Hu J: The role of pyroptosis in cancer: Pro-cancer or
pro-'host'? Cell Death Dis. 10:6502019. View Article : Google Scholar
|
|
10
|
Galluzzi L, Vitale I, Aaronson SA, Abrams
JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews
DW, et al: Molecular mechanisms of cell death: Recommendations of
the Nomenclature Committee on Cell Death. 2018.Cell Death Differ.
25:486–541. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ding J, Wang K, Liu W, She Y, Sun Q, Shi
J, Sun H, Wang DC and Shao F: Pore-forming activity and structural
autoinhibition of the gasdermin family. Nature. 535:111–116. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kuang S, Zheng J, Yang H, Li S, Duan S,
Shen Y, Ji C, Gan J, Xu XW and Li J: Structure insight of GSDMD
reveals the basis of GSDMD autoinhibition in cell pyroptosis. Proc
Natl Acad Sci USA. 114:10642–10647. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Y, Gao W, Shi X, Ding J, Liu W, He H,
Wang K and Shao F: Chemotherapy drugs induce pyroptosis through
caspase-3 cleavage of a gasdermin. Nature. 547:99–103. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shi J, Zhao Y, Wang K, Shi X, Wang Y,
Huang H, Zhuang Y, Cai T, Wang F and Shao F: Cleavage of GSDMD by
inflammatory caspases determines pyroptotic cell death. Nature.
526:660–665. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang YY, Liu XL and Zhao R: Induction of
pyroptosis and its implications in cancer management. Front Oncol.
9:9712019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rogers C, Fernandes-Alnemri T, Mayes L,
Alnemri D, Cingolani G and Alnemri ES: Cleavage of DFNA5 by
caspase-3 during apoptosis mediates progression to secondary
necrotic/pyroptotic cell death. Nat Commun. 8:141282017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Johnson DC, Taabazuing CY, Okondo MC, Chui
AJ, Rao SD, Brown FC, Reed C, Peguero E, de Stanchina E, Kentsis A
and Bachovchin DA: DPP8/DPP9 inhibitor-induced pyroptosis for
treatment of acute myeloid leukemia. Nat Med. 24:1151–1156. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang W, Liu S, Li Y, Wang Y, Deng Y, Sun
W, Huang H, Xie J, He A, Chen H, et al: Pyridoxine induces
monocyte-macrophages death as specific treatment of acute myeloid
leukemia. Cancer Lett. 492:96–105. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Z, Zhang Y and Lieberman J: Lighting
a fire: Can we harness pyroptosis to ignite antitumor immunity?
Cancer Immunol Res. 9:2–7. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang M, Qi L, Li L and Li Y: The
caspase-3/GSDME signal pathway as a switch between apoptosis and
pyroptosis in cancer. Cell Death Discov. 6:1122020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Klionsky DJ, Petroni G, Amaravadi RK,
Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cadwell K,
Cecconi F, Choi AMK, et al: Autophagy in major human diseases. EMBO
J. 40:e1088632021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tang R, Xu J, Zhang B, Liu J, Liang C, Hua
J, Meng Q, Yu X and Shi S: Ferroptosis, necroptosis, and pyroptosis
in anticancer immunity. J Hematol Oncol. 13:1102020. View Article : Google Scholar :
|
|
23
|
Galluzzi L and Green DR:
Autophagy-independent functions of the autophagy machinery. Cell.
177:1682–1699. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Frank D and Vince JE: Pyroptosis versus
necroptosis: Similarities, differences, and crosstalk. Cell Death
Differ. 26:99–114. 2019. View Article : Google Scholar
|
|
25
|
Friedmann Angeli JP, Schneider M, Proneth
B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch
A, Eggenhofer E, et al: Inactivation of the ferroptosis regulator
Gpx4 triggers acute renal failure in mice. Nat Cell Biol.
16:1180–1191. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Klobucar K, Côté JP, French S, Borrillo L,
Guo ABY, Serrano-Wu MH, Lee KK, Hubbard B, Johnson JW, Gaulin JL,
et al: Chemical screen for vancomycin antagonism uncovers probes of
the gram-negative outer membrane. ACS Chem Biol. 16:929–942. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lozzio CB and Lozzio BB: Human chronic
myelogenous leukemia cell-line with positive Philadelphia
chromosome. Blood. 45:321–334. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pertea M, Pertea GM, Antonescu CM, Chang
TC, Mendell JT and Salzberg SL: StringTie enables improved
reconstruction of a transcriptome from RNA-seq reads. Nat
Biotechnol. 33:290–295. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15:5502014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gene Ontology Consortium: Gene ontology
consortium: Going forward. Nucleic Acids Res. 43(Database Issue):
D1049–D1056. 2015. View Article : Google Scholar :
|
|
31
|
Kanehisa M, Araki M, Goto S, Hattori M,
Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T
and Yamanishi Y: KEGG for linking genomes to life and the
environment. Nucleic Acids Res. 36(Database Issue): D480–D484.
2008. View Article : Google Scholar :
|
|
32
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)). method Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
34
|
Zhao X, Zhu L, Liu D, Chi T, Ji X, Liu P,
Yang X, Tian X and Zou L: Sigma-1 receptor protects against
endoplasmic reticulum stress-mediated apoptosis in mice with
cerebral ischemia/reperfusion injury. Apoptosis. 24:157–167. 2019.
View Article : Google Scholar
|
|
35
|
Xiong Y, Hannon GJ, Zhang H, Casso D,
Kobayashi R and Beach D: p21 is a universal inhibitor of cyclin
kinases. Nature. 366:701–704. 1993. View Article : Google Scholar
|
|
36
|
Lai L, Shin GY and Qiu H: The role of cell
cycle regulators in cell survival-dual functions of
cyclin-dependent kinase 20 and p21Cip1/Waf1. Int J Mol
Sci. 21:85042020. View Article : Google Scholar
|
|
37
|
Pistritto G, Trisciuoglio D, Ceci C,
Garufi A and D'Orazi G: Apoptosis as anticancer mechanism: Function
and dysfunction of its modulators and targeted therapeutic
strategies. Aging (Albany NY). 8:603–619. 2016. View Article : Google Scholar
|
|
38
|
Nicholson DW, Ali A, Thornberry NA,
Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle
M, Lazebnik YA, et al: Identification and inhibition of the
ICE/CED-3 protease necessary for mammalian apoptosis. Nature.
376:37–43. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kavanagh E, Rodhe J, Burguillos MA, Venero
JL and Joseph B: Regulation of caspase-3 processing by cIAP2
controls the switch between pro-inflammatory activation and cell
death in microglia. Cell Death Dis. 5:e15652014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cohen GM: Caspases: The executioners of
apoptosis. Biochem J. 326(Pt 1): 1–16. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Carneiro BA and El-Deiry WS: Targeting
apoptosis in cancer therapy. Nat Rev Clin Oncol. 17:395–417. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nikoletopoulou V, Markaki M, Palikaras K
and Tavernarakis N: Crosstalk between apoptosis, necrosis and
autophagy. Biochim Biophys Acta. 1833:3448–3459. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Oakes SA and Papa FR: The role of
endoplasmic reticulum stress in human pathology. Annu Rev Pathol.
10:173–194. 2015. View Article : Google Scholar
|
|
44
|
Bortner CD and Cidlowski JA: Ions, the
movement of water and the apoptotic volume decrease. Front Cell Dev
Biol. 8:6112112020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Saliba AN, John AJ and Kaufmann SH:
Resistance to venetoclax and hypomethylating agents in acute
myeloid leukemia. Cancer Drug Resist. 4:125–142. 2021.PubMed/NCBI
|
|
46
|
Blombery P: Mechanisms of intrinsic and
acquired resistance to venetoclax in B-cell lymphoproliferative
disease. Leuk Lymphoma. 61:257–262. 2020. View Article : Google Scholar
|
|
47
|
Vanden Berghe T, Vanlangenakker N,
Parthoens E, Deckers W, Devos M, Festjens N, Guerin CJ, Brunk UT,
Declercq W and Vandenabeele P: Necroptosis, necrosis and secondary
necrosis converge on similar cellular disintegration features. Cell
Death Differ. 17:922–930. 2010. View Article : Google Scholar
|
|
48
|
Guzik K, Skret J, Smagur J, Bzowska M,
Gajkowska B, Scott DA and Potempa JS: Cigarette smoke-exposed
neutrophils die unconventionally but are rapidly phagocytosed by
macrophages. Cell Death Dis. 2:e1312011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Pozarowski P, Halicka DH and Darzynkiewicz
Z: Cell cycle effects and caspase-dependent and independent death
of HL-60 and Jurkat cells treated with the inhibitor of NF-kappaB
parthenolide. Cell Cycle. 2:377–383. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xia H, Zhang Z and You F: Inhibiting
ACSL1-related ferroptosis restrains murine coronavirus infection.
Viruses. 13:23832021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lin L, Zhang MX, Zhang L, Zhang D, Li C
and Li YL: Autophagy, pyroptosis, and ferroptosis: New regulatory
mechanisms for atherosclerosis. Front Cell Dev Biol. 9:8099552022.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lee YS, Lee DH, Choudry HA, Bartlett DL
and Lee YJ: Ferroptosis-induced endoplasmic reticulum stress:
Cross-talk between ferroptosis and apoptosis. Mol Cancer Res.
16:1073–1076. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chu B, Kon N, Chen D, Li T, Liu T, Jiang
L, Song S, Tavana O and Gu W: ALOX12 is required for p53-mediated
tumour suppression through a distinct ferroptosis pathway. Nat Cell
Biol. 21:579–591. 2019. View Article : Google Scholar :
|
|
54
|
Jiang L, Kon N, Li T, Wang SJ, Su T,
Hibshoosh H, Baer R and Gu W: Ferroptosis as a p53-mediated
activity during tumour suppression. Nature. 520:57–62. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zheng DW, Lei Q, Zhu JY, Fan JX, Li CX, Li
C, Xu Z, Cheng SX and Zhang XZ: Switching apoptosis to ferroptosis:
Metal-organic network for high-efficiency anticancer therapy. Nano
Lett. 17:284–291. 2017. View Article : Google Scholar
|
|
56
|
Rogers C, Erkes DA, Nardone A, Aplin AE,
Fernandes-Alnemri T and Alnemri ES: Gasdermin pores permeabilize
mitochondria to augment caspase-3 activation during apoptosis and
inflammasome activation. Nat Commun. 10:16892019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Newton K, Wickliffe KE, Dugger DL,
Maltzman A, Roose-Girma M, Dohse M, Kőműves L, Webster JD and Dixit
VM: Cleavage of RIPK1 by caspase-8 is crucial for limiting
apoptosis and necroptosis. Nature. 574:428–431. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Li J, McQuade T, Siemer AB, Napetschnig J,
Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, et
al: The RIP1/RIP3 necrosome forms a functional amyloid signaling
complex required for programmed necrosis. Cell. 150:339–350. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Icard P, Fournel L, Wu Z, Alifano M and
Lincet H: Interconnection between metabolism and cell cycle in
cancer. Trends Biochem Sci. 44:490–501. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Asghar U, Witkiewicz AK, Turner NC and
Knudsen ES: The history and future of targeting cyclin-dependent
kinases in cancer therapy. Nat Rev Drug Discov. 14:130–146. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ingham M and Schwartz GK: Cell-cycle
therapeutics come of age. J Clin Oncol. 35:2949–2959. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Dulić V, Kaufmann WK, Wilson SJ, Tlsty TD,
Lees E, Harper JW, Elledge SJ and Reed SI: p53-dependent inhibition
of cyclin-dependent kinase activities in human fibroblasts during
radiation-induced G1 arrest. Cell. 76:1013–1023. 1994. View Article : Google Scholar
|
|
63
|
Akino K, Toyota M, Suzuki H, Imai T,
Maruyama R, Kusano M, Nishikawa N, Watanabe Y, Sasaki Y, Abe T, et
al: Identification of DFNA5 as a target of epigenetic inactivation
in gastric cancer. Cancer Sci. 98:88–95. 2007. View Article : Google Scholar
|
|
64
|
Kim MS, Chang X, Yamashita K, Nagpal JK,
Baek JH, Wu G, Trink B, Ratovitski EA, Mori M and Sidransky D:
Aberrant promoter methylation and tumor suppressive activity of the
DFNA5 gene in colorectal carcinoma. Oncogene. 27:3624–3634. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wang CJ, Tang L, Shen DW, Wang C, Yuan QY,
Gao W, Wang YK, Xu RH and Zhang H: The expression and regulation of
DFNA5 in human hepatocellular carcinoma DFNA5 in hepatocellular
carcinoma. Mol Biol Rep. 40:6525–6531. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Van Laer L, Huizing EH, Verstreken M, van
Zuijlen D, Wauters JG, Bossuyt PJ, Van de Heyning P, McGuirt WT,
Smith RJ, Willems PJ, et al: Nonsyndromic hearing impairment is
associated with a mutation in DFNA5. Nat Genet. 20:194–197. 1998.
View Article : Google Scholar : PubMed/NCBI
|