Introduction
Malignant melanoma is one of the most aggressive
tumor types, and is characterized by high proliferation rates, poor
prognosis and low survival rate (1). The high rate of metastasis is the
main cause of mortality in patients with melanoma (2). Previous evidence has shown that
altered tumor metabolism suppresses the infiltration of immune
cells and helps melanoma cells to escape the immunity of the host
(3).
Metabolic reprogramming is a hallmark of cancer
(4), and a lipogenic phenotype is
a main characteristic of cancer. Sterol regulatory element binding
protein (SREBP) has been reported to be highly activated in tumors,
and their levels and activity are tightly controlled by endogenous
sterol levels via a negative feedback loop. Sterols regulate SREBP
by controlling its endoplasmic reticulum (ER)-to-Golgi apparatus
transport (5). SREBPs control
lipogenesis and lipid uptake, SREBP1 preferentially regulates the
lipogenic process by activating genes involved in fatty acid and
triglyceride synthesis (6),
whereas SREBP2 primarily controls cholesterol homeostasis by
activating genes required for cholesterol synthesis and uptake
(7).
Cancer cells become independent of systemic
regulation due to their autonomous generation of lipids. Cancer
cells engage in extensive de novo lipogenesis to produce
lipids (8). Three important
enzymes in de novo fatty acid synthesis, namely ATP-citrate
lyase (ACLY), acetyl coenzyme A (CoA) carboxylase (ACC) and fatty
acid synthase (FAS), are upregulated in numerous types of cancer
(9). ACLY produces acetyl CoA, in
the cytoplasm and provides the precursor of lipid synthesis, while
ACC controls the rate-limiting step of fatty acid synthesis. FAS
has been shown to play a key role in tumor malignant growth and at
the terminal step in de novo synthesis of fatty acids
(10). The FAS and
ACC genes are directly regulated by SREBP1 in fatty acid
synthesis. ACLY involved in cholesterol synthesis also participates
in fatty acid biosynthesis (11),
and is required for glucose transporter type 4 (GLUT4)-mediated
glucose-dependent fatty acid synthesis (12).
Cancer cells have alternative ways to acquire
nutrients, and certain tumors develop adaptations that allow them
to intake fatty acids directly from plasma or surrounding adipose
tissue (13). Increasing lipid
biosynthesis occurs in cancer cells, which influences the tumor
microenvironment, and SREBP1 transcription factors respond to
diverse environmental changes, such as lipid supply and
inflammatory signals (14). Cells
deal with excess lipids such as fatty acids by esterifying them to
form more inert neutral lipids, such as triacylglycerols or sterol
esters. Peroxisome proliferator-activated receptor γ (PPARγ) plays
a major role in adipogenesis and storage of fatty acids such as
triglycerides (15). The formation
of these neutral lipids occurs within membrane bilayers; however,
since bilayers are not suitable for storing large quantities of
neutral lipids, an emulsion of neutral-lipid droplets forms in the
cytoplasm (16). Lipid trafficking
is highly organized and regulated in space. The aforementioned
neutral-lipid emulsion originates in the ER, which serves as a
phospholipid reservoir in cells to generate stable emulsions
(17). When ER emulsions are
delayed, this leads to accumulation of neutral lipids in the ER and
blebbing out into inappropriate sites such as the nucleus (18). Lipids in the nucleus can sequester
transcription factors and chromatin components, as well as generate
lipoid ligands for certain nuclear receptors (17).
Tremella fuciformis-derived polysaccharide
(TFP) is extracted from Tremella fuciformis, which is a
common nutritional food in China and also a traditional Chinese
medicine due to its anti-inflammatory and anticancer properties,
and its ability to strengthen immunity, and reduce hypertension and
blood glucose (19). TFP has also
been used clinically for whitening skin, cancer and anti-aging
treatments in China. However, the protective mechanism of TFP is
not clear yet. The present study explored the function of TFP on
the lipid metabolism of B16 cells, and evaluated lipid metabolism
changes influenced the tumor growth and microenvironment in
vivo.
Materials and methods
Cell culture and treatment
The B16 mouse melanoma cells line was purchased from
the cell bank of Chinese Academy of Sciences (Shanghai, China).
Cells were cultured in DMEM medium with phenol red, supplemented
with 10% FBS, 100 U/ml penicillin, and 100 µg/ml
streptomycin in a dynamic incubation system at 37°C in a
5%-CO2 humidified atmosphere. The cells were treated in
the presence of series of dosages of TFP (0, 312.5, 625, 1,250,
2,500 and 5,000 µg/ml) for 24 h.
HPLC analysis of TFP
The content of TFP in B16 cells was detected by
high-performance lipid chromatography (HPLC). Briefly, the B16
cells with or without TFP exposure were centrifuged at 1,000 × g
for 3 min at room temperature and suspended with ultrapure water in
ice bath for 10 min, then centrifuged for 10 min. The supernatant
was collected for HPLC. HPLC separation was performed on sugar
column (water ultra-hydrogel 500) and detected using differential
detector 1200 (Agilent Technologies, Inc.). The mobile phase was
ultrapure water. The column temperature was kept at 85°C and the
flow rate was set at 0.6 ml/min.
Animal model and drug treatment
A total of 18 male C57BL/6 mice (6-8 weeks old;
weight, 25 g) were purchased from Shanghai Experimental Animal
Center, Chinese Academy of Sciences (Shanghai, China). The mice
were housed in temperature (25°C) and humidity (30-40%)-controlled
room, kept on a 12-h light/dark cycle and provided with
unrestricted amount of rodent chow and water. The animal protocol
was approved (approval no. 2020002) by the Animal Care and Use
Committee of Minnan Normal University (Zhangzhou, China).
To study the direct effects of TFP in vivo
tumor development, B16 cells were preincubated in vitro with
two dosages of TFP (2,500 and 5,000 µg/ml). After 24 h, the
cells were washed 3 times, and 1×106 B16 cells/ml
resuspended in 100 µl of PBS were inoculated subcutaneously
in the right flank of the mice. The tumor volume 2,000
mm3 calculated according to length and width was a limit
or the maximum permitted tumour burden in the present study. At the
experiment terminal, the mice were sacrificed by cervical
dislocation, sacrifice was confirmed when the mice had ceased
breathing and did not respond to stimulation. The tumors were
collected and weight was measured.
Cell proliferation assay
For determination of cell growth using Real Time
Cellular Analysis (RTCA), plate 16 assemblies were seeded with B16
cells (1×104 cells/well). The plate was then assembled
on the RTCA DP analyzer, after 24 h cells were treated with five
dosages of TFP (312.5, 625, 1,250, 2,500, 5,000 µg/ml), and
data were gathered at 5-min intervals for 120 h at 37°C in 5%
CO2.
Flow cytometric analysis of cell cycle,
apoptosis and co-localization
Cells were harvested by treatment with 0.25%
trypsin, resuspended with PBS, then stained with propidium iodide
and Annexin V. (cat. no. C1062M΄ Beyotime Institute of
Biotechnology) in dark at room temperature for 10-20 min; DNA
content of the cells was stained by DAPI at room temperature for 5
min. For co-localization, cells were stained with Cytopainter
Golgi/ER staining κit (cat. no. ab139485; Abcam), the Golgi should
exhibit green fluorescence, and ER should fluoresce red. Cell
membrane was stained by Dio fluoresce green (cat. no. C1993S;
Beyotime Institute of Biotechnology). Imaging flow cytometry (EMD
Millipore) was adjusted according to the procedure previously
described (20). Acquisition speed
was set up to low speed and the highest resolution. Cells were
acquired based on area and aspect ratio, gating out single cells
from the analysis. A total number of 10,000 cells for each sample
were acquired. Channel 2 was used to acquire Annexin V or Golgi,
cell membrane signaling, channel 4 was used to detect Nile red,
channel 7 was used to detect DAPI and channel 5 was used to detect
propidium iodide or ER. Data were analyzed in IDEAS 6.2 software
(EMD Millipore) after compensation of single-color control samples
using a compensation matrix. Co-localization of membrane, ER,
Golgi, or nucleus with lipid was analyzed using colocalization
analysis application Wizard in IDEAS software.
Nile Red staining
B16 cells cultured on confocal dish (Coverglass
Bottom Dish) were treated with different concentrations of TFP for
24 h. Then the cells were stained by Nile red (1:500) and DAPI
(1:1,000) for 30 min avoiding light, and then images were captured
using confocal laser scanning microscope (Leica Microsystems
GmbH).
Western blotting (WB)
The protein levels and phosphorylation status were
examined by WB. The cells were washed twice with ice-cold PBS and
lysed in RIPA protein lysis buffer containing 1 mM PMSF (cat. no.
R0010΄ Beijing Solarbio Science & Technology Co., Ltd.). The
samples were centrifuged at 13,000 × g for 10 min at 4°C before the
supernatant was collected and the protein concentration was
measured using a BCA kit (cat. no. P0012; Beyotime Institute of
Biotechnology). Samples were boiled for 5 min after being mixed
with a 5X loading buffer solution. The protein lysates (30
µg) were resolved in 8-10% SDS-polyacrylamide gels and
transferred to nitrocellulose membranes. The membranes were blocked
using 5% skimmed milk for 1 h at room temperature, washed three
times with TBST (0.1% Tween 20) for 10 min and then incubated
overnight at 4°C with various primary antibodies: anti-Adenosine
5′-monophosphate-activated protein kinase (AMPK; 1:2,000; cat. no.
ab32047; Abcam), anti-phosphorylated-AMP-activated protein kinase
(p-AMPK, 1:2,000; cat. no. EPR3051; Abcam), anti-SREBP-2 (1:500;
cat. no. ab30682; Abcam), anti-PPARγ (1:1,000; cat. no. C26H12;
Cell Signaling Technology, Inc.), anti-ACLY (1:1,000; cat. no.
ab40793; Abcam), anti-Lysosomal-associated membrane protein1
(LAMP1; 1:1,000; cat. no. ab24170; Abcam), anti-CD36 (1:5,000; cat.
no. ab133625; Abcam), anti-GLUT4 (1:500; cat. no. ab33780; Abcam),
anti-p-GLUT4 (1:2,000; cat. no. ab188317; Abcam), anti-FAS
(1:5,000; cat. no. ab128870; Abcam), anti-ACC (1:1,000; cat. no.
EP687Y; Abcam), anti-SREBP1 (1:1,000; cat. no. NB600-582; Novus
Biologicals, LLC). After incubation with rabbit HRP-conjugated
secondary antibody (1:8,000; cat. no. HAF008; R&D Systems,
Inc.) or mouse HRP-conjugated secondary antibody (1:8,000; cat. no.
HAF007; R&D Systems, Inc.), the immunoreactivity was detected
with using ECL solution (Beijing Solarbio Science & Technology
Co., Ltd.). Bands were scanned by OmegaLum C image system (Aplegen,
Inc.). Band signals were corrected by the Tubulin (1:1,000; cat.
no. NB600-936; Novus Biologicals, Inc.) or
glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1:2,000; cat. no.
ab9485; Abcam) signals.
Hematoxylin and eosin (H&E)
staining
Paraffin-embedded sections were cut at 3 µm
thickness, mounted on microslides and processed for H&E
staining, as previously described (21). The stained slides were dehydrated
and mounted using Cytoseal. Images of the slides were captured
(magnification, ×200) with a light microscope (Olympus
Corporation).
Immunohistochemistry
For immunohistochemistry, tissue sections were
deparaffinized with xylene and stepwise rehydrated with serial
dilutions of ethanol. After epitope retrieval with citric acid
buffer under high temperature and pressure for 1-2 min, and
endogenous horseradish peroxidase activity quenching with 3%
H2O2 for 10 min, the tissue sections were
then blocked in 5% goat serum (cat. no. C0265; Beyotime Institute
of Biotechnology) at room temperature for 1 h and incubated with
rabbit anti-mouse (ACLY, 1:100; PPARγ, 1:100; LAMP1, 1:100), and
mouse anti-mouse antibody (PDL1, 1:100, cat. no. ab238697; CD68,
1:100, cat. no. NB600-985; CD86, 1:50, cat. no. ab220188; all from
Abcam) at 4°C overnight. The primary antibody was visualized by
using anti-rabbit Alexa Fluor 647 (1:1,000; cat. no. P0180;
Beyotime Institute of Biotechnology), and anti-mouse FITC
conjugated antibody (1:1,000; cat. no. P0196; Beyotime Institute of
Biotechnology). The images were captured using Leica TCS SP8
confocal laser microscope.
RNA-seq, data processing, and gene
annotation
RNA of Model group and 5,000 µg/ml TFP group
were extracted. Total RNA was extracted using TRIzol reagent kit
(Invitrogen; Thermo Fisher scientific, Inc.). RNA quality was
assessed on an Agilent 2100 Bioanalyzer (Agilent Technologies,
Inc.). After total RNA was extracted, mRNA was enriched by Oligo
(dT) beads. Then the enriched mRNA was fragmented into short
fragments using fragmentation buffer and reversely transcribed into
cDNA by using NEBNext Ultra RNA Library Prep Kit for Illumina (New
England Biolabs). The purified double-stranded cDNA fragments were
end repaired, a base added, and ligated to Illumina sequencing
adapters. The ligation reaction was purified, ligated fragments
were subjected to size selection (200 bp) by AMPure XP Beads (1.0X)
and PCR amplification. The resulting cDNA library was sequenced
using Illumina Novaseq6000 by Gene De novo Biotechnology Co.
(Guangzhou, China).
High quality clean reads were further filtered by
fastp (version 0.18.0). The gene expression level was normalized by
using Fragments Per Kilobase of transcript per Million mapped
reads. The genes with a fold change ≥2 and P<0.05 were
considered as significant differentially expressed genes (DEGs).
Three samples were prepared from each treatment.
Gene Ontology (GO) enrichment of DEGs was performed,
the significantly enriched GO terms in DEGs was performed by
comparing to the genome background using hypergeometric test. The
biological pathways of the DEGs were enriched to the Kyoto
Encyclopedia of Genes and Genomes (KEGG) using hypergeometric test
as the above GO term enrichment (https://www.genome.jp/kegg/). Gene Set Enrichment
Analysis (GSEA) (22) was used to
identify whether a set of genes in specific GO terms\KEGG
pathways\Reactome pathways shows significant differences in two
groups. The sequences data reported in the present study was
archived in the Sequence Read Archive (SRA) with the accession
number BioProject ID PRJNA772896 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA772896/).
Statistical analysis
The data was examined using the statistical analysis
program SPSS 22.0 (IBM Corp.). The data are expressed as the mean ±
SD. Tumors weight were analyzed by using one-way Analysis of
Variance (ANOVA) followed by Fisher's least significant difference
post hoc test for comparison between two groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
TFP structure and HPLC analysis
TFP is a polysaccharide whose main chain is mannan,
which is a linear (1→3)-linked backbone, and its side chain is
composed of mannose, glucuronic acid and xylose in uncertain order,
as shown in Fig. 1A. The results
of HPLC analysis results showed that there was no obvious TFP peak,
since the TFP standard occurred at 8.183 min in B16 cells exposed
to TFP, as shown in Fig. 1B. These
data suggested that there was no TFP in B16 cells within the
detection capability of HPLC.
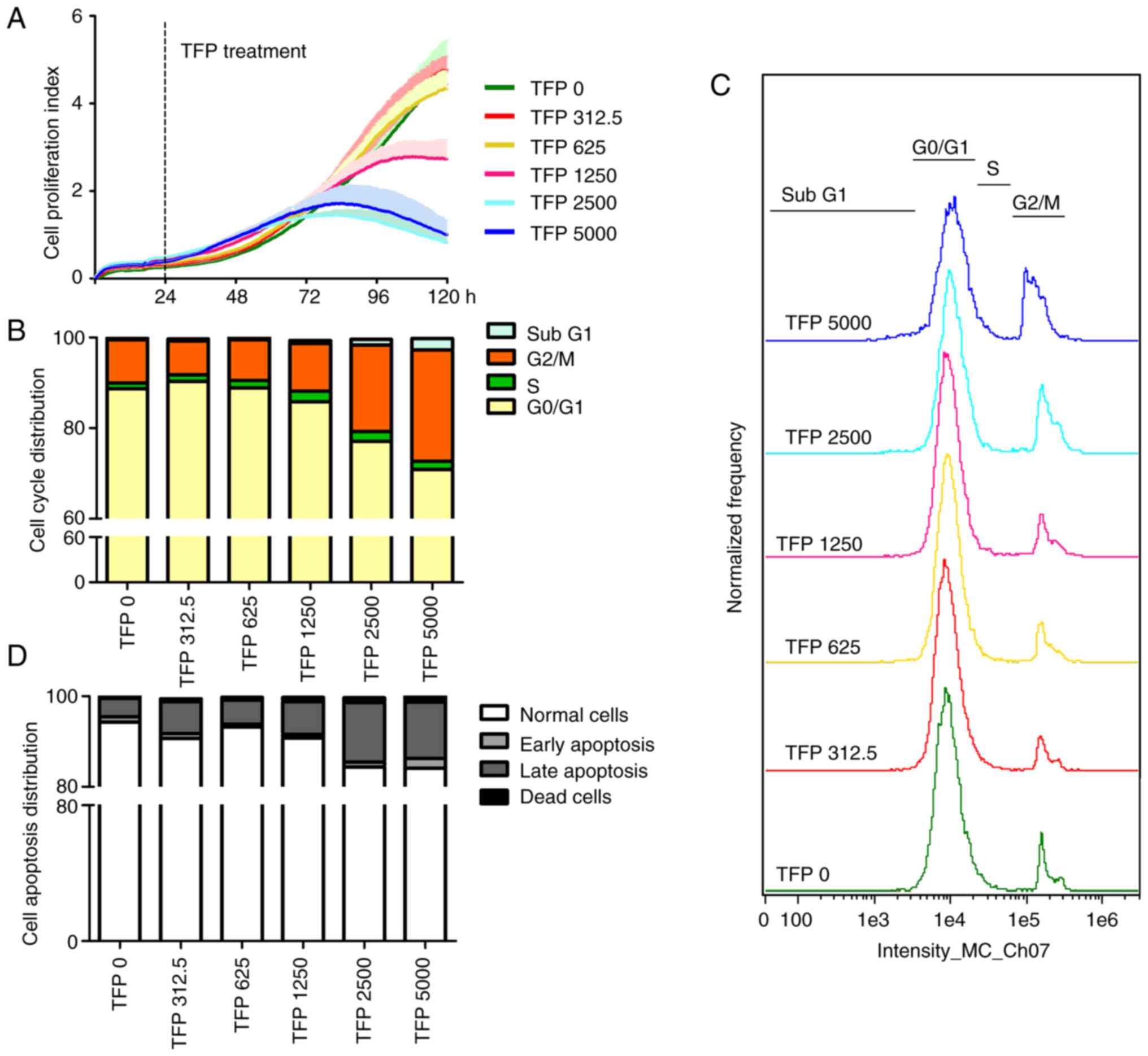
Effects of TFP on anti-proliferation,
pro-apoptosis and cell cycle arrest in vitro
The viability of B16 cells treated with TFP
decreased in a dose-dependent manner, particularly at 1,250, 2,500
and 5,000 µg/ml TFP, which significantly inhibited the rate
of B16 cell proliferation compared with the effects of other
dosages of TFP (Fig. 2A). The
effect of TFP on the relative proliferation rate of B16 cells was
measured by its IC50, which was calculated to be
1.836×107 µg/ml.
To determine whether TFP inhibits B16 cell
proliferation via cell cycle arrest, the DNA contents of the cells
were measured by flow cytometry upon staining the cell nuclei with
DAPI (Figs. 2B and C, and S1). The data showed that high dosages of
TFP increased the number of cells in the G2/M phase from
9.5% (0 µg/ml TFP) to 10.6% (1,250 µg/ml TFP), 19.1%
(2,500 µg/ml TFP) and 24.6% (5,000 µg/ml TFP), while
reducing the number of cells in the G1 phase from 88.7%
(0 µg/ml TFP) to 85.8% (1,250 µg/ml TFP), 77.1%
(2,500 µg/ml TFP) and 70.9% (5,000 µg/ml TFP). In
addition, TFP slightly increased the number of cells in S phase
from 1.27% to 1.44, 1.58, 2.32, 2.17 or 1.73% depending on the
concentration employed. These results suggested that TFP inhibited
cell proliferation mainly by inducing G2/M cell cycle
arrest without cycle progression to G1 phase through
mitosis in B16 cells.
Flow cytometry was used to evaluate the state of
cell apoptosis (Figs. 2D and
S1). The results showed that the
proportion of early apoptotic cells in untreated B16 cells was
1.13%, which slightly decreased with increasing concentration of
TFP (312.5, 625, 1,250 and 2,500 µg/ml). When the
concentration of TFP reached 5,000 µg/ml, 2.16% of cells
were markedly early apoptotic cells. B16 cells exposed to 0, 312.5,
625, 1,250, 2,500 and 5,000 µg/ml of TFP for 24 h
experienced an induction of 4.05, 7.02, 5.32, 7.19, 13.15, 12.40%
in late apoptosis, respectively. In addition, the proportion of
necrotic cells increased gradually with increasing concentrations
of TFP. These data revealed that TFP at suitable concentrations
could effectively induce B16 cell death following late apoptosis,
rather than early apoptosis, and necrosis.
Lipids uptake reprograms lipogenesis
profile in vitro
TFP markedly suppressed SREBP1 nuclear
translocation, and suppressed SREBP1 target proteins, such as ACC
and FAS, leading to reduced de novo lipogenesis (Fig. 3A). Unexpectedly, the ACLY and
plasma lipids levels increased according to increasing TFP dosages
(Fig. 3B). To explain why these
apparent contradictions coexisted in B16 treated by TFP, the levels
of GLUT4 and CD36 (lipid transport receptor) were analyzed, since
cancer cells use glucose as well as fatty acids for ATP production,
and GLUT4 and CD36 migrate together in response to either stimuli,
i.e. glucose or fatty acids (23).
The results confirmed that TFP induced high expression of GLUT4 and
CD36 (Fig. 3A), which resulted in
an increased uptake of exogenous lipids, which were present at
higher lipid fluorescence intensity in the cell membrane (Fig. 4A; panels a and b) and plasma,
including ER (Fig. 4B; panels a
and b), nucleus (Fig. 4D; panels a
and b) as compared with that of control group. However, the uptake
of lipids present in the Golgi apparatus was decreased at dosage of
TFP5000 (Fig. 4C; panels a and b).
The aforementioned data was in accordance with higher
co-localization ratios of lipids with the cellular membrane and
nucleus (Fig. 4E), and higher
lipid intensity of the whole cell compared with that of the vehicle
group (Fig. 4F).
The intracellular accumulation of lipids,
particularly in the ER, inhibited the maturation of SREBP1 and the
expression of full-length SREBP1 (Fig.
3A). Lipids are important molecules in the nucleus, there is
emerging evidence (24) for the
existence of a nuclear population of lipids, which has been
proposed to directly modulate lipid metabolism in the nucleus. In
the present study, the lipids in the nucleus may have the functions
of generating lipoid ligands for PPARγ, or cause chromosome
segregation abnormalities, or modulate exogeneous lipid metabolism
and storage.
TFP delays B16 allograft tumor growth and
reduces tumor weight
Above results have shown that TFP induces the
regulation of lipogenesis in B16 cells. To explore whether this
response is capable of preventing or slowing tumor growth,
1×106 B16 cells treated with or without TFP were
subcutaneously injected in the right flank of C57 BL/6 mice
(Fig. 5A). There were no
significant differences in body weight of TFP treated groups
referring to model group (Fig.
5B), while tumors grew more slowly and the tumor weights of the
TFP-treated groups were less than those of B16 tumor-bearing mice
(Fig. 5C). The tumor weights in
the model and TFP 2,500 and 5,000 µg/ml groups were analyzed
by one-way ANOVA followed by Fisher's least significant difference
post hoc test (Fig. 5D), which
revealed a significant difference among the three groups
[F(2,14)=13.595, P=0.001]. The tumor weight of
the TFP 2,500 µg/ml group exhibited a significant decrease
compared with that of the model group (P=0.007), as well as the TFP
5,000 µg/ml group (P=0.001).
The histological characteristics were evaluated by
(H&E) staining (Fig. 5E),
which revealed increased pigmentation located in the model tumor
section (as indicated by more white arrows), but less pigmentation
(less white arrows) and scattered vacuoles in the TFP-treated tumor
section (red arrows), which may be due to lipid accumulation, and
was dissolved by xylene during H&E staining. These data
suggested that TFP-treated B16 cells reprogrammed their lipid
metabolism profile, and TFP promoted lipid accumulation in B16
cells.
WB analysis showed upregulation of ACLY and PPARγ
(Fig. 5F and G). Upregulated ACLY
and PPARγ activity increased lipid formation. As revealed by
immunohistochemistry (Figs. 6A and
S2), TFP upregulated the
expression of ACLY and PPARγ, and promoted macrophage infiltration
(as indicated by the increase in CD86 expression). Notably, LAMP1
expression increased according to the increase in TFP dosage. In
certain areas of the TFP-treated groups, the formation of certain
strip (long and linear) and round vacuoles was observed (the round
vacuoles gathered at one end, while the strip vacuoles aligned
orderly, similar to a 'balsam pear' surface), which was accompanied
by the presence of numerous macrophages (Fig. 6B). Increased macrophage
infiltration occurred when there was no formation of these
structures. Not far from those structures, a number of bigger
vacuoles could be observed, such as lipid accumulation dissolved by
xylene in the process of staining, similar to the vacuoles noted
during H&E staining (Fig. 5E).
These data indicated that TFP controlled B16 allograft tumor growth
through lipid metabolism regulation, and enhanced the antitumor
immune response through macrophage infiltration and upregulation of
LAMP1.
RNA-sequencing (RNA-seq) analysis of
differentiated gene expression and enriched pathways
To further uncover the molecular mechanism of TFP,
tumor gene expression profile analyses were conducted with the aim
of uncovering valuable information for pharmacological research on
therapeutic targets. Gene Ontology pathway enrichment analysis
showed that negative regulation of mitotic cell cycle, regulation
of cell proliferation and cell proliferation were significantly
altered by TFP (Fig. 7A). The
enrichment in pathways of cell cycle/proliferation confirmed the
results of cell experiments.
Gene set enrichment analysis (GSEA) of the cell
cycle-related RNA-seq results (Fig.
7B) showed that TFP treatment was strongly associated with the
gene signature of cell cycle arrest, cell cycle and cycle phase
transition. Furthermore, negative regulation of cell process, cell
cycle and cell cycle phase transition were significantly positively
associated with TFP. These data indicated that cell cycle
regulation played an important role in the antitumor activity of
TFP.
To determine whether the antitumor effect of TFP
occurred through activation of lipid metabolism, gene expression
was examined. Gene microarray analysis demonstrated the
upregulation of mRNAs encoding genes involved in lipid metabolism
(Fig. 8A), including glucolipid
receptors (CD36 and Trarg1: trafficking regulator of
GLUT4), lipogenesis-related genes (ACLY), a set of
mRNAs involved in the tricarboxylic acid cycle (Fig. 8B), and a number of
lipolysis-related genes (such as LPL: lipoprotein lipase).
GSEA of regulation of lipolysis in adipocytes further provided
clues on the sources of lipids: upregulation of lipolysis of
adipocyte in tumor indicated the possible sources of more lipids
deposit in tumor (Fig. 8C). The
omics effect of TFP on lipid metabolism is consistent with the
aforementioned phenotypic results associated with increased lipid
deposits in mitochondria, nucleus, ER and membrane, as well as the
WB results related to increased glycolipid transport and increased
lipid accumulation in allografts.
Subsequently, to identify the potential reason
responsible for the activated lipid metabolism and to uncover the
underlying molecular mechanisms of TFP involved in lipid metabolic
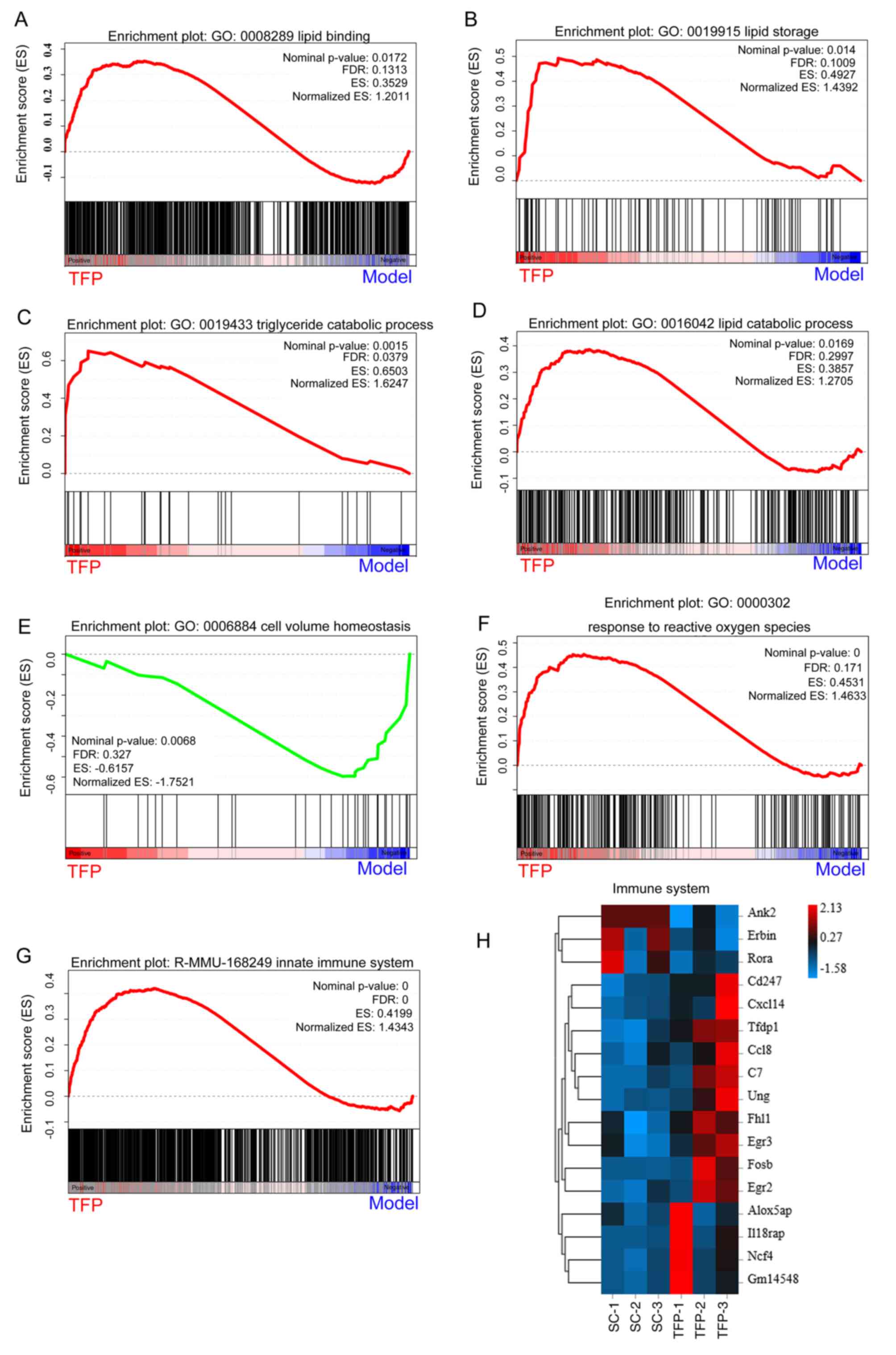
processes, GSEA was used to mine the RNA-seq data. The results
revealed that TFP treatment was associated with the gene signatures
of 'positive regulation of lipid binding' (Fig. 9A), 'positive regulation of lipid
storage' (Fig. 9B), 'triglyceride
catabolism' (Fig. 9C) and 'lipid
catabolism' (Fig. 9D). These
results indicated that TFP not only promoted lipid uptake in cells,
but also provoked lipid catabolism.
Next, it was hypothesized that metabolic changes
could induce the immune system. It was found that TFP induced the
enrichment of gene transcription signatures of 'cell volume
homeostasis' (Fig. 9E), 'response
to reactive oxygen species' (Fig.
9F) and 'innate immune system' (Fig. 9G), thus activating the immune
system (Fig. 9H). These results
confirmed the mechanisms of TFP against B16 subcutaneous
allografts, suggesting that TFP activated the immune system
disrupting cell volume homeostasis.
Discussion
Regarding the initial step of the antitumor
mechanism of TFP, it is worth noting that the high molecular weight
of TFP greatly increases the viscosity of liquids, and the
glucuronic acids in TFP have the ability to entrap cholesterol
(25). These combined TFP with
cell membrane or intercellular cholesterol were easily removed by
centrifugation in the process of intracellular fluid extraction.
This may explain why no TFP was detected in B16 cells by HPLC
analysis. The receptors of TFP have not been identified to date.
The antitumor mechanism of TFP may involve the upregulation of
glucolipid receptors, such as CD36 and GLUT4, which were markedly
elevated in vitro and in vivo in the present
study.
The current study demonstrated the influence of TFP
on the proliferation, cell cycle and apoptosis of B16 cells. TFP
inhibited cell proliferation, induced cell apoptosis and altered
cell cycle distribution. In addition, TFP downregulated SREBP,
which regulates cellular lipogenesis and lipid homeostasis
(26), and has been shown to be
involved in other cellular functions, such as regulation of cell
cycle and cell proliferation (27). TFP inhibited de novo
lipogenesis through SREBP1, thereby inhibiting cell proliferation.
TFP could prevent cells from completing mitosis, arresting them at
a 'lipogenic' G2/M phase. AMPK has been reported to
exert anti-proliferative action (28). AMPK is a cell-intrinsic regulator
of the cell cycle that coordinates cellular proliferation with
energy source availability, and regulates a number of key cellular
metabolic enzymes (29). The
inhibitor effect of TFP on phosphorylated (p)-AMPK suggested that
the anti-proliferative effect of TFP was AMPK independent. The
nutrient sensor AMPK negatively regulates SREBP to limit
lipogenesis under nutrient-limiting conditions (30). Cancer cell metabolism could be
changed if sufficient ATP and the building blocks needed to sustain
molecular synthesis were available. In the present study, the
expression levels of AMPK and particularly those of p-AMPK were
decreased by TFP, and AMPK positively regulated SREBP when CD36 and
GLUT4 were notably upregulated by TFP. Loss of AMPK or
downregulation of AMPK results in energy stress-driven apoptosis,
and slows down tumor progression (31); AMPK inhibitors rather than
activators could preferentially trigger cancer cell death in the
context of metabolic stress (32).
PPARγ is a transcription factor capable of promoting pleiotropic
effects, including the control of cell proliferation.
Pharmacological agonists of PPARγ were used in cancer therapy with
the aim of promoting cell cycle deceleration (33). TFP as AMPK inhibitor and PPARγ
agonist facilitated to control tumor growth, which was confirmed by
our present studies.
Finally, the cell cycle arrest of B16 cells treated
with TFP could be due to both the inhibition of de novo
fatty acid synthesis (causing inhibition of SREBP1) as well as the
accumulation of exogeneous lipids in the nucleus (causing induction
of PPARγ and impairment of cell proliferation through chromosome
segregation abnormalities).
The present findings indicated that TFP activated
lipid metabolism and the immune system. A balance between
lipogenesis, lipid uptake and intracellular lipolysis is required
to maintain lipid homeostasis (34). Cancer-associated adipocytes have
been found to promote lipid accumulation (35). In the case of malignant melanoma,
the tumor secreted an inhibitor of adipose LPL termed melanoma
cachexia factor, which caused the inhibition of extracellular
lipolysis (36). Activation of the
transmembrane channel for exogenous free fatty acid uptake (CD36)
and LPL (a key enzyme for extracellular lipolysis) confirmed the
disposition of extracellular lipid stores and lipolysis in cells.
The levels of free fatty acids, monoacylglycerides and
diacylglycerides were not significantly elevated (data not shown),
which suggested that lipid metabolism existed in cancer cells and
cancer-associated adipocytes.
Elevated SREBP1 in cancer cells increased de
novo lipogenic gene expression, enhanced fatty acid synthesis,
and accelerated triglyceride accumulation; however, these metabolic
abnormalities were reversed by the treatment of B16 cells with TFP.
TFP inhibited SREBP1 and AMPK, an enzyme that inhibits lipid
synthesis through the phosphorylation and inactivation of key
lipogenic enzymes. The net effect on lipogenic metabolism depends
on a balance between opposing effects. In the present study, the
net result was a decrease in SREBP1 and AMPK, while certain lipid
enzymes were reduced (FAS and ACC) and certain others activated,
particularly those participating in the tricarboxylic acid (TCA)
cycle. Importantly, several signaling molecules involved in the
processes of immune cell activation and transformation are derived
from metabolites of the TCA cycle (37). These results indicated the crucial
role of lipid metabolism in the regulation of the tumor immune
microenvironment and the feasibility of targeting lipid metabolism
to regulate tumor immunity.
Increasing evidence indicated that aberrant de
novo lipogenesis is being increasingly recognized as important
feature of malignant transformation (38), by contrast, the majority of normal
human tissues use exogenous lipids, while de novo fatty acid
synthesis is generally suppressed (39). De novo lipogenesis also
determines the sensitivity of cancer cells to oxidative
stress-induced cell death, and de novo synthesis was the
preferred mechanism for fatty acid acquisition over lipolysis or
receptor-mediated endocytosis (40). Tumor cells do not solely rely on
de novo lipogenesis, but also utilize exogenous fatty acids
for generating lipids for proliferation. Thus, despite the dominant
role of de novo lipogenesis, exogenous fatty acids may also
play an important role in cancer pathogenesis (41).
After B16 cells were exposed to TFP, their lipid
metabolism was altered. TFP treatment resulted in the
downregulation of de novo fatty acid synthesis due to
feedback suppression with concomitant increase in exogenous fatty
acid uptake. This metabolic change was facilitated by the
upregulation of several key transporters, enzymes and signaling
pathways. Specifically, CD36 and GLUT4 were remarkably upregulated;
de novo lipogenesis was completely abolished; the lipid
distribution in the cytoplasm was rearranged; and the lipid content
in the cell membrane, ER and nucleus was elevated, which was
accompanied by an increase in ACLY and PPARγ expression. The
RNA-seq data precisely depicted the effect of TFP on lipid
metabolism and processing, showing its antitumor effects. TFP
caused excess lipid metabolism and dysregulated lipid storage in
B16 cells, which resulted in the irreversible disruption of cell
volume homeostasis and activation of the immune system.
There were a number of limitations to the present
study. Firstly, functional experiments to demonstrate the effect of
TFP in differentiated adipocytes should be performed to demonstrate
a specific role in TFP augmenting lipolysis of adipocytes.
Secondly, mechanism of interactions between adipocytes and B16
cells should be explored. Thirdly, tumor cells manipulated ex
vivo may serve as a uniquely effective carriers for the
therapeutic delivery of bioactive cytokines to parental tumor by
their homing properties especially highly metastatic cells
(42). It is rarely reported that
tumor cells modified with polysaccharides are used to treat tumors.
The following antitumor effect and mechanism of B16 cells
reprogrammed by TFP in vein in lower dosage was urgently needed in
B16 subcutaneously implanted mice.
In conclusion, the present study identified the
function of TFP on B16 cells, along with its unexpected molecular
mechanism of action responsible for its antitumor effects, as
summarized in the schematic diagram of Fig. 10. TFP inhibited de novo
lipogenesis, although a larger number of lipids were present in the
cytoplasm of B16 cells. TPF caused upregulated lipid binding,
storage and catabolism with cellular response to reactive oxygen
species, thus activating the immune system via disruption of cell
volume homeostasis.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XL and YP conceived and designed the study. QS and
XL performed the experiments. XL analyzed the data and wrote the
manuscript. XL and YP confirm the authenticity of all the raw data.
All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
The present study was approved (approval no.
2020002) and supervised by Experimental research Ethics Committee
of Minna Normal University (Zhangzhou, China). All animal
experiments were performed according to the relevant regulatory
standards and were performed in accordance with the Experimental
animal research Ethics Committee of Minna Normal University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
TFP
|
Tremella fuciformis- derived
polysaccharides
|
|
SREBP
|
sterol regulatory element binding
protein
|
|
ER
|
endoplasmic reticulum
|
|
ACLY
|
ATP-citrate lyase
|
|
ACC
|
acetyl CoA carboxylase
|
|
FAS
|
fatty acid synthase
|
|
GLUT4
|
glucose transport 4
|
|
PPARγ
|
peroxisome proliferator-activated
receptor γ
|
|
RTCA
|
real time cellular analysis
|
|
DEGs
|
differentially expressed genes
|
|
GO
|
Gene Ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
GSEA
|
Gene Set Enrichment Analysis
|
|
SRA
|
Sequence Read Archive
|
|
AMPK
|
adenosine 5′-monophosphate-activated
protein kinase
|
|
pAMPK
|
phosphorylated-AMP-activated protein
kinase
|
|
GAPDH
|
glyceraldehyde-3-phosphate
dehydrogenase
|
|
ANOVA
|
Analysis of Variance
|
|
LPL
|
lipoprotein lipase
|
|
Trarg1
|
trafficking regulator of GLUT4
|
|
TCA
|
tricarboxylic acid cycle
|
|
FA
|
fatty acid
|
|
WB
|
western blotting
|
Acknowledgments
The authors would like to thank Dr Qici Wu and Bo
Leng (The Engineering Technological Center of Mushroom Industry,
Minnan Normal University, Zhangzhou, Fujian 363000, P.R. China). In
the process of HPLC analysis of TFP in the present study, they
provided numerous academic and constructive advices, and
contributed to manuscript editing.
Funding
The present study was supported by the Natural Science fund in
Fujian Province (grant no. 2021J011012), the Science and Technology
project of Zhangzhou city in Fujian Province (grant no. zz2021J44),
the Scientific Research and Nurturing Projects of Minnan Normal
University (grant no. MSPY2021) and the Records of National Science
and Technology Projects (grant no. 2021L3027).
References
|
1
|
Mizuta K, Matsubara T, Goto A, Addison WN,
Nakatomi M, Matsuo K, Tada-Shigeyama Y, Yaginuma T, Honda H,
Yoshioka I and Kokabu S: Plectin promotes tumor formation by B16
mouse melanoma cells via regulation of Rous sarcoma oncogene
activity. BMC Cancer. 22:9362022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang E, Liu Y, Xu C and Liu J:
Antiproliferative and proapoptotic activities of anthocyanin and
anthocyanidin extracts from blueberry fruits on B16-F10 melanoma
cells. Food Nutr Res. 61:13253082017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Emens LA, Ascierto PA, Darcy PK, Demaria
S, Eggermont AMM, Redmond WL, Seliger B and Marincola FM: Cancer
immunotherapy: Opportunities and challenges in the rapidly evolving
clinical landscape. Eur J Cancer. 81:116–129. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Navarro C, Ortega A, Santeliz R, Garrido
B, Chacín M, Galban N, Vera I, De Sanctis JB and Bermúdez V:
Metabolic reprogramming in cancer cells: Emerging molecular
mechanisms and novel therapeutic approaches. Pharmaceutics.
14:13032022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhao Q, Lin X and Wang G: Targeting
SREBP-1-mediated lipogenesis as potential strategies for cancer.
Front Oncol. 12:9523712022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li J, Shen H, Owens GK and Guo LW: SREBP1
regulates Lgals3 activation in response to cholesterol loading. Mol
Ther Nucleic Acids. 28:892–909. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bindesboll C, Aas A, Ogmundsdottir MH,
Pankiv S, Reine T, Zoncu R and Simonsen A: NBEAL1 controls SREBP2
processing and cholesterol metabolism and is a susceptibility locus
for coronary artery disease. Sci Rep. 10:45282020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Luo H, Chen CY, Li X, Zhang X, Su CW, Liu
Y, Cao T, Hao L, Wang M and Kang JX: Increased lipogenesis is
critical for self-renewal and growth of breast cancer stem cells:
Impact of omega-3 fatty acids. Stem Cells. 39:1660–1670. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kitamura K, Erlangga JS, Tsukamoto S,
Sakamoto Y, Mabashi-Asazuma H and Iida K: Daidzein promotes the
expression of oxidative phosphorylation- and fatty acid
oxidation-related genes via an estrogen-related receptor alpha
pathway to decrease lipid accumulation in muscle cells. J Nutr
Biochem. 77:1083152020. View Article : Google Scholar
|
|
10
|
Hu B, Lin JZ, Yang XB and Sang XT:
Aberrant lipid metabolism in hepatocellular carcinoma cells as well
as immune microenvironment: A review. Cell Prolif. 53:e127722020.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Simeone P, Tacconi S, Longo S, Lanuti P,
Bravaccini S, Pirini F, Ravaioli S, Dini L and Giudetti AM:
Expanding roles of de novo lipogenesis in breast cancer. Int J
Environ Res Public Health. 18:35752021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhao S, Torres A, Henry RA, Trefely S,
Wallace M, Lee JV, Carrer A, Sengupta A, Campbell SL, Kuo YM, et
al: ATP-citrate lyase controls a glucose-to-acetate metabolic
switch. Cell Rep. 17:1037–1052. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ramapriyan R, Caetano MS, Barsoumian HB,
Mafra ACP, Zambalde EP, Menon H, Tsouko E, Welsh JW and Cortez MA:
Altered cancer metabolism in mechanisms of immunotherapy
resistance. Pharmacol Ther. 195:162–171. 2019. View Article : Google Scholar
|
|
14
|
Welte MA: Expanding roles for lipid
droplets. Curr Biol. 25:R470–R481. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Todisco S, Santarsiero A, Convertini P, De
Stefano G, Gilio M, Iacobazzi V and Infantino V: PPAR alpha as a
metabolic modulator of the liver: Role in the pathogenesis of
nonalcoholic steatohepatitis (NASH). Biology (Basel).
11:7922022.PubMed/NCBI
|
|
16
|
Thiam AR, Farese RV Jr and Walther TC: The
biophysics and cell biology of lipid droplets. Nat Rev Mol Cell
Biol. 14:775–786. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hashemi HF and Goodman JM: The life cycle
of lipid droplets. Curr Opin Cell Biol. 33:119–124. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cartwright BR, Binns DD, Hilton CL, Han S,
Gao Q and Goodman JM: Seipin performs dissectible functions in
promoting lipid droplet biogenesis and regulating droplet
morphology. Mol Biol Cell. 26:726–739. 2015. View Article : Google Scholar :
|
|
19
|
Shen T, Duan C, Chen B, Li M, Ruan Y, Xu
D, Shi D, Yu D, Li J and Wang C: Tremella fuciformis polysaccharide
suppresses hydrogen peroxide-triggered injury of human skin
fibroblasts via upregulation of SIRT1. Mol Med Rep. 16:1340–1346.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Terrazas C, Oghumu S, Varikuti S,
Martinez-Saucedo D, Beverley SM and Satoskar AR: Uncovering
Leishmania-macrophage interplay using imaging flow cytometry. J
Immunol Methods. 423:93–98. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee YT, Lim SH, Lee B, Kang I and Yeo EJ:
Compound C Inhibits B16-F1 tumor growth in a syngeneic mouse model
via the blockage of cell cycle progression and angiogenesis.
Cancers (Basel). 11:8232019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Steinbusch LK, Schwenk RW, Ouwens DM,
Diamant M, Glatz JF and Luiken JJ: Subcellular trafficking of the
substrate transporters GLUT4 and CD36 in cardiomyocytes. Cell Mol
Life Sci. 68:2525–2538. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Romanauska A and Kohler A: The inner
nuclear membrane is a metabolically active territory that generates
nuclear lipid droplets. Cell. 174:700–715.e718. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chiu CH, Chiu KC and Yang LC: Amelioration
of obesity in mice fed a high-fat diet with uronic acid-rich
polysaccharides derived from tremella fuciformis. Polymers (Basel).
14:15142022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shao W and Espenshade PJ: Expanding roles
for SREBP in metabolism. Cell Metab. 16:414–419. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guo D, Bell EH, Mischel P and Chakravarti
A: Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr
Pharm Des. 20:2619–2626. 2014. View Article : Google Scholar :
|
|
28
|
Queiroz EA, Fortes ZB, da Cunha MA,
Barbosa AM, Khaper N and Dekker RF: Antiproliferative and
pro-apoptotic effects of three fungal exocellular beta-glucans in
MCF-7 breast cancer cells is mediated by oxidative stress,
AMP-activated protein kinase (AMPK) and the Forkhead transcription
factor, FOXO3a. Int J Biochem Cell Biol. 67:14–24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim I and He YY: Targeting the
AMP-activated protein kinase for cancer prevention and therapy.
Front Oncol. 3:1752013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li Y, Xu S, Mihaylova MM, Zheng B, Hou X,
Jiang B, Park O, Luo Z, Lefai E, Shyy JYJ, et al: AMPK
phosphorylates and inhibits SREBP activity to attenuate hepatic
steatosis and atherosclerosis in diet-induced insulin-resistant
mice. Cell Metab. 13:376–388. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rigel J, Kishton, Carson E, Cohen S,
Gerriets VA, Siska PJ, Macintyre AN, Goraksha-Hicks P, de Cubas AA,
Liu T, et al: AMPK is essential to balance glycolysis and
mitochondrial metabolism to control T-ALL cell stress and survival.
Cell Metab. 12:649–662. 2016.
|
|
32
|
Jeon SM, Chandel NS and Hay N: AMPK
regulates NADPH homeostasis to promote tumour cell survival during
energy stress. Nature. 485:661–665. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Flori E, Rosati E, Cardinali G, Kovacs D,
Bellei B, Picardo M and Maresca V: The α-melanocyte stimulating
hormone/peroxisome proliferator activated receptor-γ pathway
down-regulates proliferation in melanoma cell lines. J Exp Clin
Cancer Res. 36:1422017. View Article : Google Scholar
|
|
34
|
Cheng C, Geng F, Cheng X and Guo D: Lipid
metabolism reprogramming and its potential targets in cancer.
Cancer Commun (Lond). 38:272018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wu Q, Li B, Li Z, Li J and Sun S and Sun
S: Cancer-associated adipocytes: Key players in breast cancer
progression. J Hematol Oncol. 12:952019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Martin P: Cancer cachexia syndrome:
Reflecting on 20 years of providing cancer cachexia care as the
leader of an interdisciplinary team in an Australian cancer center.
Asia Pac J Oncol Nurs. 9:1000702022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ryan DG, Murphy MP, Frezza C, Prag HA,
Chouchani ET, O'Neill LA and Mills EL: Coupling Krebs cycle
metabolites to signalling in immunity and cancer. Nat Metab.
1:16–33. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bartolacci C, Andreani C, Vale G, Berto S,
Melegari M, Crouch AC, Baluya DL, Kemble G, Hodges K, Starrett J,
et al: Targeting de novo lipogenesis and the Lands cycle induces
ferroptosis in KRAS-mutant lung cancer. Nat Commun. 13:43272022.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li L, Che L, Tharp KM, Park HM, Pilo MG,
Cao D, Cigliano A, Latte G, Xu Z, Ribback S, et al: Differential
requirement for de novo lipogenesis in cholangiocarcinoma and
hepatocellular carcinoma of mice and humans. Hepatology.
63:1900–1913. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zaidi N, Lupien L, Kuemmerle NB, Kinlaw
WB, Swinnen JV and Smans K: Lipogenesis and lipolysis: The pathways
exploited by the cancer cells to acquire fatty acids. Prog Lipid
Res. 52:585–589. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rizzo AM, Colombo I, Montorfano G, Zava S
and Corsetto PA: Exogenous fatty acids modulate ER lipid
composition and metabolism in breast cancer cells. Cells.
10:1752021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dondossola E, Dobroff AS, Marchio S,
Cardó-Vila M, Hosoya H, Libutti SK, Corti A, Sidman RL, Arap W and
Pasqualini R: Self-targeting of TNF-releasing cancer cells in
preclinical models of primary and metastatic tumors. Proc Natl Acad
Sci USA. 113:2223–2228. 2016. View Article : Google Scholar : PubMed/NCBI
|