Introduction
Our long-term goal is to identify molecular targets
to circumvent the development of endocrine resistance and breast
cancer progression. Breast cancer is the most common cancer among
women in the United States; ~70% of breast cancers express estrogen
receptor alpha (ERα). ERα expressing tumors (ER+), which
include the luminal A and luminal B subtypes, are typically treated
with a selective estrogen receptor modulator (SERM), a selective
estrogen receptor down-regulator (SERD), or an aromatase inhibitor
(AI). These antiestrogen therapies are administered prior to and/or
following surgery, localized radiation, and/or chemotherapy
depending on stage and tumor subtype (1). Tamoxifen (TAM) has been the most
commonly used SERM (2) and still
remains the standard adjuvant treatment for pre-menopausal women
with ER+ breast cancer. However, AIs show increased
efficacy compared with TAM therapy (3) for post-menopausal women with all
stages of ER+ breast cancer and are becoming the
preferred treatment.
Regardless of the hormone used as a first-line
endocrine treatment of breast cancer, intrinsic or acquired
resistance remains a significant clinical challenge (4). Once resistance to a specific
antiestrogen modality is identified, clinical benefit is often
achieved if the patient is placed on a different antiestrogen
therapy. However, the continued development/expression of
antiestrogen resistance often persists and ER+ breast
cancers that are refractory to hormone therapy are the most common
cause of breast cancer death (5).
Multiple mechanisms of endocrine resistance have been identified
(6), primarily from pre-clinical
studies utilizing appropriate breast cancer cell models that have
been recently reviewed (7). CDK4/6
inhibitors are now commonly combined with antiestrogen treatment to
improve outcomes and reduce the development of antiestrogen
resistance for advanced breast cancer (8). Yet, resistance to CDK4/6 inhibitors
also develops leading to relapse (9) with minimal information available on
the mechanisms of resistance (10).
With the goal of improving the efficacy of
antiestrogen treatment and reducing the emergence of
antiestrogen-resistant breast cancer cells, our early studies
established that the combined treatment of an antiestrogen plus an
antiprogestin, such as mifepristone (MIF), compared with
antiestrogen treatment alone, more effectively induced apoptosis
and growth arrest of ER+ breast cancer cells (11-14).
These earlier studies were motivated by small-scale clinical trials
that showed efficacy for the antiprogestin action of MIF as a
single agent for breast cancer treatment (15). Although the efficacy of this
combined treatment was superior to that of antiestrogen
single-agent treatment, a robust apoptotic response required the
additional targeting of MEK1, the mitogen activated protein (MAP)
kinase (16). In the
aforementioned previous study, it was established by the authors
that targeting MEK1 blocked the activation of the mitogen activated
kinases MAPK1/2, also referred to as extracellular signal-regulated
protein kinases ERK1 and ERK2. Targeting MAPK1/2 resulted in
elevated cellular levels of dephosphorylated Bim extra-long
(dBimEL), a pro-apoptotic member of the BH3 family. MEK1 targeting
reduced active, phosphorylated MAPK1/2 (pMAPK1/2), established
downstream effectors of MEK1, and the reduction of pMAPK1/2
activity increased the levels of dBimEL in breast cancer cells
(16). Further, siRNA targeting
studies identified dBimEL to be required for the cytotoxic effects
of 4-hydroxytamoxifen (4-OHT) and/or MIF treatments, particularly
if these treatments were conducted under conditions of MEK1/MAPK1/2
blockade (16).
BimEL is an established pro-apoptotic member of the
BH3 family, and a major isoform encoded by the Bim gene, designated
Bcl-2-like protein 11 (BCL2L11). A key role for Bim deletion
polymorphisms may be involved in the response of patients to
kinase-targeted therapies (17).
Pre-clinical studies also identified a key role for BimEL in
mediating sensitivity to histone deacetylase inhibitors (18) and oncogene-targeted therapies,
which have previously been reviewed (19). Additionally, a previous study
identified Bim deletion polymorphisms as predictive of breast
cancer progression in young Asian women (20). To our knowledge, a role for BimEL
in the hormonal sensitivity of breast cancer in the clinic has not
been explored, but both the Raf/MEK/MAPK1/2 and PI3K/AKT
pro-survival signaling cascades that are implicated in endocrine
resistance stringently downregulate the levels of Bim in breast
cancer cells (21). In the present
study, it was hypothesized that BimEL expression and/or function
would be downregulated to facilitate the development of
antiestrogen resistance. To test this hypothesis, the
antiestrogen-sensitive ER+ MCF-7 cells and the
antiestrogen-resistant breast cancer cell model TR5 were utilized,
established by a step wise 4-OHT selection of MCF-7 cells (22). TR5 cells have been used in our
previous studies that uniquely identified a pro-survival role for
autophagy in response to antiestrogen treatment (22,23).
Autophagy is a highly conserved cellular catabolic
process that requires the expression of at least 28 autophagy
(ATG) genes. Autophagy can be induced above basal levels in
eukaryotic cells by various stressors, including reactive oxygen
species (ROS) and nutrient deprivation to protect normal and cancer
cells from death (24). The
functional unit of autophagy is the autophagosome, a
double-membrane organelle that must fuse with the lysosome
(generating the autolysosome) to allow degradation of sequestered
contents into basic macromolecules that can be released to the cell
for survival purposes. Expression of LC3-II, encoded by the
ATG8 gene, is commonly used as a marker of autophagy. LC3
was originally identified as a microtubule associated protein and
named 'microtubule-associated-protein light-chain-3'. LC3-II is a
16 kDa protein generated from a covalent conjugation of
phosphatidylethanolamine to LC3-I. LC3-I, a cytosolic 18-kDa
protein, is generated by cleavage of the LC3 and is not involved in
autophagosome membrane formation or function. LC3-II, however,
increases and becomes peripherally membrane-associated during
autophagy and is degraded with the turnover of the autolysosomal
membrane. During the initial 4-OHT selection, TR5 cells showed high
level LC3-II expression localized to autophagosomes, and siRNA
ATG6 targeting in 4-OHT-selected TR5 cells resulted in a
robust apoptotic response (22).
In the present study, further analysis of the
antiestrogen-resistant TR5 cells establishes a central role for the
EGFR/MEK1/MAPK1/2 signaling axis as a mechanism to evade hormonally
induced, dBimEL-mediated cell death and identifies pro-survival
autophagy as the overriding response to EGFR and
MEK1/MAPK1/2-targeted therapy.
Materials and methods
Cell culture
The antiestrogen-sensitive, ERα expressing MCF-7
breast adenocarcinoma cells were purchased from the American Type
Culture Collection (ATCC). The TR5 antiestrogen-resistant cell line
was established in our laboratory as previously described (22). MCF-7 and TR5 cells are routinely
checked for mycoplasma contamination using the MycoAlert Mycoplasma
Detection Kit (cat. no. LT07-418; Lonza Bioscience). During the
course of the present study and prior to manuscript submission, the
antiestrogen-resistant TR5 cells and MCF-7 parent cells were
subjected to STR profiling by the ATCC and confirmed to be a match
to HTB-22 ATCC human MCF-7 cell line. Cells were routinely cultured
in DMEM complete medium supplemented with 10% fetal bovine serum
(FBS), 2% antibiotics-antimycotics (cat. no. 15240-062; Invitrogen;
Thermo Fisher Scientific, Inc.), 1% sodium pyruvate (cat. no.
SH3023901; Thermo Fisher Scientific, Inc.), and 10 µg/ml
insulin (cat. no. I9278; MilliporeSigma). Prior to hormonal
treatments, cells were placed in DMEM-F12 medium (cat. no.
11039-021; Invitrogen; Thermo Fisher Scientific, Inc.) containing
10% dextran-coated charcoal stripped FBS, designated DCC FBS, (cat.
no. SFBU32-0500; Equitech-Bio Inc.) plus insulin (10.0
µg/ml). The serum concentration was stepped down to 5% DCC
FBS as previously described (11,22,23).
Treatments were conducted in cells seeded in the absence of
insulin. Cells were allowed to adhere to the culture vessel, which
required ~16-24 h. For all experiments, cells were plated at a
density where overcrowding did not result due to seeding densities,
minimizing effects of contact inhibition. Typically, cells were
harvested at early time points (i.e., 24 or 48 h) for analysis of
autophagy levels, and at later time points (72-144 h) for
evaluation of cell number and/or apoptosis. To be able to
reproducibly detect E2-stimulation of breast cancer cells by MTT
and cell counts, a minimum of 72-96 h was required. Treatments
were: 10 nM estradiol (E2; MilliporeSigma), 10 nM E2 plus 1
µM or 5 µM 4-OHT, 10 nM E2 plus 10 µM MIF in
the presence and absence of 4-OHT (Sigma-Aldrich; Merck KGaA),
U0126 at 5 µM or 10 µM (MilliporeSigma), chloroquine
(CQ) at 10 µM (Sigma-Aldrich; Merck KGaA), Z-VAD-FMK at 10
µM (R&D systems, Inc.), MG132 at 1 µM
(Sigma-Aldrich; Merck KGaA), erlotinib (ERL) at 5 or 10 µM,
spautin-1 at 10 µM, and compound 19 vps34 inhibitor at 1-5
µM (Selleck Chemicals).
Cell counts
Cells were seeded in triplicate at a density to
attain 50-70% confluence within 24 h. Adherent cells were treated
with drugs and/or hormones for the indicated times as described in
the figure legends. For cell counts, the adherent, monolayer cells
were released from the culture dish by trypsinization, collected,
and pooled with any detached cells collected from the culture
medium. A single cell suspension was obtained by syringing three
times with a 25 gauge 7/8″ needle. For some experiments, prior to
cell counts, trypan blue (TB; 0.08%; Sigma-Aldrich; Merck KGaA) was
added to the cell suspension for 5 min to identify non-viable cells
with compromised membrane permeability. Cell counts were conducted
with a hemocytometer or a Coulter Counter following dilution in
Isoton II.
MTT assay
Cells were seeded at a density of 7,500 cells per
well (48-72 h harvest) or 5,000 cells per well (120-144 h harvest)
in 96-well clear bottom microplates (cat. no. 3603; Corning, Inc.),
allowed to adhere for 16-24 h, and then treated in DMEM-F12 with 5%
DCC FBS as indicated in the figure legends. At the end of the
treatment period, the colorimetric MTT assay was conducted
according to the manufacturer's directions (MilliporeSigma).
Following solubilization of the purple formazan product that
correlates to the number of viable cells per well, plates were read
on a TECAN Spectrafluor Plus with a test wavelength of 570 nm and a
reference wavelength of 630 nm.
Proteolysis of long-lived proteins
Analysis of long-lived protein turnover was
conducted as previously described (23). In brief, cells were seeded in
DMEM-F12 with 5% DCC FBS plus 10 µg/ml insulin for 24 h. The
adherent cells were then incubated for 24 h at 37°C with 0.2
µCi/ml [14C (U)] L-valine (Moravek Biochemicals, Inc.).
Excess radioisotope was removed with phosphate-buffered saline, 1×
PBS (pH 7.4), prior to a 2 h incubation in DMEM/F12 media with 5%
DCC FBS, 10 mM unlabeled valine (cat. no. V0500; MilliporeSigma),
and the appropriate drug combination as described in the figure
legends. A 2 h incubation at 37°C allowed for short-lived protein
turnover, after which the medium was aspirated and cells were
placed in the same treatment medium and incubated at 37°C 24 and 48
h, allowing for long-lived protein turnover. trichloroacetic acid
precipitable and soluble radioactivity was collected from cells and
medium, respectively, quantified by liquid scintillation counting
(LS6500; Beckman Coulter, Inc.), and expressed as % long-lived
protein degradation which is the ratio of TCA-soluble radioactivity
to radioactivity in the precipitated proteins. In certain
experiments, proteolysis was measured in the absence and presence
of CQ, a lysosomotropic agent that enters the lysosome as a
protonated compound and increases intracellular pH, blocking
autolysosomal turnover along with any cellular constituents
contained within the autolysosome.
Protein harvest, immunoblotting, and
densitometry
Cell lysates were harvested for total protein as
previously described (11,12,22,23).
For all studies, with the exception of the experiments performed
for Fig. 1D, cell lysates were
derived from total cell populations (adherent and detached cells)
that were collected and combined, prior to cellular lysis. For the
studies presented in Fig. 1D, cell
lysates were harvested from adherent and detached (apoptotic) cell
populations separately. Immunoblotting was conducted according to
the manufacturer's protocol using the following primary antibodies:
BimEL (cat. no. CS2933; 1:500 dilution), GAPDH (cat. no. CS5174;
1:1,000 dilution), LC3 (cat. no. CS12741; 1:500 dilution),
cleaved-Lamin A (cat. no. CS2035; 1:1,000 dilution), phosphorylated
MAPK (cat. no. CS9101; 1:200 dilution), p62 (cat. no. CS5114;
1:1,000 dilution), cleaved-PARP (cat. no. CS9541; 1:500 dilution),
phosphorylated EGFR (cat. no. CS3777; 1:250 dilution) and EGFR
(cat. no. CS4267; 1:500 dilution), all from Cell Signaling
Technology, Inc.; ERK1 (cat. no. SC-94; 1:200 dilution), ERK2 (cat.
no. SC-154; 1:200 dilution) and ERα (cat. no. SC-8002; 1:1,000
dilution), all from Santa Cruz Biotechnology, Inc.; β-actin (cat.
no. A5441; 1:2,000 dilution; Sigma-Aldrich; Merck KGaA) and GFP
(cat. no. 2273995; 1:500 dilution; MilliporeSigma). Secondary
antibodies included anti-mouse IgG (cat. no. 715-035-150; 1:10,000)
and anti-rabbit IgG (cat. no. 711-035-152; 1:10,000) both from
Jackson ImmunoResearch Laboratories, Inc. Immunodetection was
performed using the ECL detection system (cat. no. 34080; Pierce;
Thermo Fisher Scientific, Inc.) and HyBlot CL autoradiography film
(cat. no. E3012; Denville Scientific, Inc.). Densitometry was
performed in triplicate, employing three differing levels of
exposure each analyzed using the Adobe Photoshop CS5 histogram
function to determine the intensity of signal. For each lane of the
gel, the signal intensity of the specific protein analyzed was
divided by the signal intensity of the loading control to normalize
protein loading variation per lane. For each cell population
undergoing the specified treatment, the normalized signal intensity
was divided by the normalized signal intensity of the control cells
(in lane 1 of each gel/western blot) to determine the relative fold
change of protein expression. The signal intensity in control cells
was always assigned an arbitrary value of 1 to allow direct
comparisons of signal intensity (calculated fold changes) to be
readily identified. Loading controls in the present study utilized
β-actin, GAPDH, total MAPK1/2 levels (25,26),
and/or Ponceau S (cat. no. P7170; Sigma-Aldrich; Merck KGaA)
staining for total protein normalization (27). In previous studies by the authors,
it was determined that the levels of total MAPK1/2 do not change in
response to hormonal treatments or MEK1 blockade; only the level of
phosphorylated MAPK1/2 changes with treatments (16). In some analyses, duplicate blots
were required for MAPK detection due to the interference of
residual pMAPK signal after stripping. When protein loading was
similar and signal intensity differences clearly identifiable
between the experimental groups relative to the control group,
densitometry was not performed, e.g. Fig. 1C and D.
Assessment of autophagic flux by LC3-II
turnover
Per independent experiment, cells were seeded in
duplicate at a density of 2×105 cells per 60-mm culture
dish for each treatment such that in one dish the treatment was
conducted in the presence of 10 µM CQ. The CQ-treated cell
population allowed the steady state level of LC3-II to be
determined for each treatment which was compared with the LC3-II
level in cells undergoing the same treatment under conditions of
active LC3-II flux. LC3-II signal intensities, after corrections
for protein loading variations per lane, can be compared between
cell populations undergoing the same treatment in the absence vs.
presence of CQ; CQ allows the steady state LC3-II levels to be
determined. These comparative levels of LC3-II are used to
calculate autophagic flux as previously described (28). The following formula was utilized
to approximate the flux in each treated cell population: [(+CQ
signal intensity) minus (-CQ signal intensity)]/(+CQ
signal intensity) as follows: the signal intensity of LC3-II
in the protein lysate from cells treated in the absence of CQ was
subtracted from the signal intensity of LC3-II (steady state LC3-II
levels) in duplicate cell populations undergoing the same treatment
in the presence of CQ. This value was then divided by the signal
intensity of the steady state level of LC3-II to approximate the
percent of LC3-II actually fluxed in the cell population. To graph
the relative flux indexes, the percent of LC3-II flux in the
control group, E2-treated cells were arbitrarily set equal to
1.
Transfections with BimEL cDNA
The BimEL cDNA expression vector (EX-O0071-M029) and
control vector (EX-Neg-M02) were purchased from GeneCopoeia, Inc.
The vectors/plasmids were isolated using a commercially available
midi-prep kit (cat. no. 12143; Qiagen GmbH) following the
manufacturer's protocol. For transfections, cells were seeded in
DMEM-F12 medium with 5% DCC FBS to yield ~50% confluence. Adherent
cells were transfected with plasmids (4.0 µg) by using
either x-fect reagent (cat. no. 631317; Takara Bio USA, Inc.) or
lipofectamine LTX (11668-019; Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. Following a 16-24 h
transfection at 37°C, cells were treated for various times at 37°C
and harvested for analysis as described in figure legends.
Immunocytochemistry/confocal
microscopy
Detached cells were collected via centrifugation
(300 × g for 5 min at 4°C) of treatment media removed from culture
dishes. Cell pellets were resuspended in DMEM-F12 plus 5% DCC FBS
(SFBU32-0500; Equitech-Bio, Inc.) media and aliquoted into cytospin
funnels (cat. no. 5991040; Global Medical Instrumentation, Inc.)
and centrifuged (300 × g for 5 min at 4°C) using a Shandon Elliot
Cytospin for collection on slides. Cells were fixed in 4%
paraformaldehyde (cat. no. 50980487; Thermo Fisher Scientific,
Inc.) at room temperature (RT) for 15 min, washed 3 times by gentle
agitation in 1X PBS (cat. no. 46-013-CM; Mediatech/Cellgro; Thermo
Fisher Scientific, Inc.) for 5 min at RT, and permeabilized in
ice-cold methanol at 20°C for 10 min. Cells were blocked in 15%
FBS, in 1× PBS, for 1 h at RT. Primary antibody was incubated with
cells overnight (ON) at 4°C; secondary antibody incubations were
for 1-2 h at 37°C. Antibodies diluted in Normal Antibody Diluent
(NAD) (cat. no. ABB125; ScyTek Laboratories, Inc.) included: BimEL
(1:400; CS2819; Cell Signaling Technology, Inc.) and COx IV (1:200;
cat. no. 4D11B3E8; Invitrogen; Thermo Fisher Scientific, Inc.);
AlexaFluor αRabbit (1:1,000) and Cy3 αMouse (1:800) (cat. nos.
711-545-152 and 715-165-150, respectively; both from Jackson
ImmunoResearch Laboratories, Inc.). Coverslips were mounted with
Vectashield DAPI Hard Mount (cat. no. XH-1500; Jackson
ImmunoResearch Laboratories, Inc.). Images of the slides were
captured using a Zeiss LSM 780 upright confocal microscope (Carl
Zeiss AG) equipped with 63× 1.4 NA Plan-Apochromat objective, a
digital AxioCam camera, and a motorized z-axis stage
accessory. MCF-7 cells with BimEL and COx IV antibodies were
excited using an Ar-laser: 488 nm 10 and 561 nm 8%, respectively.
Confocal Z-axis stacks were separated by 0.37 µm with
volume rendering of Z-stacks compiled using Zeiss LSM Image
Browsing software.
Electron microscopy
For EM analysis, adherent cells were collected,
washed in 2× PBS, and fixed for 1 h in ice-cold 3%
glutaraldehyde/0.1 M cacodylate buffer, pH 7.4, rinsed overnight at
4°C in 0.1 M sucrose/0.1 M cacodylate buffer; post-fixed for 1 h at
4°C in 1% OsO4/0.1 M cacodylate buffer, and embedded in
Epon. Sections (0.1 µm) were stained with uranylacetate/lead
citrate (Fluka) and examined with a JEOL 1010 transmission electron
microscope in the Electron Microscopy & Histology Core Facility
(Dept. of Cellular Biology and Anatomy, Augusta University).
Statistical analysis
All data are presented as the mean ± SD values
(n≥3). Sigma Plot 11.0 for Windows (Inpixon) was utilized to
perform statistical analyses. Statistical differences between two
groups were determined by paired Student's t-test, a one-way ANOVA
followed by a Dunnett's post hoc, or a Holm-Šídák test. For
comparisons of greater than three groups, a one-way ANOVA was
performed, followed by Tukey's multiple comparison post hoc test to
determine significance. *P<0.05 was considered to
indicate a statistically significant difference.
Results
BimEL localizes to the mitochondrial
membrane and induces apoptosis in ER+ breast cancer
cells
In a previous study by the authors, siRNA knockdown
was utilized to establish that dephosphorylated BimEL (designated
dBimEL throughout) mediated ROS-dependent apoptosis in
ER+ breast cancer cells induced by antiestrogen and
antiprogestin treatments and the direct targeting of MEK1 (16). In the present study, the
pro-apoptotic action of BimEL was further characterized. Focus was
addressed on ER+ MCF-7 breast cancer cells undergoing
treatments with E2 and E2 + 4-OHT in the absence vs. presence of
the small molecule, reversible MEK1 inhibitor U0126. Within 4 h,
treatment of MCF-7 cells with U0126 resulted in a greater than
2-fold upregulation of dBimEL; whereas dBimEL upregulation in
response to 4-OHT treatment required a minimum of 24 h. A
representative western blot showing dBimEL upregulation by MEK1
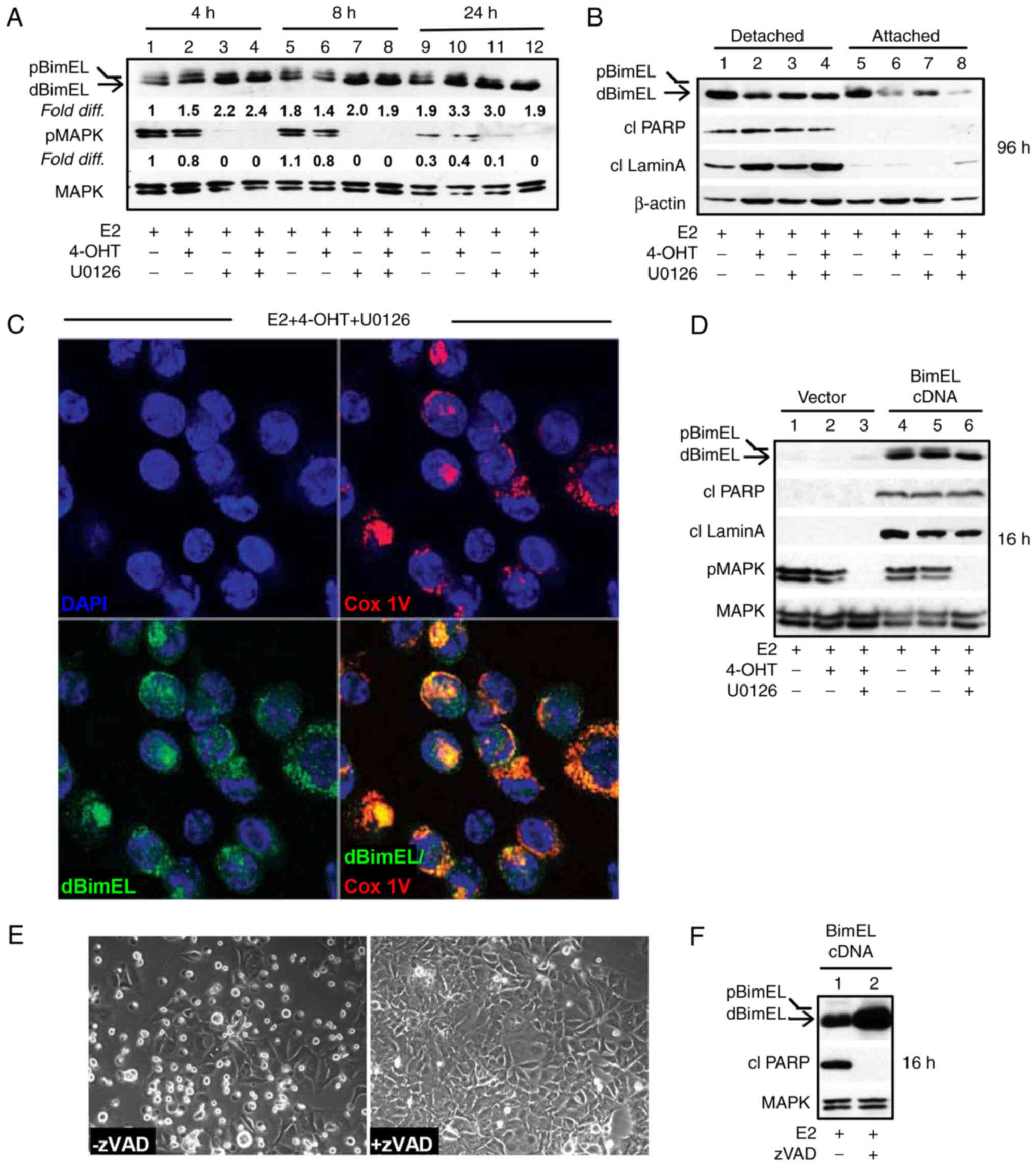
blockade (Fig. 1A) revealed the
increased signal intensity for dBimEL in U0126-treated cells (lanes
3 and 4) relative to the E2-treated control cells (lane 1). Cell
detachment due to dBimEL-dependent apoptosis, however, is not
typically detectable until ~36-48 h of treatment, with optimal
detection of the cleaved forms of PARP and lamin A detectable at
72-96 h (16). Thus, to address a
potentially selective role of dBimEL in apoptosis, the detached,
dying cells were collected separately from the adherent, viable
monolayer of MCF-7 cells after 96 h of treatment with E2, E2 +
4-OHT, and E2 + 4-OHT + U0126 for western blotting. These studies
determined that dBimEL was present at high levels in the detached,
apoptotic cells, while the adherent cells expressed predominantly
pBimEL (i.e. the non-apoptotic form of the protein). The
representative western blot in Fig.
1B shows this selective localization of dBimEL, cleaved PARP,
and cleaved lamin A in detached cells (lanes 1-4) as compared with
barely detectable levels in adherent cells (lanes 5-8). The β-actin
is present at similar levels due to equal loading of protein for
the detached and adherent cells and does not reflect the number of
detached apoptotic cells in the populations undergoing the various
treatments. For example, to accomplish equal protein loading,
detached cells were collected from five 60 mM dishes of E2-treated
MCF-7 cells for total lysate preparation. This low-level of cell
detachment and apoptosis occurs in the E2-treated cell population
as a consequence of the depletion of E2 between 72 & 96 h after
E2 supplementation. By contrast, cell detachment and apoptosis are
readily induced by treatment with 4-OHT +/- U0126, thus detached
cells collected from one 60 mM dish was sufficient. Overall, this
analysis showed cleaved PARP, cleaved lamin A, and dBimEL
selectively localized in the detached cell population undergoing
BimEL-mediated apoptosis. Furthermore, the detached, non-viable
cells showed co-localization of dBimEL with COx IV, a mitochondrial
outer membrane protein. Representative images of this
co-localization are provided from analyses of cells treated with
4-OHT + U0126 (Fig. 1C). This
co-localization was not detected in the viable adherent cells (data
not shown) and is consistent with our previous study identifying
mitochondrial membrane permeabilization and ROS production to be
dependent on BimEL expression (16).
The lag between BimEL dephosphorylation and
induction of apoptosis indicated that dBimEL is sequestered by
pro-survival members of the Bcl-2 protein family (19,29)
that require downregulation or inactivation for dBimEL to induce
apoptosis. Thus, BimEL cDNA was overexpressed to increase the ratio
of dBimEL to the intrinsically expressed anti-apoptotic BH3 family
members. Ectopic overexpression of BimEL cDNA resulted in high
level expression of dBimEL and a concomitant rapid apoptotic
response, with cleavage of PARP and lamin A detectable in
E2-treated cells within 16-24 h of transfection (Fig. 1D, lanes 4-6 compared with 1-3). The
apoptosis induced by dBimEL overexpression was caspase-mediated as
the pan-caspase inhibitor zVAD attenuated cell detachment from the
monolayer (Fig. 1E) and reduced
the levels of cleaved PARP in the cell populations at 16 h
(Fig. 1F), in a manner similar to
zVAD blockade of 4-OHT and MIF-induced apoptosis previously
described (11) and verified for
the present study (data not shown). Altogether, these studies
confirmed that dBimEL is a key effector of caspase-dependent
apoptosis in ER+ antiestrogen-sensitive breast cancer
cells.
Constitutive activation of MEK1/MAPK1/2
in antiestrogen-selected TR5 cells circumvents BimEL-dependent
apoptosis
MEK1/MAPK1/2 signaling has been implicated in
antiestrogen resistance (30);
though, it is not known whether MAPK1/2 regulation of BimEL levels
is involved. Thus, it was investigated if the MEK1/MAPK1/2/BimEL
signaling axis was altered in an antiestrogen-resistant cell line,
designated TR5, established in our laboratory (22). When initially selected, MCF-7 cells
were subjected to a stepwise drug selection that allowed cells to
adapt to increasing concentrations of 4-OHT in medium supplemented
with E2. The final step of 4-OHT selection was adaptation to 5.0
µM 4-OHT, a concentration detected in the tissues of
patients undergoing tamoxifen therapy (31). As TR5 cells were undergoing 4-OHT
selection, parent MCF-7 cells were similarly passaged in E2. For
the present study, TR5 and MCF-7 parent cells were thawed from
cryostorage. Once stably adapted to 5.0 µM 4-OHT (~2 weeks),
TR5 cells showed similar characteristics as described in our
original study, including lack of growth inhibition and induction
of apoptosis in response to 1.0 µM 4-OHT (whereas the
sensitive cells show a 30% decline in viable cell number), a
reduction in growth in 4-OHT concentrations of >4.0 µM
(Fig. 2A), and detectable
autophagosomes/autolysosomes in the cytosol (Fig. 2B, top panel). The MCF-7 parent
cells showed a 4-OHT sensitivity profile similar to that of early
passage MCF-7 cells obtained from the ATCC (Fig. 2A) and did not show elevated levels
of cytoplasmic autophagosomes (Fig.
2B, bottom panel). Immunocytochemistry studies confirmed the
presence of LC3-II puncta and showed detectable co-localization of
p62 in the autophagosomes and autolysosomes of 4-OHT selected TR5
cells. For these studies, the lysosomotropic agent CQ, was added 4
h prior to harvest to block autolysosomal turnover (Fig. 2C), which allows the identification
of autophagosomes with LC3-II and p62 localization (32). Both LC3-II and p62, encoded by the
SQSTM1 gene, are involved in autophagosome formation and
function and are commonly used as markers of functional autophagy
(33).
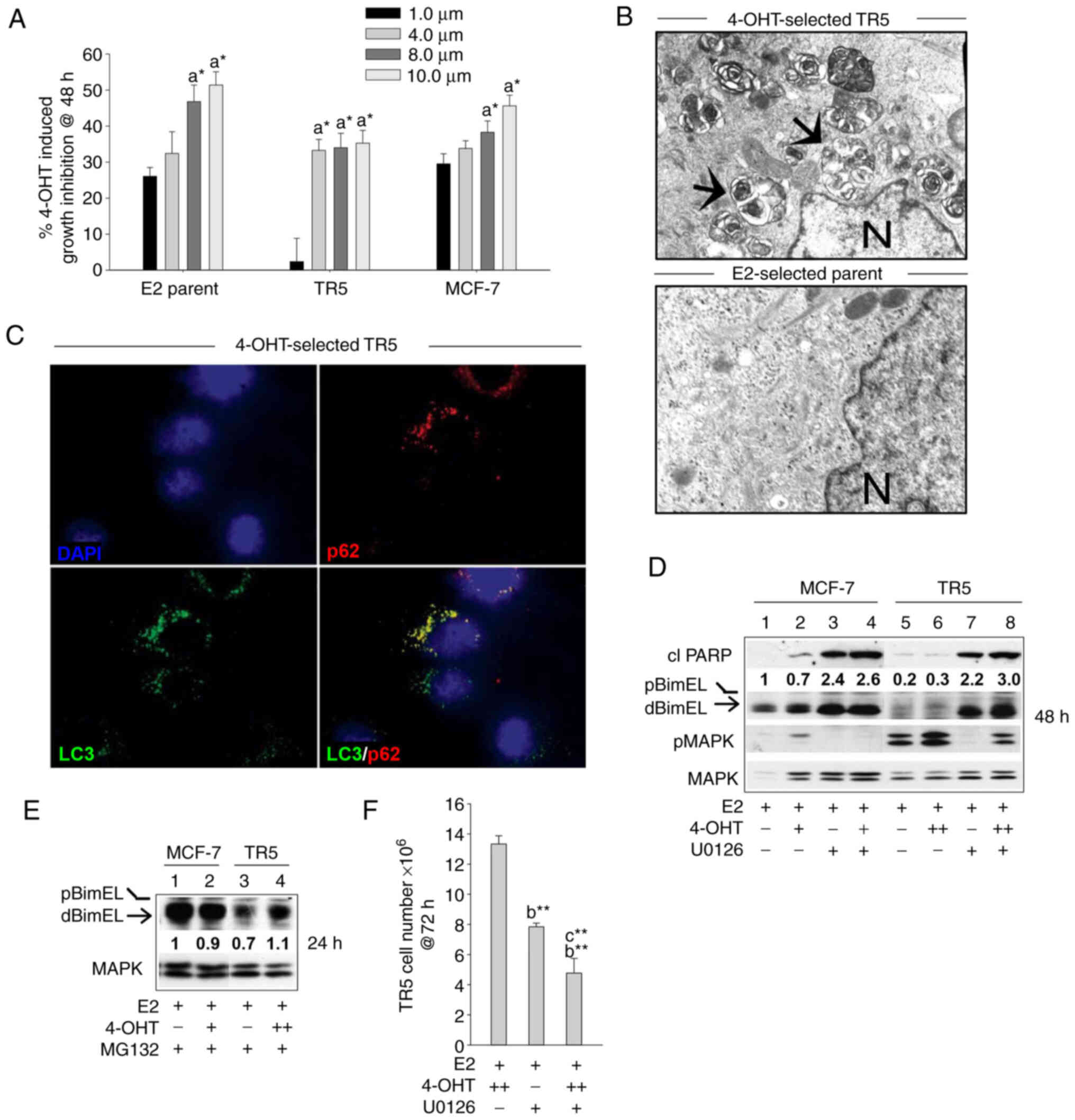 | Figure 2BimEL-dependent apoptosis is
suppressed in 4-OHT-selected, antiestrogen resistant TR5 breast
cancer cells due to upregulation of MEK1/MAPK1/2 signaling. (A)
Graphical representation of dose-dependent, 4-OHT-induced growth
inhibition of parent MCF-7 and 4-OHT resistant TR5 cells at 48 h
compared with MCF-7 ATCC early passage cells. Treatments with 4-OHT
were conducted in the presence of 10 nM E2. Percentage of growth
inhibition was determined by MTT assay. (B) High level
autophagosome production in TR5 cells continuously passaged in 5.0
µM 4-OHT as identified by electron microscopy analyses;
arrows designate autophagosomes and N, nucleus. (C)
Immunofluorescence staining shows expression of LC3 and p62 in
puncta in the cytosol of 4-OHT-selected TR5 cells; CQ was added 4 h
prior to harvest to impede autolysosome turnover to allow the
identification of LC3-II and p62 localization to autophagosomes.
(D) Targeting MEK1/MAPK1/2 with U0126 restored detectable levels of
activated dBimEL, designated by the arrow, and induced apoptosis as
evidenced by cleavage of PARP at 24 h (compare signal intensity of
dBimEL in lanes 7-8 to lanes 5-6). Total MAPK1/2 intensity was used
as loading control to allow determination of efficacy of
U026-mediated blockade of MEK (reduced pMAPK1/2 levels). (E)
Proteasome inhibition with MG132 (1 µM) restored detectable
levels of BimEL to TR5 cells undergoing 4-OHT selection; levels of
BimEL in the parent MCF-7 cells during proteasomal blockade are
shown for comparison and are similar to those identified in our
previous study (16). (F) TR5
cells show a significant reduction in cell number following 72 h
treatment with U0126 as a single agent or in combination with 5.0
µM 4-OHT. TR5 cells were seeded for 24 h in the absence of
drug selection, which is E2 plus 5.0 µM 4-OHT. The adherent
TR5 cells were then treated with E2 + 4-OHT, E2 + U0126, and E2 +
4-OHT + U0126. Cell counts were determined with a hemocytometer.
Results are expressed as the mean ± SD values. Comparisons that
were statistically significant include TR5 growth inhibition
mediated by athe designated treatment compared with the E2 (10 nM)
control cells; bthe designated treatment compared with
growth in 5.0 µM 4-OHT and cthe designated
treatment compared with growth in U0126. +, designates treatment
with 1 µM 4-OHT; ++, 5 µM 4-OHT.
*P<0.05 and **P<0.05. CQ, chloroquine;
4-OHT, 4-hydroxytamoxifen; p-, phosphorylated. |
MEK1 activity levels (i.e., phosphorylated MAPK1/2)
were next compared by western blot analyses of total protein
isolated from TR5 and MCF-7 cells treated with E2, E2 + 4-OHT, and
E2 + 4-OHT + U0126. MEK1 activity was assayed by immunoblotting
studies that compared MAPK1/2 phosphorylation levels. Elevated
levels of pMAPK1/2 and barely detectable levels of dBimEL
(pro-apoptotic form) were identified in 4-OHT-selected TR5 cells as
compared with the parent MCF-7 cells (Fig. 2D). Importantly, the targeting of
MEK1 with U0126 in 4-OHT-selected TR5 cells reduced pMAPK1/2 levels
and restored detectable BimEL expression, concomitant with
induction of apoptotic cell death at 24 h as evidenced by cleavage
of PARP (Fig. 2D; lanes 7 and 8
compared with 5 and 6). The fact that MEK1/MAPK1/2 blockade
restored detectable dBimEL levels, indicated that BimEL was
expressed primarily as pBimEL in TR5 cells and that pBimEL was
being degraded via the proteasome, a mode of pBimEL degradation
previously identified for MCF-7 cells (16). To investigate this, TR5 and MCF-7
cells were cultured in E2 or E2 plus 4-OHT in the presence of the
proteasome inhibitor MG132. Treatment with MG132 resulted in an
accumulation of pBimEL in TR5 cells, approaching levels observed in
parent MCF-7 cells (Fig. 2E).
Thus, the elevated pMAPK1/2 in TR5 cells mediated phosphorylation
of BimEL and its subsequent degradation in the proteasome. Cell
counts further showed that treatment of TR5 cells with U0126 as a
single agent or in combination with 4-OHT significantly reduced TR5
cell number by 58.7±1.9 and 63.4±4.2%, respectively (Fig. 2F). Thus, MEK1/MAPK1/2 blockade
generated pro-apoptotic dBimEL to levels that induced apoptosis and
a significant reduction in TR5 cell number.
Targeting MEK1/MAPK1/2 in
antiestrogen-resistant breast cancer cells does not effectively
block pro-survival autophagy
Because a large subpopulation (36.6±4.2%) of the
antiestrogen-resistant, 4-OHT-treated TR5 cells survived
U0126-mediated MEK1/MAPK1/2 blockade (Fig. 2F), it was hypothesized that the
surviving cells could be utilizing autophagy as a mode of survival.
To examine this hypothesis, LC3-II and p62 turnover (flux) were
analyzed as surrogate measures of functional autophagy. Due to its
association with the autophagosome membrane, LC3-II turnover occurs
concomitantly with autolysosomal flux (33,34).
Moreover, when functioning as an autophagy receptor protein that
shuttles misfolded proteins and damaged organelles to the
autophagosome, p62 is degraded during autolysosome turnover
(35). To analyze LC3-II and p62
levels and turnover (flux), hormonal treatments were conducted in
duplicate; one culture dish for each treatment contained CQ to
block autolysosomal turnover and allowed LC3-II and p62 steady
state levels to be quantified. Following treatment, cells were
harvested for total protein and western blot analyses were
performed. As revealed in Fig. 3A,
the relative steady state level of LC3-II in cell populations
treated in the presence of CQ (lanes 5-8) was compared with LC3-II
levels in the matched cell population undergoing the respective
treatment in the absence of CQ (lanes 1-4), with all signal
intensities calculated relative to the LC3-II signal intensity in
the E2-treated control cells (lane 1) assigned an arbitrary value
of 1 after corrections for loading variation per lane as described
in materials and methods. Results from multiple independent
experiments are graphically shown in Fig. 3B. These data identified: i) ~30% of
LC3-II undergoes active flux in the 4-OHT-selected TR5 cells; and
ii) the blockade of MEK1/MAPK1/2 does not attenuate the
4-OHT-induced LC3-II flux. A similar pattern of p62 flux can be
discerned in Fig. 3A, comparing
the steady state levels of p62 (lanes 5-8) to the p62 levels in
cell populations undergoing the respective treatment in the absence
of CQ (lanes 1-4). U0126 treatment effectively blocked pMAPK1/2 and
induced the accumulation of pro-apoptotic dBimEL in the cell
populations (Fig. 3A), consistent
with experiments described in Fig.
2D. Immunocytochemistry further showed co-localization of
LC3-II and p62 in cytosolic puncta in TR5 cells undergoing 4-OHT +
U0126 treatment (Fig. 3C).
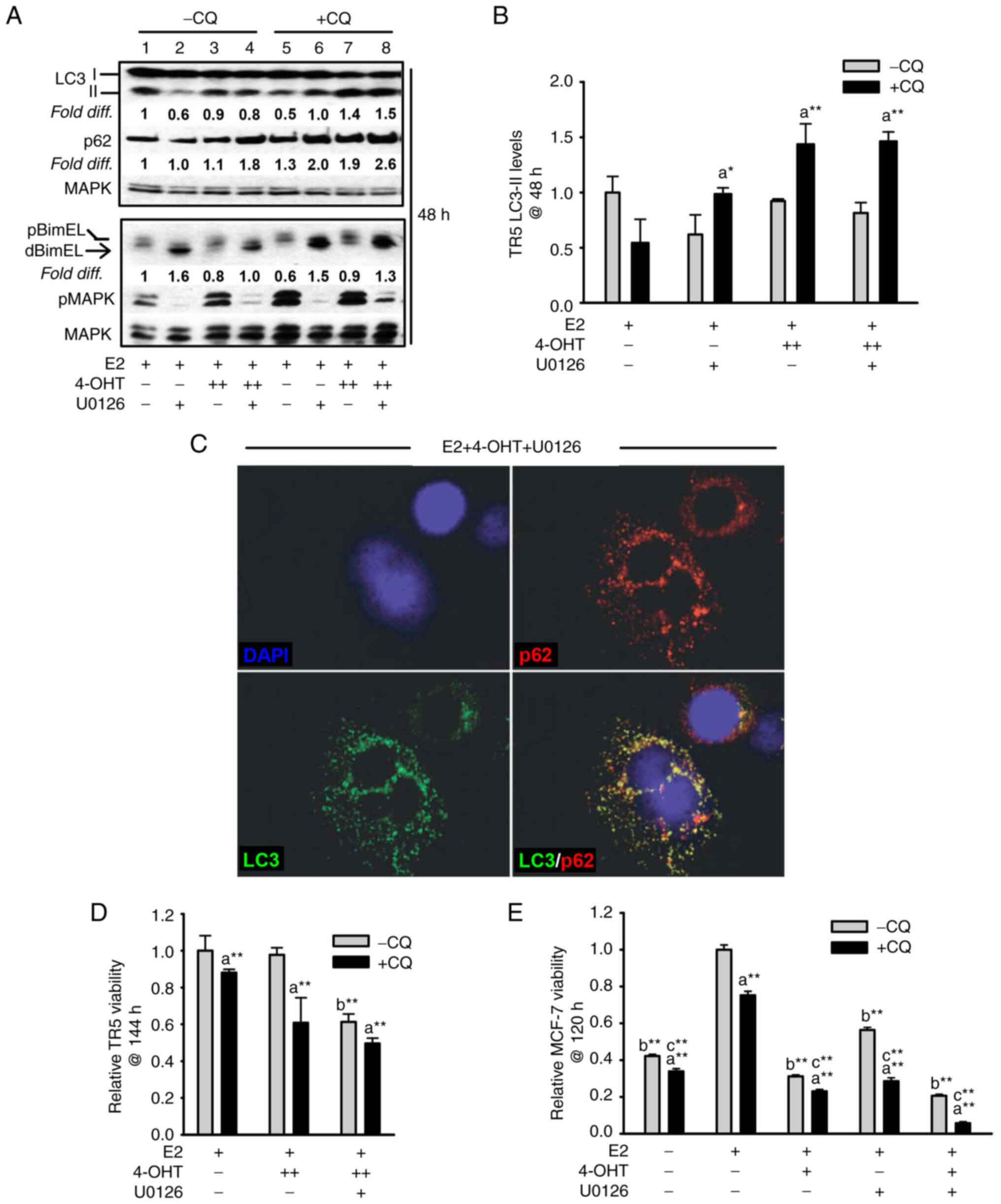 | Figure 3Targeting the MEK1/MAPK1/2 signaling
axis in antiestrogen resistant TR5 cells induces pro-survival
autophagy. (A) Immunoblot, representative of three independent
experiments, determined LC3-II levels in TR5 cells treated with
4-OHT (5 µM) and/or U0126 (10 µM) as compared with
levels in E2-treated cells. 4-OHT treatment, in the presence or
absence of U0126, increased steady-state levels of LC3-II and p62
in TR5 cells. Relative signal intensity of LC3-II, p62, and BimEL
is shown relative to their signal intensity in TR5 control cells
(passaged for 24 h in E2-supplemented DMEM/F12 medium) which was
given an arbitrary value of 1. Signal intensity of total MAPK1/2
(designated MAPK) was used to correct for loading variations per
lane and determine the efficacy of U026-mediated blockade of MEK
(reduced pMAPK1/2 levels). (B) A graphical representation of the
data shows higher steady-state LC3-II levels in the cells
undergoing treatments in the presence vs. absence of CQ, indicative
of active autolysosomal flux under conditions of MEK1/MAPK1/2
blockade. (C) MEK1/MAPK1/2 blockade by U0126 for 48 h in
4-OHT-treated TR5 cells shows functional autophagy with
co-localization of LC3-II and p62 as determined by
immunocytochemistry and confocal microscopy as described in
materials and methods. CQ was added to the treatment 4 h prior to
harvest to block autolysosomal flux. (D and E) Relative viability
of (D) antiestrogen resistant TR5 cells and (E) antiestrogen
sensitive MCF-7 cells as determined by the MTT assay is shown
following treatments with E2, and or E2 + 4-OHT in the absence or
presence of U0126, plus and minus CQ (n=3) at 144 h for TR5 cells
and 120 h for MCF-7 cells. CQ treatment reduced the viability of
TR5 and MCF-7 cells treated with 4-OHT or 4-OHT + U0126, indicating
that autophagic flux was cytoprotective under conditions of
MEK1/MAPK1/2 blockade. Results are expressed as the mean ± SD
values Comparisons that were statistically significant identify
growth inhibition mediated by aCQ relative to the
respective treatment conducted in the absence of CQ; bE2
+ 4-OHT + U0126 relative to E2 + 4-OHT; and cthe
designated treatment relative to E2. *P<0.05 and
**P<0.05. CQ, chloroquine; 4-OHT, 4-hydroxytamoxifen;
p-, phosphorylated. |
The autophagy expressed in 4-OHT-selected TR5 cells
was not cytotoxic and, therefore, not required for BimEL-dependent
apoptosis induced by MEK1/MAPK1/2 blockade. This was determined by
analyzing the effects of CQ on TR5 cell viability. It was reasoned
that if autophagic catabolism was a prerequisite for
BimEL-dependent cell death, CQ should attenuate the cytotoxic
outcome of U0126 treatment of TR5 cells. However, CQ treatment
combined with 5.0 µM 4-OHT or 5.0 µM 4-OHT + U0126
reduced TR5 cell viability (Fig.
3D), by 40±15% and significantly enhanced the cytotoxic effects
of U0126 treatment, respectively (Fig.
3D). For comparison, antiestrogen-sensitive MCF-7 cells were
similarly analyzed. The combined treatment of 4-OHT plus U0126 was
highly cytotoxic by 120 h, with a greater than 4-fold reduction in
MCF-7 cell viability as compared with a 3.2- and 1.7-fold reduction
by 4-OHT and U0126 single agent treatments, respectively (Fig. 3E). Importantly, CQ significantly
reduced cell viability in all the MCF-7 populations undergoing
treatments. These data supported the conclusion that autophagy is
protective against BimEL-dependent apoptosis in 4-OHT-selected TR5
breast cancer cells, but caution that the inhibition of autophagy
during MEK1/MAPK1/2 blockade is not an effective strategy to
eliminate the antiestrogen-resistant breast cancer cells.
Autophagy and dBimEL-dependent apoptosis
are simultaneously detected in antiestrogen-sensitive breast cancer
cells undergoing hormone treatments in the presence and absence of
MEK1/MAPK1/2
It was next sought to characterize the relationship
between autophagy and apoptosis in antiestrogen-sensitive cells
undergoing hormone treatments in the presence and absence of MEK1
blockade. In addition to treatments with E2 and 4-OHT, the
antiprogestin MIF was also used for these studies because the
combined treatment of MIF plus 4-OHT induces a robust
dBimEL-dependent apoptosis in MCF-7 cells independent of
MEK1/MAPK1/2 co-targeting (16).
It was first established that MCF-7 cells treated with 4-OHT, MIF,
or MIF + 4-OHT expressed higher levels of autophagy than the
E2-treated control cells. These studies, provided in Fig. S1, utilized EM to quantify
autophagosomes per cell, long-lived protein turnover studies
(23) as an independent read-out
of autophagic catabolism, and cell counts to identify growth
inhibition by 4-OHT ± MIF treatments relative to autophagy levels.
When hormonal treatments (E2, E2 + 4-OHT, E2 + MIF, and E2 + 4-OHT
+ MIF) were conducted for 48 h in the absence and presence of
U0126, apoptosis induced by MEK1/MAPK1/2 blockade was reproducibly
detected. Furthermore, apoptosis was most pronounced in cells
subjected to 4-OHT + MIF treatments ± U0126. This can be observed
in the representative western blot shown in Fig. 4A (top panel), showing the highest
levels of cleaved PARP and cleaved lamin A in lanes 3 and 6.
Notably, LC3-II flux was also detected in the hormonally-treated
cell populations even under conditions of MEK1/MAPK1/2 blockade at
levels comparable to LC3-II flux in the E2-treated control cells.
As revealed in Fig. 4A (bottom
panel), the relative signal intensity of LC3-II in the cell
populations undergoing the various treatments in the presence of CQ
(lanes 7-12) compared with LC3-II levels not actively fluxed in
cell populations undergoing the respective treatment in the absence
of CQ (lanes 1-6). A graphical representation of LC3-II levels for
each cell population undergoing the designated treatment in the
presence vs. absence of CQ is provided in Fig. 4B. These data demonstrated an
inability of MEK1/MAPK1/2 blockade by U0126 to effectively block
autophagy (LC3-II flux). In independent studies, long-lived protein
turnover (23) was analyzed. These
studies utilized CQ to confirm lysosome-dependent long-lived
protein turnover in the cell populations undergoing each treatment
regimen. Collectively, these studies identified active autophagic
catabolism along with detectable apoptosis in breast cancer cells
undergoing hormonal treatments in the absence and presence of
MEK1/MAPK1/2 blockade (Fig.
4C).
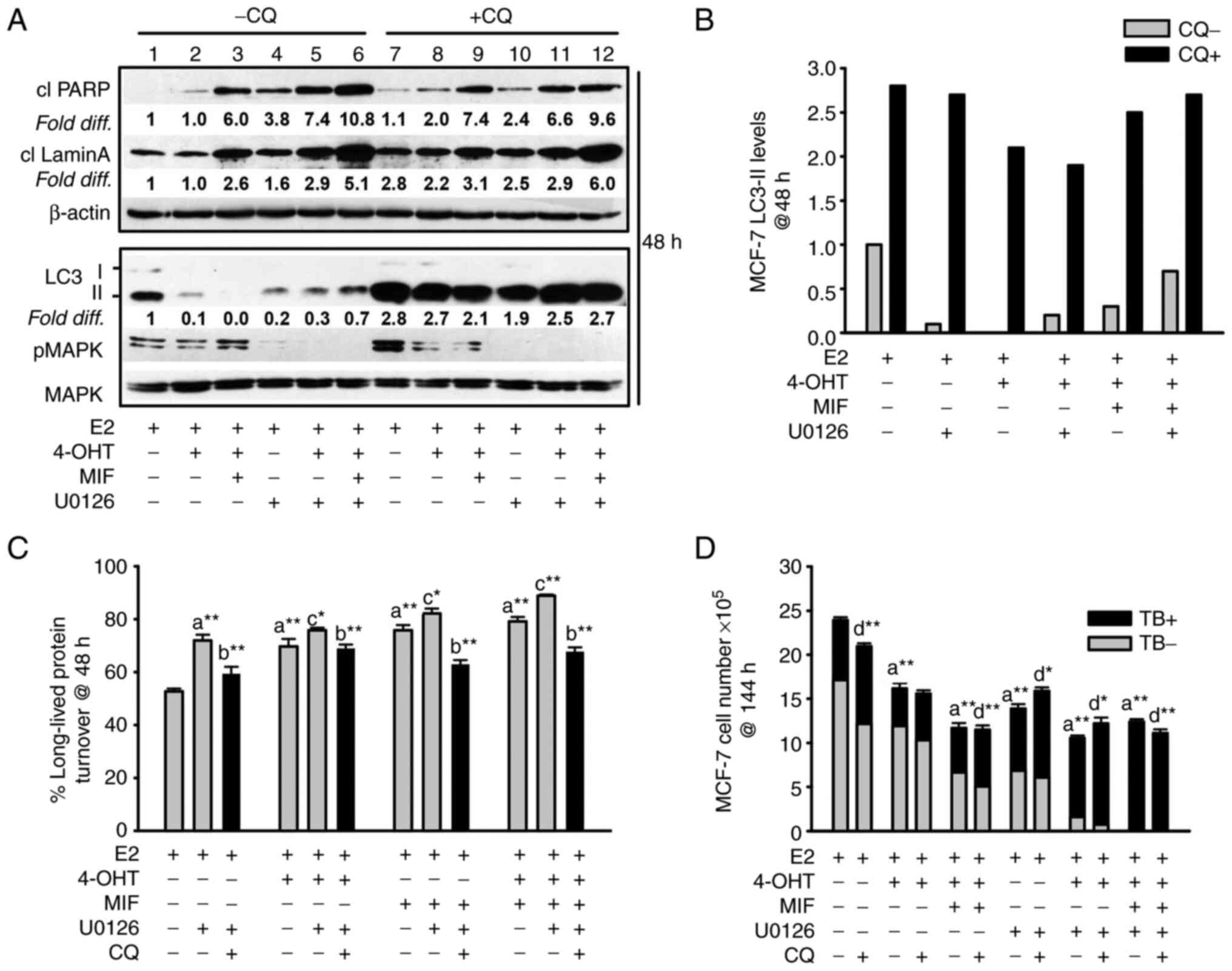 | Figure 4Apoptosis and non-cytotoxic autophagy
detected in antiestrogen sensitive breast cancer cell populations
undergoing MEK1/MAPK1/2 blockade. (A, top panel) Increased levels
of the cleaved forms of lamin A and PARP, two markers of apoptosis,
were consistently detected by 48 h of hormonal treatments conducted
in the presence of U0126 (top panel; signal intensities in lanes
4-6 were compared with lanes 1-3, with signal intensity for
E2-treated control cells in lane 1 arbitrarily set to a value of 1
after corrections were calculated per lane using β actin as the
loading control. (A, bottom panel) Increased steady-state LC3-II
levels in cells undergoing the designated treatments for 48 h in
the presence of CQ compared with the respective treatment conducted
in the absence of CQ (signal intensity in lanes 7-12 was compared
with lanes 1-6). Corrections for loading per lane were made using
total MAPK as the loading control, also allowing the efficacy
U0126-mediated blockade of MEK1/MAPK1/2 to be evaluated. (B)
Graphical representation of the relative fold difference in the
cells undergoing treatments in the presence vs. absence of CQ,
shows higher steady-state LC3-II levels in the cells undergoing
treatments in the presence of CQ, indicative of active
autolysosomal flux under conditions of MEK1/MAPK1/2 blockade. (C)
Long-lived protein catabolism (autophagic flux) was functional in
hormonally-treated breast cancer cells in the absence and presence
of MEK1/MAPK1/2 blockade by U0126 treatment, with catabolism in
4-OHT and/or MIF-treated cell populations showing increased
catabolism relative to E2-treated control cells. CQ was added to
duplicate cell populations to confirm the involvement of the
lysosome in the long-lived protein turnover measured. (D)
CQ-mediated blockade of autolysosomal flux did not increase the
number of viable (trypan blue negative, designated TB-) cells, and
modestly increased the number of non-viable cells (designated TB+)
in cell populations undergoing hormonal treatments in the absence
and presence of U0126. Results are expressed as the mean ± SD
values. Comparisons that were statistically significant include:
athe designated treatment compared with E2-treated
control cells; bthe designated treatment conducted in
the presence of CQ relative to the respective treatment conducted
in the absence of CQ; cthe designated treatment
conducted in the presence of U0126 relative to the respective
treatment conducted in the absence of U0126; dCQ
mediated inhibition of cell number or increases in trypan blue
positive cells (TB+). *P<0.05 and
**P<0.001. CQ, chloroquine; 4-OHT,
4-hydroxytamoxifen; TB+, trypan blue; MIF, mifepristone. |
The autophagy/autophagic catabolism in
antiestrogen-sensitive MCF-7 breast cancer cells undergoing the
various treatments did not appear to be required for apoptosis
induction by MEK1/MAPK1/2 blockade, previously identified as
BimEL-dependent (16). The
following data supported this conclusion: first, CQ treatment did
not attenuate the levels of cleaved PARP or cleaved lamin A in the
cell populations undergoing the various treatments (Fig. 4A, top panel, lanes 7-12 compared
with lanes 1-6 for signal intensity of cleaved PARP and cleaved
lamin A). Secondly, cell counts conducted with cell populations
undergoing the various treatments for 144 h established that
CQ-mediated blockade of autophagic flux was not cytotoxic; CQ did
not increase total cell number, nor reduce the TB positive cell
number (Fig. 4D). CQ treatment,
however, showed only modest increased MCF-7 apoptotic cell death in
response to the treatments. Thus, two other small molecule
inhibitors that target early-stage autophagy were utilized:
spautin-1, a small molecule inhibitor that targets Beclin (36) and compound 19, a selective vps34
inhibitor (37). These inhibitors
reduced cell viability in MCF-7 cell populations undergoing
MEK1/MAPK1/2 blockade in the presence and absence of antiestrogen
treatment. Spautin-1 induced a robust death response by 72 h as
evidenced by a measurable elevation in the levels of cleaved PARP
(Fig. 5A), a reduction in cell
number, and an increase in TB positive cells (Fig. 5B). In a similar manner, 72 h MTT
assays showed pronounced effects of vps34 inhibition by compound 19
on hormonally-treated MCF-7 cells during MEK1/MAPK1/2 blockade
(Fig. 5C and D). Vps34 inhibition
utilizing 1.0 µM compound 19 resulted in blockade of p62
flux and a measurable reduction in LC3-II in cells undergoing
MEK1/MAPK1/2 (Fig. 5E), consistent
with effective vps34 inhibition (37). Altogether, these studies supported
the conclusion that autophagy, when present, is protective against
apoptosis and that early-stage autophagy inhibitors are more potent
than CQ in blocking pro-survival autophagy in breast cancer cells
undergoing MEK1/MAPK1/2 blockade.
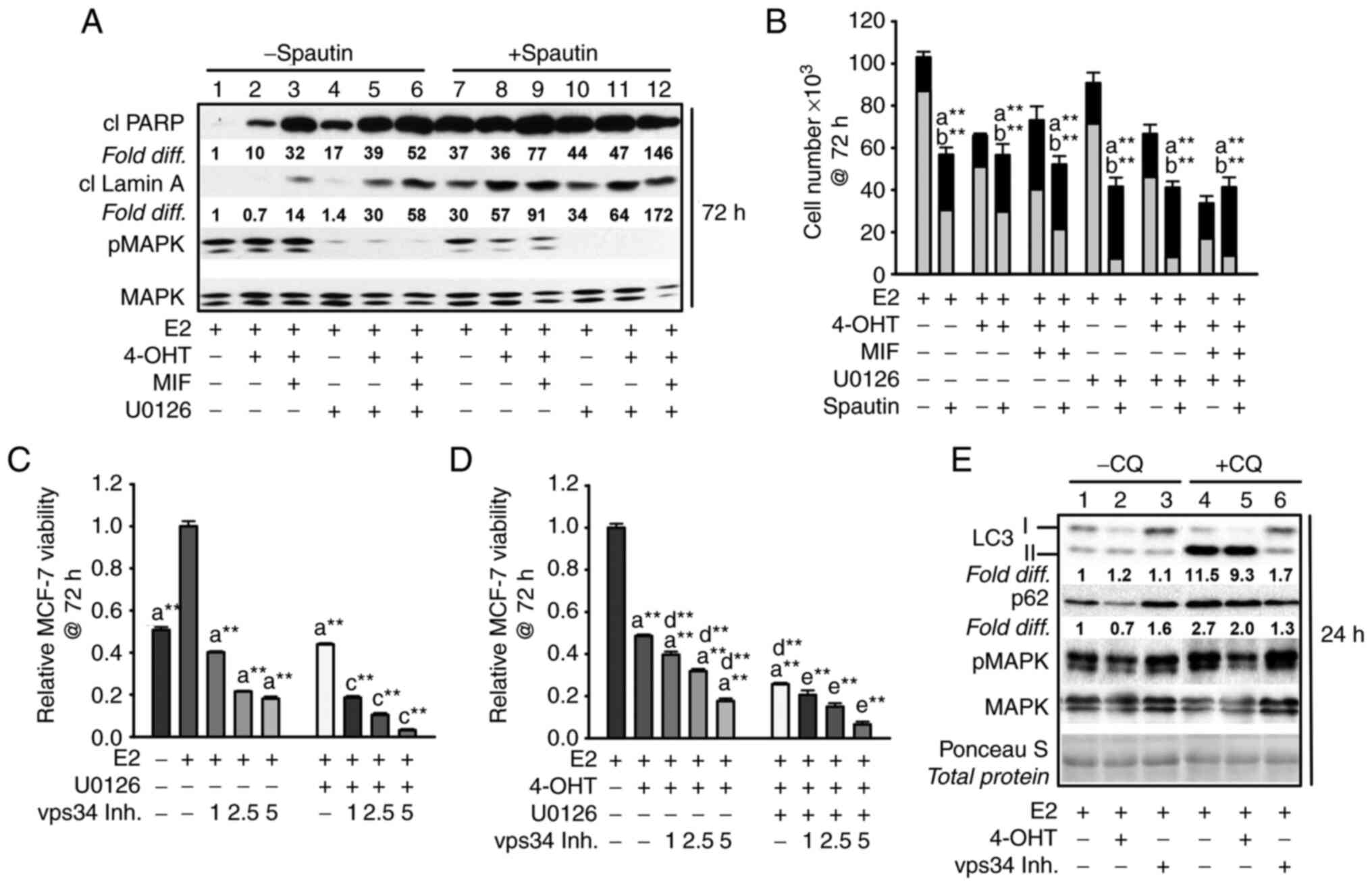 | Figure 5Inhibiting early-stage autophagy
induces apoptosis in antiestrogen sensitive breast cancer cells
undergoing MEK1/MAPK1/2 blockade. (A) Spautin-1, an inhibitor of
early autophagy, induced apoptosis by 72 h of treatment as
evidenced by increased levels of cleaved PARP and cleaved lamin A
in MCF-7 cell populations treated with hormones in the presence or
absence of U0126; signal intensities in lanes 7-12 compared with
lanes 1-6. The signal intensity was arbitrarily set to a value of
1.0 in the E2-treated control cells (lane 1), after correction for
loading variation per lane utilizing the signal intensity of total
MAPK1/2 (designated MAPK), which also allowed the level of pMAPK1/2
activation (phosphorylation) to be determined following U0126
treatment. (B) Spautin-1 decreased cell number and increased the
number of trypan blue positive (dead) cells when combined with
hormone treatments in the absence and presence of U0126. (C and D)
Vps34 inhibition by compound 19 (1.0-5.0 µM) reduced MCF-7
cell viability reproducibly detectable by 72 h in cell populations
treated with E2 (C), or E2 + 4-OHT (D) in the presence and absence
of U0126 at 10.0 and 5.0 µM for treatments in panel C and D,
respectively. (E) Within 24 h, compound 19 at 1.0 µM blocked
p62 turnover based on similar p62 levels in cells treated in the
absence vs. presence of CQ (signal intensity compared in lanes 3
and 6, with signal intensity for E2-treated control cells given an
arbitrary value of 1.0); decreased LC3-II lipidation, with higher
levels of LC3-I as compared with LC3-II detected in cells; and did
not show off target blockade of pMAPK. Total protein on the western
blot was stained with Ponceau S to verify ~ even loading per lane.
(Panel E). Comparisons that were statistically significant include:
athe designated treatment vs. E2-treated;
bthe designated treatment conducted in the presence vs.
absence of spautin-1; cthe designated treatment
conducted in the presence vs. absence of U0126; dthe
designated treatment conducted in the presence vs. absence of
4-OHT; and ethe designated treatment conducted in the
presence vs. absence of 4-OHT and U0126. *P<0.05 and
**P<0.05. CQ, chloroquine; 4-OHT, 4-hydroxytamoxifen;
MIF, mifepristone; p-, phosphorylated. |
EGFR upregulation in
antiestrogen-resistant breast cancer cells regulates MEK1/MAPK1/2
signaling in autophagy
Due to the key role of MEK1/MAPK1/2 signaling in
regulating BimEL and autophagy levels in the antiestrogen-resistant
TR5 cells, it was next sought to determine the mechanism of
MEK1/MAPK1/2 upregulation in 4-OHT-selected TR5 cells. Immunoblot
analyses determined that EGFR expression was highly upregulated,
while ERα expression was potently downregulated (Fig. 6A). The EGFR in 4-OHT-selected TR5
cells was phosphorylated at tyrosine (Tyr) 1068, and this
phosphorylation (designated pEGFR) occurred independent of
MEK1/MAPK1/2 kinase activity as treatment with U0126 did not reduce
pEGFR in TR5 cells (Fig. 6B). By
stark contrast, erlotinib (ERL), a reversible but highly selective
EGFR inhibitor (38), effectively
blocked MAPK1/2 phosphorylation in TR5 cells (Fig. 6B, lane 5; Fig. 6C, lane 4) and ERL-treated TR5 cells
showed 4- to 5-fold increases in the levels of dBimEL (Fig. 6D). It was further determined that
ERL treatment induced autophagy (Fig.
6, panels D and E), with more than a 2-fold increase in LC3-II
and p62 turnover in ERL-treated TR5 cells (Fig. 6F). This induction can be observed
by comparing LC3-II and p62 signal intensity in cells undergoing
treatments in the presence vs. absence of CQ in which steady state
levels can be directly compared between lanes 10-12 and lanes 4-6.
A graphical representation of autophagic (LC3-II) flux is shown in
Fig. 6G and was determined as
described in materials and methods. Importantly, ERL treatment
combined with 4-OHT significantly reduced TR5 cell viability, and
CQ co-treatment enhanced this reduction and also reduced cell
viability when TR5 cells were seeded in 4-OHT (Fig. 6H). Thus, autophagy induced by ERL
served a cytoprotective role. Based on these data, it was
hypothesized that the upregulation of EGFR/MEK1/MAPK1/2 facilitated
TR5 cell escape from BimEL-dependent apoptosis during the initial
stepwise 4-OHT selection and also provided proliferative capability
to TR5 cells experiencing a high-level autophagy induction. A role
of EGFR/MEK/MAPK1/2 signaling in the escape of breast cancer cells
from the action of antiestrogens was proposed in Fig. 7.
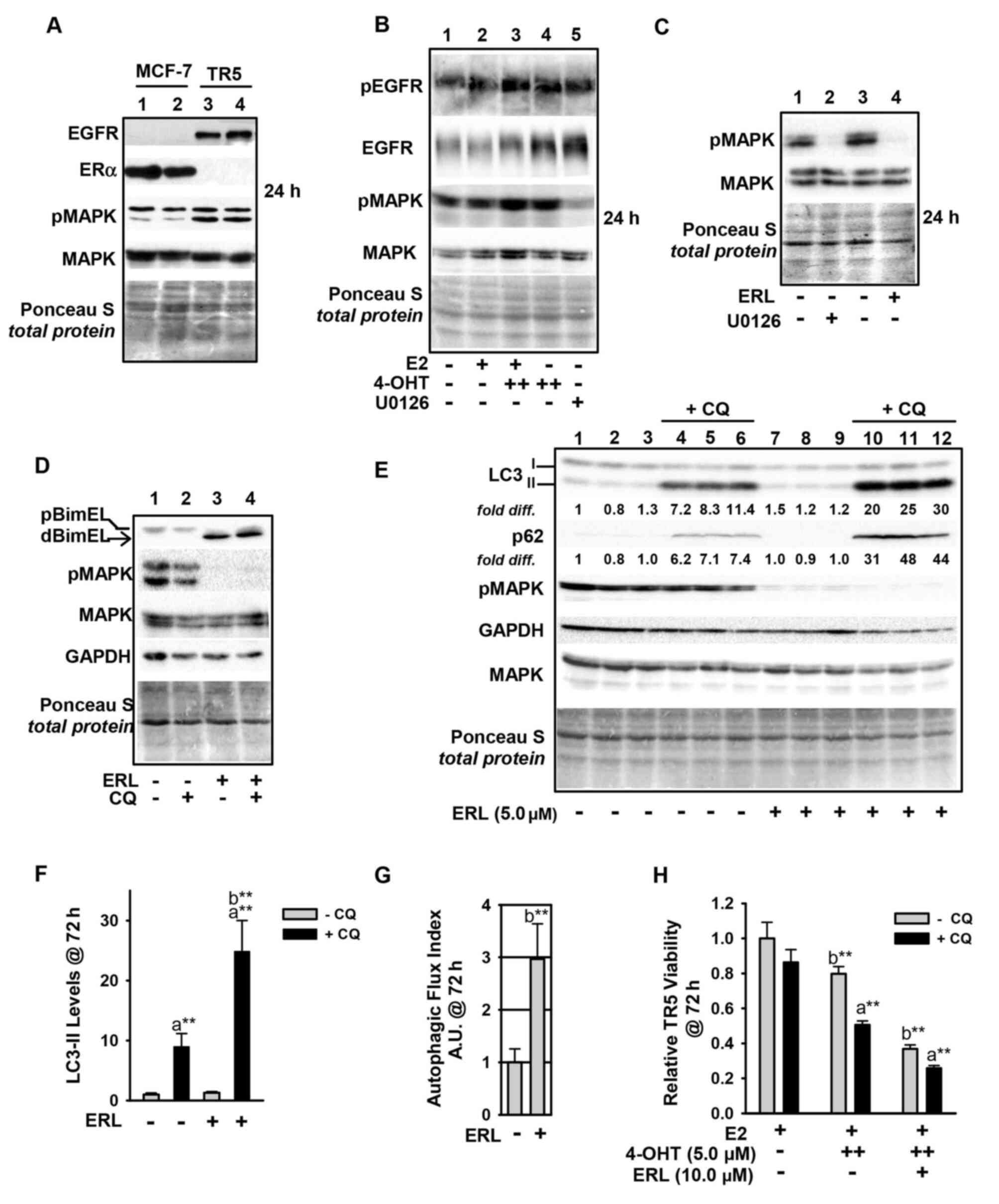 | Figure 6Upregulation of the EGFR/MEK1/MAPK1/2
signaling axis in antiestrogen resistant TR5 cells. (A) TR5 cells
express EGFR, concomitant with loss of ERα expression as shown by
immunoblotting. (B) EGFR is phosphorylated at Tyr1068 in a
MEK1/MAPK1/2 independent manner and under conditions of 4-OHT
selection at 1 and 5 µM (designated as + and ++,
respectively). (C and D) ERL, a selective tyrosine kinase inhibitor
of EGFR utilized at 5 µM, blocks pMAPK1/2 phosphorylation at
24 h as effectively as U0126-mediated blockade of MEK1/MAPK1/2
(signal intensity of pMAPK compared in lanes 2 and 4) (C), and
increases dBimEL levels in TR5 cells (D). (E) Immunoblot analyses
(n=3) shows increased autophagy in 4-OHT-selected TR5 cells treated
with 5 µM ERL for 72 h; LC3-II and p62 are elevated in
treatments conducted in the presence of CQ indicating active flux
in the ERL-treated cell populations. (F and G) A graphical
representation of LC3-II steady-state levels, (F) and flux (G) are
provided for LC3-II. Autophagic flux was calculated as described in
materials and methods. (H) MTT assay shows a decrease in the
relative viability of TR5 cells undergoing ERL treatment at 10
µM plus and minus CQ for 72 h. In panels A-E, signal
intensity of total MAPK provides the loading control, was used to
correct for loading variations to establish relative signal
intensities (D and E), and to determine efficacy of U0126-mediated
blockade of MEK (reduced pMAPK1/2 levels). Comparisons that show
statistically significant differences includea CQ
relative to the respective treatment conducted in the absence of
CQ; bthe designated treatment compared with no treatment
or growth in E2. **P<0.05. CQ, chloroquine; 4-OHT,
4-hydroxytamoxifen; p-, phosphorylated. |
Discussion
In a previous study, a key role was established for
MAPK1/2 in the phosphorylation of BimEL which led to its
proteasomal degradation in ER+ breast cancer cells and
resistance to hormonally-induced ROS-dependent apoptosis (16). In the present study, the role of
BimEL in antiestrogen sensitive and resistant breast cancer cells
was further analyzed and it was demonstrated that: i) ectopic BimEL
cDNA overexpression induced a robust and rapid apoptotic response;
ii) intrinsically expressed dBimEL co-localized with COxIV to the
outer mitochondrial membrane of apoptotic, but not viable adherent
MCF-7 cells; and iii) upregulation of the EGFR/MEK1/MAPK1/2
signaling axis blocked dBimEL-dependent apoptosis in
antiestrogen-selected, antiestrogen-resistant breast cancer cells.
The antiestrogen-resistant breast cancer cell model used in the
present study, designated TR5, was established by subjecting MCF-7
cells to a stepwise 4-OHT selection in the absence of clonal
selection (22). Altogether, these
data are consistent with a previous study by the authors
identifying dBimEL as a key effector of ROS production induced by
hormone treatments and MEK1 blockade in antiestrogen sensitive
breast cancer cells (16). Of
clinical relevance, MEK1/MAPK1/2 mediated signaling in breast
cancer is associated with increased metastasis risk (19), as well as anti-estrogen resistance
via cross-talk that occurs between the Raf/MEK/MAPK pathway and ERα
(39). Consistent with a role for
MEK1/MAPK1/2 in breast cancer metastasis, a pre-clinical study has
identified the suppression of breast cancer metastasis by
pro-apoptotic Bim (40). Thus, the
targeting of MEK1/MAPK1/2 to activate BimEL-induced apoptosis of
hormonally treated breast cancer cells has the potential to lead to
improved clinical outcomes.
To date, the targeting of MEK1/MAPK1/2 signaling in
clinical trials (41,42) has not recapitulated numerous of the
promising pre-clinical in vitro and in vivo studies
that established a role for MEK1/MAPK1/2 signaling in promoting
breast cancer cell survival and progression (19,43-45).
Even the use of a MEK inhibitor with AI has not proven effective
for the treatment of advanced-stage breast cancer (46). Understanding the survival modes of
breast cancer cells undergoing MEK1/MAPK1/2 blockade should help
identify additional molecular targets to improve the use of
selective MEK1 inhibitors in the clinic (19,41,46).
Toward this goal, the present study identified functional autophagy
in the anti-estrogen resistant TR5 cells and the antiestrogen
sensitive parent MCF-7 cells undergoing MEK1/MAPK1/2 blockade. The
autophagy was determined not to be a pre-requisite of BimEL-induced
apoptosis but provided a cytoprotective role. Thus, it was
indicated that pro-survival autophagy contributes to the lack of
efficacy of MEK1/MAPK1/2 inhibitors in the clinic and should be
considered a molecular target to improve the outcome of MEK1
targeting when utilized for the treatment of ER+ breast
cancer. It will be important to identify the underlying
mechanism(s) of autophagy in breast cancer cells that survive
MEK1/MAPK1/2 targeting considering that the Ras/MEK/MAPK pathway is
known to be upregulated in numerous breast cancers, particularly
triple negative breast cancer (TNBC) (47). Moreover, it will be important to
determine if targeting MEK1/MAPK1/2 in TNBC typically induces a
pro-survival autophagy as it was identified in TR5 cells. Such
studies are currently ongoing in our laboratories and have the
potential to identify new molecular targets for the treatment of
TNBC.
The targeting of EGFR in the antiestrogen resistant
TR5 cells with the small molecule inhibitor ERL also induced a
pro-survival autophagy, concomitant with reducing cell viability.
Thus, the co-targeting of the EGFR/MEK1/MAPK1/2 signaling axis and
autophagy along with hormonal therapy may circumvent the
development of hormonal resistance in some ER+ breast
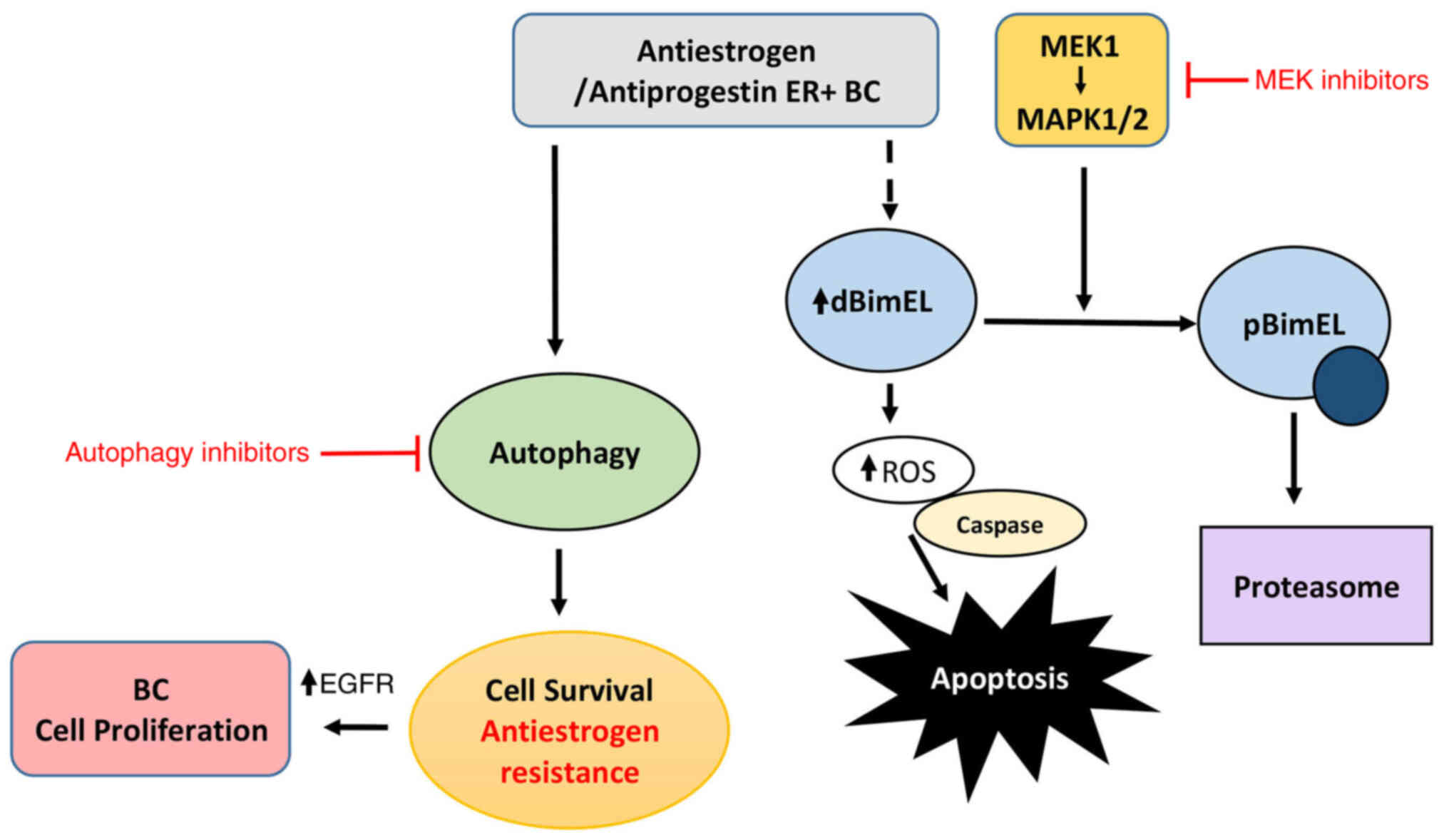
cancers if used as a first line regimen. The proposed role for
BimEL in the cytotoxic action of antiestrogens and antiprogestins
is summarized in the schematic of Fig.
7, as previously demonstrated (16) and further elaborated in the present
study; it demonstrates potential mechanisms to suppress the
cytotoxic action of BimEL that may be critical for the development
of antiestrogen resistance, i.e. upregulation of EGFR/MEK1/MAPK1/2
signaling axis and induction of autophagy. Clinically, the loss of
ERα expression is known to occur in greater than 20% of breast
cancers that relapse following endocrine therapy (48). Although there is a well-established
inverse correlation between ERα expression and elevated EGFR, the
role of EGFR in hormone status conversion in clinical breast cancer
is not well defined (48).
Importantly, EGFR has been recently identified as a therapeutic
target to increase tamoxifen sensitivity in hormone receptor
positive breast cancer (49). The
aforementioned study showed EGFR upregulation, along with ERα
downregulation, in breast cancer samples from patients with more
advanced stage and higher-grade tumors.
During the present study, an increase in
senescent-like cells in TR5 cell populations treated with
inhibitors of EGFR, and to a lesser extent MEK1 (data not shown),
was observed. These cells exhibited a flattened morphology, size
enlargement, accumulation of vacuoles, and expression of
β-galactosidase (β-gal), a marker of senescence (50). Although it was beyond the scope of
the present study to determine if the autophagy induced by ERL and
MEK inhibition facilitated the survival of the senescent-like
cells, it is interesting to hypothesize that autophagy in the
absence of MEK1/MAPK1/2 signaling is a prerequisite for a
senescence-like state (or dormancy) and that adaptive, genetic, or
epigenetic changes, such as EGFR upregulation regulate autophagy
levels and allow reversal of the senescent state in breast cancer
cells. This premise is not entirely inconsistent with the key role
for EGFR recently reported by Chen et al (51) in regulating the switch between cell
survival and cell death via autophagy regulation in cancer cells
under hypoxic condition. To further understand the role of EGFR,
our laboratories are currently investigating the role of
EGFR/MEK1/MAPK1/2 signaling in regulating senescence, apoptosis,
and autophagy in antiestrogen-resistant and sensitive
ER+ breast cancer cells.
In conclusion, the present study demonstrated that
the co-targeting of MEK1/MAPK1/2 and autophagy optimize
BimEL-mediated cytotoxic effects of hormonal treatments of
ER+ breast cancer cells. This combined treatment is
predicted to be most effective as an initial systemic treatment
approach for ER+ breast cancer because the development
of antiestrogen resistance can involve adaptive, genetic, and/or
epigenetic changes that make cells more resistant to the cytotoxic
effects of autophagy blockade. For example, it was demonstrated
that the upregulation of the EGFR/MEK1/MAPK1/2 signaling axis in an
antiestrogen-resistant MCF-7 derived model of breast cancer that
attenuates both the cytotoxic effects of BimEL and the cytostatic
effects of autophagy. Knowledge of the mechanism(s) of autophagy
induction by EGFR and/or MEK1/MAPK1/2 blockade is needed to
identify new molecular targets to improve the use of MEK1/MAPK1/2
inhibitors in the clinic.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
PVS, MT, DB and DG conceived and designed the
present study. PVS, DG and MLH wrote the manuscript. AL, CJ, MM,
SM, MLH, HC and AB conducted experiments. KL, MLH, MEM-L, DG and
MEM-L performed data analysis. CJ, MLH and DG created the figures
and diagram. JTB, WDH and PVS supervised the study. PVS and MLH
confirm the authenticity of all of the raw data. All authors read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank the Imaging Core
Facility at Augusta University, directed by Dr. Graydon Gonsalvez.
All imaging experiments were performed at the Augusta University
Imaging Core Facility.
Funding
The present study was supported by intramural finding by Augusta
University as an Extramural Success Award and an Undergraduate
Summer Student Training Research Award and by an extramural grant
to PVS from the National Cancer Institute (grant no. NIHNCI1R01
CA121438-O1A1). Research in Dr Gewirtz's laboratory is supported by
grants nos. CA268819 and CA239706 from the National Cancer
Institute/National Institutes of Health and grant no. W81XWH
19-1-0490 from the Department of Defense Congressionally Directed
Breast Cancer Research Program.
References
|
1
|
Senkus E, Kyriakides S, Ohno S,
Penault-Llorca F, Poortmans P, Rutgers E and Zackrisson S: Primary
breast cancer: ESMO clinical practice guidelines for diagnosis,
treatment and follow-up. Ann Oncol. 26(Suppl 5): v8–v30. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Early Breast Cancer Trialists'
Collaborative Group (EBCTCG); Davies C, Godwin J, Gray R, Clarke M,
Cutter D, Darby S, McGale P, Pan HC, Taylor C, et al: Relevance of
breast cancer hormone receptors and other factors to the efficacy
of adjuvant tamoxifen: Patient-level meta-analysis of randomised
trials. Lancet. 378:771–784. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Strasser-Weippl K, Badovinac-Crnjevic T,
Fan L and Goss PE: Extended adjuvant endocrine therapy in
hormone-receptor positive breast cancer. Breast. 22(Suppl 2):
S171–S175. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nardone A, De Angelis C, Trivedi MV,
Osborne CK and Schiff R: The changing role of ER in endocrine
resistance. Breast. 24(Suppl 2): S60–S66. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dodwell D, Wardley A and Johnston S:
Postmenopausal advanced breast cancer: Options for therapy after
tamoxifen and aromatase inhibitors. Breast. 15:584–594. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hartkopf AD, Grischke EM and Brucker SY:
Endocrine-resistant breast cancer: Mechanisms and treatment. Breast
Care (Basel). 15:347–354. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Clarke R, Jones BC, Sevigny CM,
Hilakivi-Clarke LA and Sengupta S: Experimental models of endocrine
responsive breast cancer: Strengths, limitations, and use. Cancer
Drug Resist. 4:762–783. 2021.PubMed/NCBI
|
|
8
|
Shah M, Nunes MR and Stearns V: CDK4/6
inhibitors: Game changers in the management of hormone
receptor-positive advanced breast cancer? Oncology (Williston
Park). 32:216–222. 2018.PubMed/NCBI
|
|
9
|
Li Z, Zou W, Zhang J, Zhang Y, Xu Q, Li S
and Chen C: Mechanisms of CDK4/6 inhibitor resistance in luminal
breast cancer. Front Pharmacol. 11:5802512020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sharifi MN, Anandan A, Grogan P and
O'Regan RM: Therapy after cyclin-dependent kinase inhibition in
metastatic hormone receptor-positive breast cancer: Resistance
mechanisms and novel treatment strategies. Cancer. 126:3400–3416.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gaddy VT, Barrett JT, Delk JN, Kallab AM,
Porter AG and Schoenlein PV: Mifepristone induces growth arrest,
caspase activation, and apoptosis of estrogen receptor-expressing,
antiestrogen-resistant breast cancer cells. Clin Cancer Res.
10:5215–5225. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schoenlein PV, Hou M, Samaddar JS, Gaddy
VT, Thangaraju M, Lewis J, Johnson M, Ganapathy V, Kallab A and
Barrett JT: Downregulation of retinoblastoma protein is involved in
the enhanced cytotoxicity of 4-hydroxytamoxifen plus mifepristone
combination therapy versus antiestrogen monotherapy of human breast
cancer. Int J Oncol. 31:643–655. 2007.PubMed/NCBI
|
|
13
|
El Etreby MF, Liang Y, Wrenn RW and
Schoenlein PV: Additive effect of mifepristone and tamoxifen on
apoptotic pathways in MCF-7 human breast cancer cells. Breast
Cancer Res Treat. 51:149–168. 1998. View Article : Google Scholar
|
|
14
|
El Etreby MF and Liang Y: Effect of
antiprogestins and tamoxifen on growth inhibition of MCF-7 human
breast cancer cells in nude mice. Breast Cancer Res Treat.
49:109–117. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Klijn JG, Setyono-Han B and Foekens JA:
Progesterone antagonists and progesterone receptor modulators in
the treatment of breast cancer. Steroids. 65:825–830. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Periyasamy-Thandavan S, Takhar S, Singer
A, Dohn MR, Jackson WH, Welborn AE, LeRoith D, Marrero M,
Thangaraju M, Huang S and Schoenlein PV: Insulin-like growth factor
1 attenuates antiestrogen- and antiprogestin-induced apoptosis in
ER+ breast cancer cells by MEK1 regulation of the BH3-only
pro-apoptotic protein Bim. Breast Cancer Res. 14:R522012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ying HQ, Chen J, He BS, Pan YQ, Wang F,
Deng QW, Sun HL, Liu X and Wang SK: The effect of BIM deletion
polymorphism on intrinsic resistance and clinical outcome of cancer
patient with kinase inhibitor therapy. Sci Rep. 5:113482015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chakraborty AR, Robey RW, Luchenko VL,
Zhan Z, Piekarz RL, Gillet JP, Kossenkov AV, Wilkerson J, Showe LC,
Gottesman MM, et al: MAPK pathway activation leads to Bim loss and
histone deacetylase inhibitor resistance: Rationale to combine
romidepsin with an MEK inhibitor. Blood. 121:4115–4125. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Adeyinka A, Nui Y, Cherlet T, Snell L,
Watson PH and Murphy LC: Activated mitogen-activated protein kinase
expression during human breast tumorigenesis and breast cancer
progression. Clin Cancer Res. 8:1747–1753. 2002.PubMed/NCBI
|
|
20
|
Lin CH, Shen CY, Lee JH, Huang CS, Yang
CH, Kuo WH, Chang DY, Hsiung CN, Kuo KT, Chen WW, et al: High
prevalence of the BIM deletion polymorphism in young female breast
cancer in an East Asian country. PLoS One. 10:e01249082015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sionov RV, Vlahopoulos SA and Granot Z:
Regulation of Bim in health and disease. Oncotarget. 6:23058–23134.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Samaddar JS, Gaddy VT, Duplantier J,
Thandavan SP, Shah M, Smith MJ, Browning D, Rawson J, Smith SB,
Barrett JT and Schoenlein PV: A role for macroautophagy in
protection against 4-hydroxytamoxifen-induced cell death and the
development of antiestrogen resistance. Mol Cancer Ther.
7:2977–2987. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Periyasamy-Thandavan S, Jackson WH,
Samaddar JS, Erickson B, Barrett JR, Raney L, Gopal E, Ganapathy V,
Hill WD, Bhalla KN and Schoenlein PV: Bortezomib blocks the
catabolic process of autophagy via a cathepsin-dependent mechanism,
affects endoplasmic reticulum stress and induces caspase-dependent
cell death in antiestrogen-sensitive and resistant ER+ breast
cancer cells. Autophagy. 6:19–35. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Towers CG, Wodetzki D and Thorburn A:
Autophagy and cancer: Modulation of cell death pathways and cancer
cell adaptations. J Cell Biol. 219:e2019090332020.
|
|
25
|
Marchetti S, Gimond C, Chambard JC,
Touboul T, Roux D, Pouysségur J and Pagès G: Extracellular
signal-regulated kinases phosphorylate mitogen-activated protein
kinase phosphatase 3/DUSP6 at serines 159 and 197, two sites
critical for its proteasomal degradation. Mol Cell Biol.
25:854–864. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jeong JE, Park JH, Kim CS, Lee SL, Chung
HL, Kim WT and Lee EJ: Neuroprotective effects of erythropoietin
against hypoxic injury via modulation of the mitogen-activated
protein kinase pathway and apoptosis. Korean J Pediatr. 60:181–188.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sander H, Wallace S, Plouse R, Tiwari S
and Gomes AV: Ponceau S waste: Ponceau S staining for total protein
normalization. Anal Biochem. 575:44–53. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Palumbo C, De Luca A, Rosato N, Forgione
M, Rotili D and Caccuri AM: c-Jun N-terminal kinase activation by
nitrobenzoxadiazoles leads to late-stage autophagy inhibition. J
Transl Med. 14:372016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ewings KE, Wiggins CM and Cook SJ: Bim and
the pro-survival Bcl-2 proteins: Opposites attract, ERK repels.
Cell Cycle. 6:2236–2240. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rani A, Stebbing J, Giamas G and Murphy J:
Endocrine resistance in hormone receptor positive breast
cancer-from mechanism to therapy. Front Endocrinol (Lausanne).
10:2452019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kisanga ER, Gjerde J, Guerrieri-Gonzaga A,
Pigatto F, Pesci-Feltri A, Robertson C, Serrano D, Pelosi G,
Decensi A and Lien EA: Tamoxifen and metabolite concentrations in
serum and breast cancer tissue during three dose regimens in a
randomized preoperative trial. Clin Cancer Res. 10:2336–2343. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tang J, Li Y, Xia S, Li J, Yang Q, Ding K
and Zhang H: Sequestosome 1/p62: A multitasker in the regulation of
malignant tumor aggression (Review). Int J Oncol. 59:772021.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shao S, Li S, Qin Y, Wang X, Yang Y, Bai
H, Zhou L, Zhao C and Wang C: Spautin-1, a novel autophagy
inhibitor, enhances imatinib-induced apoptosis in chronic myeloid
leukemia. Int J Oncol. 44:1661–1668. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Honda A, Harrington E, Cornella-Taracido
I, Furet P, Knapp MS, Glick M, Triantafellow E, Dowdle WE,
Wiedershain D, Maniara W, et al: Potent, selective, and orally
bioavailable inhibitors of VPS34 provide chemical tools to modulate
autophagy in vivo. ACS Med Chem Lett. 7:72–76. 2015. View Article : Google Scholar
|
|
38
|
Normanno N, Maiello MR and De Luca A:
Epidermal growth factor receptor tyrosine kinase inhibitors
(EGFR-TKIs): Simple drugs with a complex mechanism of action? J
Cell Physiol. 194:13–19. 2003. View Article : Google Scholar
|
|
39
|
Thomas RS, Sarwar N, Phoenix F, Coombes RC
and Ali S: Phosphorylation at serines 104 and 106 by Erk1/2 MAPK is
important for estrogen receptor-alpha activity. J Mol Endocrinol.
40:173–184. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Merino D, Best SA, Asselin-Labat ML,
Vaillant F, Pal B, Dickins RA, Anderson RL, Strasser A, Bouillet P,
Lindeman GJ and Visvader JE: Pro-apoptotic Bim suppresses breast
tumor cell metastasis and is a target gene of SNAI2. Oncogene.
34:3926–3934. 2015. View Article : Google Scholar
|
|
41
|
Rinehart J, Adjei AA, Lorusso PM,
Waterhouse D, Hecht JR, Natale RB, Hamid O, Varterasian M, Asbury
P, Kaldjian EP, et al: Multicenter phase II study of the oral MEK
inhibitor, CI-1040, in patients with advanced non-small-cell lung,
breast, colon, and pancreatic cancer. J Clin Oncol. 22:4456–4462.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Adjei AA, Cohen RB, Franklin W, Morris C,
Wilson D, Molina JR, Hanson LJ, Gore L, Chow L, Leong S, et al:
Phase I pharmacokinetic and pharmacodynamic study of the oral,
small-molecule mitogen-activated protein kinase kinase 1/2
inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers.
J Clin Oncol. 26:2139–2146. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Frogne T, Benjaminsen RV, Sonne-Hansen K,
Sorensen BS, Nexo E, Laenkholm AV, Rasmussen LM, Riese DJ II, de
Cremoux P, Stenvang J and Lykkesfeldt AE: Activation of ErbB3, EGFR
and Erk is essential for growth of human breast cancer cell lines
with acquired resistance to fulvestrant. Breast Cancer Res Treat.
114:263–275. 2009. View Article : Google Scholar :
|
|
44
|
Schiff R, Massarweh SA, Shou J, Bharwani
L, Mohsin SK and Osborne CK: Cross-talk between estrogen receptor
and growth factor pathways as a molecular target for overcoming
endocrine resistance. Clin Cancer Res. 10:331S–336S. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gee JM, Robertson JF, Ellis IO and
Nicholson RI: Phosphorylation of ERK1/2 mitogen-activated protein
kinase is associated with poor response to anti-hormonal therapy
and decreased patient survival in clinical breast cancer. Int J
Cancer. 95:247–254. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zaman K, Winterhalder R, Mamot C,
Hasler-Strub U, Rochlitz C, Mueller A, Berset C, Wiliders H, Perey
L, Rudolf CB, et al: Fulvestrant with or without selumetinib, a MEK
1/2 inhibitor, in breast cancer progressing after aromatase
inhibitor therapy: A multicentre randomised placebo-controlled
double-blind phase II trial, SAKK 21/08. Eur J Cancer.
51:1212–1220. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bartholomeusz C, Xie X, Pitner MK, Kondo
K, Dadbin A, Lee J, Saso H, Smith PD, Dalby KN and Ueno NT: MEK
inhibitor selumetinib (AZD6244; ARRY-142886) prevents lung
metastasis in a triple-negative breast cancer xenograft model. Mol
Cancer Ther. 14:2773–2781. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zattarin E, Leporati R, Ligorio F,
Lobefaro R, Vingiani A, Pruneri G and Vernieri C: Hormone receptor
loss in breast cancer: Molecular mechanisms, clinical settings, and
therapeutic implications. Cells. 9:26442020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jeong Y, Bae SY, You D, Jung SP, Choi HJ,
Kim I, Lee SK, Yu J, Kim SW, Lee JE, et al: EGFR is a therapeutic
target in hormone receptor-positive breast cancer. Cell Physiol
Biochem. 53:805–819. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Debacq-Chainiaux F, Erusalimsky JD,
Campisi J and Toussaint O: Protocols to detect
senescence-associated beta-galactosidase (SA-betagal) activity, a
biomarker of senescent cells in culture and in vivo. Nat Protoc.
4:1798–1806. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chen Y, Henson ES, Xiao W, Huang D,
McMillan-Ward EM, Israels SJ and Gibson SB: Tyrosine kinase
receptor EGFR regulates the switch in cancer cells between cell
survival and cell death induced by autophagy in hypoxia. Autophagy.
12:1029–1046. 2016. View Article : Google Scholar : PubMed/NCBI
|