Introduction
According to the 2018 global cancer statistics,
liver cancer was the sixth most common cancer and the fourth most
deadly type of cancer worldwide, with ~841,000 new cases each year
and ~782,000 deaths (1). Major
risk factors for liver cancer include hepatitis B virus infection,
hepatitis C virus infection, alcohol abuse, non-alcoholic
steatohepatitis, aflatoxin exposure and hemochromatosis (2). Liver cancer is characterized by its
high degree of invasiveness, poor clinical prognosis, frequent
recurrence and low survival rates (3). Surgical resection and transplantation
are the mainstay curative treatments for liver cancer; however,
most patients are diagnosed with advanced liver carcinoma, which
decreases the chances of survival (4). Moreover, liver cancer is resistant to
chemotherapeutic drugs, which results in cancer recurrence after
conventional treatment (5).
Therefore, developing safer and more effective drugs for liver
cancer treatment is of marked importance.
Cortex periplocae, an herbal medicine, is the dried
root bark of Periploca sepium of the Asclepiadaceae
family, and contains numerous active ingredients, including cardiac
glycosides (CGs), sterols, glycosides, esters and aldehydes that
are used clinically to treat heart failure and relieve associated
symptoms (6,7). Periplocin is one of the main and
active constituents isolated from cortex periplocae. Periplocin
undergoes hydrolysis and deglycosylation to yield periplocymarin
(PPM), which has a potent cardiotonic effect in vivo
(8,9). An increasing number of studies have
reported that CGs exert more beneficial therapeutic effects than
cardiac cardiotonics (10,11). Mounting evidence has shown that CGs
have significant anti-inflammatory, anti-tumor, and anti-senescent
effects (12-14). In terms of anti-tumor activity, CGs
inhibit the proliferation of numerous malignant cell lines and
prevent the growth of tumors in vivo (15). Moreover, PPM is considered a
natural potential anticancer agent with high permeability and
independence from P-glycoprotein influence (16). Studies on the mechanism of action
of anti-tumor agents indicate that periplocin induces DNA
double-strand breaks in liposarcoma cells and activates the
ERK1/2-Egr1 signaling pathway in gastric cancer cells, which
induces cell apoptosis (17,18).
In addition to the induction of apoptosis, several
other programmed cell death patterns can be targeted to eliminate
cancer cells. Given the pivotal roles of autophagy in tumorigenesis
and progression (19), cell death
stimulated by excessive autophagy, a concept of 'autophagic cell
death', has been the subject of intense investigation (20-22).
A recent study reported that ouabain, a representative CG, induced
autophagic cell death in non-small cell lung carcinoma via the JNK
signaling pathway (23). Newman
et al (24) reported that
oleander induced tumor cell autophagy via suppression of the
AKT/mTOR signaling pathway in a concentration and time-dependent
manner. However, recent advances in the understanding of the
molecular processes which contribute to autophagy have elucidated
the crosstalk between autophagy and apoptosis. In contrast to
autophagic cell death, autophagy exerts cytoprotective effects that
negatively modulate apoptosis in several physiological and
pathological circumstances (25).
Therefore, understanding the mechanisms which underlie the
relationship between autophagy and apoptosis is crucial for the
development of effective cancer treatments.
In the present study, HepG2 cell lines and a
xenograft mouse model were used to evaluate the antitumor effects
of PPM, by which a novel mechanism of action (MOA) was demonstrated
for PPM.
Materials and methods
Compounds
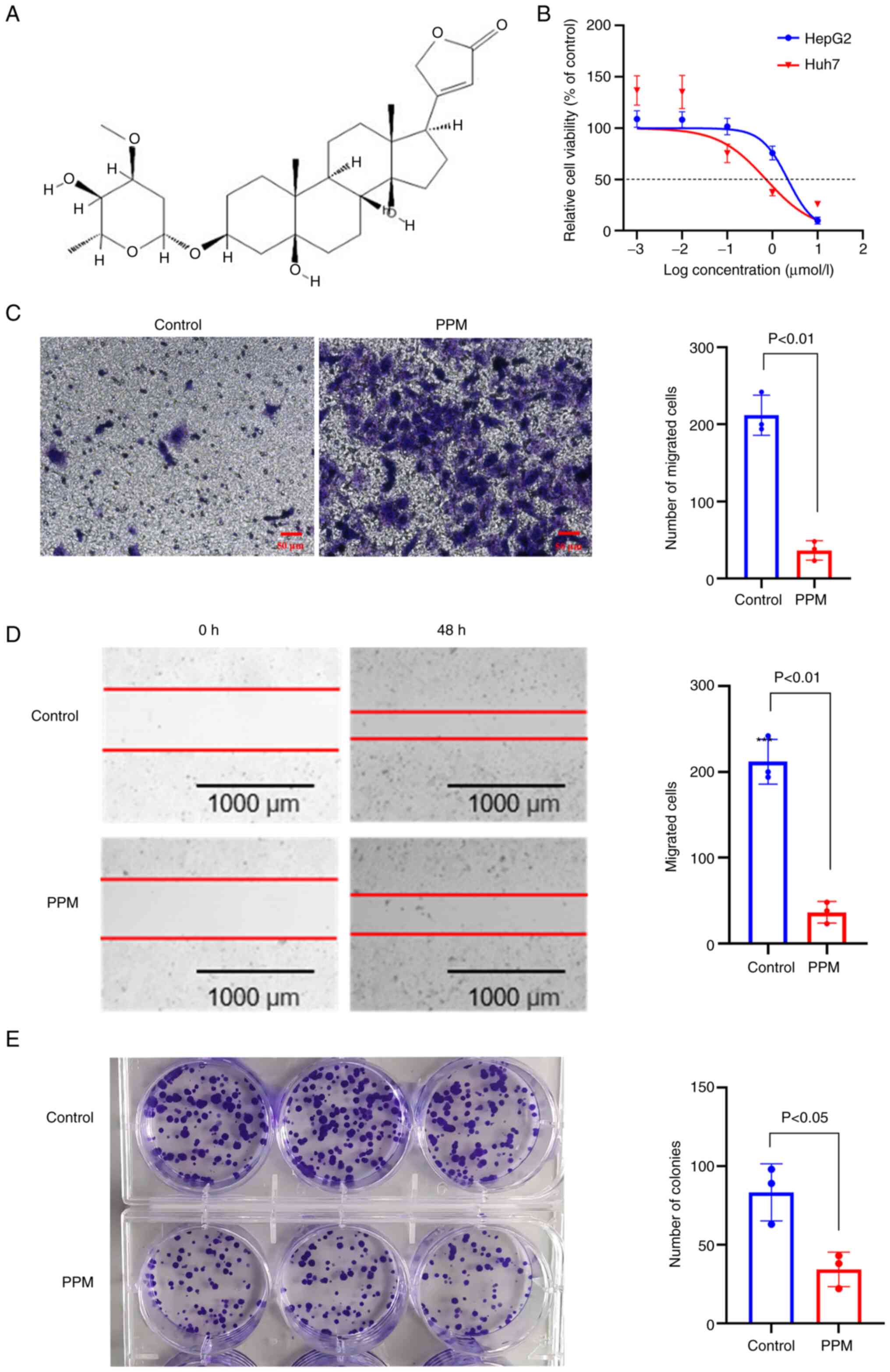
PPM (molecular formula:
C30H46O8; molecular weight:
534.3193; cat. no. 127-32-2; purity: ≥98%; Fig. 1A) was purchased from Chengdu Chroma
Biotechnology Co., Ltd.
Cell lines and culture
HepG2 human hepatoblastoma cells were purchased from
iCell Bioscience Inc. Huh7 human hepatoma cells were purchased from
Shanghai Fuheng Biotechnology Co., Ltd. HepG2 and Huh7 cell lines
were authenticated by STR profiling. Cells were cultured in DMEM
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 15% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.) and 1% (v/v)
penicillin/streptomycin at 37°C in 5% CO2. For all
experiments, 1×105 cells/well were seeded into a 96-well
tissue culture plate. After the cells reached 80% confluence, PPM
was added to the culture medium for 48 h at concentrations of
1×10−5, 1×10−6, 1×10−7,
1×10−8 and 1×10−9 mol/l, and 0 mol/l as the
control. Cell viability was evaluated using CellTiter
96® AQueous One Solution Cell Proliferation Assay (cat.
no. G3581, Promega Corporation) according to the manufacturer's
protocol.
Wound healing assay
HepG2 cells (~1×105) were seeded in
6-well plates with 2 ml of DMEM (Gibco; Thermo Fisher Scientific,
Inc.) with 15% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.) and cultured at 37°C overnight. Once the cells
reached 90% confluence, a linear scratch wound was created using a
100 µl pipette tip. At different time points (0 and 48 h),
images were captured using Cytation 5 Cell Imaging Multimode Reader
(BioTek Instruments, Inc.) and analyzed using Gen5.0 (BioTek
Instruments, Inc.). The scratch area was measured using ImageJ
(version 1.8.0; National Institutes of Health) and this was used to
calculate the wound healing percentage.
Colony formation assay
A total of 1×103 HepG2 cells/well from
both the control and test groups were plated in a six-well plate
and allowed to grow for 12 days at 37°C. The clones were then fixed
with 4% paraformaldehyde at room temperature for 20 min.
Subsequently, cells were stained with 0.5% crystal violet at room
temperature for 10 min. The number of colonies (>50 cells) was
manually counted, using light microscopy and images were captured
using a digital camera.
Cell migration assay
Migration assays were performed in 24-well, 8
µm pore, Transwell plates (cat. no. 3422, Corning, Inc.).
HepG2 cells in the logarithmic growth phase were diluted to
2.5×105 cells/ml and resuspended in 200 µl
serum-free DMEM medium (Gibco; Thermo Fisher Scientific, Inc.)
treated with PPMin the upper compartment and 660 µl DMEM
medium supplemented with 15% FBS (Gibco; Thermo Fisher Scientific,
Inc.) was added to the lower compartment. The plate was incubated
at 37°C for 48 h. Each well was then washed twice with PBS and the
inner surface of the upper chamber was carefully wiped with a
cotton swab. The migrated cells on the lower surface were imaged
using an Olympus IX70 inverted microscope (Olympus Corporation) in
three randomly selected visual fields and the migrated cells were
quantified using ImageJ (version 1.8.0; National Institutes of
Health).
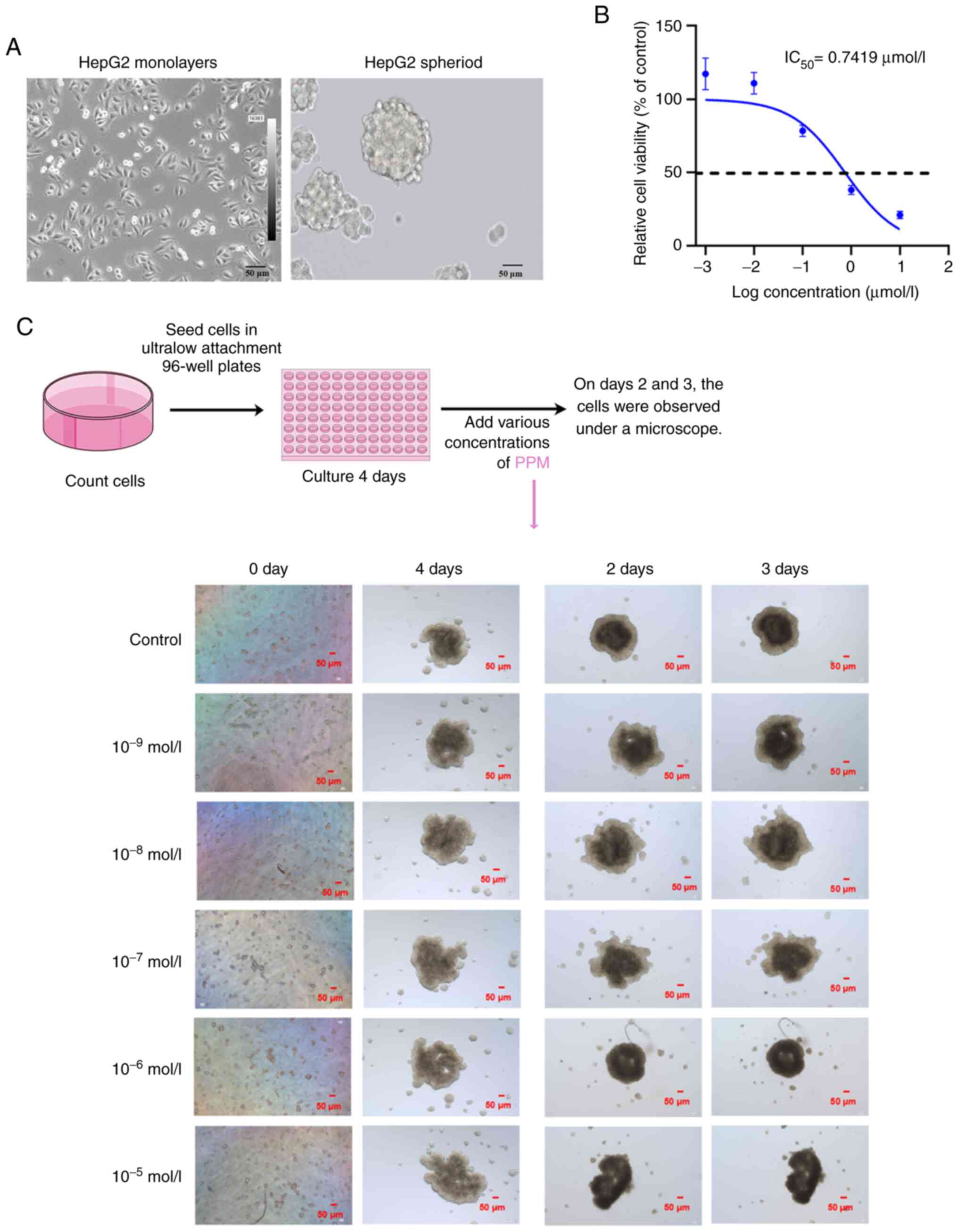
3D cell culture and cytotoxic assay
Cells were seeded (1×105 cells/well) into
ultra-low attachment 96 well microplates, centrifuged at 200 × g
for 5 min at room temperature and incubated at 37°C for 4 days to
allow the formation of cell spheroids. Half of each culture
supernatant was replaced every 2 days. Cells were continuously
cultured for 4 days in a 37°C cell incubator to observe the cell
morphology. Spheres were treated with PPM at the aforementioned
concentrations for 72 h and at 37°C. Cell viability was measured
using the CellTiter-Glo® 3D Cell Viability Assay (cat.
no. G9681, Promega Corporation) according to the manufacturer's
protocol. Cell morphology was assessed on days 2 and 3 of PPM
treatment using an Olympus IX73 light microscope (Olympus
Corporation). The experimental procedure and dosing treatment plan
were presented in Fig. 2C, which
was drawn using Figdraw (ID, RUUPO33f33; www.figdraw.com).
Mitochondrial membrane potential
detection
The mitochondrial membrane potential was assessed
using a JC-1 assay (cat. no. 10009172, Cayman Chemical Company).
Briefly, cells were incubated with JC-1 staining solution (1:10)
for 30 min at 37°C. Subsequently, the cells were washed twice with
assay buffer to remove free JC-1 and immediately imaged using a
fluorescence microscope.
Metabolic assay
HepG2 cells were cultured in Seahorse XF24 cell
culture microplates (Agilent Technologies, Inc.) and cultured at
37°C overnight. PPM (100 nmol/l) was added to the cells when they
reached 80-90% confluence and cells were incubated for 24 h at
37°C. This experiment was performed using the Seahorse XF Real-time
ATP rate assay kit (cat. no. 103592-100; Agilent Technologies,
Inc.). The experimental procedures and Seahorse assays were
performed according to the manufacturer's protocols. Wave software
version 2.4.1 (Agilent XFp Analyzer; Agilent Technologies, Inc.)
and GraphPad Prism version 9 (GraphPad Software.; Dotmatics) were
used for statistical analysis and graphical representation.
Western blotting
HepG2 cells were treated with PPM in the presence or
absence of 5 mM 3-methyladenine (3-MA, cat. no. HY-19312;
MedChemExpress) (26), 50 nmol/l
Bafilomycin A1 (Baf A1, cat. no. HY-100558; MedChemExpress)
(27) or 20 µmol/l
Z-VAD-FMK (cat. no. HY-16658B; MedChemExpress) (28) at 37°C for 24 h. Total proteins were
extracted from HepG2 cells using RIPA lysis buffer supplemented
with Pierce Phosphatase Inhibitor (cat. no. A32961, Thermo Fisher
Scientific, Inc.). The protein concentration was determined using a
BCA kit (cat. no. 23227, Thermo Fisher Scientific, Inc.). SDS-PAGE
was performed on 4-20% SDS-gels (GenScript), followed by transfer
of the resolved proteins onto nitrocellulose membranes. Membranes
were blocked in blocking buffer (cat. no. P0252, Beyotime Institute
of Biotechnology) for 20 min at room temperature. Membraned were
subsequently incubated with primary antibodies against anti-caspase
3 (1:1,000, cat. no. 14220, Cell Signaling Technology, Inc.);
anti-pro-caspase-3, (1:1,000, cat. no. ab13847, Abcam) anti-caspase
7 (1:1,000, cat. no. ab32522, Abcam), anti-caspase 9 (1:1,000, cat.
no. ab32539, Abcam), anti-Bcl-2 (1:1,000, cat. no. ab59348, Abcam),
anti-Bax (1:1,000, cat. no. ab32503, Abcam), anti-α-tubulin
(1:1,000, cat. no. ab7291, Abcam), anti-β-actin (1:1,000, cat. no.
3700, Cell Signaling Technology, Inc.), anti-microtubule-associated
protein light chain 3 (LC3; 1:1,000, cat. no. ab192890, Abcam),
anti-autophagy-related gene 4B (ATG4B; 1:1,000, cat. no ab154843,
Abcam), anti-sequestosome 1/p62 (SQSTM1/p62; 1:1,000, cat. no.
ab109012, Abcam), anti-GAPDH (1:1,000, cat. no. ab8245, Abcam),
anti-phosphorylated-Unc-51-like kinase 1 (p-ULK1, ser757; 1:1,000,
cat. no. 14202, Cell Signaling Technology, Inc.), anti-ULK1
(1:1,000, cat. no. 8054, Cell Signaling Technology, Inc.),
anti-p-AMPKα (Thr172; 1:1,000, cat. no. 2535, Cell Signaling
Technology, Inc.), anti-AMPKα (1:1,000, cat. no 5832, Cell
Signaling Technology, Inc.), anti-p-mTOR (ser2481; 1:1,000, cat.
no. 2974, Cell Signaling Technology, Inc.), anti-mTOR (1:1,000,
cat. no. 2983, Cell Signaling Technology, Inc.),
anti-lysosomal-associated membrane protein 1(LAMP1;1:1,000, cat.
no. 21997-1-AP, Proteintech Group, Inc.), anti-cathepsin D (CTSD;
1:1,000, cat. no. 21327-1-AP, Proteintech Group, Inc.), or
anti-cathepsin D (CTSB; 1:1,000, cat. no. 12216-1-AP, Proteintech
Group, Inc.) overnight at 4°C. After three rinses, the membrane was
incubated with goat anti-mouse IgG H&L (IRDye®
680RD; 1:5,000, ab216776, Abcam) or goat anti-rabbit IgG H&L
(IRDye® 800CW; 1:5,000, ab216773, Abcam) for 1 h at
37°C. A LI-COR Odyssey CLx scanning system (LI-COR Biosciences) was
used to semi-quantify the fluorescent signals, which were
normalized to β-actin, GAPDH or α-tubulin expression as
appropriate.
Red fluorescence point-green fluorescence
point-LC3 (RFP-GFP-LC3) for determining autophagic flux
HepG2 cells were cultured to ~60% confluency.
Autophagic flux was assessed using a Premo™ Autophagy Tandem Sensor
RFP-GFP-LC3B Kit (cat. no. P36239, Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocols. Confocal images were
acquired using a Carl Zeiss LSM710 confocal microscope (Zeiss
GmbH).
Annexin V apoptosis assay
Apoptosis was assessed using a FITC Annexin V
apoptosis detection kit (BD Biosciences). A total of
1×105 HepG2 cells/well were seeded in 6-well plates and
cultured at 37°C for 24 h. After treatment with 100 nmol/l PPM for
24 h at 37°C, the cells were resuspended in 100 µl binding
buffer. Incubation was performed in the dark at room temperature
for 15 min following the addition of 5 µl Annexin V-FITC and
10 µl PI. Finally, 400 µl PBS was added to each tube,
and the cells were immediately evaluated by flow cytometry using a
FACSAria™ III Cell Sorter (BD Biosciences). The percentage of
apoptotic cells was calculated based on the number of apoptotic
cells in each sample (Q2 + Q4).
Short interfering (si)RNA
transfection
ATPase Na+/K+ Transporting
Subunit α 1 (ATP1A1) siRNA and its negative control siRNA (cat. No.
siN0000001-1-5) were purchased from Guangzhou RiboBio Co., Ltd. The
sequences of the synthesized siRNA were as follows: si-ATP1A1-01,
5′-GAT TCG AAA TGG TGA GAA A-3′; si-ATP1A1-02, 5′-GTC GTC TGA TCT
TTG ATA A-3′; and si-ATP1A1-03, 5′-GAA TTT CCC TAT CGA TAA T-3′.
Cells were seeded in 6-well plates at a density of 2×105
cells/well. When the cells were 60% confluent, they were
transfected with 50 nmol/l siRNA using the RiboFECT CP Transfection
kit (Guangzhou RiboBio Co., Ltd.) according to the manufacturer's
protocols. Subsequently, analysis of protein expression levels was
performed in HepG2 cells after 24 h of treatment with PPM.
Assessment of membrane potential
HepG2 cells were placed in a recording chamber on
the stage of an Olympus IX53 inverted microscope (Olympus
Corporation). The experimental procedure was performed as
previously described (29-31). Briefly, the extracellular solution
(NaCl, 140 mmol/l; KCl, 4 mmol/l; MgCl2, 1 mmol/l;
CaCl2, 2 mmol/l; D-glucose monohydrate, 5 mmol/l;
2-[4-(2-Hydroxyethyl) piperazin-1-yl] ethanesulfonic acid, 10
mmol/l; pH, 7.4) was continuously perfused into the recording
chamber using a gravity-fed solution delivery system. The membrane
potential was recorded in a whole-cell single-electrode
current-clamp configuration using the patch-clamp technique with a
700 B amplifier (Molecular Devices, LLC). A computer equipped with
a DigiData1550B (Molecular Devices, LLC) was used to generate the
holding membrane current (I=0A) for data storage and evaluation.
The patch electrodes had a resistance of 2-5 MΩ (P-97, Sutter
Instrument Company) when filled with the internal solution (KCl, 20
mmol/l; MgCl2, 1 mmol/l; Ethylenebis
(oxyethylenenitrilo) tetraacetic acid, 5 mmol/l; phosphocreatine
disodium salt, 14 mmol/l; Na2-ATP, 5 mmol/l; pH,
7.2).
Electron microscopy
HepG2 cells were fixed in 2.5% glutaraldehyde in 0.1
M sodium cacodylate buffer at 4°C for 2 h, followed by 1% osmium
tetroxide in 0.1 M sodium cacodylate buffer for 2 h at room
temperature. Then, they were stained using 1% uranyl acetate for 1
h at room temperature, dehydrated in an increasing ethanol series
and embedded in epoxy resin. The cells were subsequently imaged
using a Hitachi-HT7700 electron microscope (Hitachi, Ltd.).
Xenograft mouse model
Animals were housed in appropriate pathogen-free
conditions. All animal experiments (approval no. 2022023) adhered
to the specifications of the Animal Care and Use Committee of Hebei
Yiling Chinese Medicine Research Institute (Shijiazhuang, China). A
total of 20 male BALB/c nude mice (5 weeks old, 18-22 g) were
purchased from Beijing Vital River Laboratory Animal Technology
Co., Ltd. (approval no. 110011220101738322). Mice were housed (n=5
per cage) in a rodent facility with a 12 h light/dark cycle, at
22°C and 40% humidity, with ad libitum access to food and
water. The 50% (v/v) Matrigel (cat. no 356234, Corning,
Inc.)-suspended HepG2 cells (2×106/mouse) were injected
subcutaneously into the flank of BALB/c nude mice. Tumor size was
measured at day 14, and mice with a similar size tumor were
selected for further experiments. When the tumor volume of the nude
mice reached 200 mm3, the mice were randomly divided
into three groups of four mice each as follows: PPM (100 mg/kg),
3-MA (2 mg/kg) + PPM (100 mg/kg), and NaCl (control). The remaining
8 mice were excluded as the tumors did not reach 200
mm3. Mice were treated daily via oral gavage for 7 days.
Tumor diameter and mouse weight were measured every 3 days. The
formula used to calculate the volume of the tumors was as follows:
Volume=(width2 × length) ×0.5. The experiments were
terminated when tumor volumes reached ~1,500 mm3.
Pentobarbital sodium (50 mg/kg) was injected intraperitoneally into
mice to anesthetize them (32).
Tumor xenografts of each mouse were removed and weighed. After
completion of the experiments, the mice were sedated with
CO2/O2 (70%/30%). Once the mice lost
consciousness, they were euthanized with 100% CO2. The
animals remained in the euthanasia chamber for 5 min and were then
observed for an additional 5 min. Breathing and heart rate were
monitored to determine death.
Immunofluorescence staining
For in vivo studies, tumor tissues were
rapidly embedded with OCT embedding agent (cat. no. G6059, Wuhan
Servicebio Technology Co., Ltd.) and snap-frozen on dry ice. Frozen
sections were cut into 10-µm-thick slices. Ki67 is a
cellular marker of proliferation (33) and the tumor cell proliferation rate
was analyzed using Ki67 staining. Tunnel staining (cat. no.
PF00006, ProteinTech Group, Inc.) was used to examine tumor cell
apoptosis, according to the manufacturer's protocol.
For in vitro studies, HepG2 cells were seeded
on 35 mm confocal dishes (Corning, Inc.) and cultured at 37°C in 5%
CO2. After treatment with 100 nmol/l PPM for 24 h at
37°C, HepG2 cells were fixed with 4% paraformaldehyde for 20 min at
room temperature and then washed twice with PBS (5 min each). HepG2
cells were permeabilized with 0.5% Triton X-100 (cat. no. P0096,
Beyotime Institute of Biotechnology) for 10 min at room
temperature, and subsequently blocked in PBS with 10% goat serum
(Beyotime Institute of Biotechnology) for 1 h at room temperature.
SQSTM1/p62 and LC3 proteins are markers for autophagosome formation
and degradation, respectively (34). HepG2 cells were subjected to
immunofluorescence staining to assess LC3 and p62/SQSTM1 protein
expression levels.
The primary antibodies used were as follows:
Anti-Ki67 (1:250, cat. no. ab16667, Abcam), anti-LC3 (1
µg/ml, cat. no. ab192890, Abcam) or anti-SQSTM1/p62 (1
µg/ml, cat. no. ab109012, Abcam). Cells were incubated in
primary antibodies overnight at 4°C, washed with 1X PBS, and then
incubated with fluorescently labeled secondary antibodies for 1 h
in the dark at 37°C. Secondary antibodies used were as follows:
donkey anti-Rabbit Alexa Fluor 555 (1:1,000; cat. nos. P0179;
Beyotime Institute of Biotechnology) and Alexa Fluor 488 goat
anti-Rabbit (1:1,000; P0176; Beyotime Institute of Biotechnology).
Each section was stained with DAPI (cat. no. S2110, Beijing
Solarbio Science & Technology Co., Ltd.). for 1 h at room
temperature and protected from light. Samples were imaged using a
LSM710 confocal microscope (Zeiss GmbH) and analyzed using Image J
(version 18.0; National Institutes of Health).
Statistical analysis
Data were presented as the mean ± standard
deviation. Data was analyzed using one way analysis of variance
with Tukey's post-hoc test. Statistical analysis was performed
using SPSS version 19.0 (IBM Corp.). P<0.05 was considered to
indicate a statistically significant difference.
Results
Effects of PPM in liver cancer cellular
behaviors
To evaluate the biological effects of PPM in liver
cancer, MTS assays were performed. The results demonstrated that
PPM exhibited a cytotoxic effect on the HepG2 and Huh7 cells after
48 h of treatment with half-maximal inhibitory concentrations
(IC50) of 2.1860 and 0.7725 µmol/l, respectively
(Fig. 1B). In the Transwell and
wound healing assays, PPM significantly decreased the migratory
ability of cells (Fig. 1C and D).
The colony formation assays demonstrated that PPM significantly
inhibited HepG2 colony formation (Fig.
1E). In the 3D cultures, HepG2 cells formed spheres with a
grape-like appearance (Fig. 2A).
Initially, the cells were diffusely distributed. With increasing
culture times, the cell volume grew continuously and by day 4,
cells appeared spherical or elliptical. After the administration of
PPM, the structure of the cancer spheroids loosened within 48 h.
These results demonstrated that PPM exhibited cytotoxic activity
after 48 h on the cancer spheroids with an IC50 of
0.7419 µmol/l (Fig. 2B and
C). PPM inhibited cell proliferation, migration and tumor
sphere-formation ability. These data demonstrated that PPM exerted
an anti-cancer effect in liver cancer cells in vitro.
PPM activates the mitochondrial apoptotic
pathway in HepG2 cells
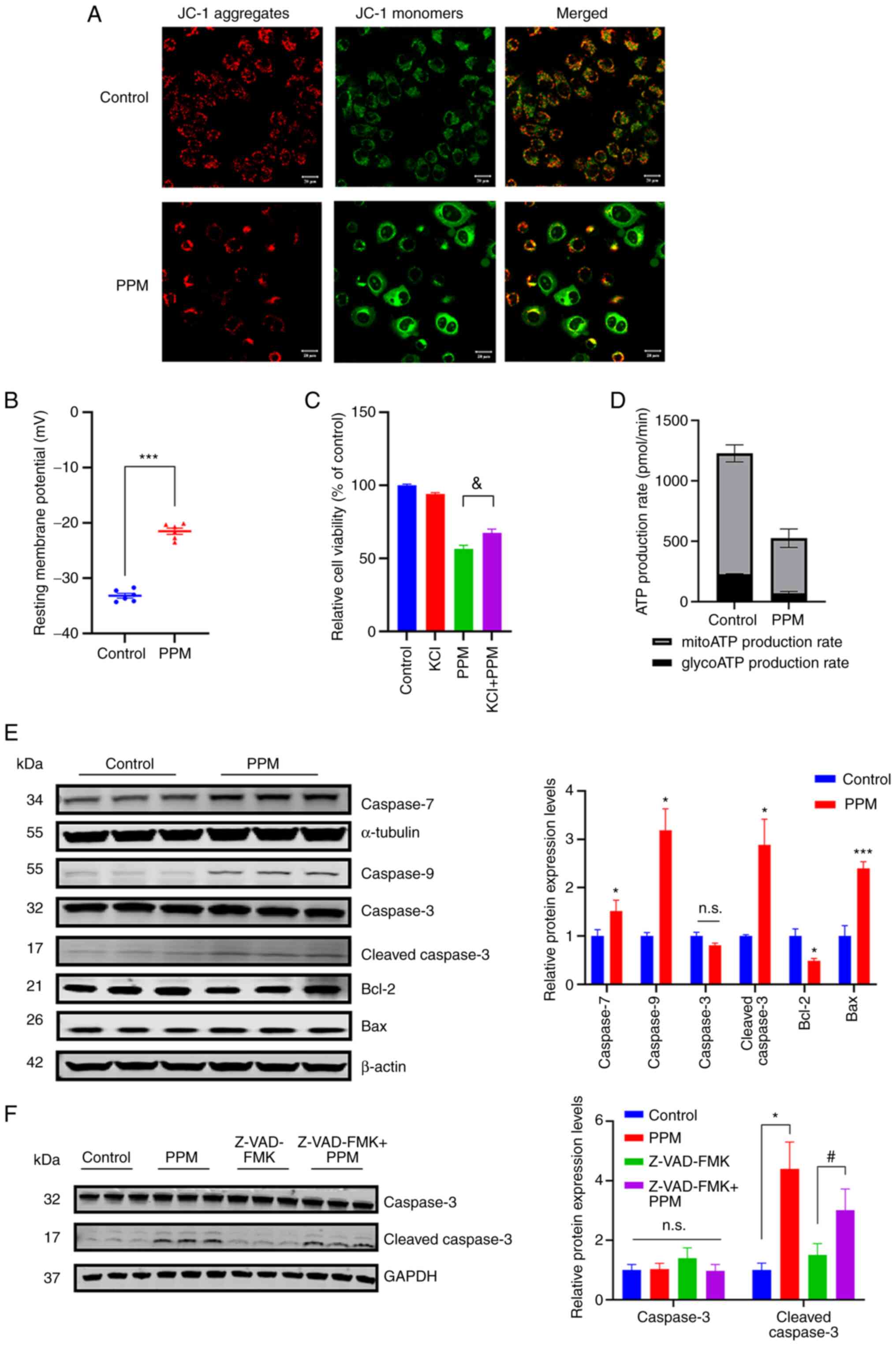
JC-1 is a critical marker of cell viability.
Exposure of HepG2 cells to PPM markedly disrupted the mitochondrial
membrane potential, based on the increase in green fluorescence
(JC-1 monomers) and decrease in red fluorescence (JC-1 aggregates;
Fig. 3A). CGs are known to exert
their action primarily by binding to the α-subunit of the
Na+/K+ATPase (NKA) pump and inhibiting the
intake of K+ coupled with the release of Na+
out of the cell (35,36). As changes in intracellular ion
concentrations can affect the resting membrane potential, the
resting membrane potential of HepG2 cells treated with PPM was
assessed. After treatment, PPM significantly depolarized the
membrane potential in the PPM-treated cells (-21.5±1.37 mV)
compared with the control (-33.2±1.05 mV; Fig. 3B). To alleviate depolarization of
the plasma membrane, KCl was added to the medium (12). Addition of KCl restored cell
viability independent of PPM inhibition (Fig. 3C). These results indicated that,
plasma membrane depolarization was involved in the anticancer
effects of PPM in liver cancer. PPM-treated HepG2 cells
demonstrated lower ATP production from both the glycolytic and
mitochondrial pathways (Fig. 3D).
The protein expression levels of caspases following PPM treatment
were assessed using western blotting. PPM significantly increased
the protein expression levels of Bax, cleaved-caspase 3, caspase 9
and caspase 7 compared with the control, and significantly reduced
the protein expression levels of Bcl-2 (Fig. 3E). To investigate the functional
involvement of caspases in PPM-induced apoptosis, Z-VAD-FMK, an
inhibitor of apoptosis, was used given its ability to function as a
pan-caspase inhibitor. The results demonstrated that PPM
significantly antagonized Z-VAD-FMK-induced inhibition of apoptosis
(Fig. 3F). These results
demonstrated that PPM activated the intrinsic mitochondrial
apoptotic pathway.
PPM selectively activates macroautophagy
via the AMPK/ULK1 and mTOR signaling pathways
LC3-II and p62/SQSTM1 are the most common markers of
autophagy. The conversion of LC3-I into LC3-II serves as the gold
standard for the detection of autophagy (37), and p62/SQSTM1, as an indicator of
autophagic flow, links LC3-II with ubiquitinated substrates for
degradation in autolysosomes (38,39).
In the present study, the distribution of the intracellular
autophagy marker LC3 was assessed to determine whether PPM could
induce autophagy in HepG2 cells. The results demonstrated that PPM
treatment significantly increased the accumulation of lipidated LC3
(conversion of LC3-I to LC3-II), which indicated the maturation of
autophagosomes (Fig. 4A). AMPK is
a known nutrient and energy sensor that maintains cellular energy
homeostasis when the ratios of intracellular AMP/ATP levels are
increased (40,41). AMPK is activated by phosphorylation
at Thr172 (p-AMPK) (42). AMPK
directly phosphorylates ULK1 at certain serine residues (S317, S555
and S777) to induce autophagy (43). To determine whether AMPK activation
was involved in PPM-induced autophagy, the protein expression
levels of p-AMPKα Thr172 and total AMPKα was assessed using western
blotting. Protein expression levels of p-AMPKα (Thr1720 were
significantly increased in HepG2 cells after PPM treatment compared
with the control, whereas the total AMPKα expression was not
significantly altered (Fig. 4B).
Furthermore, PPM significantly reduced p-mTOR, p-ULK1 (ser 757) and
total ULK1 protein expression levels compared with the control,
whereas the total mTOR protein expression level was not
significantly altered. Collectively, PPM activates macro-autophagy
via the AMPK/ULK1 and mTOR pathways. To confirm the type of
macro-autophagy, the protein expression levels of BNIP3 and PINK1,
which are indicators of mitophagy activation, were assessed. PPM
significantly decreased the protein expression levels of BNIP3 and
PINK1 compared with the control (Fig.
4D). Notably, PPM treatment significantly increased the protein
expression levels of p62/SQSTM1 and ATG4B compared with the control
group (Fig. 4A), which was
confirmed by immunofluorescence staining (Fig. 4C). Since simultaneously increased
protein levels of p62/SQSTM1 and LC3-II indicated dysfunction of
autophagy degradation (44), these
findings suggested that PPM may have exerted differential effects
at different stages of autophagy.
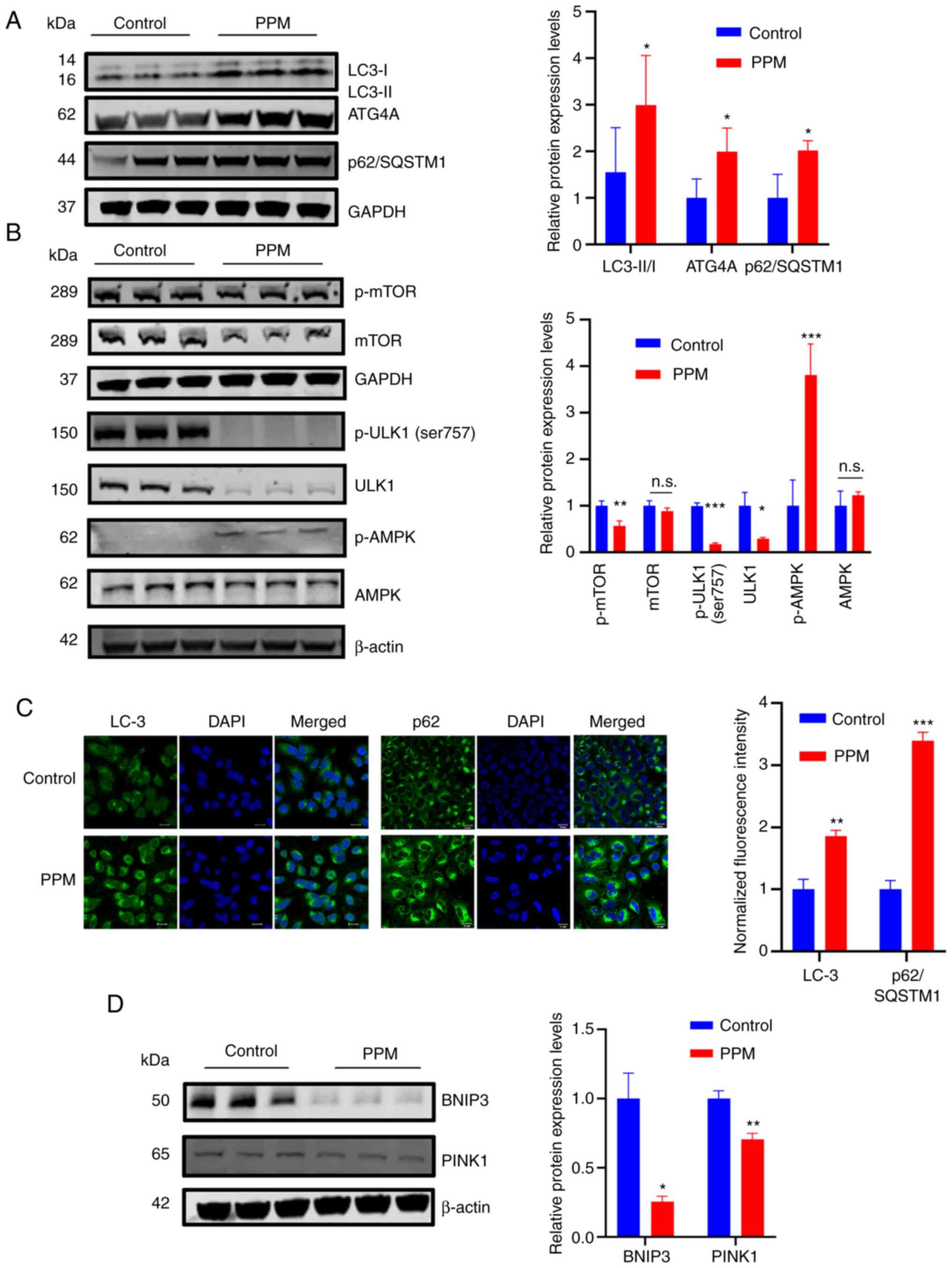 | Figure 4PPM selectively activated
macroautophagy in HepG2 cells. (A) Western blotting of LC3, ATG4A
and p62/SQSTM1 and (B) p-mTOR, mTOR, p-ULK1(Ser757), ULK1, p-AMPKα
(Thr172) and AMPKα protein expression levels after treatment with
PPM (100 nmol/l) in HepG2 cells. (C) Immunofluorescence staining of
LC3 and p62/SQSTM1. Scale bar=20 µm. (D) Western blotting of
the mitophagy markers BNIP3 and PINK1. GAPDH or β-actin was used as
a loading control. Data are presented as mean ± SD, n=3;
*P<0.05, **P<0.01 and
***P<0.001 vs. control. n.s., not significant; PPM,
periplocymarin; p, phosphorylated; LC3, microtubule-associated
protein light chain 3; ATG4A, ATG4A; p62/SQSTM1, sequestosome 1;
ULK1, Unc-51-like kinase 1; BNIP3, BCL2 Interacting Protein 3;
PINK1, PTEN induced putative kinase 1. |
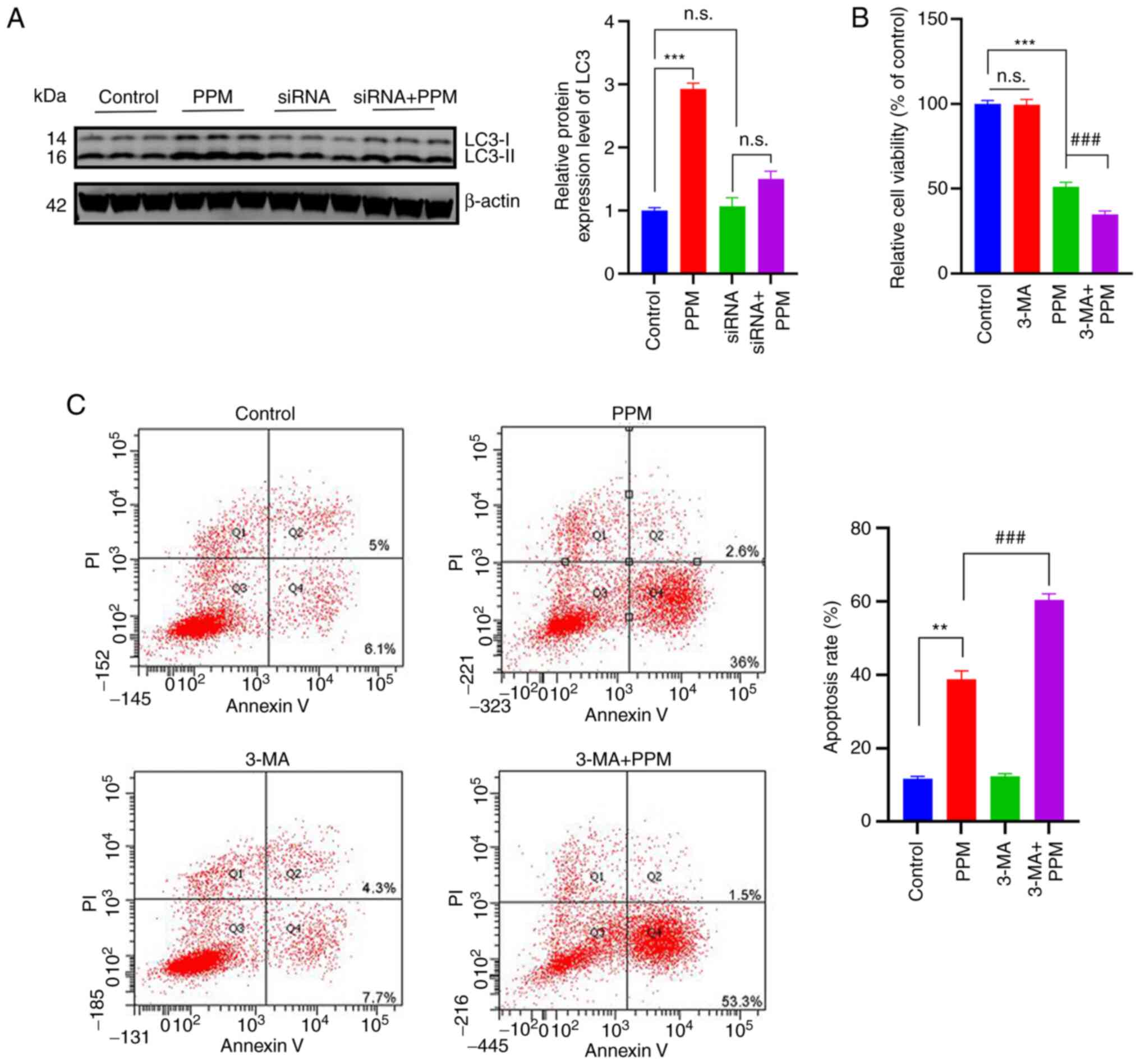
PPM activates macro-autophagy in an NKA
receptor-dependent manner to counteract the pro-apoptosis
effects
CGs work by inhibiting NKA and activating
Na+/Ca2+ exchange, which leads to an increase
in the intracellular Ca2+ concentration (45). The inhibitory effect is produced by
binding to the α-subunits of NKA (46). To confirm the activation of
autophagy in an NKA-dependent manner, HepG2 cells were transfected
with a set of three siRNAs, and ATP1A1 mRNA and protein expression
levels were significantly reduced in the si-ATP1A1-02 group 48 h
post-transfection, compared with the NC group (Fig. S1A and B). Furthermore, PPM did not
significantly induce the protein expression of LC3-II in the ATP1A1
knockdown HepG2 cells (Fig. 5A).
To evaluate the interactions between PPM-induced apoptosis and
autophagy, HepG2 cells were treated with PPM in the presence or
absence of 3-MA. The combination of 3-MA and PPM significantly
inhibited the viability of HepG2 compared with PPM alone after 24
h, which suggested that blocking autophagy with 3-MA enhanced the
cytotoxic effects of PPM on HepG2 cells (Fig. 5B). Flow cytometry analysis
demonstrated that compared with the PPM group, the combined PPM and
3-MA treatment resulted in significantly increased apoptotic cell
death (Fig. 5C).
The pro-apoptotic effects of PPM may
impair the function of autophagic lysosomes
The protein expression levels of LC3-II were
significantly increased in the combined treatment group compared
with 3-MA alone; however, the 3-MA + PPM did not demonstrate a
significant increase in LC3-II protein expression levels compared
with PPM alone (P=0.55; Fig. 6A).
Electron microscopy results demonstrated that a large number of
autophagic lysosomes were present in the PPM group, which suggested
that the degradation of autophagolysosomes may have been impaired
in HepG2 cells (Fig. 6B). The
autophagic degradation inhibitor, Baf A1 (50 nmol/l) was used to
further assess how PPM regulated autophagic degradation. LC3-II
protein expression levels were not markedly increased in the Baf A1
+ PPM group compared with that in the Baf A1 group, which
demonstrated that PPM intrinsically blocked autophagosome
degradation (Fig. 6C).
Furthermore, autophagic flux was directly monitored using the
chimeric autophagic flux reporter protein RFP-GFP-LC3. PPM markedly
increased yellow punctate fluorescence in HepG2 cells, which
suggested an impairment of autophagosome-lysosome fusion. (Fig 6D). Subsequently, lysosomal function
was evaluated in PPM-treated HepG2 cells by assessing the protein
expression levels of CTSB, CTSD and LAMP1 (14). PPM significantly upregulated the
expression of LAMP1 compared with the control and significantly
downregulated the protein expression levels of CTSB and CTSD
compared with the control, which indicated that PPM impaired
lysosomal function by inhibition of proteolytic enzyme activity
(Fig. 6E). To further investigate
whether PPM-induced apoptosis was implicated in the dysfunction of
autophagy degradation, HepG2 cells were treated with PPM (100
nmol/l) alone or in combination with Z-VAD-FMK (20 µmol/l).
The results demonstrated that PPM combined with Z-VAD-FMK
significantly reduced the protein expression levels of LC3-II and
p62/SQSTM1 compared with PPM alone (Fig. 6F).
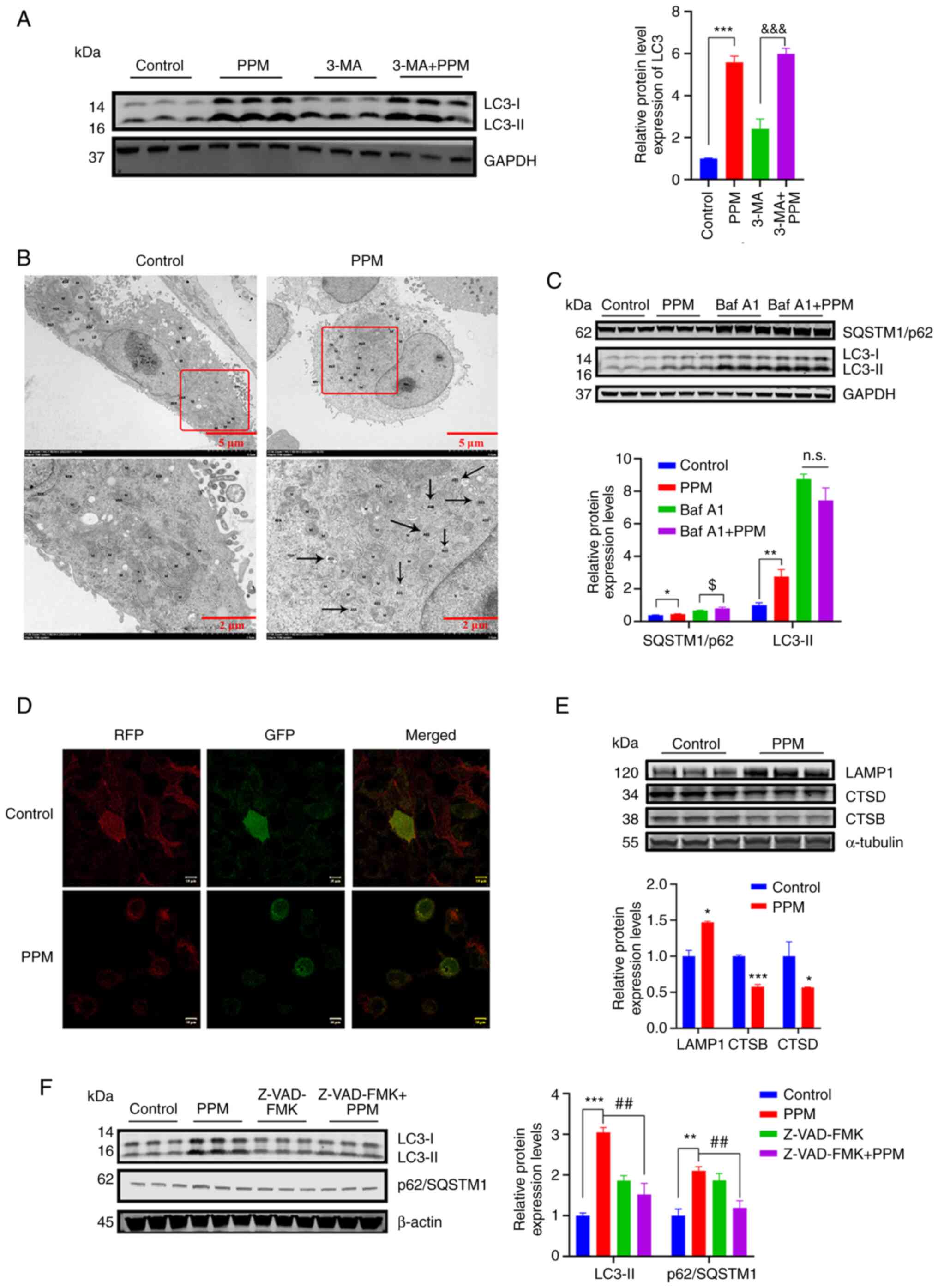 | Figure 6The pro-apoptosis effect of PPM might
impair the function of autophagic lysosomes. (A) LC3 were assessed
using western blotting. (B) Electron microscopy showed that PPM
treatment led to autolysosome increase. Black arrows indicate the
autolysosomes. (C) HepG2 cells were treated with PPM (100 nmol/l)
and/or Baf A1 (50 nmol/l) for 24 h. LC3-II and p62/SQSTM1 protein
expression levels were assessed using western blotting. (D) HepG2
cells were transfected with RFP-GFP-LC3 lentivirus and treated with
PPM (100 nmol/l) for 24 h. Cells were observed to distinguish
between autophagosome (yellow puncta) after colocalization analysis
using a confocal microscope. Scale bar=10 µm. (E) HepG2
cells were treated with PPM (100 nmol/l) for 24 h and the protein
expression levels of LAMP1, CTSB and CTSD were assessed using
western blotting. (F) HepG2 cells were treated with PPM (100 nM)
and/or 20 µmol/l Z-VAD-FMK for 24 h. LC3 and p62/SQSTM1
levels were detected by using western blot analysis. GAPDH or
β-actin were used as a loading control. Data are presented as mean
± SD, n=3; *P<0.05, **P<0.01 and
***P<0.001 vs. control.
&&&P<0.001 vs. 3-MA.
$P<0.05 vs. Baf A1. ##P<0.01 vs. PPM.
n.s., not significant; ASS, autophagolysosome; M, mitochondria;
PPM, periplocymarin; LC3, microtubule-associated protein light
chain 3; SQSTM1/p62, sequestosome 1/p62; LAMP1,
Lysosomal-associated membrane protein 1; CTSB, cathepsin B; CTSD,
cathepsin D; RFP-GFP-LC3, red fluorescence point-green fluorescence
point-LC3. |
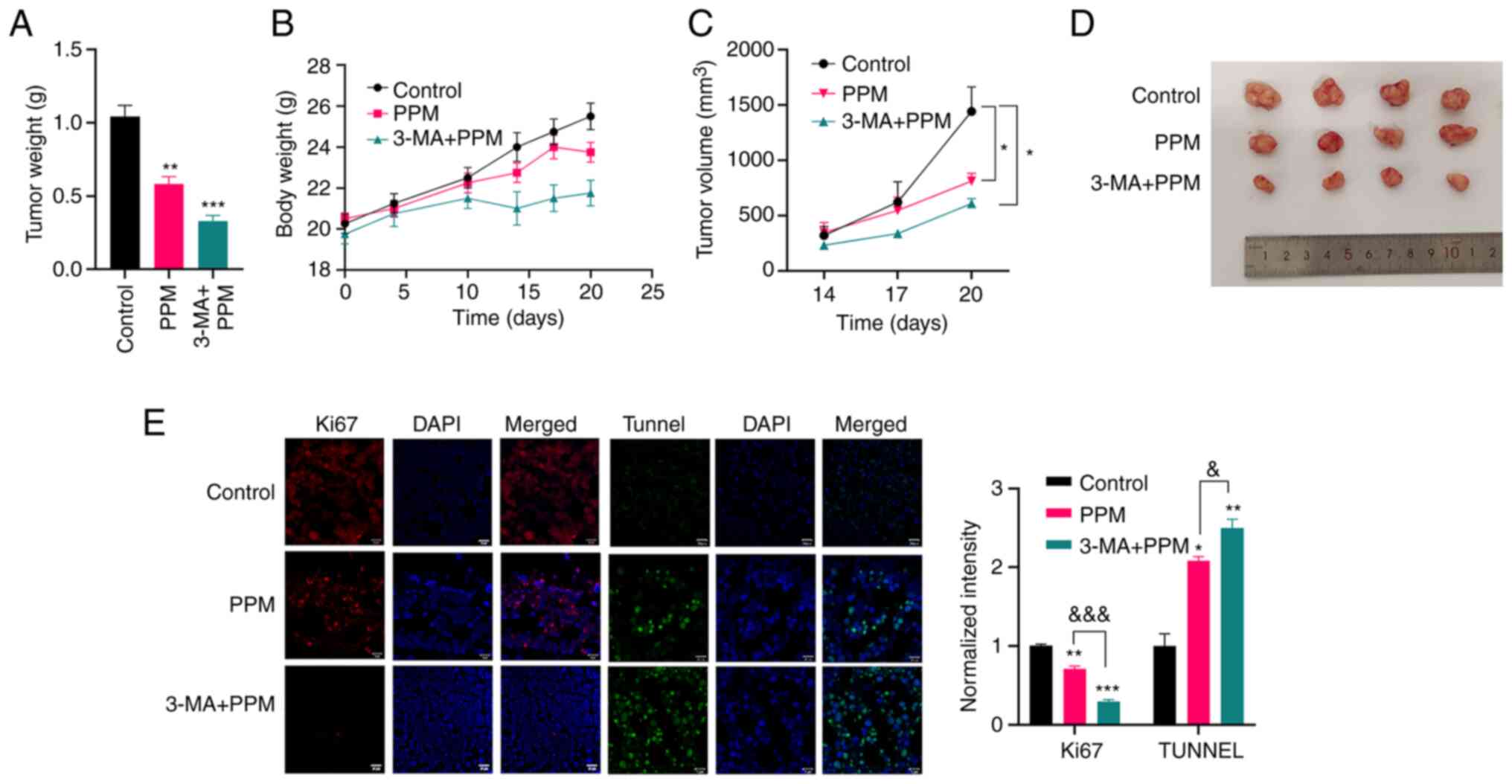
PPM suppresses tumor growth in vivo
The therapeutic effect of PPM alone or in
combination with 3-MA in HepG2 cells xenografts implanted into nude
mice was evaluated. Combined treatment with PPM and 3-MA was
significantly more effective in inhibiting tumor growth than PPM
alone (Fig. 7A-D). Furthermore,
the anti-proliferative and pro-apoptotic effects were assayed by
immunofluorescence staining for Ki67 as well as by using Tunnel
staining. PPM alone or in combination with 3-MA significantly
increased the levels of apoptosis in tumor tissues compared with
the control group (Fig. 7E).
However, the protein expression level of Ki-67 in the control group
was significantly greater than that in the PPM alone or PPM
combined with 3-MA groups. Consistently, a reduction in Ki67
staining in the combination treatment group was significantly
greater than that in the PPM group.
This demonstrated that PPM synchronously activated
the lethal apoptosis and the protective autophagy in liver cancer,
and that the autophagy counteracted the inherent pro-apoptosis
capacities and impaired the anti-cancer effects.
Discussion
Over the last decades, the emerging roles of the NKA
in a variety of critical cellular processes have indicated that NKA
inhibitors, including CGs, may have potential novel therapeutic
purposes in diseases other than heart failure (14). Notably, compelling evidence has
shown the anticancer properties of CGs in certain types of cancers
and this has highlighted their potential use in cancer therapy.
Although the anticancer effects of CGs alone or in combination with
other drugs have been verified in both preclinical studies and
clinical trials (47), the MOA of
CGs remains unclear. Early studies suggested that the increased
susceptibility of tumor cells to these CGs is dependent on the
activation of apoptosis (48,49).
CGs induce cell apoptosis by increasing the concentration of
intracellular free calcium and induce S phase cell cycle arrest by
down-regulating the expression of Cyclin A and CDK2 (50). CGs have been reported to have
downregulated the expression of the anti-apoptotic protein Bcl-2
and increased the translocation of proapoptotic protein cytochrome
C, which induced apoptosis (51).
Durmaz et al (52) reported
that CGs, particularly Lanatoside C could significantly inhibit,
PTEN protein adequate, Huh7 and, PTEN deficient, Mahlavu human
liver cancer cell proliferation by the induction of apoptosis and
G2/M arrest in the cells. As NKA has been extensively studied as a
potential anticancer target, CG-based NKA inhibitors have also been
extensively studied, and apoptosis-independent death effects have
been reported, including the involvement of the interferon
signaling pathway, endogenous interactions of volume-regulated
anion channels, activation of cancer-specific immune responses and
inhibition of protective autophagy (53).
In the present study, the roles and mechanisms
involved in the integrated regulation of macroautophagy and
apoptosis in response to PPM was assessed in HepG2 cells. To avoid
the selectivity of liver cancer cells, in vitro
proliferation inhibition was performed in Huh7 and HepG2 cells and
demonstrated IC50 values of 2.1860 µmol/l for
Huh7 and 0.7725 µmol/l for HepG2 after an incubation time of
48 h. These findings demonstrated for the first time, to the best
of our knowledge, that PPM exerted dual targeting of apoptosis and
autophagy. To evaluate the interaction between apoptosis and
protective autophagy in vitro, mutual antagonism between
apoptosis and autophagy in PPM-treated HepG2 cells was evaluated.
Briefly, macroautophagy induced by PPM counteracted apoptotic cell
death; in turn, autophagic degradation activity was impaired by
pro-apoptotic effects. Finally, the synergistic effect of the
combined application of PPM and an autophagy inhibitor in BALB/c
nude mice bearing HepG2 xenografts was demonstrated.
Although research on PPM for the treatment of liver
cancer has not been previously reported, the increased
susceptibility of other cancer cells to PPM has been reported, and
induction of apoptosis is a common outcome (10,54),
highlighting PPM as an organic potential anticancer agent (16). The present study demonstrated the
anticancer effects of PPM through the inhibition of proliferation,
migration, and clonal and spheroid formation in HepG2 cells. A
pro-apoptotic programmed cell death (PCD) pattern was evaluated,
which demonstrated that PPM treatment resulted in an elevated
membrane potential of the mitochondria and impaired mitochondrial
functions, including reduced ATP production, which indicated that
the mitochondria-mediated intrinsic apoptotic pathway was
concomitantly activated. To validate the pro-apoptotic MOA of PPM,
Z-VAD-FMK, a specific apoptosis inhibitor, was used, which reversed
the pro-apoptotic effect of PPM by significantly reducing the
protein expression level of cleaved caspase-3.
Research has suggested that high levels of autophagy
also result in increased cell death (55). The unique form of
autophagy-dependent cell death is termed 'autosis' (56), which is mediated by NKA and is
characterized by numerous autophagosomes/autolysosomes and nuclear
convolution during the early stages, and by dismantling of cellular
organelles and focal ballooning of the perinuclear space in the
later stages (57). CGs have been
shown to induce autophagy in certain cancer types (47). To investigate whether autophagic
processes could be triggered by PPM to inhibit autosis or
autophagic cell death (58), the
expression of autophagy-related genes was assessed. Inconsistent
with previous studies on the inhibition of autosis, the present
study demonstrated that PPM significantly increased LC3-II protein
expression levels, which in-turn activated macroautophagy via the
AMPK/ULK1 and mTOR signaling pathways. Moreover, the decreased
expression of BNIP3 and PINK1, biomarkers of mitophagy,
demonstrated that PPM selectively induced macroautophagy
activation. How these changes in autophagy-related genes enhanced
the anti-cancer susceptibility to PPM requires future study.
Inhibition of autophagy by 3-MA increased the pro-apoptotic effects
of PPM in HepG2 cells, which supported the evidence that autophagy
and apoptosis serve opposing roles in tumor cells (55). These data indicated that inhibition
of autophagy sensitized HepG2 cells to PPM-induced apoptosis.
However, when the increased p62/SQSTM1 protein expression levels
were assessed, new problems emerged: Autophagic degradation may be
impaired, and it may have been that the activated apoptosis
counteracted this degradation process.
The mechanisms linking autophagy and apoptosis have
not been fully delineated (59).
In the present study, the activation of autophagy elevated the
threshold of PPM-mediated apoptosis. To evaluate whether
PPM-mediated autophagy was a compensatory response, knockdown of
ATP1A1 (the NKA gene) by siRNA mostly inhibited the expression of
LC3-II in HepG2 cells treated with PPM, which indicated that
protective autophagy was triggered by PPM by direct targeting of
NKA. Electron microscopy demonstrated the accumulation of
autophagosomes/autolysosomes in PPM-treated cells. Since the
accumulation of autophagosomes/autolysosomes manifests as blockage
of autophagic degradation, we examined the effect of PPM on
autophagy flux and activity by combining PPM with Baf A1, an
autophagic degradation inhibitor. Compared with Baf A1 alone, PPM
combined with Baf A1 did not significantly increase the conversion
of LC3-I to LC3-II, which indicated that PPM is an autophagic
degradation inhibitor. Consistent with these results, it was
demonstrated that PPM increased the number of yellow puncta in the
RFP-GFP-LC3 fluorescence assay, which suggested that autophagic
degradation was blocked in HepG2 cells treated with PPM. Taken
together, these results indicated that PPM may interfere with the
fusion of autophagosomes and lysosomes. The decreased protein
expression levels of CTSD and CTSB demonstrated that PPM affected
the metabolic degradation of lysosomes to block autophagic flux in
HepG2 cells, and the protein expression level of LAMP1 was assessed
to exclude the possibility of direct damage to lysosomes by PPM.
The present study demonstrated that PPM simultaneously activated
lethal apoptosis and protective autophagy in HepG2 cells. Next,
whether the activation of apoptosis counteracted the autophagy
degradation process was assessed. The apoptosis inhibitor,
Z-VAD-FMK, combined with PPM significantly inhibited the protein
accumulation of LC3-II and p62/SQSTM1, which indicated that the
apoptosis induced by PPM was implicated in decreased autophagy
degradation. Since several clinical trials for CGs in cancer are
currently underway, their real-world benefits in liver cancer
patients and their potential self-limiting effects in vitro
remain uncertain. To validate the anti-cancer effects of PPM in
vivo and the hypothesis that a combination of an autophagy
inhibitor may be an improved strategy for the treatment of PPM in
liver cancer, nude mice were used to establish xenograft models and
it was demonstrated that the tumor volume and weight in the
combination treatment group were significantly smaller than those
in either of the single-agent treatment groups. Furthermore,
immunofluorescence studies for Ki67 demonstrated a marked reduction
in proliferation in the tissues from the mice treated with the
combination of drugs, which further indicated the regression of
tumors in the mouse models.
In conclusion, the results demonstrated that PPM, a
CG, exerts an anti-cancer effect by inducing apoptosis; that
PPM-induced autophagy activation occurred in an NKA
receptor-dependent manner and counteracted, to some extent, the
pro-apoptotic effect of PPM, and vice versa; that the abrogation of
autophagic flux resulted from decreased protein levels in the
lysosomes; and the combined application of PPM with an autophagy
inhibitor may be a good option to enhance the pro-apoptotic
activity of PPM in vitro and the anti-tumor activity in
vivo. A limitation of the present study was the small number of
animals included, which may need to be enlarged in future
investigations.
These results provide novel insights into the MOA of
PPM, and indicate PPM as a potential therapeutic agent against
liver cancer, whilst also showing activation of protective
autophagy by PPM. Finally, an improved strategy, consisting of a
combination of PPM and autophagy inhibitors, was assessed and this
showed improved anti-tumor effects.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YHo and HH designed the study. YHa completed the
first draft of this manuscript. YHa, TS and MW performed the
experiments. ToL, CZ and TiL analyzed the data and revised the
manuscript. YHa, TS and YHo confirm the authenticity of all the raw
data. All authors have read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
The study was approved by the Animal Care and Use
Committee of Hebei Yiling Chinese Medicine Research Institute
(approval no. 2022023).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This research was supported by the HaiYan Fund of Harbin Medical
University Cancer Hospital (grant no. JJMS2021-16), Research
project of China Primary Health Care Foundation (grant no.
2022-001) and S&T Program of Hebei (grant no. E2020100001).
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Massarweh NN and El-Serag HB: Epidemiology
of hepatocellular carcinoma and intrahepatic cholangiocarcinoma.
Cancer Control. 24:10732748177292452017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bruix J, Gores GJ and Mazzaferro V:
Hepatocellular carcinoma: Clinical frontiers and perspectives. Gut.
63:844–855. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guo H, Wu T, Lu Q, Li M, Guo JY, Shen Y,
Wu Z, Nan KJ, Lv Y and Zhang XF: Surgical resection improves
long-term survival of patients with hepatocellular carcinoma across
different Barcelona clinic liver cancer stages. Cancer Manag Res.
10:361–369. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cheng L, Wang C, Ma X, Wang Q, Cheng Y,
Wang H, Li Y and Liu Z: Multifunctional upconversion nanoparticles
for dual-modal imaging-guided stem cell therapy under remote
magnetic control. Advanced Functional Materials. 21:272–280. 2013.
View Article : Google Scholar
|
|
6
|
Huang M, Shen S, Luo C and Ren Y: Genus
periploca (Apocynaceae): A review of its classification,
phytochemistry, biological activities and toxicology. Molecules.
24:27492019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xie G, Sun L, Li Y, Chen B and Wang C:
Periplocin inhibits the growth of pancreatic cancer by inducing
apoptosis via AMPK-mTOR signaling. Cancer Med. 10:325–336. 2021.
View Article : Google Scholar
|
|
8
|
Yun W, Qian L, Cheng Y, Tao W, Yuan R and
Xu H: Periplocymarin plays an efficacious cardiotonic role via
promoting calcium influx. Front Pharmacol. 11:12922020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yun W, Qian L, Yuan R and Xu H:
Periplocymarin alleviates doxorubicin-induced heart failure and
excessive accumulation of ceramides. Front Cardiovasc Med.
8:7325542021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bloise E, Braca A, De Tommasi N and
Belisario MA: Pro-apoptotic and cytostatic activity of naturally
occurring cardenolides. Cancer Chemother Pharmacol. 64:793–802.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li Y, Li J, Zhou K, He J, Cao J, An M and
Chang YX: A review on phytochemistry and pharmacology of cortex
periplocae. Molecules. 21:17022016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Triana-Martínez F, Picallos-Rabina P, Da
Silva-Álvarez S, Pietrocola F, Llanos S, Rodilla V, Soprano E,
Pedrosa P, Ferreirós A, Barradas M, et al: Identification and
characterization of cardiac glycosides as senolytic compounds. Nat
Commun. 10:47312019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guerrero A, Herranz N, Sun B, Wagner V,
Gallage S, Guiho R, Wolter K, Pombo J, Irvine EE, Innes AJ, et al:
Cardiac glycosides are broad-spectrum senolytics. Nat Metab.
1:1074–1088. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Prassas I and Diamandis EP: Novel
therapeutic applications of cardiac glycosides. Nat Rev Drug
Discov. 7:926–935. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kumavath R, Paul S, Pavithran H, Paul MK,
Ghosh P, Barh D and Azevedo V: Emergence of cardiac glycosides as
potential drugs: Current and future scope for cancer therapeutics.
Biomolecules. 11:12752021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Martey ON, He X, Xing H, Deng F, Feng S,
Li C and Shi X: Periplocymarin is a potential natural compound for
drug development: Highly permeable with absence of P-glycoprotein
efflux and cytochrome P450 inhibitions. Biopharm Drug Dispos.
35:195–206. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao LM, Li L, Huang Y, Han LJ, Li D, Huo
BJ, Dai SL, Xu LY, Zhan Q and Shan BE: Antitumor effect of
periplocin in TRAIL-resistant gastric cancer cells via upregulation
of death receptor through activating ERK1/2EGR1 pathway. Mol
Carcinog. 58:1033–1045. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lohberger B, Wagner S, Wohlmuther J,
Kaltenegger H, Stuendl N, Leithner A, Rinner B, Kunert O, Bauer R
and Kretschmer N: Periplocin, the most anti-proliferative
constituent of Periploca sepium, specifically kills liposarcoma
cells by death receptor mediated apoptosis. Phytomedicine.
51:162–170. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
White E, Mehnert JM and Chan CS:
Autophagy, metabolism, and cancer. Clin Cancer Res. 21:5037–5046.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schwartz LM: Autophagic cell death during
development-ancient and mysterious. Front Cell Dev Biol.
9:6563702021. View Article : Google Scholar
|
|
21
|
Lockshin RA and Zakeri Z: Apoptosis,
autophagy, and more. Int J Biochem Cell Biol. 36:2405–2419. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Levine B and Yuan J: Autophagy in cell
death: An innocent convict? J Clin Invest. 115:2679–2688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Trenti A, Grumati P, Cusinato F, Orso G,
Bonaldo P and Trevisi L: Cardiac glycoside ouabain induces
autophagic cell death in non-small cell lung cancer cells via a
JNK-dependent decrease of Bcl-2. Biochem Pharmacol. 89:197–209.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Newman RA, Kondo Y, Yokoyama T, Dixon S,
Cartwright C, Chan D, Johansen M and Yang P: Autophagic cell death
of human pancreatic tumor cells mediated by oleandrin, a
lipid-soluble cardiac glycoside. Integr Cancer Ther. 6:354–364.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mariño G, Niso-Santano M, Baehrecke EH and
Kroemer G: Self-consumption: The interplay of autophagy and
apoptosis. Nat Rev Mol Cell Biol. 15:81–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang X, Zhou G, Liu C, Wei R, Zhu S, Xu Y,
Wu M and Miao Q: Acanthopanax versus 3-methyladenine ameliorates
sodium taurocholate-induced severe acute pancreatitis by inhibiting
the autophagic pathway in rats. Mediators Inflamm.
2016:83697042016. View Article : Google Scholar
|
|
27
|
Yang J, Wang B, Xu Q, Yang Y, Hou L, Yin
K, Guo Q, Hua Y, Zhang L, Li Y, et al: TMEM166 inhibits cell
proliferation, migration and invasion in hepatocellular carcinoma
via upregulating TP53. Mol Cell Biochem. 476:1151–1163. 2021.
View Article : Google Scholar
|
|
28
|
Liu HR, Peng XD, He HB, Wang YH, Li Y, He
GX, Liu YL, Li YL and Zeng CJ: Antiproliferative activity of the
total saponin of Solanum lyratum Thunb in Hela cells by inducing
apoptosis. Pharmazie. 63:836–842. 2008.PubMed/NCBI
|
|
29
|
Hyllienmark L and Brismar T: Effect of
metabolic inhibition on K+ channels in pyramidal cells of the
hippocampal CA1 region in rat brain slices. J Physiol. 496:155–164.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hyllienmark L and Brismar T: Effect of
hypoxia on membrane potential and resting conductance in rat
hippocampal neurons. Neuroscience. 91:511–517. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lin Z, Xing W, Gao C, Wang X, Qi D, Dai G,
Zhao W and Yan G: Inhibitory effect of vascular endothelial growth
factor on the slowly activating delayed rectifier potassium current
in guinea pig ventricular myocytes. J Am Heart Assoc.
7:e0077302018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang D, Gao JL, Zhao CY, Wang DN, Xing
XS, Hou XY, Wang SS, Liu Q and Luo Y: Cyclin G2 promotes the
formation of smooth muscle cells derived foam cells in
atherosclerosis via PP2A/NF-κB/LOX-1 pathway. Ann Transl Med.
9:4462021. View Article : Google Scholar
|
|
33
|
Scholzen T and Gerdes J: The Ki-67
protein: From the known and the unknown. J Cell Physiol.
182:311–322. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li X, Zhou Y, Zhang X, Cao X, Wu C and Guo
P: Cordycepin stimulates autophagy in macrophages and prevents
atherosclerotic plaque formation in ApoE(-/-) mice. Oncotarget.
8:94726–94737. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Medina-Ortiz K, López-Alvarez D, Navia F,
Hansen T, Fierro L and Castaño S: Identification of
Na(+)/K(+)-ATPase α/β isoforms in Rhinella marina tissues by RNAseq
and a molecular docking approach at the protein level to evaluate α
isoform affinities for bufadienolides. Comp Biochem Physiol A Mol
Integr Physiol. 254:1109062021. View Article : Google Scholar
|
|
36
|
Bers DM: Cardiac excitation-contraction
coupling. Nature. 415:198–205. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liao YX, Yu HY, Lv JY, Cai YR, Liu F, He
ZM and He SS: Targeting autophagy is a promising therapeutic
strategy to overcome chemoresistance and reduce metastasis in
osteosarcoma. Int J Oncol. 55:1213–1222. 2019.PubMed/NCBI
|
|
38
|
Zhang XJ, Chen S, Huang KX and Le WD: Why
should autophagic flux be assessed? Acta Pharmacol Sin. 34:595–599.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Seranova E, Ward C, Chipara M, Rosenstock
TR and Sarkar S: In vitro screening platforms for identifying
autophagy modulators in mammalian cells. Methods Mol Biol.
1880:389–428. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
López M and Diéguez C: Cellular energy
sensors: AMPK and beyond. Mol Cell Endocrinol. 397:1–3. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Timm KN and Tyler DJ: The role of AMPK
activation for cardioprotection in doxorubicin-induced
cardiotoxicity. Cardiovasc Drugs Ther. 34:255–269. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hardie DG, Ross FA and Hawley SA: AMPK: A
nutrient and energy sensor that maintains energy homeostasis. Nat
Rev Mol Cell Biol. 13:251–262. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kang SA, Pacold ME, Cervantes CL, Lim D,
Lou HJ, Ottina K, Gray NS, Turk BE, Yaffe MB and Sabatini DM:
mTORC1 phosphorylation sites encode their sensitivity to starvation
and rapamycin. Science. 341:12365662013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dyshlovoy SA: Blue-print autophagy in
2020: A critical review. Mar Drugs. 18:4822020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fozzard HA and Sheets MF: Cellular
mechanism of action of cardiac glycosides. J Am Coll Cardiol.
5:10A–15A. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mijatovic T, Quaquebeke V, Delest B,
Debeir O, Darro F and Kiss R: Cardiotonic steroids on the road to
anti-cancer therapy. Biochim Biophys Acta. 1776:32–57.
2007.PubMed/NCBI
|
|
47
|
Škubník J, Pavlíčková VS, Psotová J and
Rimpelová S: Cardiac glycosides as autophagy modulators. Cells.
10:33412021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cheng CF, Lu IH, Tseng HW, Sun CY, Lin LT,
Kuo ZK, Pan IH and Ko CH: Antitumor effect of periplocin in
TRAIL-resistant human hepatocellular carcinoma cells through
downregulation of IAPs. Evid Based Complement Alternat Med.
2013:9580252013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chao MW, Chen TH, Huang HL, Chang YW,
HuangFu WC, Lee YC, Teng CM and Pan SL: Lanatoside C, a cardiac
glycoside, acts through protein kinase Cδ to cause apoptosis of
human hepatocellular carcinoma cells. Sci Rep. 7:461342017.
View Article : Google Scholar
|
|
50
|
Xu ZW, Wang FM, Gao MJ, Chen XY, Shan NN,
Cheng SX, Mai X, Zala GH, Hu WL and Xu RC: Cardiotonic steroids
attenuate ERK phosphorylation and generate cell cycle arrest to
block human hepatoma cell growth. J Steroid Biochem Mol Biol.
125:181–191. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Rasheduzzaman M, Yin H and Park SY:
Cardiac glycoside sensitized hepatocellular carcinoma cells to
TRAIL via ROS generation, p38MAPK, mitochondrial transition, and
autophagy mediation. Mol Carcinog. 58:2040–2051. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Durmaz I, Guven EB, Ersahin T, Ozturk M,
Calis I and Cetin-Atalay R: Liver cancer cells are sensitive to
Lanatoside C induced cell death independent of their PTEN status.
Phytomedicine. 23:42–51. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fujii T, Shimizu T, Yamamoto S, Funayama
K, Fujita K, Tabuchi Y, Ikari A, Takeshima H and Sakai H: Crosstalk
between Na(+), K(+)-ATPase and a volume-regulated anion channel in
membrane microdomains of human cancer cells. Biochim Biophys Acta
Mol Basis Dis. 1864:3792–3804. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang HY, Xu WQ, Zheng YY, Omari-Siaw E,
Zhu Y, Cao X, Tong SS, Yu JN and Xu XM: Octreotide-periplocymarin
conjugate prodrug for improving targetability and anti-tumor
efficiency: Synthesis, in vitro and in vivo evaluation. Oncotarget.
7:86326–86338. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gordy C and He YW: The crosstalk between
autophagy and apoptosis: Where does this lead? Protein Cell.
3:17–27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Liu Y, Shoji-Kawata S, Sumpter RM Jr,
Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, Shaw SY,
et al: Autosis is a Na+, K+-ATPase-regulated form of cell death
triggered by autophagy-inducing peptides, starvation, and
hypoxia-ischemia. Proc Natl Acad Sci USA. 110:20364–20371. 2013.
View Article : Google Scholar
|
|
57
|
Liu Y and Levine B: Autosis and autophagic
cell death: The dark side of autophagy. Cell Death Differ.
22:367–376. 2015. View Article : Google Scholar :
|
|
58
|
Zhang K, Chen J, Zhou H, Chen Y, Zhi Y,
Zhang B, Chen L, Chu X, Wang R and Zhang C: PU.1/microRNA-142-3p
targets ATG5/ATG16L1 to inactivate autophagy and sensitize
hepatocellular carcinoma cells to sorafenib. Cell Death Dis.
9:3122018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mukhopadhyay S, Panda PK, Sinha N, Das DN
and Bhutia SK: Autophagy and apoptosis: Where do they meet?
Apoptosis. 19:555–566. 2014. View Article : Google Scholar : PubMed/NCBI
|