The complex and diverse group of disorders
collectively referred to as malignant cancer is characterised by
the dissemination and proliferation of aberrant cells in the body.
Physiologically normal cells undergo a systematic process of
growth, division and apoptosis, or programmed cell death, to reach
senescence. In contrast, cancer cells proliferate aggressively,
giving rise to malignant tumours that can invade and destroy
adjacent tissues and organs by spreading through the bloodstream or
lymphatic system (1). Numerous
variables can contribute to the development of malignancies,
including genetic alterations, exposure to carcinogens such as
tobacco smoke or UV radiation, and dietary and lifestyle choices
(2). While various treatment
methods, including surgery, radiation therapy, and chemotherapy,
are available for different types of malignancies, the primary
challenge lies in revolutionising the diagnosis and prognosis of
cancer at both the cellular and genetic levels (3). In the late 19th century, surgeon
William Coley observed that certain cancer patients suffering from
bacterial infections experienced spontaneous cancer remission. This
observation led Coley to inject heat-killed Streptococcus
pyogenes and S. marcescens into cancer patients to boost
their immune systems. Although the results were mixed, his work
laid the foundation for modern immunotherapy (4). In the 1890s, Paul Ehrlich delved
deeper into the concept that the immune system is capable of
detecting and eliminating malignant cells. He laid the groundwork
for active and passive immunity, a concept he later referred to as
'cancer immunity' (5). Research
at the National Cancer Institute by Rosenberg (6) explored the efficacy of interleukin-2
(IL-2) in stimulating cell growth and combating cancerous cells.
The development and advances of cancer immunotherapy gained notable
momentum following the development of the first monoclonal antibody
(mAb), rituximab, for treating non-Hodgkin lymphoma (NHL) in 1997.
Monoclonal antibodies are one of the three adoptive cell transfer
(ACT) therapies used for diagnosing malignancies and for prognostic
evaluation (7,8). For example, subcutaneous FBL3
lymphomas were lysed by infusing IL-2 intravenously. Studies also
investigated the use of genetically altered T-lymphocytes to target
tumour antigens (6,9). After a decade of studies and trials,
Eshhar et al (10)
developed a genetically engineered therapy using chimeric proteins
that could recognize and target specific malignant antigens
expressed on T-cells. This therapy is known as chimeric antigen
receptor (CAR) T-cell therapy. The process involves leukapheresis,
a procedure used to isolate T-lymphocytes from a patient's
leukocytes, followed by the infusion of genetically altered CARs
into the patient's circulatory system. Over a decade of successful
innovations, CARs have evolved, incorporating different domains and
co-stimulatory, elements that enhance their ability to bind to
cancerous cells and tissues, facilitating the lysis of cancerous
cells. This evolution has led to CAR T-cell therapy becoming a
novel therapeutic option for conditions such as multiple myeloma
(MM), B-cell acute lymphoblastic leukaemia (B-ALL), and other
aggressive tumours (11,12). Recent achievements in CAR T-cell
therapy, driven by molecular and immunological insights, provide
the foundation for its advancement as a more efficient and precise
therapeutic approach. These advances are further discussed in the
following sections.
The demand for T-cell receptors (TCRs) in curing
metastatic melanoma, was unparalleled due to their remarkable
efficacy. However, their success as a therapeutic option was
accompanied by a series of adverse side effects including
off-target toxicity, cardiac toxicity, neurological toxicities, and
various other life-threatening complications, which led to the
exploration of neoantigens as a potentially safer approach
(13,14). The introduction of CAR T-cell
therapy ushered in cutting-edge technologies across various
disciplines, including immunogenetics, molecular genetics, and
oncogenetics. This approach involves the use of protein receptors
known as chimeric T-lymphocytic receptors, which have evolved to
enable T-cells to precisely target specific antigens. Their ability
to combine components for T-cell activation and antigen binding
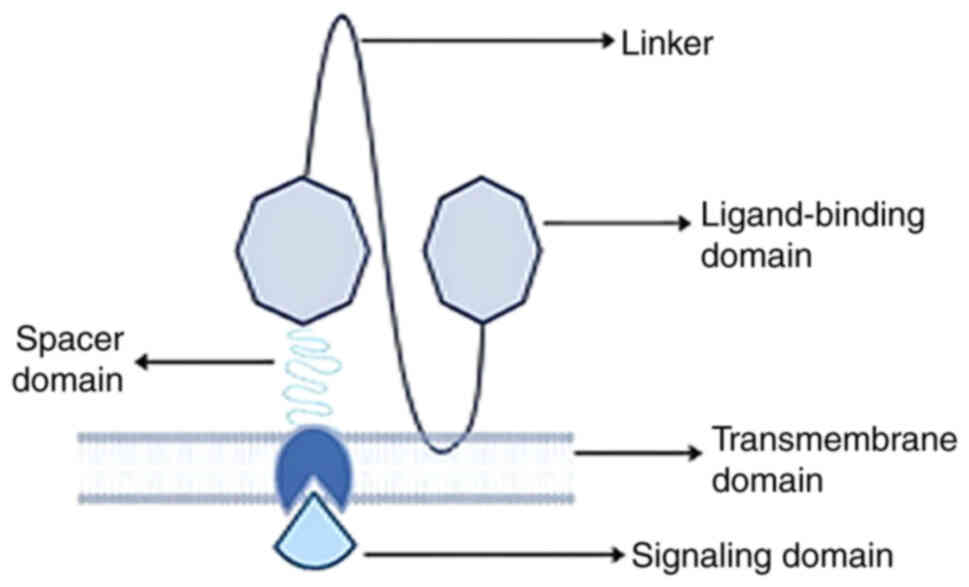
justifies the term 'chimeric' being applied to them (15,16). The basic structural composition of
CAR includes the extracellular domain (also referred to as the
target and spacer motif), the transmembrane motif, and a signalling
motif. Each of these domains significantly influences
anti-carcinogenic effectiveness and CAR T-cell production (15).
The targeting domain, also known as the
ligand-binding domain, primarily consists of a fragment called
single-chain variable fragment (ScFv) (Fig. 1), produced from non-human
sequences found in mABs capable of eliciting immunogenic responses.
Theoretically, ScFv can recognise a wide range of surface antigens
expressed on target cells (such as HER2, PSMA, and CD19) (15,17). In addition, other domains
incorporate nanobodies and receptor-cognate ligands, such as NKG2D,
IL-2R, IL-7R, T1E, and PD-1, to target multiple ligands (18,19). Certain cases have reported potent
anti-tumour activity with ScFv, which, however, can lead to
neurotoxicity and could potentially be resolved through the
optimisation of ligand-binding affinities (20).
To provide flexibility to the recognition sites of
CAR T-cells, the spacer domain connects ScFv to the transmembrane
domain. This function of the spacer domain is to determine the
impact based on its length (15).
For larger tumour sizes, the binding of epitopes with a spacer
domain of a specific length becomes necessary. However, off-target
binding can compromise the safety and effectiveness of a therapy
(21,22). For example, the interaction of
FcRs with the IgG1 Fc spacer domain on rodent macrophages can lead
to CAR-induced cell death. To address this issue, Hudecek et
al (22) proposed the
deletion of the Fc spacer CH2 domain, a critical component of
chimeric Fc-FcR interactions. Based on the results of clinical
studies, the US Food and Drug Administration (FDA) has approved
certain non-IgG-based spacer domains, such as CD8 and CD28, which
are widely used in therapy. Spacers also play a role in quantifying
and purifying CAR-positive subsets after engineering.
The transmembrane domain serves as a linker, acting
as a pivot point for transmitting ligands and recognition signals
to the signalling domain. The TCR-CD3 complex's domains play a
crucial part in organising the assembly. Cysteine residues in the
transmembrane domain of CD3 are made possible by the dimerization
of CD3ζ in the first generation of chimeric antigen T-lymphocytes.
This domain is exposed to hydrophobic conditions containing
non-polar amino acids as it is an integral part of the basic
structure of the genetically modified T-lymphocyte receptor. In the
current context, designs of this domain are borrowed from CD4 and
CD28, which are relatively independent of the TCR complex and
ensure the binding of the independent TCR to the target. According
to a review study by Cheng Zhang et al (23) and Sterner et al (24), the most widely accepted and used
transmembrane domain is CD28 or CD8α, which is chosen to optimise
receptor expression and stability.
The CAR T-cell therapy can only be partially
explained without discussing the signalling domain. The signalling
or co-stimulatory domains were engineered in the 1990s and include
a CD3ζ domain and a tyrosine-based immunoreceptor (ITAM). ITAMs are
present on the TCR-CD3 complex and serve as crucial phosphorylation
sites to recruit ZAP70, which plays a critical role in signalling
cascades (17). The importance of
signalling domains has been discussed in the previous section, with
CD28 and 4-1BB as recognised co-stimulatory domains
for various B-cell malignancies in the second generation of CARs;
this is further elaborated elsewhere (24).
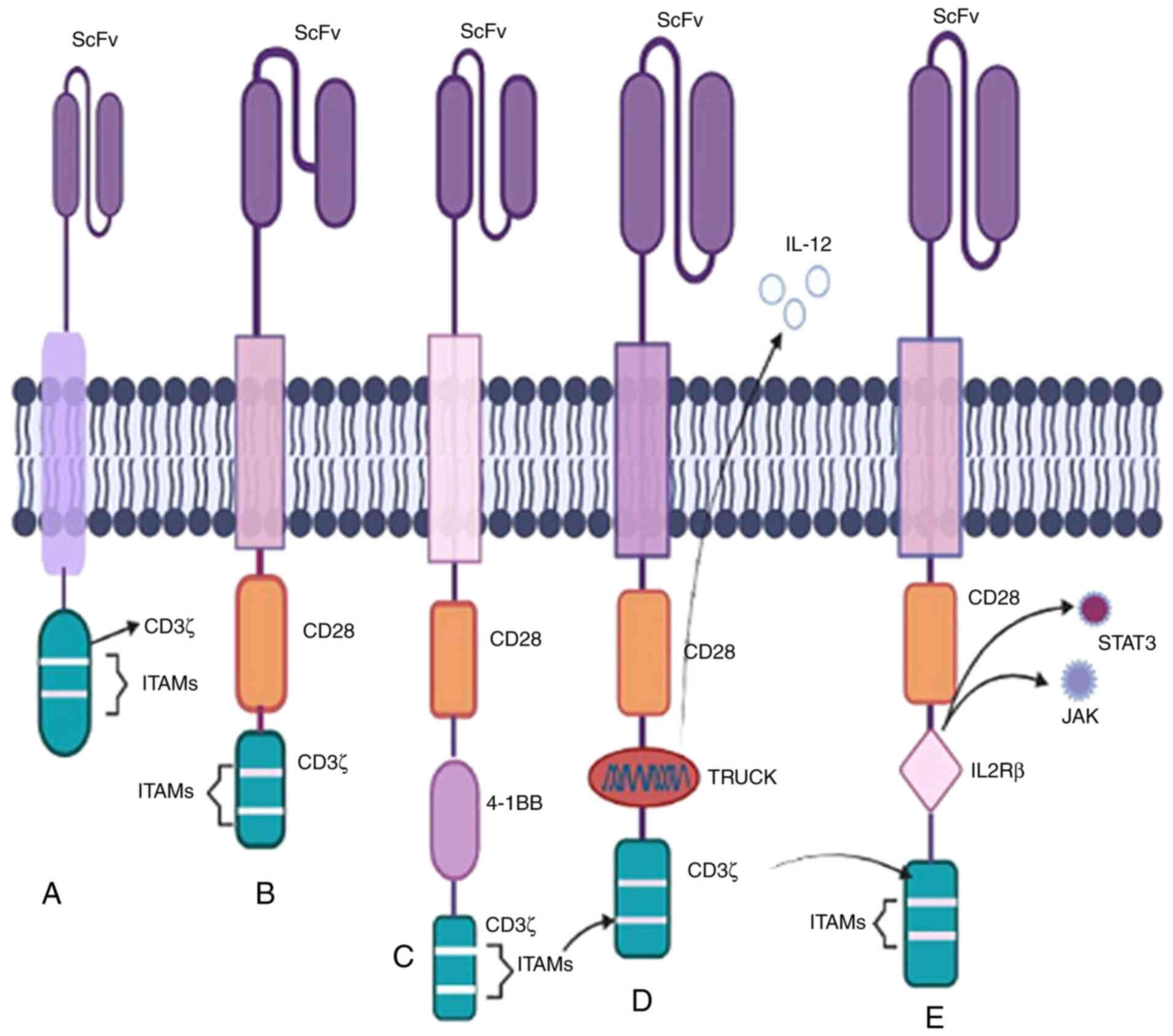
The CD3ζ chain serves as the primary stimulatory
domain in the initial CAR T-cell therapy paradigm from the 1990s.
These engineered receptors were widely accepted due to the presence
of CD3ζ and ITAMs (Fig. 2)
(25,26). Specific drugs for this generation
entered clinical trials for the management of leukaemia, ovarian
cancer, and neuroblastoma (NB) following successful preclinical
results. B-cell lymphoma (BCL) patients received infusions of
CD20-CD3ζ CAR T-cells, and several neuroblastoma patients
received treatment with ScFv-CD3ζ CAR T-cells. The
genetically altered signalling receptors, now known as CARs, were
initially referred to as the 'T body approach' model (27,28).
The success of the first generation of studies paved
the way for a second-generation therapy. The two co-stimulatory
domains that have received FDA approval are CD28 and 4-1BB, showing
substantial therapeutic benefits in several cancers including
chronic lymphoblastic leukaemia (CLL), B-ALL, and multiple myeloma
(Fig. 2) (29). Furthermore, a phosphoproteomic
mass spectroscopy study demonstrated that CARs with CD28 domains
phosphorylate more rapidly and intensely than those with 4-1BB
domains. In summary, CD28-based chimeric receptors enhance effector
T-cell proliferation responses, whereas 4-1BB-based CARs promote
T-cell accumulation (28,29).
Enhanced anti-carcinogenic efficacy is achieved by
incorporating two signalling domains. Third-generation therapies,
such as CD3-CD28-OX40 and CD3-CD28-4-1BB, boost
cytokine production and activation signals to promote prolonged
proliferation (Fig. 2) (30). Preclinical results for anti-PSMA
and anti-mesothelin CD28-4-1BB-CD3ζ CARs have shown
increased tumour eradication and persistence abilities compared
with the second-generation therapies. These two motifs have been
evaluated against several targets including CD19, PSMA, GD2,
and mesothelin (31-34).
The superiority of third-generation therapies over
second-generation therapies remain a subject of debate. For
example, CD28-4-1BB-based CARs demonstrate better preclinical
results in mouse xenografts of pancreatic cancer in the third
generation. However, second-generation therapies have still
outperformed the subsequent generations in terms of their
anti-carcinogenic potency. Third-generation therapies exhibit
improved in vitro secretion of cytokines such as IL2 and
TNFα with anti-GD2 CARs consisting of the CD28-OX40-CD3ζ domain.
This domain also shows enhanced proliferation and expansion
compared with the second- and first-generation therapies (35,36).
All previous generations of CAR T-cell therapies
exhibit a lack of anti-carcinogenic activity against solid tumours
due to the inhospitable microenvironment of solid tumours resulting
in heterogeneity and deterioration. The fourth-generation CAR
T-cell therapy is also known as T-cells redirected for universal
cytokine killing (TRUCK) or 'armoured CARs' (37,38). These armoured chimeric receptors
can express cytokines and chemokine receptors such as IL12
to enhance T-cell penetration and protect T-lymphocytes from the
oxidative stress microenvironment to enhance infiltration (Fig. 2) (39). An illustrative example involves
using antigen-negative cancer cell regulators as a target for
antagonistic antibodies such as CTLA-4 and PD-1
demonstrating that blocking PD-1 improves the regulation of
HER-2 redirected CAR T-cells leading to an enhanced immune
response in HER-2 competent transgenic mice (40).
Recently, membrane-based receptors have been
developed, incorporating an IL2Rβ domain inserted between the
co-stimulatory domains CD247 and CD28 to trigger cytokine
signalling (Fig. 2). The presence
of the YXXQ STAT3 binding motif in the IL2Rβ domain facilitates the
induction of CAR T-cells and may activate the JAK-STAT pathway to
promote cell proliferation. This generation of CART-cells has shown
better persistence in leukaemia (41,42).
CARs serve as a membrane bridge with their receptors
spanning both the cell's intracellular and extracellular matrix.
The portion protruding from the cell surface typically consists of
synthetic antibodies acting as the basic antigen recognition motif.
The choice of domains used determines the receptor's ability to
detect or bind to tumour cell antigens. Each CAR's internal region,
which includes the T-lymphocyte trigger unit and 'co-stimulatory'
domains, plays a crucial signalling role. These domains are
responsible for transmitting signals within the cell following the
interaction between the receptor and antigens. The specific domains
used can impact the overall function of the cells. Unlike
endogenous TCRs, CARs can recognise unprocessed antigens,
regardless of how major histocompatibility antigens are presented.
CARs can bind to various targets, including protein-protein
peptides, sugars, highly glycosylated proteins, and gangliosides,
thus broadening the range of potential targets. While ScFv derived
from antibodies are commonly used in the interaction between a CAR
and its target, Fab fragments (Fab) acquired from libraries and
natural ligands (also known as first-generation CARs) have also
been used (43,44).
Leukapheresis, a procedure used to isolate T-cells
from leukocytes, is the first step in the mechanism of action. In
leukapheresis, leukocytes, T-cells, and other components are
separated, following which, in vitro cloning is performed
using viruses to develop a modified gene capable of encoding the
chimeric receptors (44). The
designed CARs recognise and bind to specific antigens or proteins
found on the surface of malignant cells (45). The in vitro-engineered
T-cells are then expanded to produce tens of thousands of copies.
This process may take several weeks and involves the use of
cytokines and other growth factors to stimulate T-cell
proliferation. Following this, the engineered receptors are
introduced into the patient's bloodstream.
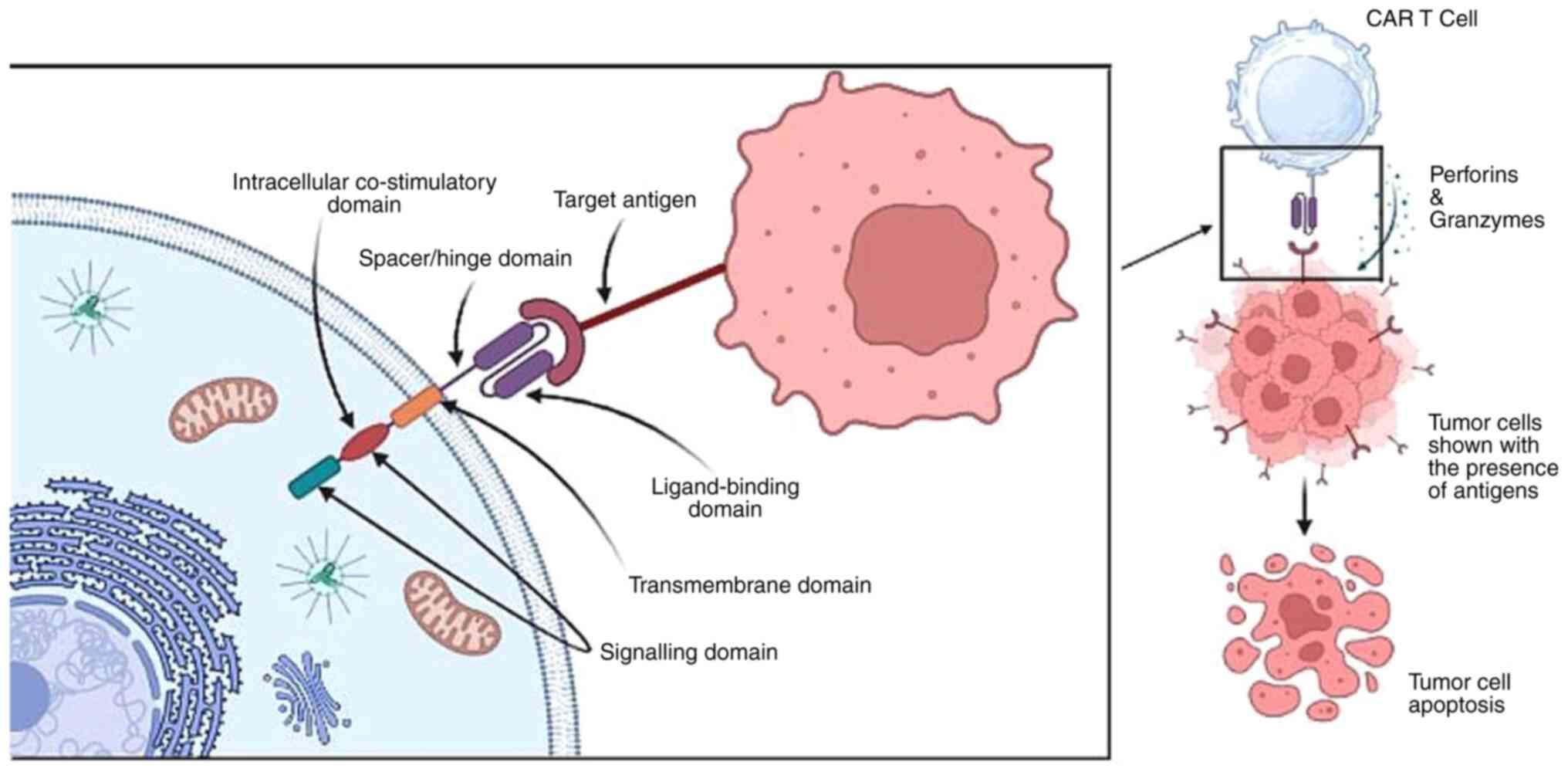
Once in the circulatory and lymphatic systems, CAR
T-cells come into contact with antigen-presenting cancerous cells.
When a CAR encounters the cancer cell expressing the target
antigen, it initiates intracellular signalling processes that
activate CAR T-cells, leading to an increase in cytokine
production. This activation allows CAR T-cells to directly attack
the tumour cells through cytotoxicity. CAR T-cells release
cytotoxic chemicals, such as perforin and granzymes, which induce
apoptosis (programmed cell death) in the cancer cells (Fig. 3) (45). Additionally, CAR T-cells can
stimulate other immune cells, such as natural killer (NK) cells and
tissue macrophages, which contribute to immune responses aimed at
destroying cancer cells or tissues. In certain rare cases, these
augmented T-cells can persist in the patient's immune system
providing ongoing protection against cancer recurrence (46).
Preclinical trials involving xenograft humanised
mouse models to investigate tumour progression have enabled
researchers to study the tumour microenvironment (TME), which
includes components such as blood vessels, lymphocytes,
fibroblasts, and the extracellular matrix (ECM). This research has
led to the development of humanised patient-derived xenograft
(hu-PDX) mouse models designed to preserve genetic profiles and
drug responses. The hu-PDX mouse model closely replicates the TME
found in humans, with cytokines and chemokines playing a critical
role in regulating angiogenesis, metastasis, and immune responses
(47,48). Humanised mouse models have been
employed in ACT T-lymphocytic therapy to enhance the recognition
and elimination of cancer cells. For example, the maturation of
transgenic Wilm's tumour (WT-1-specific TCRs in HLA-I transgenic
NSG mice transplanted with haematopoietic stem cells (HSCs)
enhances cell proliferation capacity and triggers cytokine immune
responses, which results in the amplification of anti-carcinogenic
effects (49). The success of ACT
T-lymphocytic therapies in preclinical and clinical settings paved
the way for CAR T-cell therapy in cancer treatment. The necessity
to simulate human-originated tumours within an intact immune system
has been recognized with the co-evolution of anti-tumour T-cells
and the use of CAR T-receptors on humanised (HM) and genetically
modified (GEMM) mouse models (50,51). In HM mouse models, experiments
involving humanised NRG mice treated with gp350 CAR T-cells showed
a reduction in Epstein-Barr virus (EBV) levels. This research has
stimulated further investigation into the correlation between the
spread of a virus and the incidence of cancer (52). Currently, CAR T-cell therapy has
demonstrated significant efficacy in treating various
haematological and B-cell malignancies without the restriction of
major histocompatibility complex. Chimeric receptors have been
engineered to target CD19 in the hu-BLT (bone marrow, liver,
and thymus) mouse model, with positive responses observed against
primary acute B-ALL (53). As
therapies have advanced, the introduction of co-stimulatory motifs
such as 4-1BB (CD28 and CD137) has shown
promise in enhancing anti-neoplastic responses and cancer cell
elimination in hu-SRC (SCID repopulating cell) mice (54). This review emphasises the efficacy
of CAR T-cells in BCa treatment and sheds light on various
preclinical studies aimed at better understanding their potential.
The human epidermal growth factor receptor 2 (HER2 also
known as ERBB2 belongs to the receptor tyrosine-protein
kinase family (55). When
triggered, downstream signalling pathways lead to gene
overexpression and initiate tumour metastases. In BCa, 20-30% of
the patient population exhibit HER2 amplification,
highlighting its value as a target in BCa therapy. In preclinical
studies, HER2 CAR T-cells resulted in the eradication of tumour
cells, even in trastuzumab-resistant JIMT-1 xenografts, leading to
improved survival rates in xenograft mice (56). Preclinical studies have targeted
various BCa antigens such as FRα, epidermal growth factor
receptor (EGFR), AXL, MUC1, c-Met, TEM8, and NKG2D,
to optimise the efficacy of CAR T-cell therapy in both preclinical
and clinical settings. The third generation of CAR T-cell therapy,
which includes anti-EGFR antibodies in the ScFv region, has
exhibited anti-carcinogenic responses in xenograft mouse models of
triple-negative BCa (TNBC) cell lines (57). TNBC offers several potential cell
targets, including MUC1, c-Met, AXL, NKG2D, integrin αvβ3,
and ROR1, all of which have shown promising preclinical
results in various in vitro and in vivo models of
engineered CAR T cell therapy (58). For example, CAR T-cells based on
the receptor tyrosine kinase AXL have demonstrated
significant efficacy in regulating in vitro cytotoxicity in
MDA-MB-231 xenograft mice for TNBC (59). Another surface receptor, integrin
αvβ3, which plays a role in cell adhesion between epithelial cells
and their microenvironment has been targeted using a second
generation of chimeric T-lymphocytes in preclinical stages, leading
to the release of cytokines such as IL2 and IFNγ
(60). The tyrosine kinase
receptor c-Met is also being targeted with engineered CAR
T-receptors to induce cytolysis in TNBC cells and reduce tumour
progression in TNBC xenografts with intact immune metabolism
(61). NKG2D-CAR T-cell
therapy has been used to target xenograft mouse models by inducing
pro-inflammatory responses. In second-generation therapies, these
engineered receptors, incorporating co-stimulatory motifs such as
4-1BB, have been shown to regulate anti-carcinogenic
activity in vivo (62).
Transmembrane proteins such as MUC1 are often overexpressed
in TNBC cells in glycosylated form. Therefore, tMUC1-CAR
T-cell receptors have been engineered to stimulate cytokines and
chemokines to act on mutant alleles in xenograft mice in
vitro (63). Aberrations in
breast tissues contributing to TNBC can potentially be treated with
tyrosine kinase-like orphan receptor 1 (ROR1)-CAR T-cells,
which stimulate anti-carcinogenic responses in different in
vitro models (64). Numerous
other potential cellular targets for TNBC have been explored in
preclinical studies, as demonstrated below (Table I).
Numerous clinical trials are currently underway to
develop and advance CAR T-cell immunotherapy (Table II) to assess the effectiveness of
this treatment approach for various malignancies. These trials
primarily involve patients with B-NHL, B-ALL, CLL, and
glioblastoma, as well as other solid tumours and
relapsed/refractory malignancies. It would be a remarkable
achievement if some of these trials meet the regulatory standards
set by the US FDA.
Several clinical studies for administering chimeric
antigen immunotherapy in BCa patients are presented in Table III.
Drugs for treating several refractory/relapsed
B-lymphocyte malignancies, including diffuse large cell BCL
(DLBCL), have been approved by the FDA in the United States. These
approvals stem from promising preclinical studies on mouse models
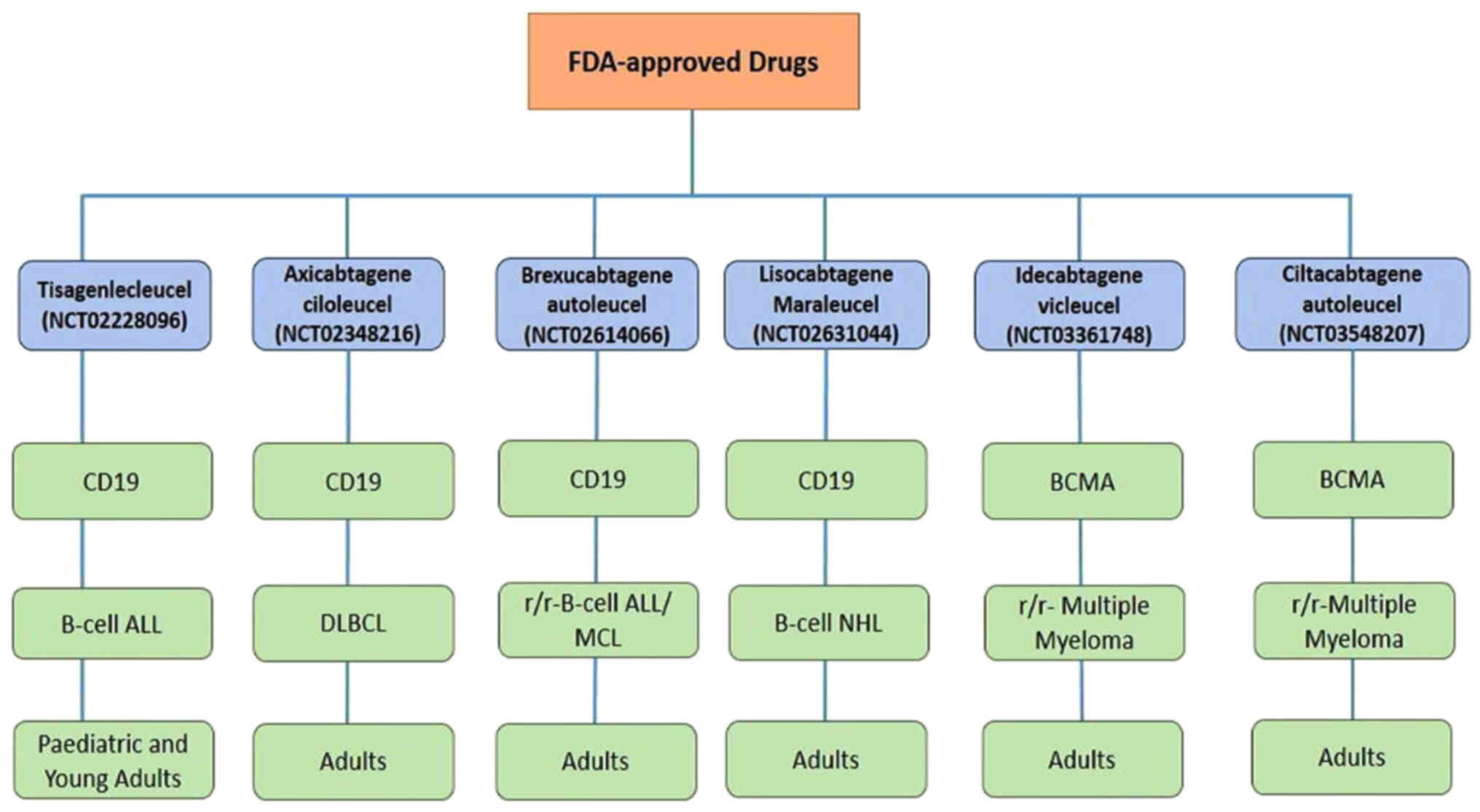
and impressive results from clinical trials. In 2018 FDA granted
approval to CTL019 (tisagenlecleucel; NCT02228096; Fig. 4) for use in paediatric B-ALL
patients who had relapsed or failed previous treatments. A
single-arm, open-label, multi-central phase II research study is
currently ongoing to evaluate CTL019's safety and efficacy
in patients with r/r-B-ALL. This treatment has shown a high
success rate, with CTL019 achieving a 3-month full remission
rate of 83% and a 6-month survival rate of 89%. Additionally, the
ZUMA-1 trial reported a total remission rate of 59% and an
overall response rate of 82% (110,111). In 2019, following the completion
of a phase II clinical study, KTE-C19 (NCT02348216;
axicabtagene ciloleucel) received FDA authorisation as an orphan
medication for the treatment of adults with r/r-DLBCL. After
undergoing Yescarta therapy, the complete remission rate was 51%
(112). In 2020, brexucabtagene
autoleucel was approved for the treatment of certain patients with
Mantle cell lymphoma (MCL) (NCT02601313). Furthermore, in October
2021, brexucabtagene autoleucel (Tecartus) received approval for
the treatment of adult patients with r/r-B-cell precursor
ALL. These trials were conducted under ZUMA-3 phase I/II
(NCT02614066), evaluating CD19-targeted CAR T-cell therapy for
adult r/r-B-ALL A phase I trial infused lisocabtagene
maraleucel (NCT02631044) to assess the drug's safety and efficacy
levels for patients with r/r-B-cell NHL. According to a
23-month trial, lisocabtagene maraleucel achieved an overall
response rate (ORR) of 73% (113). In 2021, idecabtagene vicleucel
was authorised by the US FDA for use in treating adult patients
with r/r-multiple myeloma. An ongoing phase II trial
[NCT03361748] for this drug showed ORR and CR rates of 72 and 28%,
respectively with ~65% of patients remaining in CR for a full year.
Finally, ciltacabtagene autoleucel was licensed for adult patients
with r/r-multiple myeloma in February 2022 based on the
findings of the phase II clinical study [NCT03548207]. The clinical
study reported an ORR of 97.9%, a response time of 21.8 months, and
a follow-up time of ~18 months. To identify the most precise
medications for specific types of malignancies and further enhance
treatment efficacy several clinical trials are currently underway,
with the potential for future FDA approvals (Table II).
In accordance with the intensity of their
expression, tumour antigens are categorised into three groups:
Cancer germline antigens, tumour-specific antigens (TSAs), and
tumour-associated antigens (TAAs) (114). Cancer cells display TSAs on
their surface with malignant cells being rich in TAAs such as
HER2 and CD19 (115,116). Targeting TSAs can lead to side
effects such as on-target/off-target toxicity as chimeric T-cells
attack them (117). Inhibitors
such as PARP, CDK4/6, AKT, and HER2, that affect
various carcinogenesis pathways, including the cell cycle,
metastasis, and angiogenesis, have been investigated as potential
therapeutic targets for impeding BCa proliferation. To date, four
PARP antagonists (olaparib, talazoparib, rucaparib, and
niraparib) have undergone extensive clinical research. Olaparib,
the first FDA-approved PARP inhibitor, targets the genetic
activity of the TOPBP1 and WEE1 genes. Olaparib is
administered both as combination therapy (alongside chemotherapy)
and as a monotherapy, with common side effects including anaemia
and neutropenia (NCT02000622, NCT01445418, NCT02734004,
NCT02032823, and NCT02789332) (118,119). CDK4/6 plays a crucial
role in facilitating tumour cell progression. FDA-approved CDK4/6
antagonists include palbociclib, abemaciclib, and ribociclib,
primarily targeting the FOXM1 gene. Palbociclib, the first
effective CDK4/6 inhibitor, benefits both post- and
pre-menopausal women with HER2-negative and
HR-positive BCa, with neutropenia being a common side
effect. In certain combination therapies, pulmonary embolism, back
pain, and diarrhoea were also observed (NCT02513394, NCT00141297,
NCT01037790, NCT00721409, and NCT01942135) (119,120). In the AKT/PI3K/mTOR
signalling pathway, AKT is a vital transducer affecting
genes such as PI3K, FOXO1, PIP2, PDK1, TSC1/2, and
mTOR. Activated AKT suppresses apoptotic pathways
(Bcl-2-associated death promoter) while stimulating cell
proliferation pathways. Both allosteric and ATP-competitive
inhibitors have been designed, with ATP-competitive AKT
inhibitors proving superior. MK-2206, an allosteric inhibitor, used
in combination therapy with trastuzumab, anthracycline, and
neoadjuvant chemotherapy, was particularly effective for
HER2-positive BCa. Capivasertib and ipatasertib have
demonstrated better efficacy than other ATP-competitive inhibitors
(121). Advances in
HER2-targeted malignancy treatments have led to increased survival
rates for HER2-positive BCa patients (122). CAR T-cell immunotherapy plays a
crucial role in addressing the clinical challenge of BCa
metastasis. HER2-redirected chimeric T-cell receptor
immunotherapy can trigger a remarkable immunological response in
xenograft models (123). This
third-generation CAR T-cell therapy, featuring the CD28 or
4-1BB co-stimulatory domain, enhances survival,
proliferation, and cancer cell control by CAR T-cells (124). Approximately 20-30% of patients
with HER2 amplification with adverse prognostic outcomes
have been administered HER2/ERBB2 targeting the tyrosine
kinase receptors that are responsible for activating the downstream
signalling pathways such as P13K, MEK, PKC, and
JAK/STAT once triggered, leading to tumour progression
(125). The FDA-approved mAB
trastuzumab targets HER2 receptors and has resulted in
clinical improvements for BCa patients (126). The clinical trial [NCT02792114]
identified MSLN as a prospective therapeutic target for
MSLN-specific metastatic BCa. To optimize tumour
specificity, the in vitro CAR T-cell therapy has been
developed for targeting MUC1 and ERBB2 for BCa
patients, resulting in T-cell survival within malignant tumour
cells (127). Targeting the
env gene of human endogenous retroviruses (HERV)-K with
HERV-K-targeted CAR T-cell therapy has demonstrated anti-malignant
effects, as the env protein is involved in tumour progression and
is expressed in ~70% of BCa cases (128,129). Approximately 15-30% of BCa
patients have HER1 gene amplification; primarily in cases of
TNBC (130). TNBC is resistant
to conventional (anti-HER2 and endocrine) treatments due to
the absence of EGFR, oestrogen receptors (ERs), and
progesterone receptors (PRs) (131). TNBC occurs in 45-70% of patients
(132). Several target antigens
for TNBC, such as chondroitin sulphate proteoglycan 4
(CSPG4), intracellular adhesion molecule-1 (ICAM-1),
natural killer group 2 member D ligand (NKG2DL), AXL,
tumour endothelial marker 8 (TEM8), integrin αVβ3, orphan
receptor 1 (ROR1), c-MET, folate receptor α
(FRα), EGFR, mesothelin, disialoganglioside
(GD2), mucin 1 (MUC1), and trophoblast cell-surface
antigen 2 (TROP2), have been identified (133) Atezolizumab, an anti-PD L1-based
immunotherapy, in combination with the chemotherapeutic drug
nab-paclitaxel, is used for TNBC treatment (134). Other promising target antigens
are currently in the preclinical stage. Based on the results of
nano-ultra performance liquid chromatography, five antigens have
been identified, namely, IL-32, syntenin-1, ribophorin-2,
proliferating cell nuclear antigen, and cofilin-1 (135). Targeting these tumour antigens
may replace chemotherapy treatments and serve as a future approach
for reprogramming CAR T-cells for TNBC treatment.
The remarkable success of these engineered chimeric
receptors in treating B-cell and haematological malignancies has
made them a considerable and promising therapeutic option for
B-cell tumours and haematological cancers. Despite being one of the
most advanced therapies against malignancies, CAR T-cell therapy
has several potential toxicities, including CRS, NT, on/off-target
tumour detection, and induction of anaphylaxis (159,160). Additionally, there are certain
challenges that arise during the treatment of solid tumours such as
BCa and TNBC, including antigen escape (161) and an immunosuppressive tumour
microenvironment (162).
CRS is an unfavourable inflammatory response that
can occur during CAR T-cell therapy and mAB infusions. CRS is
triggered by the activation of NK cells, B-lymphocytes,
T-lymphocytes, phagocytic cells, APCs, and certain endothelial
tissue matrix cells (163,164). CRS is witnessed following the
infusion of mAbs, IL2, and certain bispecific CAR T-cell
domains such as CD19-CD3 antibodies. The severity of CRS
depends on the tumour burden in the patient. For instance, a case
report by Teachey et al (165) noted that patients with
relapsed/refractory B-ALL who received CD19-specific CAR T-cell
immunotherapy experienced complications, including high levels of
CRS (19-43%).
Corticosteroids play a significant role in
reversing CRS symptoms without affecting the drug's anti-malignant
effect. However, it is worth noting that prolonged systemic
corticosteroid use for >14 days can have adverse effects on the
drug's anti-carcinogenic effects. To address this concern, the FDA
approved tocilizumab as a rapid reversal drug for CRS (166-169).
On/Off-target toxicity occurs when the intended
target carcinogen is primarily present in cancerous cells but also
binds to CAR T-cell target antigens in non-malignant tissues. This
toxicity has been observed in gastrointestinal and haematologic
organ systems (170). The first
instance of on/off-target toxicity was observed in renal cell
carcinoma patients who were treated with chimeric
antigen-recognising carbonic anhydrase IX (CAIX) (171).
Patients receiving CAR T-cell therapy for
CD19-specific neoplasms may experience focal neurological symptoms,
including aphasia, seizures, allodynia, and apraxia (174). Importantly, the severity of
neurological sequelae can be partially influenced by the cytokine
levels of the patient. For example, 78% of B-cell NHL patients who
received axicabtagene ciloleucel experienced NT, and 87% of B-ALL
patients treated with brexucabtagene autoleucel suffered from
neurologic toxicities. Studies have shown no clear correlation
between the proportion of modified and naturally occurring T-cells
and the presence of EEG abnormalities. It remains unclear whether
NT is specific to CD19-specific malignancies or can occur with
other antigens (175).
Anaphylaxis occurs when the body's natural defence
system overreacts and triggers an excessive inflammatory response.
The primary reason for anaphylaxis in CAR T-cell therapy is the use
of chimeric T-lymphocytic receptors derived from murine mAbs
(175,176). Mesothelin, a tumour-associated
antigen, is often overexpressed in malignancies such as malignant
pleural mesothelioma (MPM), pancreatic cancer, and ovarian cancer.
Preclinical models showed that multiple infusions of
anti-mesothelin and anti-CD19 RNA CAR T-cells had anti-tumour
effects. Based on this, human clinical trials were conducted
(NCT01355965) involving meso-RNA-CAR T therapy. However, multiple
meso-mRNA CAR T-cell infusions in a limited time frame resulted in
a patient experiencing an anaphylactic shock. To mitigate this
toxicity, T-cell infusion was terminated (177-179). The transfer of genetically
engineered T-cells requires careful monitoring, prompt recognition,
and immediate management of these side effects to reduce any
potential negative outcomes.
One major limitation when targeting BCa antigens is
the proper identification of the target antigen due to its low
expression levels in vital organs, which could lead to off-target
toxic effects (114). Another
toxic outcome when targeting these receptors on proliferating cells
in breast tissues is intratumor heterogeneity, which causes
resistance of tumour antigens to single target engineered
receptors, a phenomenon known as antigen escape. In antigen escape,
engineered CAR T-cells lose their efficacy against carcinogens. To
counteract this toxicity, several preclinical experiments have
combined dual antigens to target solid tumours, which eventually
improves treatment efficacy. For example, several tandem chimeric
receptors, including HER2 and MUC1 in BCa, have shown
enhanced anti-carcinogenic effects in preclinical models. Another
significant challenge of CAR T-cell therapy in solid tumours,
particularly in BCa, is associated with an immunosuppressive TME.
The TME is comprised of multiple immunosuppressive elements,
including carcinogenic cells, regulatory T-lymphocytes
(Treg), myeloid-derived suppressor cells (MDSCs),
cancer-associated fibroblasts, and tumour-associated macrophages
(TAMs). Additionally, various cytokines, chemokines, and
extracellular matrix components are integral parts of the TME that
help in regulating progression, angiogenesis, and metastasis by
providing necessary growth regulators, chemokines, interleukins,
transforming growth factor-β (TGF-β), indoleamine 2,3-dioxygenase
(IDO), and vascular endothelial growth factor. PD-1 and
CTLA-4 are other immunosuppressive checkpoint blockers that
affect chimeric-engineered T-receptors, hindering their
anti-carcinogenic reactions against solid tumours such as TNBC
(180-183). A strategy to combat the
immunosuppressive TME involves engineering armoured CARs that
release pro-inflammatory cytokines to favourably reshape
anti-carcinogenic responses. Interleukins such as IL-12 and
IL-18 are released to enhance anti-carcinogenic reactions by
IFNγ and Treg inhibition that triggers M1 macrophages
(184,185).
A significant barrier leading to on/off-target
tumour toxicity in solid tumours associated with TAAs is the
challenge of specifically targeting tumour cells. New CAR designs
are being developed with improved tumour selectivity and reduced
off-target effects. This includes the use of synthetic receptors
such as synNotch receptors to enhance the specificity of CAR
T-cells. Another hurdle is TME, in which solid tumours release
chemotactic cytokines such as CXCL1, CXCL12, and CXCL5,
which suppress T-cell activation (186,187). To overcome these challenges,
additional proteins such as armoured CAR T-cells are incorporated
into engineered receptors to withstand immunosuppressive responses
primarily found in TME to improve the eradication of tumours. The
incorporation of 'suicide genes' into CARs also provides an
opportunity to mitigate toxicity by deactivating the CAR T-cells
(188,189). TNBCs, which have historically
been challenging to treat and often rely on chemotherapy with low
survival rates, are now the focus of several combination therapies
in preliminary and interventional trials. Examples include
CDK7 with EGFR CAR therapy (190) and anti-PD-L1 with PARP
inhibitory therapy (190).
Researchers are continually exploring various methods to overcome
these obstacles, such as integrating CRISPR/Cas9 systems
into CAR immunotherapy for genome editing and the development of
universal CAR T-cells (190,191). Advances in multi-omics have
improved the ability to identify unique neoantigens resulting from
tumour-specific polymorphisms, potentially leading to more targeted
therapies with fewer adverse effects (192).
The potential developments in CAR T-cell therapy
are promising and marked by significant advancements. As technology
progresses, the field may witness increased safety measures,
innovative target identification, combination therapies, an
increased range of tools, and breakthroughs in manufacturing and
delivery. These developments are likely to shape the landscape of
precision immunotherapy, highlighting novel avenues for more
effective and personalised cancer treatments. The unwavering
dedication of researchers, healthcare professionals, and industry
stakeholders will pave the way for a future where CAR T-cell
immunotherapy proves highly effective in treating sarcomas,
ultimately improving patient outcomes (193).
Overall, the potential of CAR T-cells in treating
solid tumours, including BCa and TNBC, has yielded promising
results in clinical trials. The label of being 'difficult to treat'
for TNBCs may soon be erased through the effective outcomes
achievable with these engineered receptors.
Not applicable.
MZN, S, wrote the major parts of the manuscript and
prepared the figures and tables. CD and SG revised the manuscript.
HEME, FHK, AAE and NT performed the bibliographic research and
prepared the table and figures. MAK conceptualized the study and
oversaw the process. All authors helped to write the manuscript.
All authors have read and approved the final manuscript. Data
authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
This research has been funded by Scientific Research Deanship at
University of Ha'il-Saudi Arabia through project number MDR-22
038.
|
1
|
National Cancer Institute: What is cancer?
2021, https://www.cancer.gov/about-cancer/understanding/what-is-cancer.
|
|
2
|
Blackadar CB: Historical review of the
causes of cancer. World J Clin Oncol. 7:54–86. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Debela DT, Muzazu SG, Heraro KD, Ndalama
MT, Mesele BW, Haile DC, Kitui SK and Manyazewal T: New approaches
and procedures for cancer treatment: Current perspectives. SAGE
Open Med. 9:205031212110343662021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Decker WK and Safdar A: Bioimmunoadjuvants
for the treatment of neoplastic and infectious disease: Coley's
legacy revisited. Cytokine Growth Factor Rev. 20:271–281. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Valent P, Groner B, Schumacher U,
Superti-Furga G, Busslinger M, Kralovics R, Zielinski C, Penninger
JM, Kerjaschki D, Stingl G, et al: Paul Ehrlich (1854-1915) and his
contributions to the foundation and birth of translational
medicine. J Innate Immun. 8:111–120. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rosenberg SA: IL-2: The first effective
immunotherapy for human cancer. J Immunol. 192:5451–5458. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pierpont TM, Limper CB and Richards KL:
Past, present, and future of rituximab-the world's first oncology
monoclonal antibody therapy. Front Oncol. 8:163. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
June CH, O'Connor RS, Kawalekar OU,
Ghassemi S and Milone MC: CAR T cell immunotherapy for human
cancer. Science. 359:1361–1365. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rosenberg SA, Restifo NP, Yang JC, Morgan
RA and Dudley ME: Adoptive cell transfer: A clinical path to
effective cancer immunotherapy. Nat Rev Cancer. 8:299–308. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gross G, Waks T and Eshhar Z: Expression
of immunoglobulin-T-cell receptor chimeric molecules as functional
receptors with antibody-type specificity. Proc Natl Acad Sci USA.
86:10024–10028. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Graham C, Hewitson R, Pagliuca A and
Benjamin R: Cancer immunotherapy with CAR-T cells-behold the
future. Clin Med (Lond). 18:324–328. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Maus MV: A decade of CAR T cell evolution.
Nat Cancer. 3:270–271. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cameron BJ, Gerry AB, Dukes J, Harper JV,
Kannan V, Bianchi FC, Grand F, Brewer JE, Gupta M, Plesa G, et al:
Identification of a Titin-derived HLA-A1-presented peptide as a
cross-reactive target for engineered MAGE A3-directed T cells. Sci
Transl Med. 5:197ra1032013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
June CH, Riddell SR and Schumacher TN:
Adoptive cellular therapy: A race to the finish line. Sci Transl
Med. 7:280ps72015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee YH and Kim CH: Evolution of chimeric
antigen receptor (CAR) T cell therapy: current status and future
perspectives. Arch Pharm Res. 42:607–616. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Titov A, Valiullina A, Zmievskaya E,
Zaikova E, Petukhov A, Miftakhova R, Bulatov E and Rizvanov A:
Advancing CAR T-cell therapy for solid tumors: Lessons learned from
lymphoma treatment. Cancers (Basel). 12:1252020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jayaraman J, Mellody MP, Hou AJ, Desai RP,
Fung AW, Pham AHT, Chen YY and Zhao W: CAR-T design: Elements and
their synergistic function. EBioMedicine. 58:1029312020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xie YJ, Dougan M, Jailkhani N, Ingram J,
Fang T, Kummer L, Momin N, Pishesha N, Rickelt S, Hynes RO and
Ploegh H: Nanobody-based CAR T cells that target the tumor
microenvironment inhibit the growth of solid tumors in
immunocompetent mice. Proc Natl Acad Sci USA. 116:7624–7631. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Barber A, Rynda A and Sentman CL: Chimeric
NKG2D expressing T cells eliminate immunosuppression and activate
immunity within the ovarian tumor microenvironment. J Immunol.
183:6939–6947. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lynn RC, Feng Y, Schutsky K, Poussin M,
Kalota A, Dimitrov DS and Powell DJ Jr: High-affinity FRβ-specific
CAR T cells eradicate AML and normal myeloid lineage without HSC
toxicity. Leukemia. 30:1355–1364. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guest RD, Hawkins RE, Kirillova N, Cheadle
EJ, Arnold J, O'Neill A, Irlam J, Chester KA, Kemshead JT, Shaw DM,
et al: The role of extracellular spacer regions in the optimal
design of chimeric immune receptors: Evaluation of four different
scFvs and antigens. J Immunother. 28:203–211. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hudecek M, Sommermeyer D, Kosasih PL,
Silva-Benedict A, Liu L, Rader C, Jensen MC and Riddell SR: The
nonsignaling extracellular spacer domain of chimeric antigen
receptors is decisive for in vivo antitumor activity. Cancer
Immunol Res. 3:125–135. 2015. View Article : Google Scholar
|
|
23
|
Zhang C, Liu J, Zhong JF and Zhang X:
Engineering CAR-T cells. Biomark Res. 5:222017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sterner RC and Sterner RM: CAR-T cell
therapy: Current limitations and potential strategies. Blood Cancer
J. 11:692021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mohanty R, Chowdhury CR, Arega S, Sen P,
Ganguly P and Ganguly N: CAR T cell therapy: A new era for cancer
treatment (Review). Oncol Rep. 42:2183–2195. 2019.PubMed/NCBI
|
|
26
|
Zhang Q, Ping J, Huang Z, Zhang X, Zhou J,
Wang G, Liu S and Ma J: CAR-T cell therapy in cancer: Tribulations
and road ahead. J Immunol Res. 2020:19243792020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Louis CU, Savoldo B, Dotti G, Pule M, Yvon
E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, et al: Antitumor
activity and long-term fate of chimeric antigen receptor-positive T
cells in patients with neuroblastoma. Blood. 118:6050–6056. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Duong CP, Yong CS, Kershaw MH, Slaney CY
and Darcy PK: Cancer immunotherapy utilizing gene-modified T cells:
From the bench to the clinic. Mol Immunol. 67:46–57. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Qian L, Li D, Ma L, He T, Qi F, Shen J and
Lu XA: The novel anti-CD19 chimeric antigen receptors with
humanized scFv (single-chain variable fragment) trigger leukemia
cell killing. Cell Immunol. 304:49–54. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lock D, Mockel-Tenbrinck N, Drechsel K,
Barth C, Mauer D, Schaser T, Kolbe C, Al Rawashdeh W, Brauner J,
Hardt O, et al: Automated manufacturing of potent CD20-directed
chimeric antigen receptor t cells for clinical use. Hum Gene Ther.
28:914–925. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhao Z, Condomines M, van der Stegen SJC,
Perna F, Kloss CC, Gunset G, Plotkin J and Sadelain M: Structural
design of engineered costimulation determines tumor rejection
kinetics and persistence of CAR T cells. Cancer Cell. 28:415–428.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hombach A, Hombach AA and Abken H:
Adoptive immunotherapy with genetically engineered T cells:
Modification of the IgG1 Fc 'spacer' domain in the extracellular
moiety of chimeric antigen receptors avoids 'off-target'activation
and unintended initiation of an innate immune response. Gene Ther.
17:1206–1213. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhong XS, Matsushita M, Plotkin J, Riviere
I and Sadelain M: Chimeric antigen receptors combining 4-1BB and
CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and
CD8+ T cell-mediated tumor eradication. Mol Ther. 18:413–420. 2010.
View Article : Google Scholar
|
|
34
|
Quintarelli C, Orlando D, Boffa I, Guercio
M, Polito VA, Petretto A, Lavarello C, Sinibaldi M, Weber G, Del
Bufalo F, et al: Choice of costimulatory domains and of cytokines
determines CAR T-cell activity in neuroblastoma. Oncoimmunology.
7:e14335182018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Abate-Daga D, Lagisetty KH, Tran E, Zheng
Z, Gattinoni L, Yu Z, Burns WR, Miermont AM, Teper Y, Rudloff U, et
al: A novel chimeric antigen receptor against prostate stem cell
antigen mediates tumor destruction in a humanized mouse model of
pancreatic cancer. Hum Gene Ther. 25:1003–1012. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pulè MA, Straathof KC, Dotti G, Heslop HE,
Rooney CM and Brenner MK: A chimeric T cell antigen receptor that
augments cytokine release and supports clonal expansion of primary
human T cells. Mol Ther. 12:933–941. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Beatty GL and Moon EK: Chimeric antigen
receptor T cells are vulnerable to immunosuppressive mechanisms
present within the tumor microenvironment. Oncoimmunology.
3:e9700272014. View Article : Google Scholar
|
|
38
|
Chmielewski M and Abken H: TRUCKs: The
fourth generation of CARs. Expert Opin Biol Ther. 15:1145–1154.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kueberuwa G, Kalaitsidou M, Cheadle E,
Hawkins RE and Gilham DE: CD19 CAR T cells expressing IL-12
eradicate lymphoma in fully lymphoreplete mice through induction of
host immunity. Mol Ther Oncolytics. 8:41–51. 2017. View Article : Google Scholar
|
|
40
|
John LB, Devaud C, Duong CP, Yong CS,
Beavis PA, Haynes NM, Chow MT, Smyth MJ, Kershaw MH and Darcy PK:
Anti-PD-1 antibody therapy potently enhances the eradication of
established tumors by gene-modified T cells. Clin Cancer Res.
19:5636–5646. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim DW and Cho JY: Recent advances in
allogeneic CAR-T cells. Biomolecules. 10:2632020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kagoya Y, Tanaka S, Guo T, Anczurowski M,
Wang CH, Saso K, Butler MO, Minden MD and Hirano N: A novel
chimeric antigen receptor containing a JAK-STAT signaling domain
mediates superior antitumor effects. Nat Med. 24:352–359. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dai H, Wang Y, Lu X and Han W: Chimeric
antigen receptors modified T-cells for cancer therapy. J Natl
Cancer Inst. 108:djv4392016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li D, Li X, Zhou WL, Huang Y, Liang X,
Jiang L, Yang X, Sun J, Li Z, Han WD and Wang W: Genetically
engineered T cells for cancer immunotherapy. Signal Transduct
Target Ther. 4:352019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Levine BL, Miskin J, Wonnacott K and Keir
C: Global manufacturing of CAR T cell therapy. Mol Ther Methods
Clin Dev. 4:92–101. 2016. View Article : Google Scholar
|
|
46
|
Benmebarek MR, Karches CH, Cadilha BL,
Lesch S, Endres S and Kobold S: Killing mechanisms of chimeric
antigen receptor (CAR) T cells. Int J Mol Sci. 20:12832019.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Guo S and Deng CX: Effect of stromal cells
in tumor microenvironment on metastasis initiation. Int J Biol Sci.
14:2083–2093. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Morton JJ, Bird G, Keysar SB, Astling DP,
Lyons TR, Anderson RT, Glogowska MJ, Estes P, Eagles JR, Le PN, et
al: XactMice: Humanizing mouse bone marrow enables microenvironment
reconstitution in a patient-derived xenograft model of head and
neck cancer. Oncogene. 35:290–300. 2016. View Article : Google Scholar
|
|
49
|
Najima Y, Tomizawa-Murasawa M, Saito Y,
Watanabe T, Ono R, Ochi T, Suzuki N, Fujiwara H, Ohara O, Shultz
LD, et al: Induction of WT1-specific human CD8+ T cells from human
HSCs in HLA class I Tg NOD/SCID/IL2rgKO mice. Blood. 127:722–734.
2016. View Article : Google Scholar :
|
|
50
|
Yin L and Wang XJ, Chen DX, Liu XN and
Wang XJ: Humanized mouse model: A review on preclinical
applications for cancer immunotherapy. Am J Cancer Res.
10:4568–4584. 2020.
|
|
51
|
Zitvogel L, Pitt JM, Daillère R, Smyth MJ
and Kroemer G: Mouse models in on immunology. Nat Rev Cancer.
16:759–773. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Slabik C, Kalbarczyk M, Danisch S, Zeidler
R, Klawonn F, Volk V, Krönke N, Feuerhake F, Ferreira de Figueiredo
C, Blasczyk R, et al: CAR-T cells targeting Epstein-Barr virus
gp350 validated in a humanized mouse model of EBV infection and
lymphoproliferative disease. Mol Ther Oncolytics. 18:504–524. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Jin CH, Xia J, Rafiq S, Huang X, Hu Z,
Zhou X, Brentjens RJ and Yang YG: Modeling anti-CD19 CAR T cell
therapy in humanized mice with human immunity and autologous
leukemia. EBioMedicine. 39:173–181. 2019. View Article : Google Scholar :
|
|
54
|
Gulati P, Rühl J, Kannan A, Pircher M,
Schuberth P, Nytko KJ, Pruschy M, Sulser S, Haefner M, Jensen S, et
al: Aberrant Lck signal via CD28 costimulation augments
antigen-specific functionality and tumor control by redirected T
cells with PD-1 blockade in humanized mice. Clin Cancer Res.
24:3981–3993. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Roskoski R Jr: The ErbB/HER family of
protein-tyrosine kinases and cancer. Pharmacol Res. 79:34–74. 2014.
View Article : Google Scholar
|
|
56
|
Szöőr Á, Tóth G, Zsebik B, Szabó V, Eshhar
Z, Abken H and Vereb G: Trastuzumab derived HER2-specific CARs for
the treatment of trastuzumab-resistant breast cancer: CAR T cells
penetrate and eradicate tumors that are not accessible to
antibodies. Cancer Lett. 484:1–8. 2020. View Article : Google Scholar
|
|
57
|
Liu Y, Zhou Y, Huang KH, Li Y, Fang X, An
L, Wang F, Chen Q, Zhang Y, Shi A, et al: EGFR-specific CAR-T cells
trigger cell lysis in EGFR-positive TNBC. Aging (Albany NY).
11:11054–11072. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Corti C, Venetis K, Sajjadi E, Zattoni L,
Curigliano G and Fusco N: CAR-T cell therapy for triple-negative
breast cancer and other solid tumors: Preclinical and clinical
progress. Expert Opin Investig Drugs. 31:593–605. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Wei J, Sun H, Zhang A, Wu X, Li Y, Liu J,
Duan Y, Xiao F, Wang H, Lv M, et al: A novel AXL chimeric antigen
receptor endows T cells with anti-tumor effects against triple
negative breast cancers. Cell Immunol. 331:49–58. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wallstabe L, Mades A, Frenz S, Einsele H,
Rader C and Hudecek M: CAR T cells targeting
αvβ3 integrin are effective against advanced
cancer in preclinical models. Adv Cell Gene Ther. 1:e112018.
View Article : Google Scholar
|
|
61
|
Zhao X, Qu J, Hui Y, Zhang H, Sun Y, Liu
X, Zhao X, Zhao Z, Yang Q, Wang F and Zhang S: Clinicopathological
and prognostic significance of c-Met overexpression in breast
cancer. Oncotarget. 8:56758–56767. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Han Y, Xie W, Song DG and Powell DJ Jr:
Control of triple-negative breast cancer using ex vivo
self-enriched, costimulated NKG2D CAR T cells. J Hematol Oncol.
11:922018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhou R, Yazdanifar M, Roy LD, Whilding LM,
Gavrill A, Maher J and Mukherjee P: CAR T cells targeting the tumor
MUC1 glycoprotein reduce triple-negative breast cancer growth.
Front Immunol. 10:11492019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wallstabe L, Göttlich C, Nelke LC,
Kühnemundt J, Schwarz T, Nerreter T, Einsele H, Walles H, Dandekar
G, Nietzer SL and Hudecek M: ROR1-CAR T cells are effective against
lung and breast cancer in advanced microphysiologic 3D tumor
models. JCI Insight. 4:e1263452019. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zhao Z, Li Y, Liu W and Li X: Engineered
IL-7 receptor enhances the therapeutic effect of AXL-CAR-T cells on
triple-negative breast cancer. Biomed Res Int.
2020:47951712020.PubMed/NCBI
|
|
66
|
Caratelli S, Arriga R, Sconocchia T,
Ottaviani A, Lanzilli G, Pastore D, Cenciarelli C, Venditti A, Del
Principe MI, Lauro D, et al: In vitro elimination of epidermal
growth factor receptor-overexpressing cancer cells by
CD32A-chimeric receptor T cells in combination with cetuximab or
panitumumab. Int J Cancer. 146:236–247. 2020. View Article : Google Scholar
|
|
67
|
Song DG, Ye Q, Poussin M, Chacon JA,
Figini M and Powell DJ Jr: Effective adoptive immunotherapy of
triple-negative breast cancer by folate receptor-alpha redirected
CAR T cells is influenced by surface antigen expression level. J
Hematol Oncol. 9:562016. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Seitz CM, Schroeder S, Knopf P, Krahl AC,
Hau J, Schleicher S, Martella M, Quintanilla-Martinez L, Kneilling
M, Pichler B, et al: GD2-targeted chimeric antigen receptor T cells
prevent metastasis formation by elimination of breast cancer
stem-like cells. Oncoimmunology. 9:16833452019. View Article : Google Scholar
|
|
69
|
Yang Y, Vedvyas Y, McCloskey JE, Min IM
and Jin MM: ICAM-1 targeting CAR T cell therapy for triple negative
breast cancer. Cancer Res. 79(13 Suppl): S23222019. View Article : Google Scholar
|
|
70
|
Hu W, Zi Z, Jin Y, Li G, Shao K, Cai Q, Ma
X and Wei F: CRISPR/Cas9-mediated PD-1 disruption enhances human
mesothelin-targeted CAR T cell effector functions. Cancer Immunol
Immunother. 68:365–377. 2019. View Article : Google Scholar
|
|
71
|
Petrovic K, Robinson J, Whitworth K, Jinks
E, Shaaban A and Lee SP: TEM8/ANTXR1-specific CAR T cells mediate
toxicity in vivo. PLoS One. 14:e02240152019. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Byrd TT, Fousek K, Pignata A, Szot C,
Samaha H, Seaman S, Dobrolecki L, Salsman VS, Oo HZ, Bielamowicz K,
et al: TEM8/ANTXR1-specific CAR T cells as a targeted therapy for
triple-negative breast cancer. Cancer Res. 78:489–500. 2018.
View Article : Google Scholar :
|
|
73
|
Bedoya DM, King T and Posey AD: Generation
of CART cells targeting oncogenic TROP2 for the elimination of
epithelial malignancies. Cytotherapy. 21(Suppl): S11–S12. 2019.
View Article : Google Scholar
|
|
74
|
National Library of Medicine (NLM):
Genetically Modified T-cells in Treating Patients With Recurrent or
Refractory Malignant Glioma. ClinicalTrials.gov ID, NCT02208362.
NLM; Bethesda, MD: 2015, https://clinicaltrials.gov/study/NCT02208362.
|
|
75
|
National Library of Medicine (NLM):
CART-EGFRvIII + Pembrolizumab in GBM. ClinicalTrials.gov ID,
NCT03726515. NLM; Bethesda, MD: 2019, https://clinicaltrials.gov/ct2/show/NCT03726515.
|
|
76
|
National Library of Medicine (NLM):
GPC3-CAR-T Cells for Immunotherapy of Cancer With GPC3 Expression.
ClinicalTrials.gov ID, NCT03198546. NLM; Bethesda, MD: 2017,
https://clinicaltrials.gov/ct2/show/NCT03198546.
|
|
77
|
National Library of Medicine (NLM): Her2
Chimeric Antigen Receptor Expressing T Cells in Advanced Sarcoma.
ClinicalTrials.gov ID, NCT00902044. NLM; Bethesda, MD: 2010,
https://clinicaltrials.gov/ct2/show/NCT00902044.
|
|
78
|
National Library of Medicine (NLM):
CEA-Expressing Liver Metastases Safety Study of Intrahepatic
Infusions of Anti-CEA Designer T Cells (HITM). ClinicalTrials.gov
ID, NCT01373047. NLM; Bethesda, MD: 2011, https://clinicaltrials.gov/ct2/show/NCT01373047.
|
|
79
|
National Library of Medicine (NLM):
Autologous Redirected RNA Meso CAR T Cells for Pancreatic Cancer.
ClinicalTrials.gov ID, NCT01897415. NLM; Bethesda, MD: 2013,
https://clinicaltrials.gov/ct2/show/NCT01897415.
|
|
80
|
National Library of Medicine (NLM): CAR T
Cell Immunotherapy for Pancreatic Cancer. ClinicalTrials.gov ID,
NCT03323944. NLM; Bethesda, MD: 2017, https://clinicaltrials.gov/ct2/show/NCT03323944.
|
|
81
|
National Library of Medicine (NLM):
Clinical Study of CAR-CLD18 T Cells in Patients With Advanced
Gastric Adenocarcinoma and Pancreatic Adenocarcinoma.
ClinicalTrials.gov ID, NCT03159819. NLM; Bethesda, MD: 2017,
https://clinicaltrials.gov/ct2/show/NCT03159819.
|
|
82
|
National Library of Medicine (NLM): Safety
and Efficacy of CCT301 CAR-T in Adult Subjects With Recurrent or
Refractory Stage IV Renal Cell Carcinoma. ClinicalTrials.gov ID,
NCT03393936. NLM; Bethesda, MD: 2018, https://clinicaltrials.gov/ct2/show/NCT03393936.
|
|
83
|
National Library of Medicine (NLM):
PSCA-CAR T Cells in Treating Patients With PSCA+ Metastatic
Castration Resistant Prostate Cancer. ClinicalTrials.gov ID,
NCT03873805. NLM; Bethesda, MD: 2019, https://clinicaltrials.gov/ct2/show/NCT03873805.
|
|
84
|
National Library of Medicine (NLM):
CART-PSMA-TGFβRDN Cells for Castrate-Resistant Prostate Cancer.
ClinicalTrials.gov ID, NCT03089203. NLM; Bethesda, MD: 2017,
https://clinicaltrials.gov/ct2/show/NCT03089203.
|
|
85
|
National Library of Medicine (NLM):
Autologous huMNC2-CAR44 or huMNC2-CAR22 T Cells for Breast Cancer
Targeting Cleaved Form of MUC1 (MUC1*). ClinicalTrials.gov ID,
NCT04020575. NLM; Bethesda, MD: 2020, https://clinicaltrials.gov/ct2/show/NCT04020575.
|
|
86
|
National Library of Medicine (NLM):
MOv19-BBz CAR T Cells in aFR Expressing Recurrent High Grade Serous
Ovarian, Fallopian Tube, or Primary Peritoneal Cancer.
ClinicalTrials.gov ID, NCT03585764. NLM; Bethesda, MD: 2018,
https://clinicaltrials.gov/ct2/show/NCT03585764.
|
|
87
|
National Library of Medicine (NLM):
Cyclophosphamide Followed by Intravenous and Intraperitoneal
Infusion of Autologous T Cells Genetically Engineered to Secrete
IL-12 and to Target the MUC16ecto Antigen in Patients With
Recurrent MUC16ecto+ Solid Tumors. ClinicalTrials.gov ID,
NCT02498912. NLM; Bethesda, MD: 2015, https://clinicaltrials.gov/ct2/show/NCT02498912.
|
|
88
|
National Library of Medicine (NLM): T-Cell
Therapy for Advanced Breast Cancer. ClinicalTrials.gov ID,
NCT02792114. NLM; Bethesda, MD: 2016, https://clinicaltrials.gov/ct2/show/NCT02792114.
|
|
89
|
National Library of Medicine (NLM): T
Cells Expressing HER2-specific Chimeric Antigen Receptors(CAR) for
Patients With HER2-Positive CNS Tumors (iCAR). ClinicalTrials.gov
ID, NCT02442297. NLM; Bethesda, MD: 2016, https://clinicaltrials.gov/ct2/show/NCT02442297.
|
|
90
|
National Library of Medicine (NLM):
HER2-CAR T Cells in Treating Patients With Recurrent Brain or
Leptomeningeal Metastases. ClinicalTrials.gov ID, NCT03696030. NLM;
Bethesda, MD: 2018, https://clinicaltrials.gov/ct2/show/NCT03696030.
|
|
91
|
National Library of Medicine (NLM):
Malignant Pleural Disease Treated With Autologous T Cells
Genetically Engineered to Target the Cancer-Cell Surface Antigen
Mesothelin. ClinicalTrials.gov ID, NCT02414269. NLM; Bethesda, MD:
2015, https://clinicaltrials.gov/ct2/show/NCT02414269.
|
|
92
|
National Library of Medicine (NLM):
Precursor B Cell Acute Lymphoblastic Leukemia (B-ALL) Treated With
Autologous T Cells Genetically Targeted to the B Cell Specific
Antigen CD19. ClinicalTrials.gov ID, NCT01044069. NLM; Bethesda,
MD: 2010, https://clinicaltrials.gov/ct2/show/NCT01044069.
|
|
93
|
National Library of Medicine (NLM):
Treatment of Relapsed or Chemotherapy Refractory Chronic
Lymphocytic Leukemia or Indolent B Cell Lymphoma Using Autologous T
Cells Genetically Targeted to the B Cell Specific Antigen CD19.
ClinicalTrials.gov ID, NCT00466531. NLM; Bethesda, MD: 2007,
https://clinicaltrials.gov/ct2/show/NCT00466531.
|
|
94
|
National Library of Medicine (NLM): CD19
Chimeric Receptor Expressing T Lymphocytes In B-Cell Non Hodgkin's
Lymphoma, ALL & CLL (CRETI-NH). ClinicalTrials.gov ID,
NCT00586391. NLM; Bethesda, MD: 2009, https://clinicaltrials.gov/ct2/show/NCT00586391.
|
|
95
|
National Library of Medicine (NLM): CD19
Chimeric Receptor Expressing T Lymphocytes In B-Cell Non Hodgkin's
Lymphoma, ALL & CLL (CRETI-NH). ClinicalTrials.gov ID,
NCT00608270. NLM; Bethesda, MD: 2009, https://clinicaltrials.gov/ct2/show/NCT00608270.
|
|
96
|
National Library of Medicine (NLM):
Anti-CD22 Chimeric Receptor T Cells in Pediatric and Young Adults
With Recurrent or Refractory CD22-expressing B Cell Malignancies.
ClinicalTrials.gov ID, NCT02315612. NLM; Bethesda, MD: 2014,
https://clinicaltrials.gov/ct2/show/NCT02315612.
|
|
97
|
National Library of Medicine (NLM):
Re-directed T Cells for the Treatment (FAP)-Positive Malignant
Pleural Mesothelioma. ClinicalTrials.gov ID, NCT01722149. NLM;
Bethesda, MD: 2015, https://clinicaltrials.gov/ct2/show/NCT01722149.
|
|
98
|
National Library of Medicine (NLM):
Engineered Neuroblastoma Cellular Immunotherapy (ENCIT)-01.
ClinicalTrials.gov ID, NCT02311621. NLM; Bethesda, MD: 2014,
https://clinicaltrials.gov/ct2/show/NCT02311621.
|
|
99
|
National Library of Medicine (NLM): Study
of bb21217 in Multiple Myeloma. ClinicalTrials.gov ID, NCT03274219.
NLM; Bethesda, MD: 2017, https://clinicaltrials.gov/ct2/show/NCT03274219.
|
|
100
|
National Library of Medicine (NLM):
Kappa-CD28 T Lymphocytes, Chronic Lymphocytic Leukemia, B-cell
Lymphoma or Multiple Myeloma, CHARKALL (CHARKALL).
ClinicalTrials.gov ID, NCT00881920. NLM; Bethesda, MD: 2009,
https://clinicaltrials.gov/ct2/show/NCT00881920.
|
|
101
|
National Library of Medicine (NLM):
KSafety and Efficacy of ALLO-501 Anti-CD19 Allogeneic CAR T Cells
in Adults With Relapsed/Refractory Large B Cell or Follicular
Lymphoma (ALPHA). ClinicalTrials.gov ID, NCT03939026. NLM;
Bethesda, MD: 2019, https://clinicaltrials.gov/ct2/show/NCT03939026.
|
|
102
|
National Library of Medicine (NLM):
Dose-escalation, Dose-expansion Study of Safety of PBCAR0191 in
Patients With r/r NHL and r/r B-cell ALL. ClinicalTrials.gov ID,
NCT03666000. NLM; Bethesda, MD: 2019, https://clinicaltrials.gov/ct2/show/NCT03666000.
|
|
103
|
National Library of Medicine (NLM): A
Safety and Efficacy Study Evaluating CTX110 in Subjects With
Relapsed or Refractory B-Cell Malignancies (CARBON).
ClinicalTrials.gov ID, NCT04035434. NLM; Bethesda, MD: 2019,
https://clinicaltrials.gov/ct2/show/NCT04035434.
|
|
104
|
National Library of Medicine (NLM): Study
Evaluating Safety and Efficacy of UCART123 in Patients With
Relapsed/Refractory Acute Myeloid Leukemia (AMELI-01).
ClinicalTrials.gov ID, NCT03190278. NLM; Bethesda, MD: 2017,
https://clinicaltrials.gov/ct2/show/NCT03190278.
|
|
105
|
National Library of Medicine (NLM): Safety
and Efficacy of ALLO-715 BCMA Allogenic CAR T Cells in in Adults
With Relapsed or Refractory Multiple Myeloma (UNIVERSAL)
(UNIVERSAL). ClinicalTrials.gov ID, NCT04093596. NLM; Bethesda, MD:
2019, https://clinicaltrials.gov/ct2/show/NCT04093596.
|
|
106
|
National Library of Medicine (NLM):
SaalloSHRINK - Standard cHemotherapy Regimen and Immunotherapy With
Allogeneic NKG2D-based CYAD-101 Chimeric Antigen Receptor T-cells
(alloSHRINK). ClinicalTrials.gov ID, NCT03692429. NLM; Bethesda,
MD: 2018, https://clinicaltrials.gov/ct2/show/NCT03692429.
|
|
107
|
National Library of Medicine (NLM):
Adoptive Transfer of Autologous T Cells Targeted to Prostate
Specific Membrane Antigen (PSMA) for the Treatment of Castrate
Metastatic Prostate Cancer (CMPC). ClinicalTrials.gov ID,
NCT01140373. NLM; Bethesda, MD: 2010, https://clinicaltrials.gov/ct2/show/NCT01140373.
|
|
108
|
National Library of Medicine (NLM): 3rd
Generation GD-2 Chimeric Antigen Receptor and iCaspase Suicide
Safety Switch, Neuroblastoma, GRAIN (GRAIN). ClinicalTrials.gov ID,
NCT01822652. NLM; Bethesda, MD: 2013, https://clinicaltrials.gov/ct2/show/NCT01822652.
|
|
109
|
National Library of Medicine (NLM):
iC9-GD2-CARVZV-CTLs/Refractory or Metastatic GD2-positive Sarcoma
and Neuroblastoma (VEGAS). ClinicalTrials.gov ID, NCT01953900. NLM;
Bethesda, MD: 2014, https://clinicaltrials.gov/ct2/show/NCT01953900.
|
|
110
|
Almond LM, Charalampakis M, Ford SJ,
Gourevitch D and Desai A: Myeloid sarcoma: Presentation, diagnosis,
and treatment. Clin Lymphoma Myeloma Leuk. 17:263–267. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Locke FL, Neelapu SS, Bartlett NL, Siddiqi
T, Chavez JC, Hosing CM, Ghobadi A, Budde LE, Bot A, Rossi JM, et
al: Phase 1 results of ZUMA-1: A multicenter study of KTE-C19
anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol
Ther. 25:285–295. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Jain MD, Bachmeier CA, Phuoc VH and Chavez
JC: Axicabtagene ciloleucel (KTE-C19), an anti-CD19 CAR T therapy
for the treatment of relapsed/refractory aggressive B-cell
non-Hodgkin's lymphoma. Ther Clin Risk Manag. 14:1007–1017. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Abramson JS, Palomba ML, Gordon LI,
Lunning MA, Wang M, Arnason J, Mehta A, Purev E, Maloney DG,
Andreadis C, et al: Lisocabtagene maraleucel for patients with
relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001):
A multicentre seamless design study. Lancet. 396:839–852. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Abbott RC, Cross RS and Jenkins MR:
Finding the keys to the CAR: Identifying novel target antigens for
T cell redirection immunotherapies. Int J Mol Sci. 21:5152020.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Posey AD Jr, Schwab RD, Boesteanu AC,
Steentoft C, Mandel U, Engels B, Stone JD, Madsen TD, Schreiber K,
Haines KM, et al: Engineered CAR T cells targeting the
cancer-associated Tn-glycoform of the membrane mucin MUC1 control
adenocarcinoma. Immunity. 44:1444–1454. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Li X, Ding Y, Zi M, Sun L, Zhang W, Chen S
and Xu Y: CD19, from bench to bedside. Immunol Lett. 183:86–95.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Ahmed N, Brawley VS, Hegde M, Robertson C,
Ghazi A, Gerken C, Liu E, Dakhova O, Ashoori A, Corder A, et al:
Human epidermal growth factor receptor 2 (HER2)-specific chimeric
antigen receptor-modified T cells for the immunotherapy of
HER2-positive sarcoma. J Clin Oncol. 33:16882015. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Cortesi L, Rugo HS and Jackisch C: An
overview of PARP inhibitors for the treatment of breast cancer.
Target Oncol. 16:255–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Ye F, Dewanjee S, Li Y, Jha NK, Chen ZS,
Kumar A, Vishakha, Behl T, Jha SK and Tang H: Advancements in
clinical aspects of targeted therapy and immunotherapy in breast
cancer. Mol Cancer. 22:1052023. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Schoninger SF and Blain SW: The ongoing
search for biomarkers of CDK4/6 inhibitor responsiveness in breast
cancer. Mol Cancer Ther. 19:3–12. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Martorana F, Motta G, Pavone G, Motta L,
Stella S, Vitale SR, Manzella L and Vigneri P: AKT inhibitors: New
weapons in the fight against breast cancer? Front Pharmacol.
12:6622322021. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Goutsouliak K, Veeraraghavan J, Sethunath
V, De Angelis C, Osborne CK, Rimawi MF and Schiff R: Towards
personalized treatment for early stage HER2-positive breast cancer.
Nat Rev Clin Oncol. 17:233–250. 2020. View Article : Google Scholar
|
|
123
|
Tóth G, Szöllősi J, Abken H, Vereb G and
Szöőr Á: A small number of HER2 redirected CAR T cells
significantly improves immune response of adoptively transferred
mouse lymphocytes against human breast cancer xenografts. Int J Mol
Sci. 21:10392020. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Toulouie S, Johanning G and Shi Y:
Chimeric antigen receptor T-cell immunotherapy in breast cancer:
Development and challenges. J Cancer. 12:1212–1219. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Chaffer CL and Weinberg RA: A perspective
on cancer cell metastasis. Science. 331:1559–1564. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Carter P, Presta L, Gorman CM, Ridgway JB,
Henner D, Wong WL, Rowland AM, Kotts C, Carver ME and Shepard HM:
Humanization of an anti-p185HER2 antibody for human cancer therapy.
Proc Natl Acad Sci USA. 89:4285–4289. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Bou-Dargham MJ, Draughon S, Cantrell V,
Khamis ZI and Sang QA: Advancements in human breast cancer targeted
therapy and immunotherapy. J Cancer. 12:6949–6963. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Rivas SR, Valdez MJM, Govindarajan V,
Seetharam D, Doucet-O'Hare TT, Heiss JD and Shah AH: The role of
HERV-K in cancer stemness. Viruses. 14:20192022. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Zhao J, Rycaj K, Geng S, Li M, Plummer JB,
Yin B, Liu H, Xu X, Zhang Y, Yan Y, et al: Expression of human
endogenous retrovirus type K envelope protein is a novel candidate
prognostic marker for human breast cancer. Genes Cancer. 2:914–922.
2011. View Article : Google Scholar
|
|
130
|
Pegram M and Slamon D: Biological
rationale for HER2/neu (c-erbB2) as a target for monoclonal
antibody therapy. Semin Oncol. 27(Suppl 9): S13–S19. 2000.
|
|
131
|
Walsh EM, Keane MM, Wink DA, Callagy G and
Glynn SA: Review of triple negative breast cancer and the impact of
inducible nitric oxide synthase on tumor biology and patient
outcomes. Crit Rev Oncog. 21:333–351. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Tsutsui S, Ohno S, Murakami S, Hachitanda
Y and Oda S: Prognostic value of epidermal growth factor receptor
(EGFR) and its relationship to the estrogen receptor status in 1029
patients with breast cancer. Breast Cancer Res Treat. 71:67–75.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Nasiri F, Kazemi M, Mirarefin SMJ,
Mahboubi Kancha M, Ahmadi Najafabadi M, Salem F, Dashti Shokoohi S,
Evazi Bakhshi S and Safarzadeh Kozani P and Safarzadeh Kozani P:
CAR-T cell therapy in triple-negative breast cancer: Hunting the
invisible devil. Front Immunol. 13:10187862022. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Pantelidou C, Sonzogni O, De Oliveria
Taveira M, Mehta AK, Kothari A, Wang D, Visal T, Li MK, Pinto J,
Castrillon JA, et al: PARP inhibitor efficacy depends on
CD8+ T-cell recruitment via intratumoral sting pathway
activation in BRCA-deficient models of triple-negative breast
cancer. Cancer Discov. 9:722–737. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Ternette N, Olde Nordkamp MJM, Müller J,
Anderson AP, Nicastri A, Hill AVS, Kessler BM and Li D:
Immunopeptidomic profiling of HLA-A2-positive triple negative
breast cancer identifies potential immunotherapy target antigens.
Proteomics. 18:17004652018. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
National Library of Medicine (NLM):
Clinical Study of Recombinant Anti-HER2 Humanized Monoclonal
Antibody (GB221) for Injection. ClinicalTrials.gov ID, NCT04164615.
NLM; Bethesda, MD: 2019, https://clinicaltrials.gov/ct2/show/NCT04164615.
|
|
137
|
National Library of Medicine (NLM):
Concurrent WOKVAC Vaccination, Chemotherapy, and HER2-Targeted
Monoclonal Antibody Therapy Before Surgery for the Treatment of
Patients With Breast Cancer. ClinicalTrials.gov ID, NCT04329065.
NLM; Bethesda, MD: 2020, https://clinicaltrials.gov/ct2/show/NCT04329065.
|
|
138
|
National Library of Medicine (NLM):
Anti-HER2 Bispecific Antibody ZW25 Activity in Combination With
Chemotherapy With/Without Tislelizumab. ClinicalTrials.gov ID,
NCT04276493. NLM; Bethesda, MD: 2020, https://clinicaltrials.gov/ct2/show/NCT04276493.
|
|
139
|
National Library of Medicine (NLM):
Clinical Study of Recombinant Anti-HER2 Humanized Monoclonal
Antibody for Injection. ClinicalTrials.gov ID, NCT04170595. NLM;
Bethesda, MD: 2019, https://clinicaltrials.gov/ct2/show/NCT04170595.
|
|
140
|
National Library of Medicine (NLM): A
Study of RC48-ADC Administered Intravenously to Patients With
HER2-Positive Metastatic Breast Cancer With or Without Liver
Metastases. ClinicalTrials.gov ID, NCT03500380. NLM; Bethesda, MD:
2018, https://clinicaltrials.gov/ct2/show/NCT03500380.
|
|
141
|
National Library of Medicine (NLM):
Monoclonal Antibody Plus Chemotherapy in Treating Patients With
Metastatic Breast Cancer That Overexpresses HER2.
ClinicalTrials.gov ID, NCT00019812. NLM; Bethesda, MD: 2003,
https://clinicaltrials.gov/ct2/show/NCT00019812.
|
|
142
|
National Library of Medicine (NLM):
Paclitaxel Plus Monoclonal Antibody Therapy in Treating Women With
Recurrent or Metastatic Breast Cancer. ClinicalTrials.gov ID,
NCT00003539. NLM; Bethesda, MD: 2004, https://clinicaltrials.gov/ct2/show/NCT00003539.
|
|
143
|
National Library of Medicine (NLM):
Trastuzumab and Interleukin-2 in Treating Patients With Metastatic
Breast Cancer. ClinicalTrials.gov ID, NCT00006228. NLM; Bethesda,
MD: 2003, https://clinicaltrials.gov/ct2/show/NCT00006228.
|
|
144
|
National Library of Medicine (NLM):
Chemotherapy Plus Monoclonal Antibody Therapy in Treating Women
With Stage II or Stage IIIA Breast Cancer That Overexpresses HER2.
ClinicalTrials.gov ID, NCT00003992. NLM; Bethesda, MD: 2004,
https://clinicaltrials.gov/ct2/show/NCT00003992.
|
|
145
|
National Library of Medicine (NLM): Impact
of Pegfilgrastim on Trastuzumab Anti-tumor Effect and ADCC in
Operable HER2+ Breast Cancer Breast Cancer (BREASTIMMU02).
ClinicalTrials.gov ID, NCT03571633. NLM; Bethesda, MD: 2018,
https://clinicaltrials.gov/ct2/show/NCT03571633.
|
|
146
|
National Library of Medicine (NLM): A
Study of Pertuzumab in Participants With Metastatic Breast Cancer.
ClinicalTrials.gov ID, NCT02491892. NLM; Bethesda, MD: 2015,
https://clinicaltrials.gov/ct2/show/NCT02491892.
|
|
147
|
National Library of Medicine (NLM):
Trastuzumab and Pertuzumab in Treating Patients With Unresectable
Locally Advanced or Metastatic Breast Cancer That Did Not Respond
to Previous Trastuzumab. ClinicalTrials.gov ID, NCT00301899. NLM;
Bethesda, MD: 2006, https://clinicaltrials.gov/ct2/show/NCT00301899.
|
|
148
|
National Library of Medicine (NLM): A
Study of MRG002 in the Treatment of Patients With HER2-positive
Unresectable Locally Advanced or Metastatic Breast Cancer.
ClinicalTrials.gov ID, NCT04924699. NLM; Bethesda, MD: 2021,
https://clinicaltrials.gov/ct2/show/NCT04924699.
|
|
149
|
National Library of Medicine (NLM): ARX788
in HER2-positive, Metastatic Breast Cancer Subjects
(ACE-Breast-03). ClinicalTrials.gov ID, NCT04829604. NLM; Bethesda,
MD: 2021, https://clinicaltrials.gov/ct2/show/NCT04829604.
|
|
150
|
National Library of Medicine (NLM): Haplo
/ Allogeneic NKG2DL-targeting Chimeric Antigen Receptor-grafted γδ
T Cells for Relapsed or Refractory Solid Tumour. ClinicalTrials.gov
ID, NCT04107142. NLM; Bethesda, MD: 2019, https://clinicaltrials.gov/ct2/show/NCT04107142.
|
|
151
|
National Library of Medicine (NLM): A
Study to Investigate LYL797 in Adults With Solid Tumors.
ClinicalTrials.gov ID, NCT05274451. NLM; Bethesda, MD: 2022,
https://clinicaltrials.gov/ct2/show/NCT05274451.
|
|
152
|
National Library of Medicine (NLM): A
Biomarker Screening Protocol for Participants With Solid Tumors
(START). ClinicalTrials.gov ID, NCT05891197. NLM; Bethesda, MD:
2023, https://clinicaltrials.gov/ct2/show/NCT05891197.
|
|
153
|
National Library of Medicine (NLM): cMet
CAR RNA T Cells Targeting Breast Cancer. ClinicalTrials.gov ID,
NCT01837602. NLM; Bethesda, MD: 2013, https://clinicaltrials.gov/ct2/show/NCT01837602.
|
|
154
|
National Library of Medicine (NLM):
Treatment of Relapsed and/or Chemotherapy Refractory Advanced
Malignancies by CART-meso. ClinicalTrials.gov ID, NCT02580747. NLM;
Bethesda, MD: 2015, https://clinicaltrials.gov/ct2/show/NCT02580747.
|
|
155
|
National Library of Medicine (NLM): Phase
I/II Study of Anti-Mucin1 (MUC1) CAR T Cells for Patients With
MUC1+ Advanced Refractory Solid Tumor. ClinicalTrials.gov ID,
NCT02587689. NLM; Bethesda, MD: 2015, https://clinicaltrials.gov/ct2/show/NCT02587689.
|
|
156
|
National Library of Medicine (NLM):
Multi-4SCAR-T Therapy Targeting Breast Cancer. ClinicalTrials.gov
ID, NCT04430595. NLM; Bethesda, MD: 2020, https://clinicaltrials.gov/ct2/show/NCT04430595.
|
|
157
|
National Library of Medicine (NLM): EpCAM
CAR-T for Treatment of Advanced Solid Tumors. ClinicalTrials.gov
ID, NCT02915445. NLM; Bethesda, MD: 2016, https://clinicaltrials.gov/ct2/show/NCT02915445.
|
|
158
|
National Library of Medicine (NLM):
C7R-GD2.CART Cells for Patients With Relapsed or Refractory
Neuroblastoma and Other GD2 Positive Cancers (GAIL-N).
ClinicalTrials.gov ID, NCT03635632. NLM; Bethesda, MD: 2018,
https://clinicaltrials.gov/ct2/show/NCT03635632.
|
|
159
|
Bonifant CL, Jackson HJ, Brentjens RJ and
Curran KJ: Toxicity and management in CAR T-cell therapy. Mol Ther
Oncolytics. 3:160112016. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Almåsbak H, Aarvak T and Vemuri MC: CAR T
cell therapy: A game changer in cancer treatment. J Immunol Res.
2016:54746022016. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Abreu TR, Fonseca NA, Gonçalves N and
Moreira JN: Current challenges and emerging opportunities of CAR-T
cell therapies. J Control Release. 319:246–261. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Hege KM, Bergsland EK, Fisher GA,
Nemunaitis JJ, Warren RS, McArthur JG, Lin AA, Schlom J, June CH
and Sherwin SA: Safety, tumor trafficking and immunogenicity of
chimeric antigen receptor (CAR)-T cells specific for TAG-72 in
colorectal cancer. J Immunother Cancer. 5:222017. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Chen H, Wang F, Zhang P, Zhang Y, Chen Y,
Fan X, Cao X, Liu J, Yang Y, Wang B, et al: Management of cytokine
release syndrome related to CAR-T cell therapy. Front Med.
13:610–617. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Lee DW, Gardner R, Porter DL, Louis CU,
Ahmed N, Jensen M, Grupp SA and Mackall CL: Current concepts in the
diagnosis and management of cytokine release syndrome. Blood.
124:188–195. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Teachey DT, Rheingold SR, Maude SL,
Zugmaier G, Barrett DM, Seif AE, Nichols KE, Suppa EK, Kalos M,
Berg RA, et al: Cytokine release syndrome after blinatumomab
treatment related to abnormal macrophage activation and ameliorated
with cytokine-directed therapy. Blood. 121:5154–5157. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Lee DW, Kochenderfer JN, Stetler-Stevenson
M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M,
Shah NN, et al: T cells expressing CD19 chimeric antigen receptors
for acute lymphoblastic leukaemia in children and young adults: A
phase 1 dose-escalation trial. Lancet. 385:517–528. 2015.
View Article : Google Scholar
|
|
167
|
Kochenderfer JN, Dudley ME, Feldman SA,
Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes
MS, Sherry RM, et al: B-cell depletion and remissions of malignancy
along with cytokine-associated toxicity in a clinical trial of
anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood.
119:2709–2720. 2012. View Article : Google Scholar :
|
|
168
|
Brentjens RJ, Davila ML, Riviere I, Park
J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska
M, et al: CD19-targeted T cells rapidly induce molecular remissions
in adults with chemotherapy-refractory acute lymphoblastic
leukemia. Sci Transl Med. 5:177ra382013. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Grupp SA, Kalos M, Barrett D, Aplenc R,
Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, et
al: Chimeric antigen receptor-modified T cells for acute lymphoid
leukemia. N Engl J Med. 368:1509–1518. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Curran KJ, Pegram HJ and Brentjens RJ:
Chimeric antigen receptors for T cell immunotherapy: Current
understanding and future directions. J Gene Med. 14:405–415. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Lamers CH, Sleijfer S, Vulto AG, Kruit WH,
Kliffen M, Debets R, Gratama JW, Stoter G and Oosterwijk E:
Treatment of metastatic renal cell carcinoma with autologous
T-lymphocytes genetically retargeted against carbonic anhydrase IX:
First clinical experience. J Clin Oncol. 24:e20–e22. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Morgan RA, Yang JC, Kitano M, Dudley ME,
Laurencot CM and Rosenberg SA: Case report of a serious adverse
event following the administration of T cells transduced with a
chimeric antigen receptor recognizing ERBB2. Mol Ther. 18:843–851.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Marin V, Cribioli E, Philip B, Tettamanti
S, Pizzitola I, Biondi A, Biagi E and Pule M: Comparison of
different suicide-gene strategies for the safety improvement of
genetically manipulated T cells. Hum Gene Ther Methods. 23:376–386.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Rubin DB, Danish HH, Ali AB, Li K, LaRose
S, Monk AD, Cote DJ, Spendley L, Kim AH, Robertson MS, et al:
Neurological toxicities associated with chimeric antigen receptor
T-cell therapy. Brain. 142:1334–1348. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Miao L, Zhang Z, Ren Z and Li Y: Reactions
related to CAR-T cell therapy. Front Immunol. 12:6632012021.
View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Maus MV, Haas AR, Beatty GL, Albelda SM,
Levine BL, Liu X, Zhao Y, Kalos M and June CH: T cells expressing
chimeric antigen receptors can cause anaphylaxis in humans. Cancer
Immunol Res. 1:26–31. 2013. View Article : Google Scholar
|
|
177
|
Zhao Y, Moon E, Carpenito C, Paulos CM,
Liu X, Brennan AL, Chew A, Carroll RG, Scholler J, Levine BL, et
al: Multiple injections of electroporated autologous T cells
expressing a chimeric antigen receptor mediate regression of human
disseminated tumor. Cancer Res. 70:9053–9061. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Barrett DM, Zhao Y, Liu X, Jiang S,
Carpenito C, Kalos M, Carroll RG, June CH and Grupp SA: Treatment
of advanced leukemia in mice with mRNA engineered T cells. Hum Gene
Ther. 22:1575–1586. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Chang K and Pastan I: Molecular cloning of
mesothelin, a differentiation antigen present on mesothelium,
mesotheliomas, and ovarian cancers. Proc Natl Acad Sci USA.
93:136–140. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Lindau D, Gielen P, Kroesen M, Wesseling P
and Adema GJ: The immunosuppressive tumour network: Myeloid-derived
suppressor cells, regulatory T cells and natural killer T cells.
Immunology. 138:105–115. 2013. View Article : Google Scholar :
|
|
181
|
Ko HJ, Lee JM, Kim YJ, Kim YS, Lee KA and
Kang CY: Immunosuppressive myeloid-derived suppressor cells can be
converted into immunogenic APCs with the help of activated NKT
cells: An alternative cell-based antitumor vaccine. J Immunol.
182:1818–1828. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Davis RJ, Van Waes C and Allen CT:
Overcoming barriers to effective immunotherapy: MDSCs, TAMs, and
Tregs as mediators of the immunosuppressive microenvironment in
head and neck cancer. Oral Oncol. 58:59–70. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Fedorov VD, Themeli M and Sadelain M:
PD-1- and CTLA-4-based inhibitory chimeric antigen receptors
(iCARs) divert off-target immunotherapy responses. Sci Transl Med.
5:215ra1722013. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Chmielewski M, Kopecky C, Hombach AA and
Abken H: IL-12 release by engineered T cells expressing chimeric
antigen receptors can effectively Muster an antigen-independent
macrophage response on tumor cells that have shut down tumor
antigen expression. Cancer Res. 71:5697–5706. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Pegram HJ, Lee JC, Hayman EG, Imperato GH,
Tedder TF, Sadelain M and Brentjens RJ: Tumor-targeted T cells
modified to secrete IL-12 eradicate systemic tumors without need
for prior conditioning. Blood. 119:4133–4141. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Patel U, Abernathy J, Savani BN, Oluwole
O, Sengsayadeth S and Dholaria B: CAR T cell therapy in solid
tumors: A review of current clinical trials. EJHaem. 3(Suppl 1):
S24–S31. 2022. View Article : Google Scholar
|
|
187
|
Włodarczyk M and Pyrzynska B: CAR-NK as a
rapidly developed and efficient immunotherapeutic strategy against
cancer. Cancers (Basel). 15:1172022. View Article : Google Scholar
|
|
188
|
Hawkins ER, D'Souza RR and Klampatsa A:
Armored CAR T-cells: The next chapter in T-cell cancer
immunotherapy. Biologics. 15:95–105. 2021.PubMed/NCBI
|
|
189
|
Rafiq S, Hackett CS and Brentjens RJ:
Engineering strategies to overcome the current roadblocks in CAR T
cell therapy. Nat Rev Clin Oncol. 17:147–167. 2020. View Article : Google Scholar :
|
|
190
|
Xia L, Zheng Z, Liu JY, Chen YJ, Ding J,
Hu GS, Hu YH, Liu S, Luo WX, Xia NS and Liu W: Targeting
triple-negative breast cancer with combination therapy of EGFR CAR
T cells and CDK7 inhibition. Cancer Immunol Res. 9:707–722. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Zhang H, Qin C, An C, Zheng X, Wen S, Chen
W, Liu X, Lv Z, Yang P, Xu W, et al: Application of the
CRISPR/Cas9-based gene editing technique in basic research,
diagnosis, and therapy of cancer. Mol Cancer. 20:1262021.
View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Yan T, Zhu L and Chen J: Current advances
and challenges in CAR T-Cell therapy for solid tumors:
Tumor-associated antigens and the tumor microenvironment. Exp
Hematol Oncol. 12:142023. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Collins DC, Sundar R, Lim JSJ and Yap TA:
Towards precision medicine in the clinic: From biomarker discovery
to novel therapeutics. Trends Pharmacol Sci. 38:25–40. 2017.
View Article : Google Scholar
|