Introduction
Colorectal cancer has been one of the leading causes
of cancer-related deaths worldwide, especially in patients with
metastatic disease. The five-year survival rate of colorectal
cancer patients is ~60%, but it decreases to 14% when distant
metastases are present, regardless of the availability of several
combination therapies as systemic treatments (1). Various genetic abnormalities such as
wingless and int-1 (Wnt) signaling, tissue growth factor beta
(TGFβ) signaling, or TP53 signaling have been known in the
carcinogenesis of colorectal cancer (2,3).
Although patients with a few specific biomarker-defined colorectal
cancers, such as wild-type rat sarcoma viral oncogene homolog
(RAS)/v-raf murine sarcoma viral oncogene homolog B (BRAF) and
deoxyribonucleic acid mismatch repair-deficient/microsatellite
instability-high, can already receive the benefits of additional
chemotherapy to standard regimens (4-6),
the development of a novel therapeutic target and chemotherapeutic
agents for individualized treatment based on the respective genetic
abnormalities is required to improve poor prognosis.
The tumor microenvironment consists of various cell
types and fibroblasts in the tumor tissue, called cancer-associated
fibroblasts (CAFs), which play pivotal role in tumor progression
(7,8). CAFs and normal fibroblasts differ
with respect to the expression of various markers, such as fibrotic
markers, growth factors, chemokines and cytokines (9). CAFs with tumor-promoting effects
were usually activated and exhibited myofibroblast-like properties
such as the upregulation of alpha-smooth muscle actin (α-SMA)
(9). TP53 is a significant tumor
suppressor gene, and its somatic mutations are one of the most
frequent alterations in ~50% of all human cancers, including
colorectal cancer (10). The
mutational inactivation of the TP53 gene in tumor cells has been
reported to affect not only tumor cells but also the surrounding
cells in the tumor microenvironment and to promote tumor-stromal
activation and subsequent tumor growth (11,12). The secretion levels of various
proteins, reactive oxygen species, or microRNAs (miRNAs) capsulized
in exosomes have been reported as the mechanisms by which the
alteration of TP53 status in tumor cells affects surrounding cells
(11,12). These mechanisms may be involved in
the transition of normal tissue fibroblasts to CAFs; however, the
detailed mechanisms remain only partially understood.
The apelin-APJ system was first identified in 1993
as a G protein-coupled receptor, and apelin was isolated from
bovine stomach extracts in 1998 as an endogenous ligand of APJ
(13). Apelin and APJ are
ubiquitously expressed throughout the organism and play several
physiological roles (14,15). The apelin-APJ system in
fibroblasts has been reported as a mechanism for suppressing the
fibroblast-to-myofibroblast transition in non-neoplastic organ
fibrosis, including myocardial fibrosis, pulmonary fibrosis, skin
fibrosis and chronic kidney disease (16-19). It was posited by the authors that
the APJ system could inhibit tumor-promoting CAFs, especially in
acquiring myofibroblast characteristics. By contrast, the APJ
system in tumor cells, not fibroblasts, including colorectal cancer
cells, has also been reported to have a tumor-promoting effect
(20-22) in contrast to the authors'
hypothesis that the APJ system can play a role as a tumor
suppressor via the inhibition of CAF activation in the tumor
microenvironment. Although it is necessary to comprehensively view
other types of cells in the tumor microenvironment, to the best of
the authors' knowledge, no previous studies have described the
significance of the APJ system in the tumor stroma. The present
study aimed to elucidate the role of the APJ system in fibroblast
modification in the tumor microenvironment of colorectal cancer,
focusing on the p53 status of the cancer cells.
Materials and methods
Cell culture and treatment
The human colon cancer cell line HCT116 and Caco-2
exhibiting wild-type TP53 expression, SW480 and DLD-1 of
TP53 mutant cells, and non-transformed human colon
fibroblasts CCD-18Co were obtained from the American Type Culture
Collection (ATCC). Cancer cells were cultured at 37°C under 5%
CO2 in Dulbecco's Modified Eagle Medium (DMEM)
(Sigma-Aldrich; Merck KGaA) supplemented with 10% fetal bovine
serum (FBS) or Eagle's Minimum Essential Medium (EMEM) (ATCC)
supplemented with 20% FBS, and fibroblasts were cultured at 37°C
under 5% CO2 in EMEM with 10% FBS. To investigate the
effect of apelin-13 (Cayman Chemical Company) on recombinant human
TGF-β1 (rhTGF-β1)(FUJIFILM Wako Pure Chemical Corporation)-induced
mRNA and protein expression in fibroblasts, the cells were
pretreated with apelin-13 at the concentration of 10 to 1,000 nM
for 60 min and then stimulated with rhTGF-β1 for 24-48 h. ML221
(Sigma-Aldrich; Merck KGaA), the APJ antagonist, was used to
examine the effect of APJ suppression on fibroblasts at
concentrations of 0.1 to 10 µM.
Exosome isolation
Cells were cultured in DMEM with 10% FBS in 15-cm
dishes (Tissue Culture Dish VTC-D150, AS ONE CORPORATION) for 48 h,
washed with phosphate-buffered saline (PBS), replaced with DMEM
without FBS, and cultured for another 48 h, after which the culture
supernatant was collected, and sequential centrifugation was
performed. The culture supernatant was first centrifuged at 300 × g
for 10 min at 4°C, then at 2,000 × g for 10 min to precipitate the
cells. The supernatant was then centrifuged at 10,000 × g for 30
min at 4°C, and then ultra-centrifuged at 100,000 × g for 70 min at
4°C to pellet the extracellular vesicles, which were then washed
with a suspension in PBS. The final pellet was resuspended in 100
µl of PBS. The protein concentration was measured using a
bicinchoninic acid (BCA) protein assay kit (Thermo Fisher
Scientific, Inc.). Isolated exosomes were administered to
fibroblasts culture supernatant at a concentration of 100
µg/ml. The morphology of the exosomes was captured using a
transmission electron microscope (H-7600; Hitachi High-Technologies
Corporation) after preparation as described here. A total of ~5
µl of sample was placed on Parafilm. Then, a carbon-coated
400 mesh copper grid was positioned on the top of the drop for 10
sec and washed by a droplet of distilled water. The grid was
contrasted by adding a drop of 2% uranyl acetate on Parafilm and
incubating the grid on the top of the drop for 10 sec. Excess
liquid was removed by gently using an absorbent paper. After dying,
the samples were used for observation. Furthermore, the expression
of CD9, CD63 and ALIX, which are exosome markers, was confirmed by
western blotting.
Co-culture experiment
Non-contact co-cultures were performed using
Transwell inserts with 0.4-µm pores (Corning, Inc.).
Fibroblasts or cancer cells were seeded on a six-well
(2×105 cells per well) or 12-well (3×104
cells per well) plate (Corning, Inc.) in EMEM or DMEM with 10% FBS,
and cancer cells or fibroblasts were seeded on a Transwell insert
in the same conditions. After 24-48 h incubation, serum-free EMEM
was used as the co-culture medium, and each culture was incubated
for 48 h.
RNA interference
CCD-18Co cells were transfected for 48-72 h at 37°C
with small interfering (si)RNAs against APJ [Thermo Fisher
Scientific, Inc.; #1; cat. no. s223458, 5′-CAG AUG CAC GAG AAA UCC
ATT-3′ (sense) and 5′-UGG AUU UCU CGU GCA UCU GTT-3′ (antisense),
#2; s1186, 5′-UGU GGG CUA CCU ACA CGU Att-3′ (sense) and 5′-UAC GUG
UAG GUA GCC CAC Agg-3′ (antisense)], negative control siRNA (Thermo
Fisher Scientific, Inc.; cat. no. 4390843), microRNA (miR)-5703
mimic (Thermo Fisher Scientific, Inc.; cat. no. 4464066), miR-5703
inhibitor (Thermo Fisher Scientific, Inc. cat. no. 4464084), miRNA
mimic negative control (Thermo Fisher Scientific, Inc. cat. no.
4464058) and miRNA inhibitor negative control (Thermo Fisher
Scientific, Inc. cat. no. 4464076) and SW480 cells were transfected
with RAB27A siRNA [Thermo Fisher Scientific, Inc.; cat. no.
s532296; 5′-CCA GUG UAC UUU ACC AAU Att-3′ (sense) and 5′-UAU UGG
UAA AGU ACA CUG Gtc-3′ (antisense)] at a final concentration of 10
nM using the Lipofectamine® RNAiMAX Reagent (Thermo
Fisher Scientific, Inc.) according to the manufacturer's
transfection protocol in six-well or 12-well plates (Corning,
Inc.). In addition, as previously described (11), lentiviral GFP-IRES-short hairpin
(sh)RNA vectors against TP53 (cat. nos. RHS4430-101161166;
101162286; 101168779; 99365289) were obtained from Thermo Fisher
Scientific, Inc., and HCT116sh control and
HCT116sh p53 cells were generated. Cells with
stable shRNA expression were cultured with 2 µg/ml puromycin
(InvivoGen) after colony selection.
RNA/miRNA extraction and reverse
transcription-quantitative (RT-q) PCR analysis
Total RNA or miRNAs were extracted and reverse
transcribed as previously described (12). RNA was extracted from cells or
nanovesicles using an RNeasy Mini Kit (cat. no. 74106; Qiagen GmbH)
for mRNA and a miRNeasy Mini Kit (cat. no. 217084; Qiagen GmbH) for
miRNAs according to the manufacturer's instructions. qPCR was
performed using THUNDERBIRD qPCR Master Mix (Toyobo Life Science)
on the QuantStudio 6 Flex (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The thermocycling conditions of the qPCR
reaction were: 20 sec of initial denaturation at 95°C; followed by
40-60 cycles of 1 sec at 95°C for denaturation, and 20 sec at 60°C
for annealing and extension. The mRNA expression was quantified
using TaqMan gene expression assays (Applied Biosystems; Thermo
Fisher Scientific, Inc.), and data were normalized to
beta-2-microglobulin (B2M) expression levels. The miRNAs were
reverse transcribed using miRNA-specific primers (Applied
Biosystems; Thermo Fisher Scientific, Inc.), and miRNA expression
was quantified using TaqMan gene expression assays. Data were
normalized to the expression of spike-in syn-cel-miR-39 (Qiagen).
The 2−ΔΔCq method was used to analyze the relative gene
expression as previously described (23). The used primers are listed in
Table SI.
Western blotting
Protein extracts were prepared as previously
described (11). Cells were lysed
with radio-immunoprecipitation assay (RIPA) buffer containing
protease inhibitor and phosphatase inhibitor cocktail (Nacalai
Tesque, Inc.), and the proteins were collected on ice. The protein
concentration was measured by the BCA protein assay kit. Equal
amounts of protein (10 µg) were applied to each lane of 8%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) gels and run. The proteins were then transferred onto
polyvinylidene difluoride (PVDF) membranes and blocked with 4% milk
in 0.1% Tris-buffered saline with Tween-20 (TBST) at room
temperature for 1 h. Then, each membrane was subjected to overnight
incubation at 4°C with the appropriate primary antibodies for
β-actin (1:1,000; cat. no. 4970; Cell Signaling Technology, Inc.),
TP53 (1:200; cat. no. sc-126; Santa Cruz Biotechnology, Inc.), APJ
(1:1,000; cat. no. ab84296; Abcam or 1 µg/ml, cat. no.
702069; Thermo Fisher Scientific, Inc.), α-SMA (1:1,000; cat. no.
ab5694; Abcam), phosphorylated Smad2/3 (1:1,000; cat. no. 8828;
Cell Signaling Technology, Inc.) and Smad2/3 (1:1,000; cat. no.
3102, Cell Signaling Technology, Inc.). After incubation, the
membranes were incubated with horseradish peroxidase
(HRP)-conjugated secondary antibody (1:3,000; cat. no. NA934-1ML;
Cytiva) at room temperature for 60 min. Finally, the membranes were
reacted with detection reagents (Super Signal West Pico PLUS
Chemiluminescent Substrate; Thermo Fisher Scientific, Inc.) and
exposed for an appropriate time to visualize the protein bands
using the ChemiDoc MP (Bio-Rad Laboratories, Inc.). Primary
antibodies used for western blotting are listed in Table SII.
Cell proliferation assay
Cell proliferation and viability were analyzed in
12-well plates (3×104 cells per well). Water-soluble
tetrazolium (WST) assays were performed using an SF cell counting
reagent (Nacalai Tesque, Inc.). A total of 100 µl reagent
was added to 1,000 µl culture medium per well. After 30 min
of incubation at 37°C, the absorbance was measured at 450 nm with
600 nm used as a reference with a microplate reader (Thermo Fisher
Scientific, Inc.).
Cell wound healing assay
Wound healing assay was performed to assess the
migratory ability of fibroblasts. Cells seeded on a six-well plate
(2×105 cells per well) were incubated for 24 h;
thereafter, the medium was changed to serum-free, ML221 was
applied, and cells were scratched using the tip of the
200-µl sterile pipette tube. In the same manner, cells were
seeded on a six-well plate (2×105 cells per well), were
transfected with siRNA against APJ, and incubated for 24 h. Next,
the medium was changed and incubated for another 24 h, after which
cells were scratched the same as aforementioned. The wound
reduction rates were calculated by measuring the area of the wound
after 0 and 24 h in 3 microscopic fields (magnification, ×200;
BZ-X700 all-in-one fluorescence microscope; Keyence Corporation),
which were randomly selected.
Immunofluorescence staining
Fibroblasts were seeded on a 35-mm dish (Matsunami
Glass Ind., Ltd.) (2×105 cells per dish) and were
transfected with siRNA against APJ 72 h before the experiment or
were treated with ML221 (10 µM) 24 h prior to the
experiment. Cells were fixed with 100% methanol at room temperature
for 15 min. After cells were blocked with 1% bovine serum albumin
(Nacalai Tesque, Inc.), the primary antibody reaction against
anti-α-SMA (1:400; cat. no. ab5694; Abcam) was performed at room
temperature for 1 h. Following a secondary antibody reaction using
Alexa Fluor Plus 488 (1:1,000; cat. no. 4412; Cell Signaling
Technology, Inc.) at room temperature for 1 h, nuclear staining was
performed using Hoechst (1:200; cat. no. 346-07951; Dojindo
Laboratories, Inc.) at room temperature for 10 min. The images were
analyzed using a fluorescence microscope (BZ-X700 all-in-one
fluorescence microscope; Keyence Corporation). The antibody used
for fluorescent immunostaining is listed in Table SII.
Immunohistochemistry
Immunohistochemical staining for p53 and APJ in
colorectal cancers with submucosal invasion was performed using
endoscopically resected specimens at the Osaka University Hospital
between April 2015 and August 2020. The paraffin-embedded tumor
tissues were cut into 4-µm sections. The sections were
dewaxed with xylene, dehydrated with descending ethanol series,
activated with antigen retrieval citrate buffer, and incubated with
3% H2O2 at room temperature for 10 min. The
slides were blocked with serum-free phosphate buffer including
casein at room temperature for 20 min and incubated with the
primary antibodies at 4°C overnight. The primary antibodies were as
follows: anti-P53 (1:100; cat. no. sc-126; Santa Cruz
Biotechnology, Inc.) or anti-APJ (1:100; cat. no. ABD43;
Sigma-Aldrich; Merck KGaA). The sections were incubated with
HRP-conjugated secondary antibody (no dilution; cat. no. K5007;
Agilent Technologies, Inc.) at room temperature for 20 min and
counterstained with hematoxylin at room temperature for 15 sec. The
slides were captured using a light microscope (VS200; Olympus
Corporation). The expression of p53 in cancer cells, of APJ in
fibroblasts from the tumor stroma, and of α-SMA in the stroma with
HALO software were quantified (Indica Labs, Inc.). p53 expression
was assessed using a previously described scoring method (24). The p53 expression levels were
evaluated according to the positive rate of p53 staining in tumor
cells, and p53 positivity was defined as more than 10% of p53
positive staining. APJ expression levels were assessed by
calculating the H-score using the software. The area of α-SMA
staining in the stroma was calculated by the aforementioned
software. The antibodies used for immunohistochemistry in each case
are listed in Table SII.
Xenograft model
HCT116 cells (2×105) were suspended in
200 µl of PBS and co-inoculated with negative
controltransfected CCD-18Co cells (2×105) and CCD-18Co
cells with suppressed APJ expression (2×105)
subcutaneously into the left and right flank of 5-6-week-old male
BALB/c nude mice (Charles River, Yokohama, Japan), respectively
(each group n=10). A total of 20 mice were used. All mice were
housed in a specific pathogen free condition at a constant
temperature of 23±1.5°C with 45±15% humidity under a 12/12-h
light/dark cycle and were fed with MFG (Oriental Yeast Co., Ltd.)
with free access to water. In addition, gene suppression using
siRNA in the xenograft experiments was performed as previously
described. At the time of subcutaneous implantation, the mice were
anesthetized with medetomidine hydrochloride (0.75 mg/kg),
midazolam (4 mg/kg) and butorphanol tartrate (5 mg/kg). Tumor
volume was measured three times a week and calculated using the
formula: [tumor length x (tumor width)2]/2. Humane
endpoints were set at a tumor volume of 2,000 mm3 and
irreversible wasting. The duration of the experiment was 21 days.
The last measurement of the tumor volume was obtained on the 21st
day, followed by euthanasia by CO2 administration with a
fill rate of 30-70% of the chamber volume per min and death was
verified by checking for the heartbeat and breath cessation. All
mice used were euthanized.
Bioinformatics analysis
The National Center for Biotechnology Information
Gene Expression Omnibus (NCBI GEO) database and miR-DB database
(http://mirdb.org) were used in the present study. To
analyze APJ expression in colon fibroblasts, NCBI GEO database
(accession no. GSE46824; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE46824)
was used. NCBI GEO database (accession no. GSE120012; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE120012)
and miR-DB database were used to analyze miR-5703.
Statistical analysis
Data were expressed as the mean ± standard
deviation. The two-tailed, paired or unpaired Student's t-test was
used to compare two groups. A one-way analysis of variance (ANOVA)
with Tukey's post hoc test was performed to analyze the differences
among multiple groups. P<0.05 was considered to indicate a
statistically significant difference. The statistical analyses were
performed using the JMP® Pro15 software (SAS Institute
Inc.).
Study approval
The present study was approved (approval no. 20061)
by the Ethics Committee of Osaka University Graduate School of
Medicine (Osaka, Japan) for using resected human samples, and
written informed consent was obtained from all patients who
provided resected specimens. The study design was per the
principles of the Declaration of Helsinki (October 2013). The
animal experimental procedures were performed per the Osaka
University guidelines for animal experiments and were approved
(approval no. 30-015-077) by the Animal Care and Use Committee of
Osaka University Graduate School of Medicine (Osaka, Japan).
Results
Colorectal cancer cells with
p53-inactivation suppress APJ expression in fibroblasts
First, non-contact cell co-culture experiments were
conducted using a Transwell insert for fibroblasts (CCD-18Co). To
investigate the involvement of p53 inactivation in cancer cells
with APJ expression in fibroblasts, p53-suppressed colorectal
cancer cells, HCT116sh p53 cells, were
established. Successful inhibition of TP53 expression in
HCT116sh p53 cells was confirmed by RT-qPCR and
western blotting (Figs. 1A and
S1A). The expression level of
APJ in CCD-18Co cells was decreased along with increased expression
of α-SMA when co-cultured with HCT116sh p53 cells
compared with co-cultured with HCT116sh control
cells or CCD-18Co cells alone in western blotting (Figs. 1B and S1B). The mRNA levels of TGFβ1 and
VEGF-A expression in fibroblasts significantly increased when
co-cultured with HCT116sh p53 cells (Fig. 1C). In addition, the expression
levels of APJ were decreased and α-SMA were increased in
fibroblasts when co-cultured with TP53-mutant colon cancer cells
such as SW480 or DLD-1 cells, in contrast to TP53-wild colon cancer
cells such as HCT116 or Caco-2 (Figs.
1D and S1C and D). The mRNA
expression levels of TGFβ1 and VEGF-A were significantly increased
when co-cultured with SW480 and DLD-1 cells as observed in
HCT116sh p53 cells, which were also significantly
increased than those co-cultured with HCT116 and Caco-2 cells
(Fig. 1E).
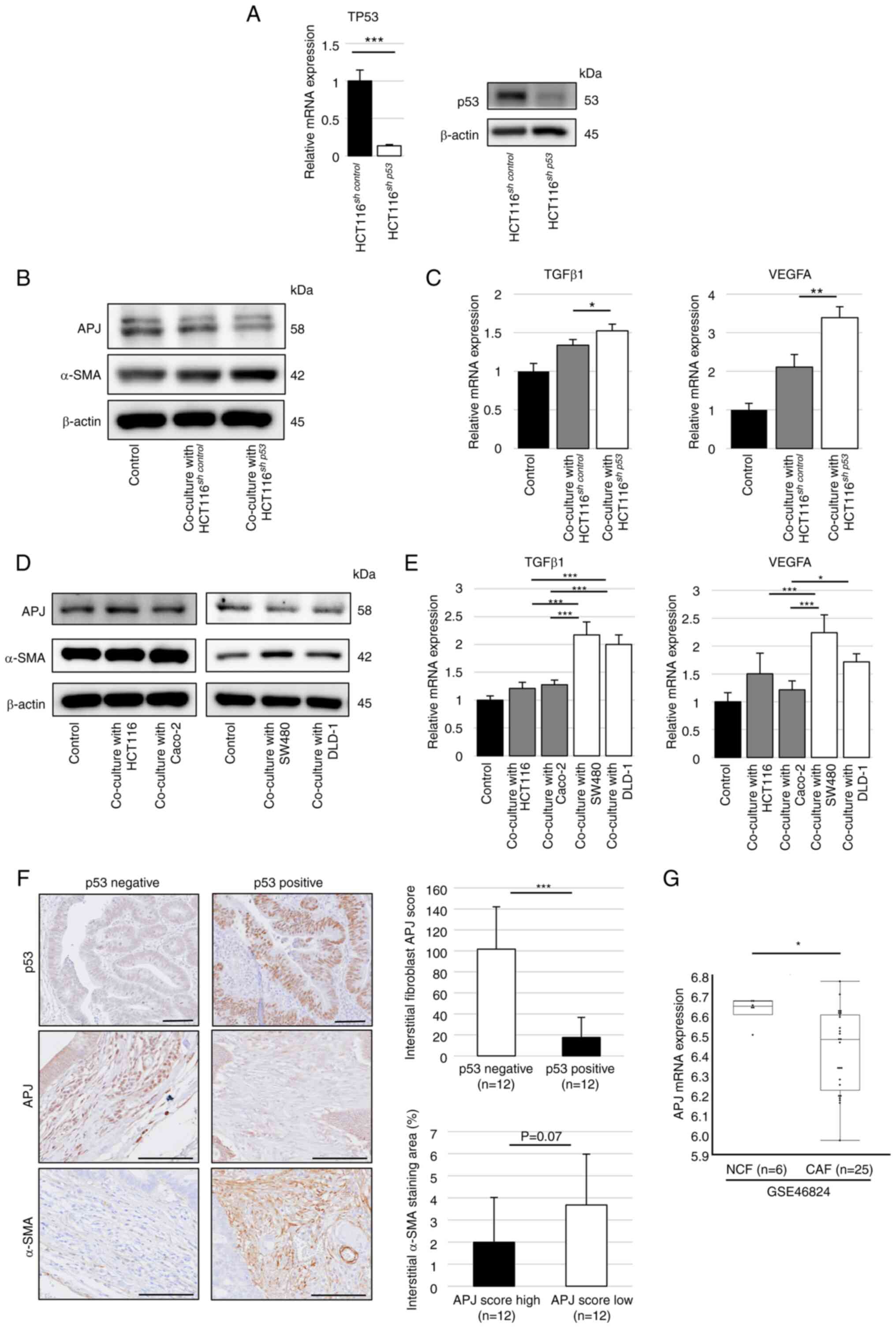 | Figure 1Colon cancer cells with deficient or
mutated TP53 function suppress APJ expression in CCD-18Co cells.
(A) RT-qPCR was performed to assess TP53 mRNA expression in
HCT116sh control or HCT116sh
p53 cells (left). Western blotting was performed for TP53
(right). (B) Western blot analysis of APJ and α-SMA in CCD-18Co
cells co-cultured with HCT116sh control or
HCT116sh p53 cells. The immunoblots were
performed three times. (C) RT-qPCR was performed to assess TGFβ1
and VEGFA mRNA expression in CCD-18Co cells co-cultured with
HCT116sh control or HCT116sh
p53 cells. (D) Western blotting was performed for APJ and
α-SMA expression in CCD-18Co cells co-cultured with HCT116 or
Caco-2 cells (left) and SW480 or DLD-1 cells (right). The
immunoblots were performed three times. (E) Relative mRNA
expression levels of TGFβ1 and VEGFA in CCD-18Co cells co-cultured
with or without HCT116, Caco-2, SW480, or DLD-1 cells. (F)
Immunohistochemical staining of APJ in colon cancer tissues.
Representative images of positive and negative p53 expression, high
and low APJ scores, and α-SMA staining. Scale bar, 100 µm.
(G) APJ expression of fibroblasts in non-tumor and colon cancer
tissues from (Gene Expression Omnibus) database. Data are presented
as the mean ± SD. A one-way ANOVA with Tukey's post hoc test was
performed to analyze the differences among multiple groups.
*P<0.05, **P<0.01 and
***P<0.001. APJ, apelin receptor; RT-qPCR, reverse
transcription-quantitative PCR; shRNA, short hairpin RNA; α-SMA,
alpha-smooth muscle actin; NCF, normal colonic fibroblasts; CAF,
cancer-associated fibroblasts. |
Next, APJ expression was examined in fibroblasts
from human colorectal cancer tissues. APJ expression levels in
cancer stromal fibroblasts were evaluated based on the p53 status
of colorectal cancer cells. Immunohistochemical staining was
performed for p53, APJ and α-SMA in endoscopically resected cancers
with submucosal invasion. The patient and lesion characteristics
are summarized in Table SIII.
The APJ staining score in cancer stromal fibroblasts was
significantly lower in p53-positive (suggesting p53 mutation)
colorectal cancers than in p53-negative (suggesting wild-type p53)
colorectal cancers. The APJ staining score was assessed separately
in the left colon (descending colon, sigmoid colon, rectum) and
right colon (cecum, ascending colon, transverse colon), but there
were no significant differences (Fig. S9). The relationship between
clinicopathological characteristics and p53 status of
endoscopic-resected cases is listed in Table SIV. Next, the included cases were
divided into two groups according to their median APJ staining
score (high or low). The area of α-SMA staining in each of these
groups was then estimated, which showed a trend towards being
larger in the low score group (Fig.
1F). mRNA expression datasets of fibroblasts extracted from
fresh surgical specimens of colorectal carcinoma (CAF group) and
normal colonic mucosa (normal colonic fibroblasts, NCF group) were
obtained using NCBI GEO database (accession no. GSE46824). The
datasets revealed that APJ expression was significantly lower in
the CAF group than in the NCF group (Fig. 1G). These results indicated that
TP53-inactivated colorectal cancer cells inhibit APJ
expression in fibroblasts and induce fibroblast modification with
myofibroblast-like properties.
APJ inhibition in fibroblasts by APJ
antagonist induces the modification into myofibroblasts-like
properties
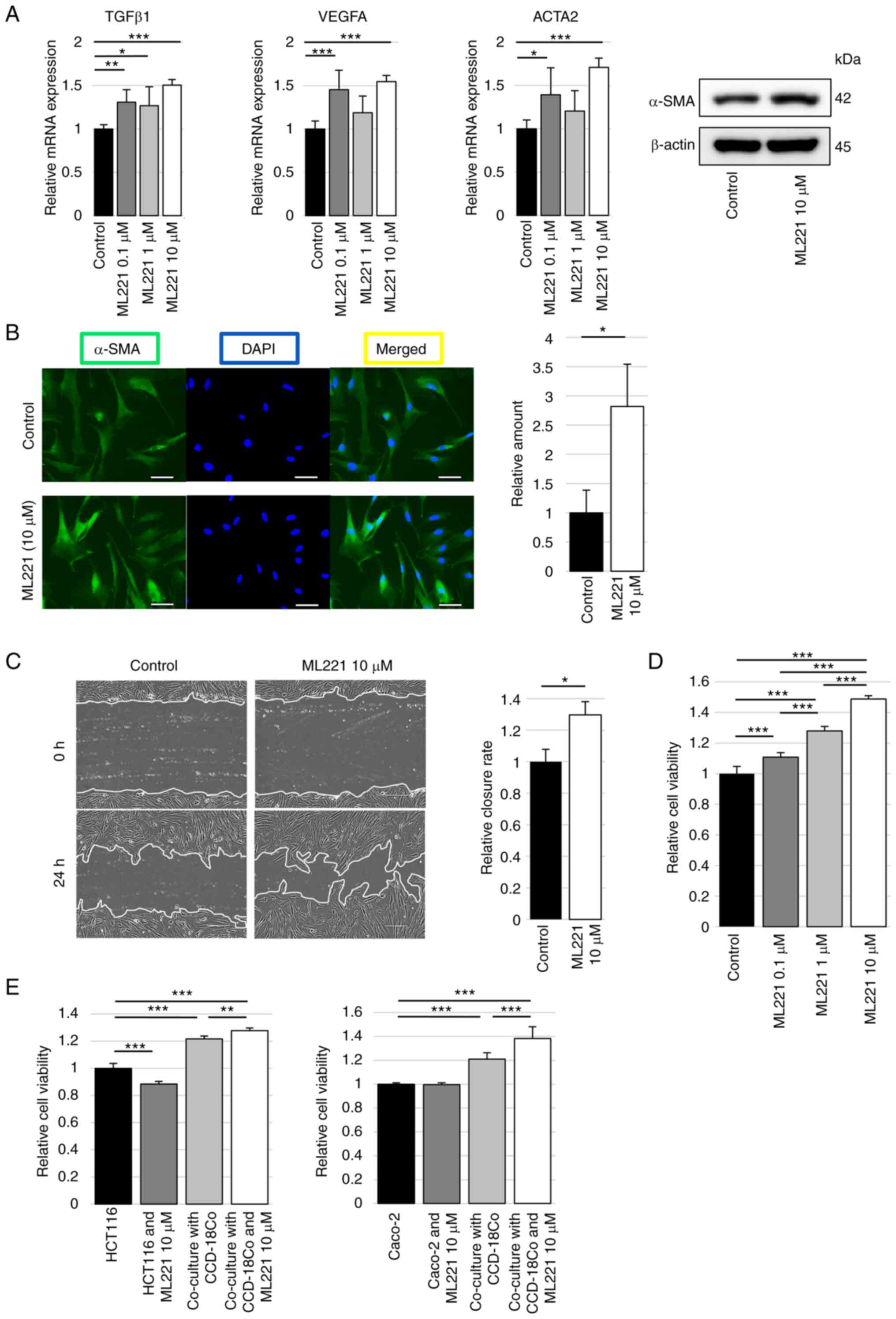
Experiments using ML221 in CCD-18Co cells were
performed to investigate the significance of APJ inhibition in
colon fibroblast modification. The relative mRNA expression levels
of TGFβ1, VEGF-A and ACTA2 were significantly increased in CCD-18Co
cells with ML221 compared with control, and the western blotting
analysis revealed increased α-SMA protein levels with ML221
(Figs. 2A and S2). α-SMA expression was next
visualized using fluorescence immunostaining, and the expression of
α-SMA was revealed to be significantly stronger in CCD-18Co cells
with ML221 than in the control (Fig.
2B). A wound healing assay of CCD-18Co cells to examine the
migration ability. The wound healing of CCD-18Co cells treated with
ML221 was significantly faster than that of the control cells
(Fig. 2C). Cell proliferation was
also examined using a WST assay. Adding ML221 significantly
increased the proliferation of CCD-18Co cells compared with the
control in ML221 dose-dependent manner (Fig. 2D). Furthermore, the proliferation
of colon cancer cells, which was increased by co-culture with
CCD-18Co cells, was significantly increased by adding ML221 to
CCD-18Co cells (Fig. 2E).
APJ suppression in fibroblasts induces
the modification into myofibroblasts-like properties
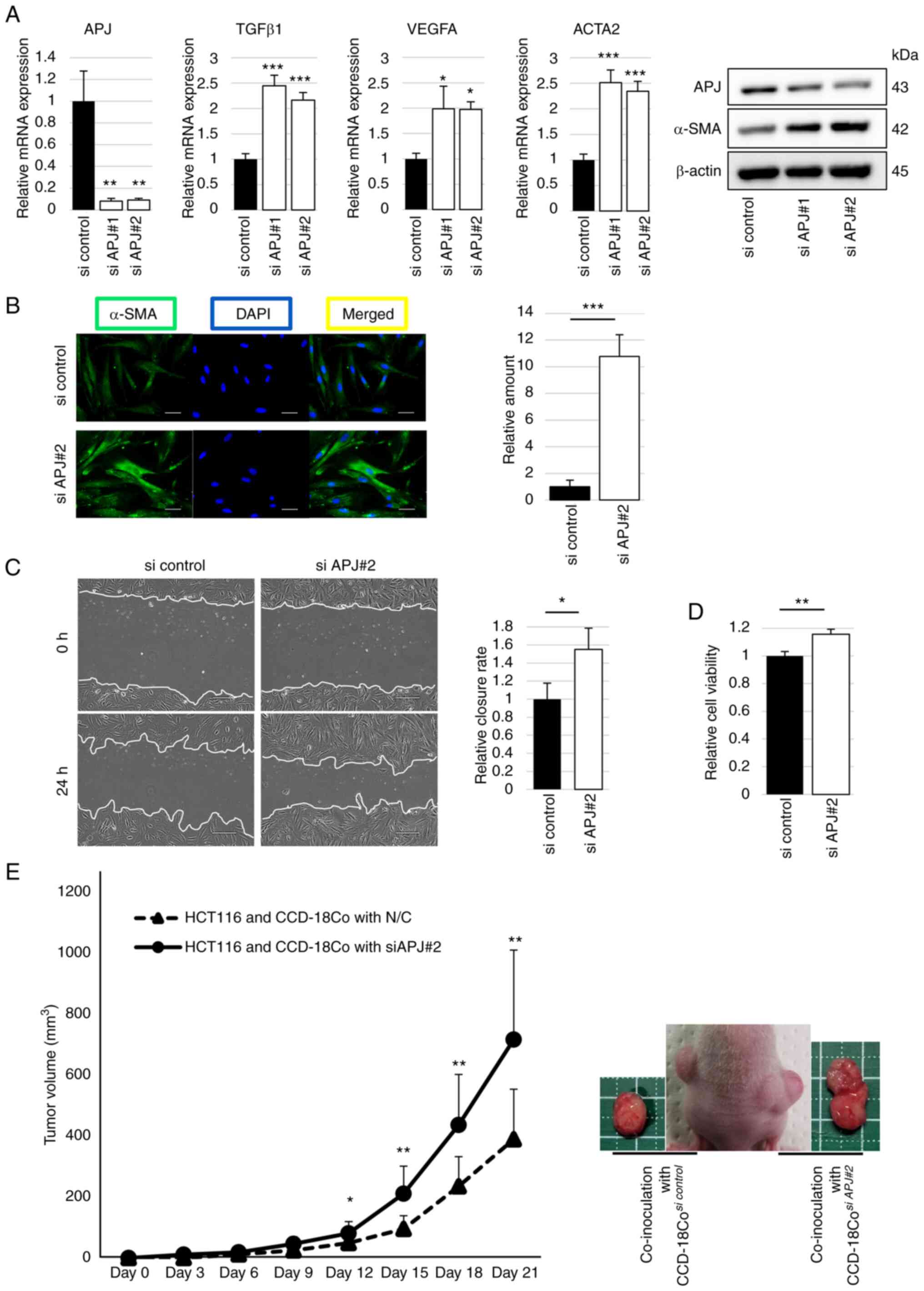
Next, APJ expression was suppressed in fibroblasts
using APJ siRNA. The APJ mRNA and protein expression levels were
successfully suppressed by siRNA with elevated α-SMA expression
levels. The relative mRNA expression levels of TGFβ1, VEGF-A and
ACTA2 were significantly increased with siRNA for APJ compared with
the control (Figs. 3A and
S3). α-SMA expression was also
visualized using fluorescence immunostaining, and α-SMA was more
strongly expressed in APJ-suppressed CCD-18Co cells than in the
control (Fig. 3B). Wound healing
assay revealed that the migration ability of APJ-suppressed
CCD-18Co cells was significantly increased compared with that of
the control (Fig. 3C). The WST
assay revealed that the viability of APJ-suppressed CCD-18Co cells
was significantly higher than that of the control (Fig. 3D).
Next, regarding fibroblast-mediated tumor growth,
the importance of APJ expression of fibroblasts was investigated
using TP53-wild cancer cells in xenograft experiments. Tumor
volumes of co-implanted HCT116 and CCD-18Co cells were
significantly higher than those of HCT116 cells alone, and APJ
suppression in CCD-18Co cells promoted faster tumor growth than the
control (Fig. 3E). These results
suggest that suppression of APJ in fibroblasts induces modification
of myofibroblast-like properties and accelerates tumor growth.
TGFβ-Smad pathway is activated in
APJ-suppressed colon fibroblasts
It has been reported that TGFβ1 induces α-SMA
production via phosphorylated Sma- and Mad-related protein 2/3
(Smad2/3); the addition of apelin inhibits the induction of
phosphorylated Smad2/3 by TGFβ1 (18,19). Next, the TGFβ-Smad pathway was
evaluated in fibroblasts as a mechanism by which the apelin-APJ
system can suppress the modification of fibroblasts into
myofibroblasts-like properties. The mRNA expression levels of
TGFβ1, VEGF-A and ACTA2 in CCD-18Co cells were increased by
rhTGF-β1 stimulation. These increased expression levels were
diminished significantly by the addition of recombinant human
apelin-13 (Fig. 4A), suggesting
the apelin-APJ system could suppress the TGFβ-induced fibroblasts
modification. Western blot analysis revealed that phosphorylated
Smad2/3 and α-SMA protein levels in CCD-18Co cells, which were
increased by rhTGF-β1 stimulation, were decreased by adding
recombinant human apelin-13 (Figs.
4B and S4). Increased
expression of phosphorylated Smad2/3 was also found in CCD-18Co
cells whose APJ expression was suppressed by ML221, siRNA or
co-cultured cancer cells (Figs.
4C and S5A-D). These results
suggested that the apelin-APJ system inhibits the TGFβ-Smad pathway
in fibroblasts.
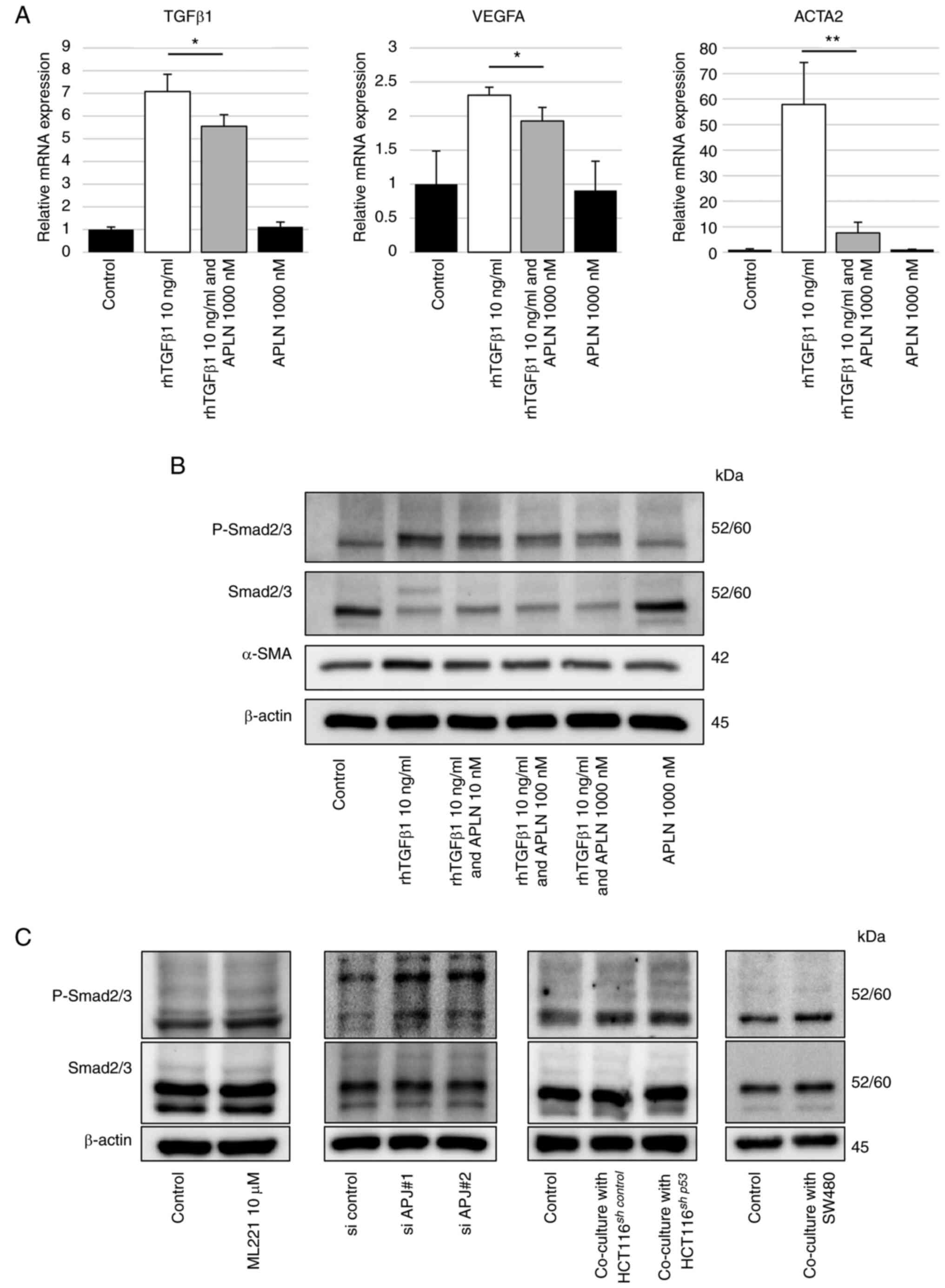 | Figure 4The APJ system inhibits TGFβ1-Smad
signaling in CCD-18Co cells. (A) Reverse transcription-quantitative
PCR was performed to assess TGFβ1, VEGFA and ACTA2 mRNA expression
in CCD-18Co cells treated with or without apelin-13 (1,000 nM)
and/or rhTGF-β1 (10 ng/ml). (B) Western blot analysis of
phosphorylated Smad2/3, Smad2/3 and α-SMA in CCD-18Co cells with or
without apelin-13 in the 10-1,000 nM range and/or rhTGF-β1 (10
ng/ml). (C) Western blotting was performed for phosphorylated
Smad2/3 and Smad2/3 in CCD-18Co cells with ML221 (10 µM),
with APJ siRNA, co-cultured with HCT116sh control
or HCT116sh p53 cells, and co-cultured with SW480
cells, compared with control, respectively. The immunoblots were
performed three times. Data are presented as the mean ± SD.
*P<0.05, and **P<0.01 vs. TGFβ1 group.
APJ, apelin receptor; rhTGF-β1, recombinant human transforming
growth factor beta 1; APLN, apelin-13; P-Smad2/3, phosphorylated
Smad2/3; α-SMA, alpha-smooth muscle actin; siRNA, short interfering
RNA; shRNA, short hairpin RNA. |
Cancer cell-derived exosomes suppress APJ
expression in fibroblasts
Cancer cell-derived exosomes and encapsulated miRNAs
are crucial communication tools in the tumor microenvironment
(12). Moreover, alterations in
TP53 expression in donor cancer cells have been reported to modify
their exosomal miRNA profiles and affect the expression of several
genes in the surrounding recipient cells (25,26). Cancer cell-derived exosomes were
used to explore the mechanisms underlying APJ suppression in colon
fibroblasts. Exosomes were collected from HCT116sh
control and HCT116sh p53 cells culture
supernatants by the ultracentrifugation method and the expression
of CD9, CD63 and ALIX, which are exosome markers, was confirmed by
western blotting. The size and morphology of isolated exosomes were
determined by transmission electron microscopy (Figs. 5A and S6). Furthermore, no significant
differences were observed in the protein levels of the pellets
isolated from HCT116sh control and
HCT116sh p53 cell culture supernatants (Fig. 5B). CCD-18Co cells were stimulated
with HCT116sh control- or HCT116sh
p53-derived exosomes to confirm the effects of the
exosomes. Western blotting demonstrated that APJ expression was
decreased in CCD-18Co cells transfected with HCT116sh
p53-derived exosomes (Figs.
5C and S7A). In addition,
the relative expression levels of TGFβ1, VEGF-A, and ACTA2 in mRNA
were significantly increased with HCT116sh
p53-derived exosomes (Fig.
5D).
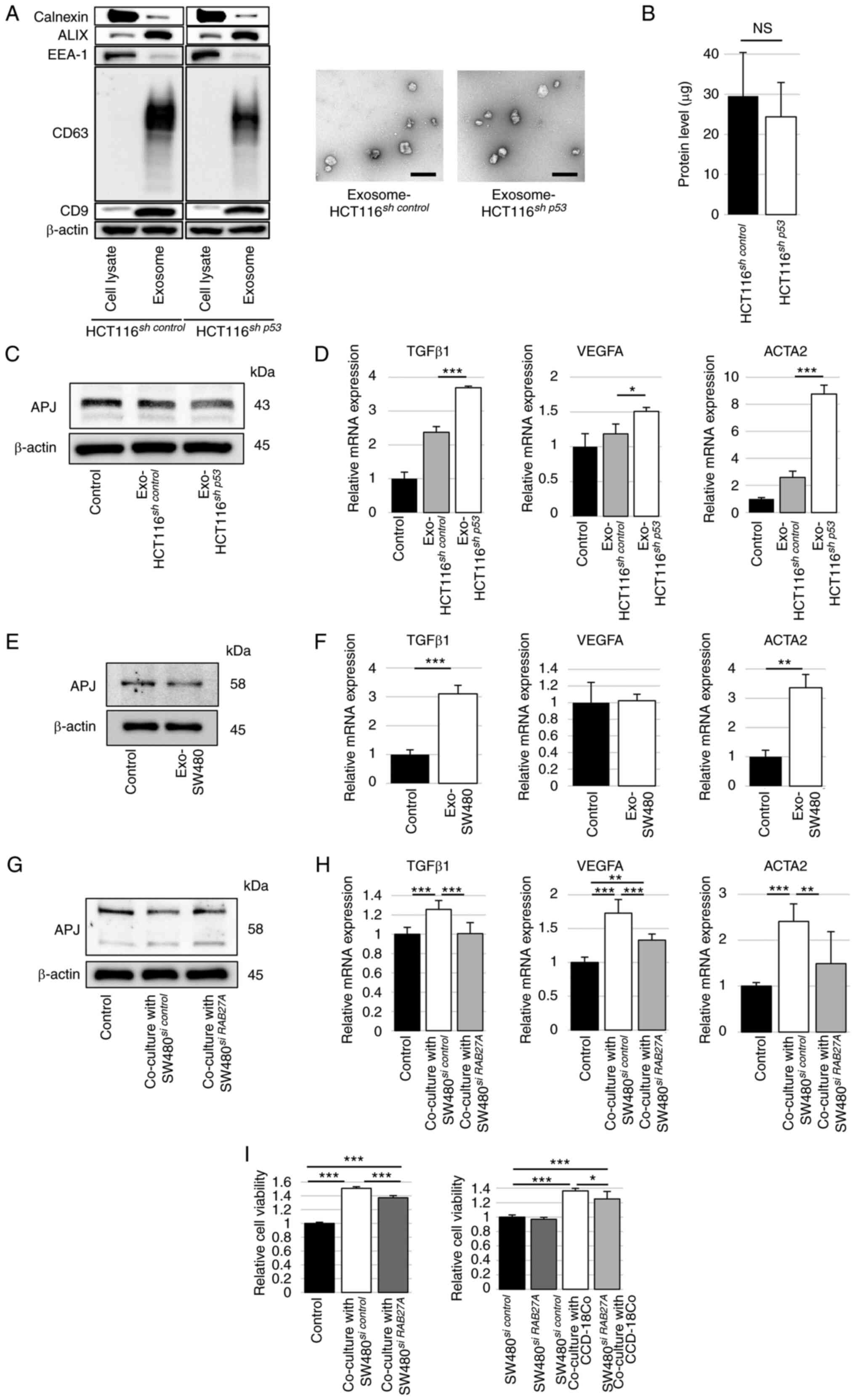 | Figure 5Exosomes derived from TP53-deficient
colon cancer cells suppress APJ expression in CCD-18Co cells. (A)
Western blot analysis was performed to assess exosomal specific
markers ALIX, CD63 and CD9 and exosome-negative proteins calnexin
and EEA-1 using cell lysates or isolated pellets from
HCT116sh control or HCT116sh
p53 cells (left). Representative images of transmission
electron microscopy for particles isolated from HCT116sh
control or HCT116sh p53 cells culture
supernatants (right). Scale bar, 200 nm. (B) Protein levels in
pellets isolated from HCT116sh control or
HCT116sh p53 cell culture supernatants. (C)
Western blot analysis of APJ in CCD-18Co cells treated with
HCT116sh control or HCT116sh
p53 cell-derived exosomes. The immunoblots were performed
three times. (D) RT-qPCR was performed to assess TGFβ1, VEGFA and
ACTA2 mRNA expression in CCD-18Co cells treated with
HCT116sh control or HCT116sh
p53 cell-derived exosomes. (E) Western blot analysis of
APJ in CCD-18Co cells treated with SW480 cell-derived exosomes. The
immunoblots were performed three times. (F) RT-qPCR was performed
to evaluate TGFβ1, VEGFA and ACTA2 mRNA expression in CCD-18Co
cells treated with SW480 cell-derived exosomes. (G) Western blot
analysis was performed for APJ in CCD-18Co cells co-cultured with
SW480 cells with or without siRNA against RAB27A. The immunoblots
were performed three times. (H) RT-qPCR was performed to
investigate TGFβ1, VEGFA and ACTA2 mRNA expression in CCD-18Co
cells co-cultured with SW480 cells with or without RAB27A siRNA.
(I) WST assay of CCD-18Co cells co-cultured with SW480 cells with
RAB27A siRNA compared with si control (left). WST assay of SW480
cells with or without siRNA against RAB27A and CCD-18Co cells
(right). Data are presented as the mean ± SD. A one-way ANOVA with
Tukey's post hoc test was performed to analyze the differences
among multiple groups. *P<0.05,
**P<0.01 and ***P<0.001. APJ, apelin
receptor; RT-qPCR, reverse transcription quantitative PCR; ALIX,
ALG-2 interacting protein X; EEA-1, early endosome antigen 1; CD63,
cluster of differentiation 63; CD9, cluster of differentiation 9;
shRNA, short hairpin RNA; NS, not significant; Exo, exosome; siRNA,
short interfering RNA. |
Next, exosomes were collected from the culture
supernatant of SW480 cells by ultracentrifugation. When the
exosomes were added to CCD-18Co cells, the protein expression of
APJ in CCD-18Co cells was decreased with SW480-derived exosomes
(Figs. 5E and S7B). The relative mRNA expression
levels of TGFβ1 and ACTA2 were significantly increased in CCD-18Co
cells with SW480-derived exosomes, as observed in HCT116sh
p53-derived exosomes (Fig.
5F). Exosome inhibition was evaluated using siRNA against
RAB27A, a regulator of exosome secretion. APJ expression in
CCD-18Co cells, suppressed by co-culture with SW480 cells, was
restored by suppressing RAB27A in SW480 cells (Figs. 5G and S7C). The mRNA expression of TGFβ1,
VEGF-A and ACTA2 in CCD-18Co cells, which were increased by being
co-cultured with SW480 cells, were significantly decreased by
suppressing RAB27A in SW480 cells (Fig. 5H). The WST assay revealed that the
viability of CCD-18Co cells, which was increased by co-culturing
with SW480 cells, was significantly decreased by the suppression of
RAB27A in SW480 cells. Furthermore, the proliferation of SW480
cells, which was increased by co-culture with CCD-18Co cells, was
reduced significantly by the suppression of RAB27A in SW480 cells
(Fig. 5I).
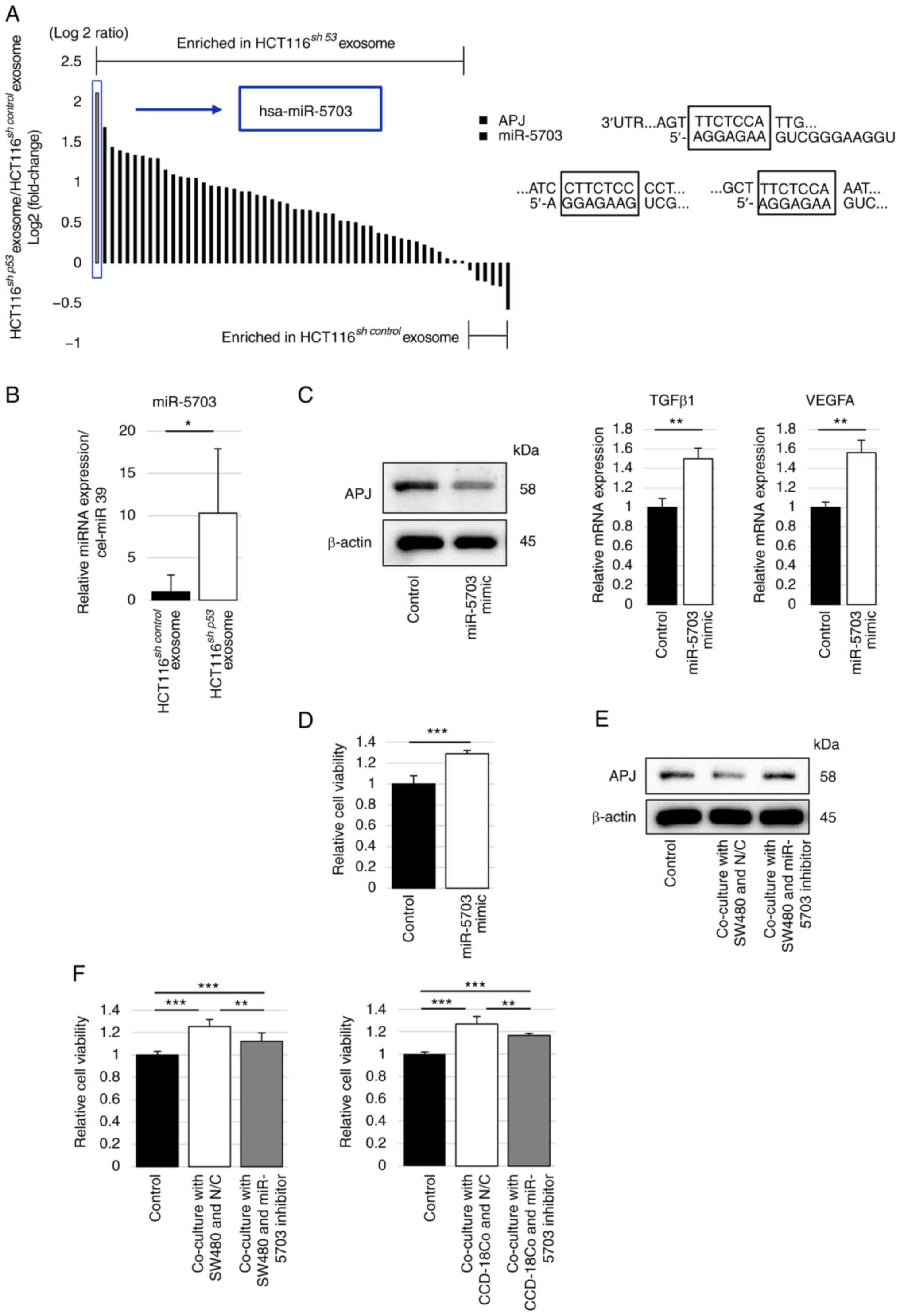
miR-5703 suppresses APJ expression in
fibroblasts
As a mechanism of APJ suppression in fibroblasts,
the role of miRNAs in cancer cell-derived exosomes was
investigated. The list of miRNAs targeting the APJ gene was created
using the miR-DB database and the miRNA profile in HCT116sh
control and HCT116sh p53-derived
exosomes was analyzed using the registered results of the miRNA
microarray analysis (NCBI GEO accession no. GSE120012). A total of
55 miRNAs targeting the APJ gene were detected in HCT116sh
control and HCT116sh p53-derived
exosomes. Of the 55 miRNAs, 49 were found to be highly expressed in
HCT116sh p53-derived exosomes compared with those
of HCT116sh control. Among them, miR-5703 was
identified as the miRNA with the highest expression in
HCT116sh p53-derived exosomes compared with those
of HCT116sh control (Fig. 6A).
Next, exosomes were collected from HCT116sh
control and HCT116sh p53 cell culture
supernatants by ultracentrifugation and the expression ratio of
miRNAs was validated using RT-qPCR analysis. The relative
expression level of miR-5703 was significantly higher in
HCT116sh p53-derived exosomes than in
HCT116sh control-derived exosomes (Fig. 6B). The miRNA mimic was transfected
into CCD-18Co cells to investigate the effect of the miRNA on APJ
expression in fibroblasts; then, the APJ protein expression of
CCD-18Co cells was suppressed and the mRNA expression levels of
TGFβ1 and VEGF-A in CCD-18Co cells were significantly increased
compared with the control (Figs.
6C and S8A). In addition,
the WST assay was performed to investigate the viability of
CCD-18Co cells treated with the miRNA mimic, which was
significantly higher than that of the control (Fig. 6D). The effects of a specific
miR-5703 inhibitor were also explored. Transfection of the miR-5703
inhibitor into CCD-18Co cells after co-culturing with SW480 cells
restored the APJ expression in CCD-18Co cells, which was suppressed
in the co-cultured SW480 cells (Figs.
6E and S8B). The WST assay
revealed that the viability of CCD-18Co cells, which was increased
by co-culturing with SW480 cells, was significantly decreased by
transfection of the miR-5703 inhibitor into CCD-18Co cells.
Finally, the effect of the miR-5703 inhibitor on
fibroblast-mediated cancer cell growth was investigated. The
miR-5703 inhibitor suppressed the proliferation of SW480 cells
co-cultured with CCD-18Co cells (Fig.
6F).
Discussion
The present study revealed that TP53-functional
deficient colorectal cancer cells induce the
fibroblast-to-myofibroblast transition via suppression of APJ
expression in co-existing fibroblasts, and APJ suppression in
fibroblasts increases proliferation and migration abilities and
accelerates tumor growth. It was also revealed that specific miRNAs
encapsulated in cancer cell-derived exosomes play a pivotal role in
the mechanism by which TP53-functional deficient cancer cells
suppress APJ expression in fibroblasts. In addition, a specific
miRNA inhibitor can restore APJ expression in fibroblasts and
suppress fibroblast-mediated tumor cell growth.
Previous studies have reported that the APJ system
in fibroblasts functioned as a mechanism of suppression of
fibroblast-to- myofibroblast transition and related non-neoplastic
organ fibrosis such as heart, lung, liver, skin, or kidney;
furthermore, one of the mechanisms by which apelin can exert a
protective effect against the fibrosis is considered to be the
suppression of the TGFβ-Smad pathway (18,19). For instance, activation of the APJ
pathway has been reported to exert diverse physiological effects,
including anti-fibrotic and anti-remodeling effects, leading to
cardiovascular protection (16).
The APJ pathway is also closely related to renal fibrosis and
improves renal interstitial fibrosis via inhibition of
phosphorylated Smad2/3 induced by TGFβ-stimulation (19). There are studies that in pulmonary
fibrosis, the apelin-APJ system inhibits the phosphorylation of
Smad2/3 and suppresses extracellular matrix production induced by
TGF-β (27), and in renal tubular
epithelial cells, it inhibits phosphorylated Smad2/3 via activation
of PKC-ε (28). It was posited by
the authors that the apelin-APJ system could inhibit
tumor-promoting CAFs, especially in acquiring myofibroblast-like
characteristics. Tumor-promoting CAFs are generally reported to be
activated by various factors and show a myofibroblast-like
phenotype represented by increased α-SMA expression (7). In addition, it was stated in the
Consensus Molecular Subtypes classification that colorectal cancer
with strong stromal response type has a poor prognosis (29). To the best of the authors'
knowledge, the present study is the first to demonstrate that the
APJ system can function as an inhibitory pathway for CAFs, and that
suppression of the APJ pathway in fibroblasts by cancer cells
promotes tumor progression.
Several studies have focused on the significance of
the APJ pathway in cancer cells but not in CAFs. Apelin signaling
in cancer cells has been reported to have a tumor-promoting effect
via the PI3k/Akt pathway or the activation of Notch3 and STAT3
(22,30). The mRNA and protein levels of
apelin and APJ in colorectal cancer are reportedly higher than in
control tissues (31). The
overexpression of apelin and APJ has also been demonstrated by
immunohistochemistry in human colon adenomas and adenocarcinomas
(20). The expression levels of
apelin and APJ have also been reported to be associated with poor
prognosis in malignant tumors of several organs, such as breast
cancer (32), cervical cancer
(33), ovarian cancer (30) and colorectal cancer (21). These data suggest that both apelin
and APJ expression concurrently increase and create an autocrine
loop, leading to cancer progression. These findings regarding the
apelin-APJ system in cancer cells differ from the suppressed
expression of APJ in fibroblasts demonstrated in the present study.
As apelin signaling in cancer cells is considered to accelerate
tumor growth, inhibition of the apelin-APJ system is a possible
therapeutic target. Indeed, Hall et al (34) demonstrated that inhibition of the
apelin-APJ axis using ML221 suppressed tumor growth in vitro
and in a xenograft model of cholangiocarcinoma cells. However,
contrary to the tumor-suppressive effect of ML221 in cancer cells,
it was revealed that APJ inhibition in fibroblasts using ML221
induced the modification of myofibroblast-like properties, and the
APJ-suppressed fibroblasts showed a tumor-promoting effect.
Therefore, it was considered that the systemic administration of an
APJ inhibitor may contribute to tumor suppression owing to its
antitumor effect on cancer cells. However, the APJ inhibitor can
have opposite tumor-promoting effects by affecting the tumor
stroma.
Several mechanisms for tumor-promoting CAFs have
been reported, including proliferation, activation,
trans-differentiation and recruitment (8). Activation of normal tissue
fibroblasts is considered one of the origins of CAFs, and diverse
types of liquid factors, such as growth factors, cytokines, or
chemokines secreted from cancer cells, have been reported to affect
normal tissue fibroblasts for the acquisition of a CAF-like
phenotype (8). Exosomes are
crucial cell-to-cell communication tools in the tumor
microenvironment (35). Exosomes
derived from donor cells capsulize various gene-expression
modulators, such as miRNAs, and can transport them to recipient
cells and modulate specific gene expression in recipient cells
(36,37). The present study revealed that
exosomes derived from cancer cells with p53 deficiency contain
higher miRNAs that suppress APJ expression in fibroblasts. Among
these, focus was addressed on the expression of miR-5703. Although
in a previous study, miR-5703 was reported to be involved in breast
cancer and bone metastasis by targeting the Runx2 pathway (38), the present study was the first to
identify that miR-5703 could suppress APJ expression in fibroblasts
and promote tumor progression. The administration of a miRNA
inhibitor restored APJ expression in fibroblasts, which was
downregulated in p53-mutant colon cancer cells. Furthermore, a
specific inhibitor suppressed fibroblast-mediated cancer cell
growth. It was considered that the apelin-APJ system in the cancer
stroma and miRNA-mediated APJ suppression in fibroblasts could be
novel therapeutic targets.
In the present study, it was revealed that
APJ-suppressed fibroblasts increased the proliferation of colon
cancer cells and accelerated tumor growth in a xenograft model.
However, the details of these mechanisms remain unclear.
Furthermore, in the histological analysis of advanced stage tumors,
numerous cancer cells harbor p53 mutations, and several different
cell types (including fibroblasts) are intermingled. Because the
interaction between cancer cells and fibroblasts in advanced stage
tumors is difficult to understand and therefore not particularly
appropriate considering the focus of the present study, the current
analysis was restricted to tissue samples corresponding to early
colorectal cancers. Further research is needed to confirm the
clinical value of the present findings regarding the miRNA-mediated
mechanisms by which normal tissue fibroblasts acquire the CAF-like
phenotype and tumor-promoting effects in clinical settings.
Developing a delivery system for the inhibitor of specific miRNAs
to target the cancer stroma is also required to use the miRNA
inhibitor as a therapeutic tool. Despite these limitations, the
present study revealed a novel mechanism of fibroblast modification
into a CAF-like phenotype. As aforementioned, the fibrotic
infiltration in the tumor stroma may be associated with
APJ-suppressed fibroblasts. As previously reported, colorectal
cancer with strong stromal response type has a poor prognosis
(29), and therefore it is
conceivable that APJ-suppressed fibroblasts may be associated with
colorectal cancer prognosis. Further research on this issue is
warranted. p53-functional deficiency in colorectal cancer cells
induces fibroblast modification via exosomal miRNAs. Targeting the
apelin-APJ system in the cancer stroma may be a novel therapeutic
strategy for p53-functional deficient colorectal cancer.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HS conceptualized the study, conducted formal
analysis and investigation, performed data visualization and wrote
the original draft; and contributed equally to data validation and
providing methodology. YH conceptualized the study and contributed
equally to formal analysis and writing-reviewing and editing the
manuscript, and supported supervision. SY conceptualized the study,
conducted investigation and contributed equally to
writing-reviewing and editing the manuscript. EK, KN, MK, RU, TI
and AS contributed equally to conducting investigation and
supported writing-reviewing and editing the manuscript. TY, YT, SS
and HI contributed equally to conducting investigation and
writing-reviewing and editing the manuscript. TT conceptualized and
supervised the study, and contributed equally to writing-reviewing
and editing the manuscript. HS and YH confirm the authenticity of
all the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved (approval no. 20061)
by the Ethics Committee of Osaka University Graduate School of
Medicine (Osaka, Japan). Written informed consent was obtained from
all patients who provided resected specimens. The study design
adhered to the principles of the Declaration of Helsinki (October
2013). The animal experimental procedures were performed per the
Osaka University guidelines for animal experiments and were
approved (approval no. 30-015-077) by the Animal Care and Use
Committee of Osaka University Graduate School of Medicine (Osaka,
Japan).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
α-SMA
|
alpha-smooth muscle actin
|
|
APJ
|
apelin receptor
|
|
ATCC
|
American Type Culture Collection
|
|
CAF
|
cancer-associated fibroblast
|
|
DMEM
|
Dulbecco's Modified Eagle Medium
|
|
EMEM
|
Eagle's Minimum Essential Medium
|
|
FBS
|
fetal bovine serum
|
|
PBS
|
phosphate-buffered saline
|
|
rhTGF-β1
|
recombinant human transforming growth
factor-beta 1
|
|
shRNA
|
short hairpin RNA
|
|
TGFβ
|
transforming growth factor beta
|
|
TP53
|
tumor protein 53
|
|
WST
|
water-soluble tetrazolium
|
Acknowledgments
Not applicable.
Funding
The present study was supported by JSPS KAKENHI (grant no.
JP21K15922) from the Ministry of Education, Culture, Sports,
Science and Technology of Japan.
References
|
1
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics. CA Cancer J Clin. 71:7–33. 2021.
|
|
2
|
Park JW, Seo MJ, Cho KS, Kook MC, Jeong
JM, Roh SG, Cho SY, Cheon JH and Kim HK: Smad4 and p53 synergize in
suppressing autochthonous intestinal cancer. Cancer Med.
11:1925–1936. 2022.
|
|
3
|
Villalba M, Evans SR, Vidal-Vanaclocha F
and Calvo A: Role of TGF-β in metastatic colon cancer: It is
finally time for targeted therapy. Cell Tissue Res. 370:29–39.
2017.
|
|
4
|
André T, Shiu KK, Kim TW, Jensen BV,
Jensen LH, Punt C, Smith D, Garcia-Carbonero R, Benavides M, Gibbs
P, et al: Pembrolizumab in microsatellite-instability-high advanced
colorectal cancer. N Engl J Med. 383:2207–2218. 2020.
|
|
5
|
Cremolini C, Loupakis F, Antoniotti C,
Lupi C, Sensi E, Lonardi S, Mezi S, Tomasello G, Ronzoni M,
Zaniboni A, et al: FOLFOXIRI plus bevacizumab versus FOLFIRI plus
bevacizumab as first-line treatment of patients with metastatic
colorectal cancer: updated overall survival and molecular subgroup
analyses of the open-label, phase 3 TRIBE study. Lancet Oncol.
16:1306–1315. 2015.
|
|
6
|
Arnold D, Lueza B, Douillard JY, Peeters
M, Lenz HJ, Venook A, Heinemann V, Van Cutsem E, Pignon JP,
Tabernero J, et al: Prognostic and predictive value of primary
tumour side in patients with RAS wild-type metastatic colorectal
cancer treated with chemotherapy and EGFR-directed antibodies in
six randomized trials. Ann Oncol. 28:1713–1729
|
|
7
|
Togo S, Polanska UM, Horimoto Y and Orimo
A: Carcinoma-associated fibroblasts are a promising therapeutic
target. Cancers (Basel). 5:149–169. 2013.
|
|
8
|
Sahai E, Astsaturov I, Cukierman E,
DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR,
Hunter T, et al: A framework for advancing our understanding of
cancerassociated fibroblasts. Nat Rev Cancer. 20:174–186. 2020.
|
|
9
|
Czekay RP, Cheon DJ, Samarakoon R, Kutz SM
and Higgins PJ: Cancer-associated fibroblasts: Mechanisms of tumor
progression and novel therapeutic targets. Cancers.
14:12312022.
|
|
10
|
Leroy B, Anderson M and Soussi T: TP53
mutations in human cancer: Database reassessment and prospects for
the next decade. Hum Mutat. 35:672–688. 2014.
|
|
11
|
Hayashi Y, Tsujii M, Kodama T, Akasaka T,
Kondo J, Hikita H, Inoue T, Tsujii Y, Maekawa A, Yoshii S, et al:
p53 functional deficiency in human colon cancer cells promotes
fibroblast-mediated angiogenesis and tumor growth. Carcinogenesis.
37:972–984. 2016.
|
|
12
|
Yoshii S, Hayashi Y, Iijima H, Inoue T,
Kimura K, Sakatani A, Nagai K, Fujinaga T, Hiyama S, Kodama T, et
al: Exosomal microRNAs derived from colon cancer cells promote
tumor progression by suppressing fibroblast TP53 expression. Cancer
Sci. 110:2396–2407. 2019.
|
|
13
|
Tatemoto K, Hosoya M, Habata Y, Fujii R,
Kakegawa T, Zou MX, Kawamata Y, Fukusumi S, Hinuma S, Kitada C, et
al: Isolation and characterization of a novel endogenous peptide
ligand for the human APJ receptor. Biochem Biophys Res Commun.
251:471–476. 1998.
|
|
14
|
Masoumi J, Jafarzadeh A, Khorramdelazad H,
Abbasloui M, Abdolalizadeh J and Jamali N: Role of apelin/APJ axis
in cancer development and progression. Adv Med Sci. 65:202–213.
2020.
|
|
15
|
Narayanan S, Harris DL, Maitra R and
Runyon SP: Regulation of the apelinergic system and its potential
in cardiovascular disease: Peptides and small molecules as tools
for discovery. J Med Chem. 58:7913–7927. 2015.
|
|
16
|
Nagpal V, Rai R, Place AT, Murphy SB,
Verma SK, Ghosh AK and Vaughan DE: MiR-125b is critical for
fibroblast-to-myofibroblast transition and cardiac fibrosis.
Circulation. 133:291–301. 2016.
|
|
17
|
Kim J: Apelin-APJ signaling: A potential
therapeutic target for pulmonary arterial hypertension. Mol Cells.
37:196–201. 2014.
|
|
18
|
Yokoyama Y, Sekiguchi A, Fujiwara C,
Uchiyama A, Uehara A, Ogino S, Torii R, Ishikawa O and Motegi SI:
Inhibitory regulation of skin fibrosis in systemic sclerosis by
apelin/APJ signaling. Arthritis Rheumatol. 70:1661–1672. 2018.
|
|
19
|
Wang LY, Diao ZL, Zhang DL, Zheng JF,
Zhang QD, Ding JX and Liu WH: The regulatory peptide apelin: A
novel inhibitor of renal interstitial fibrosis. Amino Acids.
46:2693–2704. 2014.
|
|
20
|
Picault FX, Chaves-Almagro C, Projetti F,
Prats H, Masri B and Audigier Y: Tumour co-expression of apelin and
its receptor is the basis of an autocrine loop involved in the
growth of colon adenocarcinomas. Eur J Cancer. 50:663–674.
2014.
|
|
21
|
Zuurbier L, Rahman A, Cordes M, Scheick J,
Wong TJ, Rustenburg F, Joseph JC, Dynoodt P, Casey R, Drillenburg
P, et al: Apelin: A putative novel predictive biomarker for
bevacizumab response in colorectal cancer. Oncotarget.
8:42949–42961. 2017.
|
|
22
|
Chen T, Liu N, Xu GM, Liu TJ, Liu Y, Zhou
Y, Huo SB and Zhang K: Apelin13/APJ promotes proliferation of colon
carcinoma by activating notch3 signaling pathway. Oncotarget.
8:101697–101706. 2017.
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
|
|
24
|
Wang P, Liang J, Wang Z, Hou H, Shi L and
Zhou Z: The prognostic value of p53 positive in colorectal cancer:
A retrospective cohort study. Tumour Biol.
39:10104283177036512017.
|
|
25
|
Trivedi M, Talekar M, Shah P, Ouyang Q and
Amiji M: Modification of tumor cell exosome content by transfection
with wt-p53 and microRNA-125b expressing plasmid DNA and its effect
on macrophage polarization. Oncogenesis. 5:e2502016.
|
|
26
|
Cooks T, Pateras IS, Jenkins LM, Patel KM,
Robles AI, Morris J, Forshew T, Appella E, Gorgoulis VG and Harris
CC: Mutant p53 cancers reprogram macrophages to tumor-supporting
macrophages via exosomal miR-1246. Nat Commun. 9:7712018.
|
|
27
|
Shen J, Feng J, Wu Z, Ou Y, Zhang Q, Nong
Q, Wu Q, Li C, Tan X, Ye M, et al: Apelin prevents and alleviates
crystalline silica-induced pulmonary fibrosis via inhibiting
transforming growth factor beta 1-triggered fibroblast activation.
Int J Biol Sci. 19:4004–4019. 2023.
|
|
28
|
Wang LY, Diao ZL, Zheng JF, Wu YR, Zhang
QD and Liu WH: Apelin attenuates TGF-β1-induced epithelial to
mesenchymal transition via activation of PKC-ε in human renal
tubular epithelial cells. Peptides. 96:44–52. 2017.
|
|
29
|
Guinney J, Dienstmann R, Wang X, Reynies
A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G,
Angelino P, et al: The consensus molecular subtypes of colorectal
cancer. Nat Med. 21:1350–1356. 2015.
|
|
30
|
Neelakantan D, Dogra S, Devapatla B,
Jaiprasart P, Mukashyaka MC, Janknecht R, Dwivedi SK, Bhattacharya
R, Husain S, Ding K and Woo S: Multifunctional APJ pathway promotes
ovarian cancer progression and metastasis. Mol Cancer Res.
17:1378–1390. 2019.
|
|
31
|
Podgórska M, Diakowska D,
Pietraszek-Gremplewicz K, Nienartowicz M and Nowak D: Evaluation of
apelin and apelin receptor level in the primary tumor and serum of
colorectal cancer patients. J Clin Med. 8:15132019.
|
|
32
|
Hu D, Cui Z, Peng W, Wang X, Chen Y and Wu
X: Apelin is associated with clinicopathological parameters and
prognosis in breast cancer patients. Arch Gynecol Obstet.
306:1185–1195. 2022.
|
|
33
|
Yusha C, Lin X, Zheng J, Chen J, Xue H and
Zheng X: APLN: A potential novel biomarker for cervical cancer. Sci
Prog. 104:3685042110113412021.
|
|
34
|
Hall C, Ehrlich L, Venter J, O'Brien A,
White T, Zhou T, Dang T, Meng F, Invernizzi P, Bernuzzi F, et al:
Inhibition of the apelin/apelin receptor axis decreases
cholangiocarcinoma growth. Cancer Lett. 386:179–188. 2017.
|
|
35
|
Kosaka N, Yoshioka Y, Fujita Y and Ochiya
T: Versatile roles of extracellular vesicles in cancer. J Clin
Invest. 126:1163–1172. 2016.
|
|
36
|
Valadi H, Ekström K, Bossios A, Sjöstrand
M, Lee JJ and Lötvall JO: Exosome-mediated transfer of mRNAs and
microRNAs is a novel mechanism of genetic exchange between cells.
Nat Cell Biol. 9:654–659. 2007.
|
|
37
|
Zhang Y, Liu D, Chen X, Li J, Li L, Bian
Z, Sun F, Lu J, Yin Y, Cai X, et al: Secreted monocytic miR-150
enhances targeted endothelial cell migration. Mol Cell. 39:133–144.
2010.
|
|
38
|
Pranavkrishna S, Sanjeev G, Akshaya RL,
Rohini M and Selvamurugan N: A computational approach on studying
the regulation of TGF-β1-stimulated Runx2 expression by microRNAs
in human breast cancer cells. Comput Biol Med. 137:1048232021.
|