Introduction
The evolutionary-conserved Hedgehog (Hh) signaling
pathway plays an essential role in developmental processes such as
embryonic development and cell differentiation (1-5).
In vertebrates, the canonical Hh pathway is triggered by the
interaction between a 12-pass transmembrane protein Patched
(Ptch1/2) and one of the three Hh ligands, sonic Hh (Shh), indian
Hh (Ihh) and desert Hh (Dhh), of which Shh is the most potent
ligand for the Hh pathway. In the absence of Hh ligands, Ptch is
localized in the base of the primary cilium (PC) and suppresses the
activation of GPCR-like transmembrane protein Smoothened (Smo) by
indirectly blocking Smo localization to the PC, while full-length
GLI is sequestered by Sufu in the cytosol and phosphorylated by
PKA, GSK3β and CK1. Hyperphosphorylated GLI2/3 undergoes partial
proteasomal degradation via β-TRCP-mediated ubiquitination, and the
resulting truncated forms of GLI (GLI2R, GLI3R) translocate into
the nucleus, where they then act as transcriptional repressors.
Upon Hh ligand binding, Ptch is internalized and
degraded via Smurf-mediated ubiquitination. This causes Smo to be
liberated and activated through phosphorylation by CK1 and GRK2,
then translocated to the PC. At this point, Smo releases GLI
transcription factors from suppressive interaction with Sufu, thus
bypassing the phosphorylation and subsequent GLI2/3 cleavage. The
active forms of GLI (GLI1 and GLI2A) shuffle to the nucleus and
turn on the transcriptions of an array of downstream genes,
including those related to the Hh pathway (for example, GLI1, Smo
and Ptch1), proliferation (such as cyclinD1 and c-Myc), survival
(for example, Bcl-2) and stem cell self-renewal (including Nanog
and Sox2) (6).
The Hh pathway is associated with important
phenotypes of cancers, such as cancer cell growth, self-renewal of
cancer stem cells and anticancer drug resistance. Uncontrolled
canonical Hh signaling is associated with the development of
various cancer types. Almost all cases of sporadic basal cell
carcinomas (BCCs) are attributable to Hh pathway activation. Loss
of function mutation of Ptch1 accounts for ~90% of patients with
BCC, while R562Q (Smo-M1) and W535L (Smo-M2) are the main
activating Smo mutations found in the remaining BCC population
(7). In the case of breast
cancers, the levels of Smo and GLI1 are significantly higher in
triple-negative breast cancer (TNBC) than they are in other
subtypes of breast cancers, and the expression levels are
correlated with tumor stages of TNBC (8). Hh ligands are highly expressed in
most patients with colorectal cancer (CRC) (9), and the expression levels of Ptch1
and Smo gradually increase with the progression of CRC (10). Loss of heterozygosity or somatic
mutation of Ptch1 or Sufu also occurs in numerous other cancers,
including bladder cancer, esophageal squamous cell carcinoma,
medulloblastoma and rhabdomyosarcoma.
Moreover, a number of studies using various cancer
models have revealed that GLI transcription factors can be
modulated by oncogenic signaling pathways such as Raf/MEK/ERK and
PI3K/Akt signaling, independent of Hh ligand or PTCH/Smo. This
so-called 'non-canonical Hh pathway' activation also plays an
important role in cancer development. For example, in pancreatic
ductal adenocarcinoma (PDCA), mutant K-Ras mediated Gli1
transcriptional activation is needed for cell proliferation and
PDCA formation in vivo (11). In esophageal adenocarcinoma,
mTOR-S6K1 signaling increases both the phosphorylation and
transcriptional activity of GLI in a Smo-independent manner
(12). In melanoma cells, the
stability and activity of Gli1 is upregulated by the oncogenic WIP1
phosphatase (13).
Hence, the Hh pathway is an important cancer
therapeutic target. Among the oncogenic components of Hh pathway
components, Smo has represented the primary drug target, presumably
because it has a large drug-binding pocket. Various classes of Smo
antagonists have been developed, among which vismodegib (GDC-0449)
and sonidegib (LDE225) have been FDA-approved as cancer drugs for
advanced and metastatic BCC. Despite the favorable clinical
efficacy of the Smo inhibitors, the patients' tumor tissues showed
the emergence of drug resistance through several mechanisms,
including the acquisition of drug-resistant Smo mutations, the
amplification of GLI2 and its downstream genes, and the
upregulation of non-canonical GLI activation signaling. This
situation warrants the development of next-generation inhibitors
targeting the drug-resistant mutant Smo or targeting the
upregulated non-canonical GLI pathway (5).
To identify new class Hh pathway inhibitors, several
groups have conducted small molecule screenings using
GLI1-dependent reporter assays and identified compounds that
inhibit GLI1-mediated transcription with novel modes of action.
GANT-58 and GANT-61, both of which inhibit GLI1 activity in the
nucleus, were identified, and GANT-61 was shown to act by
interfering with GLI-DNA binding (14,15). Furthermore, a previous study
involving a small molecule screening of 120,000 compounds
identified four compounds (HPI-1 to 4), each with a distinct
mechanism of action but relatively low potency (16). In the present study, an
FDA-approved drug library was screened using GLI-luciferase
reporter assay in HCT116 cells, and it was revealed that
daunorubicin inhibits GLI1-dependent transcription. In the present
study, it was reported for the first time, to the best of the
authors' knowledge, that the anticancer activity of daunorubicin is
mediated in part by the suppression of the non-canonical Hh
pathway. The current study is expected to provide a rationale for
expanding the clinical applicability of daunorubicin.
Materials and methods
Cell culture
The NIH3T3 cell line stably expressing
GLI-luciferase reporter construct was purchased from BPS Bioscience
Inc., and maintained in DMEM (GenDEPOT, LLC) plus 10% fetal calf
serum (Gibco; Thermo Fischer Scientific, Inc.) and 1%
penicillin/streptomycin. Human CRC cell lines HCT116, SNU283, HCT8
(colon; colorectal) and HT29, DLD-1 (colon) were purchased from
Korea Cell Line Bank (Seoul, Korea) and analyzed by STR profiling.
HCT116 p53 knockout cell line was provided from the Keck School of
Medicine of USC (Professor LIN ZHANG). Cells were cultured in
RPMI-1640 medium (GenDEPOT, LLC) with 10% fetal bovine serum
(Gibco; Thermo Fischer Scientific, Inc.), 1 mM L-glutamine, 26 mM
sodium bicarbonate and 1% P/S (penicillin/streptomycin). All cells
were cultured at 37°C in a 5% CO2 incubator. All cell
lines were confirmed not to be infected with mycoplasma using a
mycoplasma detection kit (cat. no. SMD0173; BIOMAX).
Reagents and antibodies
The FDA-approved drug library (1,018 drugs),
vismodegib and daunorubicin were purchased from Selleck Chemicals.
BODIPY-cyclopamine was obtained from BioVision, Inc. Oxaliplatin
and Irinotecan were purchased from Sigma-Aldrich; Merck KGaA.
Z-VAD-FMK (Promega Corporation) was treated at 25 μM as a
caspase inhibitor. Anti-SMO/Smoothened (1:1,000; cat. no.
sc-166685;), anti-Lamin B (1:1,000; cat. no. sc-6216), Anti-Bcl-2
(1:1,000; cat. no. sc-509), anti-Bcl-xL (1:1,000; cat. no.
sc-8392), anti-Bax (1:1,000; cat. no. sc-20067), anti-survivin
(1:1,000; cat. no. sc-17779), anti-p53 (1:1,000; cat. no. sc-126),
anti-p21 (1:1,000; cat. no. sc-397), anti-p300 (1:1,000; cat. no.
sc-32244) and anti-PCAF (1:1,000; cat. no. sc-13124) antibodies
were all purchased from Santa Cruz Biotechnology, Inc.
Anti-PTCH/Patched (1:1,000; cat. no. 2468S), anti-β-tubulin
(1:1,000; cat. no. 2126S), anti-Mcl-1 (1:1,000; cat. no. 4572S),
anti-Bak (1:1,000; cat. no. 12105S), Anti-XIAP (1:1,000; cat. no.
2042S), anti-Bid (1:1,000; cat. no. 2002S), anti-Puma (1:1,000;
cat. no. 4976S), anti-NOXA (1:1,000; cat. no. 14766S), Anti-BIM
(1:1,000; cat. no. 2933S), anti-GLI1 (1:500; cat. no. 3538S),
anti-GLI2 (1:1,000; cat. no. 2585S), anti-cleaved poly (ADP-ribose)
polymerase (c-PARP) (1:1,000; cat. no. 9541S), anti-PARP (1:1,000;
cat. no. 9542S), anti-c-caspase-3 (1:1,000; cat. no. 9664S),
Anti-caspase-3 (1:1,000; cat. no. 9662S), anti-caspase-9 (1:1,000;
cat. no. 9502S), anti-c-caspase-8 (1:1,000; cat. no. 9496S),
anti-caspase-8 (1:1,000; cat. no. 4790T), anti-β-TrCP (1:1,000;
cat. no. 4394S) and horseradish peroxidase (HRP)-conjugated
anti-rabbit (1:2,000; cat. no. 7074S) were purchased from Cell
Signaling Technology, Inc. Mouse IgG secondary antibodies (1:2,000;
cat. no. 170-6516) were purchased from Bio-Rad Laboratories, Inc.
Anti-GLI3 (1:1,000; cat. no. A303-417) was purchased from Bethyl
Laboratories, Inc. Anti-β-actin antibody (1:20,000; cat. no. A5316)
was purchased from Sigma-Aldrich; Merck KGaA. Human phospho-kinase
antibody array (cat no. ARY003B) was acquired from R&D Systems,
Inc. Scrambled small interfering (si)RNA and AKT siRNA, ERK siRNA,
PCAF siRNA and β-TrCP siRNA were purchased from Santa Cruz
Biotechnology, Inc. Lipofectamine RNAi Max reagent (Thermo Fisher
Scientific, Inc.) was used for siRNA transfection.
GLI-luciferase reporter gene assay
NIH3T3 cell lines stably expressing GLI-luciferase
reporter construct were seeded at a rate of 25,000 cells/100
μl per well in 96-well tissue culture-treated plates. After
overnight 37°C incubation, 500 nl of pre-plated compounds dissolved
in DMSO were pin-transferred to the cells using a Janus automated
liquid handler workstation (PerkinElmer, Inc.) and incubated at
37°C for 24 h. Bright-Glo luciferase assay reagent (Promega
Corporation) was added to the cells according to the manufacturer's
instructions, and the emitting luminescence (560 nm) was read using
an Envision plate reader (PerkinElmer, Inc.).
Nuclear cytosol fractionation assay
The cytoplasmic and nuclear fractions were extracted
using NE-PER™ Nuclear and Cytoplasmic Extraction
Reagents (Thermo Fisher Scientific, Inc.). The harvested cells were
washed with trypsin-EDTA, then centrifuged at 500 × g for 5 min at
4°C. Cold CER I was added to the pellet, vortex followed, and then
incubation on ice for 10 min. CER II was added, followed by brief
vortex, and the pellet was centrifuged at 16,000 × g for 5 min at
4°C to transfer the supernatant (cytoplasmic extract) to a new EP
tube. NER was added in the tube with the pellet and placed it on
ice for 40 min, with vortex performed every 10 min. After
centrifugation at 16,000 × g for 10 min at 4°C, the supernatant
(nuclear extract) was then transferred to a new tube. Following
quantification, western blot analysis was performed.
MTT cell viability assay
Cells (5,000 cells/100 μl per well) were
seeded in 96-well tissue culture-treated plates. After one day, 500
nl of each pre-plated serially diluted daunorubicin (7 point,
5-fold serial dilution from 4 mM in DMSO) was pin-transferred to
the assay plate. After 24 h, MTT reagents (Promega Corporation)
were added as per the manufacturer's instructions and dissolved in
100 μl of DMSO after 4 h and the resulting absorption
signals were read at 570 nM using Envision plate reader
(PerkinElmer, Inc.). The GI50s were calculated using
Prism8 software (GraphPad Software Inc.; Dotmatics).
Western blotting
Cells were lysed with RIPA buffer containing 50 mM
Tris-HCl pH 7.4, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, and 1%
sodium deoxycholate, protease and phosphatase inhibitor cocktail
(Sigma-Aldrich; Merck KGaA). The BCA protein assay was used to
measure protein concentration with a NanoDrop 2000c
spectrophotometer (Software: NanoDrop 2000/2000c, version: 1.6.198,
Thermo Fisher Scientific, Inc.) Thermo Fisher Scientific, Inc.)
Equal amounts (50 μg) of proteins were loaded on 8, 10 and
12% SDS-PAGE according to size and then relocated to nitrocellulose
membranes (Cytiva). At this point, the membranes were blocked with
5% skim milk with TBS containing 0.1% Tween 20 for 2 h at 4°C.
After blocking the membranes, they were incubated with the primary
antibodies overnight at 4°C. After being incubated overnight at
4°C, they were incubated with HRP-labeled secondary antibodies for
2 h at 4°C. ECL western blotting substrate (DoGenBio) was added,
and the resulting signals were detected using X-ray films.
Transfection
Con siRNA (cat. no. sc-37007), GLI1 siRNA (cat. no.
sc-37911), AKT siRNA (cat. no. sc-43609), ERK siRNA (cat. no.
sc-29307, sc-35335), PCAF siRNA (cat. no. sc-36198) and β-TrCP
siRNA (cat. no. sc-37178) were all purchased from Santa Cruz
Biotechnology, Inc (The siRNA sequence is unavailable by Santa Cruz
Biotechnology, Inc). HCT116 cells (2×106) were incubated
with 200 nM siRNA using Lipofectamine RNA iMAX for 6 h at 37°C.
After 6 h, HCT116 cells were harvested after treatment with
daunorubicin for 24 h.
Immunoprecipitation
Cells were lysed with 300 μl of lysis buffer
(1 mM PMSF, protease inhibitors and phosphatase inhibitors) and
analyzed for their content of bicinchoninic acid. A total of 50
μl protein G PLUS-Agarose beads were added to the
supernatant for 1 h after the 2 μg primary antibodies (GLI1,
ITCH, β-TrCP and PCAF) were incubated with it overnight at 4°C.
Immunoprecipitants were washed and isolated by centrifugation at
17,000 × g for 10 min and heated with a 2X sample buffer. Lastly,
western blotting was used to analyze the supernatant.
Colony formation assay
A colony is defined to consist of at least 50 cells.
HCT-116 cells were seeded at a density of 5×102
cells/well in six-well plates and then incubated at 37°C with a 5%
CO2 incubator. The medium was changed two to three times
per week. After two weeks, cells were washed with DPBS (Dulbecco's
phosphate buffered saline), fixed with 4% paraformaldehyde for 30
min, then stained with 0.5% crystal violet for 30 min at room
temperature (RT) for visualization and cell counting.
Flow cytometric analysis of cell
apoptosis
As an apoptosis marker, the translocation flipping
of phosphatidylserine from the inner membrane to the outer leaflet
of the plasma membrane was detected through the binding of
fluorescein isothiocyanate (FITC)-conjugated annexin V. Briefly,
HCT116 cells (5×105) were either untreated or treated
with daunorubicin, then resuspended in the binding buffer of the
Annexin V-PI Apoptosis Detection Kit (cat. no. 556547; BD
Biosciences). Cells were then mixed with annexin V-FITC and
propidium iodide (PI) before being incubated at 4°C for 30 min in
the dark. At this point, flow cytometry was immediately used to
evaluate the cells (Navios EX flow cytometer; Beckman Coulter,
Inc.).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNAs were extracted using TRIzol reagent
(Molecular Research Center, Inc.). The amplification of transcripts
was executed using a reverse transcriptase PCR kit (Thermo Fisher
Scientific, Inc.) in accordance with the manufacturer's
instructions. RT-qPCR was carried out on a Applied
Biosystems® QuantStudio™ 6 Flex Real-Time PCR using
gene-specific Taqman™ probes (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The 45 cycle PCR stage consisted of 2 steps:
Denaturation step, 15 sec at 95°C; combined annealing and extension
step, 1 min at 60°C. The Taqman™ probes were GAPDH (Hs99999905_m1),
GLI1 (HS01110766_m1), Cyclin D1 (Hs00765553_m1), Snail
(Hs00195591_m1), and Myc (Hs00153408_m1). (The sequence is
unavailable by Thermo Fisher Scientific, Inc.) The mRNA expression
was normalized to expression values of GAPDH. The relative gene
expression ratios were analyzed using the 2-ΔΔCq method
(17).
Immunofluorescence staining
HCT-116 cells (1×105) were cultured on
glass coverslips within a 12-well plate and fixed with 3.7%
formaldehyde for 30 min at RT. With 0.5% Triton X-100, cells were
permeable for 30 min at RT, then blocked using 1% bovine serum
albumin (Thermo Fisher Scientific, Inc.) for 2 h. After blocking,
HCT-116 cells were incubated with primary antibodies overnight at
4°C. Next, the cells were washed for 30 min at RT and incubated
with Alexa Fluor® 594-conjugated secondary antibody
(1:200; cat. no. A-11005; Invitrogen; Thermo Fisher Scientific,
Inc.). DAPI (1 μg/ml) was then used to counterstain the
nuclei. Lastly, the cells were covered with VECTASHIELD mounting
medium (Vector Laboratories, Inc.) and then visualized through
fluorescence microscopy.
Caspase 3/7 activity measurement
Cells were plated in a 96-well white-wall plate at
8×103 cells per well, in triplicate. After being treated
with daunorubicin (0, 0.5 and 1 μM) for 24 h, 100 μl
of Caspase-Glo 3/7 reagent (Promega Corporation) was added to each
well. After 2 h of incubation in the dark, the caspase-3/7
activities were measured using a Varioskan Lux reader (Thermo
Fisher Scientific, Inc.). Differences in caspase-3/7 activity
between the drug-treated cells and untreated cells were expressed
in terms of fold-change in luminescence.
MG132 and leupeptin treatment
HCT-116 cells were seeded at a density of
7×103 cells/60 mm culture dish and then incubated at
37°C in a 5% CO2 incubator. The next day, cells were
treated with 1 μM daunorubicin. After 18 h of treatment,
cells were incubated with 2 μM MG132 (cat. no. 474790) or
100 rM Leupeptin (cat. no. L5793; both from Sigma-Aldrich; Merck
KGaA) for 6 h. The cells were harvested after 6 h by centrifugation
at 17,000 × g for 10 min at 4°C.
Cycloheximide (CHX) assay
HCT-116 cells were seeded at a density of
7×103 cells/60 mm culture dish and then incubated at
37°C in a 5% CO2 incubator. The next day, cells were
pre-treated with 1 μM daunorubicin. And then, cells were
harvested at 0, 1, 4 and 7 h after the following treatment with 200
μg/ml CHX (Sigma-Aldrich; Merck KGaA).
Tumor xenograft experiment
Animal experiments were implemented according to the
animal care guidelines approved (approval no. KOREA-2020-0174) by
the Korea University Institutional Animal Care and Use Committee
(Seoul, Korea). Four-week-old female BALB/c nude mice (n=10; body
weight, ±15 g) were purchased from Orient Bio, Inc. and contained
in a specific pathogen-free environment. Prior to the experiment,
the animals were housed for a week for acclimation and given free
access to food and water. The temperature was maintained at
20-24°C, with a relative humidity of 45-65% and a 12/12-h
light/dark cycle. A total of 100 μl of HCT116 cells
(2×106) in culture medium were implanted subcutaneously
in the dorsal flank of 5-week-old BALB/c nude female mice. The
tumor size was measured every two days. After the tumors reached a
size of 100 mm3, the nude mice were injected
intraperitoneally with either vehicle (DMSO) or daunorubicin (2
mg/kg) every two days for 15 days. The experiment was terminated
before the tumor size reached 1,000 mm3, and the mice
were sacrificed by inhaling 30% CO2 (4.5 l/min) for 2
min in a CO2 gas chamber. A total of 5 min later, after
confirming that rigor mortis had occurred, the tumor tissue was
removed.
Immunohistochemistry (IHC) staining
Sections (4-μm thickness) of formalin-fixed,
paraffin-embedded tumor tissue were deparaffinized in xylene and
hydrated in a graded alcohol series. To block endogenous
peroxidase, 3% hydrogen peroxide was used for 15 min, and for
antigen retrieval, heating at 100°C for 20 min was used. A
universal blocking solution was applied to the tissue slides for 15
min at RT, followed by an overnight incubation with primary
antibodies at 4°C. At RT, the samples were incubated with
peroxidase-conjugated anti-goat IgG for 1 h. The EnVision Plus
system (Dako; Agilent Technologies, Inc.) was used to visualize IHC
responses using diaminobenzidine staining. Antibodies used in this
method (including supplier, catalogue number and dilution) are
listed in Table I. The staining
intensity was divided into 5 grades, as shown in Table II.
 | Table IAntibodies used for
immunohistochemical staining. |
Table I
Antibodies used for
immunohistochemical staining.
| Antibody | Supplier | Catalogue
number | Dilution |
|---|
| GLI1 | Novus Biologicals,
LLC | NB600-600 | 1:400 |
| p53 | Santa Cruz
Biotechnology, Inc. | SC-126 | 1:600 |
| Table IIImmunohistochemical scoring. |
Table II
Immunohistochemical scoring.
| Percentage
score | Observation | Intensity
score | Observation |
|---|
| 1 | 0-5% | 0 | None |
| 2 | 6-25% | 1 | White brown |
| 3 | 26-50% | 2 | Brown |
| 4 | 51-75% | 3 | Dark brown |
| 5 | 76-100% | | |
TUNEL staining
Paraffin-embedded tumor tissues were deparaffinized
in xylene and hydrated in a graded alcohol series (100% xylene for
3 min × 3, 100% ethanol for 1 min × 2, 95% ethanol for 1 min × 2,
70% ethanol for 1 min). Using the in situ cell death
detection kit TMR red (cat. no. 12156792910; Roche Diagnostics) in
accordance with the manufacturer's instructions, TUNEL staining was
carried out. After TUNEL staining and mounting, a solution
containing DAPI was applied (ProLongTM Gold antifade reagent with
DAPI; cat. no. REFP36935; Invitrogen; Thermo Fisher Scientific,
Inc.). Following mounting, the slides were examined using a
fluorescence microscope (HC-010_LSM900/Super-Resolution laser
Scanning Microscope) with excitation wavelengths of 488 and 640 nm.
The observation process was performed at the top, middle, and
bottom of each slide and the average value was recorded.
Combination index (CI) and statistical
analysis
The CI method of Chou-Talalay (18,19) was used to analyze drug effects in
order to determine whether the cytotoxic interactions of
daunorubicin and irinotecan were synergistic, additive, or
antagonistic in CRC cells. All statistics were analyzed using the
GraphPad InStat 8 software (GraphPad Software Inc.; Dotmatics).
One-way ANOVA was used for group comparisons, followed by Tukey's
post hoc tests. Unpaired t-tests were used to determine
significance between two groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
Daunorubicin inhibits GLI1 activity in
CRC cells
In an attempt to identify a new antagonist of the Hh
pathway, an FDA-approved drug library comprised of 1,018 drugs was
screened against the commercially available NIH3T3 cell line that
stably expresses the GLI-dependent firefly luciferase reporter. As
a result, it was found that 10 drugs inhibited GLI luciferase
activity (Fig. S1A). As shown in
Fig. S1B, among the 10 drugs
examined, only daunorubicin and pralatrexate were demonstrated to
decrease GLI1 in HCT116, a CRC cell. It was discovered that
daunorubicin suppresses the activity of GLI-driven luciferase
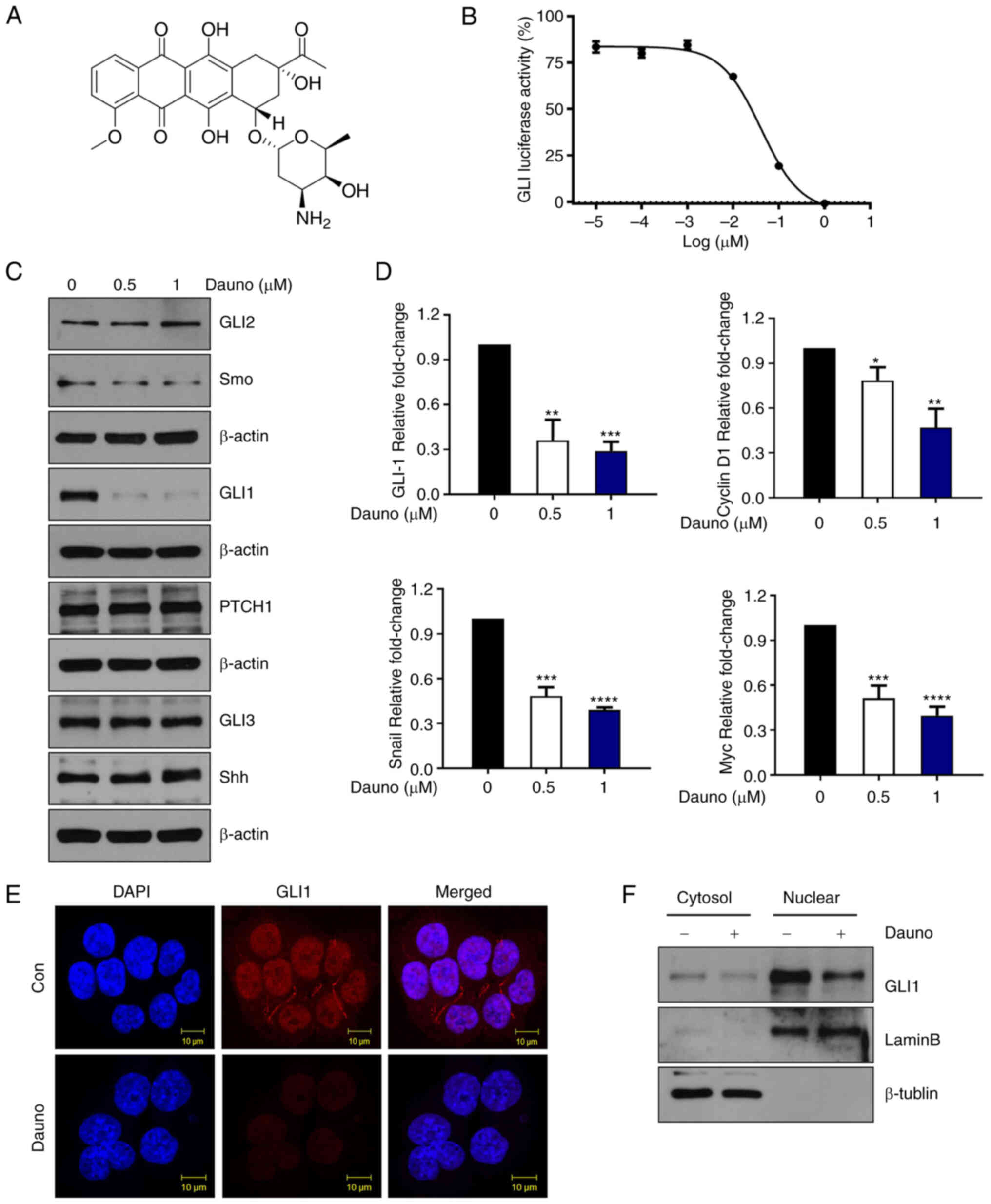
(Fig. 1A and B). Next, HCT116
cells were treated with daunorubicin (0, 0.5 and 1 μM) for
24 h and the protein levels of the components involved in the
canonical Hh pathway were examined. None of SHH, GLI2, GLI3, Smo
and Ptch1 protein levels were significantly changed by daunorubicin
(Figs. 1C-E and S2A). Moreover, daunorubicin was shown
to downregulate GLI1 protein levels in other CRC cells, including
HT29 and SNU283 cells (Fig.
S2B). The mRNA expression level of GLI1, cyclin D1, Snail and
Myc was significantly downregulated (Fig. 1D). Western blot analysis of the
cytoplasmic and nuclear extractions of HCT116 cells revealed that
daunorubicin treatment downregulated nuclear expression of GLI1
when compared with the control (Figs.
1F and S2C). These results
indicated that daunorubicin inhibits GLI1 expression.
Daunorubicin induces caspase-dependent
apoptosis in HCT116 cells
First, to examine the cytotoxicity of daunorubicin
in CRC cells (Fig. 2A), various
CRC cell lines were cultured with different concentrations (0-20
μM) of daunorubicin for 24 h. The survival of CRC cell lines
HCT116, HT29 and SNU283 with high expression of GLI1 was reduced by
daunorubicin in a dose-dependent manner. On the other hand, DLD-1
and HCT8 with low GLI1 expression showed less reduction by
daunorubicin. Furthermore, to confirm the daunorubicin reduced cell
proliferation in CRC cells, a colony forming assay was performed in
HCT116 cells. As demonstrated in Fig.
2B, colony formation was strongly inhibited in HCT116 cells.
Moreover, to examine whether the cell death induced by the
treatment promoted apoptosis, Annexin V-PI staining was conducted
along with the treatment of daunorubicin at 0, 0.5 and 1 μM
for 24 h of HCT116 cells in a dose-dependent manner in a flow
cytometric analysis (Fig. 2C).
Caspase-3/7 activity was also augmented in a dose-dependent manner
(Fig. 2D). Daunorubicin increased
the levels of c-PARP, caspase-8, caspase-9 and caspase-3, which
were suppressed by co-treatment of a pan-caspase inhibitor
z-VAD-fmk, thus indicating that daunorubicin induced
caspase-dependent apoptosis in HCT116 cells (Figs. 2E and S3A). To determine whether cell death is
affected by the expression of GLI1, GLI1 was knocked down in the
HCT116 cells. As revealed in Fig. 2F
and G, when GLI1 was knocked down, daunorubicin-induced cell
death and c-PARP, caspase-8, caspase-9 and caspase-3 were not
increased (Figs. 2F, 2G and S3B). Therefore, the aforementioned
results suggested that the daunorubicin induces caspase-dependent
apoptosis through inhibition of GLI1 in CRC.
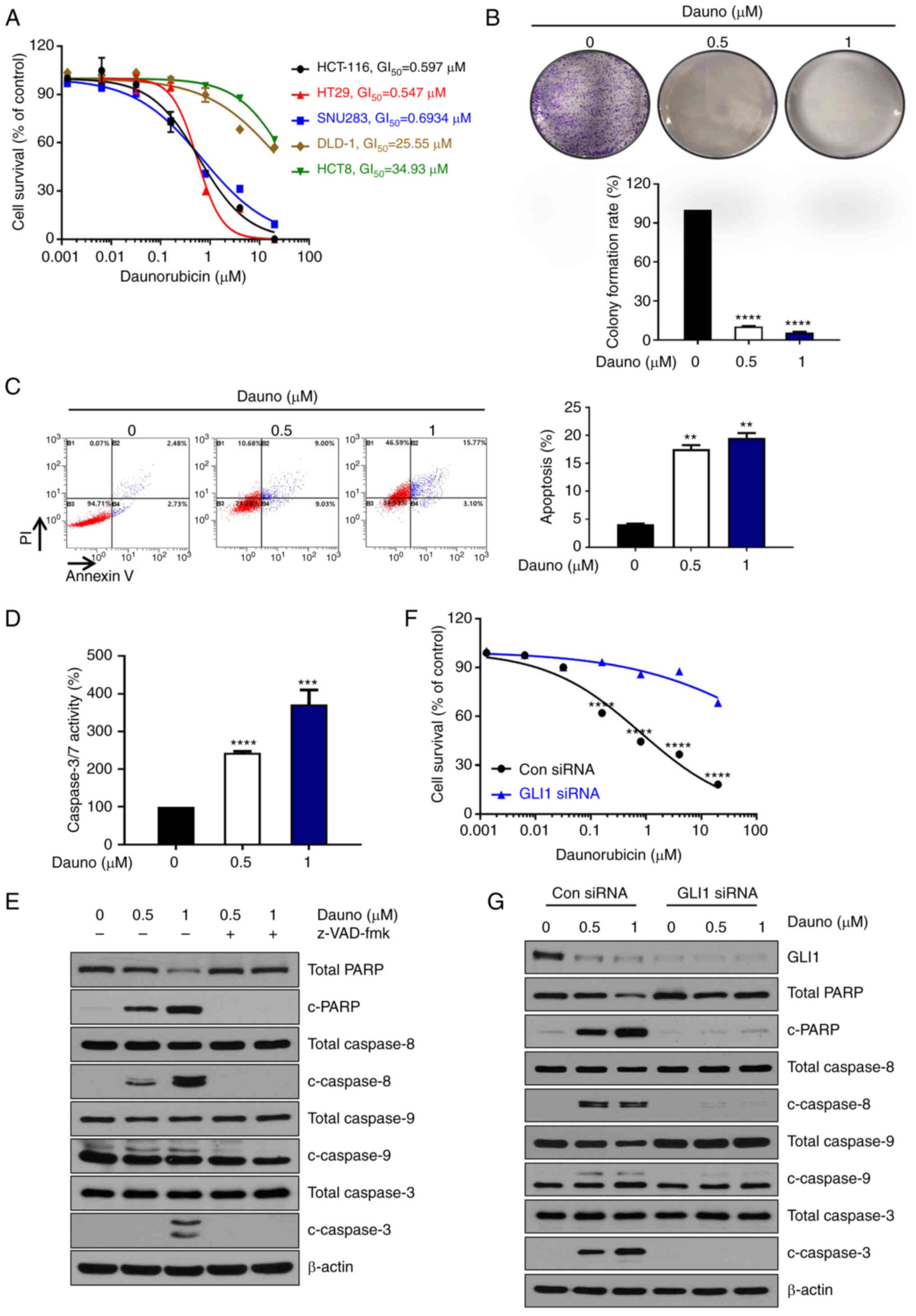 | Figure 2Daunorubicin-induced
caspase-dependent apoptosis in HCT116 cells. (A) Daunorubicin
decreased the proliferation of HCT116, HT29, SNU283, DLD-1 and HCT8
cells with GI50 of 0.597, 0.547, 0.6934, 25.55 and 34.93
μM, respectively. (B) Colony formation assay using HCT116
cells after treatment of daunorubicin (0, 0.5 and 1) and
quantitation. (C) Treatment of daunorubicin (0, 0.5 and 1
μM) for 24 h-induced apoptosis of HCT116 cells in a
dose-dependent manner. Quantitation of cell death is plotted on the
right. The graph was drawn by combining the B2 and B4 quadrants.
(D) Treatment of daunorubicin (0, 0.5 and 1 μM) for 24 h led
to a dose-dependent increase in caspase3/7 activity. (E) HCT116
cells were pretreated with 25 μM z-VAD-fmk for 30 min and
then treated with daunorubicin (0, 0.5 and 1 μM). Western
blotting was used to measure the expression levels of c-PARP,
caspase3, caspase9 and caspase8. (F) After GLI1 knockdown using
GLI1 siRNA, cell survival induced by daunorubicin was detected. (G)
After GLI1 knockdown using GLI1 siRNA, western blotting was used to
measure the expression levels of c-PARP, caspase3, caspase9 and
caspase8. The data are expressed as the mean of 3 independent
experiments. **P<0.005, ***P<0.001 and
****P<0.0001. siRNA, small interfering RNA; c-PARP,
cleaved poly (ADP-ribose) polymerase. |
Daunorubicin induces p53-mediated
apoptosis and GLI1 downregulation in HCT-116 cells
To determine whether GLI1 inhibition by daunorubicin
occurs by the canonical or non-canonical pathway, it was attempted
to demonstrate this by inhibiting AKT or ERK, which are
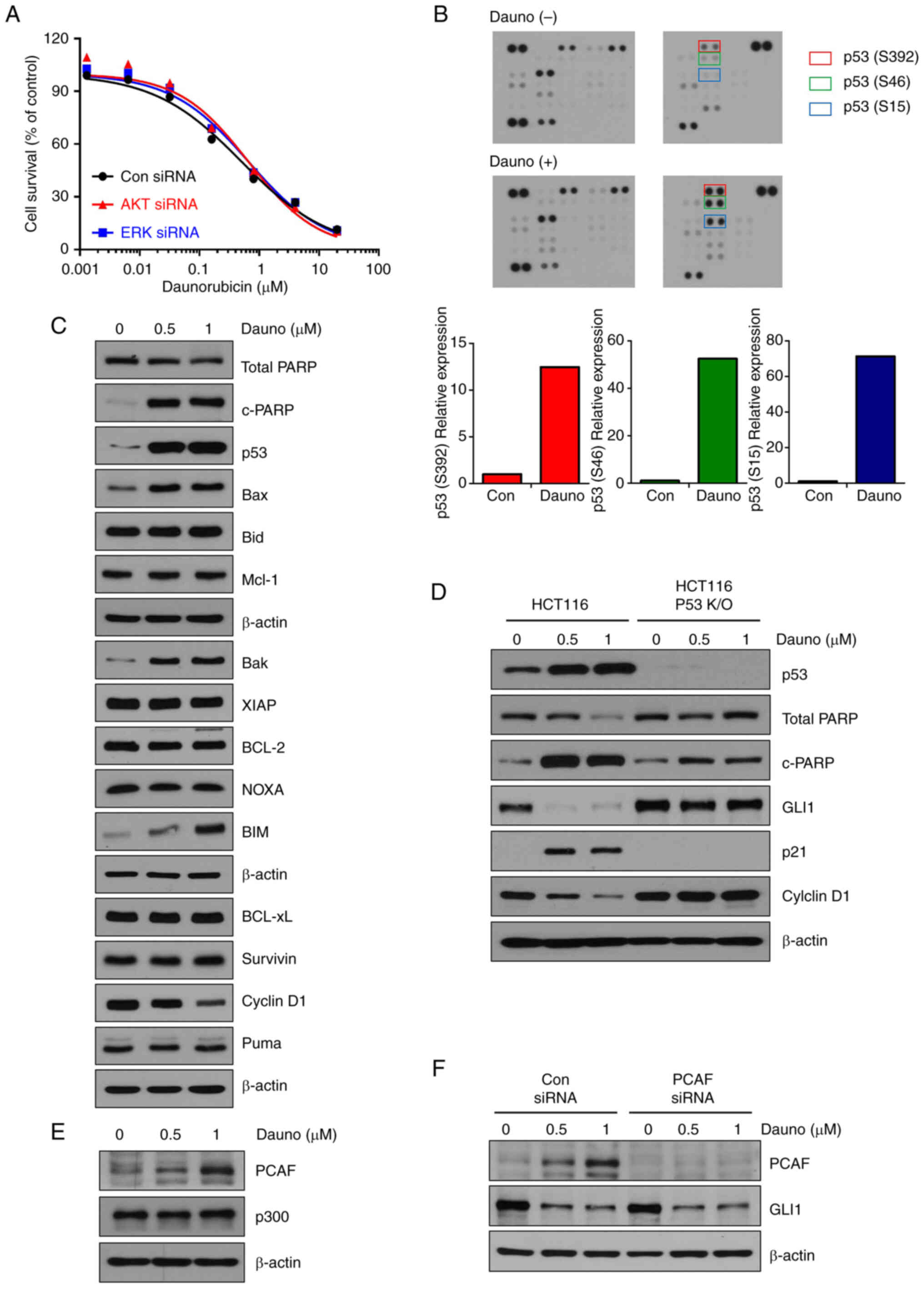
representative non-canonical Hh signaling pathways. When inhibiting
AKT or ERK, decreased cell survival and inhibition of GLI1
expression by daunorubicin was observed (Figs. 3A, S4A and B). These results indicated that
inhibition of GLI1 by daunorubicin occurs through the canonical Hh
pathway.
A human phospho-kinase antibody array was then used
to investigate which pathways are regulated by daunorubicin (1
μM). Interestingly, among the phosphorylation on the protein
array, the p53 phosphorylation (S15, S46 and S392) was shown to be
markedly increased by daunorubicin treatment (Fig. 3B). These results indicated the
existence of daunorubicin-induced p53 activation. Furthermore,
daunorubicin also enhanced the phosphorylation of Chk2 (T68)
(20) and p27 (T198) (21), both of which are involved in cell
cycle arrest (Fig. 3B). The
levels of apoptosis-related marker proteins were also examined.
Consistent with the results provided in Fig. 3B, daunorubicin increased the
levels of pro-apoptotic proteins such as p53, Bim, Bak, Bax and
c-PARP, while it decreased the levels of anti-apoptotic Cyclin D1
(Figs. 3C and S5A). In the HCT116 p53 knockout cell
line, daunorubicin-induced PARP cleavage and p21 did not increase,
while the decreased expression of GLI1 and CyclinD1 was
significantly reversed compared with the HCT116 wild-type cells.
This suggested that daunorubicin induces apoptosis through p53
activation (Figs. 3D and S5B). The activation of p300 and PCAF is
necessary for the p53-dependent response to genotoxic stress,
according to a common mechanism (22). As previously reported, it has been
demonstrated that upregulation of PCAF induced by p53 is required
for the suppression of GLI1 in response to DNA damage (23). It was investigated whether the
expression levels of PCAF and P300 were changed by daunorubicin.
Daunorubicin increased the expression of PCAF in a dose-dependent
manner (Figs. 3E and S5C). To confirm whether the
PCAF-dependent suppression of GLI1 occurred, PCAF was knocked down
using PCAF siRNA. By demonstrating that GLI1 was still reduced by
daunorubicin despite PCAF suppression, these results proved that
the inhibition of GLI1 by daunorubicin was not dependent on PCAF
(Figs. 3F and S5D).
Daunorubicin promotes GLI1 ubiquitination
and proteasomal degradation
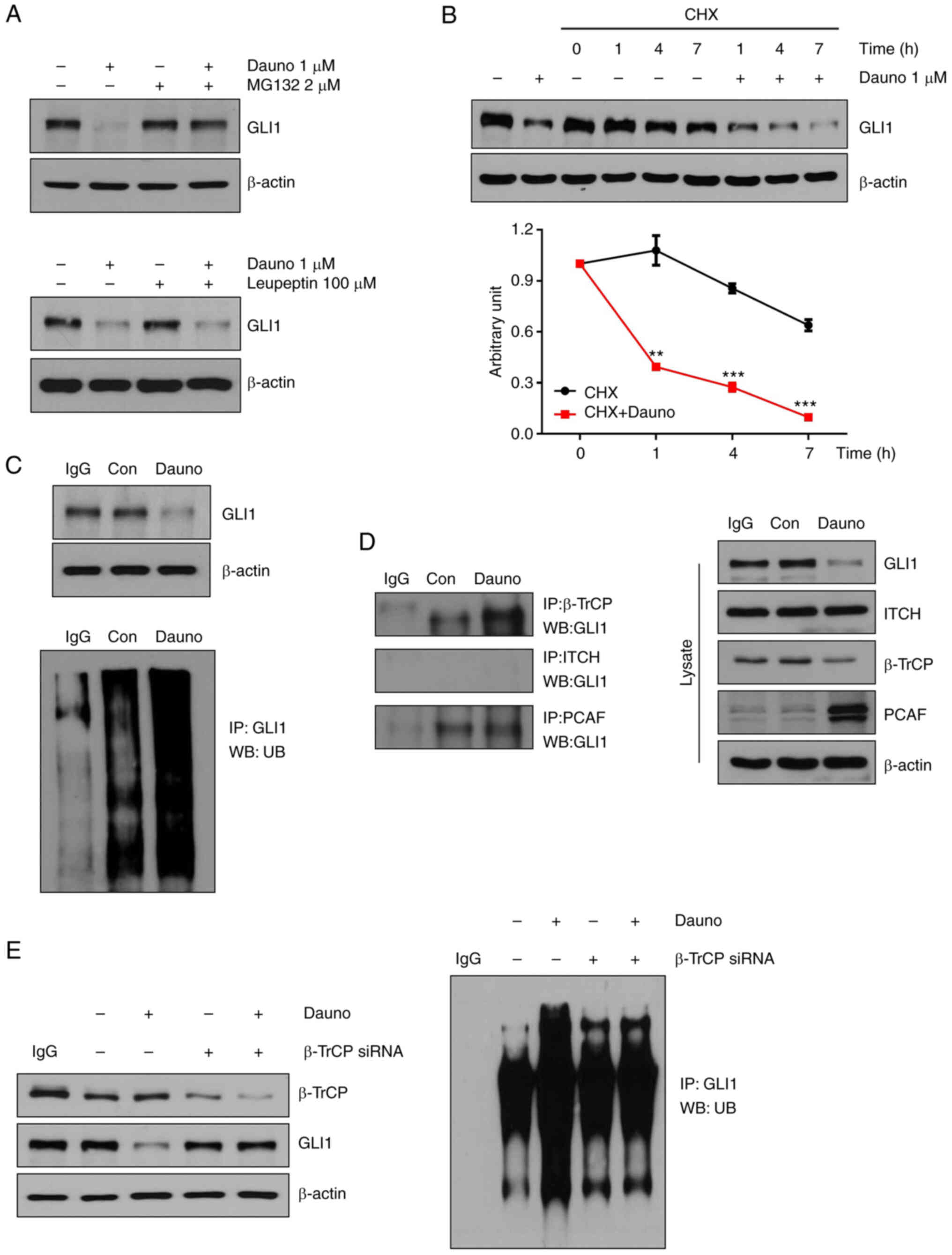
Daunorubicin did not displace the BODIPY-cyclopamine
from Smo in HCT116 cells, indicating that daunorubicin does not
bind to Smo (Fig. S6);
therefore, it was investigated how it regulates the downstream
GLI1. Treatment with MG132, a proteasome inhibitor, and leupeptin,
a lysosome inhibitor, was administered to investigate whether the
reduction of GLI1 was attributable to proteasomal or lysosomal
degradation. The GLI1 expression decreased by daunorubicin was
increased by MG132 and not changed by leupeptin (Figs. 4A and S7A). These data demonstrated that the
reduction of GLI1 by daunorubicin was caused by proteasome
degradation. Moreover, HCT116 cells were treated with CHX, a
protein synthesis inhibitor. The expression of GLI1 was
significantly reduced when HCT116 cells were exposed to
daunorubicin and CHX compared with when they were exposed to CHX
alone, thus suggesting that daunorubicin decreased the level of
GLI1 through ubiquitin-proteasome degradation (Fig. 4B). These results confirmed that
daunorubicin promotes the ubiquitination of GLI1 (Figs. 4C and S7B). To investigate what E3 ligase is
responsible for the GLI1 ubiquitination, immunoprecipitation was
performed to measure the interaction between GLI1 and E3 ligase. As
shown in Fig. 4D, daunorubicin
increased interaction between GLI1 and β-TrCP (Figs. 4D and S7C). To investigate whether
β-TrCP-dependent GLI1 ubiquitination occurred, β-TrCP was knocked
down using β-TrCP siRNA. Knockdown of β-TrCP restored the
daunorubicin-induced reduction of GLI1, and the ubiquitination of
GLI1 was also unchanged (Figs. 4E
and S7D). These data suggested
that daunorubicin induces GLI1 ubiquitination via β-TrCP.
Daunorubicin inhibits Hh pathway and
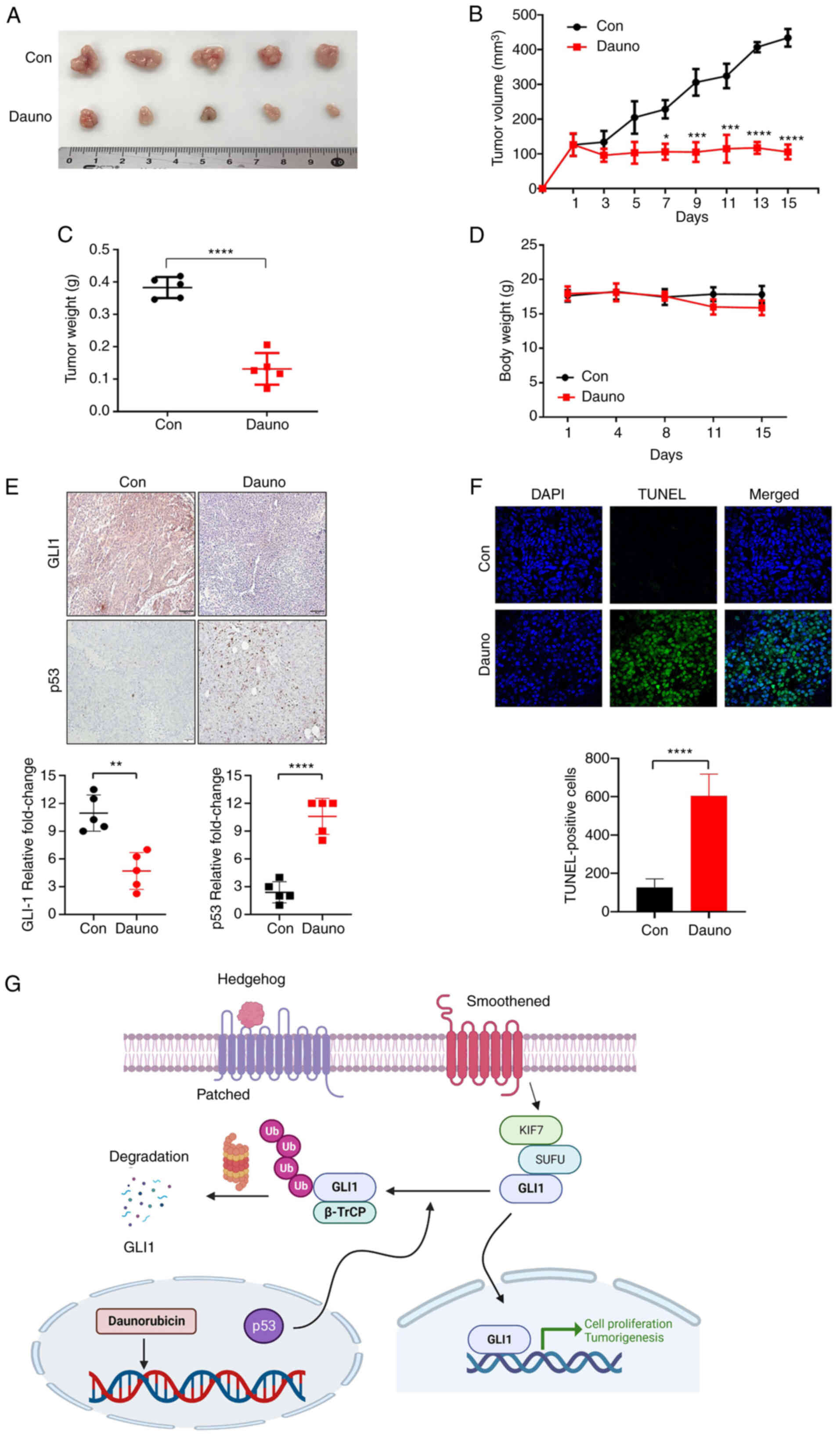
induces apoptosis in HCT116 xenograft in vivo mouse model
HCT116 cells (2×106) were subcutaneously
injected into BALB/c nude mice. When the size of the tumors was 100
mm3, nude mice were treated with daunorubicin (2 mg/kg)
every two days for 15 days by intraperitoneal injection. The tumor
growth and weight were repressed in the daunorubicin-treated group
compared with the control group (Fig.
5A-C), while body weight did not significantly differ between
the control and daunorubicin-treated groups (Fig. 5D). These data indicated that
daunorubicin induced apoptosis. Consistent with the in vitro
results, IHC showed significant suppression of GLI1 levels in the
daunorubicin-treated group in addition to increased p53 (Fig. 5E). A TUNEL assay indicated
daunorubicin-induced apoptosis in tumor tissues (Fig. 5F). Taken together, these results
indicated that daunorubicin exhibited anticancer effects in the CRC
xenograft mice, in part through GLI1 downregulation.
In addition, it was confirmed whether daunorubicin
had a combined effect with oxaliplatin or irinotecan, which are
representative anticancer drugs used for CRC. As demonstrated in
Fig. S8, a combined effect of
daunorubicin and irinotecan was observed. Additional research is
needed to confirm the possibility of future combinatory use.
Discussion
Daunorubicin is used as the first-line treatment for
leukemia, and it is typically administrated in combination with
other chemotherapeutic agents such as cytarabine (24). It has also been reported to have
anticancer effects in monotherapy or combination therapy in solid
tumors. It is well-established that the ability of daunorubicin to
intercalate DNA and interfere with DNA replication process is the
main mode of action for its anticancer effect. The results of the
present study demonstrated that daunorubicin exerts anticancer
activity in CRC in part through its ability to suppress the
non-canonical Hh pathway.
From the screening using FDA-approved drug library,
daunorubicin was identified as an inhibitor of Hh signaling. The
results of the present study revealed that daunorubicin suppressed
the Shh/Smo-independent non-canonical Hh pathway in CRC cells. It
was found that daunorubicin suppresses GLI1 through (i)
upregulating p53 and (ii) promoting β-TrCP-mediated GLI1
ubiquitination and the subsequent proteasomal degradation, which
suppresses GLI1 function. It has been previously reported that p53
negatively regulates GLI1 function. A previous study revealed that
p53 elevates the PCAF level, which consequently increases GLI1
ubiquitination and proteasomal degradation (23). However, in the present study, as
demonstrated in Figs. 3 and
4, it was identified for the
first time that daunorubicin suppresses GLI1 through B-TrCP, not
through PCAF-induced reduction of GLI1.
In the present experimental condition, daunorubicin
was not found to downregulate the protein levels of Ptch1 or Smo.
Daunorubicin did not displace the BODIPY-cyclopamine from Smo in
cells, implying that daunorubicin does not bind to Smo (Fig. S6). These results suggested that
daunorubicin does not target Smo but inhibits Hh signaling through
GLI1. Previous studies using patient samples revealed that the
nuclear Smo is found in nearly 50% of patients with pancreatic
cancer and CRC (25,26). Moreover, Rahman et al
(27) identified that nuclear Smo
acts as a Smo-independent Hh activation mechanism and confers
resistance to Smo inhibitors. The present study demonstrated that
daunorubicin strongly suppresses Hh signaling while also enhancing
Smo in the nucleus. The daunorubicin-induced suppression of the
non-canonical Hh pathway might overwhelm the Hh pathway activation
exerted by the nuclear Smo. However, further research is needed to
inspect whether the nuclear Smo induced by daunorubicin is
functionally identical to the oncogenic nuclear Smo in cancer cells
(27,28). In an in vivo study using a
mouse xenograft of HCT116 cells, administration of daunorubicin (2
mg/kg, ip) was found to greatly suppress the tumor progress and
GLI1 level in tumor tissues (Fig.
5).
Daunorubicin has been shown to have no effect in
patients with advanced colorectal carcinoma in clinical trial II
(29). However, the procedure was
performed without selecting a patient group, and it is considered
that daunorubicin would be effective in a patient group in which
GLI1 was overexpressed.
Collectively, the present results revealed that
daunorubicin suppresses Hh pathway in CRC. To the best of the
authors' knowledge, this is the first study on the anticancer
mechanism of daunorubicin with regard to the Hh pathway. The
current study disclosed a new mode of daunorubicin's anticancer
effect, and it may provide a rationale for expanding the clinical
applicability of daunorubicin.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
BRK, DYK and NLT performed most of the experiments
and wrote the manuscript. BGK provided technical assistance. SIL,
BYM and SHK contributed to conceptualization of the study and
interpretation of results. SCO and WYH supervised the projects,
provided funding and helped writing the manuscript. All authors
read and approved the final manuscript. BRK and SCO confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
Animal experiments were implemented according to the
animal care guidelines approved (approval no. KOREA-2020-0174) by
the Korea University (Seoul, Korea) Institutional Animal Care and
Use Committee (IACUC).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present was supported by the Basic Science Research Program
through the National Research Foundation of Korea (NRF) funded by
the Ministry of Education (grant no. RS-2023-00238158), the
National Research Foundation of Korea (NRF) grant funded by the
Korea government (MSIT) (grant no. NRF-2018M3A9G1075 561) and the
Korea Institute of Science and Technology (grant no. 2E33131).
References
|
1
|
Girardi D, Barrichello A, Fernandes G and
Pereira A: Targeting the hedgehog pathway in cancer: Current
evidence and future perspectives. Cells. 8:1532019.
|
|
2
|
Niyaz M, Khan MS and Mudassar S: Hedgehog
signaling: An achilles' heel in cancer. Transl Oncol. 12:1334–1344.
2019.
|
|
3
|
Pak E and Segal RA: Hedgehog signal
transduction: Key players, oncogenic drivers, and cancer therapy.
Dev Cell. 38:333–344. 2016.
|
|
4
|
Sari IN, Phi LTH, Jun N, Wijaya YT, Lee S
and Kwon HY: Hedgehog signaling in cancer: A prospective
therapeutic target for eradicating cancer stem cells. Cells.
7:2082018.
|
|
5
|
Peer E, Tesanovic S and Aberger F:
Next-generation hedgehog/GLI pathway inhibitors for cancer therapy.
Cancers (Basel). 11:5382019.
|
|
6
|
Katoh Y and Katoh M: Hedgehog target
genes: Mechanisms of carcinogenesis induced by aberrant hedgehog
signaling activation. Curr Mol Med. 9:873–886. 2009.
|
|
7
|
Barnes EA, Heidtman KJ and Donoghue DJ:
Constitutive activation of the shh-ptc1 pathway by a patched1
mutation identified in BCC. Oncogene. 24:902–915. 2005.
|
|
8
|
Tao Y, Mao J, Zhang Q and Li L:
Overexpression of hedgehog signaling molecules and its involvement
in triple-negative breast cancer. Oncol Lett. 2:995–1001. 2011.
|
|
9
|
van den Brink GR, Bleuming SA, Hardwick
JC, Schepman BL, Offerhaus GJ, Keller JJ, Nielsen C, Gaffield W,
van Deventer SJ, Roberts DJ and Peppelenbosch MP: Indian hedgehog
is an antagonist of Wnt signaling in colonic epithelial cell
differentiation. Nat Genet. 36:277–282. 2004.
|
|
10
|
Yoshikawa K, Shimada M, Miyamoto H,
Higashijima J, Miyatani T, Nishioka M, Kurita N, Iwata T and Uehara
H: Sonic hedgehog relates to colorectal carcinogenesis. J
Gastroenterol. 44:1113–1117. 2009.
|
|
11
|
Rajurkar M, De Jesus-Monge WE, Driscoll
DR, Appleman VA, Huang H, Cotton JL, Klimstra DS, Zhu LJ, Simin K,
Xu L, et al: The activity of Gli transcription factors is essential
for Kras-induced pancreatic tumorigenesis. Proc Natl Acad Sci USA.
109:E1038–E1047. 2012.
|
|
12
|
Wang Y, Ding Q, Yen CJ, Xia W, Izzo JG,
Lang JY, Li CW, Hsu JL, Miller SA, Wang X, et al: The crosstalk of
mTOR/S6K1 and hedgehog pathways. Cancer Cell. 21:374–387. 2012.
|
|
13
|
Pandolfi S, Montagnani V, Penachioni JY,
Vinci MC, Olivito B, Borgognoni L and Stecca B: WIP1 phosphatase
modulates the Hedgehog signaling by enhancing GLI1 function.
Oncogene. 32:4737–4747. 2013.
|
|
14
|
Lauth M, Bergström A, Shimokawa T and
Toftgard R: Inhibition of GLI-mediated transcription and tumor cell
growth by small-molecule antagonists. Proc Natl Acad Sci USA.
104:8455–8460. 2007.
|
|
15
|
Agyeman A, Jha BK, Mazumdar T and Houghton
JA: Mode and specificity of binding of the small molecule GANT61 to
GLI determines inhibition of GLI-DNA binding. Oncotarget.
5:4492–4503. 2014.
|
|
16
|
Hyman JM, Firestone AJ, Heine VM, Zhao Y,
Ocasio CA, Han K, Sun M, Rack PG, Sinha S, Wu JJ, et al:
Small-molecule inhibitors reveal multiple strategies for Hedgehog
pathway blockade. Proc Natl Acad Sci USA. 106:14132–14137.
2009.
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
|
|
18
|
Chou TC: Theoretical basis, experimental
design, and computerized simulation of synergism and antagonism in
drug combination studies. Pharmacol Rev. 58:621–681. 2006.
|
|
19
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984.
|
|
20
|
Garcia-Santisteban I, Llopis A, Krenning
L, Vallejo-Rodríguez J, van den Broek B, Zubiaga AM and Medema RH:
Sustained CHK2 activity, but not ATM activity, is critical to
maintain a G1 arrest after DNA damage in untransformed cells. BMC
Biol. 19:352021.
|
|
21
|
De Vita F, Riccardi M, Malanga D, Scrima
M, De Marco C and Viglietto G: PKC-dependent phosphorylation of p27
at T198 contributes to p27 stabilization and cell cycle arrest.
Cell Cycle. 11:1583–1592. 2012.
|
|
22
|
Sakaguchi K, Herrera JE, Saito S, Miki T,
Bustin M, Vassilev A, Anderson CW and Appella E: DNA damage
activates p53 through a phosphorylation-acetylation cascade. Genes
Dev. 12:2831–2841. 1998.
|
|
23
|
Mazzà D, Infante P, Colicchia V, Greco A,
Alfonsi R, Siler M, Antonucci L, Po A, De Smaele E, Ferretti E, et
al: PCAF ubiquitin ligase activity inhibits Hedgehog/Gli1 signaling
in p53-dependent response to genotoxic stress. Cell Death Differ.
20:1688–1697. 2013.
|
|
24
|
Murphy T and Yee KWL: Cytarabine and
daunorubicin for the treatment of acute myeloid leukemia. Expert
Opin Pharmacother. 18:1765–1780. 2017.
|
|
25
|
Li T, Liao X, Lochhead P, Morikawa T,
Yamauchi M, Nishihara R, Inamura K, Kim SA, Mima K, Sukawa Y, et
al: SMO expression in colorectal cancer: Associations with
clinical, pathological, and molecular features. Ann Surg Oncol.
21:4164–4173. 2014.
|
|
26
|
Maréchal R, Bachet JB, Calomme A, Demetter
P, Delpero JR, Svrcek M, Cros J, Bardier-Dupas A, Puleo F, Monges
G, et al: Sonic hedgehog and Gli1 expression predict outcome in
resected pancreatic adenocarcinoma. Clin Cancer Res. 21:1215–1224.
2015.
|
|
27
|
Rahman MM, Hazan A, Selway JL, Herath DS,
Harwood CA, Pirzado MS, Atkar R, Kelsell DP, Linton KJ, Philpott MP
and Neill GW: A novel mechanism for activation of GLI1 by nuclear
SMO that escapes anti-SMO inhibitors. Cancer Res. 78:2577–2588.
2018.
|
|
28
|
Niyaz M, Khan MS, Wani RA, Shah OJ, Besina
S and Mudassar S: Nuclear localization and overexpression of
smoothened in pancreatic and colorectal cancers. J Cell Biochem.
120:11941–11948. 2019.
|
|
29
|
Harvey J, Bonnem E, Grady K, Goodman A and
Schein P: Phase II study of daunorubicin in previously untreated
patients with advanced colorectal carcinoma. Med Pediatr Oncol.
13:30–31. 1985.
|