Non-steroidal anti-inflammatory drugs (NSAIDs) are
primarily used as analgesics, antipyretics and anti-inflammatory
agents. NSAIDs mainly act by inhibiting prostaglandin (PG)
production. A number of experimental, epidemiological and clinical
studies have revealed the antitumour properties of NSAIDs,
particularly of cyclooxygenase (COX)-2 inhibitors (1, 2).
NSAIDs have been shown to inhibit malignant transformation in
several cancer cell lines. Moreover, the frequent use of NSAIDs has
been associated with a reduced risk of colorectal,
gastrointestinal, breast, prostate and lung cancer (3–6). The
mechanism underlying the antitumour activity of NSAIDs has not been
fully elucidated; however, it may involve the inhibition of COXs or
other non-COX enzymatic pathways.
The use of herbal extracts containing salicylates
dates back thousands of years. In 1874, Maclagan successfully used
salicylic acid isolated from willow bark for the treatment of the
inflammation associated with rheumatic fever (7). A more effective and tolerable
synthetic acetylated form of salicylic acid was introduced by Felix
Hoffman in 1897; this derivative was named aspirin (8, 9).
Over time, several other drugs with the same antipyretic, analgesic
and anti-inflammatory properties were introduced, including
antipyrine, acetaminophen, phenylbutazone, naproxen and
indomethacin. As these drugs share a similar mechanism of action
and are clearly distinct from other groups of drugs used in the
treatment of inflammation (glucocorticoids), they were collectively
named non-steroidal anti-inflammatory drugs (NSAIDs) (10, 11).
The main mechanism through which NSAIDs exert their
effects is the inhibition of PG biosynthesis. PGs have been
implicated in a number of physiological and pathological disorders,
such as inflammation, pain, pyrexia, cancer, osteoporosis,
cardiovascular diseases and asthma (12, 13). Following exposure to physiological
and pathological stimuli, polyunsaturated fatty acids, including
arachidonic acid (AA), are released from membrane phospholipids
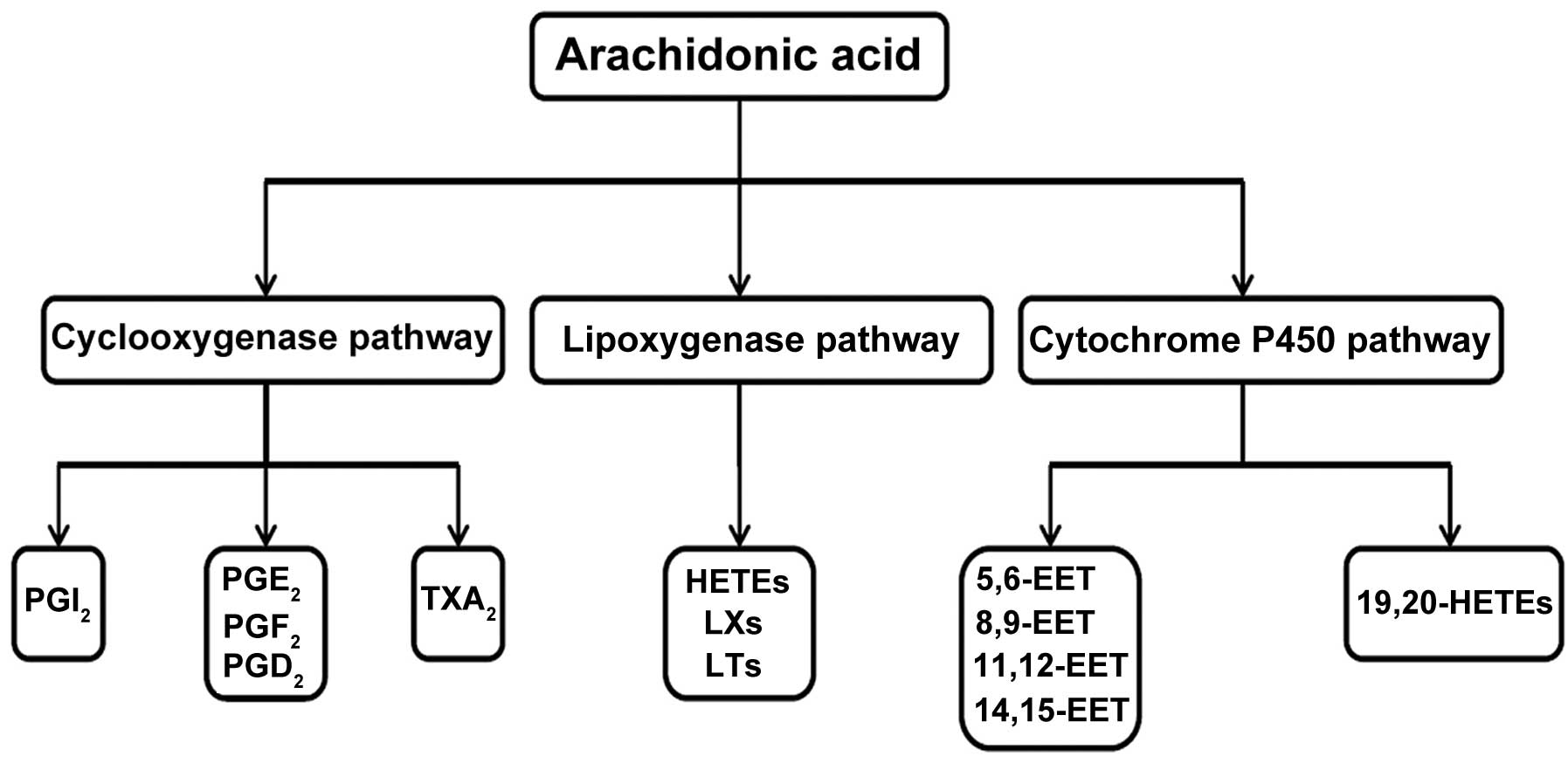
through the action of phospholipase A2 enzymes. Free AA is
subsequently converted via one of three enzymatic pathways
(14–16) (Fig.
1): In the COX pathway, AA is converted to PGs, prostacyclins
(PCs) and thromboxanes (TXs); in the lipoxygenase (LOX) pathway, AA
is converted to hydroxyeicosatetraenoic acids (HETEs), leukotrienes
(LTs) and lipoxins (LXs); lastly, in the cytochrome P450 (CYP450)
monooxygenase pathway, AA release leads to the production of HETEs
and epoxyeicosatrienoic acids (EETs). Additionally, in a
non-enzymatic pathway, AA release results in the synthesis of
isoprostanes. The products of these metabolic pathways are referred
to as eicosanoids. Eicosanoids represent important intercellular
and intracellular signalling molecules that participate in a wide
range of physiological processes, such as the regulation of smooth
muscle tone, vascular permeability, platelet aggregation,
transporter proteins and proliferation. In addition, eicosanoids
are involved in inflammation, autoimmunity, angiogenesis, allergic
diseases and cancer (17–20). Extensive research has been focused
on PGs and other COX-derived metabolites. However, a number of
studies suggested that LOX-derived products also affect the
development and progression of several malignancies (21–25).
There are 3 COX isoforms, commonly referred to as
COX-1, COX-2 and COX-3. COX-1, also referred to as PGH synthase, is
the key enzyme responsible for the oxidation of AA to PGG2 and
PGH2. COX-1 is constitutively expressed, with its levels remaining
constant under most physiological and pathological conditions. By
contrast, the expression of COX-2 is highly inducible in response
to mitogenic and inflammatory stimuli, such as fibroblast growth
factor (26), transforming growth
factor β (27), epidermal growth
factor (28), vascular endothelial
growth factor, tumour necrosis factor α and interleukins 1α and 1β
(29). The function of COX-3
remains unclear (30–32). An aberrant constitutive expression
of COX-2 has been demonstrated during the early stages of
carcinogenesis (33, 34). There is compelling evidence
supporting a role for COX-2 in tumour development. COX-2 expression
has been shown to be elevated in several human tumours, including
colorectal (35, 36), gastric (37) and pancreatic cancer (38), oesophageal adenocarcinoma (39), lung (40) and breast cancer (41). The tumour-promoting effect of COX-2
may be a consequence of the numerous effects that COX-2 exerts on
cells. COX-2 may promote proliferation, angiogenesis and
invasiveness, prevent apoptosis and enhance cell adhesion and
motility (42). Treatment with
COX-2-specific inhibitors results in a wide range of cellular
effects, including induction of apoptosis, reduction of cell
proliferation, inhibition of angiogenesis and enhanced anticancer
drug-induced cytotoxicity (43–46).
These findings suggest that NSAIDs may exert their anticancer
effects through COX-2 inhibition. Although the significance of
COX-2 inhibitors is well established, the mechanism underlying
their chemopreventive and chemotherapeutic actions is largely
unknown. Indeed, there is evidence suggesting that the antitumour
effect of NSAIDs may not only be mediated by the inhibition of
COX-2 activity, but that other cellular targets may also play a
role (46). This hypothesis is
supported by the observation that NSAID treatment reduced cell
survival in COX-2-overexpressing as well as COX-deficient cancer
cell lines (47–49).
Information regarding the role of LOXs in the
promotion of cancer growth is limited. The identification of LOX
isoforms in cancer, stromal and immune cells has led to the
hypothesis that these enzymes may contribute to tumour development
and growth (50), with interest
mainly focused on 5-LOX, 12-LOX and 15-LOX. Under physiological
conditions, the expression of 5-LOX is limited to immune cells
(51, 52). 5-LOX may directly control tumour
cell function or indirectly affect the tumour microenvironment.
Increased 5-LOX activity has been demonstrated to play a role in
the early stages of colon cancer (53) and in carcinogenesis in human oral
cavity tissues (54). It was also
reported that 5-LOX expression may be involved in the development
of BCR-ABL-induced chronic myeloid leukaemia (55). Moreover, the 5-LOX pathway may be
involved in the metastatic process of pancreatic, intestinal and
prostate cancers (56, 57). The inhibition of 5-LOX expression
and activity promotes cell apoptosis and tumour growth arrest.
Additionally, 5-LOX inhibition affects epithelial-to-mesenchymal
transition in certain cancer cell lines and suppresses metastasis
in pancreatic cancer. These effects are likely due to the
upregulation of E-cadherin and paxillin (58–61).
The finding that 12-LOX is overexpressed in murine lung carcinoma
and human prostate cancer cells suggests a possible role for this
enzyme in cancer development (22,
62). The 12-LOX inhibitor
baicalein induces apoptosis in cancer cells. This induction is
mediated through the regulation of the B-cell lymphoma-2 (Bcl-2)
protein (63–65). Furthermore, 12-LOX controls
G1/S-phase arrest by inhibiting Akt and mitogen-activated protein
kinases and regulating the expression of nuclear factor (NF)-κB
(66). A proangiogenic function
for 12-LOX products has also been suggested. The downregulation of
15-LOX expression has been shown in breast and prostate cancer and
colorectal adenocarcinomas (67–70).
The 15-LOX-2 isoform suppresses cell cycle progression and promotes
cell senescence (70–72). Taken together, these findings
suggest that LOXs may be potential targets for anticancer
therapy.
CYP450s are monooxygenases that catalyse a variety
of reactions. These enzymes have variable substrates, including
fatty acids, steroids and xenobiotics. CYP450 enzymes are localised
to the mitochondria and the endoplasmic reticulum. Mitochondrial
CYP450s metabolise endogenous substrates, whereas microsomal
CYP450s are involved in the metabolic reactions of exo- and
endogenous substrates. Significant attention has been focused on
the roles of COX- and LOX-derived products in carcinogenesis;
however, little is known regarding the role of CYP450-derived
products in this process. CYP450 activity in cancer cells may lead
to the deactivation of antitumour drugs, thereby limiting
therapeutic efficacy. The CYP1, CYP2 and CYP3 families are
important enzymes that metabolise a significant number of
clinically important drugs (73).
Aberrant CYP450 enzymatic activity has been detected in a variety
of human cancer cell lines and has been shown to contribute to
neoangiogenesis, cancer cell migration, tumour growth and
metastasis (74–78).
Drug resistance is considered to be a major
hindrance to the success of chemotherapeutic treatment. Multidrug
resistance (MDR) is a multifactorial phenomenon and is often
associated with the overexpression of ATP-binding cassette (ABC)
transporter proteins (79,
80). Accumulating evidence
indicates that NSAIDs exert a chemosensitising effect; however, the
exact mechanism underlying this action remains unknown, although
several molecular mechanisms have been suggested.
NSAIDs, particularly COX-2 inhibitors, may supress
MDR by inhibiting ABC transporters and sensitise cancer cells to
the antiproliferative effects of anticancer drugs. These effects of
NSAIDs have been demonstrated in several different malignancies
(81–85). Permeability glycoprotein (P-gp),
which acts on a broad substrate range, is one of the most
extensively investigated and best characterised transporter
proteins. NSAIDs have been shown to suppress the expression and
function of this transporter in a variety of cancer cell types.
Zatelli et al (85)
demonstrated that treatment with the selective COX-2 inhibitor
NS-398 resulted in significantly increased doxorubicin accumulation
and sensitivity in chemoresistant MCF7 breast cancer cells. Those
effects depended on the inhibition of P-gp expression and function.
By contrast, it was suggested that NSAIDs are not involved in the
regulation of P-gp activity and function and that their
chemosensitising effect is mediated through different mechanisms
(86). However, the majority of
the studies contradict this hypothesis. Awara et al
(87) reported an enhancement of
doxorubicin antitumour activity with celecoxib-induced P-gp
inhibition. This was demonstrated by a significant reduction in the
efflux of the P-gp substrate Rhodamine 123. Similar findings were
reported by other research groups (82, 85,
88, 89). Indomethacin and a COX-2 selective
inhibitor, SC236, sensitised HepG2 human hepatocellular carcinoma
cells to the cytotoxic effects of doxorubicin. This effect was the
result of increased intracellular retention and accumulation of
doxorubicin via the inhibition of P-gp and MDR associated protein 1
(MRP1) expression and activity (90). Kang et al (91) detected an inhibition of the MRP1
efflux pump and enhanced doxorubicin cytotoxicity with celecoxib
treatment. Similar results were obtained by Ko et al
(92), where celecoxib not only
reverted MRP1-related drug resistance, but also inhibited the
function of breast cancer resistance protein (BCRP). Due to its
expression in malignant hematopoietic and lymphoid cells, BCRP
potentially plays an important role in drug resistance, not only in
breast cancer, but also in hematological malignancies. Furthermore,
BCRP is expressed in leukaemic stem cells, contributing to the
resistance of these cancers to chemotherapy or targeted therapy
(93). The drugs used to treat
these cancers are often BCRP substrates. Little is known regarding
the effects of NSAIDs on antitumour drug cytotoxicity in
hematological malignancies. Accumulating evidence indicates a
positive effect of NSAIDs on chemotherapeutic drug action in
BCRP-overexpressing solid tumours. Co-treatment with mitoxantrone
and indomethacin sensitised resistant MCF-7/MX cells to
mitoxantrone (94). Studies that
combined NSAIDs with cisplatin-based chemotherapy have yielded
opposing results. A recent study revealed that celecoxib and SC-236
antagonised the cytotoxicity of cisplatin in human gastric cells,
whereas indomethacin and nimesulid exerted no effects (95). By contrast, the use of another
COX-2 selective inhibitor, JTE-522, in combination with cisplatin,
resulted in synergistic antitumour activity in a gastric cancer
cell line (96). In other cancer
cell lines, celecoxib potentiated the cytotoxicity of cisplatin
(97, 98). The discrepancy regarding the
effects of NSAIDs on cisplatin action may be partially explained by
the different chemical structures of the utilised NSAIDs and by the
different tumour cell types employed (95).
Apart from ABC transporter inhibition, other
mechanisms have been suggested to explain the chemosensitising
effect of NSAIDs, including the inhibition of several
transcriptional factors, varying functions of COX-2 in cancer
cells, ceramide production and DNA hypermethylation (Table I). NF-κB inhibition may play a role
in NSAID-enhanced antitumour drug cytotoxicity (99). NF-κB has been shown to be involved
in chemoresistance in different cancer types. The constitutive
expression of this transcription factor in tumours protects against
apoptotic stimuli. Moreover, the inhibition of NF-κB activity may
affect intracellular drug accumulation and transport. The enhanced
accumulation of doxorubicin in MDA-MB-231 human breast cancer cells
upon celecoxib treatment was not mediated by changes in COX-2
enzyme activity or through P-gp, MRP1 or BCRP inhibition, but
rather due to the inhibition of NF-κB. Xia et al also
demonstrated that NSAIDs may sensitise cancer cells to antitumour
drugs by inducing DNA hypermethylation (100). The ability of celecoxib to
modulate DNA methylation has also been demonstrated (101). The expression of the MDR1
gene, which codes for the P-gp protein, is regulated through the
methylation of CpG islands located within the MDR1 promoter
(102–104). Xia et al observed that
treatment with celecoxib significantly enhanced CpG island
methylation, which led to the suppression of P-gp expression
(100). The ability of celecoxib
to repress the activity of the transcription factor Sp1 was
previously demonstrated (105).
The MDR1 gene promoter contains a binding side for this
factor. This binding site may be susceptible to celecoxib-induced
hypermethylation, thereby limiting the ability of Sp1 to bind DNA.
Celecoxib, in combination with the 5-LOX inhibitor MK-886, exerted
a significant additive cytotoxic effect on Caco-2 and HT-29 cancer
cells, which was, in part, mediated by ceramide-induced apoptosis
(106). El-Awady et al
(107) demonstrated the diverse
effects of celecoxib on the anticancer activity of etoposide,
cisplatin, 5-fluorouracil (5-FU) and doxorubicin in five cancer
cell lines, namely the HeLa, HCT-116, HepG2, MCF7 and U251. In the
MCF7 breast cancer cell line, the interaction of celecoxib with
these four chemotherapeutics was antagonistic, indicating that
celecoxib is of little value when used in combination with
antitumour drugs in the treatment of breast cancer. By contrast,
other data indicate that celecoxib enhances the cytotoxicity of
anticancer drugs in breast cancer cells (99, 108). The interaction of celecoxib with
etoposide, cisplatin and 5-FU was shown to be dependent on the
cancer cell line employed, the drug type used and the incubation
schedule. The combination of celecoxib and the same antitumour drug
also exerted different effects on different cell lines. One
plausible explanation for this finding may be that COX-2 has
different roles in different cancer types (107). In cancers where COX-2 increases
tumour growth and progression (109), COX-2 inhibitors may be of
therapeutic benefit. However, in other malignancies, COX-2 has been
reported to exert proapoptotic and tumour-suppressing effects
(110–112). In such cancer types, COX-2
inhibition may lead to enhanced tumour growth, inhibition of
apoptosis and decreased efficacy of anticancer drugs (107). Several studies reported a direct
association between COX-2 expression and the ABC transporters P-gp
and MRP1. Patel et al (113) demonstrated that the
overexpression of COX-2 led to increased P-gp expression and
activity, whereas the COX-2 inhibitor NS398 was able to block this
increase. In colon cancer, a resistance to cisplatin resulted from
COX-2 overexpression, which induced MRP1 expression (114). A positive correlation between the
expression of COX-2 and P-gp was also reported by studies on
hepatocellular carcinoma, breast and ovarian cancer (115–117). COX-2 was found to be involved in
the regulation of P-gp, MRP1 and BCRP transporter expression via
the COX-2/PGE2/PGE receptor 4/phosphatidyl inositol 3-kinase
pathway (116, 118).
Synergistic effects of NSAIDs with hypericin
(HY)-mediated photodynamic therapy (PDT) have also been reported
(119–122). The specific inhibition of COX,
LOX and CYP450 activity increased the efficacy of HY-PDT in the
HT-29 cancer cell line (121). An
important role for the MRP1 and BCRP transporters in HY efflux was
also demonstrated (119).
Proadifen, a P450 monooxygenase inhibitor, was shown to inhibit
these transport proteins, resulting in a significant increase in
intracellular HY accumulation in HT-29 cells and MRP1 and
BCRP-overexpressing cells.
Taken together, the abovementioned findings indicate
that the mechanism through which NSAIDs affect the action and
effectiveness of cytotoxic drugs varies. The exact mechanism may
depend on the cancer cell line, the structures of the NSAIDs and
chemotherapeutics, the specific interactions between the drugs and
the incubation schedule. The mechanism underlying the NSAID-induced
increase in antitumour drug cytotoxicity may be one of the
abovementioned processes. However, more than one mechanisms are
likely involved.
A growing amount of evidence from various animal
models suggests positive effects of NSAID use in combination with
antitumour drugs (87, 123–129) (Table II). However, the exact mechanism
through which this combined treatment results in improved
antitumour activity in in vivo models is not clearly
understood. Given the complexity of animal models in comparison to
in vitro systems, the effects of the tumour
microenvironment, tumour angiogenesis, the immune system and
pharmacokinetic processes must be taken into consideration
(123, 126, 130). As NSAIDs may alter ABC
transporter expression or activity in cancer cell lines, this
mechanism may also be involved in vivo. Awara et al
(87) reported that the inhibition
of P-gp activity by NSAIDs is likely responsible for the enhanced
antitumour effects of doxorubicin. It was suggested that NSAIDs
exert their growth-inhibitory functions and synergistic effects
with chemotherapeutics through multiple pathways. Neoangiogenesis
plays a key role in tumour promotion and progression. Certain
studies demonstrated the ability of NSAIDs, particularly selective
COX-2 inhibitors, to suppress tumour growth by inhibiting
angiogenesis and cell proliferation (131, 132). Although the suppression of
angiogenesis that occurs with NSAID treatment alone may not be
sufficient to inhibit tumour growth, NSAIDs may enhance the
antiangiogenic and antiproliferative effects of certain antitumour
drugs (123, 125, 127). As shown by Irie et al
(125), celecoxib alone did not
significantly inhibit tumour growth, although it did exhibit a
certain antiangiogenic activity. However, in combination with 5-FU,
celecoxib enhanced the antitumour effect of 5-FU and significantly
suppressed angiogenesis and tumour growth, likely via the
inhibition of VEGF and the induction of IFN-γ (125). Treatment with celecoxib in
combination with doxorubicin and irinotecan was also found to be
effective in decreasing tumour growth through the inhibition of
cell proliferation and the suppression of tumour vasculature
(127). A number of intracellular
signalling proteins are involved in cell proliferation, survival
and apoptosis. Several lines of evidence suggest that COX-2 may
elevate the levels of the antiapoptotic proteins Bcl-2 and Mcl-1
through mitogen-activated protein kinase activation, which results
in an inhibition of the cytochrome c pathway (133–135). Moreover, a study by Zhang et
al (129) revealed an
improved therapeutic benefit of 5-FU via celecoxib addition, which
occurred through the induction of the cytochrome c-dependent
apoptotic pathway, as well as a possible role for 5-FU in the
celecoxib-mediated inhibition of COX-2 expression. As previously
mentioned, the antiproliferative, antiangiogenic and antitumour
effects of NSAIDs may be, to a certain extent, COX-2-independent.
Consistent with these findings, piroxicam was able to exert its
effect via a COX/PGE2-independent mechanism (128). Moreover, piroxicam enhanced
cisplatin-induced cytotoxicity via the upregulation of endogenous
drug effectors and the inhibition of certain cell growth
regulators. In vitro studies demonstrated that NSAIDs may
mediate their antitumour effects through modulation of the NF-κB
signalling pathway (99, 136). NF-κB, with its dual anti- and
proapoptotic functions, plays an important role in regulating
cellular proliferation and apoptotic cell death. The inhibition of
NF-κB activity may be responsible for the celecoxib-induced
doxorubicin cytotoxicity that results in decreased tumour volume
(99). By contrast, certain
studies suggested that NSAIDs may activate NF-κB, thereby inducing
apoptosis (137, 138). Apart from enhancing the
cytotoxicity of chemotherapeutic drugs, the addition of NSAIDs may
also reduce the severity of chemotherapy-associated adverse
effects, such as late diarrhoea and cachexia (139).
Due to the limited effectiveness of certain cancer
treatments, it is necessary to establish a novel treatment strategy
that improves patient response to chemotherapy. A large number of
studies have demonstrated that COX-2 may be involved in the
development of several cancer types. COX-2 may positively affect
multiple processes, including tumour cell growth, migration and
invasiveness, but may also downregulate apoptosis and angiogenic
stimulation (35–38). Moreover, the overexpression of
COX-2 may also reduce the response of cancer cells to cytotoxic
therapy (140). Preclinical
studies suggested that treatment with NSAIDs, particularly COX-2
inhibitors, may affect the outcome of chemotherapy through various
mechanisms, including the inhibition of neoangiogenesis and the
induction of apoptosis (130,
131, 141). Despite promising preclinical
results with NSAIDs in combination with antitumour drugs, little is
known regarding the effects of this combination on humans. The
currently available clinical results are contradictory and mainly
disappointing (142–145) (Table III). For example, several
combinations did not appear to improve therapy outcome, including
celecoxib and docetaxel (144,
146); celecoxib and 5-FU
(142, 143); rofecoxib, 5-FU and leucovorin
(147); celecoxib and
transtuzumab (148); rofecoxib,
cisplatin and gemcitabine (149);
celecoxib, docetaxel and carboplatin (150); and celecoxib and platinum
derivates (151). However,
certain phase II studies have yielded encouraging results. In the
case of non-small-cell lung carcinoma (NSCLC), the combination of
celecoxib and chemotherapy was associated with increased overall
survival (152, 153). In the case of heavily pretreated
recurrent ovarian cancer, the administration of celecoxib in
combination with carboplatin-based chemotherapy also yielded
promising results (154). The
discrepancy in various results may be due to multiple factors, such
as complex pharmacodynamic interactions between NSAIDs and the
cytotoxic drugs and the varying levels of intratumoural COX-2 and
ABC transporters (155–159). The role of COX-2 expression in
the response of cancer cells to combined NSAID and antitumour drug
therapy was demonstrated by Edelman et al and has been
supported by other studies (155,
160). Patients with
COX-2-overexpressing tumours who did not receive combined
celecoxib/chemotherapy treatment exhibited a significantly worse
outcome. Possible adverse effects in patients with
COX-2-non-expressing tumours that received celecoxib treatment were
also demonstrated (160).
NSAIDs are potent antitumour drugs, capable of
inhibiting tumour angiogenesis, proliferation, invasion and
motility, as well as of inducing apoptosis. Furthermore, a number
of experimental and preclinical studies indicated that combining
NSAIDs with antitumour drugs may improve outcome. The exact
mechanism underlying this synergistic effect has not yet been fully
elucidated, but may involve several diverse processes, including
the inhibition of COX-2 expression, ABC transporter activity or
NF-κB. However, the results of combined therapy in clinical trials
are mainly disappointing. Despite significant efforts to determine
the exact mechanism through which NSAIDs modulate the efficacy of
anticancer drugs, there remain several unanswered questions.
This study was supported by the Cancer Research
Foundation (contact no. O-12-102/0001-00).
|
1
|
Hanif R, Pittas A, Feng Y, Koutsos MI,
Qiao L, Staiano-Coico L, Shiff SI and Rigas B: Effects of
nonsteroidal anti-inflammatory drugs on proliferation and on
induction of apoptosis in colon cancer cells by a
prostaglandin-independent pathway. Biochem Pharmacol. 52:237–245.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Souza RF, Shewmake K, Beer DG, Cryer B and
Spechler SJ: Selective inhibition of cyclooxygenase-2 suppresses
growth and induces apoptosis in human esophageal adenocarcinoma
cells. Cancer Res. 60:5767–5772. 2000.PubMed/NCBI
|
|
3
|
Dai Y and Wang WH: Non-steroidal
anti-inflammatory drugs in prevention of gastric cancer. World J
Gastroenterol. 12:2884–2889. 2006.PubMed/NCBI
|
|
4
|
DuBois RN and Smalley WE: Cyclooxygenase,
NSAIDs, and colorectal cancer. J Gastroenterol. 31:898–906. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rao CV and Reddy BS: NSAIDs and
chemoprevention. Curr Cancer Drug Targets. 4:29–42. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Winde G, Schmid KW, Brandt B, Muller O and
Osswald H: Clinical and genomic influence of sulindac on rectal
mucosa in familial adenomatous polyposis. Dis Colon Rectum.
40:1156–1169. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maclagan T: The treatment of acute
rheumatism by salicin and salicylic acid. Lancet. 113:875–877.
1879. View Article : Google Scholar
|
|
8
|
Dugowson CE and Gnanashanmugam P:
Nonsteroidal anti-inflammatory drugs. Phys Med Rehabil Clin N Am.
17347–354. (vi)2006. View Article : Google Scholar
|
|
9
|
Vane JR and Botting RM: The mechanism of
action of aspirin. Thromb Res. 110:255–258. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Flower RJ: Drugs which inhibit
prostaglandin biosynthesis. Pharmacol Rev. 26:33–67. 1974.
|
|
11
|
Vane JR and Botting RM: Anti-inflammatory
drugs and their mechanism of action. Inflamm Res. 47 (Suppl
2):S78–S87. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Marnett LJ, Rowlinson SW, Goodwin DC,
Kalgutkar AS and Lanzo CA: Arachidonic acid oxygenation by COX-1
and COX-2. Mechanisms of catalysis and inhibition. J Biol Chem.
274:22903–22906. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rao P and Knaus EE: Evolution of
nonsteroidal anti-inflammatory drugs (NSAIDs): cyclooxygenase (COX)
inhibition and beyond. J Pharm Pharm Sci. 11:S81–S110.
2008.PubMed/NCBI
|
|
14
|
Dubois RN, Abramson SB, Crofford L, et al:
Cyclooxygenase in biology and disease. FASEB J. 12:1063–1073.
1998.PubMed/NCBI
|
|
15
|
Rigas B and Shiff SJ: Nonsteroidal
anti-inflammatory drugs (NSAIDs), cyclooxygenases, and the cell
cycle. Their interactions in colon cancer. Adv Exp Med Biol.
470:119–126. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang D, Mann JR and DuBois RN: The role of
prostaglandins and other eicosanoids in the gastrointestinal tract.
Gastroenterology. 128:1445–1461. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Capdevila JH, Falck JR and Harris RC:
Cytochrome P450 and arachidonic acid bioactivation. Molecular and
functional properties of the arachidonate monooxygenase. J Lipid
Res. 41:163–181. 2000.PubMed/NCBI
|
|
18
|
Gerritsen ME: Physiological and
pathophysiological roles of eicosanoids in the microcirculation.
Cardiovasc Res. 32:720–732. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harder DR, Campbell WB and Roman RJ: Role
of cytochrome P-450 enzymes and metabolites of arachidonic acid in
the control of vascular tone. J Vasc Res. 32:79–92. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Harizi H, Corcuff JB and Gualde N:
Arachidonic-acid-derived eicosanoids: roles in biology and
immunopathology. Trends Mol Med. 14:461–469. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen YQ, Duniec ZM, Liu B, et al:
Endogenous 12(S)-HETE production by tumor cells and its role in
metastasis. Cancer Res. 54:1574–1579. 1994.PubMed/NCBI
|
|
22
|
Gao X, Grignon DJ, Chbihi T, et al:
Elevated 12-lipoxygenase mRNA expression correlates with advanced
stage and poor differentiation of human prostate cancer. Urology.
46:227–237. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Honn KV, Tang DG, Gao X, et al:
12-lipoxygenases and 12(S)-HETE: role in cancer metastasis. Cancer
Metastasis Rev. 13:365–396. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tang DG and Honn KV: 12-Lipoxygenase,
12(S)-HETE, and cancer metastasis. Ann N Y Acad Sci. 744:199–215.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Timar J, Raso E, Fazakas ZS, Silletti S,
Raz A and Honn KV: Multiple use of a signal transduction pathway in
tumor cell invasion. Anticancer Res. 16:3299–3306. 1996.PubMed/NCBI
|
|
26
|
Kage K, Fujita N, Oh-hara T, Ogata E,
Fujita T and Tsuruo T: Basic fibroblast growth factor induces
cyclooxygenase-2 expression in endothelial cells derived from bone.
Biochem Biophys Res Commun. 254:259–263. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fong CY, Pang L, Holland E and Knox AJ:
TGF-beta1 stimulates IL-8 release, COX-2 expression, and PGE(2)
release in human airway smooth muscle cells. Am J Physiol Lung Cell
Mol Physiol. 279:L201–L207. 2000.PubMed/NCBI
|
|
28
|
Saha D, Datta PK, Sheng H, et al:
Synergistic induction of cyclooxygenase-2 by transforming growth
factor-beta1 and epidermal growth factor inhibits apoptosis in
epithelial cells. Neoplasia. 1:508–517. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Diaz A, Chepenik KP, Korn JH, Reginato AM
and Jimenez SA: Differential regulation of cyclooxygenases 1 and 2
by interleukin-1 beta, tumor necrosis factor-alpha, and
transforming growth factor-beta 1 in human lung fibroblasts. Exp
Cell Res. 241:222–229. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chandrasekharan NV, Dai H, Roos KL, et al:
COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and
other analgesic/antipyretic drugs: cloning, structure, and
expression. Proc Natl Acad Sci USA. 99:13926–13931. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cui JG, Kuroda H, Chandrasekharan NV, et
al: Cyclooxygenase-3 gene expression in Alzheimer hippocampus and
in stressed human neural cells. Neurochem Res. 29:1731–1737. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kis B, Snipes JA and Busija DW:
Acetaminophen and the cyclooxygenase-3 puzzle: sorting out facts,
fictions, and uncertainties. J Pharmacol Exp Ther. 315:1–7. 2005.
View Article : Google Scholar
|
|
33
|
Cerella C, Sobolewski C, Chateauvieux S,
et al: COX-2 inhibitors block chemotherapeutic agent-induced
apoptosis prior to commitment in hematopoietic cancer cells.
Biochem Pharmacol. 82:1277–1290. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Surh YJ and Kundu JK: Signal transduction
network leading to COX-2 induction: a road map in search of cancer
chemopreventives. Arch Pharm Res. 28:1–15. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Eberhart CE, Coffey RJ, Radhika A,
Giardiello FM, Ferrenbach S and DuBois RN: Up-regulation of
cyclooxygenase 2 gene expression in human colorectal adenomas and
adenocarcinomas. Gastroenterology. 107:1183–1188. 1994.PubMed/NCBI
|
|
36
|
Sano H, Kawahito Y, Wilder RL, et al:
Expression of cyclooxygenase-1 and −2 in human colorectal cancer.
Cancer Res. 55:3785–3789. 1995.
|
|
37
|
Ristimaki A, Honkanen N, Jankala H,
Sipponen P and Harkonen M: Expression of cyclooxygenase-2 in human
gastric carcinoma. Cancer Res. 57:1276–1280. 1997.PubMed/NCBI
|
|
38
|
Yip-Schneider MT, Barnard DS, Billings SD,
et al: Cyclooxygenase-2 expression in human pancreatic
adenocarcinomas. Carcinogenesis. 21:139–146. 2000.
|
|
39
|
Wilson KT, Fu S, Ramanujam KS and Meltzer
SJ: Increased expression of inducible nitric oxide synthase and
cyclooxygenase-2 in Barrett's esophagus and associated
adenocarcinomas. Cancer Res. 58:2929–2934. 1998.
|
|
40
|
Wolff H, Saukkonen K, Anttila S,
Karjalainen A, Vainio H and Ristimaki A: Expression of
cyclooxygenase-2 in human lung carcinoma. Cancer Res. 58:4997–5001.
1998.PubMed/NCBI
|
|
41
|
Hwang D, Scollard D, Byrne J and Levine E:
Expression of cyclooxygenase-1 and cyclooxygenase-2 in human breast
cancer. J Natl Cancer Inst. 90:455–460. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cao Y and Prescott SM: Many actions of
cyclooxygenase-2 in cellular dynamics and in cancer. J Cell
Physiol. 190:279–286. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hida T, Kozaki K, Muramatsu H, et al:
Cyclooxygenase-2 inhibitor induces apoptosis and enhances
cytotoxicity of various anticancer agents in non-small cell lung
cancer cell lines. Clin Cancer Res. 6:2006–2011. 2000.PubMed/NCBI
|
|
44
|
O'Kane SL, Eagle GL, Greenman J, Lind MJ
and Cawkwell L: COX-2 specific inhibitors enhance the cytotoxic
effects of pemetrexed in mesothelioma cell lines. Lung Cancer.
67:160–165. 2010.PubMed/NCBI
|
|
45
|
Sinha-Datta U, Taylor JM, Brown M and
Nicot C: Celecoxib disrupts the canonical apoptotic network in
HTLV-I cells through activation of Bax and inhibition of PKB/Akt.
Apoptosis. 13:33–40. 2008. View Article : Google Scholar
|
|
46
|
Totzke G, Schulze-Osthoff K and Janicke
RU: Cyclooxygenase-2 (COX-2) inhibitors sensitize tumor cells
specifically to death receptor-induced apoptosis independently of
COX-2 inhibition. Oncogene. 22:8021–8030. 2003. View Article : Google Scholar
|
|
47
|
Elder DJ, Halton DE, Hague A and Paraskeva
C: Induction of apoptotic cell death in human colorectal carcinoma
cell lines by a cyclooxygenase-2 (COX-2)-selective nonsteroidal
anti-inflammatory drug: independence from COX-2 protein expression.
Clin Cancer Res. 3:1679–1683. 1997.
|
|
48
|
Grosch S, Tegeder I, Niederberger E,
Brautigam L and Geisslinger G: COX-2 independent induction of cell
cycle arrest and apoptosis in colon cancer cells by the selective
COX-2 inhibitor celecoxib. FASEB J. 15:2742–2744. 2001.PubMed/NCBI
|
|
49
|
Zhang X, Morham SG, Langenbach R and Young
DA: Malignant transformation and antineoplastic actions of
nonsteroidal antiinflammatory drugs (NSAIDs) on cyclooxygenase-null
embryo fibroblasts. J Exp Med. 190:451–459. 1999. View Article : Google Scholar
|
|
50
|
Pidgeon GP, Lysaght J, Krishnamoorthy S,
et al: Lipoxygenase metabolism: roles in tumor progression and
survival. Cancer Metastasis Rev. 26:503–524. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Radmark O, Werz O, Steinhilber D and
Samuelsson B: 5-Lipoxygenase: regulation of expression and enzyme
activity. Trends Biochem Sci. 32:332–341. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Werz O and Steinhilber D: Therapeutic
options for 5-lipoxygenase inhibitors. Pharmacol Ther. 112:701–718.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wasilewicz MP, Kolodziej B, Bojulko T, et
al: Overexpression of 5-lipoxygenase in sporadic colonic adenomas
and a possible new aspect of colon carcinogenesis. Int J Colorectal
Dis. 25:1079–1085. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Metzger K, Angres G, Maier H and Lehmann
WD: Lipoxygenase products in human saliva: patients with oral
cancer compared to controls. Free Radic Biol Med. 18:185–194. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chen Y, Hu Y, Zhang H, Peng C and Li S:
Loss of the Alox5 gene impairs leukemia stem cells and prevents
chronic myeloid leukemia. Nat Genet. 41:783–792. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hennig R, Ventura J, Segersvard R, et al:
LY293111 improves efficacy of gemcitabine therapy on pancreatic
cancer in a fluorescent orthotopic model in athymic mice.
Neoplasia. 7:417–425. 2005.PubMed/NCBI
|
|
57
|
Paruchuri S, Broom O, Dib K and Sjolander
A: The pro-inflammatory mediator leukotriene D4 induces
phosphatidylinositol 3-kinase and Rac-dependent migration of
intestinal epithelial cells. J Biol Chem. 280:13538–13544. 2005.
View Article : Google Scholar
|
|
58
|
Hayashi T, Nishiyama K and Shirahama T:
Inhibition of 5-lipoxygenase pathway suppresses the growth of
bladder cancer cells. Int J Urol. 13:1086–1091. 2006.
|
|
59
|
Meng Z, Cao R, Yang Z, Liu T, Wang Y and
Wang X: Inhibitor of 5-lipoxygenase, zileuton, suppresses prostate
cancer metastasis by upregulating E-cadherin and paxillin.
Urology. 82(1452): e7–e14. 2013.PubMed/NCBI
|
|
60
|
Schroeder CP, Yang P, Newman RA and Lotan
R: Simultaneous inhibition of COX-2 and 5-LOX activities augments
growth arrest and death of premalignant and malignant human lung
cell lines. J Exp Ther Oncol. 6:183–192. 2007.
|
|
61
|
Shin VY, Jin HC, Ng EK, Sung JJ, Chu KM
and Cho CH: Activation of 5-lipoxygenase is required for nicotine
mediated epithelial-mesenchymal transition and tumor cell growth.
Cancer Lett. 292:237–245. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Hagmann W, Gao X, Zacharek A,
Wojciechowski LA and Honn KV: 12-Lipoxygenase in Lewis lung
carcinoma cells: molecular identity, intracellular distribution of
activity and protein, and Ca2+-dependent translocation from cytosol
to membranes. Prostaglandins. 49:49–62. 1995.PubMed/NCBI
|
|
63
|
Pidgeon GP, Kandouz M, Meram A and Honn
KV: Mechanisms controlling cell cycle arrest and induction of
apoptosis after 12-lipoxygenase inhibition in prostate cancer
cells. Cancer Res. 62:2721–2727. 2002.PubMed/NCBI
|
|
64
|
Tang DG, Chen YQ and Honn KV: Arachidonate
lipoxygenases as essential regulators of cell survival and
apoptosis. Proc Natl Acad Sci USA. 93:5241–5246. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wong BC, Wang WP, Cho CH, et al:
12-Lipoxygenase inhibition induced apoptosis in human gastric
cancer cells. Carcinogenesis. 22:1349–1354. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Terada N, Shimizu Y, Kamba T, et al:
Identification of EP4 as a potential target for the treatment of
castration-resistant prostate cancer using a novel xenograft model.
Cancer Res. 70:1606–1615. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Jiang WG, Watkins G, Douglas-Jones A and
Mansel RE: Reduction of isoforms of 15-lipoxygenase (15-LOX)-1 and
15-LOX-2 in human breast cancer. Prostaglandins Leukot Essent Fatty
Acids. 74:235–245. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Shappell SB, Boeglin WE, Olson SJ, Kasper
S and Brash AR: 15-lipoxygenase-2 (15-LOX-2) is expressed in benign
prostatic epithelium and reduced in prostate adenocarcinoma. Am J
Pathol. 155:235–245. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Shureiqi I, Wu Y, Chen D, et al: The
critical role of 15-lipoxygenase-1 in colorectal epithelial cell
terminal differentiation and tumorigenesis. Cancer Res.
65:11486–11492. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Tang DG, Bhatia B, Tang S and
Schneider-Broussard R: 15-Lipoxygenase 2 (15-LOX2) is a functional
tumor suppressor that regulates human prostate epithelial cell
differentiation, senescence, and growth (size). Prostaglandins
Other Lipid Mediat. 82:135–146. 2007. View Article : Google Scholar
|
|
71
|
Bhatia B, Tang S, Yang P, et al:
Cell-autonomous induction of functional tumor suppressor
15-lipoxygenase 2 (15-LOX2) contributes to replicative senescence
of human prostate progenitor cells. Oncogene. 24:3583–3595. 2005.
View Article : Google Scholar
|
|
72
|
Tang S, Bhatia B, Maldonado CJ, et al:
Evidence that arachidonate 15-lipoxygenase 2 is a negative cell
cycle regulator in normal prostate epithelial cells. J Biol Chem.
277:16189–16201. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Brown CM, Reisfeld B and Mayeno AN:
Cytochromes P450: a structure-based summary of biotransformations
using representative substrates. Drug Metab Rev. 40:1–100. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Cheranov SY, Karpurapu M, Wang D, Zhang B,
Venema RC and Rao GN: An essential role for SRC-activated STAT-3 in
14,15-EET-induced VEGF expression and angiogenesis. Blood.
111:5581–5591. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Jiang JG, Ning YG, Chen C, et al:
Cytochrome p450 epoxygenase promotes human cancer metastasis.
Cancer Res. 67:6665–6674. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Webler AC, Michaelis UR, Popp R, et al:
Epoxyeicosatrienoic acids are part of the VEGF-activated signaling
cascade leading to angiogenesis. Am J Physiol Cell Physiol.
295:C1292–C1301. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Webler AC, Popp R, Korff T, et al:
Cytochrome P450 2C9-induced angiogenesis is dependent on EphB4.
Arterioscler Thromb Vasc Biol. 28:1123–1129. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Yan G, Chen S, You B and Sun J: Activation
of sphingosine kinase-1 mediates induction of endothelial cell
proliferation and angiogenesis by epoxyeicosatrienoic acids.
Cardiovasc Res. 78:308–314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Gottesman MM: Mechanisms of cancer drug
resistance. Annu Rev Med. 53:615–627. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Turk D and Szakacs G: Relevance of
multidrug resistance in the age of targeted therapy. Curr Opin Drug
Discov Devel. 12:246–252. 2009.PubMed/NCBI
|
|
81
|
Arico S, Pattingre S, Bauvy C, et al:
Celecoxib induces apoptosis by inhibiting
3-phosphoinositide-dependent protein kinase-1 activity in the human
colon cancer HT-29 cell line. J Biol Chem. 277:27613–27621. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Arunasree KM, Roy KR, Anilkumar K, Aparna
A, Reddy GV and Reddanna P: Imatinib-resistant K562 cells are more
sensitive to celecoxib, a selective COX-2 inhibitor: role of COX-2
and MDR-1. Leuk Res. 32:855–864. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Roy KR, Reddy GV, Maitreyi L, et al:
Celecoxib inhibits MDR1 expression through COX-2-dependent
mechanism in human hepatocellular carcinoma (HepG2) cell line.
Cancer Chemother Pharmacol. 65:903–911. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Yu L, Wu WK, Li ZJ, Liu QC, Li HT, Wu YC
and Cho CH: Enhancement of doxorubicin cytotoxicity on human
esophageal squamous cell carcinoma cells by indomethacin and
4-[5-(4-chlorophenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide
(SC236) via inhibiting P-glycoprotein activity. Mol Pharmacol.
75:1364–1373. 2009.
|
|
85
|
Zatelli MC, Luchin A, Tagliati F, et al:
Cyclooxygenase-2 inhibitors prevent the development of
chemoresistance phenotype in a breast cancer cell line by
inhibiting glycoprotein p-170 expression. Endocr Relat Cancer.
14:1029–1038. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
de Vries EF, Doorduin J, Vellinga NA, van
Waarde A, Dierckx RA and Klein HC: Can celecoxib affect
P-glycoprotein-mediated drug efflux? A microPET study. Nucl Med
Biol. 35:459–466. 2008.PubMed/NCBI
|
|
87
|
Awara WM, El-Sisi AE, El-Sayad ME and Goda
AE: The potential role of cyclooxygenase-2 inhibitors in the
treatment of experimentally-induced mammary tumour: does celecoxib
enhance the anti-tumour activity of doxorubicin? Pharmacol Res.
50:487–498. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Yan YX, Li WZ, Huang YQ and Liao WX: The
COX-2 inhibitor celecoxib enhances the sensitivity of KB/VCR oral
cancer cell lines to vincristine by down-regulating P-glycoprotein
expression and function. Prostaglandins Other Lipid Mediat.
97:29–35. 2011. View Article : Google Scholar
|
|
89
|
Zrieki A, Farinotti R and Buyse M:
Cyclooxygenase inhibitors down regulate P-glycoprotein in human
colorectal Caco-2 cell line. Pharm Res. 25:1991–2001. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ye CG, Wu WK, Yeung JH, et al:
Indomethacin and SC236 enhance the cytotoxicity of doxorubicin in
human hepatocellular carcinoma cells via inhibiting P-glycoprotein
and MRP1 expression. Cancer Lett. 304:90–96. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Kang HK, Lee E, Pyo H and Lim SJ:
Cyclooxygenase-independent down-regulation of multidrug
resistance-associated protein-1 expression by celecoxib in human
lung cancer cells. Mol Cancer Ther. 4:1358–1363. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Ko SH, Choi GJ, Lee JH, Han YA, Lim SJ and
Kim SH: Differential effects of selective cyclooxygenase-2
inhibitors in inhibiting proliferation and induction of apoptosis
in oral squamous cell carcinoma. Oncol Rep. 19:425–433. 2008.
|
|
93
|
Natarajan K, Xie Y, Baer MR and Ross DD:
Role of breast cancer resistance protein (BCRP/ABCG2) in cancer
drug resistance. Biochem Pharmacol. 83:1084–1103. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Elahian F, Kalalinia F and Behravan J:
Evaluation of indomethacin and dexamethasone effects on
BCRP-mediated drug resistance in MCF-7 parental and resistant cell
lines. Drug Chem Toxicol. 33:113–119. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Chen M, Yu L, Gu C, Zhong D, Wu S and Liu
S: Celecoxib antagonizes the cytotoxic effect of cisplatin in human
gastric cancer cells by decreasing intracellular cisplatin
accumulation. Cancer Lett. 329:189–196. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Sugiura T, Saikawa Y, Kubota T, et al:
Combination chemotherapy with JTE-522, a novel selective
cyclooxygenase-2 inhibitor, and cisplatin against gastric cancer
cell lines in vitro and in vivo. In Vivo. 17:229–233. 2003.
|
|
97
|
Kim SH, Kim SH, Song YC and Song YS:
Celecoxib potentiates the anticancer effect of cisplatin on vulvar
cancer cells independently of cyclooxygenase. Ann N Y Acad Sci.
1171:635–641. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Li WZ, Wang XY, Li ZG, Zhang JH and Ding
YQ: Celecoxib enhances the inhibitory effect of cisplatin on
Tca8113 cells in human tongue squamous cell carcinoma in vivo and
in vitro. J Oral Pathol Med. 39:579–584. 2010.PubMed/NCBI
|
|
99
|
van Wijngaarden J, van Beek E, van Rossum
G, et al: Celecoxib enhances doxorubicin-induced cytotoxicity in
MDA-MB231 cells by NF-kappaB-mediated increase of intracellular
doxorubicin accumulation. Eur J Cancer. 43:433–442. 2007.
|
|
100
|
Xia W, Zhao T, Lv J, et al: Celecoxib
enhanced the sensitivity of cancer cells to anticancer drugs by
inhibition of the expression of P-glycoprotein through a
COX-2-independent manner. J Cell Biochem. 108:181–194. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Pereira MA, Tao L, Wang W, et al:
Modulation by celecoxib and difluoromethylornithine of the
methylation of DNA and the estrogen receptor-alpha gene in rat
colon tumors. Carcinogenesis. 25:1917–1923. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Ellinger J, Bastian PJ, Jurgan T, et al:
CpG island hypermethylation at multiple gene sites in diagnosis and
prognosis of prostate cancer. Urology. 71:161–167. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Enokida H, Shiina H, Igawa M, et al: CpG
hypermethylation of MDR1 gene contributes to the pathogenesis and
progression of human prostate cancer. Cancer Res. 64:5956–5962.
2004. View Article : Google Scholar
|
|
104
|
Qiu YY, Mirkin BL and Dwivedi RS: MDR1
hypermethylation contributes to the progression of neuroblastoma.
Mol Cell Biochem. 301:131–135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Wei D, Wang L, He Y, Xiong HQ, Abbruzzese
JL and Xie K: Celecoxib inhibits vascular endothelial growth factor
expression in and reduces angiogenesis and metastasis of human
pancreatic cancer via suppression of Sp1 transcription factor
activity. Cancer Res. 64:2030–2038. 2004. View Article : Google Scholar
|
|
106
|
Cianchi F, Cortesini C, Magnelli L, et al:
Inhibition of 5-lipoxygenase by MK886 augments the antitumor
activity of celecoxib in human colon cancer cells. Mol Cancer Ther.
5:2716–2726. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
El-Awady RA, Saleh EM, Ezz M and Elsayed
AM: Interaction of celecoxib with different anti-cancer drugs is
antagonistic in breast but not in other cancer cells. Toxicol Appl
Pharmacol. 255:271–286. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Chen C, Shen HL, Yang J, Chen QY and Xu
WL: Preventing chemoresistance of human breast cancer cell line,
MCF-7 with celecoxib. J Cancer Res Clin Oncol. 137:9–17. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Fosslien E: Molecular pathology of
cyclooxygenase-2 in neoplasia. Ann Clin Lab Sci. 30:3–21.
2000.PubMed/NCBI
|
|
110
|
Bol DK, Rowley RB, Ho CP, et al:
Cyclooxygenase-2 overexpression in the skin of transgenic mice
results in suppression of tumor development. Cancer Res.
62:2516–2521. 2002.PubMed/NCBI
|
|
111
|
Nakopoulou L, Mylona E, Papadaki I, et al:
Overexpression of cyclooxygenase-2 is associated with a favorable
prognostic phenotype in breast carcinoma. Pathobiology. 72:241–249.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Xu Z, Choudhary S, Voznesensky O, et al:
Overexpression of COX-2 in human osteosarcoma cells decreases
proliferation and increases apoptosis. Cancer Res. 66:6657–6664.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Patel VA, Dunn MJ and Sorokin A:
Regulation of MDR-1 (P-glycoprotein) by cyclooxygenase-2. J Biol
Chem. 277:38915–38920. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Saikawa Y, Sugiura T, Toriumi F, et al:
Cyclooxygenase-2 gene induction causes CDDP resistance in colon
cancer cell line, HCT-15. Anticancer Res. 24:2723–2728.
2004.PubMed/NCBI
|
|
115
|
Surowiak P, Materna V, Matkowski R, et al:
Relationship between the expression of cyclooxygenase 2 and
MDR1/P-glycoprotein in invasive breast cancers and their prognostic
significance. Breast Cancer Res. 7:R862–R870. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Surowiak P, Pawelczyk K, Maciejczyk A, et
al: Positive correlation between cyclooxygenase 2 and the
expression of ABC transporters in non-small cell lung cancer.
Anticancer Res. 28:2967–2974. 2008.PubMed/NCBI
|
|
117
|
Ziemann C, Schafer D, Rudell G, Kahl GF
and Hirsch-Ernst KI: The cyclooxygenase system participates in
functional MDR1b overexpression in primary rat hepatocyte cultures.
Hepatology. 35:579–588. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Liu B, Qu L and Tao H: Cyclo-oxygenase 2
up-regulates the effect of multidrug resistance. Cell Biol Int.
34:21–25. 2010.PubMed/NCBI
|
|
119
|
Jendzelovsky R, Mikes J, Koval J, et al:
Drug efflux transporters, MRP1 and BCRP, affect the outcome of
hypericin-mediated photodynamic therapy in HT-29 adenocarcinoma
cells. Photochem Photobiol Sci. 8:1716–1723. 2009. View Article : Google Scholar
|
|
120
|
Kleban J, Mikes J, Horvath V, et al:
Mechanisms involved in the cell cycle and apoptosis of HT-29 cells
pre-treated with MK-886 prior to photodynamic therapy with
hypericin. J Photochem Photobiol B. 93:108–118. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Kleban J, Mikes J, Szilardiova B, et al:
Modulation of hypericin photodynamic therapy by pretreatment with
12 various inhibitors of arachidonic acid metabolism in colon
adenocarcinoma HT-29 cells. Photochem Photobiol. 83:1174–1185.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Kleban J, Szilardiova B, Mikes J, et al:
Pre-treatment of HT-29 cells with 5-LOX inhibitor (MK-886) induces
changes in cell cycle and increases apoptosis after photodynamic
therapy with hypericin. J Photochem Photobiol B. 84:79–88. 2006.
View Article : Google Scholar
|
|
123
|
Hida T, Kozaki K, Ito H, et al:
Significant growth inhibition of human lung cancer cells both in
vitro and in vivo by the combined use of a selective cyclooxygenase
2 inhibitor, JTE-522, and conventional anticancer agents. Clin
Cancer Res. 8:2443–2447. 2002.
|
|
124
|
Hossain MA, Kim DH, Jang JY, et al:
Aspiri. induces apoptosis in vitro and inhibits tumor growth
of human hepatocellular carcinoma cells in a nude mouse xenograft
model. Int J Oncol. 40:1298–1304. 2012.PubMed/NCBI
|
|
125
|
Irie T, Tsujii M, Tsuji S, et al:
Synergistic antitumor effects of celecoxib with 5-fluorouracil
depend on IFN-gamma. Int J Cancer. 121:878–883. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Knapp DW, Glickman NW, Widmer WR, et al:
Cisplatin versus cisplatin combined with piroxicam in a canine
model of human invasive urinary bladder cancer. Cancer Chemother
Pharmacol. 46:221–226. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Ponthan F, Wickstrom M, Gleissman H, et
al: Celecoxib prevents neuroblastoma tumor development and
potentiates the effect of chemotherapeutic drugs in vitro and in
vivo. Clin Cancer Res. 13:1036–1044. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Spugnini EP, Cardillo I, Verdina A, et al:
Piroxicam and cisplatin in a mouse model of peritoneal
mesothelioma. Clin Cancer Res. 12:6133–6143. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Zhang DQ, Guo Q, Zhu JH and Chen WC:
Increase of cyclooxygenase-2 inhibition with celecoxib combined
with 5-FU enhances tumor cell apoptosis and antitumor efficacy in a
subcutaneous implantation tumor model of human colon cancer. World
J Surg Oncol. 11(16)2013. View Article : Google Scholar
|
|
130
|
Tsujii M, Kawano S, Tsuji S, Sawaoka H,
Hori M and DuBois RN: Cyclooxygenase regulates angiogenesis induced
by colon cancer cells. Cell. 93:705–716. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Leahy KM, Ornberg RL, Wang Y, Zweifel BS,
Koki AT and Masferrer JL: Cyclooxygenase-2 inhibition by celecoxib
reduces proliferation and induces apoptosis in angiogenic
endothelial cells in vivo. Cancer Res. 62:625–631. 2002.PubMed/NCBI
|
|
132
|
Patel MI, Subbaramaiah K, Du B, et al:
Celecoxib inhibits prostate cancer growth: evidence of a
cyclooxygenase-2-independent mechanism. Clin Cancer Res.
11:1999–2007. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Sakamoto T, Uozaki H, Kondo K, et al:
Cyclooxygenase-2 regulates the degree of apoptosis by modulating
bcl-2 protein in pleomorphic adenoma and mucoepidermoid carcinoma
of the parotid gland. Acta Otolaryngol. 125:191–195. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Tjiu JW, Liao YH, Lin SJ, et al:
Cyclooxygenase-2 overexpression in human basal cell carcinoma cell
line increases antiapoptosis, angiogenesis, and tumorigenesis. J
Invest Dermatol. 126:1143–1151. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Wang F, Sun GP, Zou YF, et al: Expression
of COX-2 and Bcl-2 in primary fallopian tube carcinoma:
correlations with clinicopathologic features. Folia Histochem
Cytobiol. 49:389–397. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Stark LA, Din FV, Zwacka RM and Dunlop MG:
Aspirin-induced activation of the NF-kappaB signaling pathway: a
novel mechanism for aspirin-mediated apoptosis in colon cancer
cells. FASEB J. 15:1273–1275. 2001.
|
|
137
|
Park IS, Jo JR, Hong H, et al: Aspirin
induces apoptosis in YD-8 human oral squamous carcinoma cells
through activation of caspases, down-regulation of Mcl-1, and
inactivation of ERK-1/2 and AKT. Toxicol In Vitro. 24:713–720.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Stark LA, Reid K, Sansom OJ, et al:
Aspirin activates the NF-kappaB signalling pathway and induces
apoptosis in intestinal neoplasia in two in vivo models of human
colorectal cancer. Carcinogenesis. 28:968–976. 2007. View Article : Google Scholar
|
|
139
|
Trifan OC, Durham WF, Salazar VS, et al:
Cyclooxygenase-2 inhibition with celecoxib enhances antitumor
efficacy and reduces diarrhea side effect of CPT-11. Cancer Res.
62:5778–5784. 2002.PubMed/NCBI
|
|
140
|
Altorki NK, Port JL, Zhang F, et al:
Chemotherapy induces the expression of cyclooxygenase-2 in
non-small cell lung cancer. Clin Cancer Res. 11:4191–4197. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Masferrer JL, Leahy KM, Koki AT, et al:
Antiangiogenic and antitumor activities of cyclooxygenase-2
inhibitors. Cancer Res. 60:1306–1311. 2000.PubMed/NCBI
|
|
142
|
Kohne CH, De Greve J, Hartmann JT, et al:
Irinotecan combined with infusional 5-fluorouracil/folinic acid or
capecitabine plus celecoxib or placebo in the first-line treatment
of patients with metastatic colorectal cancer. EORTC study 40015.
Ann Oncol. 19:920–926. 2008. View Article : Google Scholar
|
|
143
|
Maiello E, Giuliani F, Gebbia V, et al:
Gruppo Oncologico dell'Italia Meridionale, FOLFIRI with or without
celecoxib in advanced colorectal cancer: a randomized phase II
study of the Gruppo Oncologico dell'Italia Meridionale (GOIM). Ann
Oncol. 17 (Suppl 7):vii55–59. 2006.
|
|
144
|
Schneider BJ, Kalemkerian GP, Kraut MJ, et
al: Phase II study of celecoxib and docetaxel in non-small cell
lung cancer (NSCLC) patients with progression after platinum-based
therapy. J Thorac Oncol. 3:1454–1459. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Skapek SX, Anderson JR, Hill DA, et al:
Safety and efficacy of high-dose tamoxifen and sulindac for desmoid
tumor in children: results of a Children's Oncology Group (COG)
phase II study. Pediatr Blood Cancer. 60:1108–1112. 2013.PubMed/NCBI
|
|
146
|
Csiki I, Morrow JD, Sandler A, et al:
Targeting cyclooxygenase-2 in recurrent non-small cell lung cancer:
a phase II trial of celecoxib and docetaxel. Clin Cancer Res.
11:6634–6640. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Becerra CR, Frenkel EP, Ashfaq R and
Gaynor RB: Increased toxicity and lack of efficacy of rofecoxib in
combination with chemotherapy for treatment of metastatic
colorectal cancer: a phase II study. Int J Cancer. 105:868–872.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Dang CT, Dannenberg AJ, Subbaramaiah K, et
al: Phase II study of celecoxib and trastuzumab in metastatic
breast cancer patients who have progressed after prior
trastuzumab-based treatments. Clin Cancer Res. 10:4062–4067. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Gridelli C, Gallo C, Ceribelli A, et al:
Factorial phase III randomised trial of rofecoxib and prolonged
constant infusion of gemcitabine in advanced non-small-cell lung
cancer: the GEmcitabine-COxib in NSCLC (GECO) study. Lancet Oncol.
8:500–512. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Groen HJ, Sietsma H, Vincent A, et al:
Randomized, placebo-controlled phase III study of docetaxel plus
carboplatin with celecoxib and cyclooxygenase-2 expression as a
biomarker for patients with advanced non-small-cell lung cancer:
the NVALT-4 study. J Clin Oncol. 29:4320–4326. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Koch A, Bergman B, Holmberg E, et al
Swedish Lung Cancer Study Group: Effect of celecoxib on survival in
patients with advanced non-small cell lung cancer: a double blind
randomised clinical phase III trial (CYCLUS study) by the Swedish
Lung Cancer Study Group. Eur J Cancer. 47:1546–1555. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Altorki NK, Keresztes RS, Port JL, et al:
Celecoxib, a selective cyclo-oxygenase-2 inhibitor, enhances the
response to preoperative paclitaxel and carboplatin in early-stage
non-small-cell lung cancer. J Clin Oncol. 21:2645–2650. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Nugent FW, Mertens WC, Graziano S, et al:
Docetaxel and cyclooxygenase-2 inhibition with celecoxib for
advanced non-small cell lung cancer progressing after
platinum-based chemotherapy: a multicenter phase II trial. Lung
Cancer. 48:267–273. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Legge F, Paglia A, D'Asta M, Fuoco G,
Scambia G and Ferrandina G: Phase II study of the combination
carboplatin plus celecoxib in heavily pre-treated recurrent ovarian
cancer patients. BMC Cancer. 11(214)2011. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Altorki NK, Christos P, Port JL, et al:
Preoperative taxane-based chemotherapy and celecoxib for carcinoma
of the esophagus and gastroesophageal junction: results of a phase
2 trial. J Thorac Oncol. 6:1121–1127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
An Y and Ongkeko WM: ABCG2: the key to
chemoresistance in cancer stem cells? Expert Opin Drug Metab
Toxicol. 5:1529–1542. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Huang WZ, Fu JH, Wang DK, et al:
Overexpression of cyclooxygenase-2 is associated with
chemoradiotherapy resistance and prognosis in esophageal squamous
cell carcinoma patients. Dis Esophagus. 21:679–684. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Robey RW, To KK, Polgar O, et al: ABCG2: a
perspective. Adv Drug Deliv Rev. 61:3–13. 2009. View Article : Google Scholar
|
|
159
|
Szczuraszek K, Materna V, Halon A, et al:
Positive correlation between cyclooxygenase-2 and ABC-transporter
expression in non-Hodgkin's lymphomas. Oncol Rep. 22:1315–1323.
2009.PubMed/NCBI
|
|
160
|
Edelman MJ, Watson D, Wang X, et al:
Eicosanoid modulation in advanced lung cancer: cyclooxygenase-2
expression is a positive predictive factor for celecoxib +
chemotherapy - Cancer and Leukemia Group B Trial 30203. J Clin
Oncol. 26:848–855. 2008.PubMed/NCBI
|