Introduction
Renal cell carcinoma (RCC) is among the most
resistant variety of cancer showing resistance to conventional
cytotoxic chemotherapy. Cytokine therapy was the standard treatment
for RCC until 2006. However, development of various
molecular-targeted drugs, such as vascular endothelial growth
factor (VEGF) receptor (VEGFR) tyrosine kinase inhibitors (TKIs)
and mammalian target of rapamycin (mTOR) inhibitors, significantly
improved metastatic RCC treatment (1-3).
Axitinib, an oral second-generation multitargeted
TKI targeting VEGFR-1, -2, and -3 was approved by the US Food and
Drug Administration in 2012 (4-6).
Currently, it is used as a second-line treatment for metastatic RCC
(7-9);
however, information on its third-line or later treatment use
remains scarce. Approximately one-third of patients with RCC
exhibit TKI resistance in clinical trials (10). Drug resistance can develop in
initially responsive patients typically one year after treatment,
thereby complicating advanced RCC management with TKIs (11). Therefore, understanding the
mechanism underlying axitinib in the second-line and later settings
is important for effective treatment.
Targeted therapy resistance is of two types:
intrinsic and acquired. Intrinsic resistance refers to the
immediate ineffectiveness of therapeutic agents, often due to
pre-existing resistant tumour clones formed via inherited
resistance or evolutionary clonal selection. In contrast, acquired
resistance is observed when tumours regrow following initial
regression despite continued therapy. Although the precise
mechanisms of resistance to targeted therapies remain unclear, both
laboratory and clinical studies have identified several factors
contributing to intrinsic and acquired resistance (12).
We previously established everolimus-resistant
papillary RCC (PRCC) cells exhibiting cross-resistance to other
mTOR inhibitors, decreased mTOR activity, and downregulated mRNA
levels of DNA damage-inducible transcript 4 (DDIT4), DEP
domain-containing mTOR-interacting protein (DEPTOR),
hypoxia-inducible factor 1 subunit alpha (HIF1A), and
phospholipase D1 (PLD1), which possibly contributed to
everolimus resistance (13). mRNA
levels of ATP-binding cassette (ABC) transporter ABCB1 mRNA
are downregulated in everolimus-resistant PRCC cells after
long-term everolimus exposure (14). However, effects of long-term
exposure to axitinib remain unknown. Therefore, in this study, we
aimed to elucidate axitinib resistance mechanisms using PRCC cells
by generating axitinib-resistant PRCC cells and comparing their
molecular characteristics with those of parental PRCC cells.
Materials and methods
Chemicals
Axitinib, sunitinib, temsirolimus, and
5-fluorouracil were purchased from Sigma-Aldrich; Merck KGaA.
Everolimus and rapamycin were obtained from Selleck Chemicals, LLC
and LC Laboratories, respectively. Erlotinib, sorafenib, and SN-38
(active metabolite of irinotecan hydrochloride) were purchased from
LKT Laboratories Inc. Carboplatin, cisplatin, doxorubicin
hydrochloride, etoposide, paclitaxel, vinblastine sulphate, and
vincristine sulphate were purchased from FUJIFILM Wako Pure
Chemical Corp. Water-soluble tetrazolium salt (WST)-1 and 1-methoxy
phenazinium methylsulphate (PMS) were obtained from Dojindo
Laboratories.
Cells and cell culture
Caki-2 cells (RRID:CVCL_0235; DS Pharma Biomedical)
were used as human PRCC model cells (13-15).
Short tandem repeat-polymerase chain reaction-PCR profiling using
the PowerPlex 16 System (Promega Corp.) confirmed that the cell
used in this study was the same as the cell registered in DSMZ
(ACC-54 CAKI-2), and the cell registered in ATCC (HTB-47 Caki-2),
by the comparison with the database of JCRB Cell Bank. The cells
were subsequently cultured in the Roswell Park Memorial Institute
(RPMI)-1640 medium (Invitrogen, Life Technologies) supplemented
with 10% heat-inactivated foetal bovine serum (Invitrogen, Life
Technologies) and 100 IU/ml penicillin + 100 µg/ml streptomycin
(Invitrogen, Life Technologies) in a humidified atmosphere
containing 95% air and 5% CO2 at 37˚C. The cells were
sub-cultured every 3-4 d using 0.05% trypsin-0.02%
ethylenediaminetetraacetic acid (Invitrogen, Life
Technologies).
Establishment of axitinib-resistant
sublines
Clinically achievable plasma concentration of
axitinib at 10 mg is approximately 30 ng/ml (equivalent to
approximately 0.08 µM) (16-18).
Caki-2 cells were cultured in RPMI-1640 medium supplemented with
0.1 µM axitinib. After three months, the cells tolerating 0.1 µM
axitinib were isolated and cloned, and the selected clones were
named as Caki/AX cells. Caki/AX cells were maintained under
conditions similar to those used for Caki-2 cells, except that the
medium contained 0.1 µM axitinib.
Cell growth assay
Caki-2 and Caki/AX cell growth was evaluated using
growth curves. On day 0, the cells (1,000 cells/well) were seeded
in a 96-well plate in a culture medium without axitinib and counted
from day 0 to 12. The cell counts were determined via WST-1
colorimetric assay based on the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay,
as previously described (13,19,20).
Three hours after the addition of the WST-1 reagent solution,
absorbance at 450 nm and a reference wavelength of 630 nm was
determined using the Spectra Fluor microplate reader (Tecan Group,
Ltd.), according to the manufacturer's instructions. Preliminary
experiments revealed a proportional relationship between the
absorbance and cell number. Log phase doubling time of Caki-2 and
Caki/AX cells were calculated as previously described (13,19,20).
Growth inhibition assay
Effects of molecular targeted and cytotoxic
anticancer drugs on Caki-2 and Caki/AX cell growth were evaluated
using the WST-1 assay as previously described (13,14,19-22).
To examine the effects of molecular targeted drugs, the cells were
initially seeded at a density of 500 cells/well in a 96-well plate
without any drugs. After 24 h of pre-culture, the medium was
replaced with a fresh medium containing various concentrations of
the tested molecular targeted drugs. After 168 h of incubation,
cell counts were determined via WST-1 assay.
To examine the effects of cytotoxic anticancer
drugs, the cells were seeded at a density of 1,000 cells/well, and
drug exposure time was 72 h. All other experimental conditions were
identical to those described above. Subsequently, 50% inhibitory
concentration (IC50) values of the tested drugs were
estimated using the sigmoid inhibitory effect model, as previously
described (13,14,19-22).
Reverse transcription-quantitative PCR
(RT-qPCR)
Next, mRNA expression levels of the ABC
transporters, ABCB1 and ABCG2, were measured via
RT-qPCR. Total RNA was extracted from Caki-2 and Caki/AX cells
using the GenElute Mammalian Total RNA Miniprep kit (Sigma-Aldrich;
Merck KGaA), and an aliquot (500 ng) was used for reverse
transcription using the PrimeScript RT reagent kit (Takara Bio
Inc.). Reverse transcription reaction was performed at 37˚C for 15
min and terminated via heating at 85˚C for 5 sec, followed by
cooling at 4˚C.
Real-time PCR was performed using the 7500 Fast
Real-Time PCR system (Applied Biosystems) and SYBR Premix Ex Taq
(Takara Bio). PCR cycling conditions were as follows: 95˚C for 30
sec, followed by 40 cycles of 95˚C for 3 sec and 60˚C for 30 sec.
Dissociation curve analysis was performed via heating at 95˚C for
15 sec followed by 60˚C for 1 min, and 95˚C for 15 sec. All PCR
primers used in this study are listed in Table SI (13,14,20).
Primers were synthesised by GeneDesign, Inc. β-actin was used as an
internal standard. The comparative Cq method was used to determine
the relative target mRNA levels (13,14,20,23,24).
PCR array
PCR array was performed using the RT2
Profiler PCR Array (catalogue No. PAHS-091Z; Qiagen), as previously
described (13). Total RNA was
extracted as described above, and an aliquot (500 ng of total RNA)
was used for reverse transcription with the RT2 First
Strand kit (Qiagen), according to the manufacturer's instructions.
Real-time PCR was performed using the 7500 Fast Real-Time PCR
System (Applied Biosystems) and RT2 SYBR-Green Master
Mix (Qiagen). PCR conditions were as follows: 95˚C for 10 min,
followed by 40 cycles of 95˚C for 15 sec, and 60˚C for 1 min.
Dissociation was initiated at 95˚C for 15 sec, followed by 60˚C for
1 min, and 95˚C for 15 sec. The data were analysed using the
2-ΔΔCq method (25).
Statistical analyses
Two groups were compared using an unpaired Student's
t-test with the JMP Pro 15.2.0. software (SAS Institute Japan
Ltd.). P<0.05 (two-tailed) was considered to indicate a
statistically significant difference.
Results
Caki-2 and Caki/AX cell growth
curves
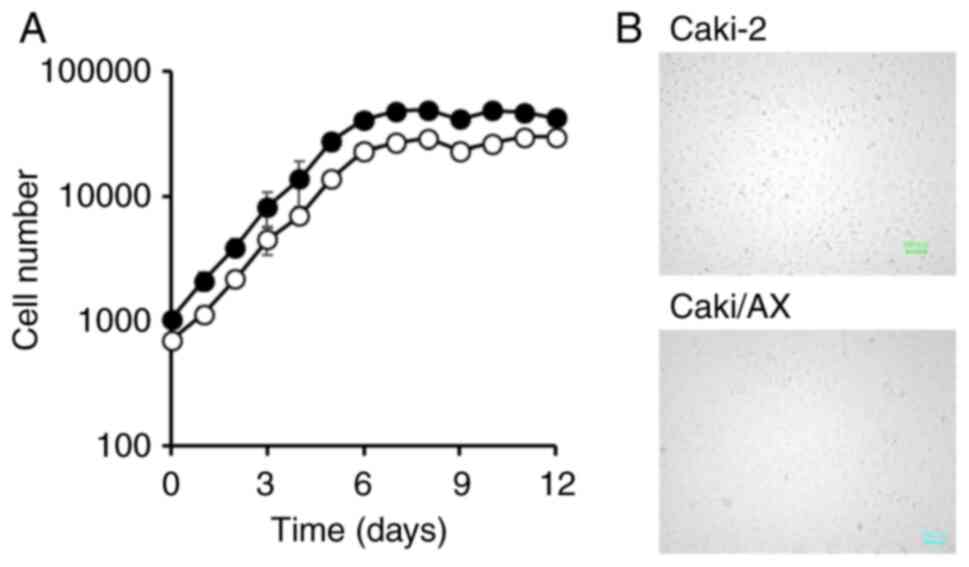
Growth curves of Caki-2 and Caki/AX cells revealed a
logarithmic phase that continued for at least six days after cell
seeding (Fig. 1). The cell
doubling time were approximately 24.2 and 24.4 h for Caki-2 and
Caki/AX cells, respectively. Notably, growth rates were comparable
in both cell types.
Cell sensitivities to TKIs and mTOR
inhibitors
Table I shows the
IC50 values of the tested TKIs in Caki-2 and Caki/AX
cells. IC50 value of axitinib was significantly high in
Caki/AX cells, showing 2.83-fold resistance. Similarly,
IC50 value of sunitinib was significantly higher in
Caki/AX cells than in Caki-2 cells, with Caki/AX cells showing a
1.2-fold higher resistance than Caki-2 cells. In contrast,
IC50 values of sorafenib and erlotinib were lower in
Caki/AX cells than in Caki-2 cells; however, the difference was not
significant.
 | Table ISensitivities of Caki-2 and Caki/AX
cells to tyrosine kinase inhibitors. |
Table I
Sensitivities of Caki-2 and Caki/AX
cells to tyrosine kinase inhibitors.
| | IC50
value, µM | |
|---|
| Drug | Caki-2 | Caki/AX | R.R. |
|---|
| Axitinib | 3.92±1.39 |
11.1±4.27a | 2.83 |
| Sorafenib | 3.65±0.24 |
3.34±0.07a | 0.92 |
| Sunitinib | 2.84±0.22 |
3.33±0.35a | 1.17 |
| Erlotinib | 0.44±0.30 | 0.36±0.11 | 0.81 |
IC50 values of the tested mTOR inhibitors
in Caki-2 and Caki/AX cells are presented in Table II. Their IC50 values
were higher in Caki/AX cells than in Caki-2 cells, with the
relative resistance to mTOR inhibitors being approximately
10-fold.
| Table IISensitivities of Caki-2 and Caki/AX
cells to mammalian target of rapamycin inhibitors. |
Table II
Sensitivities of Caki-2 and Caki/AX
cells to mammalian target of rapamycin inhibitors.
| | IC50
value, nM | |
|---|
| Drug | Caki-2 | Caki/AX | R.R. |
|---|
| Everolimus | 52.6±51.0 |
653±620a | 12.4 |
| Temsirolimus | 6.20±11.0 |
92.2±84.3a | 14.9 |
| Rapamycin | 62.5±55.6 |
814±487b | 13.0 |
Cell sensitivities to cytotoxic
anticancer drugs
IC50 values of the tested cytotoxic
anticancer drugs in Caki-2 and Caki/AX cells are presented in
Table III. Vinblastine,
vincristine, paclitaxel, and doxorubicin sensitivities were lower
in Caki/AX cells than in Caki-2 cells. SN-38 sensitivity was
decreased, but not significantly, in Caki/AX cells. Notably,
etoposide, 5-fluorouracil, cisplatin, and carboplatin sensitivities
were comparable between Caki-2 and Caki/AX cells.
| Table IIISensitivities of Caki-2 and Caki/AX
cells to cytotoxic anticancer drugs. |
Table III
Sensitivities of Caki-2 and Caki/AX
cells to cytotoxic anticancer drugs.
| | IC50
value | |
|---|
| Drug | Caki-2 | Caki/AX | R.R. |
|---|
| Vinblastine,
nM | 9.16±2.86 |
29.1±9.00a | 3.18 |
| Vincristine,
nM | 13.5±2.06 |
44.7±3.54a | 3.31 |
| Paclitaxel, nM | 6.34±2.09 |
26.6±8.00a | 4.20 |
| Doxorubicin,
nM | 149±110 |
349±188a | 2.34 |
| Etoposide, µM | 5.00±3.65 | 4.75±2.85 | 0.95 |
| SN-38, nM | 36.3±26.0 | 72.2±47.9 | 1.99 |
| 5-fluorouracil,
µM | 20.4±12.1 | 19.4±12.9 | 0.95 |
| Cisplatin, µM | 1.99±0.63 | 2.52±0.53 | 1.27 |
| Carboplatin,
µM | 25.2±14.1 | 17.4±12.1 | 0.69 |
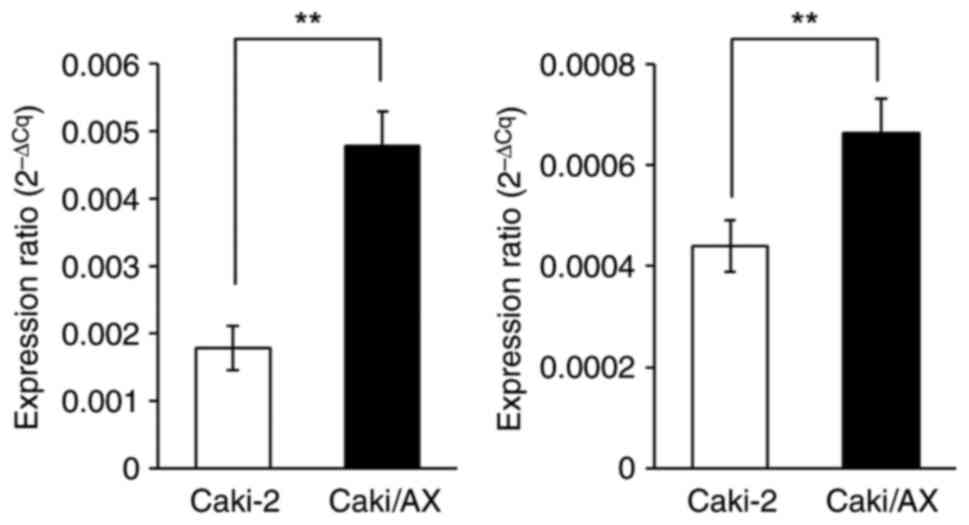
ABCB1 and ABCG2 mRNA expression
levels
ABCB1 and ABCG2 mRNA levels were
significantly higher in Caki/AX cells than in Caki-2 cells
(Fig. 2).
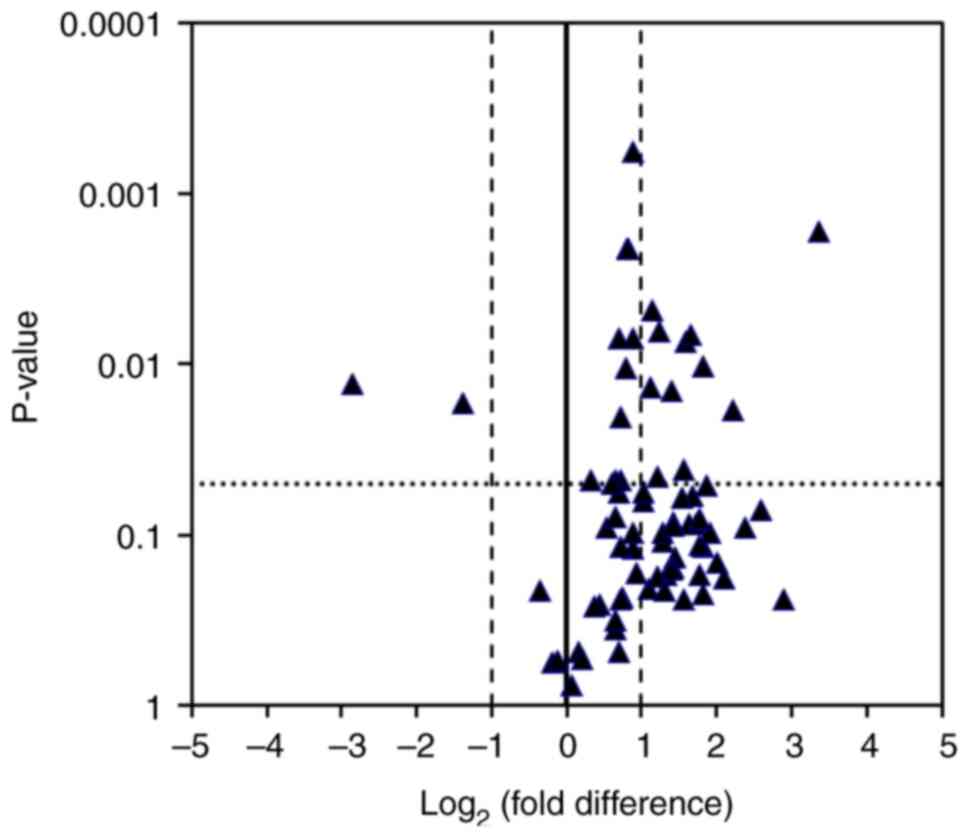
PCR array
Next, mRNA levels of molecules associated with
VEGF-related signalling pathways were analysed using a PCR array
(Fig. 3). Volcano plot showed the
mRNAs up- or downregulated in Caki/AX cells compared to those in
Caki-2 cells. Notably, mRNA levels of FIGF (also known as
VEGFD) and sphingosine kinase 1 (SPHK1) were
upregulated ≥ 2-fold, whereas those of Rac family small GTPase 2
(RAC2) were downregulated >2-fold in Caki/AX cells
(Table IV).
| Table IVChanges in the expression levels of
vascular endothelial growth factor signalling pathway-related
mRNAs. |
Table IV
Changes in the expression levels of
vascular endothelial growth factor signalling pathway-related
mRNAs.
| A, Increased in
Caki/AX cells |
|---|
| Gene | Description | Log2
(fold-difference)a |
|---|
| FIGF | C-fos-induced
growth factor (vascular endothelial growth factor D) | 2.21 |
| NFATC3 | Nuclear factor of
activated T-cells, cytoplasmic, calcineurin-dependent 3 | 1.21 |
| NFATC4 | Nuclear factor of
activated T-cells, cytoplasmic, calcineurin-dependent 4 | 0.82 |
| PDGFC | Mitogen-activated
protein kinase 3 | 1.13 |
| PIK3CA | Mitogen-activated
protein kinase-activated protein kinase 3 | 1.15 |
| PIK3CB | Nuclear factor of
activated T-cells 5, tonicity-responsive | 1.24 |
| PIK3R1 | Nuclear factor of
activated T-cells, cytoplasmic, calcineurin-dependent 3 | 1.58 |
| PLCG2 | Phospholipase C,
gamma 2 (phosphatidylinositol-specific) | 1.40 |
| PPP3R2 | Protein phosphatase
3, regulatory subunit B, beta | 1.82 |
| SH2D2A | SH2 domain
containing 2A | 1.66 |
| SPHK1 | Sphingosine kinase
1 | 3.35 |
| B, Decreased in
Caki/AX cells |
| Gene | Description | Log2
(fold-difference)a |
| RAC2 | Ras-related C3
botulinum toxin substrate 2 (rho family, small GTP binding protein
Rac2) | -2.86 |
| VEGFC | Vascular
endothelial growth factor C | -1.39 |
Additionally, mRNA levels of VEGFA (Fig. S1) and cadherin 1 (Fig. S2), which encodes the
calcium-dependent cell-cell adhesion protein E-cadherin, were
significantly lower in Caki/AX cells than in Caki-2 cells.
Discussion
Target gene mutations reducing the drug affinity for
target molecules are involved in TKI resistance mechanisms.
Activation of bypass signalling pathways also contributes to TKI
resistance. Other resistance mechanisms include lysosomal
sequestration of TKIs, activation of angiogenic switches, and
involvement of ABC transporters (12). However, the specific mechanisms
underlying axitinib resistance remain unclear. Therefore, in this
study, axitinib-resistant PRCC cells were generated and molecularly
compared with their parental cells to elucidate the underlying
resistance mechanisms.
Axitinib-resistant Caki/AX cells exhibited cell
growth comparable to that of their parental Caki-2 cells, with
equivalent doubling time of 24.2 and 24.4 h for Caki-2 and Caki/AX
cells, respectively. Moreover, continuous exposure to axitinib did
not affect cell growth, suggesting that changes in cell growth do
not contribute to the development of drug resistance in Caki/AX
cells.
Caki/AX cells exhibited significantly lower
sensitivity to axitinib than Caki-2 cells, with approximately
3-fold resistance. This suggests that long-term exposure to 0.1 µM
axitinib, equivalent to the plasma concentrations achieved with
clinical dosing, induces resistance in these cells. Additionally,
Caki/AX cells showed cross-resistance to sunitinib, but not
sorafenib and erlotinib. They exhibited altered sensitivity to
TKIs; however, the specific factors responsible for this could not
be identified in this study. Sensitivity to mTOR inhibitors was
also reduced in Caki/AX cells, indicating the development of
cross-resistance to mTOR inhibitors.
Caki/AX cells were resistant to vinblastine,
vincristine, paclitaxel, and doxorubicin. These cytotoxic
anticancer drugs are substrates of ABCB1, also known as
P-glycoprotein (19,21). ABCB1 mRNA levels were higher
in Caki/AX cells than in Caki-2 cells, suggesting that drug
resistance is induced by ABCB1 mRNA upregulation. Axitinib
and sunitinib also act as ABCB1 substrates (26-29);
therefore, resistance to these drugs was partially due to the
upregulation of ABCB1 mRNA levels in this study.
Furthermore, moderate resistance to SN-38, an active irinotecan
metabolite, was observed, possibly due to ABCG2 mRNA
upregulation because SN-38 is a substrate of ABCG2(22).
Volcano plot revealed increased FIGF and
SPHK1 mRNA levels and decreased RAC2 mRNA levels in
Caki/AX cells. FIGF encodes a c-fos-induced growth factor,
also known as VEGFD. Lieu et al (30) reported increased VEGFD levels after
bevacizumab chemotherapy, suggesting their association with
bevacizumab chemotherapy resistance. Therefore, axitinib resistance
may be partially due to the upregulation of FIGF levels,
despite bevacizumab being a human monoclonal VEGFA-neutralising
antibody. Here, mRNA levels of VEGFA and VEGFC were
decreased in Caki/AX cells, indicating an association between
reduced VEGF levels and axitinib resistance in Caki/AX cells.
SPHK1, encoded by SPHK1, acts as a
proto-oncogenic factor synthesizing sphingosine-1 phosphate (S1P).
Tumour cells often exhibit elevated levels of S1P and its receptor,
S1PR1, which promotes drug resistance. Signalling through S1P
via its receptor, S1PR1, facilitates cancer cell survival by
activating anti-apoptotic pathways (31). Therefore, targeting S1P and its
receptors can potentially inhibit cancer cell proliferation and
overcome drug resistance (31).
Bao et al (32) reported
that SPHK1 overexpression is associated with RCC development and
resistance to antiangiogenic agents. Elevated SPHK1 levels predicts
poor outcomes and resistance to angiogenic agents in patients with
RCC. These findings indicate the potential role of SPHK1 in
axitinib resistance in Caki/AX cells.
RAC2 is a small GTPase contributing to B-cell
receptor (BCR)-activated calcium mobilization via phospholipase Cγ2
(33-35).
It acts as a regulator of cell adhesion, linking BCR signalling
pathways to cellular adhesion processes (33-35).
Its diverse functions are crucial for fundamental cellular
physiological processes and immune responses (33,35).
Shaffer et al (35)
reported the involvement of RAC2 in the resistance to the Bruton
tyrosine kinase inhibitor, ibrutinib. Wu et al (36) revealed that the reduction of RAC2
expression using RNA interference and clustered regularly
interspaced palindromic repeat technology impairs cell adhesion and
that its overexpression reverses ibrutinib-induced cell adhesion
impairment. RNA-sequencing analysis has shown that
ibrutinib-resistant cells exhibit higher RAC2 levels than their
parental cells (35). RAC2
knockdown significantly reduces the levels of the signatures
associated with the activated B-cell-diffuse large B-cell lymphoma
identity, B-cell-specific genes repressed by B-lymphocyte-induced
maturation protein-1, and genes induced by nuclear factor-κB,
signal transducer and activator of transcription 3, and interferon
regulatory factor 4. Although the direct involvement of RAC2 in
axitinib resistance remains unclear, its downregulation possibly
contributes to axitinib resistance.
Overall, this study showed that Caki/AX cells
develop drug resistance via various mechanisms, including
ABCB1 mRNA upregulation. Our findings suggest that the
upregulation of FIGF and SPHK1 mRNA levels and
downregulation of RAC2 mRNA levels contribute to the
acquired axitinib resistance of Caki/AX cells. These changes align
with mechanisms previously reported in the literature, and
particularly, the involvement of FIGF and SPHK1 suggests the
possibility of resistance acquisition via angiogenic and
sphingolipid-related pathways. Specifically, the decreased RAC2
expression is a novel finding of this study with limited prior
documentation, indicating a potential cell line-specific molecular
alteration. This investigation is based on a single cell line and
remains a preliminary exploration. However, other factors may also
be involved in axitinib resistance, warranting further gene
expression analyses via next-generation sequencing. Additionally,
mRNA levels of cadherin 1 were significantly lower in Caki/AX cells
than in Caki-2 cells, suggesting the induction of
epithelial-mesenchymal transition, consistent with a previous
report (37). This study used only
a few cell lines in vitro, which possibly limits the
generalisability and warrants the cautious interpretation of our
findings. Therefore, further research incorporating additional
models, including animal and clinical specimens, is necessary to
fully validate and extend our observations.
Supplementary Material
Vascular endothelial growth factor
mRNA levels in Caki-2 and Caki/AX cells. Relative target gene level
is expressed as 2 ΔCq. ΔCq was calculated by subtracting
the Cq of the internal standard (β-actin) from that of the target
gene. Open (□) and closed (■) bars indicate the Caki-2 and Caki/AX
cells, respectively. Each bar represents the mean ± standard
deviation (n=4). **P<0.01 (unpaired Student's
t-test).
Cadherin 1 mRNA levels in Caki-2 and
Caki/AX cells. Relative target gene expression is expressed as
2-ΔCq. ΔCq was calculated by subtracting the Cq of the
internal standard (β-actin) from that of the target gene. Open (□)
and closed (■) bars indicate the Caki-2 and Caki/AX cells,
respectively. Each bar represents the mean ± standard deviation
(n=4). **P<0.01 (unpaired Student's t-test).
Primer sequences for reverse
transcription-quantitative polymerase chain reaction.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YN conceptualized the study, performed the
investigation, compiled the data, generated the figures, and wrote
the original draft. AI and KY generated the figures, collected and
organized the data, and reviewed and edited the manuscript. KT
conceptualized the study, curated all of the data, reviewed and
edited the manuscript, and supervised the study. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interest
The authors declare that they have no competing
interests.
References
|
1
|
Hsieh JJ, Purdue MP, Signoretti S, Swanton
C, Albiges L, Schmidinger M, Heng DY, Larkin J and Ficarra V: Renal
cell carcinoma. Nat Rev Dis Primers. 3(17009)2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ljungberg B, Albiges L, Abu-Ghanem Y,
Bensalah K, Dabestani S, Fernández-Pello S, Giles RH, Hofmann F,
Hora M, Kuczyk MA, et al: European association of urology
guidelines on renal cell carcinoma: The 2019 update. Eur Urol.
75:799–810. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Saliby RM, Saad E, Labaki C, Xu W, Braun
DA, Viswanathan SR and Bakouny Z: Novel targeted therapies for
renal cell carcinoma: Building on the successes of vascular
endothelial growth factor and mTOR inhibition. Hematol Oncol Clin
North Am. 37:1015–1026. 2023.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Escudier B and Gore M: Axitinib for the
management of metastatic renal cell carcinoma. Drugs RD.
11:113–126. 2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Bellesoeur A, Carton E, Alexandre J,
Goldwasser F and Huillard O: Axitinib in the treatment of renal
cell carcinoma: Design, development, and place in therapy. Drug Des
Devel Ther. 11:2801–2811. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Tiako Meyo M, Chen J, Goldwasser F, Hirsch
L and Huillard O: A profile of avelumab plus axitinib in the
treatment of renal cell carcinoma. Ther Clin Risk Manag.
18:683–698. 2022.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Hoshi S, Numahata K, Kanno H, Sato M,
Kuromoto A, Nezu K, Sakai T, Konno C, Ishizuka Y, Izumi H, et al:
Updated recommendation on molecular-targeted therapy for metastatic
renal cell cancer. Mol Clin Oncol. 7:591–594. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Jakobsson M, Strambi A, Nilsson F,
Arpegård J and Dalén J: Real-world experience of second-line
axitinib in metastatic renal cell carcinoma: Analysis of the
Swedish population. Future Oncol. 20:1385–1392. 2024.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Powles T, Albiges L, Bex A, Comperat E,
Grünwald V, Kanesvaran R, Kitamura H, McKay R, Porta C, Procopio G,
et al: Renal cell carcinoma: ESMO Clinical Practice Guideline for
diagnosis, treatment and follow-up. Ann Oncol. 35:692–706.
2024.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Figlin RA, Kaufmann I and Brechbiel J:
Targeting PI3K and mTORC2 in metastatic renal cell carcinoma: New
strategies for overcoming resistance to VEGFR and mTORC1
inhibitors. Int J Cancer. 133:788–796. 2013.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Sweeney PL, Suri Y, Basu A, Koshkin VS and
Desai A: Mechanisms of tyrosine kinase inhibitor resistance in
renal cell carcinoma. Cancer Drug Resist. 6:858–873.
2023.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Ou X, Gao G, Habaz IA and Wang Y:
Mechanisms of resistance to tyrosine kinase inhibitor-targeted
therapy and overcoming strategies. MedComm (2020).
5(e694)2024.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Nakayama Y, Enomoto D, Yamamoto K and
Takara K: Molecular characteristics of everolimus-resistant renal
cell carcinoma cells generated by continuous exposure to
everolimus. Anticancer Res. 43:4349–4357. 2023.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Nakayama Y, Ino A, Yamamoto K and Takara
K: Down-regulation of ABCB1 in everolimus-resistant renal cell
carcinoma cells. Anticancer Res. 44:2871–2876. 2024.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Fujita M, Tohji C, Honda Y, Yamamoto Y,
Nakamura T, Yagami T, Yamamori M and Okamura N: Cytotoxicity of
15-deoxy-Δ(12,14)-prostaglandin J(2) through PPARγ-independent
pathway and the involvement of the JNK and Akt pathway in renal
cell carcinoma. Int J Med Sci. 9:555–566. 2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Chen Y, Tortorici MA, Garrett M, Hee B,
Klamerus KJ and Pithavala YK: Clinical pharmacology of axitinib.
Clin Pharmacokinet. 52:713–725. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Rini BI, Garrett M, Poland B, Dutcher JP,
Rixe O, Wilding G, Stadler WM, Pithavala YK, Kim S, Tarazi J and
Motzer RJ: Axitinib in metastatic renal cell carcinoma: Results of
a pharmacokinetic and pharmacodynamic analysis. J Clin Pharmacol.
53:491–504. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
18
|
Fukudo M, Tamaki G, Azumi M, Kakizaki H,
Matsumoto S and Tasaki Y: Absorption of the orally active
multikinase inhibitor axitinib as a therapeutic index to guide dose
titration in metastatic renal cell carcinoma. Invest New Drugs.
39:595–604. 2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Takara K, Sakaeda T, Yagami T, Kobayashi
H, Ohmoto N, Horinouchi M, Nishiguchi K and Okumura K: Cytotoxic
effects of 27 anticancer drugs in HeLa and MDR1-overexpressing
derivative cell lines. Biol Pharm Bull. 25:771–778. 2002.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Nakayama Y, Ino A, Yamamoto K and Takara
K: Involvement of everolimus-induced ABCB1 downregulation in
drug-drug interactions. Biomed Rep. 21(184)2024.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Takara K, Obata Y, Yoshikawa E, Kitada N,
Sakaeda T, Ohnishi N and Yokoyama T: Molecular changes to HeLa
cells on continuous exposure to cisplatin or paclitaxel. Cancer
Chemother Pharmacol. 58:785–793. 2006.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Takara K, Kitada N, Yoshikawa E, Yamamoto
K, Horibe S, Sakaeda T, Nishiguchi K, Ohnishi N and Yokoyama T:
Molecular changes to HeLa cells on continuous exposure to SN-38, an
active metabolite of irinotecan hydrochloride. Cancer Lett.
278:88–96. 2009.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Kitada N, Takara K, Minegaki T, Itoh C,
Tsujimoto M, Sakaeda T and Yokoyama T: Factors affecting
sensitivity to antitumor platinum derivatives of human colorectal
tumor cell lines. Cancer Chemother Pharmacol. 62:577–584.
2008.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Minegaki T, Takara K, Hamaguchi R,
Tsujimoto M and Nishiguchi K: Factors affecting the sensitivity of
human-derived esophageal carcinoma cell lines to 5-fluorouracil and
cisplatin. Oncol Lett. 5:427–434. 2013.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Poller B, Iusuf D, Sparidans RW, Wagenaar
E, Beijnen JH and Schinkel AH: Differential impact of
P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)
on axitinib brain accumulation and oral plasma pharmacokinetics.
Drug Metab Dispos. 39:729–735. 2011.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Thomas-Schoemann A, Blanchet B, Bardin C,
Noé G, Boudou-Rouquette P, Vidal M and Goldwasser F: Drug
interactions with solid tumour-targeted therapies. Crit Rev Oncol
Hematol. 89:179–196. 2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Sato H, Siddig S, Uzu M, Suzuki S, Nomura
Y, Kashiba T, Gushimiyagi K, Sekine Y, Uehara T, Arano Y, et al:
Elacridar enhances the cytotoxic effects of sunitinib and prevents
multidrug resistance in renal carcinoma cells. Eur J Pharmacol.
746:258–266. 2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Beretta GL, Cassinelli G, Pennati M, Zuco
V and Gatti L: Overcoming ABC transporter-mediated multidrug
resistance: The dual role of tyrosine kinase inhibitors as
multitargeting agents. Eur J Med Chem. 142:271–289. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Lieu CH, Tran H, Jiang ZQ, Mao M, Overman
MJ, Lin E, Eng C, Morris J, Ellis L, Heymach JV, et al: The
association of alternate VEGF ligands with resistance to anti-VEGF
therapy in metastatic colorectal cancer. PLoS One.
8(e77117)2013.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Alkafaas SS, Elsalahaty MI, Ismail DF,
Radwan MA, Elkafas SS, Loutfy SA, Elshazli RM, Baazaoui N, Ahmed
AE, Hafez W, et al: The emerging roles of sphingosine 1-phosphate
and SphK1 in cancer resistance: A promising therapeutic target.
Cancer Cell Int. 24(89)2024.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Bao JM, Zhi X, Yuan HY, Chen TY, Chen MK,
Zhou JH, Xue KY, Yang JK and Liu CD: Overexpression of SPHK1
associated with targeted therapy resistance in predicting poor
prognosis in renal cell carcinoma. Transl Cancer Res. 12:572–584.
2023.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Walliser C, Hermkes E, Schade A, Wiese S,
Deinzer J, Zapatka M, Désiré L, Mertens D, Stilgenbauer S and
Gierschik P: The phospholipase Cγ2 mutants R665W and L845F
identified in ibrutinib-resistant chronic lymphocytic leukemia
patients are hypersensitive to the Rho GTPase Rac2 protein. J Biol
Chem. 291:22136–22148. 2016.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Pasqualucci L: Epigenetic rewiring of BCR
signaling as a novel mechanism of ibrutinib resistance in
ABC-DLBCL. Blood Cancer Discov. 2:555–558. 2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Shaffer AL III, Phelan JD, Wang JQ, Huang
D, Wright GW, Kasbekar M, Choi J, Young RM, Webster DE, Yang Y, et
al: Overcoming acquired epigenetic resistance to BTK inhibitors.
Blood Cancer Discov. 2:630–647. 2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Wu W, Wang W, Franzen CA, Guo H, Lee J, Li
Y, Sukhanova M, Sheng D, Venkataraman G, Ming M, et al: Inhibition
of B-cell receptor signaling disrupts cell adhesion in mantle cell
lymphoma via RAC2. Blood Adv. 5:185–197. 2021.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Lin WH, Cooper LM and Anastasiadis PZ:
Cadherins and catenins in cancer: Connecting cancer pathways and
tumor microenvironment. Front Cell Dev Biol.
11(1137013)2023.PubMed/NCBI View Article : Google Scholar
|