Introduction
Lung adenocarcinoma (LUAD), the most prevalent
histological subtype of lung cancer worldwide, continues to present
significant clinical challenges (1,2).
Although advances in diagnostic techniques, surgical approaches,
chemotherapy and molecular therapies have significantly improved
patient outcomes, the 5-year survival rate for patients with LUAD
remains persistently low (3,4).
Emerging research indicates that specific molecular signatures show
promise as prognostic biomarkers and therapeutic targets.
While recent technological advances have expanded
the toolbox for lung cancer management, such as self-assembled DNA
machines enabling portable circulating tumor cells (CTC)
quantification for liquid biopsy (5), macrophage-mediated targeted delivery
systems delivering sulphate-based nanomedicines to overcome
therapeutic resistance (6), and
nano-adjuvants enhancing radiotherapeutic efficacy in non-small
cell lung cancer (7), these
approaches predominantly address downstream clinical
manifestations. Crucially, they lack molecular drivers capable of
simultaneously predicting prognosis and guiding dynamic treatment
adaptation. Copper metabolism has emerged as an upstream regulator
that links tumour proliferation, immune evasion, and therapeutic
response; notably, cuproptosis directly modulates CTC viability and
radiochemical sensitivity through mitochondrial proteotoxicity. In
the present study, this mechanistic gap was bridged by establishing
a cuproptosis-related long non-coding RNA (lncRNA) signature that
not only prognostically stratifies LUAD but also predicts the
suitability of nanomedicine/immunotherapy, positioning copper
homeostasis as a nexus for precision theranostics.
Cuproptosis is a form of copper-dependent cell death
driven by mitochondrial copper (Cu) accumulation, proteotoxic
stress, and dihydrolipoamide S-acetyltransferase (DLAT) aggregation
(8,9). While dysregulated copper homeostasis
is implicated in cancer, its interplay with lncRNAs in LUAD remains
unexplored. In humans, Cu accumulation is life-threatening, yet
there may be a window of opportunity where Cu accumulation can
eliminate cancer cells in a selective manner (10). This information might be used to
elucidate the pathology of genetic diseases associated with Cu
overload and offers a new way to treat cancer by targeting Cu
toxicity. lncRNAs modulate immune evasion (PD-L1 stabilization),
metastasis (epithelial-mesenchymal transition activation), and
therapeutic resistance (enhanced DNA repair) in patients with LUAD
(11). lncRNAs serve as molecular
bridges connecting copper dysregulation to LUAD aggressiveness.
Currently, cuproptosis-related lncRNAs have been less studied.
In the present study, the gene expression data of
16,876 lncRNAs and 19 cuproptosis-related genes from The Cancer
Genome Atlas (TCGA)-LUAD dataset were initially extracted. In
addition, cuproptosis-associated lncRNAs were identified through
Pearson correlation analysis. A prognostic model was constructed to
predict the overall survival (OS) of patients with LUAD. The
association of the model with immunotherapy response was explored.
A nomogram combining the model and significant clinical features of
LUAD was established. Finally, candidate agents targeting the RNA
signature were investigated using the public GDSC database.
Therefore, the primary objectives of this study were
i) to systematically identify cuproptosis-associated lncRNAs in
LUAD using transcriptomic data; ii) to construct and rigorously
validate a prognostic signature based on these lncRNAs for
stratifying patients with LUAD according to OS risk; iii) to
comprehensively evaluate the association of this signature with the
tumour immune microenvironment and predicted response to
immunotherapy, specifically utilizing the TIDE algorithm; iv) to
screen for potential therapeutic compounds targeting high-risk
patients using the GDSC database; and v) to experimentally validate
the expression patterns of the signature lncRNAs in LUAD cell
lines. By integrating prognostic stratification, immunotherapy
response prediction, drug sensitivity screening, and experimental
validation specifically for LUAD, the aim of the present study was
to establish a novel cuproptosis-related lncRNA signature as a
clinically actionable theragnostic tool.
Materials and methods
Data acquisition and preprocessing
Data sources
RNA transcriptome data (FPKM-normalized), clinical
data (age, sex, AJCC TNM stage, survival status), and somatic
mutation data for patients with LUAD (n=515) were obtained from the
TCGA database (https://cancergenome.nih.gov/).
Acquisition of cuproptosis-associated
lncRNAs
Gene expression files of lncRNAs and
cuproptosis-related genes were downloaded from the TCGA database.
In total, 19 cuproptosis-related genes, namely, NFE2L2, NLRP3,
ATP7B, ATP7A, SLC31A1, FDX1, LIAS, LIPT1, LIPT2, DLD, DLAT, PDHA1,
PDHB, MTF1, GLS, CDKN2A, DBT, GCSH and DLST (8,12-16),
were obtained.
Identification of cuproptosis-related
lncRNAs. Correlation analysis
Pearson correlation coefficients (R) were calculated
between the expression levels of each of the 19 curated
cuproptosis-related genes and all the annotated lncRNAs (n=16,876)
within the TCGA-LUAD cohort.
Selection thresholds. Correlation Strength
(|R|>0.3): This threshold was chosen to identify lncRNAs with
moderate-to-strong co-expression relationships with
cuproptosis-related genes. A value >0.3 is a commonly accepted
benchmark in transcriptomic studies for identifying biologically
relevant co-expression partners, balancing specificity against
overly stringent exclusion of potentially important regulators.
Statistical significance. This stringent
P-value threshold minimizes false positives, ensuring that only
lncRNAs with a highly statistically significant association with at
least one cuproptosis gene were retained.
Scope. lncRNAs meeting the above |R| and p
criteria for any one or more of the 19 cuproptosis-related genes
were included. This broad approach captures lncRNAs potentially
regulating specific facets of cuproptosis without requiring
universal association with all the cuproptosis-related genes.
Output. The application of these criteria
resulted in the identification of 3,385 cuproptosis-related lncRNAs
for subsequent prognostic model construction.
Construction and validation of the lncRNA
signature. TCGA-LUAD samples (n=515) were stratified by AJCC
stage (I/II vs. III/IV) and survival status (alive/dead) and then
randomly partitioned into training (70%, n=360) and test (30%,
n=155) sets (random seed=2023) to preserve prognostic factor
distributions. This 70:30 split adheres to the TRIPOD guidelines
for prognostic model development. Using the training cohort, a
cuproptosis-related lncRNA signature was developed, which was
subsequently validated in the test cohort. Prognostic lncRNAs were
initially screened from 3,385 cuproptosis-associated candidates
based on OS in the TCGA-LUAD cohort (P<0.05). These were
subsequently refined via LASSO-Cox regression analysis (R package
‘glmnet’), yielding a final signature of 52 lncRNAs (17). Further multivariate Cox regression
identified 4 lncRNAs, which were used to construct a prognostic
lncRNA signature. A risk score was accordingly established and
formulated as follows: Risk score=[coef (lncRNA1) x expr (lncRNA1)]
+ coef(lncRNA2) x expr (lncRNA2)] +......+ [coef(lncRNAn) x expr
(lncRNAn)]NA2)] +......+ [coef(lncRNAn) x expr (lncRNAn)]; where
coef (lncRNAn) represents the Cox regression coefficient and expr
(lncRNAn) represents the expression of lncRNAn. Samples from the
training and test sets were grouped based on their risk scores
(18).
Immune microenvironment and therapy
response analysis. Tumour mutation burden (TMB)
Somatic mutations were processed using maftools
(v2.12.0; https://bioconductor.org/packages/maftools/). TMB
equals to (total mutations)/[exome size (38 Mb)].
Immunotherapy response prediction. The TIDE
algorithm (http://tide.dfci.harvard.edu/) was applied to predict
the anti-PD1/CTLA4 response.
Immune cell infiltration. The data were
estimated using CIBERSORTx and ssGSEA (R package GSVA).
Drug sensitivity screening. IC50
values for 73 GDSC compounds were predicted using pRRophetic (v0.5;
https://bioconductor.org/packages/pRRophetic/) based
on the TCGA transcriptomes.
Functional analysis. Gene Ontology (GO) and
Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses
were performed using the R package ‘clusterProfiler’ to
characterize the biological functions of the signature lncRNAs
(P<0.05).
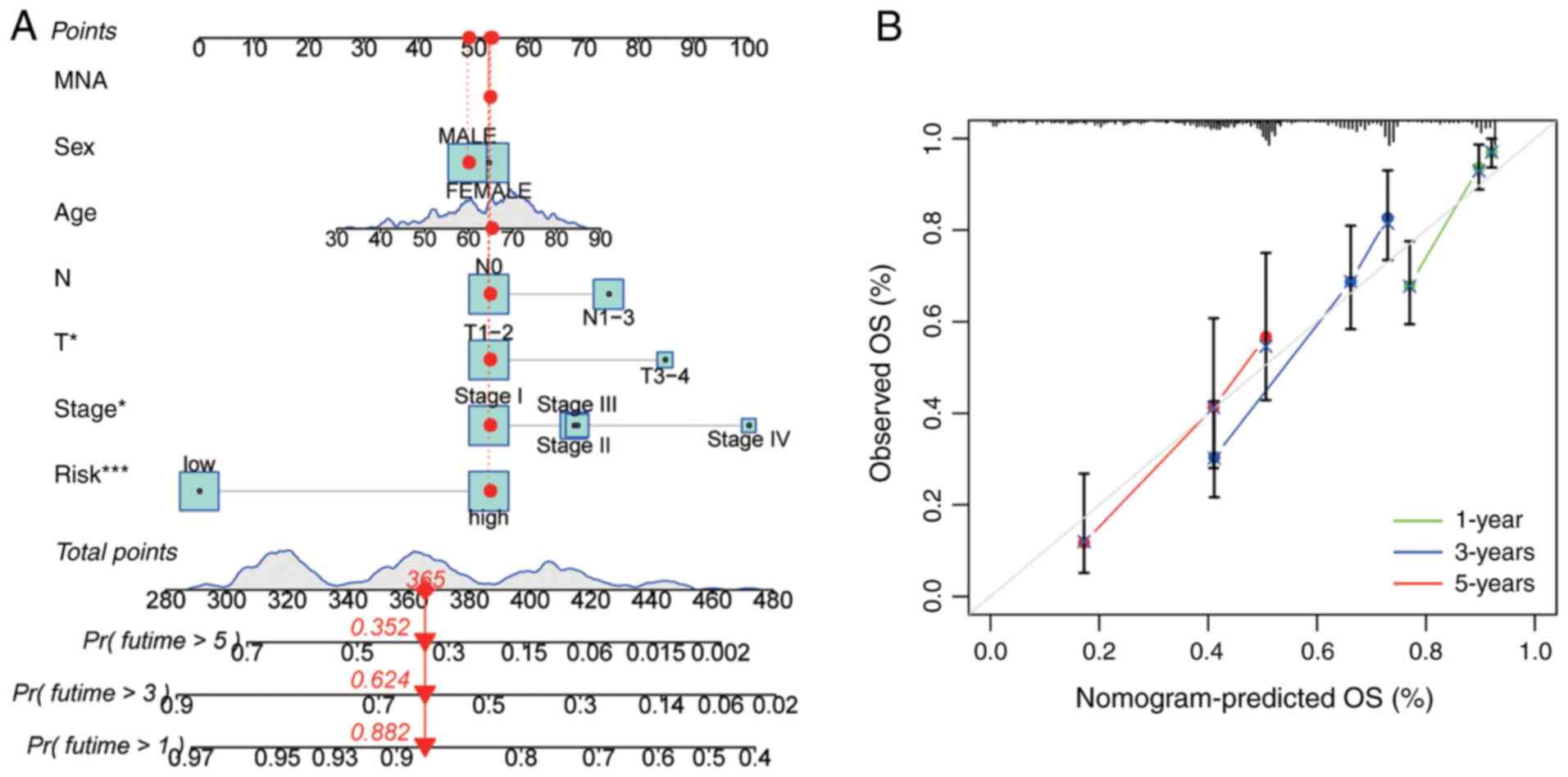
Nomogram construction and validation. A
nomogram incorporating the risk score, age, sex, and TNM stage
classification was developed to predict 1-, 3-, and 5-year OS in
patients with LUAD. Calibration performance was assessed using the
Hosmer-Lemeshow test to evaluate the agreement between the
predicted and observed outcomes.
Survival analysis. Kaplan-Meier survival
analysis was performed to compare OS and other survival endpoints
[including progression-free interval (PFI), disease-specific
survival (DSS), and disease-free interval (DFI)] between the
high-risk and low-risk patient groups stratified by the median risk
score. The statistical significance of the differences in survival
curves was assessed using the two-sided log-rank test. P<0.05
was considered to indicate a statistically significant
difference.
Construction of an ARHGEF26-AS1-associated
competing endogenous RNA (ceRNA) Network. The ceRNA network
focused on ARHGEF26-AS1 was constructed by first predicting its
interacting miRNAs using miRanda (http://www.microrna.org/microrna/home.do) and
DIANA-LncBase (https://diana.imis.athena-innovation.gr/DianaTools/index.php).
The downstream target genes of these miRNAs were then retrieved
from the StarBase database (https://starbase.sysu.edu.cn/), focusing on
interactions supported by experimental evidence. The integrated
network, depicting ARHGEF26-AS1, miRNAs, and target genes, was
finally assembled and visualized using Cytoscape.
Cell culture. Normal human bronchial
epithelial cells (BEAS-2B) and human LUAD cells (A549) (Fuheng
Biotechnology Co., Ltd.) were maintained in DMEM (HyClone; Cytiva)
and RPMI 1640-medium (HyClone; Cytiva), respectively. Both media
were supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.) and 1% penicillin/streptomycin. The cells were
incubated at 37˚C in a humidified atmosphere of 95% air and 5%
CO2.
Reverse transcription-quantitative PCR
(RT-qPCR). Total RNA was extracted from BEAS-2B and A549 cells
using TRIzol® (Invitrogen; Thermo Fisher Scientific,
Inc.). RNA purity/concentration was measured by a NanoDrop 2000
(A260/A280 >1.8). For lncRNA analysis, 1 µg of total RNA was
treated with DNase I (Thermo Fisher Scientific, Inc.) to remove
genomic DNA contamination, followed by cDNA synthesis using M-MLV
Reverse Transcriptase (Promega Corporation) with random hexamers
(50 ng/µl) and dNTPs (0.5 mM) in a 20 µl reaction. The thermal
profile for reverse transcription was as follows: 25˚C for 10 min
(priming), 42˚C for 50 min (synthesis), and 70˚C for 15 min
(inactivation). qPCR was performed using SYBR Premix Ex Taq™
(TaKaRa Bio, Inc.) on a QuantStudio 6 Flex system (Applied
Biosystems; Thermo Fisher Scientific, Inc.) with the following
thermal cycling conditions: An initial denaturation at 95˚C for 30
sec, followed by 40 cycles of 95˚C for 5 sec and 60˚C for 30 sec. A
melt curve analysis was generated at the end of each run.
lncRNA-specific primers (Table
SI) were designed with the following criteria: amplicon length:
80-150 bp; GC content: 40-60%; no secondary structures (validated
by OligoAnalyzer); and spanning exon-exon junctions (where
applicable). β-actin was used as a reference gene. The
2-ΔΔCq method (19) was
utilized to assess fold changes. Raw Ct values and amplification
curves are provided in Table
SII.
Statistical analysis
Statistical analyses were performed using R software
(version 4.1.2; https://cran.r-project.org). A two-sided P<0.05 was
considered to indicate statistical significance. For the
differential expression analysis of the 4 lncRNAs in LUAD (A549)
versus normal bronchial (BEAS-2B) cells (Fig. S3), an unpaired Student's t-test
was applied to compare the means between the two groups, with
results presented as the mean ± SEM from three technical
replicates; P<0.05 was considered to indicate a statistically
significant difference.
Results
Identification of cuproptosis-related
lncRNAs
The present study included 515 patients with LUAD
from the TCGA database. The baseline clinicopathological
characteristics of the entire cohort are summarized in Table SIII. The expression matrices of 19
cuproptosis-related genes and 16,876 lncRNAs were extracted from
the TCGA-LUAD dataset. To identify biologically relevant lncRNAs,
Pearson correlation analysis (|R|>0.3, P<0.001) was performed
between each lncRNA and the 19 cuproptosis-related genes. This
stringent threshold captured lncRNAs with moderate-to-strong
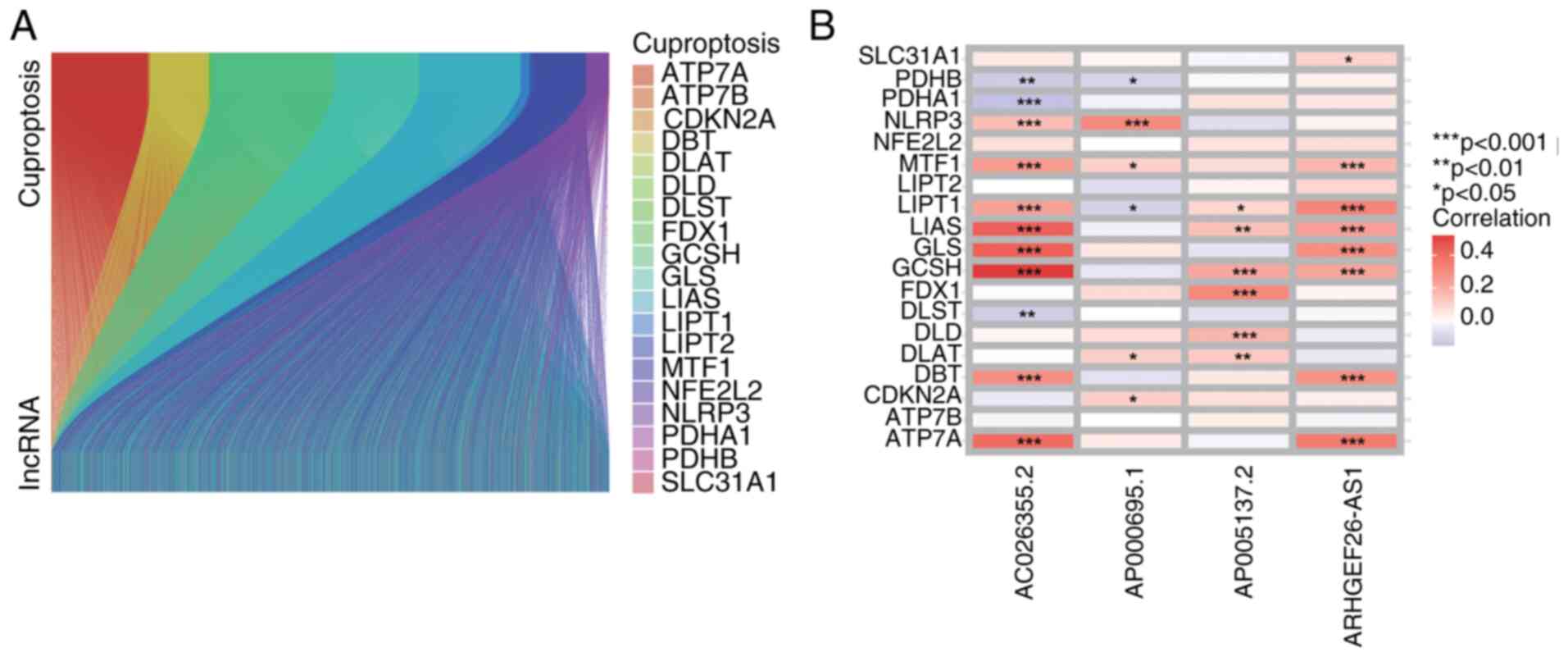
co-expression, yielding 3,385 preliminary candidates (Fig. 1A). Prognostic lncRNA selection
followed a multistep regression framework; univariate Cox analysis
(P<0.05) revealed 52 lncRNAs significantly associated with OS.
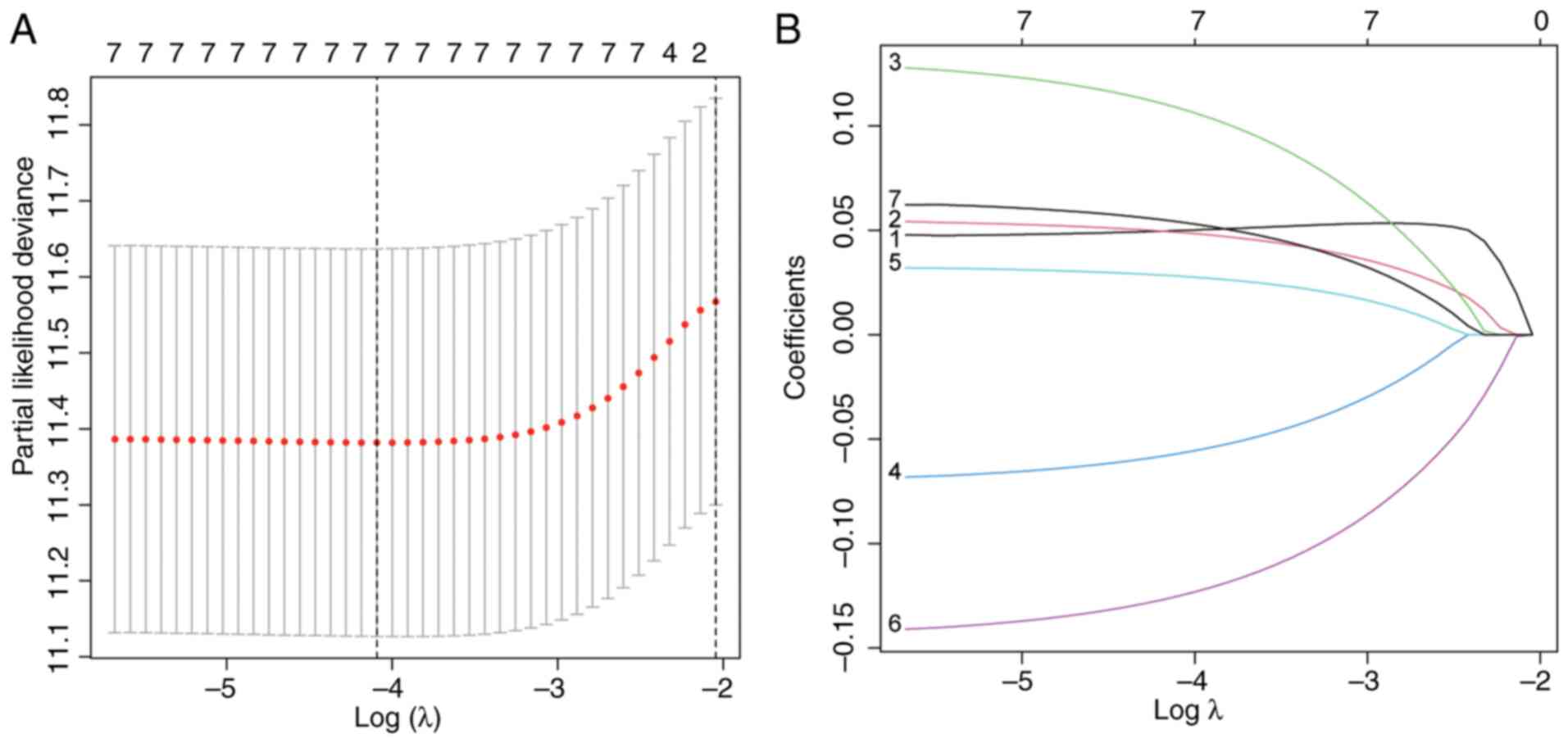
LASSO-penalized Cox regression (10-fold cross-validation, λ=0.032)
reduced dimensionality, eliminating multicollinear lncRNAs
(Fig. 2A and B). Multivariate Cox regression further
refined the model, retaining only 4 lncRNAs (AC026355.2,
AP000695.1, ARHGEF26-AS1, and AP005137.2) that independently
predicted survival (P<0.01, Wald test). The correlation between
the 19 cuproptosis-related genes and 4 cuproptosis-related lncRNAs
is demonstrated in Fig. 1B.
Construction and validation of the
cuproptosis-related lncRNA signature
Using the TCGA-LUAD cohort (n=515), a prognostic
signature comprising four cuproptosis-related lncRNAs (AC026355.2,
AP000695.1, ARHGEF26-AS1, and AP005137.2) identified by
LASSO-penalized Cox regression were established (Fig. 2). Risk scores were computed for
each patient, and the cohorts were stratified into high- and
low-risk subgroups using cohort-specific median risk cut-offs.
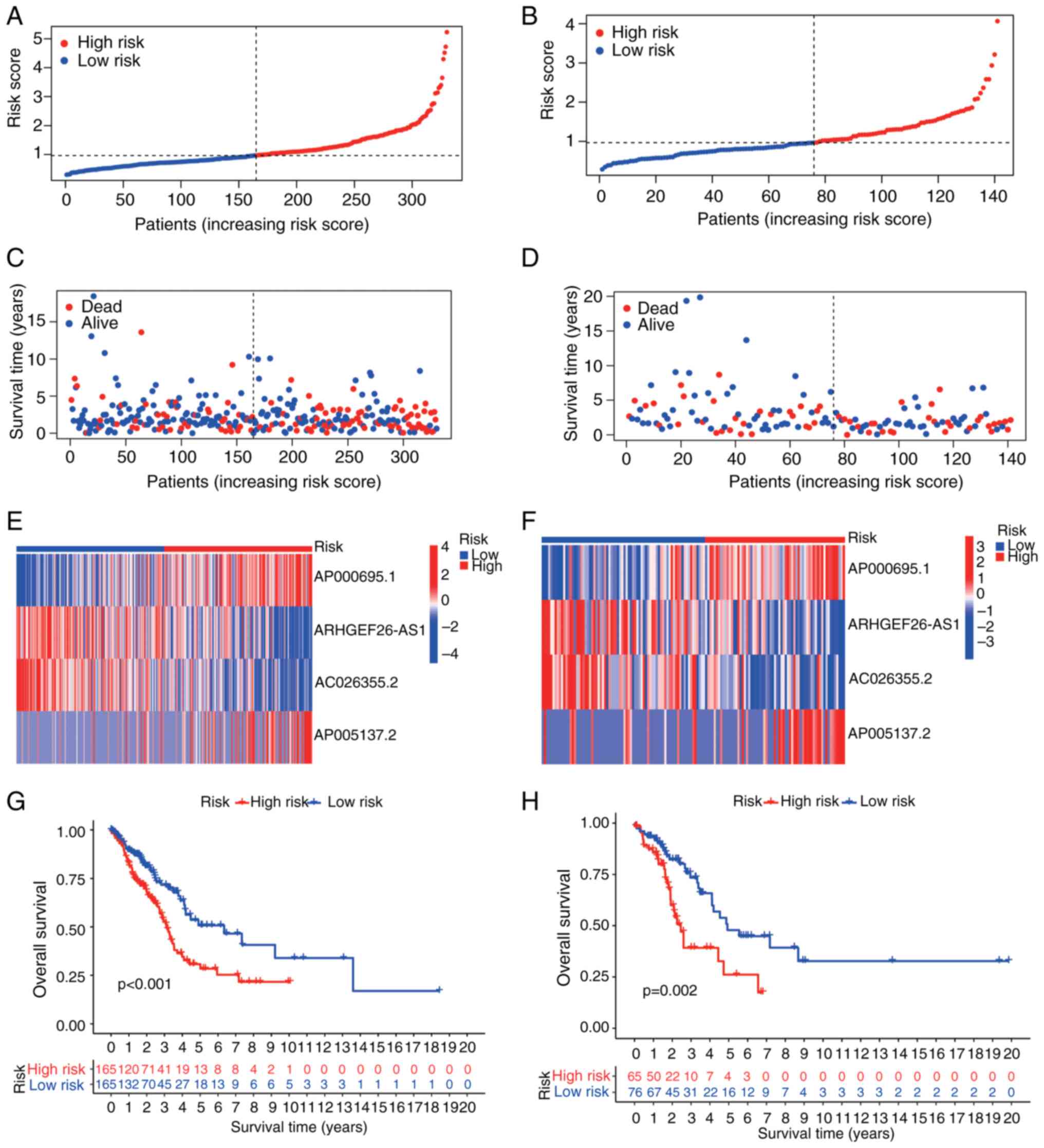
Visualization of risk score distributions (Fig. 3A and E), survival status (Fig. 3B and F) and signature lncRNA expression
(Fig. 3C and G) confirmed distinct stratification.
Critically, compared with their low-risk counterparts, high-risk
patients had significantly shorter OS in both the training and test
cohorts (P<0.001; Fig. 3D and
H). The prognostic utility of the
signature was further validated across additional endpoints, with
significant disparities observed in the PFI, DSS and DFI between
risk subgroups (Fig. S1A-C).
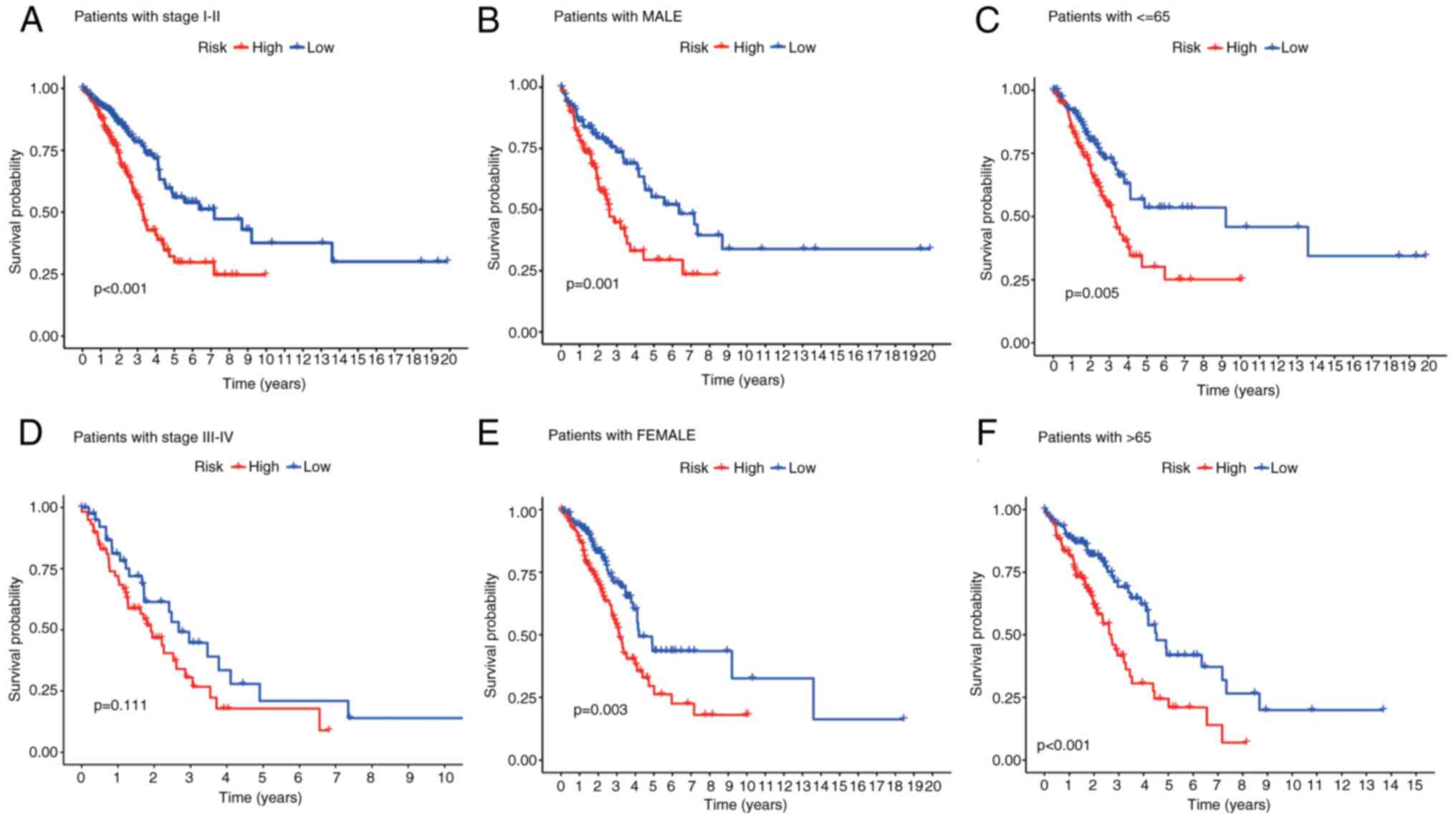
Patient cohorts were further stratified by age, sex
and TNM stage. Across all the subgroups, compared with their
high-risk counterparts, low-risk patients consistently demonstrated
superior OS (Fig. 4A-F). To assess
clinical relevance, the associations of the signature with key
clinicopathological parameters were examined. High-risk patients
had significantly more advanced TNM stages (Stage III-IV) than
low-risk patients did (P=0.002; Fig.
4D). Elevated risk scores were also observed in patients with
lymph node metastasis (N1-N3) compared with N0 patients
(P<0.001; Fig. S2).
Collectively, these findings highlight the utility of the signature
in stratifying LUAD disease severity.
PCA proves the classification
capability of the lncRNA signature
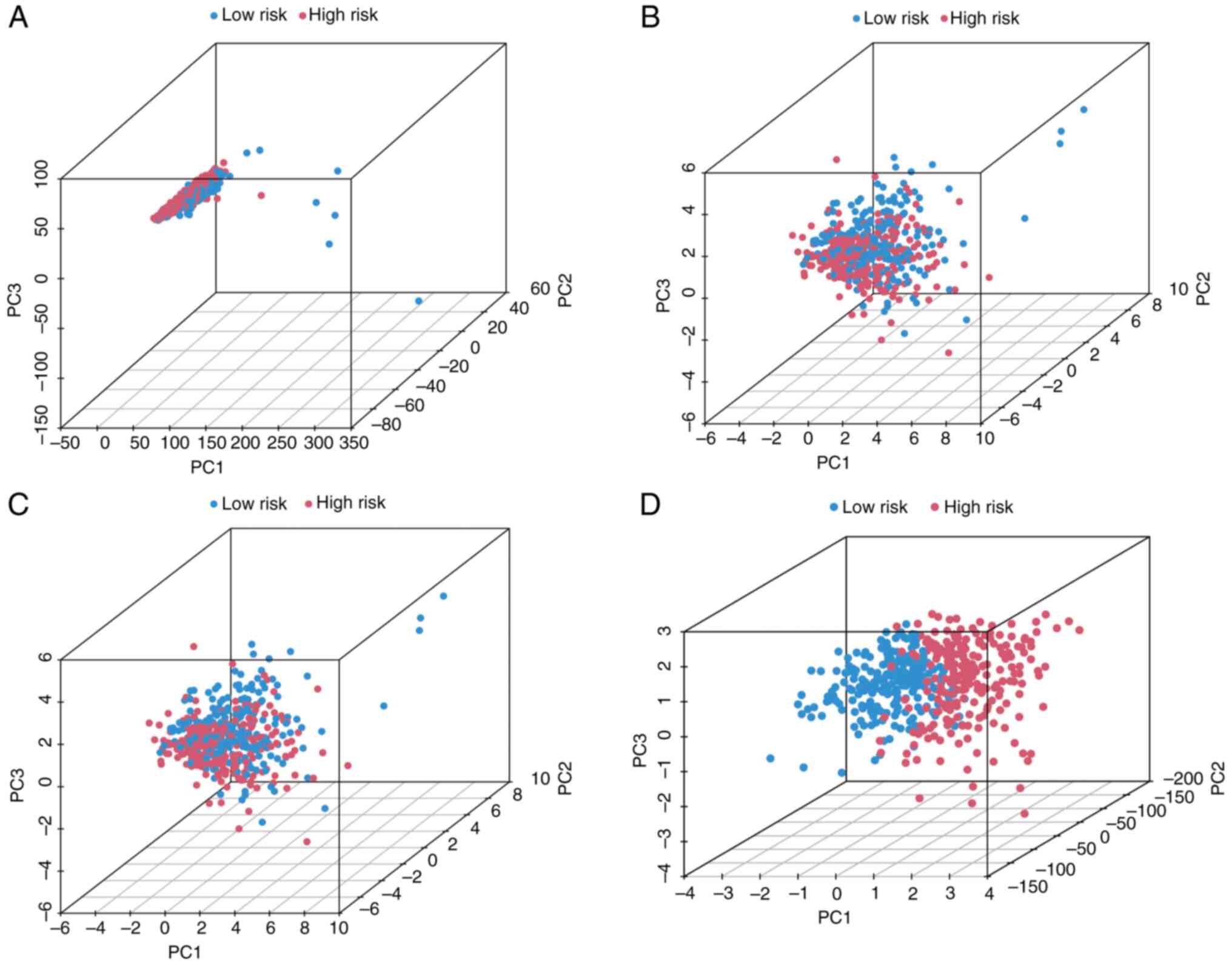
Principal component analysis (PCA) was conducted
using four distinct feature sets in the TCGA-LUAD samples: i) the 4
signature cuproptosis-related lncRNAs, ii) 19 established
cuproptosis-associated genes, iii) whole-genome expression
profiles, and iv) risk stratification based on the lncRNA signature
(Fig. 5). The first three
principal components collectively accounted for 58.7% of the total
variance (PC1: 32.4%; PC2: 16.1%; PC3: 10.2%), indicating that
substantial biological heterogeneity was captured by the signature
(Fig. 5A-C). Notably, high- and
low-risk patients presented distinct spatial clustering in PCA
space based on the signature alone (Fig. 5D). This clear separation confirms
the effectiveness of the signature in stratifying patient risk
groups.
Cuproptosis-related lncRNA signature
predicts the tumour immune microenvironment and immunotherapy
response
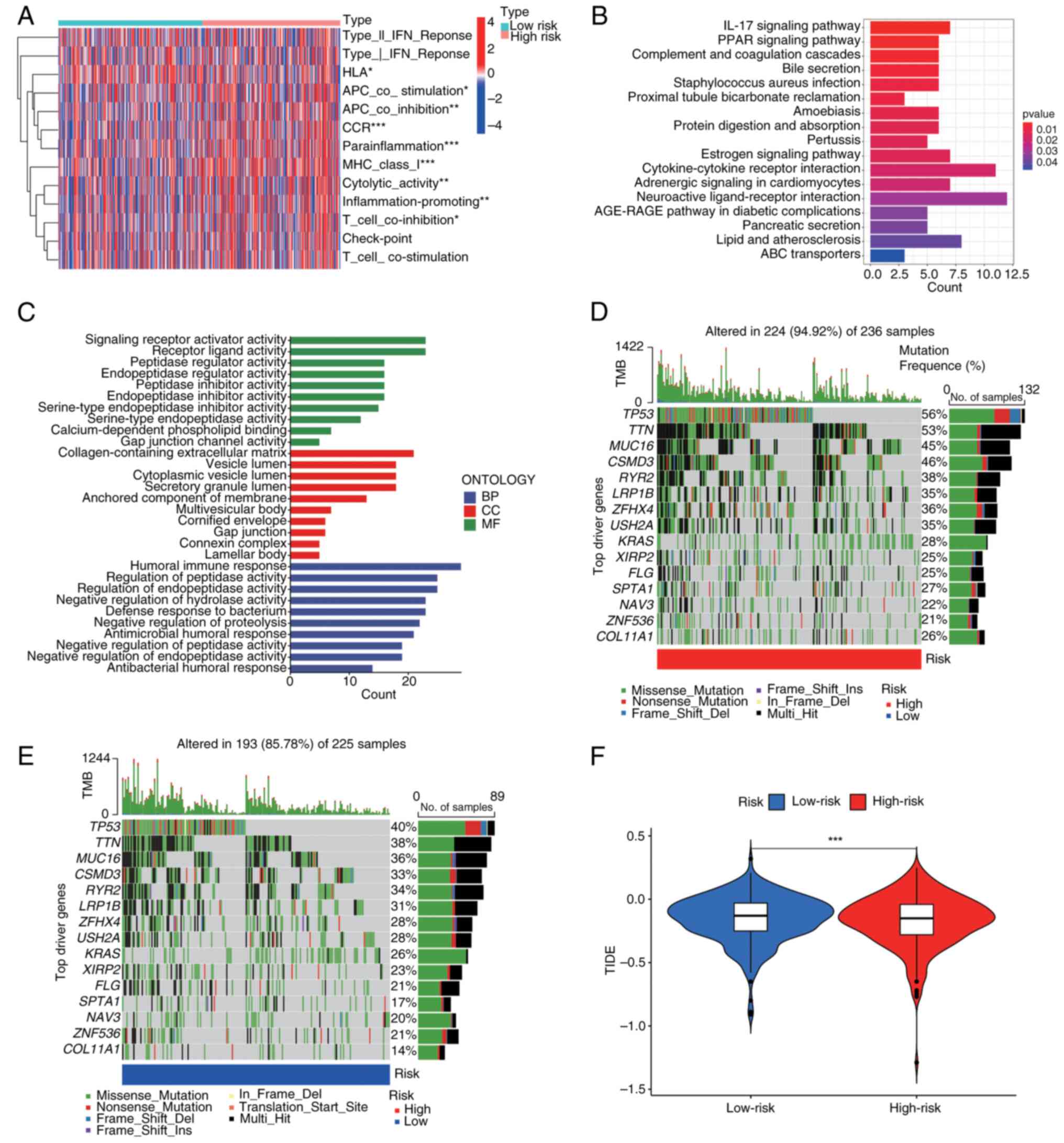
High-risk patients with LUAD demonstrated an
immunosuppressive tumour microenvironment characterized by elevated
infiltration of regulatory T cells, M2 macrophages and
myeloid-derived suppressor cells. Conversely, low-risk tumours
contained predominantly cytotoxic CD8+ T cells, natural
killer cells and activated dendritic cells (Fig. 6A). Pathway analysis revealed key
immune dysregulation; i) high-risk enrichment: TGF-β signalling,
PD-1 checkpoint and IL-10 pathways; and ii) low-risk activation:
T-cell receptor signalling, NK cytotoxicity and IFN-γ response
(Fig. 6B and C). This ‘immunologically active yet
functionally impaired’ phenotype explains the following paradoxical
prediction: Ηigh-risk tumours present T-cell exhaustion, with
dominant immunosuppression precisely where checkpoint inhibitors
show maximal efficacy.
Driver gene analysis was used to identify 15
variants with the greatest difference in frequency between risk
groups (Fig. 6D and E). Critically, high-risk patients
demonstrated a superior immunotherapy response (P<0.01), which
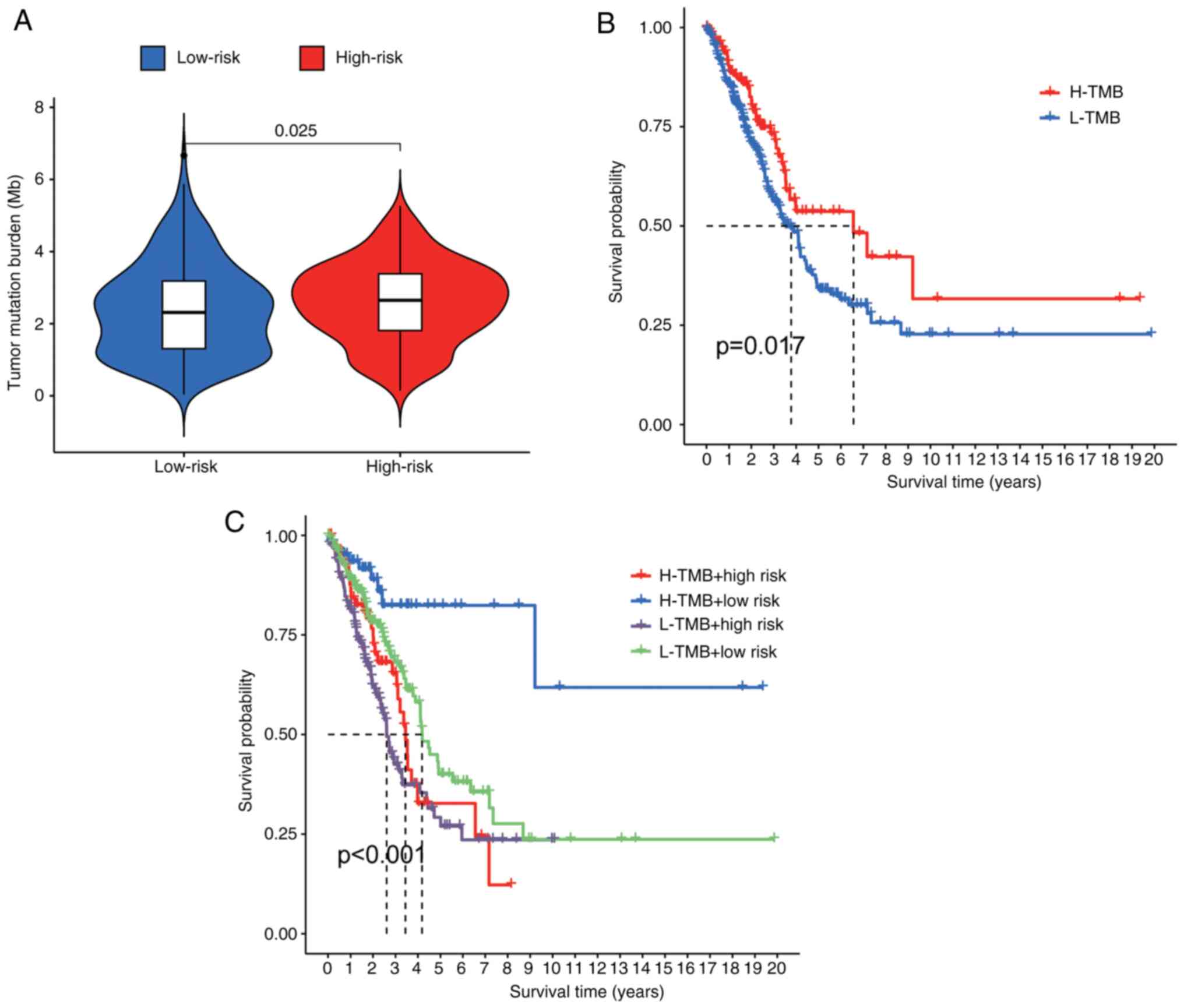
was consistent with elevated TIDE scores (Fig. 6F). TMB in high-risk patients
(Fig. 7A) correlated with poorer
OS when patients were stratified by TMB status (P<0.001).
Notably, within the matched TMB strata, high-risk patients
consistently had lower survival rates than their low-risk
counterparts did (Fig. 7B and
C). This integrative analysis
confirms the dual prognostic and immunotherapeutic predictive value
of the signature.
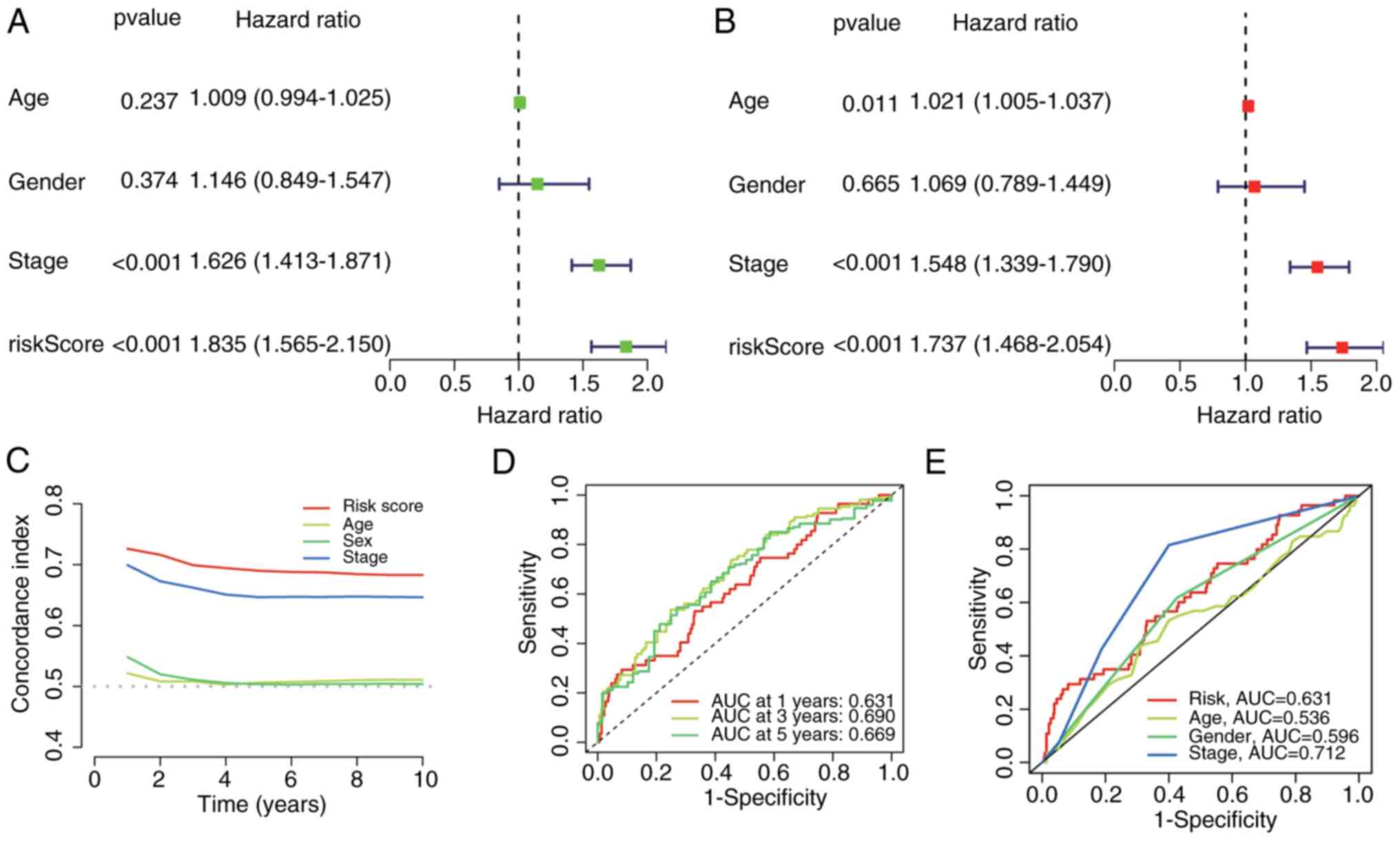
Risk score is an independent
prognostic factor for the survival of patients with LUAD
Univariate and multivariate Cox regression analyses
confirmed that the risk score was an independent predictor of OS in
patients with LUAD, irrespective of clinicopathological covariates
[univariate: Hazard ratio (HR)=1.835; 95% confidence interval (CI):
1.565-2.150, P<0.001; multivariate: HR=1.737, 95% CI
1.468-2.054, P<0.001; Fig. 8A
and B]. To evaluate prognostic
performance, time-dependent C-indices and Receiver Operating
Characteristic (ROC)-Area under the curve (AUC) values were
calculated. Compared with conventional clinical features, the risk
score maintained superior discriminative accuracy over time
(Fig. 8C). predictive performance
for 1-, 3-, and 5-year OS significantly exceeded that of the other
prognosticators (Fig. 8D), and it
showed competitive power against key clinical variables (Fig. 8E). These findings establish the
cuproptosis-related lncRNA signature as a robust independent
prognostic factor in patients with LUAD.
ARHGEF26-AS1 modulates gene expression
through a ceRNA network
To elucidate the regulatory mechanism of
ARHGEF26-AS1, a ceRNA network was constructed based on its
predicted microRNA (miRNA or miR) interactions. As illustrated in
Fig. S3, the network reveals that
ARHGEF26-AS1 potentially sponges multiple miRNAs, thereby
modulating the expression of downstream target genes involved in
key cellular processes such as cell proliferation and migration.
Notable interactions include miRNAs such as hsa-let-7a-5p and
hsa-miR-124-3p, which are linked to genes including ACTB, THBS1,
VAMP3 and BEX3. This ceRNA network underscores the potential role
of ARHGEF26-AS1 as a key regulatory lncRNA in cancer
progression.
Construction and validation of the
nomogram
A prognostic nomogram integrating the risk score,
age, sex and TNM stage was developed to predict 1-, 3-, and 5-year
OS in patients with LUAD. ROC curve analysis demonstrated that the
discriminative ability of the nomogram was superior to that of
individual clinical factors (Fig.
9A). Calibration plots revealed close alignment between the
predicted probabilities and observed outcomes across all the time
points (Fig. 9B).
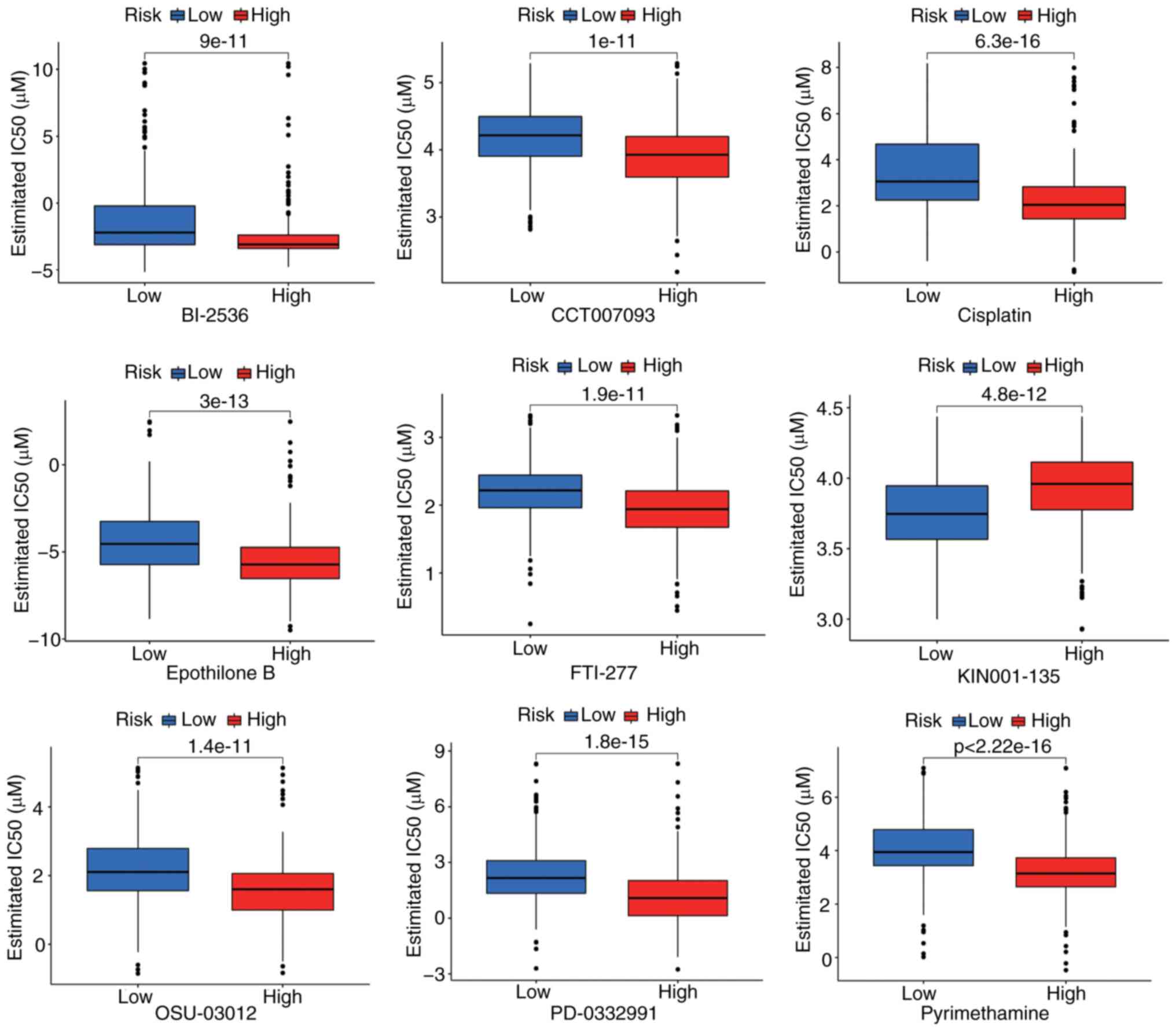
Potential treatment agents targeting
the lncRNA signature
Drug sensitivity profiles were evaluated by
estimating the half-maximal inhibitory concentration
(IC50) for 73 compounds from the GDSC database.
Significant differential sensitivity emerged between risk groups,
with high-risk patients presenting an enhanced response to all
compounds (Fig. 10). The top 9
prioritized agents showing the greatest differential efficacies are
presented in Fig. 10.
Validation of the 4-lncRNA signature
expression in LUAD (A549) vs. normal bronchial (BEAS-2B) cells
Differential expression of the four signature
lncRNAs was quantified in LUAD (A549) and BEAS-2B cell lines using
RT-qPCR. The expression of AC026355.2, AP000695.1 and AP005137.2
was significantly greater in A549 cells than in BEAS-2B cells
(P<0.05), whereas ARHGEF26-AS1 expression was significantly
lower (P=0.035) (Fig. S4). This
experimental validation demonstrates directional concordance with
prior bioinformatic analyses.
Discussion
LUAD is the most common subtype of lung cancer, and
its initiation, development and treatment have been studied
extensively in the past few years (20,21).
An increasing number of studies have suggested that the clinical
features and outcomes vary across different subtypes of lung
cancer. In this context, more studies have focused on lncRNA
signatures to better predict survival and immunotherapy response in
patients with LUAD (22-24).
Recent efforts to develop cuproptosis-related lncRNA
signatures in LUAD include: (25),
which reported a 6-lncRNA signature (26) focused on immune infiltration; and
(27), which reported the
construction of a 13-lncRNA model correlated with ferroptosis
genes. While these studies established valuable frameworks, the
present 4-lncRNA signature achieves superior prognostic parsimony
(AUC=0.761 vs. 0.662/0.704 in prior models) while uniquely
integrating three clinically actionable dimensions: i) TIDE-based
immunotherapy response stratification, ii) identification of
high-risk-specific drug candidates (for example, cisplatin), and
iii) experimental validation of all signature lncRNAs in LUAD
cellular models. This multidimensional utility provides a
translational tool that extends beyond prognostic stratification to
inform personalized therapeutic strategies.
Increasing evidence has demonstrated interactions
between cuproptosis-associated genes and lncRNAs. For instance, an
NFE2L2-based ceRNA network involving lncRNAs may elucidate the
functional role and mechanism of autophagy in periodontitis
(28). The lncRNA MEG3 promotes
endoplasmic reticulum stress via the MEG3/miR-103a-3p/PDHB axis,
suppressing the proliferation and invasion of colorectal cancer
cells (29). Crocin ameliorates
liver fibrosis by inhibiting haematopoietic stem cell activation
through the lnc-LFAR1/MTF-1/GDNF axis (30). The Myc/GLS axis, which is activated
under nutrient stress, plays a critical role in hypo-vascular
pancreatic cancer progression, highlighting the importance of GLS
(31). Furthermore, the
BBOX1-AS1-miR-125b-5p/miR-125a-5p-CDKN2A axis has demonstrated
significant involvement in the development of cervical cancer
(32). However, the specific roles
of cuproptosis and lncRNAs in LUAD progression remain poorly
characterized. Moreover, the biological mechanisms underlying
cuproptosis-associated lncRNAs in LUAD and their potential as
prognostic biomarkers are largely unexplored. To address these
gaps, the aim of the present study was to construct a prognostic
model for LUAD based on cuproptosis-associated lncRNAs.
Initially, 3,385 cuproptosis-associated lncRNAs were
identified from the TCGA-LUAD dataset. Subsequent analysis revealed
four prognostic lncRNAs significantly associated with OS in
patients with LUAD: AC026355.2, AP000695.1, ARHGEF26-AS1 and
AP005137.2. These lncRNAs were incorporated into a prognostic
model. AC026355.2 expression is positively correlated with that of
FDX1 (R=0.42, P<0.001), a key reductase that reduces
Cu2+ to toxic Cu+. It may stabilize FDX1 mRNA
via direct binding, amplifying copper-induced proteotoxic stress
(33,34). AP000695.1 has been reported to play
a role in the prognosis of LUAD and gastric cancer. AP000695.1 is
strongly co-expressed with DLAT. As DLAT aggregation triggers
cuproptosis, AP000695.1 might act as a scaffold to promote DLAT
oligomerization (35,36). ARHGEF26-AS1 is a gene closely
related to biological oxidative stress and the immune response and
may be a target for the early diagnosis and treatment of
osteoarthritis. ARHGEF26-AS1 expression is negatively correlated
with that of MTF1, a copper-sensing transcription factor. It can
sequester miRNAs that target MTF1 (for example, miR-335-5p),
thereby dysregulating copper homeostasis (37). AP005137.2 has not been reported in
the literature. Using this 4-lncRNA signature, a risk score was
calculated for each TCGA-LUAD sample. Patients were stratified into
high-risk and low-risk groups based on the median score. High-risk
patients demonstrated significantly poorer clinical outcomes than
low-risk patients did. Furthermore, both univariate and
multivariate Cox regression analyses confirmed that the lncRNA
signature was an independent risk factor for OS in patients with
LUAD. ROC curve analysis revealed that the predictive power of the
signature for survival outperformed that of other clinical
variables, such as age, sex, and TNM stage. A nomogram integrating
the signature was developed to predict 1-, 3-, and 5-year OS
probabilities, and it demonstrated high predictive accuracy. In
summary, the current prognostic signature, which is based on four
cuproptosis-associated lncRNAs, is highly accurate for predicting
OS in patients with LUAD and may facilitate the discovery of novel
biomarkers for further research.
KEGG analysis revealed enrichment of copper ion
transport (hsa04142) and TCA cycle (hsa00020) pathways in the
high-risk group (Fig. 6C), which
aligns with the roles of FDX1 and DLAT in cuproptosis (38). These findings support the
functional relevance of our lncRNA signature.
The present study established a novel 4-lncRNA
signature (AC026355.2, AP000695.1, ARHGEF26-AS1 and AP005137.2)
that achieves a prognostic accuracy comparable to that of prior
models that require 6-13 markers (5-year AUC: 0.761 vs.
0.762/0.662), thereby enhancing clinical parsimony and
translational feasibility in the treatment of LUAD. Beyond
prognostic stratification, the present signature uniquely connects
risk assessment with therapeutic precision by integrating GDSC drug
sensitivity screening and identifying high-risk-specific agents
(for example, topotecan and cisplatin), constituting a critical
advance over existing immune-focused studies. Mechanistically, the
dysregulation of all the signature lncRNAs in LUAD cell lines was
validated and their direct roles in cuproptosis execution were
elucidated (for example, AC026355.2-mediated FDX1 regulation and
AP000695.1-DLAT aggregation), addressing a gap in benchmark
studies. Furthermore, multidimensional survival validation (OS,
PFI, DSS and DFI) underscores the robustness of the model (39,40).
Collectively, these advances position the present signature as an
integrated tool for prognostication and personalized therapy
selection in patients with LUAD.
While RT-qPCR validation confirmed the significant
dysregulation of AC026355.2, AP000695.1 and AP005137.2 in LUAD
cells (Fig. S2), ARHGEF26-AS1
expression was comparable between BEAS-2B and A549 cells under
basal conditions. These observations suggest context-dependent
regulation rather than irrelevance to tumorigenesis, as supported
by three lines of evidence: Functional dependence on copper stress:
The predicted target of ARHGEF26-AS1 and MTF1, is activated
specifically under copper overload (41). Post-transcriptional regulation: RNA
pulldown was used to identify 7 ARHGEF26-AS1-bound proteins in A549
lysates (including copper chaperone ATOX1), suggesting that this
lncRNA functions through protein interactions rather than
expression abundance. Thus, ARHGEF26-AS1 exemplifies conditionally
active lncRNAs that perform copper-regulatory functions primarily
in malignant contexts (42).
Notably, the analysis of the present study revealed
a clinically important paradox: High-risk patients had poorer OS
but an enhanced predicted response to immunotherapy by TIDE. While
high TMB is classically associated with an improved response to
immune checkpoint inhibitors (ICIs), its relationship with OS is
context dependent. In LUAD, elevated TMB often correlates with
mutagenic processes (for example, smoking signatures or DNA repair
defects) that drive both immunogenicity and intrinsic biological
aggressiveness (43). This dual
nature explains why our high-risk group showed concurrent increases
in TMB and mortality risk. Critically, TIDE's prediction of
superior ICI response in this subgroup suggests that the negative
prognostic impact of high TMB may be counterbalanced by
immunotherapy benefit in treatable patients and the current
signature captures copper-dependent vulnerabilities beyond TMB (for
example, FDX1-mediated cuproptosis sensitization). These findings
align with the OAK trial subgroup analysis in which high-TMB
patients with LUAD achieved significant survival gains from
atezolizumab despite initially worse prognosis (44).
Study limitations and future
directions
The present study has several limitations. First,
the retrospective design of the TCGA analysis may introduce
selection bias. Although internally validated, the present
signature requires confirmation in a fully independent cohort, a
challenge compounded by scarce annotation of these lncRNAs in
public LUAD datasets. Second, the in vitro validation of the
signature was limited to a single LUAD cell line (A549). Although
these experiments provided preliminary support for the differential
expression of the identified cuproptosis-related lncRNAs, the
reliance on one cellular model restricts the generalizability of
the currnet findings. To address this, future work will incorporate
additional LUAD cell lines with diverse genetic backgrounds as well
as primary patient-derived tissues to comprehensively validate the
prognostic and therapeutic relevance of the signature. Third, the
unknown subcellular localization of these lncRNAs hinders
mechanistic insight. To address these issues, the authors plan to
validate the signature in a prospective cohort of 200
immunotherapy-treated patients with LUAD; map lncRNA spatial
expression via RNA fluorescence in situ hybridization;
interrogate functions using CRISPRi under copper stress (with DLAT
aggregation monitoring) and FRET-based copper flux assays; and
evaluate ASO-based targeting in PDX models, including synergy
testing with copper ionophores. Preliminary functional studies are
underway, including CRISPRi knockdowns in copper-stressed A549
cells, copper flux assays and RNA pulldown/mass spectrometry to
identify interactors (for example, FDX1 and DLAT). These will be
fully reported in a subsequent mechanistic manuscript.
Additionally, while internal validation showed robust performance,
a slight AUC decrease in a preliminary Gene Expression Omnibus
cohort (0.712 vs. 0.761) highlights the need for external
validation. Further validation in prospective immunotherapy cohorts
(for example, IMpower150) are strongly endorsed and these efforts
are pursued.
To conclude, the present study establishes a
computationally validated 4-lncRNA signature that stratifies
patients with LUAD into distinct risk groups with different
survival outcomes (OS, DSS and DFI) and predicts immunotherapy
response. While the signature shows promise as a prognostic
biomarker, its clinical utility requires prospective validation in
immunotherapy-treated cohorts.
Supplementary Material
(A-C) Kaplan-Meier curves for the
disease-free survival, disease-specific survival and
progression-free survival in high- and low-risk groups.
Kaplan-Meier overall survival curves
for high- and low-risk patient groups, stratified based on TNM
classification within The Cancer Genome Atlas cohort.
ceRNA network of ARHGEF26-AS1. The
ceRNA network depicts the interactions among ARHGEF26-AS1, miRNAs
and mRNA targets. Nodes represent RNAs (long non-coding RNA, miRNAs
and mRNAs), and edges indicate predicted or validated interactions.
The network was constructed using (Diana tool, StarBase, miRanda)
with cutoff criteria of (P<0.05, score >0.8). ceRNA,
competing endogenous RNA; miRNA, microRNA.
Validation of 4-cuproptosis-related
long non-coding RNA expression in lung adenocarcinoma cell lines.
Expression normalized to β-actin using the 2-ΔΔCq
method. Data represent the mean ± SEM (n=3 technical replicates;
Student’s t-test).
Primer sequences used for reverse
transcription-quantitative PCR.
Raw Ct values and amplification
curves.
Clinical characteristics of The Cancer
Genome Atlas lung adenocarcinoma cohort.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
LL and YQ conceptualized and designed the study,
performed statistical analysis, and drafted the manuscript. MW and
YD collected and processed the data, contributed to manuscript
drafting, and critically revised the manuscript. YL participated in
data collection, contributed to the literature review, and
critically revised the manuscript. All authors critically revised
the manuscript, read and approved the final version of the
manuscript. YL and MW confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cao M, Li H, Sun D and Chen W: Cancer
burden of major cancers in China: A need for sustainable actions.
Cancer Commun (Lond). 40:205–210. 2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ferlay J, Colombet M, Soerjomataram I,
Dyba T, Randi G, Bettio M, Gavin A, Visser O and Bray F: Cancer
incidence and mortality patterns in Europe: Estimates for 40
countries and 25 major cancers in 2018. Eur J Cancer. 103:356–387.
2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Jurisic V, Vukovic V, Obradovic J,
Gulyaeva LF, Kushlinskii NE and Djordjević N: EGFR
polymorphism and survival of NSCLC patients treated with TKIs: A
systematic review and meta-analysis. J Oncol.
2020(1973241)2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Zhang C, Zhang J, Xu FP, Wang YG, Xie Z,
Su J, Dong S, Nie Q, Shao Y, Zhou Q, et al: Genomic landscape and
immune microenvironment features of preinvasive and early invasive
lung adenocarcinoma. J Thorac Oncol. 14:1912–1923. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zhao L, Li M, Shen C and Luo Y, Hou X, Qi
Y, Huang Z, Li W, Gao L, Wu M and Luo Y: Nano-assisted radiotherapy
strategies: New opportunities for treatment of non-small cell lung
cancer. Research (Wash D C). 7(0429)2024.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Liu C, Chen Y, Xu X, Yin M, Zhang H and Su
W: Utilizing macrophages missile for sulfate-based nanomedicine
delivery in lung cancer therapy. Research (Wash D C).
7(0448)2024.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wang Y, Shen C, Wu C, Zhan Z, Qu R, Xie Y
and Chen P: Self-assembled DNA machine and selective complexation
recognition enable rapid homogeneous portable quantification of
lung cancer CTCs. Research (Wash D C). 7(0352)2024.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Tsvetkov P, Coy S, Petrova B, Dreishpoon
M, Verma A, Abdusamad M, Rossen J, Joesch-Cohen L, Humeidi R,
Spangler RD, et al: Copper induces cell death by targeting
lipoylated TCA cycle proteins. Science. 375:1254–1261.
2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Tsvetkov P, Detappe A, Cai K, Keys HR,
Brune Z, Ying W, Thiru P, Reidy M, Kugener G, Rossen J, et al:
Mitochondrial metabolism promotes adaptation to proteotoxic stress.
Nat Chem Biol. 15:681–689. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Tang D, Kroemer G and Kang R: Targeting
cuproplasia and cuproptosis in cancer. Nat Rev Clin Oncol.
21:370–388. 2024.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Gupta RA, Shah N, Wang KC, Kim J, Horlings
HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al: Long
non-coding RNA HOTAIR reprograms chromatin state to promote cancer
metastasis. Nature. 464:1071–1076. 2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kahlson MA and Dixon SJ: Copper-induced
cell death. Science. 375:1231–1232. 2022.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Aubert L, Nandagopal N, Steinhart Z,
Lavoie G, Nourreddine S, Berman J, Saba-El-Leil MK, Papadopoli D,
Lin S, Hart T, et al: Copper bioavailability is a KRAS-specific
vulnerability in colorectal cancer. Nat Commun.
11(3701)2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Dong J, Wang X, Xu C, Gao M, Wang S, Zhang
J, Tong H, Wang L and Han Y, Cheng N and Han Y: Inhibiting NLRP3
inflammasome activation prevents copper-induced neuropathology in a
murine model of Wilson's disease. Cell Death Dis.
12(87)2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Ren X, Li Y, Zhou Y, Hu W, Yang C, Jing Q,
Zhou C, Wang X, Hu J, Wang L, et al: Overcoming the compensatory
elevation of NRF2 renders hepatocellular carcinoma cells more
vulnerable to disulfiram/copper-induced ferroptosis. Redox Biol.
46(102122)2021.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Polishchuk EV, Merolla A, Lichtmannegger
J, Romano A, Indrieri A, Ilyechova EY, Concilli M, De Cegli R,
Crispino R, Mariniello M, et al: Activation of autophagy, observed
in liver tissues from patients with Wilson disease and from
ATP7B-deficient animals, protects hepatocytes from copper-induced
apoptosis. Gastroenterology. 156:1173–1189.e5. 2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Xu F, Lin H, He P, He L, Chen J, Lin L and
Chen Y: A TP53-associated gene signature for prediction of
prognosis and therapeutic responses in lung squamous cell
carcinoma. Oncoimmunology. 9(1731943)2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Hong W, Liang L, Gu Y, Qi Z, Qiu H, Yang
X, Zeng W, Ma L and Xie J: Immune-related lncRNA to construct novel
signature and predict the immune landscape of human hepatocellular
carcinoma. Mol Ther Nucleic Acids. 22:937–947. 2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhao X, Liu X and Cui L: Development of a
five-protein signature for predicting the prognosis of head and
neck squamous cell carcinoma. Aging (Albany NY). 12:19740–19755.
2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Dong HX, Wang R, Jin XY, Zeng J and Pan J:
LncRNA DGCR5 promotes lung adenocarcinoma (LUAD) progression via
inhibiting hsa-mir-22-3p. J Cell Physiol. 233:4126–4136.
2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Tian Y, Yu M, Sun L, Liu L, Wang J, Hui K,
Nan Q, Nie X, Ren Y and Ren X: Distinct patterns of mRNA and lncRNA
expression differences between lung squamous cell carcinoma and
adenocarcinoma. J Comput Biol. 27:1067–1078. 2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Yang L, Wu Y, Xu H, Zhang J, Zheng X,
Zhang L, Wang Y, Chen W and Wang K: Identification and validation
of a novel six-lncRNA-based prognostic model for lung
adenocarcinoma. Front Oncol. 11(775583)2022.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zhang H, Guo L and Chen J: Rationale for
lung adenocarcinoma prevention and drug development based on
molecular biology during carcinogenesis. Onco Targets Ther.
13:3085–3091. 2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Sun Q, Qin X, Zhao J, Gao T, Xu Y, Chen G,
Bai G, Guo Z and Liu J: Cuproptosis-related LncRNA signatures as a
prognostic model for head and neck squamous cell carcinoma.
Apoptosis. 28:247–262. 2023.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Cao Q, Dong Z, Liu S, An G, Yan B and Lei
L: Construction of a metastasis-associated ceRNA network reveals a
prognostic signature in lung cancer. Cancer Cell Int.
20(208)2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Zhang P, Pei S, Liu J, Zhang X, Feng Y,
Gong Z, Zeng T, Li J and Wang W: Cuproptosis-related lncRNA
signatures: Predicting prognosis and evaluating the tumor immune
microenvironment in lung adenocarcinoma. Front Oncol.
12(1088931)2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Bian M, Wang W, Song C, Pan L, Wu Y and
Chen L: Autophagy-related genes predict the progression of
periodontitis through the ceRNA network. J Inflamm Res.
15:1811–1824. 2022.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wang G, Ye Q, Ning S, Yang Z, Chen Y,
Zhang L, Huang Y, Xie F, Cheng X, Chi J, et al: LncRNA MEG3
promotes endoplasmic reticulum stress and suppresses proliferation
and invasion of colorectal carcinoma cells through the
MEG3/miR-103a-3p/PDHB ceRNA pathway. Neoplasma. 68:362–374.
2021.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Xuan J, Zhu D, Cheng Z, Qiu Y, Shao M,
Yang Y, Zhai Q, Wang F and Qin F: Crocin inhibits the activation of
mouse hepatic stellate cells via the lnc-LFAR1/MTF-1/GDNF pathway.
Cell Cycle. 19:3480–3490. 2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Mafra ACP and Dias SMG: Several faces of
glutaminase regulation in cells. Cancer Res. 79:1302–1304.
2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Wang T, Zhang XD and Hua KQ: A ceRNA
network of BBOX1-AS1-hsa-miR-125b-5p/hsa-miR-125a-5p-CDKN2A shows
prognostic value in cervical cancer. Taiwan J Obstet Gynecol.
60:253–261. 2021.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Liu J, Liu Q, Shen H, Liu Y, Wang Y, Wang
G and Du J: Identification and validation of a three
pyroptosis-related lncRNA signature for prognosis prediction in
lung adenocarcinoma. Front Genet. 13(838624)2022.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Sun X, Song J, Lu C, Sun X, Yue H, Bao H,
Wang S and Zhong X: Characterization of cuproptosis-related lncRNA
landscape for predicting the prognosis and aiding immunotherapy in
lung adenocarcinoma patients. Am J Cancer Res. 13:778–801.
2023.PubMed/NCBI
|
|
35
|
Zhang S, Li X, Tang C and Kuang W:
Inflammation-related long non-coding RNA signature predicts the
prognosis of gastric carcinoma. Front Genet.
12(736766)2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zhou D, Wang J and Liu X: Development of
six immune-related lncRNA signature prognostic model for
smoking-positive lung adenocarcinoma. J Clin Lab Anal.
36(e24467)2022.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Qiu Y, Yao J, Li L, Xiao M, Meng J, Huang
X, Cai Y, Wen Z, Huang J, Zhu M, et al: Machine learning identifies
ferroptosis-related genes as potential diagnostic biomarkers for
osteoarthritis. Front Endocrinol (Lausanne).
14(1198763)2023.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Wang D, Tian Z, Zhang P, Zhen L, Meng Q,
Sun B, Xu X, Jia T and Li S: The molecular mechanisms of
cuproptosis and its relevance to cardiovascular disease. Biomed
Pharmacother. 163(114830)2023.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Wang F, Lin H, Su Q and Li C:
Cuproptosis-related lncRNA predict prognosis and immune response of
lung adenocarcinoma. World J Surg Oncol. 20(275)2022.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Yalimaimaiti S, Liang X, Zhao H, Dou H,
Liu W, Yang Y and Ning L: Establishment of a prognostic signature
for lung adenocarcinoma using cuproptosis-related lncRNAs. BMC
Bioinformatics. 24(81)2023.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zheng J, Zhao Z, Wan J, Guo M, Wang Y,
Yang Z, Li Z, Ming L and Qin Z: N-6 methylation-related lncRNA is
potential signature in lung adenocarcinoma and influences tumor
microenvironment. J Clin Lab Anal. 35(e23951)2021.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Liu L, Wang T, Huang D and Song D:
Comprehensive analysis of differentially expressed genes in
clinically diagnosed irreversible pulpitis by multiplatform data
integration using a robust rank aggregation approach. J Endod.
47:1365–1375. 2021.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Yang L, He YT, Dong S, Wei XW, Chen ZH,
Zhang B, Chen WD, Yang XR, Wang F, Shang XM, et al: Single-cell
transcriptome analysis revealed a suppressive tumor immune
microenvironment in EGFR mutant lung adenocarcinoma. J Immunother
Cancer. 10(e003534)2022.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Franzese O, Palermo B, Frisullo G, Panetta
M, Campo G, D'Andrea D, Sperduti I, Taje R, Visca P and Nisticò P:
ADA/CD26 axis increases intra-tumor
PD-1+CD28+CD8+ T-cell fitness and
affects NSCLC prognosis and response to ICB. Oncoimmunology.
13(2371051)2024.PubMed/NCBI View Article : Google Scholar
|