Introduction
Hepatocellular carcinoma (HCC) is estimated to be
the fifth most common cause of cancer-related mortality worldwide
(1). Although ~80% of cases are
reported in developing countries, where the prevalence of hepatitis
is high, HCC is one of the few types of cancer whose incidence is
on the increase in developed countries (2,3).
Although chemotherapy has provided significant survival benefits
for HCC patients, such drugs are associated with marked tissue
toxicity, and drugs or alternative therapies that target tumor
cells without compromising normal tissue function are required
(4). Increased concentrations of
cytotoxic drugs and higher doses of radiation often fail to improve
the health of liver cancer patients, and may cause resistance to
apoptosis. An anticancer agent with lower toxicity that
preferentially induces apoptosis in human cancer cells while
creating an internal oxidative environment would be useful.
Ursolic acid (UA), a pentacyclic triterpenoid, has
been identified in various natural products, such as vegetables and
medicinal herbs (5). UA may
inhibit cell growth and induce apoptosis in certain tumors
(6,7) through multiple pathways, including
inhibiting DNA replication, activating caspases and downregulating
anti-apoptotic genes (8,9). UA specifically inhibits tumorigenesis
(10), tumor progression (11), angiogenesis and tumor invasion
(12).
Viili, a Nordic traditional fermented dairy product
containing lactobacillus, yeast and filamentous fungi,
generates large quantities of extracellular polysaccharide (EPS)
(13). Viili exopolysaccharides
(VEPS) reportedly have antioxidant properties (14), regulate immunity function and lower
cholesterol (15). Astragalus,
particularly A. membraneuse, is a common traditional Chinese
medicine; its polysaccharides [or Astragalus polysaccharides (APS)]
reportedly improve immune function (16), modulate the immune system and
promote tumor cell apoptosis (17).
Cyclo-oxidase (COX)-2 is a key enzyme that catalyzes
arachidonic acid into prostaglandins (18,19).
COX-2 is not expressed in the majority of organs under normal
physiological conditions, but it is expressed in the majority of
cancer cells (20). COX-2 is
believed to inhibit cancer cell apoptosis (21), thus causing resistance to
chemotherapy as COX-2 selective inhibitors suppress tumor cell
proliferation and induce apoptosis (22). For these reasons, naturally derived
COX-2 inhibitors have been used to study chemotherapy and
chemoprevention. In this study we analyzed the synergistic effect
of UA in combination with VEPS and APS, on cell proliferation,
morphologic change, anti-oxidation and COX-2 expression.
Materials and methods
Chemicals
Ursolic acid (>99.8%) was purchased from Sigma
(St. Louis, MO, USA). VEPS (>78%) and APS (>80%) were
extracted in our laboratory. DMEM and the RevertAid First Strand
cDNA Synthesis kit were purchased from Thermo Fisher Scientific
Inc. (St. Louis, MO, USA). Fetal bovine serum (FBS) was purchased
from Gibco (Milan, Italy). Penicillin streptomycin solution,
trypsin, phosphate-buffered saline (PBS), DMSO,
3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide (MTT)
and cell lysis solution were purchased from Solarbio (Beijing,
China). Anti-COX-2 and anti-β-actin antibodies were purchased from
Bioworld Technology (St. Louis Park, MN, USA) and goat anti-rabbit
IgG antibody (H+L) was purchased from Thermo Fisher Scientific Inc.
(Rockford, IL, USA). Superoxide dismutase (SOD) and malondialdehyde
(MDA) test kits were purchased from Nanjing Biological Engineering
(Nanjing, China), human COX-2 and human prostaglandin E2 (PGE2)
enzyme-linked immunosorbent assay (ELISA) kits were purchased from
Bio-Swamp (Shanghai, China). The RNeasy Mini kit was purchased from
Qiagen (Hilden, Germany), and the SYBR Premix Ex Taq II PCR Master
Mix kit was purchased from Takara Biotechnology (Dalian, China).
The West Pico Mouse IgG Detection kit was purchased from Thermo
Fisher Scientific.
Cell culture and reagents
The HepG2 human HCC cell line was a gift from the
Academy of Military Science (Beijing, China). Cells were maintained
in DMEM medium supplemented with 10% FBS, 100 U/ml of penicillin
and 100 μg/ml of streptomycin, and incubated in a humidified 5%
CO2 incubator at 37°C. The culture medium was changed
every two days and the cells were subcultured every fifth day.
Cells in the mid-log phase were used for experiments.
Stock solutions of UA, VEPS and APS were prepared in
DMSO and diluted with medium. The final concentration of DMSO was
<0.1%, which demonstrated no effect on cell viability and DNA
fragmentation. DMSO was also used in the controls. For all assays,
the HepG2 cells (5×104/ml) were treated with UA at 0.1,
1, 2.5, 5 or 10 μg/ml; or VEPS or APS at 10, 20, 40, 50 or 100
μg/ml, respectively. For combined treatments, UA and VEPS 1:1, or
UA and APS 1:1 were applied and incubated for 24, 48 and 72 h,
respectively, at 37°C.
Cell viability/proliferation assay
HepG2 cells were plated at 5×103
cells/well in 96-well plates, and incubated with varying
concentrations of UA, VEPS and APS at different time points. The
MTT solution was added to the culture medium at a final
concentration of 0.5 mg/ml for 4 h, and dark blue formazan crystals
were dissolved with 150 μl DMSO. The absorbance value was measured
at 570 nm using a multiwell spectrophotometer (Bio-Rad, Hercules,
CA, USA). Experiments were performed in quadruplicate and repeated
three times. The percentage of cell inhibition was calculated using
the formula: inhibitory rate (%)=[1−(absorbanceexperiment
well)/(absorbancecontrol well)] × 100.
Observations of apoptosis morphology
using a scanning electronic microscope (SEM)
HepG2 cells (1×105 cells/well) were grown
on cover slips in 6-well plates in a CO2 incubator, then
treated with UA (5 μg/ml), VEPS (50 μg/ml), APS (50 μg/ml), UA/VEPS
1:1 in combination, or UA and APS 1:1 in combination. After 48 h,
the cells were washed with PBS three times, and subsequently fixed
for 2 h at 4°C with 2.5% glutaraldehyde in 0.2 M cacodylate buffer,
pH 7.4. Dehydration was performed with gradients of 30, 50, 70, 90
and 100% ethanol, at 10-min intervals. HepG2 cell surfaces were
coated with a gold spray and examined by SEM (Su1510, Hitachi,
Japan). Images were collected in TIFF files (PC-SeM, Hitachi) and
edited using Photoshop; no artifacts were added.
Flow cytometric analysis
HepG2 cells (1×106 cells/ml) were treated
with varying concentrations of UA (5 μg/ml), VEPS (50 μg/ml), APS
(50 μg/ml), UA/VEPS 1:1 in combination, and UA/APS 1:1 in
combination during the exponential growth phase. Cells were
collected after being cultured for 48 h. Collected cells were
washed twice with cold PBS, fixed with 70% pre-cooled ethanol, and
subsequently incubated overnight in the dark at 4°C. Cell cycle
analysis was performed by flow cytometer (BD Biosciences, Franklin
Lakes, NJ, USA).
Determination of intracellular SOD
activity
HepG2 cells were plated at 5×103
cells/well in 96-well plates, and incubated with varying
concentrations of UA, VEPS, APS, combined 1:1 UA and VEPS, or
combined 1:1 UA and APS for 48 h. The cells were washed twice with
ice-cold PBS, lysed for 10 min in lysis buffer, and centrifuged for
5 min at 1,400 × g. Cellular SOD activity was measured in
non-protein cell lysates using a commercially available SOD assay
kit according to the manufacturer’s instructions.
Determination of intracelluar MDA
content
HepG2 cells were plated at 5×103
cells/well in 96-well plates, and incubated with different
concentrations of UA, VEPS, APS, combined 1:1 UA and VEPS, or
combined 1:1 UA and APS for 48 h. The cells were washed twice with
ice-cold PBS, lysed for 10 min in lysis buffer, and centrifuged for
5 min at 1,400 × g. Cellular MDA content was measured in
non-protein cell lysates using a commercially available MDA assay
kit according to the manufacturer’s instructions.
Reverse-transcription polymerase chain
reaction (RT-PCR)
After HepG2 cells were exposed for 48 h to 5 μg/ml
of UA, 50 μg/ml of VEPS, 50 μg/ml APS, combined 1:1 UA and VEPS, or
combined 1:1 UA and APS, total RNA was extracted from the cells
using RNeasy Mini kit. First strand cDNA was generated via reverse
transcription of 2 μg of the total RNA using a RevertAid First
Strand cDNA Synthesis kit (Fermentas, USA). The standard PCR
conditions for COX-2 were: 94°C for 4 min, then 30 cycles at 94°C
for 45 sec, 56°C for 45 sec and 72°C for 1 min, followed by 10 min
at 72°C. β-actin, a housekeeping gene, was selected as an internal
standard to account for variability in amplification due to
differences in the starting mRNA concentrations. The PCR conditions
were as follows: 94°C for 3 min, then 35 cycles at 94°C for 30 sec,
57°C for 45 sec and 72°C for 30 sec, followed by 10 min at 72°C.
The correct fragment of PCR was confirmed by a commercial
sequencing service company (BGI, Beijing, China). Primer sequences
for COX-2 and β-actin are listed in Table I.
 | Table IPrimer sequences used for PCR. |
Table I
Primer sequences used for PCR.
| Genes | Primers (5′-3′) | Primers (5′-3′) | Size (bp) | Accession |
|---|
| COX-2 |
TGAAACCCACTCCAAACACAG |
TCATCAGGCACAGGAGGAAG | 232 | NM_000963 |
| β-actin |
AAATCTGGCACCACACCTT |
AGCACTGTGTTGGCGTAGAG | 646 | NG_007992 |
COX-2 expression by western blotting
HepG2 cells were incubated with 5 μg/ml UA, 50 μg/ml
VEPS, 50 μg/ml APS, combined 1:1 UA and VEPS, or combined 1:1 UA
and APS for 48 h. Cells were collected and washed twice with
ice-cold PBS. Lysates were incubated for 10 min on ice, sonicated
and centrifuged for 15 min at 12,000 × g. After protein
concentrations were determined using the Bradford assay, the
samples were boiled for 10 min, equal amounts of protein (20
μl/lane) were separated by SDS-PAGE, transferred to nitrocellulose
membranes, and immunoblotted with a 1:1000 dilution of primary
antibody against COX-2 and 1:4000 dilution of primary antibody
against β-actin at 4°C overnight. The secondary antibody was goat
anti-rabbit IgG antibody (H+L) diluted 1:5,000 in blocking solution
for 1 h at room temperature. Immunoreactivity was detected using
West Pico Mouse IgG Detection kit and visualized by
autoradiography.
Measurement of intracellular COX-2 by
ELISA
HepG2 cells were plated at 5×103
cells/well in 96-well plates, and incubated with varying
concentrations of UA, VEPS, APS, combined 1:1 UA and VEPS, or
combined 1:1 UA and APS for 48 h. Cells were washed twice with
ice-cold PBS and lysed for 10 min in lysis buffer. COX-2
concentration was measured in plates coated with purified human
COX-2 antibody, using a commercially available human COX-2 ELISA
kit, according to the manufacturer’s instructions.
Measurement of PGE2 levels by ELISA
HepG2 cells were plated at 5×103
cells/well in 96-well plates, and incubated with varying
concentrations of UA, VEPS, APS, combined 1:1 UA and VEPS, or
combined 1:1 UA and APS for 48 h. PGE2 levels in the culture media
were analyzed in plates coated with purified human PGE2 antibody,
using a commercially available human PGE2 ELISA kit, according to
the manufacturer’s instructions.
Statistical analysis
Data are reported as the means ± SD. Statistical
analyses used analysis of variance (ANOVA) tests. Differences among
means were determined by the least significance difference test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
UA, VEPS, APS and combined treatments
inhibited HepG2 cell proliferation
UA, VEPS, APS and combination treatments induced
HepG2 cell deaths in a time- and dose-dependent manner (Fig. 1). Incubation with varying doses of
UA, VEPS, APS and combined treatments for different time periods
(24, 48, or 72 h) resulted in the significant inhibition of cell
proliferation (P<0.05). However, inhibition at 48 h was stronger
than that at 24 h or 72 h (P<0.05). Furthermore, there was
greater inhibition by the combined treatments than with individual
treatments (P<0.05).
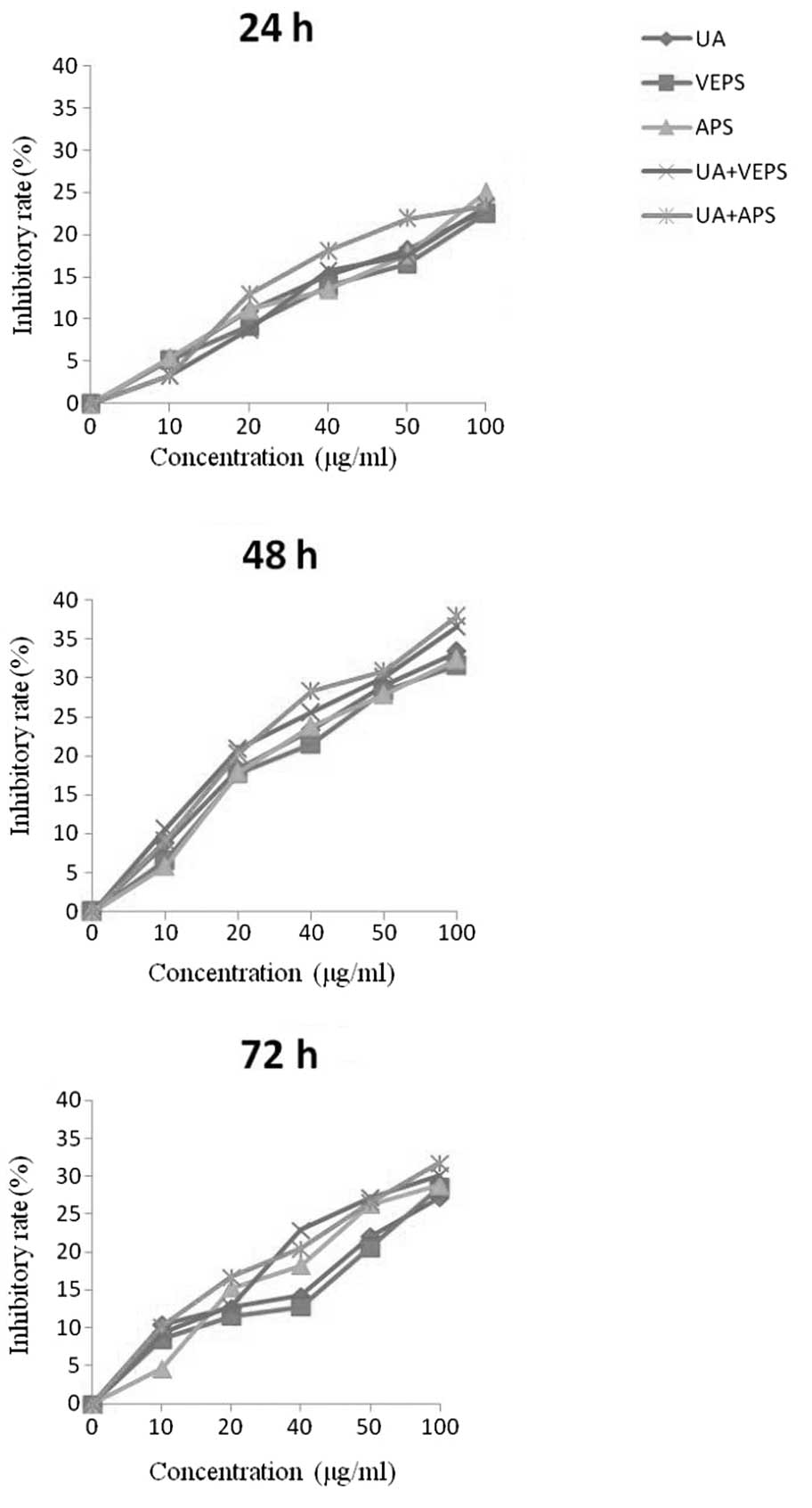 | Figure 1Inhibitory effect of different
compounds on HepG2 cell proliferation. HepG2 cells were incubated
at different concentration of UA (0.1, 1, 2.5, 5, or 10 μg/ml),
VEPS/APS (10, 20, 40, 50, or 100 μg/ml), or combined treatments for
different time periods (24, 48, or 72 h). UA, ursolic acid; VEPS,
viili exopolysaccharides; APS, Astragalus polysaccharide. |
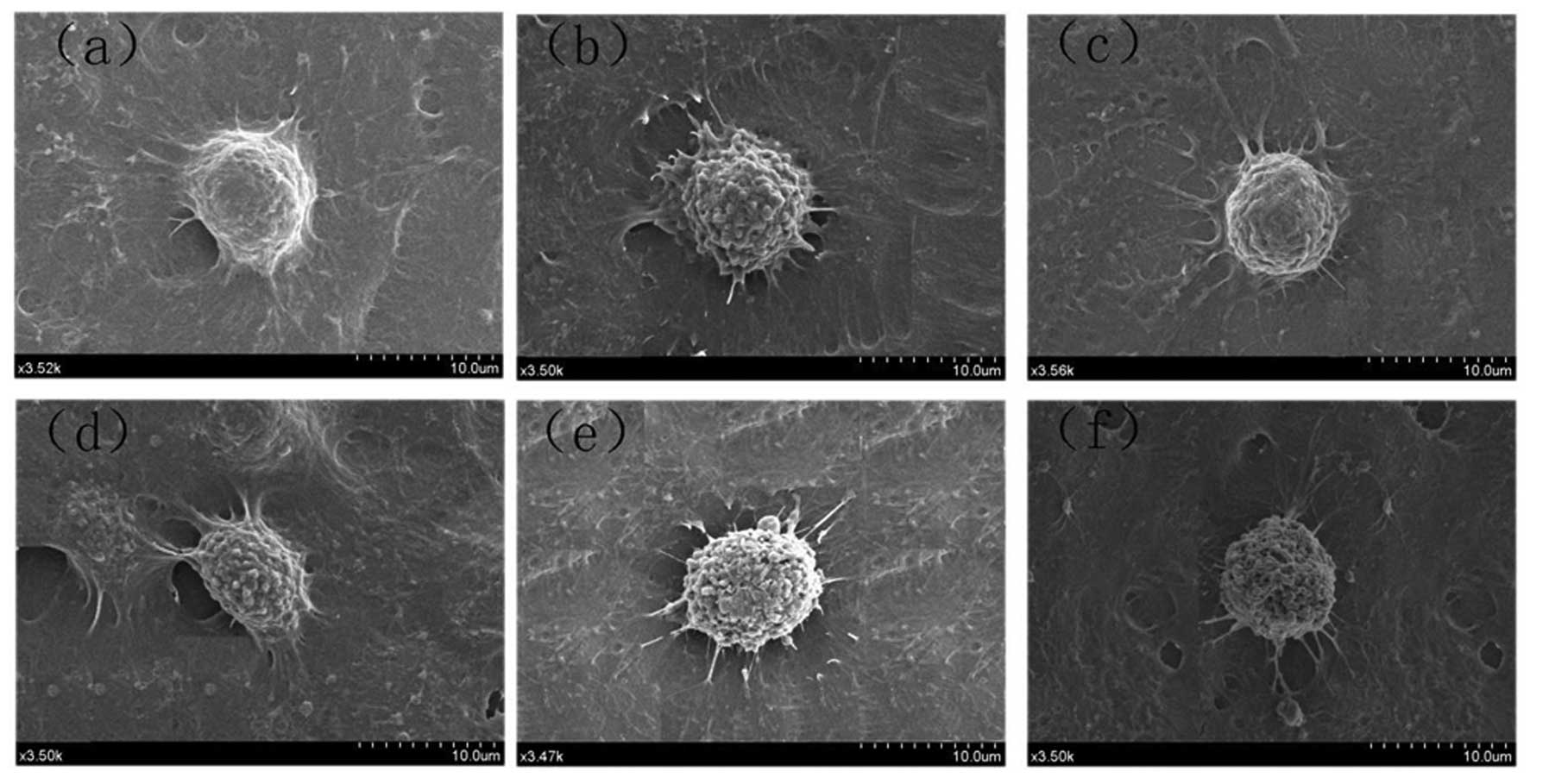
UA, VEPS, APS and combined treatments
induced apoptotic blebs in HepG2 cells
Morphological changes to cells following exposure to
UA (5 μg/ml), VEPS (50 μg/ml), APS (50 μg/ml), and combined
treatments for 48 h were visualized under a SEM (Fig. 2). Following treatment with UA,
VEPS, or APS, HepG2 cells demonstrated characteristic apoptotic
features, with shrinkage, nuclear condensation and DNA
fragmentation.
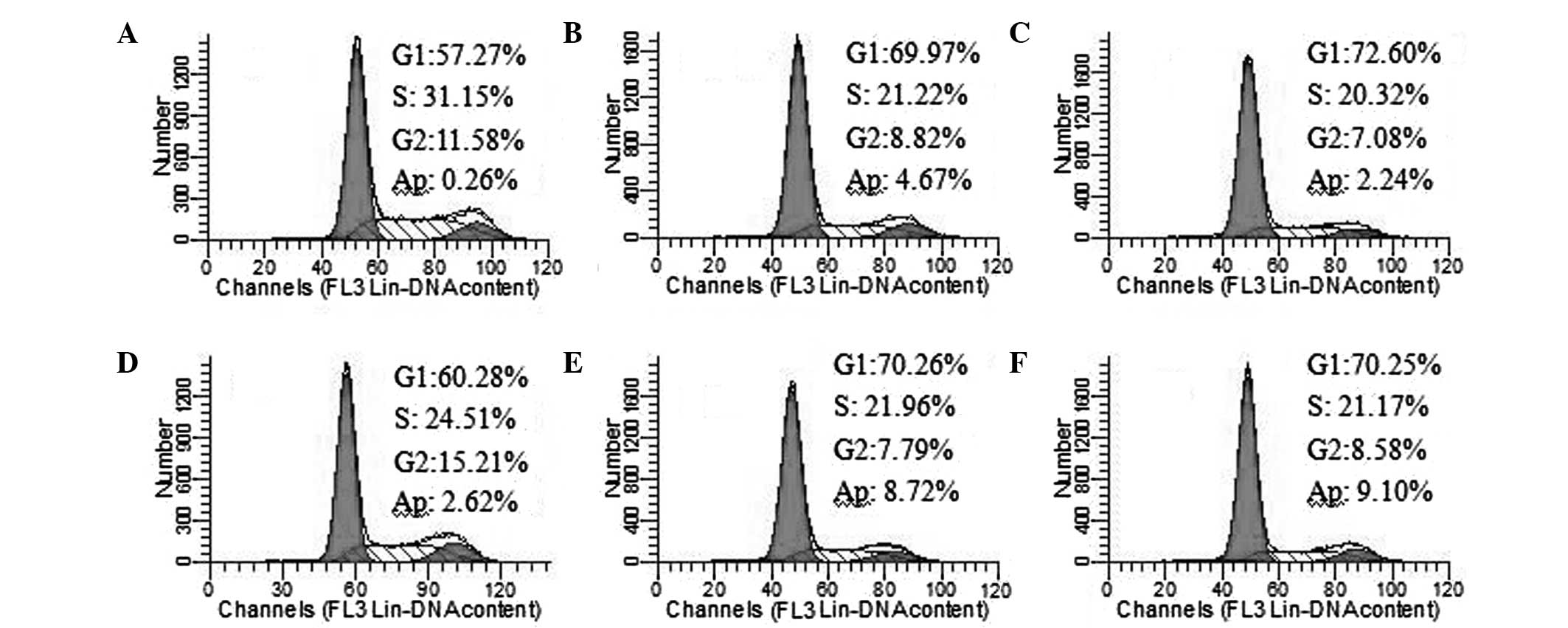
UA, VEPS, APS and combined treatments
increased cell arrest in the HepG2 cell cycle
Following the exposure of HepG2 cells to UA (5
μg/ml), VEPS (50 μg/ml), APS (50 μg/ml), and combined treatments
for 48 h, the cell cycle and apoptosis were monitored by flow
cytometry (Fig. 3). The percentage
of cells increased in the G1- and S-phases. A sub-G1 peak
(apoptosis peak) was also observed. Cell apoptosis increased when
HepG2 cells were treated with UA and combined compounds. The
results indicate that UA, VEPS and APS are capable of inducing cell
cycle arrest.
UA, VEPS, APS and combined treatments
reduced intracellular ROS activity
SOD is a critical antioxidant enzyme that cleans up
cytosolic ROS efficiently. As the drug concentrations increased,
SOD activity was significantly downregulated in HepG2 cells
(Fig. 4; P<0.05). SOD activity
was markedly affected at concentrations of 5 μg/ml UA, 50 μg/ml
VEPS, 50 μg/ml APS and by the combined treatments (P<0.01).
Similarly, MDA is an indicator for intracellular ROS, as the
accumulation of MDA is normally increased in cancer cells. Results
of our assay showed that the MDA content increased at a low
concentration, but decreased at a high concentration of VEPS and
APS in HepG2 cells (Fig. 5,
P<0.05), However, MDA content was significantly decreased by UA
from 0.1 to 10 μg/ml, and when combined with 40, 50 and 100 μg/ml
VEPS and APS (P<0.01), respectively.
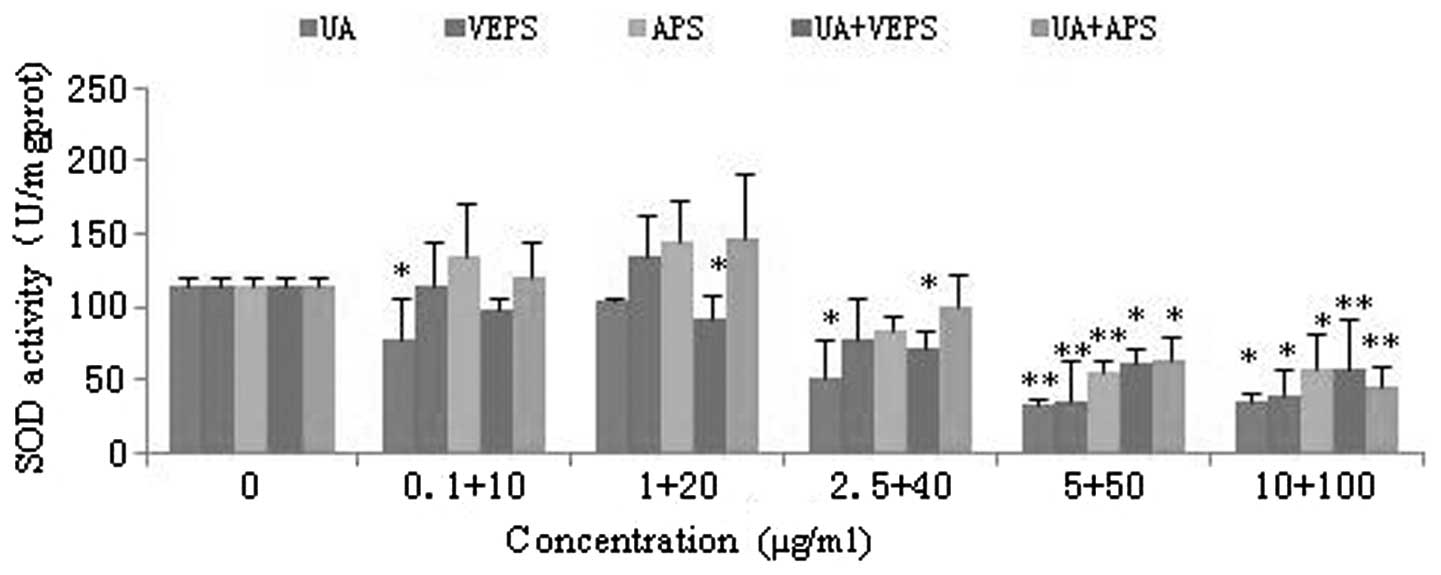 | Figure 4Effects of different compounds on SOD
activity in HepG2 cells. HepG2 cells were treated with various
doses of UA (0.1, 1, 2.5, 5, or 10 μg/ml), VEPS/APS (10, 20, 40,
50, or 100 μg/ml), or combined treatments for 48 h.
*P<0.05, **P<0.01, compared with
control group. SOD, superoxide dismutase; UA, ursolic acid; VEPS,
viili exopolysaccharides; APS, Astragalus polysaccharide. |
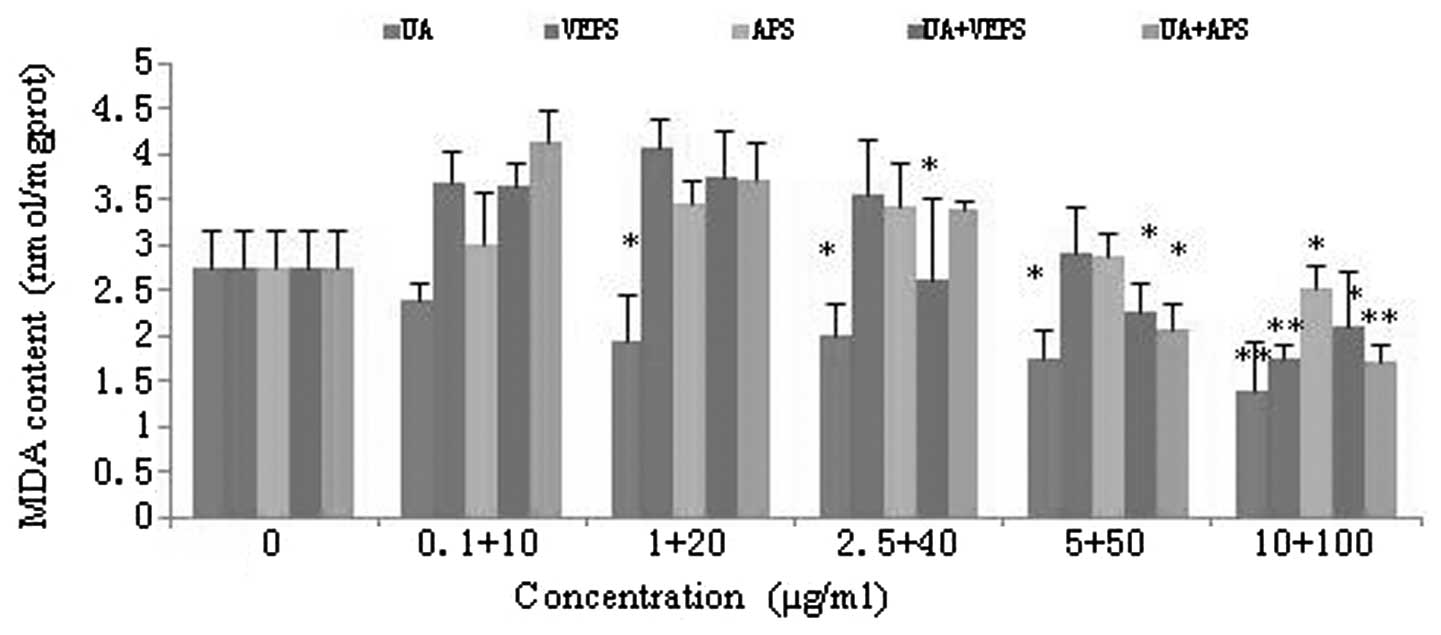 | Figure 5Effects of different compounds on MDA
content in HepG2 cells. HepG2 cells were treated with different
doses of UA (0.1, 1, 2.5, 5, or 10 μg/ml), VEPS/APS (10, 20, 40,
50, or 100 μg/ml), or combined treatments for 48 h.
*P<0.05, **P<0.01, compared with the
control group. MDA, malondialdehyde; UA, ursolic acid; VEPS, viili
exopolysaccharides; APS, Astragalus polysaccharide. |
UA, VEPS, APS and combined treatments
reduced COX-2 expression in HepG2 cells
RT-PCR analysis results (Fig. 6A, P<0.05) revealed that COX-2
mRNA expression in HepG2 cells was higher in the control group, and
downregulated by various compounds (Fig. 6B, P<0.05). For HepG2 cells
treated with 5 μg/ml of UA, COX-2 mRNA expression was markedly
downregulated compared with the controls (P<0.05), and COX-2
mRNA expression decreased further when treated with combinations of
UA and VEPS or APS compared with 50 μg/ml of VEPS or APS alone,
compared with the controls (P<0.05). Western blotting results
(Fig. 6C) revealed that COX-2
protein expression was higher in the control compared with the
experimental groups (Fig. 6D).
Additionally, COX-2 protein expression following treatment with 5
μg/ml UA or either combination of UA and VEPS or APS was markedly
reduced, compared with the controls (P<0.01). However, no clear
change was observed in the VEPS group, and reduction with 50 μg/ml
of APS was less significant when compared with the controls
(P<0.05).
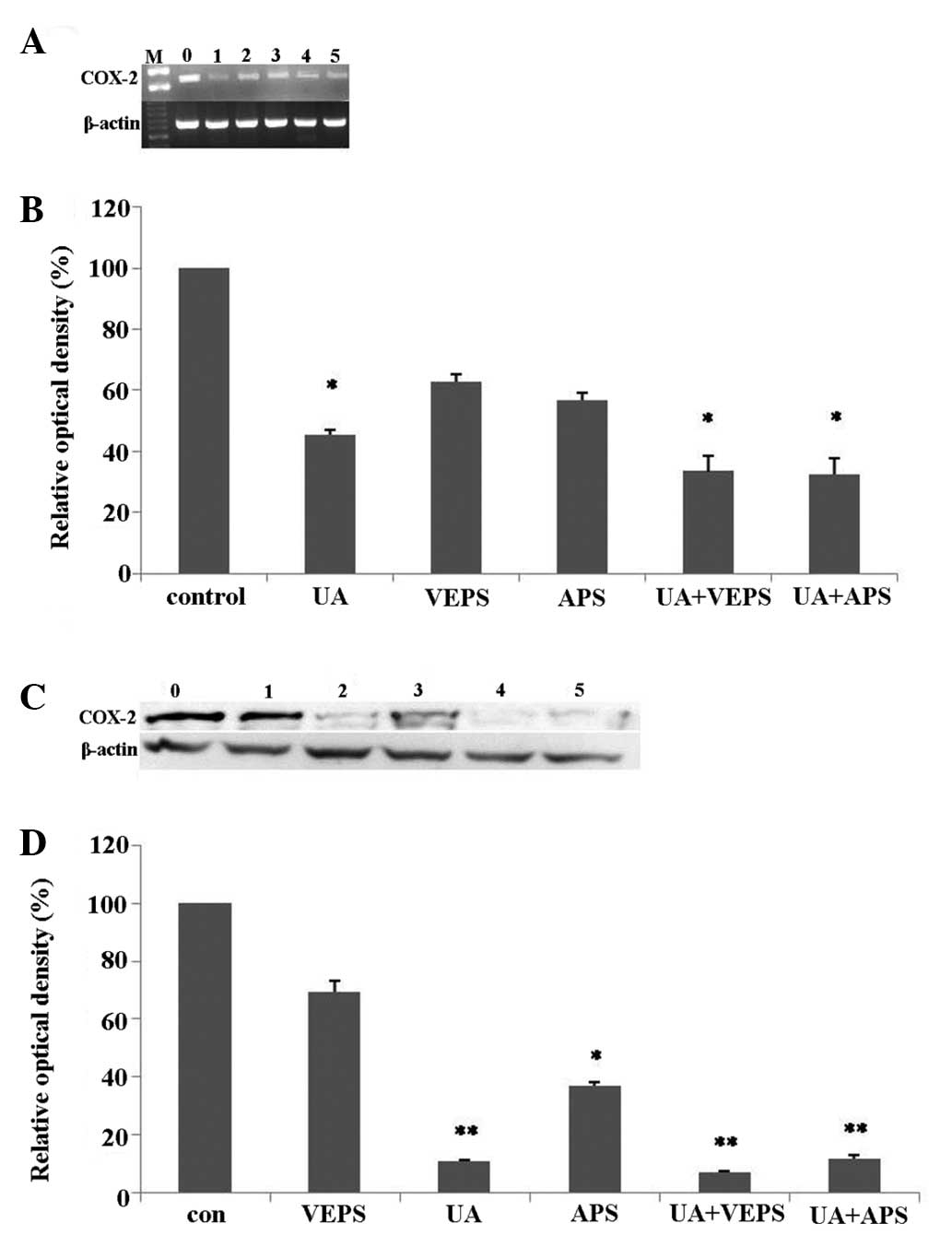 | Figure 6Inhibitory effects of different
compounds on (COX)-2 expression in HepG2 cells. HepG2 cells were
treated with doses of UA (5 μg/ml), VEPS (50 μg/ml), APS (50 μg/ml)
or combined treatments for 48 h. (A) RT-PCR analysis of COX-2
genes. Lane 0, control; lane 1, UA (5 μg/ml); lane 2, VEPS (50
μg/ml); lane 3, APS (50 μg/ml), lane 4, UA+VEPS; lane 5, UA+APS for
48 h. (B) Quantitative analysis of protein levels.
*P<0.05, **P<0.01, compared with
control group. (C) Western blot analysis of protein. Lane 0,
control; lane 1, VEPS (50 μg/ml); lane 2, UA (5 μg/ml); lane 3, APS
(50 μg/ml), lane 4, UA+VEPS; lane 5, UA+APS for 48 h. (D)
Quantitative analysis of protein levels. *P<0.05
compared with control group. COX-2, cyclooxygenase-2; RT-PCR,
reverse-transcription polymerase chain reaction; UA, ursolic acid;
VEPS, viili exopolysaccharides; APS, Astragalus polysaccharide. |
UA, VEPS, APS and combined treatments
reduced COX-2 and PGE2 in HepG2 cells by ELISA
Based on the standard curve, as the concentrations
of the different tested compounds increased, the COX-2
concentration decreased (Table
II), although the effects varied among these treatments; 5
μg/ml UA, 50 μg/ml VEPS, 50 μg/ml APS and the combined treatments
significantly reduced COX-2 concentration in HepG2 cells compared
with the controls (P<0.05). Similarly, based on the standard
PGE2 curve, with increasing concentrations of the various tested
compounds, PGE2 production decreased (Table III). Although effects varied
among these treatments, 5 and 10 μg/ml UA significantly reduced the
PGE2 titer, while the combined treatments; 5 μg/ml UA with 50 μg/ml
VEPS or with 50 μg/ml APS, significantly reduced the PGE2
concentration in HepG2 cells compared with the controls
(P<0.01). However, the reduction with 50 μg/ml VEPS or APS alone
was less significant, compared with the controls (P<0.05).
| Table IIInhibitory effects of different
compounds on cyclooxygenase (COX)-2 concentration in HepG2 cells at
48 h. |
Table II
Inhibitory effects of different
compounds on cyclooxygenase (COX)-2 concentration in HepG2 cells at
48 h.
| COX-2
concentration |
|---|
|
|
|---|
| Samples | 0 (μg/ml) | 10 (μg/ml) | 20 (μg/ml) | 40 (μg/ml) | 50 (μg/ml) | 100 (μg/ml) |
|---|
| UA | 11.10±0.66 | 8.63±0.47 | 8.75±1.45 | 6.90±0.72 | 4.33±0.20b | 5.49±1.13a |
| VEPS | 11.10±0.66 | 9.51±0.58 | 7.94±1.01 | 6.62±0.31 | 5.73±0.47a | 6.22±0.98 |
| APS | 11.10±0.66 | 9.47±0.87 | 8.95±1.95 | 8.01±0.69 | 5.59±0.94a | 6.08±1.56 |
| UA+VEPS | 11.10±0.66 | 7.39±0.25 | 8.11±0.85 | 5.77±0.55 | 4.65±0.69b | 5.27±1.67a |
| UA+APS | 11.10±0.66 | 7.90±0.94 | 8.82±0.40 | 6.45±1.51 | 4.81±0.55b | 5.37±0.49a |
| Table IIIInhibitory effects of different
compounds on PGE2 concentration in HepG2 cells at 48 h. |
Table III
Inhibitory effects of different
compounds on PGE2 concentration in HepG2 cells at 48 h.
| | PGE2
concentration | |
|---|
| |
| |
|---|
| Samples | 0 (μg/ml) | 10 (μg/ml) | 20 (μg/ml) | 40 (μg/ml) | 50 (μg/ml) | 100 (μg/ml) |
|---|
| UA | 120.67±7.31 | 78.10±16.79 | 51.44±10.04 | 39.13±5.46 | 17.85±3.35b | 21.69±3.53b |
| VEPS | 120.67±7.31 | 109.13±6.17 | 61.69±7.34 | 52.46±8.32 | 30.67±6.17 | 35.28±11.42 |
| APS | 120.67±7.31 | 94.00±12.66 | 63.74±10.01 | 50.15±4.80 | 34.77±10.85 | 42.72±13.08 |
| UA+VEPS | 120.67±7.31 | 84.00±8.57 | 61.95±7.47 | 46.56±6.54 | 22.21±5.40b | 28.62±2.77a |
| UA+APS | 120.67±7.31 | 88.36±21.92 | 58.62±2.77 | 37.59±8.15 | 18.87±6.98b | 26.82±10.04a |
Discussion
Although the occurrence and development of tumors
and malignancies are complex, cancer events are not unusual
processes. Potentially cancerous cells are constantly produced, but
are usually eliminated in a healthy environment. However,
carcinogenesis may occur if the body’s internal antioxidant or
anti-inflammatory environment changes entirely, or the mechanisms
that inhibit abnormal cell proliferation disappear. Previous
studies have revealed that antioxidation and anti-inflammation may
increase the risk of cancer in certain environments (23,24),
where antioxidants are over-enriched, due to the internal
antioxidative system, or ARE genes, may be triggered, which
eventually increase cell proliferation (25,26).
Thus an antioxidant environment may not curb cancer, particularly
in cancerous organisms, where COX-2 is overexpressed. COX-2, a
double-edged molecule, participates in inflammatory and
anti-inflammatory processes. As a downstream product of COX-2, PGE2
frequently affects inflammation, and may be further derived into
several so-called electrophilic oxo-derivative (EFOX) molecules
with short life-cycles that strongly regulate cell proliferation
through the Nrf2/keap1/ARE pathways (27).
In this study we have demonstrated that UA, VEPS,
APS and their combined treatments markedly reduced COX-2
expression, and reduced the concentration of PGE2 in HepG2 cells,
as shown by RT-PCR, western blot and ELISA analyses. The inhibition
of COX-2 in cancer cells may increase oxidative stress due to
decreased levels of EFOXs molecules that mediate the gene
expression of SOD and other AREs (28). This is due to the fact that the ARE
family, including SOD, create an antioxidative and
anti-inflammatory protective environment in order to increase cell
proliferation (29). Increased MDA
levels, metabolic products of fatty acids, also indicate an
oxidative environment. UA is a potent antioxidant with multiple
functions, which reduced MDA significantly, while the inhibition of
MDA and cell proliferation occurred only with a high concentration
of VEPS and APS. Complications in in vivo metabolism occur
when the metabolites of fatty acids accumulate. However, it is
possible to ignore MDA as it minimizes the metabolism of fatty
acids in vitro. Thus it is possible that the inhibition of
HepG2 cell proliferation by UA, VEPS, APS and the combined
treatments may be attributed to the inhibition of COX-2 and the
associated decreases in SOD activity, which increase the oxidative
environment and induce apoptosis, as shown by the higher rate of
apoptotic blebs which were observed under the SEM, and the
cell-cycle arrest detected by flow cytometry.
These compounds, particularly VEPS, may also have
cancer-preventive roles in vivo through the innate immune
system (30–32), as VEPS are capable of activating
macrophages and lymphocytes without causing severe inflammation or
other diseases, and a correlation between its dietary use and
cancer epidemiology has been suggested (33).
In conclusion, this study has demonstrated that UA,
VEPS, APS, and their combined treatments inhibit HepG2 cell growth.
The mechanism for this inhibition may be through the inhibition of
COX-2, which in turn reduces EFOXs that activate the internal
anti-oxidative elements, including SOD, thus leading HepG2 cancer
cells into an over-oxidative environment, causing apoptosis and
retarding cell proliferation.
Acknowledgements
This study was supported by an initial fund from
Tianjin City Government for the ‘1000 Talents Plan’ program to C.L.
The authors would like to thank Dr Changlu Wang, Dr Mianhua Chen
and Dr Yurong Wang, of the Tianjin University of Science and
Technology for their tireless aid. The authors are also thankful to
Dr Wentian Liu, Department of Gastroenterology, Tianjin Medical
University’s General Hospital, Tianjin, China for his critical
comments on the preparation of this manuscript.
References
|
1
|
Lodato F, Mazzella G, Festi D, Azzaroli F,
Colecchia A and Roda E: Hepatocellular carcinoma prevention: a
worldwide emergence between the opulence of developed countries and
the economic constraints of developing nations. World J
Gastroenterol. 12:7239–7249. 2006.
|
|
2
|
El-Serag HB: Epidemiology of
hepatocellular carcinoma. Clin Liver Dis. 5:87–107. 2001.
View Article : Google Scholar
|
|
3
|
Goodgame B, Shaheen NJ, Galanko J and
El-Serag HB: The risk of end stage liver disease and hepatocellular
carcinoma among persons infected with hepatitis C virus:
publication bias? Am J Gastroenterol. 98:2535–2342. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Simonetti RG, Cammà C, Fiorello F, Politi
F, D’Amico G and Pagliaro L: Hepatocellular carcinoma: a worldwide
problem and the major risk factors. Dig Dis Sci. 36:962–972. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu J: Oleanolic acid and ursolic acid:
research perspectives. J Ethnopharmacol. 100:92–94. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Aggarwal BB and Shishodia S: Molecular
targets of dietary agents for prevention and therapy of cancer.
Biochem Pharmacol. 71:1397–1421. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hsu YL, Kuo PL and Lin CC: Proliferative
inhibition, cell-cycle dysregulation, and induction of apoptosis by
ursolic acid in human non-small cell lung cancer A549 cells. Life
Sci. 75:2303–2316. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Choi BM, Park R, Pae HO, et al: Cyclic
adenosine monophosphate inhibits ursolic acid-induced apoptosis via
activation of protein kinase A in human leukaemic HL-60 cells.
Pharmacol Toxicol. 86:53–58. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Choi YH, Baek JH, Yoo MA, et al: Induction
of apoptosis by ursolic acid through activation of caspases and
down-regulation of c-IAPs in human prostate epithelial cells. Int J
Oncol. 17:565–571. 2000.PubMed/NCBI
|
|
10
|
Huang MT, Ho CT, Wang ZY, et al:
Inhibition of skin tumorigenesis by rosemary and its constituents
carnosol and ursolic acid. Cancer Res. 54:701–708. 1994.PubMed/NCBI
|
|
11
|
Nishino H, Nishino A, Takayasu J, et al:
Inhibition of the tumor-promoting action of
12-O-tetradecanoylphorbol-13-acetate by some oleanane-type
triterpenoid compounds. Cancer Res. 48:5210–5215. 1988.PubMed/NCBI
|
|
12
|
Cha HJ, Bae SK, Lee HY, et al:
Anti-invasive activity of ursolic acid correlates with the reduced
expression of matrix metalloproteinase-9 (MMP-9) in HT1080 human
fibrosarcoma cells. Cancer Res. 56:2281–2284. 1996.PubMed/NCBI
|
|
{
label needed for ref[@id='b13-mmr-09-06-2505']
}
|
[13] Saxelin ML,
Nurmiaho-Lassila EL, Meriläinen VT and Forsén RI: Ultrastructure
and host specificty of bacteriophages of Streptococcus
cremoris, Streptococcus lactis subsp diacetylactis, and
Leuconostoc cremoris from Finnish fermented milk ‘viili’.
Appl Environ Microbiol. 52:771–777. 1986.PubMed/NCBI
|
|
14
|
Liu L, Wu J, Zhang J, et al: A
compatibility assay of ursolic acid and foodborne microbial
exopolysaccharides by antioxidant power and anti-proliferative
properties in hepatocarcinoma cells. J Food Agric Environ.
10:111–114. 2012.
|
|
15
|
Kitazawa H, Yamaguchi T and Itoh T: B-cell
mitogenic activity of slime products produced from slime-forming,
encapsulated Lactococcus lactis ssp cremoris. J Dairy Sci.
75:2946–2951. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shao BM, Xu W, Dai H, et al: A study on
the immune receptors for polysaccharides from the roots of
Astragalus membranaceus, a Chinese medicinal herb. Biochem
Biophys Res Commun. 320:1103–1111. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ross R: Atherosclerosis - an inflammatory
disease. N Eng J Med. 340:115–126. 1999. View Article : Google Scholar
|
|
18
|
Akhtar M, Cheng Y, Magno RM, et al:
Promoter methylation regulates Helicobacter
pylori-stimulated cyclooxygenase-2 expression in gastric
epithelial cells. Cancer Res. 61:2399–2403. 2001.
|
|
19
|
Kim H, Lim JW and Kim KH: Helicobacter
pylori-induced expression of interleukin-8 and cyclooxygenase-2
in AGS gastric epithelial cells: mediation by NF-kappaB. Scand J
Gastroenterol. 36:706–716. 2001.
|
|
20
|
Shariat SF, Kim JH, Ayala GE, et al:
Cyclooxygenase-2 is highly expressed in carcinoma in situ
and T1 transitional cell carcinoma of the bladder. J Urol.
169:938–942. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Leng J, Han C, Demetris AJ, et al:
Cyclooxygenase-2 promotes hepatocellular carcinoma cell growth
through Akt activation: evidence for Akt inhibition in
celecoxib-induced apoptosis. Hepatology. 38:756–768. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Baek JY, Hur W, Wang JS, et al: Selective
COX-2 inhibitor, NS-398, suppresses cellular proliferation in human
hepatocellular carcinoma cell lines via cell cycle arrest. World J
Gastroenterol. 13:1175–1181. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mantovani A, Allavena P, Sica A and
Balkwill F: Cancer-related inflammation. Nature. 454:436–444. 2008.
View Article : Google Scholar
|
|
24
|
Coussens LM and Werb Z: Inflammation and
cancer. Nature. 420:860–867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Oztürk HS, Karayvaz M, Kaçmaz M, et al:
Activities of the enzymes participating in purine and free-radical
metabolism in cancerous human colorectal tissues. Cancer Biochem
Biophs. 16:157–168. 1998.PubMed/NCBI
|
|
26
|
Eapen CE, Madesh M, Balasubramanian KA, et
al: Mucosal mitochondirial function and antioxidation defences in
patients with gastric carcinoma. Scand J Gastroenterol. 33:975–981.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Luo C, Urgard E, Vooder T and Metspalu A:
The role of COX-2 and Nrf2/ARE in anti-inflammation and
antioxidative stress: Ageing and anti-ageing. Med Hypotheses.
77:174–178. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen C: COX-2’s new role in inflammation.
Nat Chem Biol. 6:401–402. 2010.
|
|
29
|
Groeger AL, Cipollina C, Cole MP, Woodcock
SR, et al: Cyclooxygenase-2 generates anti-inflammatory mediators
from omega-3 fatty acids. Nat Chem Biol. 6:433–441. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kerr JF, Winterford CM and Harmon BV:
Apoptosis. Its significance in cancer and cancer therapy. Cancer.
73:2013–2026. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Elstein KH and Zucker RM: Comparison of
cellular and nuclear flow-cytometric techniques for discriminating
apoptotic subpopulations. Exp Cell Res. 211:322–331. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lebeer S, Claes IJ, Verhoeven TL, et al:
Exopolysaccharides of Lactobacillus rhamnosus GG form a
protective shield against innateimmune factors in the intestine.
Microb Biotechnol. 4:368–374. 2011.
|
|
33
|
Goodman MT, Wu AH, Tung KH, et al:
Association of dairy products, lactose, and calcium with the risk
of ovarian cancer. Am J Epidemiol. 156:148–157. 2002. View Article : Google Scholar : PubMed/NCBI
|