Introduction
Trichosanthin (TCS) is a protein isolated from the
root of the tuber Trichosanthes kirilowii, which has been
widely used in traditional Chinese medicine. TCS, a 27 kDa protein
consisting of 247 amino acid residues, is a member of the type I
ribosome-inactivating protein family (1). Pharmacological studies have
demonstrated that TCS exhibits a broad spectrum of biological and
pharmacological activities, including abortifacient, anti-human
immunodeficiency virus (HIV), immunomodulatory and antitumor
activities (2–5).
TCS has been demonstrated to be effective in
treating chorionic epithelioma in vitro (3). Cancer cell line studies have
indicated that TCS interferes with cell growth and causes apoptosis
in cervical carcinoma HeLa cells, leukemia K562 and HL60 cells,
gastric carcinoma MKN-45 cells and nasopharyngeal cancer CNE-1
cells (2,4). Several studies have also demonstrated
that TCS treatment induces apoptosis and inhibits metastasis in
human epithelial type 2 (HEp-2) human laryngeal epidermoid
carcinoma cells (5,6). However, the mechanism by which TCS
exerts its antitumor effects remains to be fully elucidated.
Although certain studies have reported that TCS irreversibly
inactivates ribosomes and inhibits protein synthesis in carcinoma
to inhibit malignant tumor proliferation (7,8).
Others have observed that TCS induces apoptosis in tumors either
through caspase-3 activation or by the disruption of cytoskeletal
configurations (9,10). It is possible that the antitumor
activity of TCS, demonstrated by the in vitro inhibition of
cell viability, is mediated by multiple mechanisms, including cell
cycle arrest and apoptosis.
Mitogen-activated protein kinases (MAPKs) are
signaling molecules, which convert extracellular stimuli into a
wide range of cellular responses (8). c-Jun N-terminal protein kinase (JNK)
is an MAPK subfamily with three isoforms (JNK1, 2 and 3) and a
splicing variant. JNK1 and JNK2 are ubiquitously expressed, whereas
JNK3 is primarily expressed in neuronal and heart tissues (11). JNKs and p38 are activated by
environmental and genotoxic stresses and are important in
inflammation and tissue homeostasis through the control of cell
proliferation, differentiation, survival and migration of specific
cell types (8). The JNK inhibitor,
CEP-11004, reduces the anti-HIV activity of TCS, suggesting that
TCS may specifically target JNK (12). TCS has also been reported to
suppress the elevation of p38 MAPK, impairing the viral replication
in Vero cells or inhibit HeLa cell proliferation through
suppression of the protein kinase c/MAPK signaling pathway
(13,14). These findings suggest that the MAPK
family may be responsible for the antiviral activity of TCS
(12).
In the present study, the mechanism by which TCS
exerts antitumor activity in the HEp-2 and AMC-HN-8 human laryngeal
epidermoid carcinoma cell lines was investigated by examining the
potential role of JNK in inhibiting cell viability and inducing
apoptosis in response to TCS treatment.
Materials and methods
Materials
TCS was purchased from the Jinshan Pharmaceutical
Co, Ltd. (Shanghai, China). Cisplatin was obtained from the Jingang
Pharmaceutical Co Ltd. (Nanjing, China). The HEp-2 and AMC-HN-8
human laryngeal epidermoid carcinoma cell lines were provided by
the Cell Bank of the Chinese Institute of Biochemistry and Cell
Biology (Shanghai, China). The cell counting kit-8 (CCK-8) assay
and Hochest33258 were purchased from Dojindo (Kumamoto, Japan). The
JNK inhibitor, SP600125, was purchased from Sigma-Aldrich (St.
Louis, MO, USA). The CytoTox 96 non-radioactive cytotoxicity assay
was obtained from Promega Corportation (Madison, WI, USA).
Polyclonal rabbit anti-human antibodies against caspase-3,
caspase-9, JNK, phosphorylated (phospho)-JNK (Thr183/Tyr185),
phospho-extracellular signal-regulated kinase (ERK)1/2
(Thr202/Tyr204) and phospho-p38 (Thr180/Tyr182) were obtained from
Cell Signaling Technology, Inc. (Beverly, MA, USA).
Cell culture conditions
All the cells were maintained in RPMI-1640 medium,
which was purchased from Gibco-BRL (Grand island, CA, USA)
supplemented with 10% heat-inactivated fetal bovine serum (FBS;
Gibco-BRL), penicillin (100 units/ml; Beyotime, Jiangsu, China) and
streptomycin (100 μg/ml; Beyotime) in a 5% CO2 incubator
at 37°C. For the present study, the cells were incubated in either
the presence or absence of different concentrations of TCS (1, 5, 9
or 13 μg/ml), with or without 3 μg/ml cisplatin for 5 days.
Cell viability and cell proliferation
assays
The inhibitory effects of the various treatments on
HEp-2 and AMC-HN-8 cell viability were determined by CCK-8 assay.
The cells, in the exponential growth phase, were seeded into
96-well plates with 100 μl (1×104/ml) per well.
Subsequently, different concentrations of TCS (1, 5, 9 and 13
μg/ml) were added. Each treatment was performed in triplicate wells
and a control group consisting of cells grown in culture medium
containing no drugs was included. The plates were placed in an
incubator in a humidified atmosphere at 37°C for 5 days. At the end
of the exposure, 10 μl CCK-8 was added to each well and the plates
were incubated at 37°C for 1 h. The absorbance of each well was
then measured using a standard enzyme-linked immunosorbant assay at
450 and 650 nm and the absorbance values were labeled A450 and
A650, respectively. The inhibitory rate was calculated as follows:
HEp-2 or AMC-HN-8 cells (100%) = 1 − (Atreat490 −
Atreat650)]/(Acontrol490 − Acontrol650) ×100% and the results were
measured in triplicate. SPSS 16.0 software (SPSS, Inc., Chicago,
IL, USA) was used to calculate the 50% inhibitory concentration for
each group of drugs and the cell proliferation was analyzed by
counting the number of cells with a hemocytometer (Hausser
Scientific, Horsham, PA, USA) using trypan blue (Sangon Biotech,
Shanghai, China) exclusion as the criterion for cell viability.
Cell necrosis assays
Following treatment of the HEp-2 and AMC-HN-8 cells
with different concentrations of TCS (final concentrations of 1, 5,
9 and 13 μg/ml) for 48 h, the culture supernatant was harvested by
centrifugation at 670 × g for 10 min. The supernatant (50 μl) from
each well of the assay was transferred to the corresponding well of
a flat-bottomed 96-well enzymatic assay plate with 50 μl
reconstituted substrate mix (CytoTox 96® non-radioactive
cytotoxicity assay; Promega Corporation, Madison, WI, USA). The
assay plate was then incubated at room temperature and protected
from light for 30 min. The absorbance of each well was then
measured at 490 nm following the addition of 50 μl stop solution
(Promega Corportation).
Hoechst 33258 staining
The cells were cultured on cover slides and
incubated in the presence of 5 μg/ml TCS for 48 h. Following
fixation with 1% glutaraldehyde (Sangon Biotech) for 30 min, the
cells were stained with 10 μg/ml Hoechst 33258 at room temperature
for 10 min. For each cover slide, 1,500 cells were observed under a
Leica fluorescence microscope (magnification, ×200; model, Laser
355,375, Leica Microsystems, Wetzlar, Germany) and images were
captured using a digital camera (Leica Microsystems). Apoptosis, a
form of cell death, is characterized by morphological changes,
including chromatin condensation and apoptotic body formation. The
percentage of cells undergoing apoptosis was determined (15).
Annexin V/propodium iodide (PI) staining
assay
The cells were treated either with different
concentrations of TCS or 3 μg/ml cisplatin for 48 h. The cells were
then trypsinized, harvested by centrifugation at 536 × g for 5 min,
washed twice and resuspended in 100 μl binding buffer (eBioscience,
San Diego, CA, USA). The cell density was adjusted to
1×105/ml and the cells were incubated with 5 μl annexin
V-fluorescein isothiocyanate (FITC) and 5 μl PI for 15 min at room
temperature. The samples were analyzed using a
fluorescence-activated cell sorting (FACS)scan flow cytometer
(Becton-Dickinson, New York, NY, USA). Early apoptosis and necrosis
were determined as the percentage of annexin V positive/PI negative
or the percentage of annexin V positive/PI positive cells.
Analysis of cell cycle phase distribution
by flow cytometry
The present study synchronized the cells in the
G0-phase through serum deprivation prior to treatment. After 3 days
culture in medium containing 5 μg/ml TCS, the HEp-2 and AMC-HN-8
cells were collected by trypsinization, washed with
phosphate-buffered saline (PBS; Sangon Biotech, Shanghai, China)
and fixed in ice-cold 70% ethanol for 24 h. The cells were then
stained with 1 ml PI (0.1 mg/ml with 0.1% Triton X-100; Sangon
Biotech) and incubated in the dark for 30 min. The samples were
analyzed by flow cytometric analysis using FACSCalibur
(Becton-Dickinson). PI is a highly water soluble, fluorescent
compound that cannot pass through intact membranes and is generally
excluded from viable cells. PI binds to DNA by intercalating
between the bases in double-stranded nucleic acids in exposed
nuclei (16).
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR) assays
The total RNA was extracted from the HEp-2 and
AMC-HN-8 cells using aguanidium thiocyanate hot phenol-chloroform
method (17). This total RNA (100
ng) was used for the synthesis of first-strand cDNA by RT in a
reaction mixture (20 μl) containing 5 μM random hexamers, 2.5 μM
oligo (dT) primer and 0.1 μM specific primer (Takara Bio, Inc.,
Shiga, Japan). The reaction was performed at 37 °C for 15 min and
at 85°C for 5 sec.
The RT-qPCR reactions were performed using an ABI
PRISM 7500 RT-qPCR system (Applied Biosystems, Foster City, CA,
USA). For data analysis, the 2−ΔCT (18) method was used to calculate the fold
change. GADPH was considered to be unaffected by the treatment
conditions in the present study and was used as a reference gene
for normalization of the threshold cycle. The following primer
sequences for the genes were used: p21, forward
5′-TGTCCGTCAGAACCCATGC-3′ and reverse 5′-AAAGTCGAAGTTCCATCGCTC-3′;
p27, forward 5′-ATCACAAACCCCTAGAGGGCA-3′ and reverse
5′-GGGTCTGTAGTAGAACTCGGG-3′; Ki67, forward
5′-AGAAGAAGTGGTGCTTCGGAA-3′ and reverse
5′-AGTTTGCGTGGCCTGTACTAA-3′; p53, forward
5′-ACAGCTTTGAGGTGCGTGTTT-3′ and reverse
5′-CCCTTTCTTGCGGAGATTCTCT-3′ and PCNA, forward
5′-ACACTAAGGGCCGAAGATAACG-3′ and reverse
5′-CAGCATCTCCAATATGGCTGAG-3′. The following conditions were used: 1
cycle at 95°C for 30 sec, 40 cycles as 95°C for 5 sec, 34 seconds
at 60°C, 1 cycle at 95° for 15 sec, 60 sec at 60°C and 15 sec at
95°C.
Western blot analysis
The whole cell extracts were prepared by directly
dissolving the cells in 1X SDS loading buffer (Beyotime Institute
of Biotechnology, Shanghai, China). Equal quantities
(1×105 cells/lane) of protein were separated by 12%
SDS-polyacrylamide gel electrophoresis (15% SDS-polyacrylamide gel
electrophoresis for caspase-9 and caspase-3), transferred onto
polyvinylidene fluoride membranes (Millipore Corporation, Bedford,
MA, USA) and inhibited at room temperature for 30 min with 5%
non-fat milk in Tris-buffered saline containing Tween 20 (TBST;
Sangon Biotech) containing 20 mm Tris-HCl (pH 7.6), 500 mm NaCl and
0.1% (v/v) Tween 20. The blots were incubated overnight at 4°C with
the following primary antibodies diluted in TBST buffer: JNK,
phospho-JNK (Thr183/Tyr185), p38, phospho-p38 (Thr180/Tyr182),
caspase-9 or caspase-3 primary antibodies (1:5,000). Following
washing with TBST, the membranes were incubated with horseradish
peroxidase-conjugated goat anti-rabbit secondary antibodies
(1:4,000; KPL, Gaithersburg, MD, USA) and were visualized using an
enhanced chemiluminescence detection kit (Thermo Scientific, Inc.,
Fair Lawn, NJ, USA) according to the manufacturer’s
instructions.
Statistical analysis
SPSS 16.0 software (SPSS, Inc.) was used for data
analysis. All data are expressed as the mean ± standard deviation
from at least two independent experiments. A two-sided Student’s
t-test was used to compare continuous variables between two groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
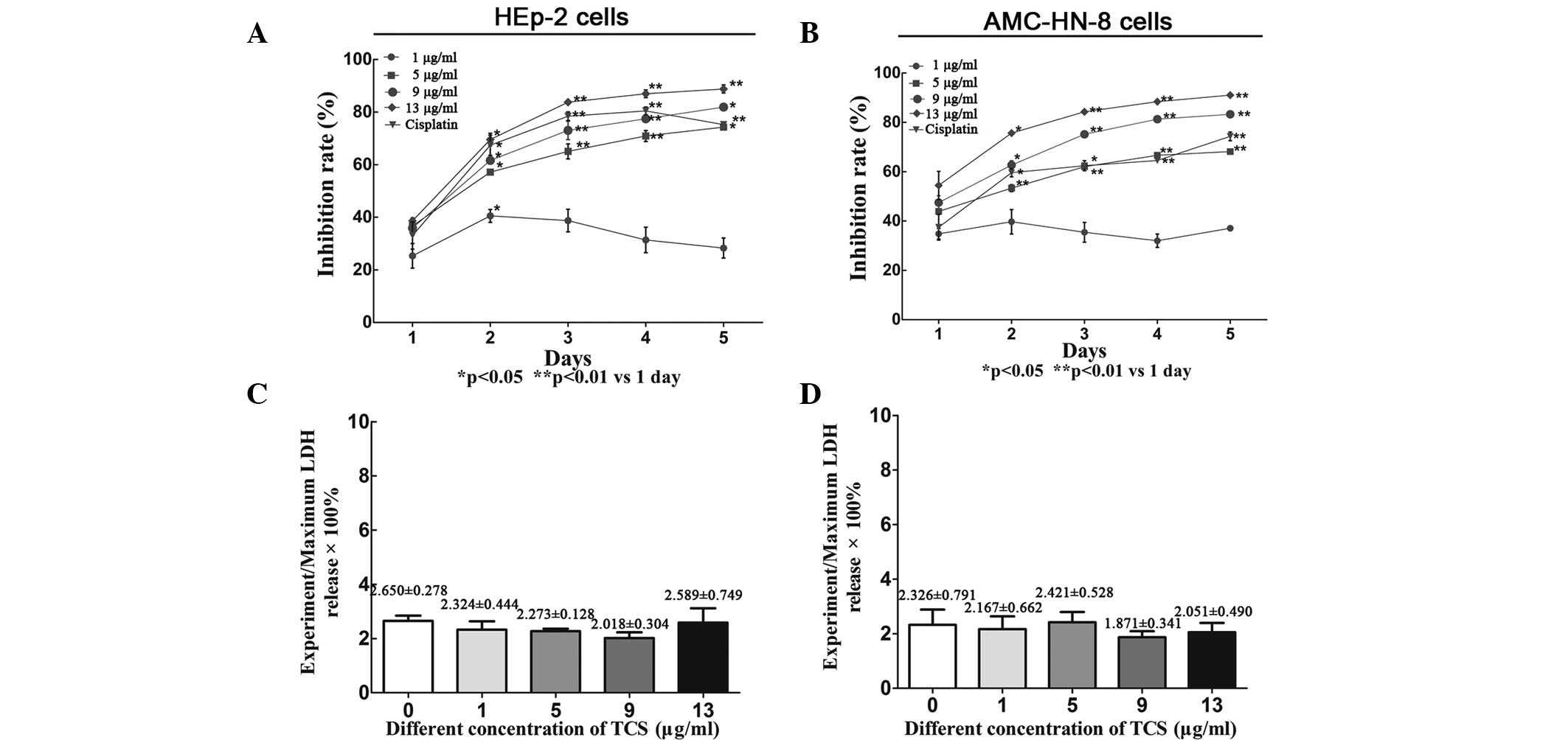
TCS inhibits the viability of HEp-2 and
AMC-HN-8 cells independently of necrosis
To validate the suppressive effect of TCS on cell
viability, the HEp-2 and AMC-HN-8 cells were treated with a range
of concentrations (0–13 μg/ml) of TCS for 5 days followed by CCK-8
assay analysis. TCS inhibited the viability of the HEp-2 and
AMC-HN-8 cells in a dose-and time-dependent manner (Fig. 1A and B). According to the slope of
growth inhibition, the most significant effects of TCS were
observed after 2 days of treatment. Lactate dehydrogenase (LDH) is
a stable cytosolic enzyme, which can be released during the
necrosis process (19). However,
no differences in LDH levels were observed in the culture
supernatant between the day 2 experiment group and the control
group (P>0.05), suggesting that the suppressive activity of TCS
was independent of necrosis (Fig. 1C
and D). LDH is released upon cell lysis; however, necrosis is
characterized by cell swelling and rapid loss of membrane
integrity, causing LDH to be released into the cell culture
supernatant, which was assessed in the present study using a
CytoTox 96® non-radioactive cytotoxicity assay (20). During apoptosis, cells undergo
nuclear and cytoplasmic shrinkage, chromatin is partitioned into
multiple fragments and the cells are fragmented into multiple
membrane-surrounded apoptotic bodies, however the integrity of the
cell membrane is retained during this process. Therefore, TCS
significantly suppresses the viability of HEp-2 and AMC-HN-8 cells
through apoptosis or another method that is independent of necrosis
(15).
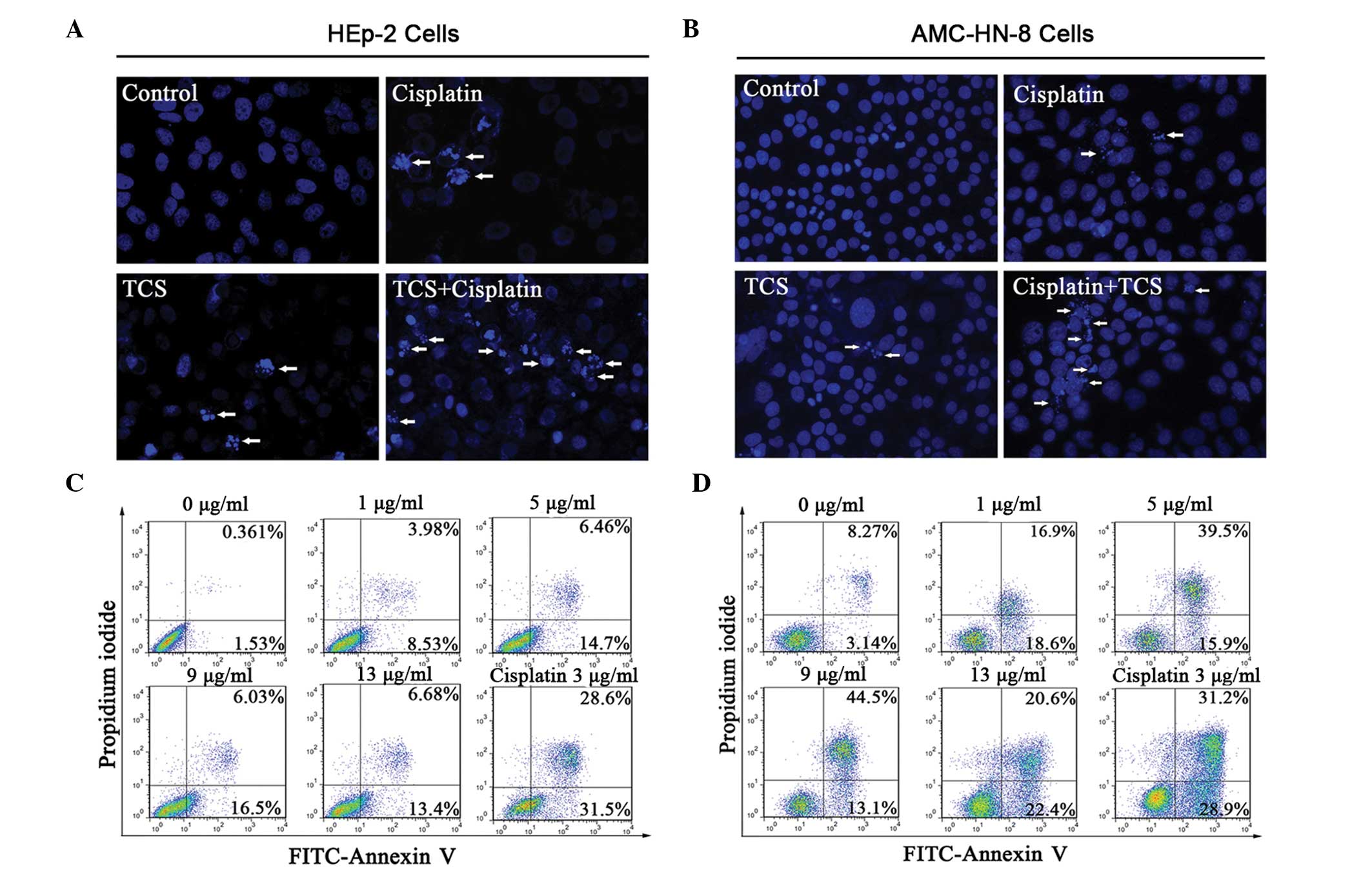
TCS induces apoptosis in HEp-2 and
AMC-HN-8 cells
The HEp-2 and AMC-HN-8 cells were treated with TCS
(5 μg/ml), cisplatin (3 μg/ml) or the two combined for 2 days
followed by staining with Hoechst 33258. The morphological features
of apoptosis, including the primary feature of apoptotic body
formation, were then visualized by fluorescence microscopy
(Fig. 2A and B). The results
revealed significantly more apoptotic cells in the combination
group compared with either the TCS- or cisplatin-only group when
the total number of apoptotic cells were counted on a 1
cm2 cover slip (P<0.05, supplemental Fig. 1). The apoptotic HEp-2 and AMC-HN-8
cells were also examined following treatment with different
concentrations of TCS (0–13 μg/ml) for 48 h using flow cytometry.
The lower left quadrant of each panel in Fig. 2 indicated viable cells, which
excluded PI and were negative for annexin V-FITC binding (Fig. 2C and D). The upper right quadrants
indicated non-viable, necrotic cells, which were positive for
annexin V-FITC binding and PI uptake (Fig. 2C and D). The percentage of annexin
V-FITC positive/PI negative cells, which indicated cells with an
exposed phospholipid and intact plasma membrane, increased between
1.53 and 13.4% in the HEp-2 group and between 3.14 and 22.4% in the
AMC-HN-8 group in the presence of 0–13 μg/ml TCS for 48 h.
Collectively, these data confirmed that TCS induced apoptosis in
the HEp-2 and AMC-HN-8 human laryngeal epidermoid carcinoma cells
in a dose-dependent manner. However, it remains to be elucidated
whether 13 μg/ml TCS and 3 μg/ml cisplatin shared similar
inhibitory effects on the HEp-2 cells (69.64±3.1% and 67.38±5.0%,
respectively) and AMC-HN-8 cells (75.66±4% and 59.58±3.3%,
respectively) on the second day (Fig.
1A and B). The cisplatin-induced apoptotic rate was markedly
higher, suggesting that the increased suppressive effect of TCS on
the HEp-2 and AMC-HN-8 cells was partly independent of
apoptosis.
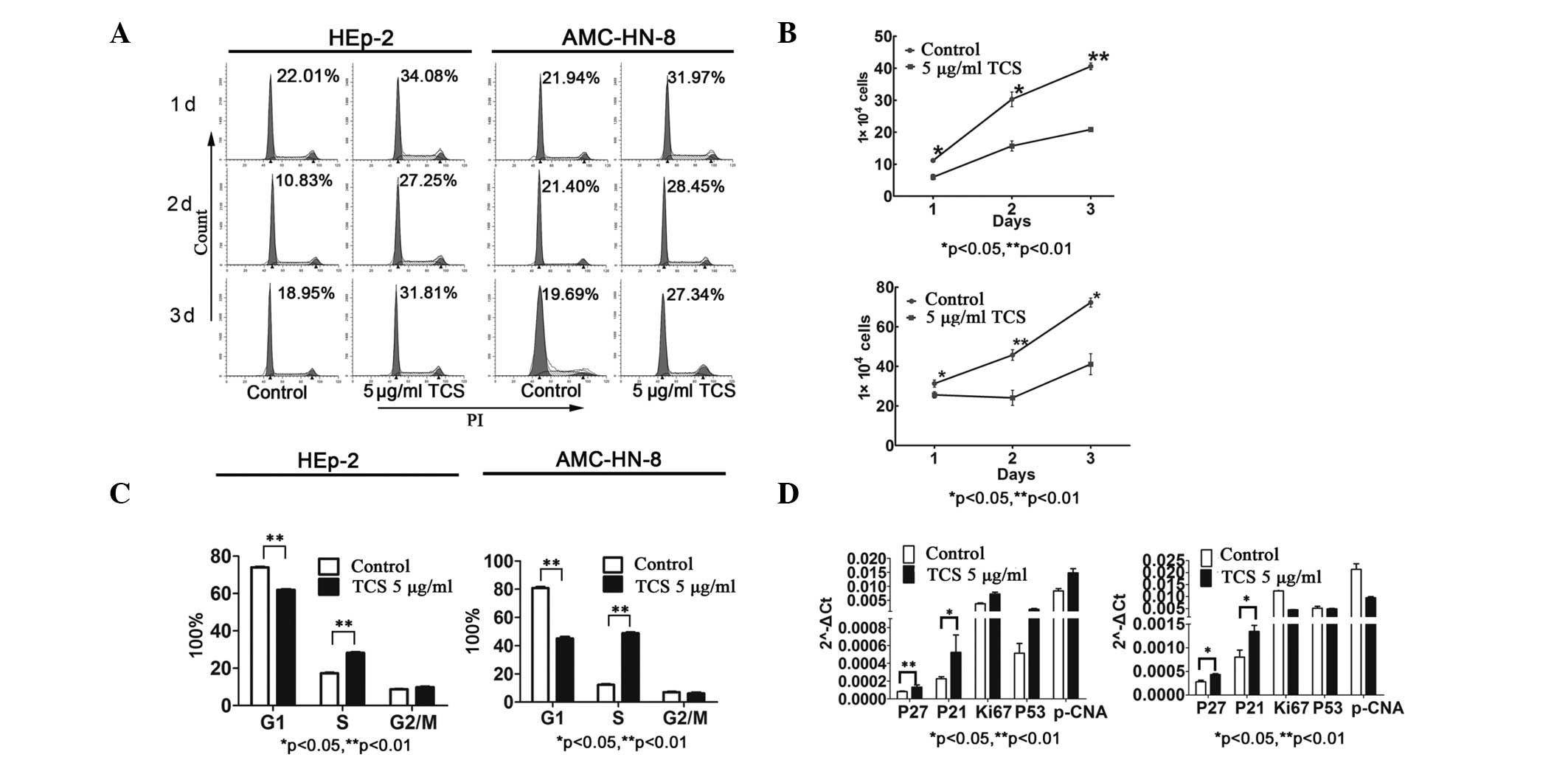
TCS induces cell cycle arrest in human
laryngeal epidermoid carcinoma cells
To investigate the potential suppressive effects of
TCS on the HEp-2 and AMC-HN-8 cells not caused by apoptosis, the
cell cycle was analyzed. Cell cycle analysis indicated that culture
with TCS for 48 h induced cell cycle arrest, as revealed by changes
in the percentages of cells in each cell cycle sub-phase (Fig. 3A and B). After 3 days, the majority
of the TCS-treated HEp-2 and AMC-HN-8 cells exhibited a prolonged
S-phase (Fig. 3A). On the second
day, the percentage of cells in the S-phase was significantly
increased and the percentage of cells in the G1-phase was
significantly decreased compared with the control (P<0.01;
Fig. 3B). Based on this
observation, it was hypothesized that TCS may impair DNA
replication in the HEp-2 and AMC-HN-8 cells or may activate
cyclin-dependent kinase (CDK) inhibitors (CKIs) to arrest the cells
in the early S-phase. Consistent with this hypothesis, the cell
proliferation assay indicated that TCS treatment resulted in a
significant reduction in the number of growing cells compared with
the untreated cells (Fig. 3C). To
further determine the molecular mechanism underlying the
TCS-induced S-phase cell cycle arrest, RT-qPCR was performed. Cell
cycle-regulatory genes, including p27kip1 and
p21Wafl/cip1, were examined by RT-qPCR in the
cells pre-treated with 5 μg/ml TCS for 48 h. As expected, the mRNA
expression levels of p27kip1 and
p21Wafl/cip1 were significantly upregulated
following TCS stimulation for 48 h. The expression levels of the
p21Wafl/cip1 family CKIs,
p21Wafl/cip1 and p27kip1, are
important in the precise regulation of CDK activity, which is
essential for normal cell cycle progression (21).
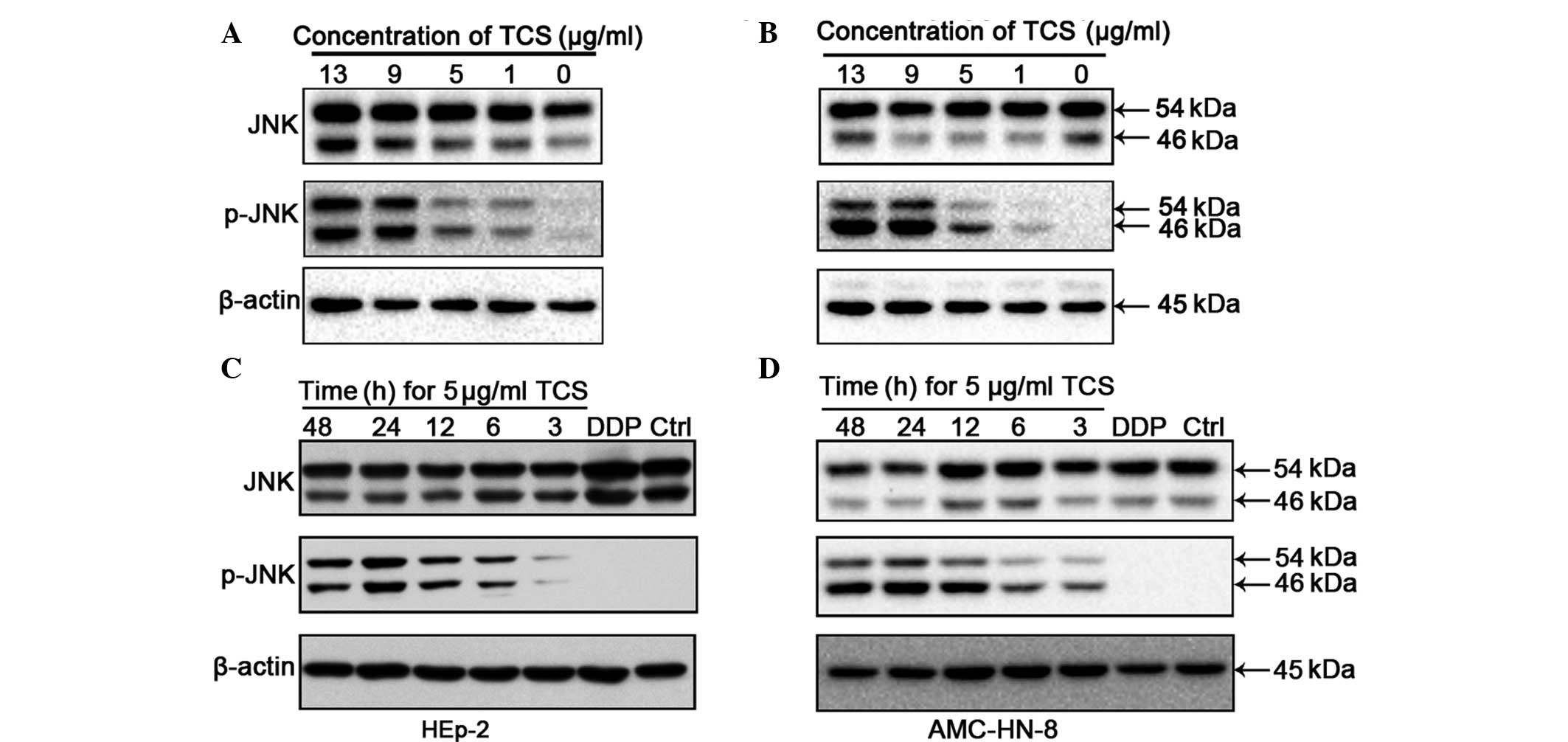
JNK is activated by TCS treatment
The activation of JNK, which occurs through
phosphorylation at Thr183 and Tyr185, was investigated by western
blot analysis of the HEp-2 and AMC-HN-8 cells following treatment
with different concentrations of TCS for different durations.
Compared with the control group, the increased concentrations of
TCS and the increased treatment durations caused upregulation in
the levels of 54/46 kDa phospho-JNK fragments (Fig. 4). However, no significant
differences were observed in the protein levels of JNK between the
different experimental groups. In these experiments, βactin was
used as a control to confirm equal loading. JNK/MAPK is important
in cell proliferation, differentiation and apoptosis and the
present study hypothesized that the antitumor activity of TCS may
be mediated by the activation of JNK/MAPK.
Inhibitory effects on the cell viability
and apoptotic activity of TCS is suppressed by the SP600125
JNK/MAPK inhibitor
In general, TCS treatment reduced the viability of
the HEp-2 and AMC-HN-8 cells, as discussed previously. Compared
with HEp-2 cells treated with 5 μg/ml TCS alone, co-administration
with SP600125 and TCS increased the cell viability between
52.2±9.3% and 63.58±3.5%, respectively, and suppressed the
inhibitory effect of TCS on the HEp-2 cells. This result was also
observed in the AMC-HN-8 cells. Increasing the concentration of
SP600125 caused further increases in cell viability, however, the
magnitude of the effect differed (Fig.
5A and B). In addition, SP600125 decreased the apoptotic rate
(Fig. 5; upper right + lower right
quadrants) of the HEp-2 cells between 13.56% and 4.67% and AMC-HN-8
cells between 21.8 and 5.67%. To investigate the involvement of
various caspase pathways during TCS-induced apoptosis, western blot
analysis of the TCS-treated cells was performed using specific
antibodies against caspase-3 and caspase-9. The cells were treated
with 5 μg/ml TCS and different concentrations of SP600125 (0–10 μM)
for 48 h followed by protein extract preparation and SDS-PAGE
fractionation. SP600125 is a known JNK inhibitor, which inhibits
JNK/MAPK phosphorylation (22),
however, SP600125 did not affect p38/MAPK or ERK/MAPK
phosphorylation (Fig. 5D and E).
Furthermore, the TCS-induced caspase-9 and caspase-3 cleavage was
inhibited by SP600125 in a dose-dependent manner. These results
suggested that JNK may be important in TCS-induced apoptosis and
the inhibition of cell viability.
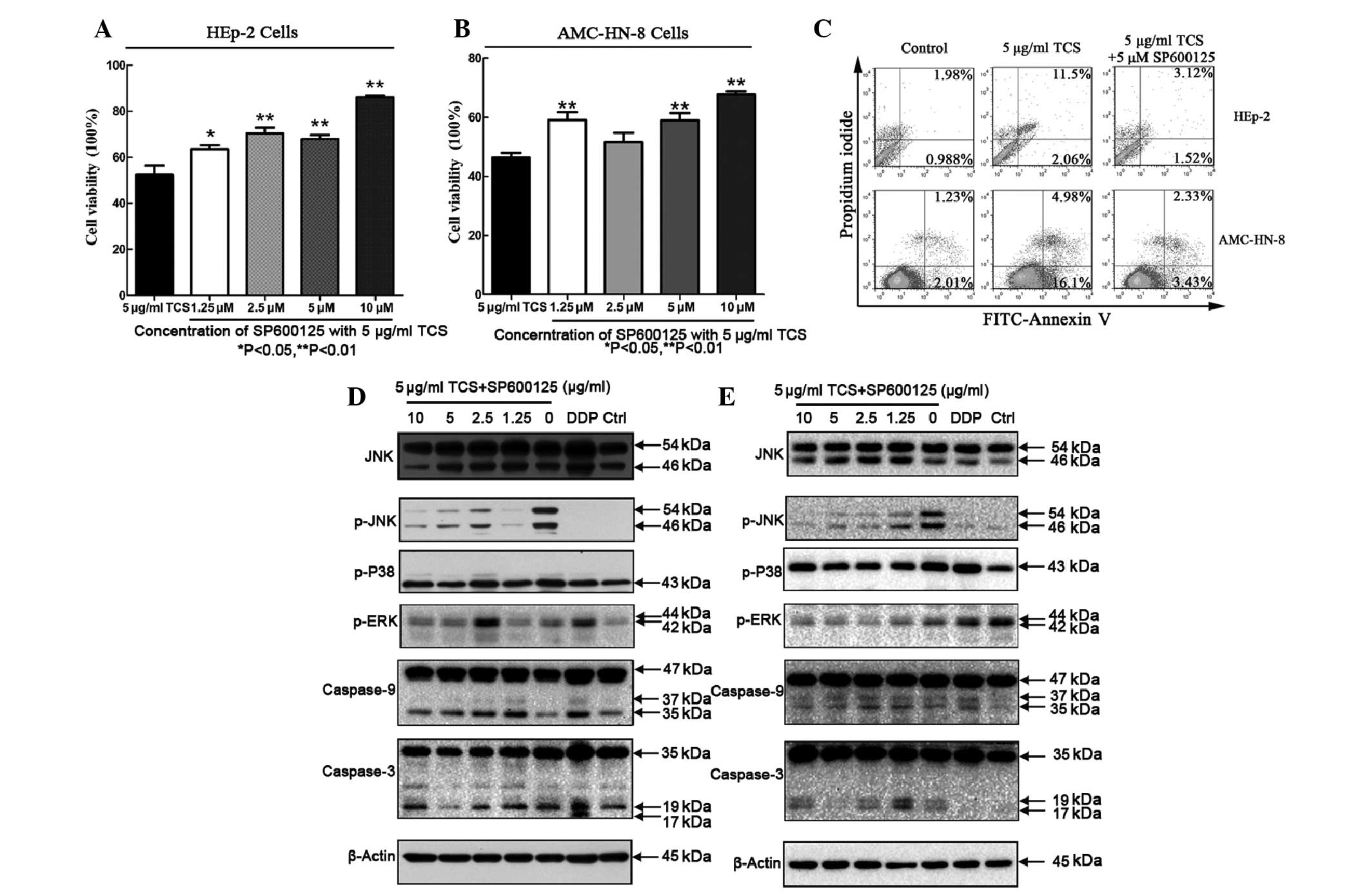 | Figure 5Cell growth inhibitory and apoptotic
activities of TCS can be suppressed by SP600125. (A, B and C) HEp-2
and AMC-HN-8 cells were treated with 5 μg/ml TCS and different
concentrations of SP600125, a JNK/MAPK inhibitor for 48 h. A cell
counting kit-8 assay and flow cytometric analysis were performed to
detect the growth inhibition and apoptotic rate among the
experimental groups. The experiment was repeated three times. The
results are expressed as the mean ± standard deviation. Statistical
analyses of the data was performed using Student’s t-test and
**P<0.01. SP600125 inhibited the TCS-induced
activation of the apoptosis-associated proteins caspase-3 and
caspase-9 in the (D) HEp-2 and (E) AMC-HN-8 cells. SP600125
decreased the levels of p-JNK in a dose-dependent manner, but did
not affect p-p38/MAPK or p-ERK/MAPK activation. TCS, trichosanthin;
HEp-2, human epithelial type 2; JNK, c-Jun N-terminal protein
kinase; MAPK, mitogen-activated protein kinase; ERK, extracellular
signal-regulated kinase; p-, phosphorylated-; FITC, fluorescein
isothiocyanate; PI, propidium iodide; DDP, cisplatin. |
Discussion
In the present study, TCS inhibited the viability of
HEp-2 and AMC-HN-8 human laryngeal epidermoid carcinoma cell lines
independently of necrosis. Furthermore, flow cytometric analysis
using annexin V/PI demonstrated that TCS induced early and late
apoptosis in the HEp-2 and AMC-HN-8 cells in a dose-dependent
manner. In accordance with this observation, stereotypical
apoptotic features were detected (Fig.
2A and B). In previous studies, a concentration of ~100 μg/ml
TCS has been used to investigate its antitumor activity. Apoptosis
is considered to be the primary antitumor mechanism of TCS
(9, 10,23).
However, high concentrations of TCS are cytotoxic, not only to
tumor cells, but also to normal cells, suggesting that TCS is
unsuitable for in vivo experiments. By contrast, lower
concentrations of TCS (1, 5, 9 and 13 μg/ml) were used in the
present study and, although TCS-mediated apoptosis was less evident
compared with that following 3 μg/ml cisplatin treatment, for which
the apoptosis mechanism has been clearly demonstrated, TCS did
inhibit HEp-2 and AMC-HN-8 cell growth (CCK-8; Fig. 1A). Thus, a low concentration of TCS
can be used in combination chemotherapy regimens with other
cytotoxic drugs.
In the present study, the inhibitory effect of 13
μg/ml TCS on the viability of the HEp-2 and AMC-HN-8 cells was not
lower than that of 3 μg/ml cisplatin, which is an important
chemotherapeutic agent for head and neck squamous cell carcinoma
(24). However, the apoptotic rate
caused by 3 μg/ml cisplatin was higher compared with that caused by
13 μg/ml TCS (Fig. 2C and D). This
observation suggested that, in addition to apoptosis, TCS may
inhibit cell viability through other mechanisms. Following
treatment with 5 μg/ml TCS for 3 days, the cells were stained with
a trypan blue solution and counted using a hemocytometer. The cell
proliferation assay indicated that TCS caused a significant
reduction in the number of growing cells compared with the
untreated cells (P<0.05). Consistent with this result, TCS
treatment induced cell cycle arrest as changes were observed in the
percentage of cells in S-phase. In the cells pretreated with 5
μg/ml TCS for 2 days, the percentage of cells in the S-phase was
significantly increased compared with the control (Fig. 3A and C). Similarly, the important
CKIs, p21Wafl/cip1 and p27kip1,
were prominently upregulated following TCS stimulation for 48 h
(Fig. 3D). The findings of the
present study and those of a previous study supported the
hypothesis that treatment of uninfected HEp-2 cells with TCS
prolongs the S-phase (6). The
present study hypothesized that TCS may impair DNA replication
through the induction of DNA damage. The DNA damage caused by TCS
may be detected by the G2 checkpoint, which prevents cells from
entering mitosis and results in S-phase or G2/M-phase arrest to
provide time for the repair or initiation of the cell death pathway
(25). However, the precise
mechanism by which TCS induces S-phase arrest requires further
investigation.
In the present study, a low concentration of TCS led
to activation of the phospho-JNK in the HEp-2 and AMC-HN-8 cells in
a dose- and time-dependent manner (Fig. 4A and B). JNK activation leads to
the phosphorylation of cytoplasmic and nuclear targets, including
the AP-1 transcription factor Jun and cell cycle regulators,
including Cdc25, Aurora B and Cdh1, involved in modulating cell
cycle, cell growth and cell death (26). Therefore, the antitumor activity of
TCS may be associated with its activity in activating the JNK/MAPK
pathway. SP600125, a known JNK inhibitor, specifically inhibited
phospho-JNK activation in a dose-dependent manner without affecting
other MAPK family members, including phospho-ERK or phospho-p38
(Fig. 4D and E). Compared with 5
μg/ml TCS alone, the cell viability was significantly increased
following addition of different concentrations of SP600125 to the
culture medium. In addition, the protein levels of caspase-3 and
caspase-9 protein, key cysteine proteases that are essential for
apoptosis in eukaryotic cells (15), were downregulated by SP600125 in a
dose-dependent manner. Caspase-3 activation has been detected in
several TCS-treated tumor cell lines, including HL-60, K562, HeLa
and choriocarcinoma cells (30).
Caspase-9 is the primary executor of the mitochondrial apoptoti
pathway. Inhibition of caspase-9 with z-LEHD-FMK inhibits the
cellular apoptotic processes induced by TCS, suggesting that the
caspase-9-mediated mitochondrial pathway is involved in TCS-induced
apoptosis (2). As the cytoplasmic
injection of cytochrome c rescues the apoptotic defects of
JNK-deficient fibroblasts, the pro-apoptotic functions of JNK may
be mediated through the mitochondrial pathway (8). SP600125-mediated inhibition of the
TCS-induced JNK pathway activation may reduce the ability of TCS to
inhibit cell viability and induce apoptosis in the HEp-2 and
AMC-HN-8 cells. The inhibition of cell viability and the apoptotic
effect of TCS appear to be associated with phospho-JNK
activation.
The present study demonstrated that low
concentrations of TCS, compared with the higher concentrations of
TCS used in previous studies (10,23,31)
assessing antitumor activity suppressed human laryngeal epidermoid
carcinoma cell viability and induced apoptosis via the induction of
JNK pathway activation. The TCS-induced S-phase cell cycle arrest
was partially attributed to the ability of TCS to inhibit cell
viability. Therefore, TCS treatment offers a potential novel
chemotherapy regimen for laryngocarcinoma, which can be used with
other cytotoxic drugs in combination chemotherapy regimens.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (nos. 30972691 and 30801283), the
Technology Project of Shanghai (nos. 11JC1410802, 09QA1401000 and
10QA1405900) and Shanghai’s Health System of Talents Training Plan
(nos. XYQ2011055 and XYQ2011015).
Abbreviations:
|
TCS
|
trichosanthin
|
|
MAPK
|
mitogen-activated protein kinase
|
|
JNK
|
c-Jun N-terminal protein kinase
|
|
LDH
|
lactate dehydrogenase
|
References
|
1
|
Shaw PC, Chan WL, Yeung HW and Ng TB:
Minireview: trichosanthin - a protein with multiple pharmacological
properties. Life Sci. 55:253–262. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li M, Li X and Li JC: Possible mechanisms
of trichosanthin-induced apoptosis of tumor cells. Anat Rec
(Hoboken). 293:986–992. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tsao SW, Ng TB and Yeung HW: Toxicities of
trichosanthin and alpha-momorcharin, abortifacient proteins from
Chinese medicinal plants, on cultured tumor cell lines. Toxicon.
28:1183–1192. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fang EF, Ng TB, Shaw PC and Wong RN:
Recent progress in medicinal investigations on trichosanthin and
other ribosome inactivating proteins from the plant genus
Trichosanthes. Curr Med Chem. 18:4410–4417. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
He D, Yau K, He X, et al: Conversion of
trichosanthin-induced CD95 (Fas) type I into type II apoptotic
signaling during Herpes simplex virus infection. Mol Immunol.
48:2000–2008. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
He D, Zheng Y and Tam S: The anti-herpetic
activity of trichosanthin via the nuclear factor-kappaB and p53
pathways. Life Sci. 90:673–681. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sha O, Yew DT, Ng TB, Yuan L and Kwong WH:
Different in vitro toxicities of structurally similar type I
ribosome-inactivating proteins (RIPs). Toxicol In Vitro.
24:1176–1182. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang P, Xu J and Zhang C: CREB, a possible
upstream regulator of Bcl-2 in trichosanthin-induced HeLa cell
apoptosis. Mol Biol Rep. 37:1891–1896. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang P and Li JC: Trichosanthin-induced
specific changes of cytoskeleton configuration were associated with
the decreased expression level of actin and tubulin genes in
apoptotic Hela cells. Life Sci. 81:1130–1140. 2007. View Article : Google Scholar
|
|
11
|
Liu J and Lin A: Role of JNK activation in
apoptosis: a double-edged sword. Cell Res. 15:36–42. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ouyang DY, Chan H, Wang YY, et al: An
inhibitor of c-Jun N-terminal kinases (CEP-11004) counteracts the
anti-HIV-1 action of trichosanthin. Biochem Biophys Res Commun.
339:25–29. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang H, Chan H, Wang YY, et al:
Trichosanthin suppresses the elevation of p38 MAPK, and Bcl-2
induced by HSV-1 infection in Vero cells. Life Sci. 79:1287–1292.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang P, Chen LL, Yan H and Li JC:
Trichosanthin suppresses HeLa cell proliferation through inhibition
of the PKC/MAPK signaling pathway. Cell Biol Toxicol. 25:479–488.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kanduc D, Mittelman A, Serpico R, et al:
Cell death: apoptosis versus necrosis (review). Int J Oncol.
21:165–170. 2002.PubMed/NCBI
|
|
16
|
Gao LL, Feng L, Yao ST, et al: Molecular
mechanisms of celery seed extract induced apoptosis via s phase
cell cycle arrest in the BGC-823 human stomach cancer cell line.
Asian Pac J Cancer Prev. 12:2601–2606. 2011.PubMed/NCBI
|
|
17
|
Chirgwin JM, Przybyla AE, MacDonald RJ and
Rutter WJ: Isolation of biologically active ribonucleic acid from
sources enriched in ribonuclease. Biochemistry. 18:5294–5299.
1979.PubMed/NCBI
|
|
18
|
Perez IC, Le Guiner C, Ni W, Lyles J,
Moullier P and Snyder RO: PCR-based detection of gene transfer
vectors: application to gene doping surveillance. Anal Bioanal
Chem. 405:9641–9653. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Korzeniewski C and Callewaert DM: An
enzyme-release assay for natural cytotoxicity. J Immunol Methods.
64:313–320. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Decker T and Lohmann-Matthes ML: A quick
and simple method for the quantitation of lactate dehydrogenase
release in measurements of cellular cytotoxicity and tumor necrosis
factor (TNF) activity. J Immunol Methods. 115:61–69. 1988.
View Article : Google Scholar
|
|
21
|
Lu Z and Hunter T: Ubiquitylation and
proteasomal degradation of the p21(Cip1), p27(Kip1) and p57(Kip2)
CDK inhibitors. Cell Cycle. 9:2342–2352. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bennett BL, Sasaki DT, Murray BW, et al:
SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase.
Proc Natl Acad Sci USA. 98:13681–13686. 2001.PubMed/NCBI
|
|
23
|
Xu J, Gao DF, Yan GL and Fan JM: Induced
apoptotic action of recombinant trichosanthin in human stomach
adenocarcinoma MCG803 cells. Mol Biol Rep. 36:1559–1564. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lefebvre JL: Candidates for larynx
preservation: the next step? Oncologist. 15(Suppl 3): 30–32. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen T, Stephens PA, Middleton FK and
Curtin NJ: Targeting the S and G2 checkpoint to treat cancer. Drug
Discov Today. 17:194–202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Miotto B and Struhl K: JNK1
phosphorylation of Cdt1 inhibits recruitment of HBO1 histone
acetylase and blocks replication licensing in response to stress.
Mol Cell. 44:62–71. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li J, Xia X, Nie H, Smith MA and Zhu X:
PKC inhibition is involved in trichosanthin-induced apoptosis in
human chronic myeloid leukemia cell line K562. Biochim Biophys
Acta. 1770:63–70. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li J, Xia X, Ke Y, et al: Trichosanthin
induced apoptosis in HL-60 cells via mitochondrial and endoplasmic
reticulum stress signaling pathways. Biochim Biophys Acta.
1770:1169–1180. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang P, Yan H and Li JC: CREB-mediated
Bcl-2 expression in trichosanthin-induced Hela cell apoptosis.
Biochem Biophys Res Commun. 363:101–105. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang C, Gong Y, Ma H, et al: Reactive
oxygen species involved in trichosanthin-induced apoptosis of human
choriocarcinoma cells. Biochem J. 355:653–661. 2001.PubMed/NCBI
|
|
31
|
Li M, Chen F, Liu CP, et al: Dexamethasone
enhances trichosanthin-induced apoptosis in the HepG2 hepatoma cell
line. Life Sci. 86:10–16. 2010. View Article : Google Scholar : PubMed/NCBI
|