Introduction
Cervical cancer is the most common gynecological
malignancy, and is the second leading cause of cancer-associated
mortality in females worldwide (1,2). One
reason for the high levels of prevalence of this cancer is the lack
of awareness and early detection approaches (3,4).
Therefore, understanding the underlying molecular mechanisms of
cervical cancer, and establishing more effective therapies are
important areas of ongoing research.
The identification and characterization of key
microRNAs (miRNAs) that participate in cervical cancer, is
essential for determining the underlying mechanisms of this disease
and establishing novel therapeutic strategies. miRNAs are 20–24 nt
RNAs that are derived from distinct hairpin precursors in animals,
plants and fungi, which bind to complementary sequences on target
mRNAs (5,6). miRNAs regulate gene expression by
cleaving target mRNAs, and by translational suppression at the
post-transcriptional level (7).
Previous studies have shown that miRNAs have important roles in
various biological and metabolic processes, including cell growth,
apoptosis, viral infection, differentiation, signal transduction
and cancer (8–11). Numerous studies have demonstrated
that miRNAs are involved in the initiation and progression of
cancer, and may be potential biomarkers for the diagnosis and
prognosis of tumors, in addition to functioning as potential
therapeutic targets (12–14). Therefore, it may be beneficial to
identify novel miRNAs to act as diagnostic and therapeutic
biomarkers, or therapeutic targets, in cervical cancer.
Recently, molecular network analysis technology,
combined with gene expression profile data, has exhibited potential
in a number of areas, including classification of diseases and the
identification of novel therapeutic targets (15,16).
In the present study a microarray dataset of healthy and malignant
cervical samples was downloaded from the Gene Expression Omnibus
(GEO) database. Differentially expressed genes (DEGs) were
identified between these groups. Based on the TarBase v5.0
database, regulatory networks were constructed from selected miRNAs
and their corresponding target genes from the identified DEGs. Key
miRNAs, which may be used as potential biomarkers or therapeutic
targets in cervical cancer, were subsequently identified.
Materials and methods
Affymetrix microarray data
A gene expression profile generated by Zhai et
al (17) was used in the
present study, which was deposited in the GEO database (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE7803).
This gene expression profile is based on the GPL96 platform
(Affymetrix Human Genome U133A Array). A total of 38 samples were
available, including 21 invasive squamous cell cervical carcinoma
(SCC) samples, ten normal squamous cervical epithelium (NE) samples
and seven high-grade squamous intraepithelial cervical lesion
(HSIL) samples.
Screening of DEGs
In order to identify the DEGs, the original GSE7803
dataset was converted into an identifiable expression form and was
normalized. Probe sets were mapped to the National Centers of
Biotechnology Information genes (http://www.ncbi.nlm.nih.gov). Probe sets that
corresponded to numerous genes or to no genes were removed from
subsequent analyses. For genes that corresponded with numerous
probe sets and had a plurality of expression values, the expression
values were averaged. Subsequently, the SAMR package (18) in R and a significance analysis of
microarray (SAM) were used to identify the DEGs between the samples
(19). SAM software is a practical
tool used for detecting significantly expressed genes, and for
controlling the proportion of falsely detected genes. In the
present study, genes with a fold-change >1.2 and a false
discovery rate (FDR) <0.05 were selected as DEGs. In addition,
the identified DEGs were divided into two groups: DEGs from the NE
and HSIL samples were considered pre-invasive DEGs, whereas DEGs
from the HSIL and invasive SCC samples were considered invasive
DEGs.
Functional enrichment analysis of
DEGs
The Database for Annotation, Visualization and
Integrated Discovery (DAVID; http://david.abcc.ncifcrf.gov/) is a web-accessible
program that provides a comprehensive set of functional annotation
tools, which may be used by investigators to understand the
underlying biological functions of large lists of genes (20). The present study used DAVID to
perform a Gene Ontology (GO) enrichment analysis of the identified
DEGs. Based on hypergeometric distribution, GO terms were enriched,
and numerous testing corrections were conducted using the
Benjamini-Hochberg method (21).
An FDR<0.05 was set as the cut-off value.
Construction of regulatory networks
TarBase is a database that contains a manually
curated collection of experimentally supported miRNA targets from a
animal, pant and viral species of central scientific interest
(22). TarBase v5.0 is the updated
and extended version of the TarBase database, with >1,300
experimentally supported miRNA-target interactions (MTIs). It
contains 1,094 human MTIs between 285 miRNAs and 1,721 target
genes.
In the present study, human miRNA target gene data
were downloaded from the TarBase v5.0 database (http://diana.cslab.ece.ntua.gr/tarbase/). miRNAs that
interacted with the identified DEGs were then selected.
Subsequently, MTIs regulatory networks were constructed from these
selected miRNAs and their corresponding target genes within the
DEGs. The MTIs regulatory networks were visualized by Cytoscape
(23). In addition, the MTIs
regulatory networks were divided into two groups: The regulatory
network constructed from the selected miRNAs and the pre-invasive
DEGs was termed the pre-invasive regulatory network, whereas the
regulatory network constructed from the selected miRNAs and the
invasive DEGs was termed the invasive regulatory network.
Comparison of the regulatory
networks
In order to determine the differences between the
pre-invasive and invasive stages of cervical cancer, regulatory
networks were constructed and compared. Regulatory networks may be
characterized by topological properties, such as degree (24). Degree is defined as the number of
edges per node, which indicates the number of interacting partners.
The present study used Freeman’s degree centrality to analyze the
degree of the regulatory networks (25). Freeman’s degree centrality consists
of ingoing (in-degree) and outgoing degree (out-degree). In-degree
refers to the number of links a node receives from other nodes,
whereas out-degree refers to the number of links originating from a
particular node.
Results
DEG analysis
The original GSE7803 dataset was downloaded from the
GEO database, and the DEGs were identified using SAM. Genes with a
fold-change >1.2 and an FDR <0.05 were classed as DEGs. A
total of 1,160 pre-invasive and 756 invasive DEGs were identified.
In addition, 2,001 DEGs were identified from the NE and invasive
SCC samples.
GO analysis of DEGs
In order to study the DEGs that contributed to
cervical cancer, a GO enrichment analysis for the pre-invasive and
invasive DEGs was performed using DAVID software. The pre-invasive
DEGs (e.g. PSMB10, POU2AF1, ST6GAL1, CLU, SERPING1 and APOL2) were
predominantly involved in the immune response, such as the acute
inflammatory response (FDR=3.06E-04; Table I). By contrast, the invasive DEGs
(e.g. TTK, AURKA, BRCA2, PSMC3IP, CDK10 and TUBG1) were
predominantly involved in the regulation of the cell cycle, such as
Cell Cycle (FDR=1.25E-19; Table
II).
 | Table IFunctional enrichment results of
differentially expressed genes from NE samples and HSIL
samples. |
Table I
Functional enrichment results of
differentially expressed genes from NE samples and HSIL
samples.
| Category | ID | Description | FDR | Count | Gene |
|---|
| GOTERM_BP_FAT | GO:0002526 | Acute inflammatory
response | 3.06E-04 | 23 | CLU, SERPING1,
APOL2… |
| GOTERM_CC_FAT | GO:0031975 | Envelope | 0.008979 | 69 | HCCS, CYP24A1,
S100A6… |
| GOTERM_CC_FAT | GO:0005783 | Endoplasmic
reticulum | 0.009737 | 96 | TUSC3, VAPB,
LMAN2L… |
| GOTERM_BP_FAT | GO:0006959 | Humoral immune
response | 0.014053 | 18 | PSMB10, POU2AF1,
ST6GAL1… |
| GOTERM_BP_FAT | GO:0006956 | Complement
activation | 0.014251 | 13 | C4A, C3, C4B… |
| GOTERM_BP_FAT | GO:0006259 | DNA metabolic
process | 0.01695 | 59 | XRCC4, CTCF,
PTTG1… |
| GOTERM_BP_FAT | GO:0002541 | Activation of
plasma proteins involved in acute inflammatory response | 0.018628 | 13 | C4A, C3, CFB… |
| GOTERM_CC_FAT | GO:0031967 | Organelle
envelope | 0.015595 | 68 | HCCS, CYP24A1,
S100A6… |
| GOTERM_CC_FAT | GO:0031090 | Organelle
membrane | 0.021051 | 105 | HCCS, CYP24A1,
TUSC3… |
| GOTERM_CC_FAT | GO:0044432 | Endoplasmic
reticulum part | 0.02565 | 44 | ARL6IP1, DERL2,
TUSC3… |
| GOTERM_BP_FAT | GO:0051605 | Protein maturation
by peptide bond cleavage | 0.045871 | 18 | C4A, CFB, C4B… |
| GOTERM_BP_FAT | GO:0016064 | Immunoglobulin
mediated immune response | 0.046239 | 14 | XRCC4, C4A,
MSH2… |
| GOTERM_BP_FAT | GO:0019724 | B cell mediated
immunity | 0.070007 | 14 | XRCC4, C4A,
MSH2… |
| GOTERM_CC_FAT | GO:0005740 | Mitochondrial
envelope | 0.066884 | 49 | HCCS, CYP24A1,
COX5A… |
| Table IIFunctional enrichment results of
differentially expressed genes from invasive SCC of the cervix
samples and HSIL samples. |
Table II
Functional enrichment results of
differentially expressed genes from invasive SCC of the cervix
samples and HSIL samples.
| Category | ID | Description | FDR | Count | Gene |
|---|
| GOTERM_CC_FAT | GO: 0031981 | Nuclear lumen | 7.07E-20 | 131 | PNMA3, STK38,
PKMYT1… |
| GOTERM_BP_FAT | GO: 0007049 | Cell cycle | 1.25E-19 | 98 | TTK, AURKA,
BRCA2… |
| GOTERM_CC_FAT | GO: 0070013 | Intracellular
organelle lumen | 1.06E-17 | 145 | CDKN2A, OIP5,
SRRM2… |
| GOTERM_CC_FAT | GO: 0043233 | Organelle
lumen | 1.20E-17 | 147 | SRRM2, SMARCD1,
SRRM1… |
| GOTERM_CC_FAT | GO: 0031974 | Membrane-enclosed
lumen | 2.83E-17 | 148 | TFDP2, POLQ,
WDHD1… |
| GOTERM_BP_FAT | GO: 0022403 | Cell cycle
phase | 4.80E-16 | 64 | PSMC3IP, CDK10,
TUBG1… |
| GOTERM_CC_FAT | GO: 0005654 | Nucleoplasm | 2.79E-15 | 90 | MCM3, UBN1,
WEE1… |
| GOTERM_BP_FAT | GO: 0000279 | M phase | 6.61E-15 | 55 | CDCA3, STAG1,
CDC6… |
| GOTERM_BP_FAT | GO: 0022402 | Cell cycle
process | 7.36E-15 | 74 | INHBA, REC8,
PA2G4… |
| GOTERM_CC_FAT | GO: 0005694 | Chromosome | 4.77E-13 | 59 | BLM, HIST1H2AG,
NEK2… |
| GOTERM_CC_FAT | GO: 0043228 |
Non-membrane-bounded organelle | 7.88E-13 | 173 | KNTC1, TTK,
AURKA… |
| GOTERM_CC_FAT | GO: 0043232 | Intracellular
non-membrane-bounded organelle | 7.88E-13 | 173 | RAD9A, ACTN3,
MYH9… |
| GOTERM_BP_FAT | GO: 0000278 | Mitotic cell
cycle | 6.72E-12 | 54 | NCAPH, NCAPG,
ZWILCH… |
| GOTERM_BP_FAT | GO: 0051301 | Cell division | 8.94E-11 | 46 | ESPL1, MYH9,
CDC25C… |
| GOTERM_BP_FAT | GO: 0048285 | Organelle
fission | 1.25E-10 | 40 | RAD21, NCAPG,
OIP5… |
Construction of regulatory networks
Based on human MTIs data, pre-invasive and invasive
regulatory networks were constructed. The pre-invasive regulatory
network consisted of 80 pairs of regulatory interactions between 18
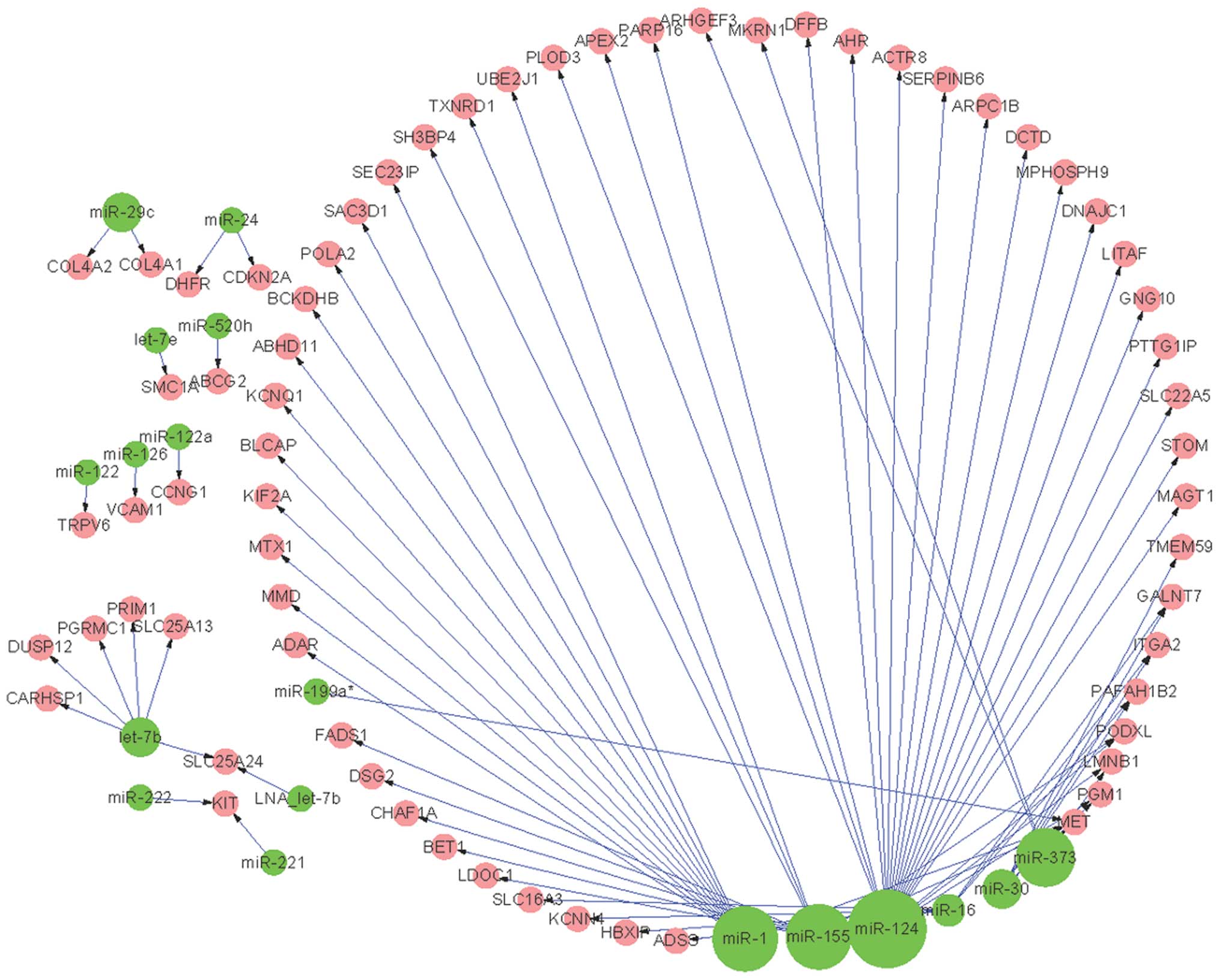
miRNAs and 66 pre-invasive DEGs (Fig.
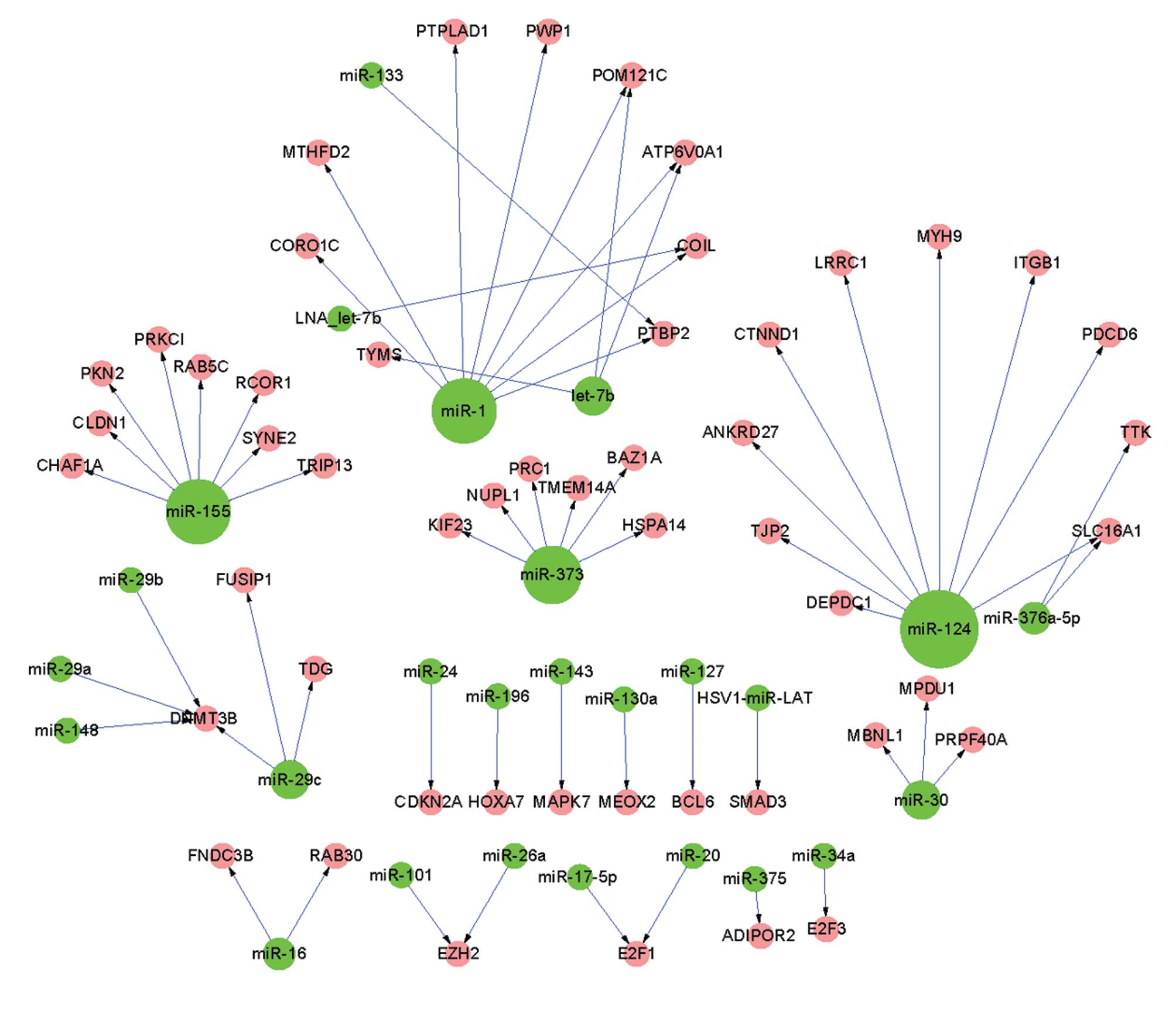
1). The invasive regulatory network consisted of 64 pairs of
regulatory interactions between 26 miRNAs and 51 invasive DEGs
(Fig. 2). The highest out-degree
was observed in miR-124, in the pre-invasive as well as the
invasive regulatory networks.
Comparisons between the regulatory
networks
Based on the topological properties of the networks,
the similarities and differences between the pre-invasive and
invasive regulatory networks were identified. The invasive
regulatory network (Fig. 2)
consisted of many smaller sub-networks and the out-degree of miRNAs
was decreased, compared with those in the pre-invasive regulatory
network (Fig. 1). For example,
there were 14 DEGs associated with miR-1, and 21 DEGs associated
with miR-124 in the pre-invasive regulatory network (Fig. 1). However, only eight and nine DEGs
were associated with miR-1 and miR-124 in the invasive regulatory
network, respectively (Fig.
2).
A total of 10 common miRNAs were identified in the
regulatory networks. Three miRNAs: miR-1, miR-124 and miR-16, had a
degree change >5. In addition, there were eight miRNAs that were
only detected in the pre-invasive regulatory network (Fig. 1), including miR-126 and miR-199a.
By contrast, there were 16 miRNAs that were only detected in the
invasive regulatory network (Fig.
2), including miR-127, miR-143, miR-17-5p, miR-26a, miR-29a,
miR-34a and miR-375.
Discussion
Malignant transformation during tumor progression
results from a series of genetic alterations (26). In order to gain a better
understanding of the genetic changes that occur during the
progression of cervical cancer, a gene expression profile (GSE7803)
was analyzed using a bioinformatics approach. In the present study,
a total of 756 invasive DEGs, 1,160 pre-invasive DEGs, and 2,001
DEGs from invasive SCC and NE samples, were identified. These
findings are in accordance with those of previous studies, which
have consistently shown that the expression of genes is markedly
altered in invasive tumor cells, compared with that of noninvasive
and normal cells (27,28). Furthermore, the results of a GO
enrichment of the identified DEGs, indicated that the expression of
key genes differs between the pre-invasive and invasive stages of
cervical cancer.
Clusterin (CLU) was initially identified as a
secreted glycoprotein that has a cytoprotective role. However,
numerous intracellular CLU variants have recently been identified
in diverse pathological conditions (29–31).
Furthermore, recent studies have shown that CLU is involved in
various biological functions, such as cell death, tumor progression
and neuro-degenerative disorders (32,33).
A previous study used DNA microarray data to identify novel
candidate molecular markers for cervical cancer diagnosis and
therapy, and observed the downregulation of human C1 inhibitor
(SERPING1) in invasive cervical carcinoma cells (34). In addition, a recent genomic study
demonstrated that apolipoprotein L2 (APOL2) is markedly upregulated
in cervical cancer (35). These
findings, as well as the results of the present study, indicate
that CLU, SERPING1 and APOL2 may have important roles in the
progression of cervical cancer.
TTK has been shown to be associated with metastasis
via chromosomal instability, in a previous study, which aimed to
identify genes associated with the progression and metastasis of
advanced cervical cancer following radiotherapy (36). Furthermore, genetic variants of
Aurora A kinase (AURKA) have been shown to be associated with a
radiotherapy-induced early adverse reaction in patients with
cervical cancer (37). Previous
studies have demonstrated that both BRCA1 and BRCA2 participate in
a common DNA damage response pathway, and are involved in the
activation of homologous recombination and double-strand break
repair (38). By contrast, Narayan
et al (39) reported the
downregulation of BRCA1 in a small subset of patients with cervical
cancer. These previous findings and the results of the present
analysis suggest that TTK, AURKA and BRCA2 may participate in the
progression of cervical cancer.
In order to obtain the upstream regulatory
information of the DEGs, two regulatory networks were constructed
based on the TarBase v5.0 database. These regulatory networks were
then compared, and the common and specific miRNAs were identified.
The miRNA with the highest out-degree was shown to be miR-124, in
the pre-invasive as well as the invasive regulatory networks.
miR-124 has previously been shown to be the most abundant miRNA
expressed in neuronal cells (40).
Furthermore, previous studies have shown that the upregulation of
miR-124 induces neuronal differentiation of various tumor cell
lines in mice (41-43). Wilting et al (44) previously demonstrated that the
silencing of miR-124 expression, by methylation, inhibited the
development of cervical carcinoma. These results suggest that
miR-124 may be a potential therapeutic target for cervical cancer
therapy.
Of the eight miRNAs specific to the pre-invasive
regulatory network, miR-126 has previously been reported to be
downregulated in cervical cancer tissues (45), and miR-199a has previously been
suggested as a potential therapeutic target for cervical cancer
therapy (46). miR-126 is a human
miRNA that is expressed only in endothelial cells, throughout
capillaries as well as in larger blood vessels (47), and acts upon various transcripts in
order to control angiogenesis (48). miR-126 has been identified as a
tumor suppressor and as an oncogene, depending on the type of
cancer involved. Inhibition of cancer progression by miR-126 is
achieved through the negative control of proliferation, migration,
invasion and cell survival. However, miR-126 may also support
cancer progression through the promotion of blood vessel formation
and inflammation at the site of activation (49). According to these previous findings
and the results of the present study, miR-126 may be a potential
biomarker for the diagnosis of cervical cancer, and a therapeutic
target for the pre-invasive stage of this disease.
Of the 16 miRNAs specific to the invasive regulatory
network, seven have been reported in previous studies and described
as being upregulated or downregulated in cervical cancer. These
include miR-127, miR-143, miR-17-5p, miR-26a, miR-29a, miR-34a and
miR-375 (45,50–54).
Lee et al (46)
demonstrated that the expression of miR-127 was significantly
increased in patients with invasive squamous cell carcinoma, which
had metastasized to the lymph nodes. The results of the present
study are in accordance with those of previous studies, which
indicate that miR-127 may be a marker for lymph node metastasis in
invasive cervical cancer. miR-143 is highly conserved in
vertebrates (55) and changes in
miR-143 expression have frequently been implicated in cancer
(56–58). Furthermore, the upregulation of
miR-143 has previously been observed in a hepatocellular carcinoma
model during tumor metastasis, through repression of FNDC38
(59). However, reduced expression
of miR-143 has also been observed in a range of cancer stages,
including at very early stages (60). The results of previous studies and
of the present study indicate that miR-143 may be involved in tumor
progression, and may be a candidate for RNA-targeted treatment of
tumors (61). Wang et al
(52) previously reported that
miR-375 is downregulated in squamous cervical cancer, and inhibits
cell migration and invasion by targeting the transcription factor,
SP1. This finding indicates that deregulation of miR-375 may have
an important role in the malignant transformation of cervical
cancer cells. However, the elucidation of the underlying molecular
mechanisms of miR-17-5p, miR-26a, miR-29a and miR-34a in the
progression of cervical cancer, and the use of other miRNAs
screened in the present study as biomarkers or therapeutic targets
in cervical cancer require further investigation.
In conclusion, a total of 1,160 and 756 DEGs were
identified in the pre-invasive and invasive stages of cervical
cancer, respectively. The GO enrichment analysis demonstrated that
the DEGs were primarily involved in the immune response and
regulation of the cell cycle, in the pre-invasive and invasive
stages, respectively. These findings indicate that the expression
of key genes differs between the pre-invasive and invasive stages
of cervical cancer progression. Based on the analysis of the
regulatory networks, a total of 18 and 26 key miRNAs were screened
in the pre-invasive and invasive stages, respectively. It is
hypothesized that these miRNAs are involved in the malignant
transformation of cervical cancer cells. In addition, these miRNAs
may have a function as novel biomarkers in cervical cancer
diagnosis and detection, and as therapeutic targets in this
disease. Further studies in independent patient cohorts are
required, in order to validate the potential roles of these
miRNAs.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Estimating the world cancer burden: Globocan 2000. Int J Cancer.
94:153–156. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Colomiere M, Ward AC, Riley C, et al:
Cross talk of signals between EGFR and IL-6R through JAK2/STAT3
mediate epithelial-mesenchymal transition in ovarian carcinomas. Br
J Cancer. 100:134–144. 2009. View Article : Google Scholar :
|
|
4
|
Ono K, Tanaka T, Tsunoda T, et al:
Identification by cDNA microarray of genes involved in ovarian
carcinogenesis. Cancer Res. 60:5007–5011. 2000.PubMed/NCBI
|
|
5
|
Rhoades MW, Reinhart BJ, Lim LP, Burge CB,
Bartel B and Bartel DP: Prediction of plant microRNA targets. Cell.
110:513–520. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lu Y, Zhou Y, Qu W, Deng M and Zhang C: A
Lasso regression model for the construction of microRNA-target
regulatory networks. Bioinformatics. 27:2406–2413. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ying SY, Chang DC and Lin SL: The microRNA
(miRNA): Overview of the RNA genes that modulate gene function. Mol
Biotech. 38:257–268. 2008. View Article : Google Scholar
|
|
8
|
Pritchard CC, Kroh E, Wood B, et al: Blood
cell origin of circulating microRNAs: a cautionary note for cancer
biomarker studies. Cancer Prev Res (Phila). 5:492–497. 2012.
View Article : Google Scholar
|
|
9
|
Farazi TA, Hoell JI, Morozov P and Tuschl
T: MicroRNAs in human cancer. Adv Exp Med Biol. 774:1–20.
2013.PubMed/NCBI
|
|
10
|
Balch C, Naegeli K, Nam S, et al: A unique
histone deacetylase inhibitor alters microRNA expression and signal
transduction in chemoresistant ovarian cancer cells. Cancer Biol
Ther. 13:681–693. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Aydoğdu E, Katchy A, Tsouko E, et al:
MicroRNA-regulated gene networks during mammary cell
differentiation are associated with breast cancer. Carcinogenesis.
33:1502–1511. 2012. View Article : Google Scholar
|
|
12
|
Lu J, Getz G, Miska EA, et al: MicroRNA
expression profiles classify human cancers. Nature. 435:834–838.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Garzon R, Fabbri M, Cimmino A, Calin GA
and Croce CM: MicroRNA expression and function in cancer. Trends
Mol Med. 12:580–587. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cho WC: MicroRNAs: potential biomarkers
for cancer diagnosis, prognosis and targets for therapy. Int J
Biochem Cell Biol. 42:1273–1281. 2010. View Article : Google Scholar
|
|
15
|
Wu SF, Qian WY, Zhang JW, et al: Network
motifs in the transcriptional regulation network of cervical
carcinoma cells respond to EGF. Arch Gynecol Obstet. 287:771–777.
2013. View Article : Google Scholar
|
|
16
|
Higareda-Almaraz JC, Enríquez-Gasca Mdel
R, Hernández-Ortiz M, Resendis-Antonio O and Encarnación-Guevara S:
Proteomic patterns of cervical cancer cell lines, a network
perspective. BMC Sys Biol. 5:962011. View Article : Google Scholar
|
|
17
|
Zhai Y, Kuick R, Nan B, et al: Gene
expression analysis of preinvasive and invasive cervical squamous
cell carcinomas identifies HOXC10 as a key mediator of invasion.
Cancer Res. 67:10163–10172. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tusher VG, Tibshirani R and Chu G:
Significance analysis of microarrays applied to the ionizing
radiation response. Proc Natl Acad Sci USA. 98:5116–5121. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Larsson O, Wahlestedt C and Timmons JA:
Considerations when using the significance analysis of microarrays
(SAM) algorithm. BMC Bioinformatics. 6:1292005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dennis G Jr, Sherman BT, Hosack DA, et al:
DAVID: Database for annotation, visualization and integrated
discovery. Genome Biol. 4:P32003. View Article : Google Scholar
|
|
21
|
Benjamini Y and Hochberg Y: Controlling
the false discovery rate: A practical and powerful approach to
multiple testing. J R Stat Soc Series B Stat Methodol. 57:289–300.
1995.
|
|
22
|
Papadopoulos GL, Reczko M, Simossis VA,
Sethupathy P and Hatzigeorgiou AG: The database of experimentally
supported targets: a functional update of TarBase. Nucleic Acids
Res. 37:D155–D158. 2009. View Article : Google Scholar :
|
|
23
|
Shannon P, Markiel A, Ozier O, et al:
Cytoscape: a software environment for integrated models of
biomolecular interaction networks. Genome Res. 13:2498–2504. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang S, Jin G, Zhang XS and Chen L:
Discovering functions and revealing mechanisms at molecular level
from biological networks. Proteomics. 7:2856–2869. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Freeman LC: Centrality in social networks
conceptual clarification. Soc Networks. 1:215–239. 1979. View Article : Google Scholar
|
|
26
|
Fearon ER and Vogelstein B: A genetic
model for colorectal tumorigenesis. Cell. 61:759–767. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ryu B, Jones J, Hollingsworth MA, Hruban
RH and Kern SE: Invasion-specific genes in malignancy: serial
analysis of gene expression comparisons of primary and passaged
cancers. Cancer Res. 61:1833–1838. 2001.PubMed/NCBI
|
|
28
|
Kitahara O, Furukawa Y, Tanaka T, et al:
Alterations of gene expression during colorectal carcinogenesis
revealed by cDNA microarrays after laser-capture microdissection of
tumor tissues and normal epithelia. Cancer Res. 61:3544–3549.
2001.PubMed/NCBI
|
|
29
|
Reddy KB, Jin G, Karode MC, Harmony JA and
Howe PH: Transforming growth factor beta (TGF beta)-induced nuclear
localization of apolipoprotein J/clusterin in epithelial cells.
Biochemistry. 35:6157–6163. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang CR, Leskov K, Hosley-Eberlein K, et
al: Nuclear clusterin/XIP8, an x-ray-induced Ku70-binding protein
that signals cell death. Proc Natl Acad Sci USA. 97:5907–5912.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
O’Sullivan J, Whyte L, Drake J and
Tenniswood M: Alterations in the post-translational modification
and intracellular trafficking of clusterin in MCF-7 cells during
apoptosis. Cell Death Differ. 10:914–927. 2003. View Article : Google Scholar
|
|
32
|
Klokov D, Leskov K, Araki S, et al: Low
dose IR-induced IGF-1-sCLU expression: a p53-repressed expression
cascade that interferes with TGFbeta1 signaling to confer a
pro-survival bystander effect. Oncogene. 32:479–490. 2013.
View Article : Google Scholar
|
|
33
|
Choi I, Kim J, Park JY and Kang SW:
Cotransin induces accumulation of a cytotoxic clusterin variant
that cotranslationally rerouted to the cytosol. Exp Cell Res.
319:1073–1082. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Santin AD, Zhan F, Bignotti E, et al: Gene
expression profiles of primary HPV16- and HPV18-infected early
stage cervical cancers and normal cervical epithelium:
identification of novel candidate molecular markers for cervical
cancer diagnosis and therapy. Virology. 331:269–291. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ahn WS, Bae SM, Lee JM, et al: Searching
for pathogenic gene functions to cervical cancer. Gynecol Oncol.
93:41–48. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Harima Y, Ikeda K, Utsunomiya K, et al:
Identification of genes associated with progression and metastasis
of advanced cervical cancers after radiotherapy by cDNA microarray
analysis. Int J Radiat Oncol. 75:1232–1239. 2009. View Article : Google Scholar
|
|
37
|
Ishikawa A, Suga T, Shoji Y, et al:
Genetic Variants of NPAT-ATM and AURKA are associated with an early
adverse reaction in the gastrointestinal tract of patients with
cervical cancer treated with pelvic radiation therapy. Int J Radiat
Oncol. 81:1144–1152. 2011. View Article : Google Scholar
|
|
38
|
Chen JJ, Silver D, Cantor S, Livingston DM
and Scully R: BRCA1, BRCA2 and Rad51 operate in a common DNA damage
response pathway. Cancer Res. 59(Suppl 7): 1752–1756. 1999.
|
|
39
|
Narayan G, Arias-Pulido H, Nandula SV, et
al: Promoter hyper-methylation of FANCF disruption of Fanconi
Anemia-BRCA pathway in cervical cancer. Cancer Res. 64:2994–2997.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lagos-Quintana M, Rauhut R, Meyer J,
Borkhardt A and Tuschl T: New microRNAs from mouse and human. RNA.
9:175–179. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Makeyev EV, Zhang J, Carrasco MA and
Maniatis T: The MicroRNA miR-124 promotes neuronal differentiation
by triggering brain-specific alternative pre-mRNA splicing. Mol
Cell. 27:435–448. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Smirnova L, Gräfe A, Seiler A, Schumacher
S, Nitsch R and Wulczyn FG: Regulation of miRNA expression during
neural cell specification. Eur J Neurosci. 21:1469–1477. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Krichevsky AM, King KS, Donahue CP,
Khrapko K and Kosik KS: A microRNA array reveals extensive
regulation of microRNAs during brain development. RNA. 9:1274–1281.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wilting SM, van Boerdonk RA, Henken FE, et
al: Methylation-mediated silencing and tumour suppressive function
of hsa-miR-124 in cervical cancer. Mol Cancer. 9:1672010.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang X, Tang S, Le SY, et al: Aberrant
expression of oncogenic and tumor-suppressive microRNAs in cervical
cancer is required for cancer cell growth. PloS one. 3:e25572008.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lee JW, Choi CH, Choi JJ, et al: Altered
MicroRNA expression in cervical carcinomas. Clin Cancer Res.
14:2535–2542. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
van Solingen C, Seghers L, Bijkerk R, et
al: Antagomir-mediated silencing of endothelial cell specific
microRNA-126 impairs ischemia-induced angiogenesis. J Cell Mol Med.
13:1577–1585. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang S, Aurora AB, Johnson BA, et al: The
endothelial-specific microRNA miR-126 governs vascular integrity
and angiogenesis. Dev Cell. 15:261–271. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Meister J and Schmidt MH: miR-126 and
miR-126*: new players in cancer. Scientific World Journal.
10:2090–2100. 2010. View Article : Google Scholar
|
|
50
|
Sahasrabuddhe VV, Luhn P and Wentzensen N:
Human papillomavirus and cervical cancer: biomarkers for improved
prevention efforts. Future Microbiol. 6:1083–1098. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Liu L, Yu X, Guo X, et al: miR-143 is
downregulated in cervical cancer and promotes apoptosis and
inhibits tumor formation by targeting Bcl-2. Mol Med Rep.
5:753–760. 2012.
|
|
52
|
Wang F, Li Y, Zhou J, et al: miR-375 is
down-regulated in squamous cervical cancer and inhibits cell
migration and invasion via targeting transcription factor SP1. Am J
Pathol. 179:2580–2588. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wei Q, Li YX, Liu M, Li X and Tang H:
MiR-17-5p targets TP53INP1 and regulates cell proliferation and
apoptosis of cervical cancer cells. IUBMB life. 64:697–704. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Pang RT, Leung CO, Ye TM, et al:
MicroRNA-34a suppresses invasion through downregulation of Notch1
and Jagged1 in cervical carcinoma and choriocarcinoma cells.
Carcinogenesis. 31:1037–1044. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Trakooljul N, Hicks JA and Liu HC:
Identification of target genes and pathways associated with chicken
microRNA miR-143. Anim Genet. 41:357–364. 2010.PubMed/NCBI
|
|
56
|
Gao W, Yu Y, Cao H, Shen H, Li X, Pan S
and Shu Y: Deregulated expression of miR-21, miR-143 and miR-181a
in non small cell lung cancer is related to clinicopathologic
characteristics or patient prognosis. Biomed Pharmacother.
64:399–408. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ahmad I, Singh LB, Yang ZH, et al: Mir143
expression inversely correlates with nuclear ERK5 immunoreactivity
in clinical prostate cancer. Br J Cancer. 108:149–154. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Deftereos G, Corrie SR, Feng Q, et al:
Expression of mir-21 and mir-143 in cervical specimens ranging from
histologically normal through to invasive cervical cancer. PLoS
One. 6:e284232011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhang H, Cai X, Wang Y, Tang H, Tong D and
Ji F: microRNA-143, down-regulated in osteosarcoma, promotes
apoptosis and suppresses tumorigenicity by targeting Bcl-2. Oncol
Rep. 24:1363–1369. 2010.PubMed/NCBI
|
|
60
|
Slaby O, Svoboda M, Fabian P, et al:
Altered expression of miR-21, miR-31, miR-143 and miR-145 is
related to clinicopathologic features of colorectal cancer.
Oncology. 72:397–402. 2007. View Article : Google Scholar
|
|
61
|
Kitade Y and Akao Y: MicroRNAs and their
therapeutic potential for human diseases: microRNAs, miR-143
and-145, function as anti-oncomirs and the application of
chemically modified miR-143 as an anti-cancer drug. J Pharmacol
Sci. 114:276–280. 2010. View Article : Google Scholar
|