Introduction
Nasopharyngeal carcinoma (NPC) is a type of
malignant neoplasm, which is prevalent in Southeast Asia and
Southern China, with incidence rates of 20–30 per 100,000 males and
15–20 per 100,000 females. In addition, NPC accounts for ~50,000
mortalities annually worldwide and ~80,000 novel cases are
diagnosed each year (1,2). NPC is a radiosensitive neoplasm, for
which radiotherapy (RT) is the primary treatment option. RT
treatment of early stage NPC is effective in 85% of patients;
however, for loco-regional advanced NPC, current treatments are
ineffective (3). Radioresistance
remains to be an obstacle for the successful treatment of NPC in
numerous cases (4,5) and accounts for the majority of
treatment failures, resulting in relapse and metastasis in NPC
patients following RT (6).
Autophagy involves the degradation of long-lived
proteins and cytoplasmic organelles, the products of which are
recycled for the production of macromolecules and adenosine
triphosphate (ATP) in order to maintain cellular homeostasis
(7). Therefore, autophagy is an
important survival mechanism in response to several types of
stresses, including nutrient starvation, hypoxia, overcrowding,
high temperatures as well as the accumulation of damaged or
expendable organelles and cytoplasmic components (7,8).
Microtubule-associated protein 1 light chain 3 (LC3)-II is a marker
of autophagy (9); increased LC3-II
levels indicate that the autophagy has been initiated. During
autophagic degradation, LC3-II is converted back to LC3-I through
protease cleavage (10). Previous
studies have demonstrated that autophagy is initiated in various
types of cancer cells in response to anticancer therapies (11–17).
Autophagy was reported to be induced by ionizing radiation (IR) in
certain types of cancer cells, including malignant glioma cells
(11–13). In addition, genetic knockout
studies of autophagy-associated genes were demonstrated to enhance
the development of spontaneous malignancies, whereas mice deficient
in autophagy-associated genes showed increased sensitivity to
radiotherapy (14–17). Therefore, elucidating the
mechanisms involved in autophagy may provide effective strategies
for improving the radiation sensitization of NPC.
Poly [adenosine diphosphate (ADP)-ribose]
polymerase-1 (PARP-1) is a nuclear enzyme which binds DNA by two
zinc finger motifs and transfers chains of ADP-ribosyl moieties
(PARs) from nicotinamide-adenine-dinucleotide (NAD+) to
chromatin-associated acceptor proteins (18). This post-translational modification
has an important role in facilitating DNA repair through releasing
PARP-1 from DNA and allowing for the recruitment of proteins
involved in both base excisional repair and homologous
recombination (18,19). Irradiation is known to induce DNA
damage and activate PARP-1 (20).
PARP-1 and DNA-dependent protein kinase-deficient cell lines were
reported to be 4-fold more sensitive to IR; in addition, these
cells demonstrated reduced potentially lethal damage recovery
(PLDR) in G0 cells compared with that of their
proficient counterparts (21). A
previous study demonstrated that PARP-1 mediated IR-induced
autophagy and PARP-1 inhibition resulted in the radiation
sensitization of CNE-2 cells (22). In addition, the liver kinase B1
(LKB1)/adenosine monophosphate (AMP)-activated protein kinase
(AMPK)/mammalian target of rapamycin (mTOR) signaling pathway has
been reported to link cellular metabolism and energy status to the
signal transduction pathways involved in cell growth, proliferation
and autophagy (23,24). Mouse embryonic fibroblasts (MEFs)
with Bax/Bak double-knockout
(Bax−/−Bak−/−) is a well-established model for
studying necrotic cell death; these cells were used to determine a
novel regulatory function of PARP-1 in autophagy via the activation
of serine/threonine protein kinase LKB1 and AMPK as well as the
subsequent suppression of mTOR. These results indicated that
autophagy served as a cell survival mechanism to counteract
reactive oxygen species-mediated necrosis (25).
The LKB1/AMPK/mTOR signaling pathway was extensively
studied in metabolic disorders and evidence has suggested its
implication in cancer cell biology (23,24).
In addition, PARP-1 was reported to be a crucial for the repair of
radiation-induced single-strand DNA breaks (26). PARP inhibitors have demonstrated
promising results for use in cancer therapy; however, certain
limitations have arisen which require further investigation. The
chemical structures of PARP inhibitors are highly varied; however,
they have short half-lives and so require frequent administration,
which contributes to poor patient compliance (27). In addition, long-term inhibition of
PARP activity may result in novel mutations or other unknown
adverse effects; therefore, the long-term effects of these drugs
require verification (28,29). It was hypothesized that the
regulation of autophagy in CNE-2 cells following IR occurred via
the PARP-1/LKB1/AMPK/mTOR pathway. Thus, the elucidation of this
mechanism may provide evidence for the use of PARP-1, LKB1 or AMPK
inhibitors and mTOR activators as adjuvant therapies for the
treatment of NPC. The present study aimed to investigate whether
the mechanism of PARP-1-mediated IR-induced autophagy in CNE-2
cells proceeded via activation of the LKB1/AMPK/mTOR signaling
pathway. In addition, the effect of PARP-1 and AMPK inhibition on
the radiation sensitization of CNE-2 cells was investigated.
Pharmacological or genetic regulation of these pathways may be a
potential strategy to enhance radiosensitivity of NPC.
Materials and methods
Cell culture
CNE-2 human nasopharyngeal carcinoma cells were
purchased from the Cancer Hospital of Shanghai Fudan University
(Shanghai, China) (30). Cells
were cultured in RPMI-1640 medium (HyClone Laboratories, Inc.,
Logan, UT, USA) supplemented with 10% fetal bovine serum
(Gibco-BRL, Carlsbad, CA, USA), penicillin (100 U/ml), streptomycin
(100 U/ml) (North China Pharmaceutical Group Corp., Shijiazhuang,
China) and were maintained in a humidified 5% CO2
atmosphere at 37°C.
Reagents
5-amino-1-β-D-ribofuranosyl-1H-imidazole-4-car
boxamide (AICAR), an activator of AMPK, was purchased from Cayman
Chemical (Ann Arbor, MI, USA). 6-[4-(2-Piperidin-1
-yl-ethoxy)-phenyl)]-3-pyridin-4-yl-pyrrazolo [1,5-a]-pyrimidine
(Compound C), an inhibitor of AMPK, was purchased from Merck
Millipore Calbiochem (Darmstadt, Germany). Rabbit polyclonal
anti-PARP-1 (1:1,000; cat. no. 9542S), rabbit monoclonal
anti-phospho-LKB1-Ser428 (p-LKB1; 1:1,000; cat. no. 3482S), rabbit
monoclonal anti-phospho-AMPK-Thr172 (p-AMPK; 1:2,000; cat. no.
4188S) and rabbit polyclonal anti-phospho-P70S6K-T421/S424
(p-P70S6K; 1:1,000; cat. no. 9204S) primary antibodies were
purchased from Cell Signaling Technology (Danvers, MA, USA). Rabbit
polyclonal anti-LC3-II primary antibody (1:1,000; cat. no. L7543)
was purchased from Sigma-Aldrich (Shanghai, China). Rabbit
polyclonal GAPDH primary antibody (1:5,000; cat. no. 10494-1-AP)
was purchased from Proteintech Group, Inc. (Chicago, IL, USA) and
the fluorescent-labeled goat anti-rabbit immunoglobulin G (IgG)
secondary antibody (1:15,000; cat. no. 7054) was purchased from
Cell Signaling Technology. Lysis buffer, a mixture of
radioimmunoprecipitation assay and phenyl-methylsulfonyl fluoride,
was purchased from Beyotime Institute of Biotechnology (Shanghai,
China). A PhosSTOP tablet (Roche, Basel, Switzerland) was also
dissolved in lysis buffer. 3-(4,
5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
dimethyl sulfoxide (DMSO), 100% methanol and Giemsa solution were
purchased from Solarbio Science and Technology Co., Ltd (Beijing,
China).
Fluorescence microscopy
PARP-1 gene silencing of CNE-2 cells was
established as previously described (31). For the lentiviral infection, CNE-2
cells were cultured in 6-well plates. Subsequently, the
PARP-1-shRNA-expressing lenti-virus (Shanghai Genechem
Biotechnology, Shanghai, China) was added, with a multiplicity of
infection of 20 in the CNE-2 cells for 8 h. The transduction
efficiency was determined using an inverted fluorescence microscope
(IX71; Olympus Corp., Beijing, China).
Irradiation of CNE-2 cells
IR was performed using 6-MV X-rays with a linear
accelerator (Precise 1120, Elekta Instrument AB, Stockholm,
Sweden), at a dose rate of 220 cGy/min (source-to-surface distance,
100 cm).
Western blot analysis
CNE-2 cells were washed with ice-cold
phosphate-buffered saline (PBS) twice and lysed with lysis buffer
at 4°C for 30 min. The lysates were then centrifuged at 4°C for 15
min at a centrifugal acceleration of 18,500 x g. Protein content in
the supernatants was determined using the Bicinchoninic Acid
Protein Assay kit (Beyotime Institute of Biotechnology). In order
to detect PARP-1, equal amounts of protein (50 µg) were
loaded onto a 7% SDS-polyacrylamide gel, for p -LK B1-Ser428, p-A M
PK-T h r172 and p-P70S6K-T421/S424 detection, equal amounts of
protein were loaded onto a 10% SDS-polyacrylamide gel. A 15%
SDS-polyacrylamide gel was used to detect equal amounts of LC3-II
protein. Following electrophoresis, proteins were transferred onto
polyvinylidene fluoride membranes (Merck Millipore, Billerica, MA,
USA). Membranes were then blocked with milk for 1 h and then were
incubated with following primary antibodies at 4°C overnight:
Anti-PARP-1 (1:1,000), anti-p-LKB1-Ser428 (1:1,000),
anti-p-AMPK-T172 (1:2,000), anti-p-P70S6K-T421/S424 (1:1,000),
anti-GAPDH (1:5,000) and anti-LC3-II (1:1,000). Following washing
and incubating with fluorescent-labeled goat anti-rabbit IgG
secondary antibody (1:15,000) at room temperature for 1 h, the
fluorescence intensities of the blots were detected using the
Odyssey Infrared Imaging system version 3.0.X (LI-COR Biosciences,
Lincoln, NE, USA).
MTT assay
Cells were prepared at a concentration of
1×104 cells/ml and seeded onto 96-well plates at 100
µl/well. Following 24 h of adherence, the cells were treated
with Compound C for 24 h, then incubated for 0, 1, 2, 3 or 4 days.
An MTT assay was performed by adding 20 µl MTT (5 mg/ml) to
wells and incubating for 3 h in the dark. Subsequently, the
supernatants were removed. A total of 150 µl DMSO was added
to each well and after 15 min the absorbance value (optical
density, OD) of each well was measured using a microplate reader
(MK3; Thermo Fisher Scientific, Shanghai, China) at 492 nm. The MTT
assay procedure was used to determine the proliferation rate of
PARP-1-silenced CNE-2 cells as well as control IR-treated
and untreated CNE-2 cells. All experiments were performed in
triplicate.
Plate clone formation assay
Cells were seeded at a density of 5×103
cells/well into 6-well plates. Following 24 h of adherence, the
cells were treated with Compound C (10 µM) for 24 h, then
incubated at 37°C for 10 days. Cells were then washed twice with
PBS and fixed in 100% methanol for 30 min, prior to staining with
Giemsa solution for 30 min. The number of colonies containing ≥50
cells were counted under a microscope (IX71). The clone formation
assay was also applied to PARP-1-silenced CNE-2 cells as
well as control IR-treated and untreated CNE-2 cells. All
experiments were performed in triplicate.
Statistical analysis
Values are expressed as the mean ± standard
deviation. Data were analyzed using the one-way analysis of
variance and least significant difference tests with SPSS 16.0
software (SPSS, Inc., Chicago, IL, USA) to determine statistical
significance. P<0.05 was considered to indicate a statistically
significant difference between values.
Results
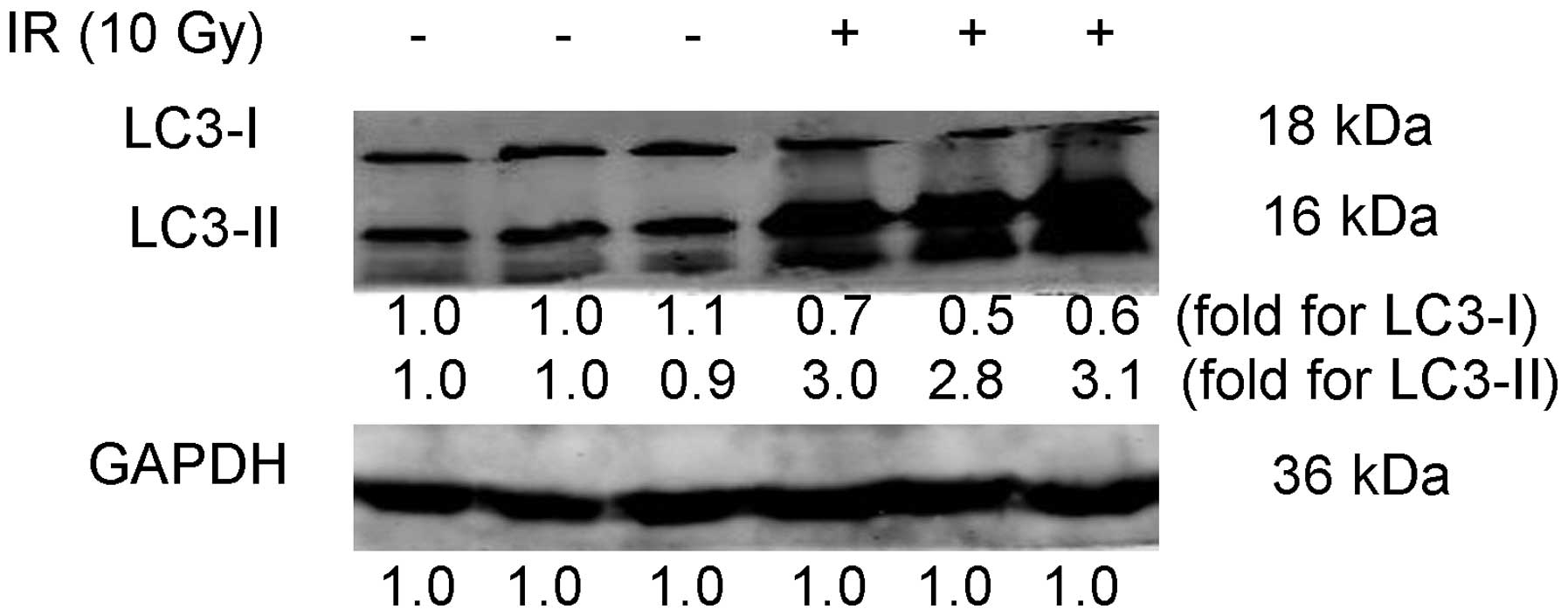
IR induces autophagy in CNE-2 cells
LC3-II is a marker of autophagy, the accumulation of
which suggests the process of autophagy; therefore, the autophagic
response was measured through the conversion of LC3-I to LC3-II
(32). In the present study,
protein expression levels of LC3-I and LC3-II in CNE-2 cells
treated with 10 Gy IR were compared with those in the untreated
control group CNE-2 cells. At 48 h post IR, CNE-2 cells were
collected and subjected to western blot analysis. The relative
density of LC3-I and LC3-II to GAPDH levels were then determined
and the mean fold change of three independent experiments was
calculated. As shown in Fig. 1,
following IR, the relative density of LC3-I (0.6±0.1) was
significantly decreased compared with that of the untreated control
group (1.0±0.2; P<0.05). In addition, the relative density of
LC3-II in cells treated with IR (3.0±0.2) was significantly
increased compared with that of the untreated control group
(1.0±0.1; P<0.05). This implied that LC3-I was converted to
LC3-II following IR, therefore indicating that IR induced autophagy
in CNE-2 cells.
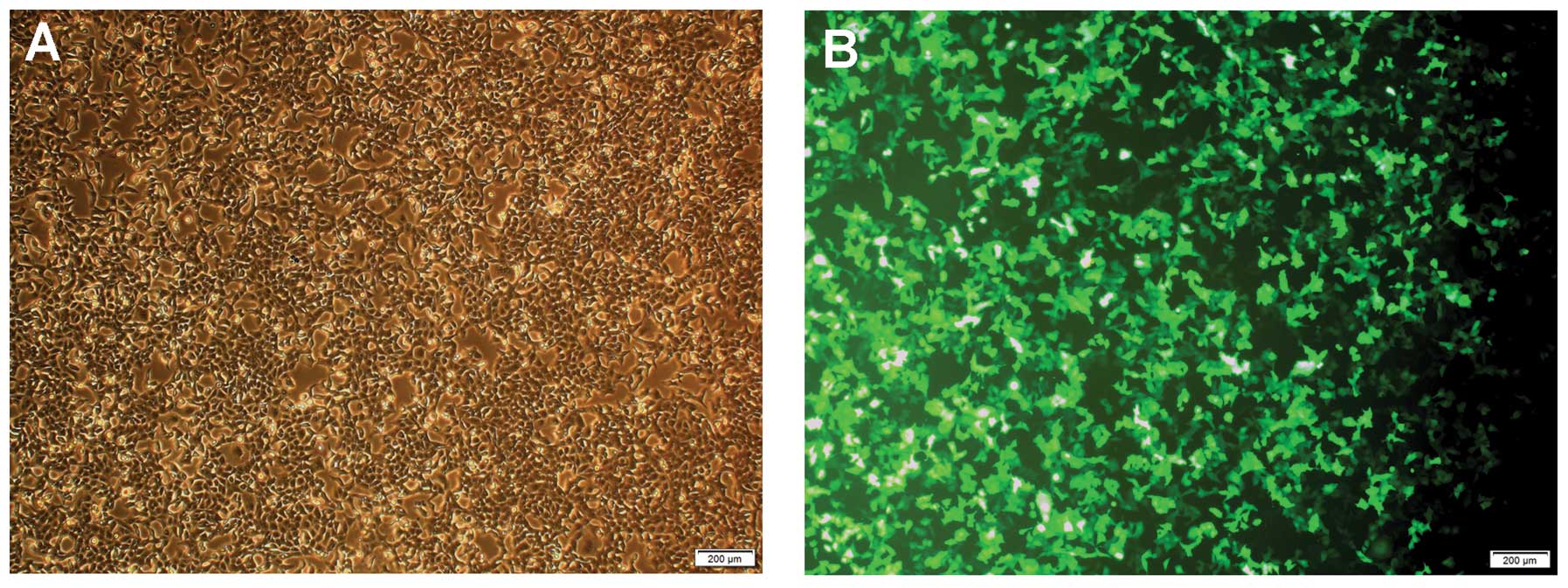
Expression of PARP-1 in CNE-2 cells
CNE-2 cells were transfected with LV-shRNA in order
to silence the PARP-1 gene. The lentivirus contained a gene
encoding green fluorescent protein; therefore, effectively
transfected CNE-2 cells would appear green under an inverted
fluorescence microscopy. As shown in the photomicrographs in
Fig. 2, the majority of CNE-2
cells were effectively transfected with LV-shRNA. The protein
expression levels of PARP-1 were then determined using western blot
analysis. As shown in Fig. 3A, the
relative density of PARP-1 in IR-treated PARP-1-silenced
cells (3.5±0.5) was significantly decreased compared with that of
the IR-only treatment group (11.0±0.5; P<0.05); this therefore
confirmed that LV-shRNA effectively silenced PARP-1 in CNE-2
cells. In addition, the relative density of PARP-1 in IR-treated
cells (11.0±0.5) was markedly increased compared with that of the
untreated CNE-2 cells (1.0±0.4; P<0.05), therefore indicating
that IR promoted the activation of PARP-1. Furthermore, following
IR, CNE-2 cells were treated with an AMPK activator (2.0 mM AICAR)
or inhibitor (10 µM Compound C). The results revealed that
there were no significant differences in the relative density of
PARP-1 expression between the AICAR (10.5±0.3) or Compound C
(11.3±0.4) treatment groups and the IR-only treatment group
(11.0±0.5; P>0.05). This therefore indicated that the activation
or inhibition of AMPK did not effect the expression of PARP-1.
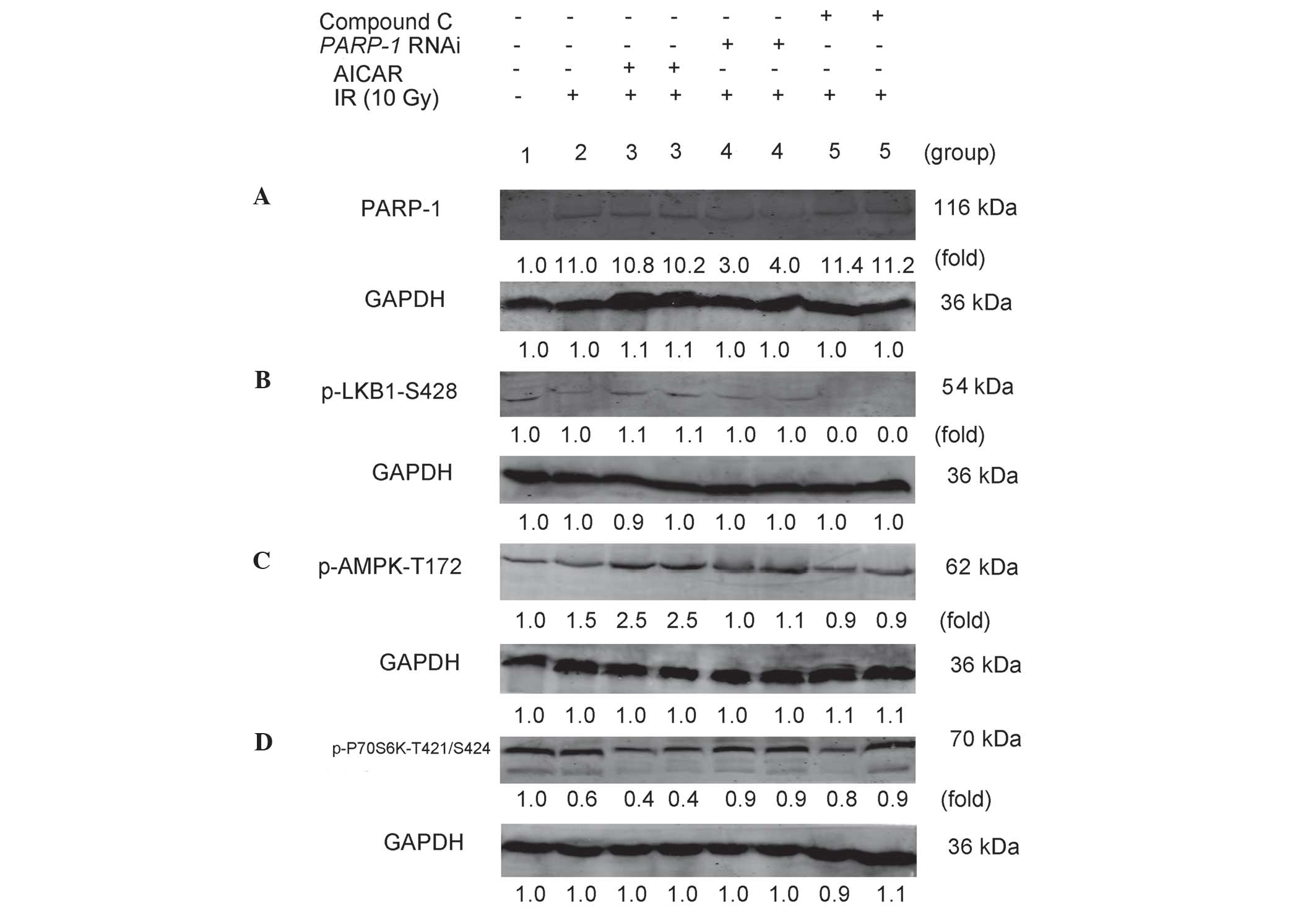 | Figure 3PARP-1 promotes autophagy through the
AMPK/mammalian target of rapamycin pathway in CNE-2 cells following
IR. CNE-2 cells in each group were treated as follows: 1, untreated
control group); 2, 10 Gy IR; 3, pretreatment with AICAR (2.0 mM)
for 2 h + 10 Gy IR; 4, PARP-1 RNAi using
lentivirus-delivered small-hairpin RNA transfection + 10 Gy IR; 5,
pretreatment with Compound C (10 µM) for 2 h + 10 Gy IR. (A)
After 48 h, CNE-2 cells were collected and subjected to western
blot analysis for the detection of PARP-1. After 30 min, CNE-2
cells were collected and subjected to western blot for detection of
(B) p-LKB1-S428, (C) p-AMPK-Thr172 and (D) p-P70S6K-T421/S424.
GAPDH was used as the loading control. Protein expression levels of
PARP-1, p-LKB1-Ser428, p-AMPK-Thr172 and p-P70S6K-T421/S424 were
then quantified and the mean fold change relative to GAPDH for
three independent experiments. PARP-1, poly-(adenosine
diphosphate-ribose) polymerase-1; AMPK, adenosine
monophosphate-activated protein kinase; IR, ionizing radiation;
AICAR, 5-amino-1-β-D-ribofuranosyl-1H-imidazole-4-carboxamide;
RNAi, RNA interference; Compound C,
6-[4-(2-Piperidin-1-yl-ethoxy)-phenyl)]-3-pyridin-4-yl-pyrrazolo
[1,5-a]-pyrimidine; LKB1, liver kinase B1, p-, phosphorylated. |
Expression of p-LKB1-Ser428 in CNE-2
cells
As shown in Fig.
3B, dim bands were observed for p-PKB1-Ser428 in untreated
CNE-2 cells, IR-treated CNE-2 cells in the presence and absence of
AICAR as well as IR-treated PARP-1-silenced cells. These
bands were too dim to detect any changes, which indicated that
p-LKB1-Ser428 was expressed in CNE-2 cells whether they were
treated with IR or not and PARP-1 gene silencing did not
decrease p-LKB1-Ser428. No bands were detected for the IR-treated
cells with Compound C, which indicated that Compound C may block
the expression of p-LKB1-Ser428 in CNE-2 cells.
Expression of p-AMPK-Thr172 in CNE-2
cells
As shown in Fig.
3C, following IR, the relative density of p-AMPK-Thr172 in
CNE-2 cells (1.5±0.1) was increased compared with that of the
untreated cells (1.0±0.1; P<0.05). This indicated that IR
promoted the expression of AMPK in CNE-2 cells. The relative
density of p-AMPK in AICAR-treated cells following IR (2.5±0.2) was
significantly increased compared with that of the IR-only treatment
group (1.5±0.1; P<0.05), confirming that AICAR promoted AMPK
expression in IR-treated cells. Following PARP-1 silencing,
the relative density of IR-treated cells (1.1±0.1) was markedly
decreased compared with that of the IR-only treatment group
(1.5±0.1; P<0.05). This indicated that PARP-1 gene
silencing reduced the expression of AMPK following IR. Furthermore,
p-AMPK-Thr172 expression in Compound C-treated cells following IR
(0.9±0.2) was significantly decreased compared with that of the
IR-only treatment (1.5±0.1; P<0.05).
Expression of p-P70S6K-T421/S424 in CNE-2
cells
mTOR inhibits autophagy predominantly through
activating the downstream molecule p70S6K (33). As shown in Fig. 3D, following IR, the relative
density of p-p70S6K in CNE-2 cells (0.6±0.2) was decreased compared
with that of the untreated cells (1.0±0.1; P<0.05), suggesting
that IR attenuated the expression of p-P70S6K in CNE-2 cells. The
relative density of p-P70S6K in AICAR-treated cells following IR
(0.4±0.1) was significantly decreased compared with that of the
IR-only treatment group (0.6±0.2; P<0.05). This indicated AICAR
reduced mTOR activity in CNE-2 cells following IR. By contrast,
PARP-1 silencing markedly increased the relative density of
p-P70S6K in IR-treated cells (0.9±0.1) compared with that of the
IR-only treatment group (0.6±0.2; P<0.05), which suggested that
PARP-1 gene silencing induced mTOR activity following IR.
The relative density of p-P70S6K in Compound C-treated cells
following IR was significantly increased compared with that of the
IR-only treatment group (0.6±0.2; P<0.05), indicating that AMPK
inhibition enhanced mTOR activity following IR.
Expression of LC3-II in CNE-2 cells
As shown in Fig. 4,
following IR, AICAR treatment markedly increased the expression of
LC3-II (2.0±0.2) compared with that of IR-only-treated CNE-2 cells
(1.0±0.1; P<0.05). This indicated that increased AMPK levels
promoted the expression of LC3-II in CNE-2 cells following IR The
relative density of LC3-II in PARP-I-silenced cells
following IR (0.7±0.1) was significantly decreased compared with
that of IR-only-treated CNE-2 cells (1.0±0.1; P<0.05). In
addition, Compund C treatment following IR (0.6±0.2) markedly
decreased LC3-II expression compared with that of the IR-only
treatment group (1.0±0.1; P<0.05), suggesting that inhibition of
AMPK reduced the expression of LC3-II following IR.
 | Figure 4Expression of LC3-II in IR-treated
CNE-2 cells. CNE-2 cells in each group were treated as follows: 2,
10 Gy IR; 3, pretreatment with AICAR (2.0 mM) for 2 h + 10 Gy IR;
4, PARP-1 RNA interference using lentivirus-delivered
small-hairpin RNA transfection + 10 Gy IR; 5, pretreatment with
Compound C (10 µM) for 2 h + 10 Gy IR. After 48 h, CNE-2
cells were collected and subjected to western blot analysis for
detection of LC3-II. GAPDH was used as the loading control. Protein
expression levels of LC3-II were then quantified and the mean fold
change relative to GAPDH for three independent experiments. LC3,
micro-tubule-associated protein 1 light chain 3; IR, ionizing
radiation; AICAR,
5-amino-1-β-D-ribofuranosyl-1H-imidazole-4-carboxamide; PARP-1,
poly-(adenosine diphosphate-ribose) polymerase-1; Compound C,
6-[4-(2-Piperidin-1-yl-ethoxy)-phenyl)]-3-pyridin-4-yl-pyrrazolo
[1,5-a]-pyrimidine. |
Effect of PARP-1 and AMPK expression on
the proliferation rate of CNE-2 cells
As shown Figs. 5
and 6, PARP-1 gene
silencing following IR significantly attenuated the proliferation
of CNE-2 cells (P<0.05), demonstrating sensitization to IR. In
addition, the AMPK inhibitor Compound C sensitized CNE-2 cells to
IR, as indicated by the inhibition of proliferation (P<0.05).
Furthermore in Compound C-treated cells without IR, CNE-2 cell
proliferation was also significantly reduced compared with the
control group (P<0.05).
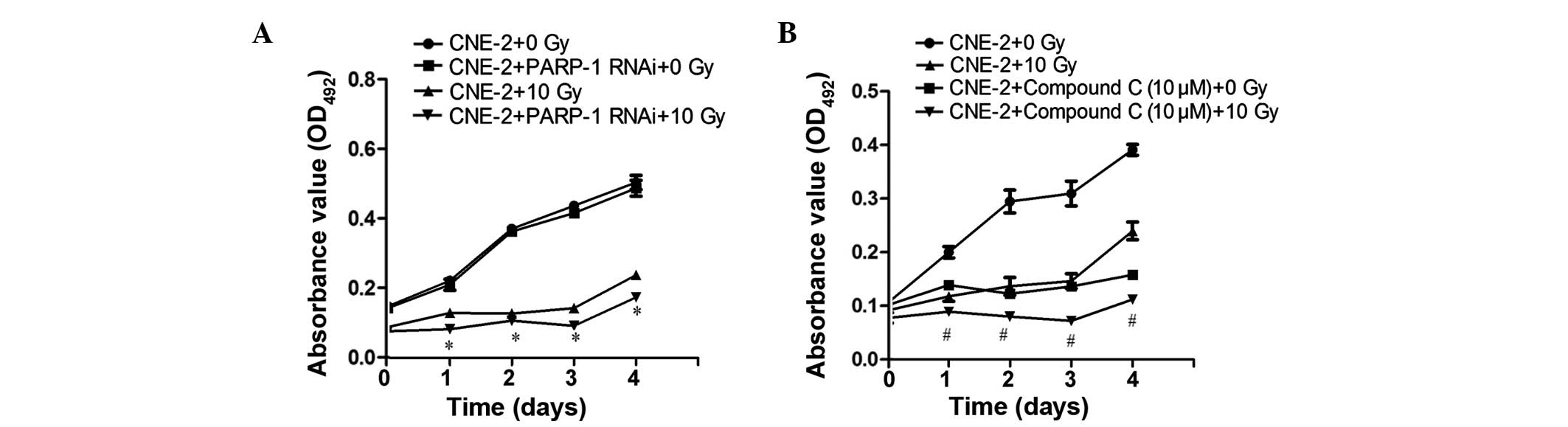 | Figure 5PARP-1 or AMPK inhibition
attenuates the proliferation of CNE-2 cells following 10 Gy IR, as
determined using an MTT assay. (A) The PARP-1 gene was
silenced by RNAi using lentivirus-delivered small-hairpin RNA
transfection. (B) Cells were treated with 10 µM Compound C,
an AMPK inhibitor, for 24 h. An MTT assay was then used to
determine cell proliferation rates in the presence or absence of 10
Gy IR following incubation for 0, 1, 2, 3 and 4 days. Values are
presented as the mean ± standard deviation. *P<0.05,
vs. CNE-2 and PARP-1 RNAi + 0 Gy IR; #P<0.05,
vs. CNE-2 + Compound C (10 µM) + 0 Gy IR. IR, ionizing
radiation; PARP-1, poly-(adenosine diphosphate-ribose)
polymerase-1; Compound C,
6-[4-(2-Piperidin-1-yl-ethoxy)-phenyl)]-3-pyridin-4-yl-pyrrazolo
[1,5-a]-pyrimidine; AMPK, adenosine monophosphate-activated protein
kinase; OD, optical density. |
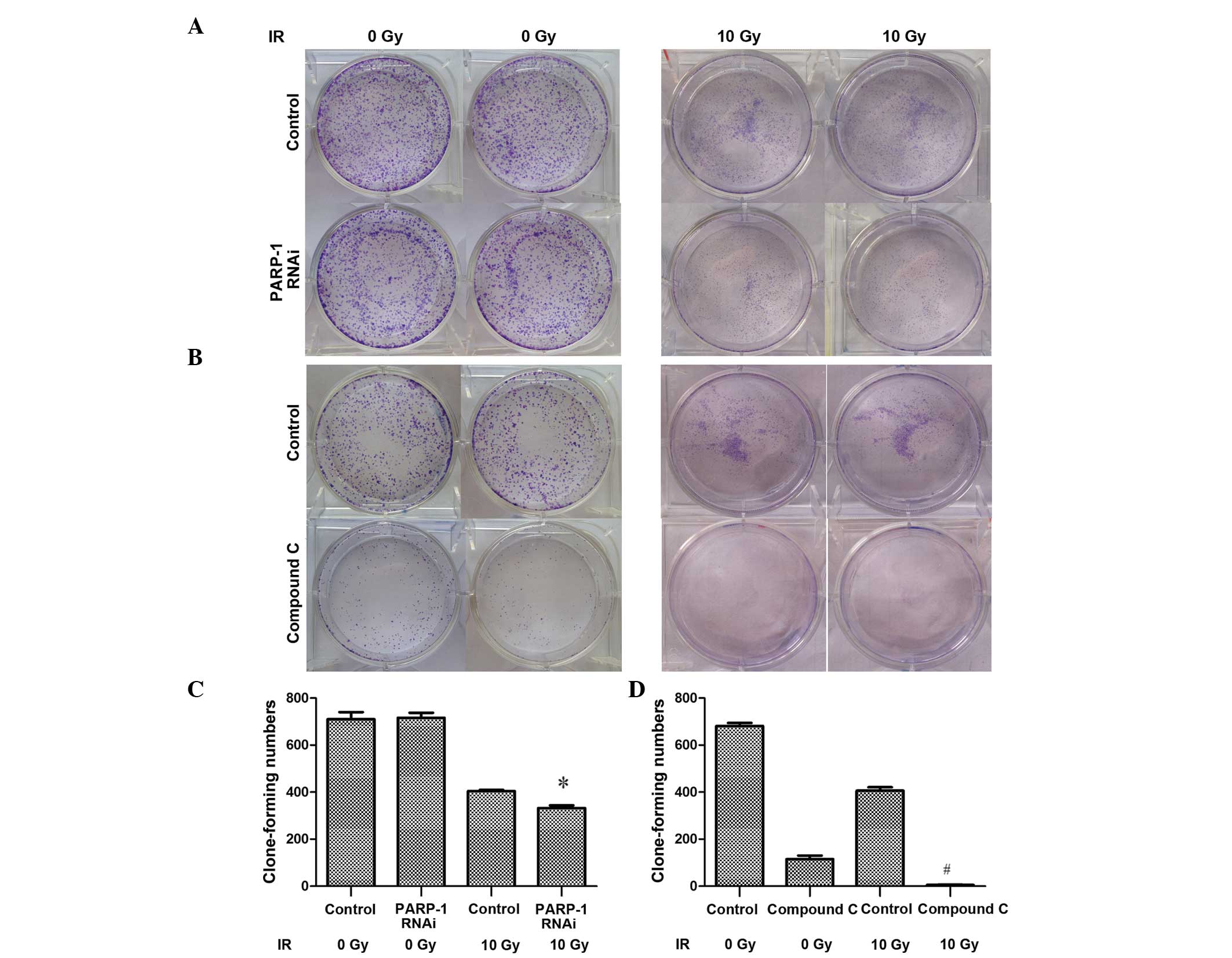 | Figure 6PARP-1 or AMPK inhibition
attenuates the proliferation of CNE-2 cells following 10 Gy IR, as
determined using a clone formation assay. (A) The PARP-1
gene was silenced by RNAi using lentivirus-delivered small-hairpin
RNA transfection. (B) Cells were treated with 10 µM Compound
C, an AMPK inhibitor, for 24 h. A colony formation assay was used
to determine cell proliferation rates in the presence or absence of
10 Gy IR, cells were seeded onto plates as a density of
5×103. The number of colonies containing ≥50 cells were
counted under a microscope to quantify the proliferation of (C)
PARP-1 RNAi-silenced cells and (D) Compound C-treated cells.
Values are presented as the mean ± standard deviation.
*P<0.05, vs. CNE-2 + PARP-1 RNAi with 0 Gy IR;
#P<0.05, vs. CNE-2 + Compound C (10 µM) with 0
Gy IR. IR, ionizing radiation; PARP-1, poly-(adenosine
diphosphate-ribose) polymerase-1; RNAi, RNA interference; Compound
C, 6-[4-(2-Piperidin-1-yl-ethoxy)-phenyl)]-3-pyridin-4-yl-pyrrazolo
[1,5-a]-pyrimidine; AMPK, adenosine monophosphate-activated protein
kinase. |
Discussion
Autophagy has been reported to be associated with
cancer processes as well as protection against cellular stress.
Autophagy, is a cellular degradation process which occurs at low
basal levels in the majority of cell types for the maintenance of
cellular homeostasis. It is important for the degradation and
recycling of damaged proteins, organelles and other cytoplasmic
constituents. Autophagy is induced following types of metabolic
stress, including oxidative stress, nutrient starvation or
endoplasmic reticulum stress, in order to supply nutrients and
energy for promoting cell survival (34). Song et al (35) reported that autophagy acts as a
protective mechanism response to the apoptosis induced by IR. In
addition, it was suggested that tumors utilize autophagy as a
survival mechanism to overcome the stresses imposed during cancer
progression and those caused by radiation. However, when these
stresses reach a critical point, autophagy was hypothesized to
revert to mediating cell death (36). Several studies have indicated that
pharmacologic or genetic inhibition of autophagy may enhance the
effectiveness of cancer treatments by sensitizing cancer cells to
radiation (22,37). The present study aimed to
investigate the expression of upstream molecules of autophagy,
including PARP-1, AMPK and mTOR in CNE-2 cells following IR. The
results confirmed that PARP-1 regulated autophagy in CNE-2 cells
following IR through activation of AMPK and the subsequent
suppression of mTOR. In addition, it was demonstrated that Compound
C inhibited the expression of p-LKB1-Ser428 following IR.
Therefore, there are three key aspects of the present study which
require discussion: The induction of autophagy through irradiation;
the role of PARP-1, AMPK and mTOR as upstream molecules of
autophagy; and the inhibition of p-LKB1-Ser428 expression by
Compound C.
A previous study confirmed that 10 Gy IR induced
autophagy and served as a cell survival mechanism against
IR-induced cell death (22);
therefore, 10 Gy IR was used in the present study. The results
demonstrated that following IR the expression of LC3-I decreased in
CNE-2 cells compared with that of the untreated control group; in
addition, LC3-II levels were increased compared with those of the
untreated control group. This indicated that LC3-I converted to
LC3-II following IR, thus inducing autophagy.
Huang et al (25) identified the novel role of PARP-1
in autophagy, the mechanism of which proceeded via the
LKB1/AMPK/mTOR pathway in order to enhance cell survival following
H2O2 -induced oxidative stress in
Bax−/−Bak−/− MEFs. In the present study,
CNE-2 cells were exposed to IR. p70S6 kinase is a downstream
molecule of the mTOR pathway; therefore, the p70S6K expression may
be used to indicate mTOR activity. In the present study western
blot analysis was used to assess the protein expression of PARP-1,
p-LKB1, p-AMPK and p-p70S6K following IR in different experimental
groups, including cell transfected with LV-shRNA to silence
PARP-1 as well as treatment with the AMPK activator AICAR or
the AMPK inhibitor Compound C. The results demonstrated that IR
promoted the expression of PARP-1; however, AICAR and Compound C
exhibited no significant effects on the expression of PARP-1. In
addition, it was revealed that whether CNE-2 cells are treated with
IR or not, they express p-LKB1-Ser428; notably, Compound C
inhibited the expression of p-LKB1-Ser428. IR was demonstrated to
promote the expression of AMPK in CNE-2 cells, which was further
enhanced following AICAR treatment. By contrast, PARP-1 gene
silencing and Compound C treatment reduced the expression of AMPK
following IR. Furthermore, IR reduced the expression of p-P70S6K in
CNE-2 cells, which was further attenuated by AICAR. However,
PARP-1 gene silencing and Compound C induced the expression
of p-P70S6K following IR. Subsequently, it was demonstrated that IR
promoted the expression of LC3-II in CNE-2 cells, which was further
enhanced following AICAR treatment. PARP-1 gene silencing as
well as Compound C treatment were found to reduce the expression of
LC3-II following IR. Consequently, it was concluded that IR induced
the activation of PARP-1, p-AMPK and LC3-II as well as inhibited
the expression of p-P70S6K. Activation of AMPK had no impact on
PARP-1 expression, whereas it was demonstrated to promote the
expression of AMPK and LC3-II as well as inhibit p-P70S6K
expression. By contrast, PARP-1 gene silencing inhibited the
expression of AMPK and LC3-II as well as activated the expression
of p-P70S6K. AMPK inhibition reduced the expression of AMPK and
LC3-II as well as activated p-P70S6K expression, while it exhibited
no impact on PARP-1 expression. Overall, these results implied that
PARP-1 promoted autophagy through the AMPK/mTOR pathway in CNE-2
cells following 10 Gy IR.
DNA damage has been shown to be generated by
irradiation (38); such damage may
lead to PARP-1 activation. The activation of PARP-1 initiates in
the production of large quantities of PAR polymers and the
subsequent translocation of apoptosis-inducing factors from the
mitochondria to the nucleus, resulting in programmed necrotic cell
death (39,40). In turn, the transient accumulation
of PAR and its metabolism induces a reduction in ATP and
NAD+ levels as well as an increase in cellular AMP.
Furthermore, AMPK activation results in the inactivation of the
mTOR complex 1 pathway via Raptor phosphorylation and P70S6K
inhibition. Therefore, mTOR complex 1 inhibition contributes
towards the establishment of autophagy (41).
Compound C, a cell-permeable pyrrazolopyrimidine
compound, was reported to have an inhibitory effect on kinase
insert domain receptor/vascular endothelial growth factor receptor
2, activin receptor-like kinase (ALK)-2/bone morphogenetic protein
receptor-I and AMPK kinase activity, while further exhibiting an
attenuating effect on ALK-5/transforming growth factor β receptor
type 1, ZAPK, spleen tyrosine kinase, protein kinase Cθ (PKCθ),
protein kinase A (PKA) and Janus kinase 3. The results of the
present study demonstrated that Compound C blocked the expression
of p-LKB1-Ser428. The are several potential reasons for this. LKB1
phosphorylation at Ser-428 by ribosomal protein S6 kinase A1 and/or
PKA is required to inhibit cell growth (42) and since Compound C reduces PKA it
may reduce or inhibit the expression of p-LKB1-Ser428
concomitantly. In addition, activation of PKC induces LKB1-S428
phosphorylation (43); therefore,
the expression of p-LKB1-Ser428 may be due to the Compound
C-induced inhibition of PKCθ. Furthermore, Compound C may promote
enzyme activity to degrade p-LKB1. LKB1 is a known tumor suppressor
gene, which is an upstream molecule of AMPK kinase involved in the
regulation of energy metabolism (44). In the present study, limited
expression of p-LKB1-Ser428 was observed in CNE-2 cells, which was
too small to detect the changes in expression between groups. IR
did not appear to influence the expression of p-LKB1-Ser428 and
PARP-1 silencing had no observable impact on LKB1-Ser428
expression. However, LKB1 is phosphorylated at numerous sites in
human cells, including Thr-363, Ser-428 and Ser334; the present
study only detected the site of Ser428, which may account for the
limited expression of p-LKB1.
In the present study, MTT and clone formation assays
demonstrated that PARP-1 or AMPK inhibition attenuated the
proliferation of CNE-2 cells. This therefore indicated that
inhibition of autophagy contributed to the radiation sensitization
of CNE-2 cells. In addition, it was observed that CNE-2 cells
treated with Compound C without IR exposure demonstrated reduced
proliferation compared with that of the control group. This may be
due to the fact that autophagy occurs at low basal levels in the
majority of cell types in order to maintain cellular homeostasis
(34). Without IR, CNE-2 cells
retain low basal levels of autophagy and the artificial
downregulation of autophagy contributes to cell death. A previous
study demonstrated that Compound C had a slight, but significant,
anti-proliferative and anti-migratory actions on endothelial cells
(45).
In conclusion, the results of the present study
revealed that PARP-1 promoted autophagy through the AMPK/mTOR
pathway in CNE-2 cells following IR. In addition, it was determined
that the inhibition of PARP-1 or AMPK contributed to the radiation
sensitization of CNE-2 cells. LKB1 expression was limited and it
was not possible to detect the changes in LKB1 protein levels in
CNE-2 cells; therefore, the results did not determine that PARP-1
promoted autophagy through the LKB1/AMPK/mTOR pathway in CNE-2
cells following IR. However, using ELISA or mass spectrometry in
combination with western blot analysis may solve this issue for
future studies.
Acknowledgments
The authors would like to thank Professor Xin Huang
for her help in the preparation of this manuscript. The study was
supported by grants from the Natural Science Foundation of China
(nos. 81160285 and 81260346) and the Guangxi Natural Science
Foundation (nos. 2010gxnsfa013240 and 2013jjBA40201).
References
|
1
|
Yoshizaki T, Ito M, Murono S, Wakisaka N,
Kondo S and Endo K: Current understanding and management of
nasopharyngeal carcinoma. Auris Nasus Larynx. 39:137–144. 2012.
View Article : Google Scholar
|
|
2
|
Daker M, Bhuvanendran S, Ahmad M, Takada K
and Khoo AS: Deregulation of lipid metabolism pathway genes in
nasopharyngeal carcinoma cells. Mol Med Rep. 3:731–741. 2013.
|
|
3
|
Qiu S, Lin S, Tham IW, Pan J, Lu J and Lu
JJ: Intensity-modulated radiation therapy in the salvage of locally
recurrent nasopharyngeal carcinoma. Int J Radiat Oncol Biol Phys.
83:676–683. 2012. View Article : Google Scholar
|
|
4
|
Feng XP, Yi H, Li MY, et al:
Identification of biomarkers for predicting nasopharyngeal
carcinoma response to radiotherapy by proteomics. Cancer Res.
70:3450–3462. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guo Y, Zhu XD, Qu S, et al: Identification
of genes involved in radioresistance of nasopharyngeal carcinoma by
integrating gene ontology and protein-protein interaction networks.
Int J Oncol. 40:85–92. 2012.
|
|
6
|
Li G, Qiu Y, Su Z, et al: Genome-wide
analyses of radioresistance-associated miRNA expression profile in
nasopharyngeal carcinoma using next generation deep sequencing.
PLoS One. 8:e844862013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Levine B and Klionsky DJ: Development by
self-digestion: molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Levine B and Yuan J: Autophagy in cell
death: an innocent convict? J Clin Invest. 115:2679–2688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Giménez-Xavier P, Francisco R, Santidrián
AF, Gil J and Ambrosio S: Effects of dopamine on LC3-II activation
as a marker of autophagy in a neuroblastoma cell model.
Neurotoxicology. 30:658–665. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Holt SV, Wyspianska B, Randall KJ, James
D, Foster JR and Wilkinson RW: The development of an
immunohistochemical method to detect the autophagy-associated
protein LC3-II in human tumor xenografts. Toxicol Pathol.
39:516–523. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Paglin S, Hollister T, Delohery T, et al:
A novel response of cancer cells to radiation involves autophagy
and formation of acidic vesicles. Cancer Res. 61:439–444.
2001.PubMed/NCBI
|
|
12
|
Yao KC, Komata T, Kondo Y, Kanzawa T,
Kondo S and Germano IM: Molecular response of human glioblastoma
multiforme cells to ionizing radiation: cell cycle arrest,
modulation of the expression of cyclin-dependent kinase inhibitors
and autophagy. J Neurosurg. 98:378–384. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ito H, Daido S, Kanzawa T, Kondo S and
Kondo Y: Radiation-induced autophagy is associated with LC3 and its
inhibition sensitizes malignant glioma cells. Int J Oncol.
26:1401–1410. 2005.PubMed/NCBI
|
|
14
|
Dalby KN, Tekedereli I, Lopez-Berestein G
and Ozpolat B: Targeting the prodeath and prosurvival functions of
autophagy as novel therapeutic strategies in cancer. Autophagy.
6:322–329. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Apel A, Herr I, Schwarz H, Rodemann HP and
Mayer A: Blocked autophagy sensitizes resistant carcinoma cells to
radiation therapy. Cancer Res. 68:1485–1494. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Marx J: Autophagy: is it cancer’s friend
or foe? Science. 312:1160–1161. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu WK, Coffelt SB, Cho CH, et al: The
autophagic paradox in cancer therapy. Oncogene. 31:939–953. 2012.
View Article : Google Scholar
|
|
18
|
Weaver AN and Yang ES: Beyond DNA repair:
additional functions of PARP-1 in cancer. Front Oncol. 3:2902013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Houtgraaf JH, Versmissen J and van der
Giessen WJ: A concise review of DNA damage checkpoints and repair
in mammalian cells. Cardiovasc Revasc Med. 7:165–172. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cho EA, Kim EJ, Kwak SJ and Juhnn YS: cAMP
signaling inhibits radiation-induced ATM phosphorylation leading to
the augmentation of apoptosis in human lung cancer cells. Mol
Cancer. 13:362014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Veuger SJ, Curtin NJ, Richardson CJ, Smith
GC and Durkacz BW: Radiosensitization and DNA repair inhibition by
the combined use of novel inhibitors of DNA-dependent protein
kinase and poly (ADP-ribose) polymerase-1. Cancer Res.
63:6008–6015. 2003.PubMed/NCBI
|
|
22
|
Zhou ZR, Zhu XD, Zhao W, et al: Poly
(ADP-ribose) polymerase-1 regulates the mechanism of
irradiation-induced CNE-2 human nasopharyngeal carcinoma cell
autophagy and inhibition of autophagy contributes to the radiation
sensitization of CNE-2 cells. Oncol Rep. 29:2498–2506.
2013.PubMed/NCBI
|
|
23
|
Shackelford DB and Shaw RJ: The LKB1-AMPK
pathway: metabolism and growth control in tumour suppression. Nat
Rev Cancer. 9:563–575. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Green AS, Chapuis N, Lacombe C, Mayeux P,
Bouscary D and Tamburini J: LKB1/AMPK/mTOR signaling pathway in
hematological malignancies: from metabolism to cancer cell biology.
Cell Cycle. 10:2115–2120. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang Q, Wu YT, Tan HL, Ong CN and Shen
HM: A novel function of poly (ADP-ribose) polymerase-1 in
modulation of autophagy and necrosis under oxidative stress. Cell
Death Differ. 16:264–277. 2009. View Article : Google Scholar
|
|
26
|
Miura K, Sakata K, Someya M, et al: The
combination of olaparib and camptothecin for effective
radiosensitization. Radiat Oncol. 7:622012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Powell C, Mikropoulos C, Kaye SB, et al:
Pre-clinical and clinical evaluation of PARP inhibitors as
tumour-specific radiosensitisers. Cancer Treat Rev. 36:566–575.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Damia G and D’Incalci M: Targeting DNA
repair as a promising approach in cancer therapy. Eur J Cancer.
43:1791–1801. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Woon EC and Threadgill MD: Poly
(ADP-ribose) polymerase inhibition-where now? Curr Med Chem.
12:2373–2392. 2005. View Article : Google Scholar
|
|
30
|
Gu SY, Tang WP, Zeng Y, et al: An
epithelial cell line established from poorly differentiated
nasopharyngeal carcinoma. Ai Zheng. 2:70–72. 1983.In Chinese.
|
|
31
|
Lu XD: The relation between PARP-1 and
autophagy in CNE-2 cells related to the radiation sensitization.
Guangxi Yi Ke Da Xue Xue Bao. 2014.In Chinese.
|
|
32
|
Vallejo D, Crespo I, San-Miguel B, et al:
Autophagic response in the Rabbit Hemorrhagic Disease, an animal
model of virally-induced fulminant hepatic failure. Vet Res.
45:152014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhuang W, Qin Z and Liang Z: The role of
autophagy in sensitizing malignant glioma cells to radiation
therapy. Acta Biochim Biophys Sin (Shanghai). 41:341–351. 2009.
View Article : Google Scholar
|
|
34
|
Galluzzi L, Vicencio JM, Kepp O, Tasdemir
E, Maiuri MC and Kroemer G: To die or not to die: that is the
autophagic question. Curr Mol Med. 8:78–91. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Song L, Liu H, Ma L, Zhang X, Jiang Z and
Jiang C: Inhibition of autophagy by 3-MA enhances endoplasmic
reticulum stress-induced apoptosis in human nasopharyngeal
carcinoma cells. Oncol Lett. 6:1031–1038. 2013.PubMed/NCBI
|
|
36
|
Hu YL, Jahangiri A, Delay M and Aghi MK:
Tumor cell autophagy as an adaptive response mediating resistance
to treatments such as antiangiogenic therapy. Cancer Res.
72:4294–4299. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bae H and Guan JL: Suppression of
autophagy by FIP200 deletion impairs DNA damage repair and
increases cell death upon treatments with anticancer agents. Mol
Cancer Res. 9:1232–1241. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shim HJ, Lee EM, Nguyen LD, Shim J and
Song YH: High-dose irradiation induces cell cycle arrest, apoptosis
and developmental defects during drosophila oogenesis. PLoS One.
9:e890092014. View Article : Google Scholar
|
|
39
|
Yu SW, Wang H, Poitras MF, et al:
Mediation of poly (ADP-ribose) polymerase-1-dependent cell death by
apoptosis-inducing factor. Science. 297:259–263. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yu SW, Andrabi SA, Wang H, et al:
Apoptosis-inducing factor mediates poly (ADP-ribose) (PAR)
polymer-induced cell death. Proc Natl Acad Sci USA.
103:18314–18319. 2006. View Article : Google Scholar
|
|
41
|
Ethier C, Tardif M, Arul L and Poirier GG:
PARP-1 modulation of mTOR signaling in response to a DNA alkylating
agent. PLoS One. 7:e479782012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Karuman P, Gozani O, Odze RD, et al: The
Peutz-Jegher gene product LKB1 is a mediator of p53-dependent cell
death. Mol Cell. 7:1307–1319. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Memmott RM and Dennis PA: LKB1 and
mammalian target of rapamycin as predictive factors for the
anticancer efficacy of metformin. J Clin Oncol. 27:e2262009.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lee JH, Koh H, Kim M, et al:
Energy-dependent regulation of cell structure by AMP-activated
protein kinase. Nature. 447:1017–1020. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nilufar E, Yadollah S, Benroz N, et al:
Effect of metformin on the proliferation, migration and MMP-2 and-9
expression of human umbilical vein endothelial cells. Mol Med Rep.
5:1068–1074. 2012.
|